
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel cyclopeptides containing heterocyclic skeletons†
Fatima Hamdan
 a,
Fatemeh Tahoori
b and
Saeed Balalaie
*ac
a,
Fatemeh Tahoori
b and
Saeed Balalaie
*ac
aPeptide Chemistry Research Center, K. N. Toosi University of Technology, P. O. Box 15875-4416, Tehran, Iran. E-mail: Balalaie@kntu.ac.ir
bRazi Vaccine and Serum Research Institute, Agricultural Research, Education and Extension Organization (AREEO), Karaj, Iran
cMedical Biology Research Center, Kermanshah University of Medical Sciences, Kermanshah, Iran
First published on 3rd October 2018
Abstract
Cyclopeptides can be considered as naturally biologically active compounds. Over the last several decades, many attempts have been made to synthesize complex naturally occurring cyclopeptides, and great progress has been achieved to advance the field of total synthesis. Moreover, cyclopeptides containing heterocyclic skeletons have been recently developed into powerful reactions and approaches. This review aims to highlight recent advances in the synthesis of cyclopeptides containing heterocyclic skeletons such as triazole, oxazole, thiazole, and tetrazole.
 Fatima Hamdan | Fatima Hamdan was born in Lebanon in 1982 and is currently a PhD student. She received her M.Sc. in Chemistry from the Doctoral School of Science and Technology, Lebanese University, Lebanon, in 2011. She moved to K. N. Toosi University of Technology for her PhD studies in 2013. She is working on the synthesis of novel anti-cancer peptides containing RGD backbones. |
 Fatemeh Tahoori | Fatemeh Tahoori was born in Iran in 1983; she received her PhD at K. N. Toosi University of Technology in 2013 under the supervision of Prof. Saeed Balalaie, where her work focused on the synthesis of cyclopeptides containing triazole moieties. Currently, she is working as an assistant professor at Razi Vaccine and Serum Research Institute. |
 Saeed Balalaie | Saeed Balalaie was born in Iran in 1965; he received his BSc degree from the University of Tehran in 1989, his MSc from Shahid Beheshti University, Iran, in 1991, and his PhD degree from Sharif University of Technology, Iran, in 1996. He started his career as an assistant professor at K. N. Toosi University of Technology in Iran, Tehran, Iran, in 1997; he was promoted to associate professor in 2003 and full professor in 2007 at the aforementioned university. He received an Alexander von Humboldt research fellowship in 2001 and worked with Professor Rolf Gleiter at the University of Heidelberg, Germany. His research interests focus on the synthesis of organic compounds through designing novel domino reactions as well as the synthesis of peptides and cyclopeptides. |
1. Introduction
Previous reviews of this subject discussed the natural existence of cyclopeptides but failed to provide important knowledge about the synthesis of cyclopeptides containing heterocyclic skeletons. Due to the rapid development of this field, we believe it is worthwhile to update this subject. Selected examples from recent reports will be discussed, with emphasis on approaches that are very interesting and will open new portals for science to exploit these recent methods for the synthesis of these cyclopeptides.A key step in chemistry and biology is molecular recognition. Many cellular processes are controlled by the binding of hormones and peptides to receptors. Due to their therapeutic abilities, peptides have received growing interest in recent years. To date, more than 60 peptide drugs have been released to the market for the benefit of patients, and hundreds of novel therapeutic peptides are in preclinical and clinical development. The reason behind this success is the potent and specific, yet safe, mode of action of peptides.1 Despite their high potential, peptides have limitations, such as short half-life, rapid metabolism, and poor oral bioavailability. However, different types of modifications can be employed to improve the pharmacokinetic properties of peptides. For several key reasons, cyclization of linear peptides is often used as an attractive modification. First, it is anticipated that by obtaining fixed geometries, cyclic peptides can bind more efficiently to their respective receptors. It is additionally hoped that these conformationally constrained cyclic peptides will be selective to explicit receptor subtypes, resulting in higher receptor binding affinities compared with their linear analogues. Moreover, cyclic peptides are often chosen over their linear analogues due to their enhanced enzymatic stability and provide enhanced membrane permeability, which result in improved bioavailability of the cyclopeptides; they also possess entropic advantages in molecular recognition.2 Therefore, they are sought as promising lead compounds for drug discovery and the development of new peptide analogues; simulated peptides, which can provide biological targets of relevant receptors and adjust their properties, are the most important areas of medicine, chemistry, and biology.3–9 Due to the unique, promising properties and biological significance of cyclopeptides in addition to their additional level of synthetic complexity, naturally occurring cyclic peptides have been attracting the attention of synthetic chemists. In the past decades, different cyclopeptides containing heterocyclic motifs have been developed. This review aims to highlight novel, recently developed peptide cyclizations with heterocyclic skeleton approaches.3
Over 100 cyclopeptides have been isolated and characterized from plants, marine organisms,10,11 fungi12 and microorganisms. Due to the widespread occurrence of these compounds, they represent an important class of natural products. These peptides have certain advantages over chemical drugs, such as relatively simple structures, fewer side effects, and good absorption.
