
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical studies on the parasitic plant Thonningia sanguinea Vahl†
Ama Kyeraa Thomford ab,
Reda Fouad Ahmed Abdelhameedac and
Koji Yamada*a
ab,
Reda Fouad Ahmed Abdelhameedac and
Koji Yamada*a
aGraduate School of Biomedical Sciences, Nagasaki University, Bunkyo-machi 1-14, Nagasaki 852-8521, Japan. E-mail: kyamada@nagasaki-u.ac.jp; Fax: +81-95-819-2462; Tel: +81-95-819-2462
bDepartment of Biomedical Sciences, School of Allied Health Sciences, University of Cape Coast, Ghana
cDepartment of Pharmacognosy, Faculty of Pharmacy, Suez Canal University, 41522 Ismailia, Egypt
First published on 7th June 2018
Abstract
Thonningia sanguinea Vahl plays an important role in traditional medicine in many African cultures. A study of the n-hexane fraction of the whole plant of T. sanguinea led to the isolation of two glucocerebroside molecular species TSC-1 and TSC-2, one β-sitosteryl-3β-D-glucopyranoside-6′-O-fatty acid ester molecular species, TSS-1, and seven known triterpenes (1–7). The ethyl acetate fraction also afforded five known lignans (8–12) and one known flavanone (13). Their structures were elucidated by means of chemical and spectroscopic methods (methanolysis, NMR and mass spectrometry). Spectral analyses of the glucocerebrosides revealed mainly sphingosine-type (TSC-1) and phytosphingosine-type (TSC-2) cerebrosides, with both possessing mainly 2-hydroxy fatty acid and β-D-glucopyranose moieties. TSS-1 was found to be a β-sitosterol-type with fatty acid methyl esters and β-D-glucopyranoside moieties. The classes of compounds isolated from this plant are well known for their interesting biological activities including antimicrobial, antioxidant, anticancer, antiinflammatory and analgesic. They may therefore be responsible in part or in whole for these activities, hence validating the traditional uses of the plant. To the best of our knowledge, this is the first report on the isolation of all these compounds from T. sanguinea.
1. Introduction
Thonningia sanguinea Vahl is a flowering plant in the monotypic genus Thonningia of the family Balanophoraceae. Synonyms for the plant include T. angolensis, T. coccinea Mangenot, T. dubia Hemsl., T. elegans Hemsl., and T. ugandensis Hemsl. It is a fleshy subterranean herb growing from an underground tuber. It is a parasitic plant growing on the terminal roots of host plants such as Hevea brasiliensis, Phoenix dactylifera and Theobroma cacao.1 The flowering stem produces a bright red or pink inflorescence containing male and female flowers. The plant grows in Tropical Africa, from Senegal to Ethiopia, south to Angola, Zambia and Tanzania. It is commonly found in rain-forests, gallery forests and adjacent woodland.1 Commonly known in English as “ground pineapple”, it is also known as “kwaebedwaa” in the local Akan language of Ghana.T. sanguinea is best known for its use in traditional medicine in many African countries. In Ghanaian traditional medicine for instance, this plant is used in treatment of bronchial asthma sometimes for prophylaxis,2 sexually transmitted diseases and as an aphrodisiac. It is used to treat diarrhoea and worm infestation in Cote d'Ivoire and Congo. It is also mixed with Capsicum to produce a topical cream for treating haemorrhoids and torticollis. It is also used to treat dysentery, sore throat, skin infections, abscesses, dental caries, gingivitis, fever, malaria, heart disease, rickets, and rheumatism.1,3,4
Chemically, an aqueous and hydroalcoholic flower extract of T. sanguinea revealed the presence of alkaloids, catechin tannins, flavonoids, saponins, quinones and polyphenols on preliminary screening.5 Brevifolin carboxylic acid (BCA),6 gallic acid (GA)6 and two ellagitannins: thonningianins A (Th A) and Th B7 are the only four compounds reported to have been isolated from the plant. N'guessan et al. reported that both GA and BCA, isolated from T. sanguinea demonstrated moderate antibacterial activity against Salmonella enteritidis, Salmonella typhimurium, and Salmonella abony in the disc diffusion method, with significant antioxidant activity in the DPPH radical scavenging activity assay.6 GA is well known as a potent antioxidant phenolic compound, with numerous biological activities including antitumor, antimicrobial and antimelanogenic.6 BCA has been shown to inactivate HBsAg and inhibit hepatitis B virus replication and tumour growth.8 The ellagitannins isolated from the plant also possessed hepatoprotective actions, potent antimicrobial effects4,6,7,9 and significant free radical scavenging activity against DPPH by ESR analysis.7 Th A effectively inhibited of the proliferation of HepG-2 human hepatocellular carcinoma cells by inducing apoptosis. This was observed as an increase in the sub-G1 cell population, DNA fragmentation, and increase in the content of reactive oxygen species.10 Th A was also shown to be a potent in vitro inhibitor of rat liver crude glutathione S-transferases (GSTs) and hGSTP1-1 activity.11
Biological study on T. sanguinea plant extracts (in vivo and in vitro assays) indicated the prophylactic potential of the aqueous extract and its n-butanolic fraction in bronchial asthma. The active agents extracted into n-butanol and may be flavonoids and/or phenolic in nature.12 The anticoccidial activity of the extract against Eimeria sp. sporazoites,13 antibacterial effects against some multidrug resistant strains of Salmonella enterica16 and their effects on extended spectrum-β-Lactamases (ESBL) producing Escherichia coli and Klebsiella pneumoniae strains3 have been reported. The significant antimalarial effects of T. sanguinea root extracts against Plasmodium falciparum (in vitro),14 Plasmodium berghei and Plasmodium chabaudi (in vivo)1 have also been reported. The aqueous extract of T. sanguinea exhibited hepatoprotective activity towards a variety of different toxicants including galactosamine, carbon tetrachloride and aflatoxin B1.2,15 It also suppressed CYP3A2 and CYP1A2 expression at the level of transcription,16 protects against aflatoxin B1 acute hepatotoxicity in Fischer 344 rats2 and also inhibits the liver drug metabolising enzymes of rats.17
The aqueous decoction of T. sanguinea has been used for more than thirty five years as a mono-herbal product produced by the Centre for Plant medicine Research, Akuapem-Mampong, Ghana (CPMR) under the registered names CAMPA-T® and NINGER®. Due to the reported clinical effectiveness of these herbal formulations at the CPMR clinic, they have been approved for use as standardised herbal medicines by the Food and Drug Authority. CAMPA-T® and NINGER® are now part of the Essential Herbal Medicines List recommended for use in pilot herbal clinics under the Integrated Herbal Medicine Services Programme in public hospitals. These T. sanguinea-based products are prescribed for the management of arthritic pain, sexual weakness, male and female infertility, dysmenorrhoea, amenorrhoea and uterine fibroids. Therefore, T. sanguinea is an important medicinal plant that is contributing immensely to public health care in Ghana.