Cyclic peptides have established many successes, e.g. octreotide (Novartis); integrilin, a cyclic peptide heptapeptide Gp IIb/IIIa inhibitor (Cor Pharmaceuticals); and the naturally occurring cyclosporine A for immunosuppression. However, the cost of cyclic peptide synthesis is high because the required reagents are expensive. Meanwhile, many attempts have been made to optimize cyclization yields; the use of inexpensive reagents and chromatographic separation has pushed the balance toward the synthesis of cyclosporine A compared to its isolation from its natural source.13
The synthesis of cyclic peptides that simulate natural compounds is of great interest to chemists. Various approaches and methods to form rings of peptide sequences have been suggested and provided. One of these methods is head-to-tail loop forming for shorter sequences, performed on resins. The type of reagents used for each sequence depends on various parameters, such as the size of the ring.13,14
Other methods of ring formation require the use of different chemical reactions, such as the creation of lactam bridges;15 disulfide bridges, which are most commonly used for sequences with two cysteine amino acids;16,17 click chemistry;18,19 metathesis;20,21 and the Ugi,22,23 Hantszch,24,25 Horner–Wadsworth–Emmons (HWE),26 Mannich27 and Heck28 reactions.
Also, new methods have been used in the past decade to cyclize linear peptides through the use of enzymes29 and microwaves.8,30