Despite the interesting clinical usage in Ghana, as well as the reported pharmacological activities of the extracts of T. sanguinea, there is no detailed study on the chemical composition of the plant. In this report, we seek to provide a chemical profile for T. sanguinea to better understand these reported pharmacological activities and give credence to its use in traditional medicine.
2. Experimental
2.1. General experimental information
NMR spectra were recorded in chloroform-d, methanol-d4 and pyridine-d5 (Nacalai Tesque, Inc., Kyoto, Japan) with Varian Unity Plus 400 spectrometer (Palo Alto, CA, USA) operating at 400 MHz for 1H and 100 MHz for 13C, and with a JEOL JNM-AL 300 spectrometer (JEOL Ltd, Tokyo, Japan) at 300 MHz for 1H NMR. The UV spectra were recorded using a double beam Shimadzu UV-visible spectrophotometer (model UV-1601 PC, Kyoto City, Japan). IR spectra were recorded using a Jasco FT/IR-410 K spectrometer (Jasco Co. Ltd., Tokyo, Japan) with a range of 400–4000 cm−1. FAB-MS spectra were recorded on a JMS 700N spectrometer (JEOL Ltd., Tokyo, Japan) in positive ion mode, with glycerol or m-nitrobenzyl alcohol, with or without NaCl, as the matrix. The optical rotation measurements were done using a Jasco P-1020 polarimeter (Jasco Co. Ltd., Tokyo, Japan). Extraction and isolation of compounds were done the following solvents: acetone, acetonitrile, n-butanol, chloroform, ethyl acetate, n-hexane and methanol (Nacalai Tesque, Inc., Kyoto, Japan). Column chromatography (CC) was performed using Sephadex LH-20 (25–100 mm, GE Healthcare UK Ltd., Buckinghamshire HP7 9NA, UK), silica gel Purasil 60 Å, 230–400 mesh (Whatman, Sanford, ME, USA) and Cosmosil 140C18-PREP silica gel 90 Å, 40–63 mesh (Nacalai Tesque, Inc., Kyoto, Japan). TLC was performed on 0.25 mm thick, precoated silica gel 60 F254 and silica gel RP-18 F254 plates (Merck, Darmstadt, Germany). Prep. TLC was performed on 2 mm thick PLC silica gel 60 F254 glass plates (Merck, Darmstadt, Germany). Spots were developed with 5% H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH and detected by illumination under a short wavelength UV (254 nm). Analytical HPLC was performed on a Cosmosil 5C18-AR-II 4.6 mm × 250 mm column (Nacalai Tesque, Inc., Kyoto, Japan) with methanol (Nacalai Tesque, Inc., Kyoto, Japan) at a flow rate of 0.8 mL min−1 and Cosmosil-sugar-D 4.6 ID × 250 mm, 1 mL min−1, refractive index (RI) detector using 95% acetonitrile. Preparative HPLC was performed on a Develosil 5C18 4.6 mm × 150 mm column (Nacalai Tesque, Inc., Kyoto, Japan) using 100% MeOH as solvent, at a flow rate of 0.5 mL min−1 on a Jasco DG-2080-53 Plus degasser, Jasco PU-2080 Plus pump, Jasco AS-2055 Plus auto sampler, Jasco CO-2065 Plus column oven (maintained at 35 °C) and Jasco MD-2018 Plus PDA detector (Jasco Co. Ltd., Tokyo, Japan).
:MeOH and detected by illumination under a short wavelength UV (254 nm). Analytical HPLC was performed on a Cosmosil 5C18-AR-II 4.6 mm × 250 mm column (Nacalai Tesque, Inc., Kyoto, Japan) with methanol (Nacalai Tesque, Inc., Kyoto, Japan) at a flow rate of 0.8 mL min−1 and Cosmosil-sugar-D 4.6 ID × 250 mm, 1 mL min−1, refractive index (RI) detector using 95% acetonitrile. Preparative HPLC was performed on a Develosil 5C18 4.6 mm × 150 mm column (Nacalai Tesque, Inc., Kyoto, Japan) using 100% MeOH as solvent, at a flow rate of 0.5 mL min−1 on a Jasco DG-2080-53 Plus degasser, Jasco PU-2080 Plus pump, Jasco AS-2055 Plus auto sampler, Jasco CO-2065 Plus column oven (maintained at 35 °C) and Jasco MD-2018 Plus PDA detector (Jasco Co. Ltd., Tokyo, Japan).