However, in this review, we decided to investigate the insertion of heterocycle rings in cyclopeptides. Heterocycles, which are an extensively published subject, include triazole,18 imidazole,31–33,37 oxazole25,33–35 and thiazole rings.13,24,25,34,36 These rings endow peptide structures with rigidity, and the interactions of these structures with receptors can be studied more readily and accurately (Fig. 1).
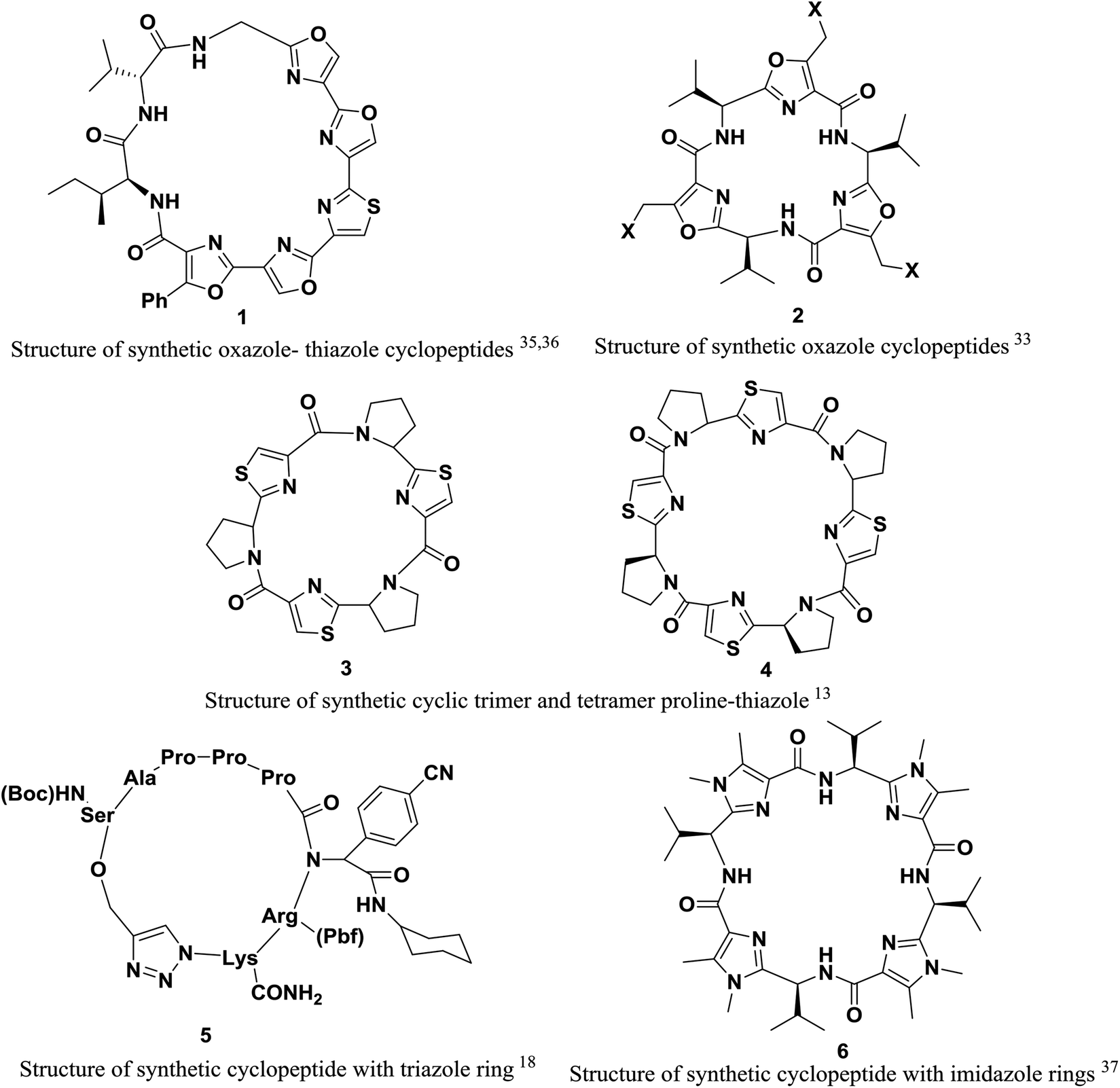 | ||
| Fig. 1 Structures of some synthetic cyclopeptides. | ||
2. Cyclopeptides with triazole moieties
In the last decade, the synthesis of 1,2,3-triazole units has been of great interest. 1,2,3-Triazoles represent a class of heterocycles with extended properties for employment in peptide sciences because they are considered to be amide bond isosteres while being stable to enzymatic degradation. Due to these characteristics, 1,2,3-triazoles are promising candidates for the development of novel peptidomimetics with possibly improved biological activities. Only a few examples of triazole-based peptidomimetics can be found in the literature.38The first to study and investigate 1,2,3-triazoles was Huisgen in 1960; he described the Huisgen 1,3-dipolar cycloaddition from the reaction of an alkyne and an azide to afford 1,4- and 1,5-disubstituted triazole regioisomers. Several methodologies using transition metals have been investigated to control the regioselectivity and to improve the reaction conditions for the formation of 1,2,3-triazoles. Basically, Sharpless and Meldal developed the copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC), which