2.2. Plant collection and identification
Thonningia sanguinea whole plant was collected by the staff of the Centre for Plant Medicine Research from the eastern region of Ghana in the month of January, 2015 and authenticated by the curator of their herbarium. A voucher specimen with the number CSRPM no. 140 was assigned to the sample.2.3. Extraction and isolation
The whole plant of T. sanguinea was shade-dried for seven days and pulverised. The dried powdered plant material (3.5 kg) was extracted by cold maceration with MeOH (3 × 10 L for 3 days), followed by MeOH:CHCl3 (1:1; 3 × 10 L for 3 days). The filtrates were pooled together and concentrated in vacuo using the rotary evaporator. The methanol/chloroform crude extract (423 g) was dissolved in distilled water and serially partitioned between n-hexane, ethyl acetate, n-butanol solvents to obtain the n-hexane (20 g), ethyl acetate (260 g), n-butanol (88 g) and aqueous (30 g) fractions. The n-hexane fraction (20 g) was subjected to silica gel CC (800 g) using n-hexane:EtOAc (9:1–1:9), CHCl3:MeOH (8:2–6:4) and 80% acetone to give sixteen sub-fractions (H-1–H-16). Fraction H-4 (500 mg) was subjected to repeated silica gel CC using n-hexane:EtOAc (8.5:1.5–1:1) to afford compound 6 (200 mg). Fraction H-5 (330 mg) was subjected to repeated silica gel CC using CHCl3:MeOH (1:0–1:1) to afford seven fractions. H-5-6 (138 mg) was further chromatographed on a C18 RP silica gel column (10 g) using MeOH:H2O (8:2–1:0) and acetone:H2O (1:1–1:0) to afford compound 7 (22 mg). Fraction H-10 (600 mg) was chromatographed on silica gel (20 g) using CHCl3:MeOH (9.8:0.2–1:1) to afford eleven sub-fractions (H-10-1–H-10-11). H-10-4 (350 mg) was further chromatographed on a C18 RP silica gel column (10 g) using 100% MeOH to afford compound 1 (28 mg), TSS-1 (94 mg) and a mixture purified by preparative HPLC using 100% MeOH to afford compound 2 (40 mg) and 4 (15 mg). Fraction H-13 (2.3 g) was chromatographed on silica gel (100 g) using CHCl3:MeOH (9.8:0.2–2:8) to afford five sub-fractions H-13-1–H-13-5. H-13-4 (340 mg) was subjected to repeated silica gel CC using CHCl3:MeOH (9.5:0.5–1:1) to afford TSC-1 (30 mg), TSC-2 (45 mg) and a mixture purified by preparative HPLC using 100% MeOH to afford compound 3 (13 mg) and 5 (8 mg). The ethyl acetate fraction (70 g) was subjected to silica gel CC (240 g) using CHCl3:MeOH (9.8:0.2–3:7) and 80% acetone to afford twenty two sub-fractions (E-1–E-22). Fraction E-2 (1.08 g) was subjected to silica gel CC (10 g) using n-hexane:EtOAc (8:2–2:8) to give fourteen sub-fractions (E-2-1–E-2-14) which afforded compound 8 (100 mg) and 9 (200 mg). Fraction E−9 (500 mg) was subjected to silica gel CC (15 g) using CHCl3:MeOH (9.9:0.1–9.6:0.4) to give sixteen sub-fractions (E-9-1–E-9-16). E-9-7 (175 mg) was subjected to repeated silica gel CC (10 g) using n-hexane:EtOAc (8:2–1:1) and CHCl3:MeOH (9.7:0.3–1:1) to afford compound 10 (14 mg) and 11 (23 mg). E-9-8 (168 mg) was chromatographed on a C18 RP silica gel column (10 g) using MeOH:H2O (3:7–4:6) to afford compound 12 (117 mg) and 13 (18 mg).