afforded regioselective formation of the 1,4-regioisomer; meanwhile, ruthenium-catalyzed cycloaddition afforded the 1,5-regioisomer (Scheme 1). To date, many examples of the application of the click chemistry reaction in chemical biology and medicinal chemistry have been reported. Thus, ‘click’ has been successfully applied to the macrocyclization of peptides.2,39,40
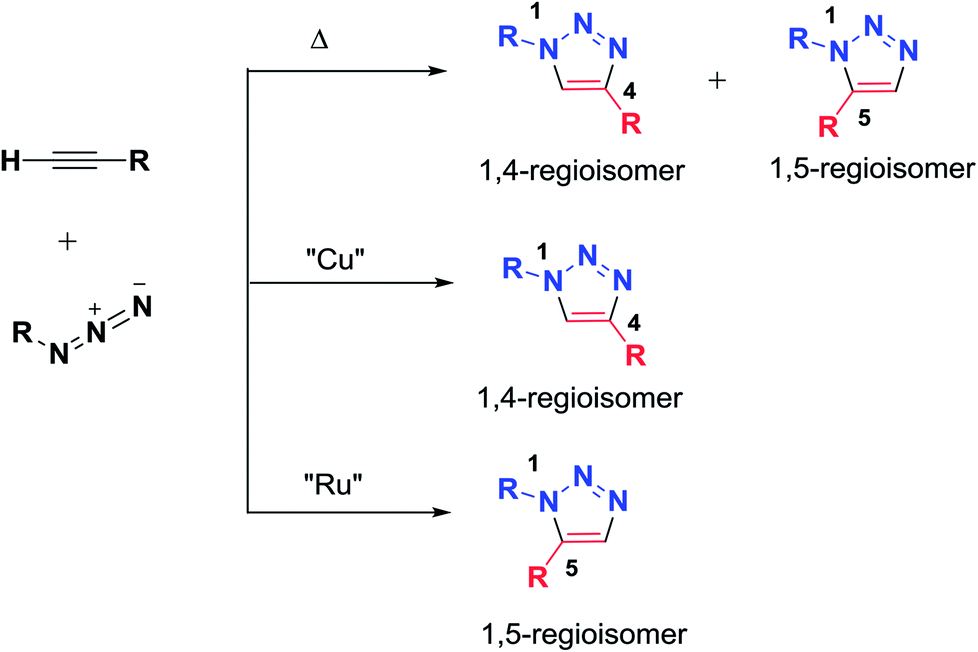 | ||
| Scheme 1 Synthesis of 1,4- and 1,5-disubstituted 1,2,3-triazoles by heat, CuAAc, and RuAAC. | ||
Azido- and alkyne building blocks can be built into the backbones of peptides by classical methods of solid-phase or solution peptide chemistry.41,42 The triazole moiety can be incorporated by different approaches. For example, the synthesis of small cyclic triazolo-peptides is usually attained by solution-phase synthesis of the precursor followed by cyclisation employing Cu-AAC (Scheme 2A). For the synthesis of longer triazole peptides, the triazole can be incorporated during peptide elongation by coupling of a triazole-containing dipeptide mimic by classical solid phase peptide synthesis (SPPS) methods (Scheme 2B).43 Alternatively, the heterocycle can be installed on a solid support by Cu-AAC of an alkyne precursor with an immobilized azide (Scheme 2C).44 Finally, peptide fragments functionalized with azides and alkynes can be bonded by Cu-AAC in solution, a strategy that has been successfully used as a ligation method for the assembly of larger proteins by Cu-AAC45 (Scheme 2D).
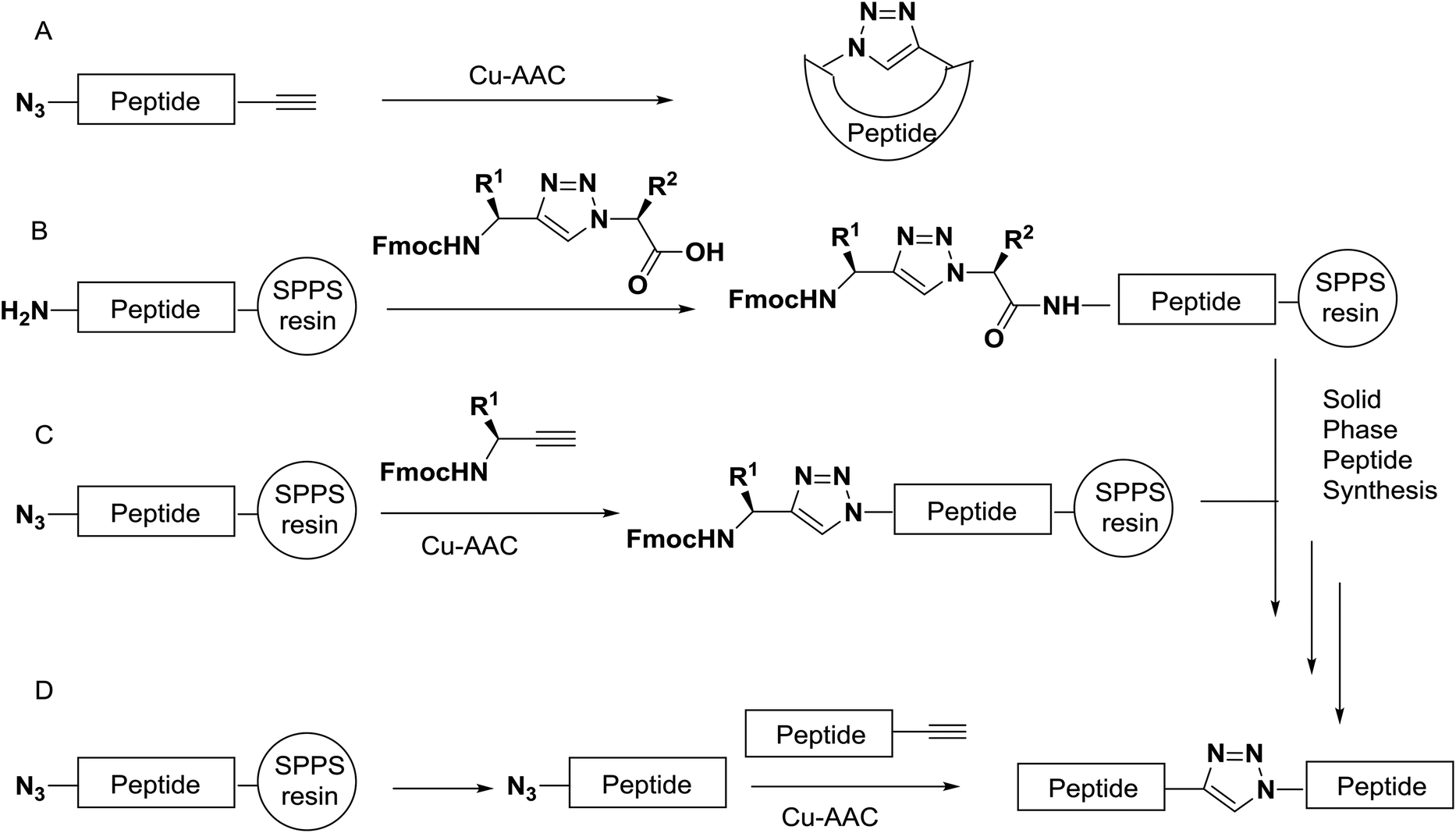 | ||
| Scheme 2 Schematic examples of the synthesis of triazole peptides. | ||
Various experimental reaction conditions have been established for Cu-AAC in solution and on solid supports. Cu-AAC is a metal-catalyzed, stepwise reaction that requires the presence of an active copper(I) species, an azide, an alkyne, and a proton acceptor.46 The reaction is very robust, provided that all the aforementioned reactants are maintained in solution and that the copper(I) is not oxidized or disproportionated. In particular, the copper(I) source should be chosen in accordance with the solvent system employed. In general, copper(I) halides or [Cu(CH3CN)PF6] are the preferred Cu(I) sources to perform Cu-AAC in organic solvents such as THF, DMSO, and toluene. Employment of these salts often requires an equivalent of an amine base, such as triethylamine, diisopropylethylamine, or piperidine.46,47 For Cu-AAC conjugations in aqueous media, in situ generation of the Cu(I) catalyst from Cu(II) salts in the presence of a reducing agent, e.g. sodium ascorbate, has become the method of choice. Water-soluble sources of copper(I) have found frequent application because Cu(II) salts are often less expensive and available in higher purity than copper halides. The combination of CuSO4 and sodium ascorbate is particularly suited for Cu-AACs in polar solvents, such as water, alcohols, acetonitrile, DMF, or mixtures thereof. If the substrates of Cu-AAC possess potential coordination or chelation sites for copper species (e.g. carboxylates, phosphates, thiols, imidazoles, or amines) it is recommended to add a stabilizing ligand to prevent segregation of the copper catalyst.48 In addition, the presence of such