| Position | TSC-1 | TSC-2 | ||
|---|---|---|---|---|
| 1H | 13C | 1H | 13C | |
| a Spectra were acquired at 23 °C. Chemical shifts are given in δ (ppm) and are referenced to internal solvent signals for C5D5N at 7.19 (δH) and 123.5 (δC) ppm. X and y are olefinic signals (double bond location). | ||||
| NH | 8.35 (1H, d, J = 8.8 Hz) | 8.55 (1H, d, J = 8.8 Hz) | ||
| 1a | 4.21 (dd, J = 10.0, 5.0 Hz, 1H) | 70.8 | 4.29 (dd, J = 10.0, 5.0 Hz, 1H) | 69.8 |
| 1b | 4.77 (1H, m) | 4.70 (d, J = 5.0 Hz, 1H) | ||
| 2 | 4.77 (1H, m) | 54.0 | 4.70 (1H, m) | 51.1 |
| 3 | 4.71 (1H, m) | 71.8 | 4.59 (1H, m) | 71.8 |
| 4 | 5.97 (1H, m) | 131.4 | 4.49 (1H, m) | |
| 5 | 5.97 (1H, m) | 131.4 | ||
| x | 5.48 (1H, m) | 129.9 | 5.48 (1H, m) | 129.5 |
| y | 5.49 (1H, m) | 129.9 | 5.48 (1H, m) | 130.0 |
| 1′ | 175.0 | 175.0 | ||
| 2′ | 4.59 (1H, m) | 71.8 | 4.29 (1H, m) | 71.8 |
| –CH3 | 0.86 (3H, t) | 13.6 | 0.83 (3H, t) | 13.6 |
| 1′′ | 4.89 (1H, d, J = 8.0 Hz) | 105.5 | 4.93 (1H, d, J = 8.0 Hz) | 104.9 |
| 2′′ | 4.03 (1H, m) | 74.5 | 4.00 (1H, m) | 75.2 |
| 3′′ | 4.23 (1H, m) | 78.0 | 4.18 (1H, m) | 77.9 |
| 4′′ | 4.21 (1H, m) | 71.6 | 4.18 (1H, m) | 70.8 |
| 5′′ | 3.90 (1H, m) | 78.0 | 3.85 (1H, m) | 77.9 |
| 6a′′ | 4.36 (1H, m) | 62.0 | 4.29 (1H, m) | 62.0 |
| 6b′′ | 4.59 (1H, m) | 4.56 (1H, m) | ||
:1) (0.2 mL) at 70 °C for 8 h in a sealed small-volume vial followed by evaporation in vacuo to dryness. The mixture was separated using preparative TLC to afford the LCB, LCB acetates and LCB glucoacetates. 1H, 13C NMR and FAB-MS analyses were performed on the FAMEs and the LCB products.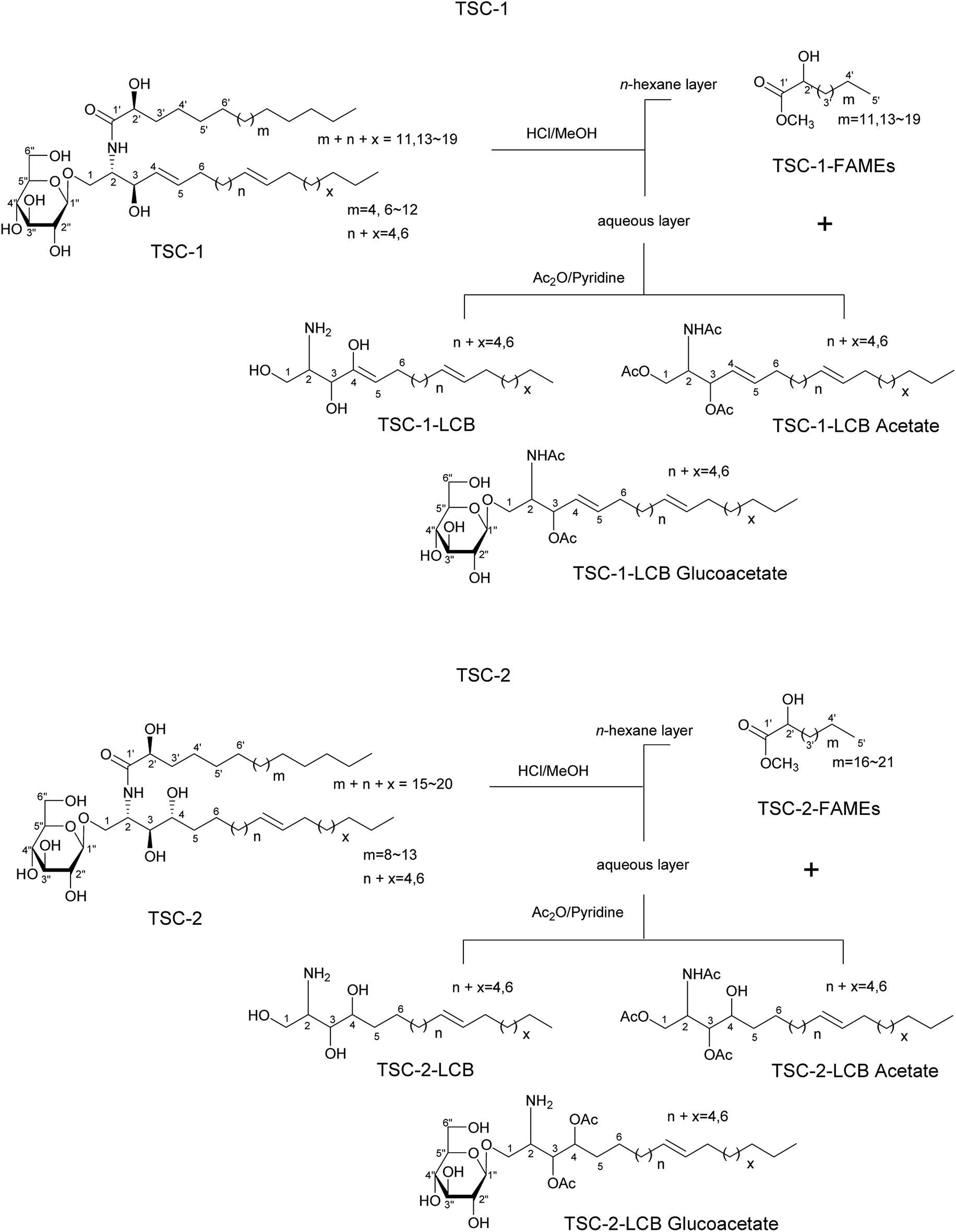 | ||
| Fig. 1 Structures of TSC-1 and TSC-2. | ||
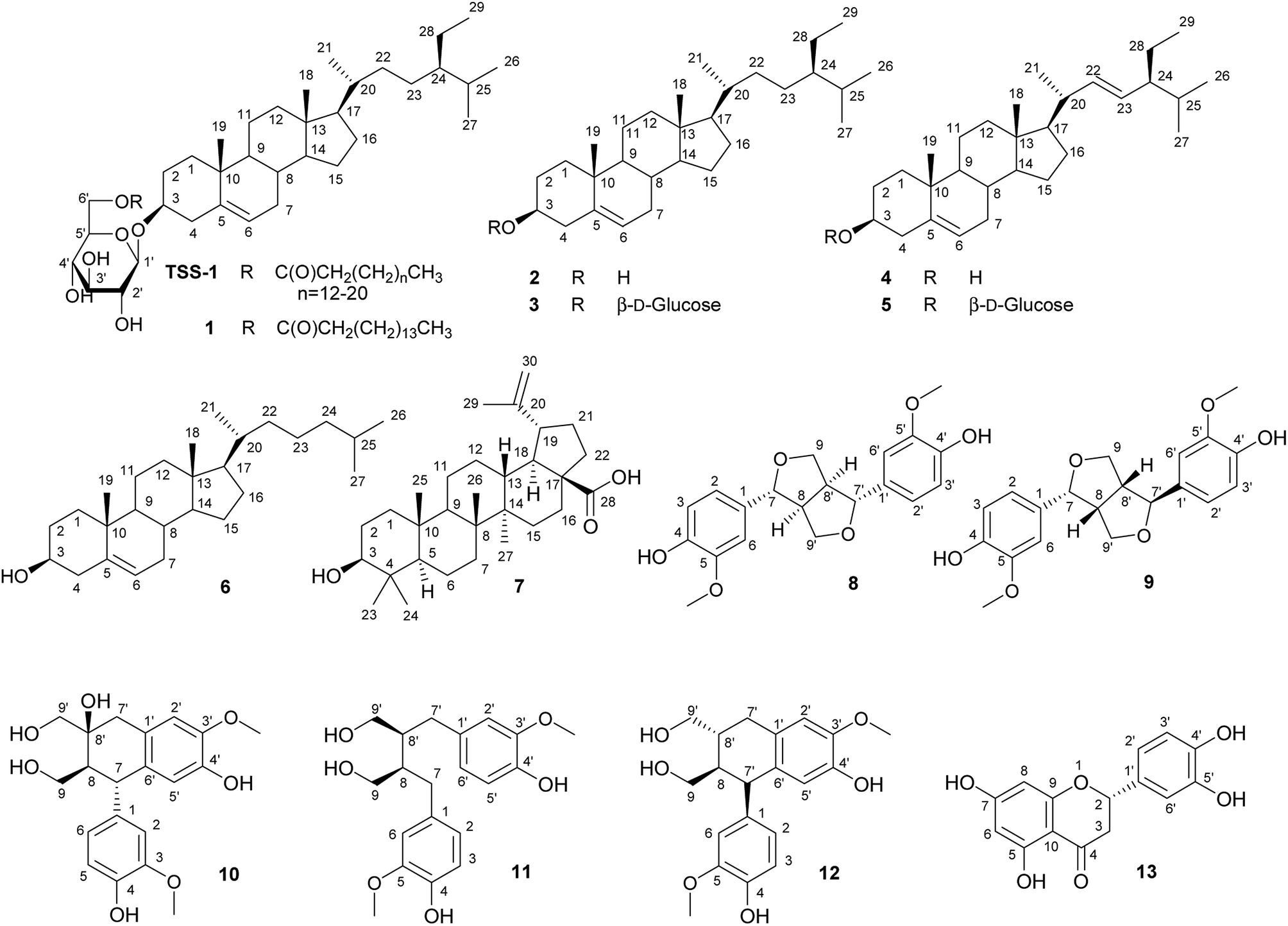 | ||
| Fig. 2 Structure of TSS-1 and comp. 1–13. | ||
2.4. Spectral data
3. Results and discussion
Chromatographic separation of the n-hexane fraction, from the chloroform/methanol crude extract of T. sanguinea whole plant lead to the isolation of two glucocerebroside molecular species TSC-1 and TSC-2, and one β-sitosteryl-3β-D-glucopyranoside-6′-O-fatty acid ester molecular species TSS-1, together with seven known triterpenes: β-sitosteryl-3β-D-glucopyranoside-6′-O-palmitate 1, β-sitosterol 2, β-sitosterol-3β-D-glucopyranoside 3, β-stigmasterol 4, β-stigmasterol-3β-D-glucopyranoside 5, cholesterol 6 and betulinic acid 7. Five known lignans: (+)-epipinoresinol 8, (+)-pinoresinol 9, (+)-cycloolivil, 10, (+)-secoisolariciresinol 11 and (+)-isolariciresinol 12 and one known flavanone (+)-eriodictyol 13, were also isolated from the ethyl acetate fraction of T. sanguinea whole plant. The known compounds were identified by comparison with authentic samples or reported spectral and physical data. See Fig. 1 and 2; Spectral data.3.1. Chemistry