ligands often accelerates the reaction and prevents oxidation of the Cu(I) catalyst.49,50 A large number of stabilizing ligands for Cu(I) have been reported, of which polytriazole-based systems are the most frequently employed. Among these readily accessible and structurally diverse ligands,51,52 tris-(benzyltriazolylmethyl)amine (TBTA) is commercially available and, thus, most commonly used. Although some general procedures and reaction conditions can be defined for performing Cu-AAC with peptidyl substrates, it should be noted that individual optimizations may be required in certain cases in order to achieve optimal results.45
Due to hydrogen bonding and dipole interactions, triazole products can function as more than passive linkers and can readily interact with biological targets.53,54 The cycloaddition of an N-terminal azide and a C-terminal alkyne allows the synthesis of triazole-containing cyclopeptide analogues55 (Scheme 3a). Basically, the tyrosinase inhibitory activity of these isosteres was retained.56 Lokey and co-workers described the utility of this reaction as a macrocyclization tool.57 They achieved the cyclization of leucine-rich tetra-, penta-, hexa- and heptapeptides on solid supports. However, the formation of dimeric and trimeric by-products has been noted in many copper-catalyzed azide-alkyne cycloadditions.26 A tandem dimerization–macrocyclization approach enabled by several tandem click reactions was demonstrated by Ghadiri and co-workers;58 they synthesized C2-symmetric cyclic peptide scaffolds that self-assembled into peptide nanotubes.59 This approach was more successful than a conventional macrolactamization of a linear precursor already containing both triazole units (Scheme 3b).