The NMR spectral data of TSS-1 in CDCl3 showed resonances of a carboxylic acid group (δC 174.8), a long methylene chain centered at δH 1.25, (δC 29.1–29.9) and overlapped methyls at δH 0.68–0.85 (δC 14.1), indicating normal type terminal methyls of the fatty acids. The characteristic signals of a sitosterol skeleton were determined as follows: a methine proton at δH 3.55 (1H, m, H-3, δC 79.5) and an olefinic proton signal at δH 5.38 (1H, m, H-5, δC 140.3) were assigned as C-3 and C-5 respectively. Two angular methyl protons at δH 0.68 (3H, s) and 1.01 (3H, s), corresponding to δC 11.8 and 19.3 were assigned as C-18 and C-19 respectively. The proton signals at δH 0.87 (3H, s, δC 19.0; C-26) and 0.88 (3H, s, δC 19.8; C-27) indicated the presence of an isopropenyl group in the molecular structure. The proton signal at δH 0.84 (3H, t, J = 7.8 Hz; δC 11.9) was assigned as C-29. All other NMR assignments were in agreement with known β-sitosteryl-3β-D-glucopyranoside-6′-O-fatty acid methyl esters.19 Characteristic signals indicative of a presence of a monosaccharide moiety at δH 3.38–4.53, 6H with an anomeric proton signal at δH 4.37 (1H, d, J = 7.8 Hz; δC 101.2) were observed. See Fig. 1 and Spectral data.
To identify the sugar moiety in TSS-1, the methylated sugar moiety in the aqueous layer after methanolysis was analysed by HPLC against standard sugars (glucose and galactose) and identified as glucose (glucose tR = 14.11 min, galactose tR = 13.27 min). The coupling constant of the anomeric proton at δH 4.38 (1H, d, J = 8.0 Hz) and the chemical shift of the anomeric carbon δC (101.2) confirmed the β-configuration of the glucopyranoside moiety (α-glucopyranoside: J = 3.7 Hz; δC 98.5).18 The absolute configuration of the sugar moiety was determined by the Tanaka et al. method.18 Direct HPLC analysis of the reaction mixture of the sugar moiety exhibited a peak at tR = 18.7 min, which were coincided with the aryl-isothiocyanate derivative of D-glucose, confirming the absolute configuration of the sugar moiety (L-glucose tR = 19.22 min).18
The positive FAB-MS spectral data showed a series of molecular ion peaks at m/z: 851, 865, 879, 893, 907, 921, 935, 949, 963 and 977 [M + Na]+. Therefore, TSS-1 is presumed to be a molecular species consisting of β-sitosteryl-3β-D-glucopyranoside-6′-O-fatty acid methyl ester possessing mainly a hydroxy fatty acid moiety (normal type terminal methyl groups at δC 14.2) and a β-D-glucopyranose moiety. The core structure of the β-sitosterol-3β-D-glucopyranose skeleton of TSS-1 was characterized by comparison of its 13C NMR spectral data with that of known β-sitosterol-3β-D-glucopyranose-6-O-fatty acid esters.19 See Fig. 2 and Spectral data.
The NMR spectral data of TSC-1 in C5D5N showed resonances of a secondary amide proton doublet at δH 8.35 (1H, d, J = 8.8 Hz), a long methylene chain, centered at δH 1.26, (δC 29.1–29.3) and overlapped methyls at δH 0.86 (δC 13.6), indicating the presence of a sphingolipid skeleton. Characteristic signals indicative of a monosaccharide moiety at δH 3.90–4.89 (6H), with the anomeric proton signal at δH 4.89 (1H, d, J = 8.0 Hz; δC 105.5) were observed. The characteristic resonances for the 2-amino-1,3,2′-triol region of the hydrocarbon chain were observed at δH 4.77 (1H, m, H-2), 4.59 (1H, m, H-2′), 4.77 (1H, m, H-1b), 4.21 (1H, m, H-1a), 4.71 (1H, m, H-3) corresponding to the following 13C NMR data: δC 54.0 (C-2), 71.8 (C-2′), 70.8 (C-1), 71.8 (C-3), and an amide carbonyl signal at δC 175.0 (C-1′). See Fig. 1 and Table 1.
The presence of two disubstituted double bonds at δC 131.4 (2CH, C-4, C-5) and 129.9 (2CH) were observed. The E geometry for the double bonds was supported from the characteristic chemical shift of the allylic carbons at δC 32.2, 32.3 and 34. (Z geometry = δC 27.0).
The positive FAB-MS spectral data showed a series of molecular ion peaks due to [M + Na]+ at m/z: 736, 764, 778, 792, 806, 820, 834 and 848. TSC-1 is hence presumed to be a molecular species consisting of a sphingosine-type cerebroside possessing mainly a 2-hydroxy fatty acid moiety (normal type terminal methyls at δC 13.6) and a β-D-glucopyranose moiety.
The sphingosine skeleton was characterized by comparison of its 1H and 13C NMR spectral data (Fig. 1 and Table 1) with that of known cerebrosides.27 The relative stereochemistry of the ceramide moiety is presumed to be (2S,3R,4E,2′R) since the characteristic 13C NMR signals (C-1, 2, 3, 4, 1′ and 2′) in addition to the optical rotation value of ([α]20D = −31.3) are in good agreement with those of the sphingosine-type glucocerebroside molecular species possessing a 2S,3R,4E,2′R configuration.27 The analysis of the methylated sugar against standard sugars (glucose and galactose) using HPLC indicated that the sugar moiety in TSC-1 was glucose (glucose tR = 14.11 min, galactose tR = 13.27 min). The coupling constant of the anomeric proton at δH 4.89 (1H, d, J = 8.0 Hz) and the chemical shift of the anomeric carbon δC (105.5) confirmed the β-configuration of the glucopyranoside moiety (α-glucopyranoside: J = 3.7 Hz; δC 98.5).18 The absolute configuration of the sugar moiety was determined using the Tanaka et al. method.18 HPLC analysis of the reaction mixture exhibited a peak at tR = 18.68 min, which were coincided with the aryl-isothiocyanate derivative of D-glucose, confirming the absolute configuration of the sugar moiety (L-glucose tR = 19.22 min).
To determine the length of the FAMEs and LCB in the glucocerebrosides, the methanolysis products of TSC-1 were subjected to 1H, 13C NMR, and FAB-MS analyses. Molecular ion peaks at 286, 314, 327, 343, 357, 371, 385, 399 and 413 [M + H]+ indicated the presence of C-16, C-18–C-25 fatty acid methyl esters, possessing normal terminal methyl groups (δC 13.6). The LCB mixture showed molecular ion peaks at 395 [M+] (LCB) and 424 [M + H]+ (LCB acetate) indicating the presence of a C-16 LCB, while 652 [M + Na]+ (LCB glucoacetate) indicated the presence of a C-18 LCB in TSC-1. See Fig. 1.
The NMR spectra of TSC-2 in C5D5N showed resonances for a secondary amide proton doublet at δH 8.55 (1H, d, J = 8.0 Hz), protons of a long methylene chain, centered at δH 1.25, (δC 29.1–29.8) and overlapped methyls at δH 0.83 (δC 13.6), indicating the presence of a sphingolipid skeleton. The proton signals at δH 3.85–4.93 (6H) and the anomeric proton signal at δH 4.93 (1H, d, J = 8.0 Hz; δC 104.9) confirmed the presence of a monosaccharide moiety.
TSC-2 showed characteristic resonances for the 2-amino-1,3,4,2′-tiol region of hydrocarbon chain with proton signals at δH 4.70 (1H, m, H-2), 4.29 (1H, m, H-2′), 4.70 (1H, m, H-1b), 4.29 (1H, m, H-1a), 4.59 (1H, m, H-3), and 4.49 (1H, m, H-4), corresponding to the following 13C NMR data: δC 51.1 (C-2), 71.8 (C-2′), 69.8 (C-1), 71.8 (C-3) and an amide carbonyl signal at δC 175.0 (C-1′), see Table 1. A disubstituted double bond in the side chain of the base was observed at δC 129.5 and 130. The E geometry for the double bond was supported from the chemical shift of the allylic carbons at δC 32.3 and 34.9 (Z geometry = δC 27.0).
HPLC analysis of the methylated sugar moiety indicated the sugar moiety to be glucose (glucose tR = 14.11 min, galactose tR = 13.27 min). The coupling constant of the anomeric proton at δH 4.93 (1H, d, J = 8.0 Hz) and the chemical shift of the anomeric carbon δC (104.9) confirmed the β-configuration of the glucopyranoside moiety (α-glucopyranoside: J = 3.7 Hz; δC 98.5).18 The absolute configuration of the sugar moiety was determined using the Tanaka et al. method.18 HPLC analysis of the reaction mixture exhibited a peak at tR = 18.5 min, which were coincided with the aryl-isothiocyanate derivative of D-glucose, confirmed the absolute configuration of the sugar moiety (L-glucose tR = 19.22 min).