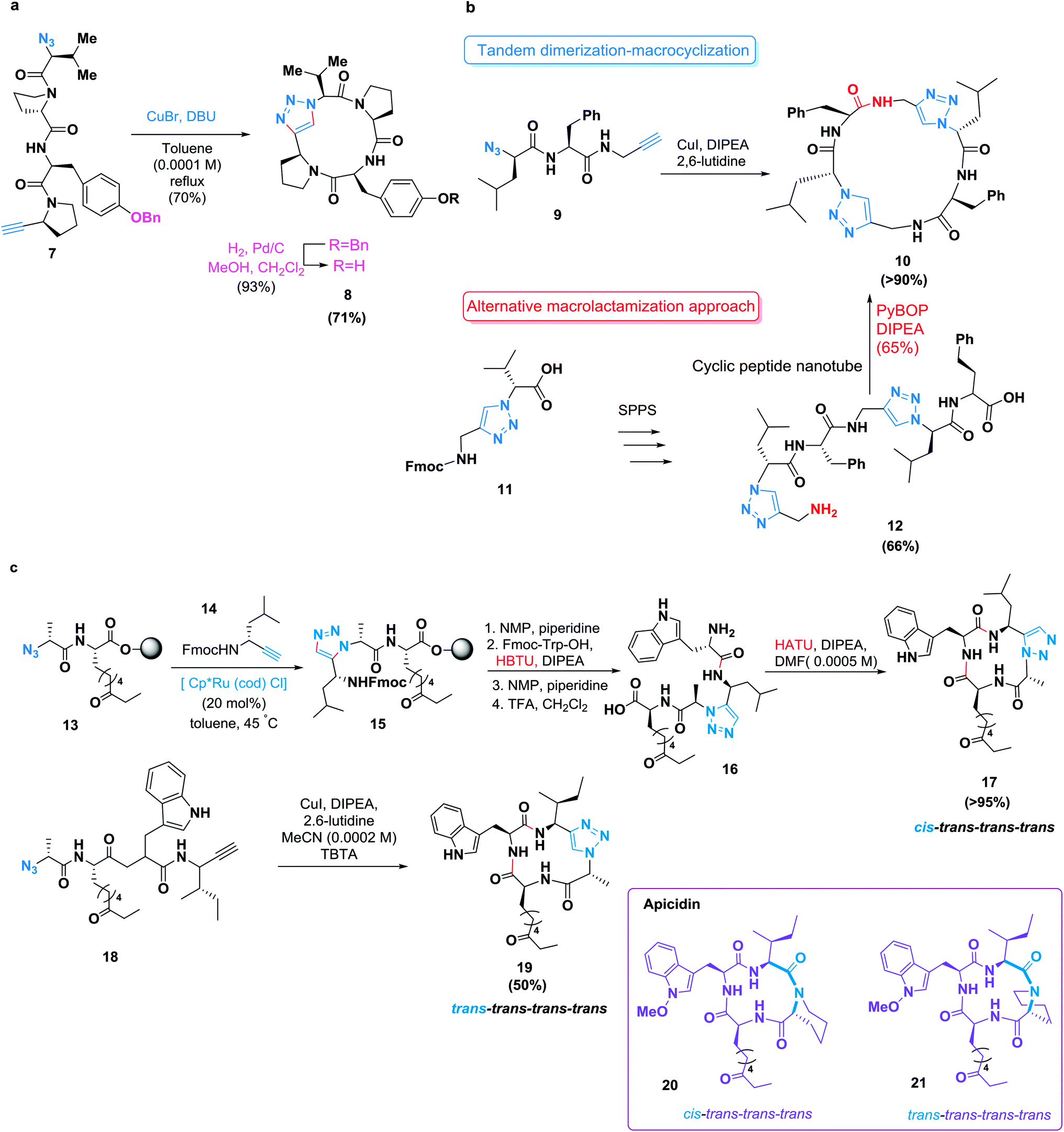 | ||
| Scheme 3 Azide-alkyne cycloadditions in the synthesis of peptide macrocycles. (a) A click-mediated macrocyclization of Tyr-Pro-Val-Pro. (b) Synthesis of a cyclic peptide nanotube through either a high-yielding tandem dimerization-macrocyclization approach by two tandem click reactions of an azido-dipeptide alkyne (top) or through a less efficient conventional macrolactamization approach (bottom). (c) The synthesis of triazole-modified analogues of the cyclic tetrapeptide apicidin. Top: ruthenium-catalyzed formation of a 1,5-disubstituted 1,2,3-triazole on a solid phase is followed by macrolactamization to yield an analogue resembling the biologically active conformation of apicidin. Bottom: a Cu(I)-catalyzed intramolecular azide–alkyne cycloaddition to yield an analogue of apicidin resembling its predominant conformation in solution. | ||
Meanwhile, 1,5-disubstituted 1,2,3-triazoles are also known as surrogates for trans-amide bonds. In another approach, Ghadiri and co-workers proved that the incorporation of one or two of these moieties into cyclic tetrapeptides resulted in a stronger binding affinity for the somatostatin (SST) receptor.60a In addition, Ghadiri and co-workers successfully fused these heterocycles as cis-amide isosteres of a naturally occurring cyclic tetrapeptide. This approach was accomplished through initial ruthenium(II)-catalyzed formation of the 1,5-disubstituted 1,2,3-triazole moiety in the linear peptide on the resin surface, followed by a conventional macrolactamization of the pseudotetrapeptide.60b The synthesized analogue, incorporating a 1,5-disubstituted 1,2,3-triazole, displayed similar biological activity to that of the naturally occurring apicidin.