In the positive FAB-MS spectral of TSC-2, a series of molecular ion peaks due to 810, 824, 838, 852, 866 and 880 [M + Na]+ were observed. Therefore, TSC-2 is presumed to be a molecular species consisting of a phytosphingosine-type cerebroside possessing mainly a 2-hydroxy fatty acid moiety (normal methyls at δC 13.6) and a β-D-glucopyranose moiety.
The core structure of the phytosphingosine skeleton in TSC-2 was characterized by comparison of its 1H and 13C NMR spectral data (Fig. 1 and Table 1) with that of known cerebrosides.28 The relative stereochemistry of the ceramide moiety is presumed to be (2S,3R,4R,2′R) since the characteristic 13C NMR signals (C-1, 2, 3, 4, 1′ and 2′) and the optical rotation value ([α]22D = +29.4) are in good agreement with those of the phytosphingosines-type glucocerebroside molecular species possessing a 2S,3R,4R,2′R configuration.28
In the FAB-MS analyses of the methanolysis products of TSC-2, the FAME mixture showed molecular ion peaks at 343, 357, 371, 383 and 413 [M + H]+, indicating the presence of C-20–C-25 fatty acid methyl esters, possessing normal terminal methyl groups (δC 13.6). The LCB also indicated the presence of a C-16 and C-18 long chain base identified from the corresponding molecular ion peaks at of the LCB acetates at m/z 455 [M + H]+ and 484 [M + H]+ respectively. See Fig. 1 and Table 1.
3.2. Discussion
A sphingosine-type (TSC-1) and phytosphingosine-type (TSC-2) cerebrosides, with both containing mainly a 2-hydroxy fatty acids and β-D-glucopyranose moieties were isolated from the n-hexane fraction of T. sanguinea in this study. Cerebrosides and ceramides have received a lot of interest in their isolation and characterization due to their significant biological activities such as immunomodulatory antioxidant, antitumour, antiinflammatory and antiviral.29,30Seven triterpenes were isolated from the n-hexane fraction of plant: β-sitosteryl-3β-D-glucopyranoside-O-fatty acid methyl esters molecular species TSS-1, β-sitosterol-3β-D-glucopyranoside-6′-O-palmitate 1, β-sitosterol 2, β-sitosterol-3β-D-glucopyranoside 3, β-stigmasterol 4, β-stigmasterol-3β-D-glucopyranoside 5, cholesterol 6 and betulinic acid 7. Biological functions of plant sterols include antihelmintic, antidiabetic, antiinflammatory, antiapoptotic, antinociceptive, antioxidant, immunomodulatory and neuroprotective in neurodegenerative disorders like Alzheimer's disease.31,32
Betulinic acid exhibits a variety of biological and medicinal properties such as inhibition of HIV, antibacterial, antimalarial, antiinflammatory, antihelmintic, antinociceptive, anti-HSV-1 and anticancer activities.33
The five lignans isolated from the ethyl acetate fraction of T. sanguinea: (+)-epipinoresinol 8, (+)-pinoresinol 9, (+)-cycloolivil 10, (+)-secoisolariciresinol 11 and (+)-isolariciresinol 12 are reported to also have antiviral, antifungal, antimicrobial antifeedant and insecticidal properties and are probably related to plant defense against various pathogens and pests. They also have significant biological activities including antitumour, antiinflammatory, immunosuppression, cardiovascular, antioxidant and antiviral.34 Pinoresinol and secoisolariciresinol are mammalian lignan precursors which are converted into enterodiol (END) and enterolactone (ENL) by the intestinal microflora.22,35 These enterolignans afford protection against osteoporosis, cardiovascular diseases, liver diseases, hyperlipidemia, breast cancer, colon cancer, prostate cancer and menopausal syndrome.25,36,37 The flavanone (+)-eriodictyol 13, also isolated from the ethyl acetate fraction is reported to have significant antiinflammatory, anticancer, neurotrophic, and antioxidant effects.38
4. Conclusions
In summary, this paper describes the isolation of two glucocerebrosides, TSC-1 and TSC-2, one molecular species TSS-1, seven triterpenes 1–7, five lignans 8–12, and one flavanone 13, from the n-hexane and ethyl acetate fractions of whole plant of T. sanguinea. To the best of our knowledge, all the isolated compounds from T. sanguinea in this study are being reported for the first time. These compounds have a wide range of biological activities and may act individually or in synergy to produce these biological effects. They may therefore be partly, or wholly responsible for these biological actions, giving credence to the use of T. sanguinea in traditional medicine. Further work to increase the amount of isolated cerebrosides molecular species to determine the double bond location in the long chain bases as well as purify and characterize the individual pure cerebrosides is underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Centre for Plant Medicine Research, Akuapem-Mampong, Ghana, for proving the plant material for this study. We are also grateful to Mr M. Inada and Dr N. Tsuda of the Scientific Support Section of Joint Research Center, Nagasaki University, for 1H, 13C NMR and MS measurements. This work was supported by Ministry of Science, Culture, Technology and Sports (MEXT), Japan, which is gratefully acknowledged.References
- A. A. Jigam, U. T. Abdulrazaq, H. A. Mahmud and F. O. Tijani, J. Appl. Pharm. Sci., 2012, 2, 47–51 Search PubMed.
- M. A. Gyamfi and Y. Aniya, Hum. Exp. Toxicol., 1998, 17, 418–423 Search PubMed.
- J. D. N'guessan, M. R. Dinzedi, N. Guessennd, A. Coulibaly, M. Dosso, A. J. Djaman and F. Guede-Guina, Trop. J. Pharm. Res., 2007, 6, 779–783 Search PubMed.
- F. C. Ohiri and V. C. Uzodinma, Fitoterapia, 2000, 71, 176–178 CrossRef PubMed.
- A. V. Kouakou, J. D. N'guessan, A. K. M. Kra and F. Guéde-Guina, J. Soc. Ouest-Afr. Chim., 2006, 22, 21–25 Search PubMed.
- J. D. N'guessan, A. P. Bidié, B. N. Lenta, B. Weniger, P. André and F. Guédé-Guina, Afr. J. Biotechnol., 2007, 6, 1685–1689 Search PubMed.
- I. I. Ohtani, N. Gotoh, J. Tanaka, T. Higa, M. A. Gyamfi and Y. Aniya, J. Nat. Prod., 2000, 63, 676–679 CrossRef.
- C. C. Chang, Y. C. Lien, K. C. S. C. Liu and S. S. Lee, Phytochemistry, 2003, 63, 825–833 CrossRef PubMed.
- M. A. Gyamfi and Y. Aniya, Toxicology, 2001, 164, 171 Search PubMed.
- T.-T. Zhang, L. Yang and J.-G. Jiang, Food Funct., 2015, 6, 2588–2597 Search PubMed.
- M. A. Gyamfi, I. I. Ohtani, E. Shinno and Y. Aniya, Food Chem. Toxicol., 2004, 42, 1401–1408 CrossRef PubMed.
- A. K. Nyarko and M. E. Addy, J. Ethnopharmacol., 1994, 41, 45–51 CrossRef PubMed.
- K. S. Konan, A. Toure, K. Ouattara, A. J. Djaman and J. D. N’guessan, Afr. J. Microbiol. Res., 2012, 6, 6247–6251 Search PubMed.
- N. L. Etkins, Trop. Doct., 1997, 27, 12–16 CrossRef PubMed.
- M. A. Gyamfi, M. Yonamine and Y. Aniya, Gen. Pharmacol., 1999, 32, 661–667 CrossRef PubMed.
- M. A. Gyamfi, T. Tanaka and Y. Aniya, Life Sci., 2004, 74, 1723–1737 CrossRef PubMed.
- M. A. Gyamfi, N. Hokama, K. Oppong-Boachie and Y. Aniya, Hum. Exp. Toxicol., 2000, 19, 623–631 Search PubMed.
- T. Tanaka, T. Nakashima, K. Ueda, K. Tomii and I. Kouno, Chem. Pharm. Bull., 2007, 55, 899–901 CrossRef PubMed.
- V. A. S. Ng, E. M. G. Agoo, C.-C. Shen and C. Y. Ragasa, Int J Pharm Clin Res., 2015, 7, 643–646 Search PubMed.
- M. Arora and A. N. Kalia, Int. J. Pharm. Pharm. Sci., 2013, 5, 245–249 Search PubMed.
- S. P. Sawan, T. L. James, L. D. Gruenke and J. C. Craig, J. Magn. Reson., 1979, 35, 409–413 Search PubMed.
- A. Ikuta and H. Itokawa, Phytochemistry, 1988, 27, 2813–2815 CrossRef.
- E. Okuyama, K. Suzumura and M. Yamazaki, Chem. Pharm. Bull., 1995, 43, 2200–2204 CrossRef PubMed.
- R. Ghogomu-Tih, B. Bodo, B. Nyasse and B. L. Sondengam, Planta Med., 1985, 51, 464 CrossRef PubMed.
- A. H. Banskota, T. Usia, Y. Tezuka, K. Kouda, N. T. Nguyen and S. J. Kadota, J. Nat. Prod., 2002, 65, 1700–1702 CrossRef.
- L. A. L. da Silva, L. G. Faqueti, F. H. Reginatto, A. D. C. dos Santos, A. Barison and M. W. Biavatti, Rev. Bras. Farmacogn., 2015, 25, 375–381 CrossRef.
- R. Higuchi, M. Inagaki, K. Togawa, K. T. Miyamoto and T. Komori, Justus Liebigs Ann. Chem., 1994, 635–658 Search PubMed.
- R. Higuchi, J. X. Jhou, K. Inukai and T. Komori, Liebigs Ann. Chem., 1991, 745–752 CrossRef.
- K. Yamada, R. Matsubara, M. Kaneko, T. Miyamoto and R. Higuchi, Chem. Pharm. Bull., 2001, 49, 447–452 CrossRef PubMed.
- R. Abdelhameed, M. S. Elgawish, A. Mira, A. K. Ibrahim, S. A. Ahmed, K. Shimizu and K. Yamada, RSC Adv., 2016, 6, 20422–20430 RSC.
- C. Shi, F. Wu, X. Zhu and J. Xu, Biochim. Biophys. Acta, 2013, 1830, 2538–2544 CrossRef PubMed.
- S. Saeidnia, A. Manayi, A. R. Gohari and M. Abdollahi, Eur. J. Med. Plants, 2014, 4, 590–609 CrossRef.
- G. M. Moghaddam, B. H. A. Faujan and S.-K. Alireza, Pharmacol. Pharm., 2012, 3, 119–123 CrossRef.
- J.-Y. Pan, S.-L. Chen, M.-H. Yang, J. Wu, J. Sinkkonen and K. Zoud, Nat. Prod. Rep., 2009, 26, 1251–1292 RSC.
- S. Heinonen, T. Nurmi, K. Liukkonen, K. Poutanen, K. Wahala, T. Deyama, S. Nishibe and H. Adlercreutz, J. Agric. Food Chem., 2001, 49, 3178–3186 CrossRef PubMed.
- H. Adlercreutz, Crit. Rev. Clin. Lab. Sci., 2007, 44, 483–525 CrossRef PubMed.
- J. M. Landete, Food Res. Int., 2012, 46, 410–424 CrossRef.
- S. E. Lee, H. Yang, G. W. Son, H. R. Park, C.-S. Park, Y.-H. Jin and Y. S. Park, Int. J. Mol. Sci., 2015, 16, 14526–14539 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and FAB-MS data for TSC-1, TSC-2, TSS-1 and comp. 1–13 are available. See DOI: 10.1039/c8ra03913e |
| This journal is © The Royal Society of Chemistry 2018 |