Rademann and co-workers successfully reported the first cyclization of azido-alkynyl peptides to produce 1,5-disubstituted triazole-containing macrocycles. They developed an on-solid-support peptide synthesis strategy. In their approach, cyclization and cleavage from the solid support were carried out in the same chemical reaction.61 Using this strategy, simple purification of the synthesized cyclic peptide can be achieved because the open-chain by-product oligomers remain attached to the solid support. This approach includes a dipolar cycloaddition of polymer-bound azidopeptidylphosphoranes which is metal-free and does not require amino acid alkynes (Scheme 4).62
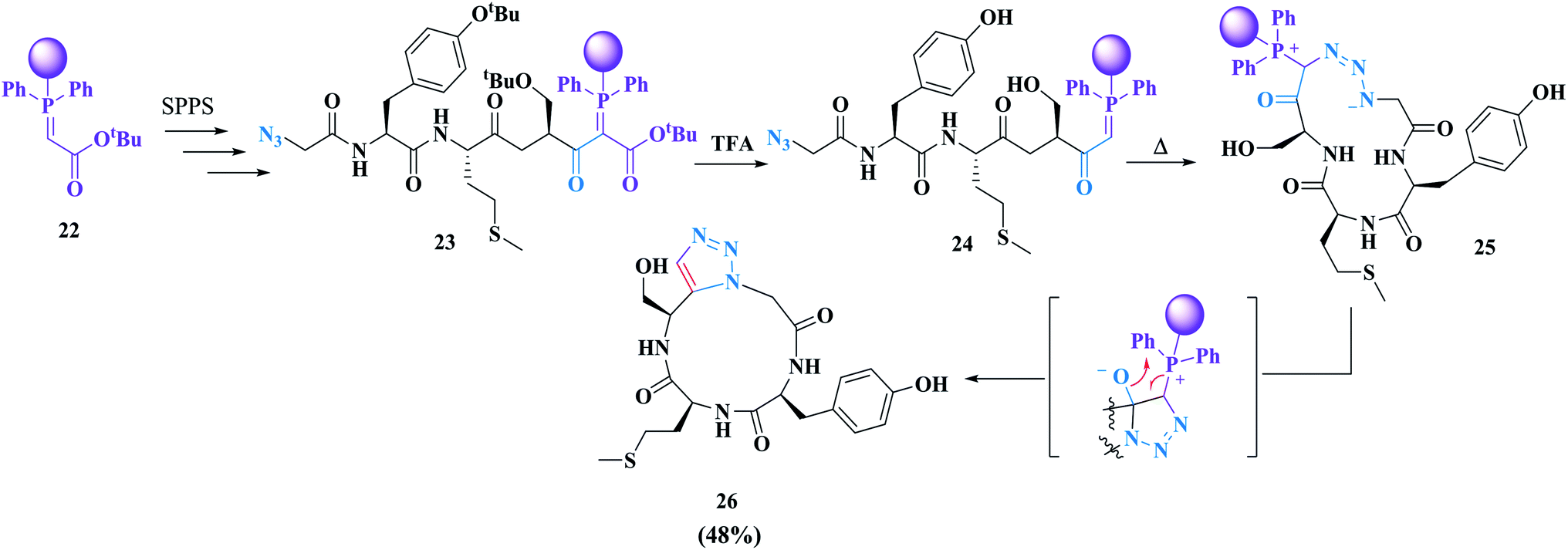 | ||
| Scheme 4 Synthesis of a cyclic tetrapeptide analogue containing a 1,5-disubstituted 1,2,3-triazole through intramolecular cyclative cleavage of a solid-support-bound azidopeptidylphosphorane. HATU, 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; NMP, N-methyl-2-pyrrolidone; TBTA, tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl] amine. | ||
One of the major applications of click chemistry is the preparation of small cyclic peptide analogues that are too strained for closure via lactamization. The synthesis of cyclo-[(L)-Pro-(L)-Tyr-(L)Pro-(L)Val], a potent tyrosine inhibitor, is difficult to achieve using traditional methods due to its unfavorable transition states. In order to perform this cyclization, Bock and co-workers designed and synthesized its triazole analogue.55 The desired cyclic peptide was isolated in 70% yield after CuI-catalyzed cycloaddition, as shown in Scheme 5. All the resulting triazole-peptides 30–32 were as active as the parent cyclopeptide 29 with regard to the inhibition of tyrosinase51,52 (Fig. 2). This approach allows this strategy to be used as a general method for preparing conformational constrained cyclic peptidomimetics in solution phase.63
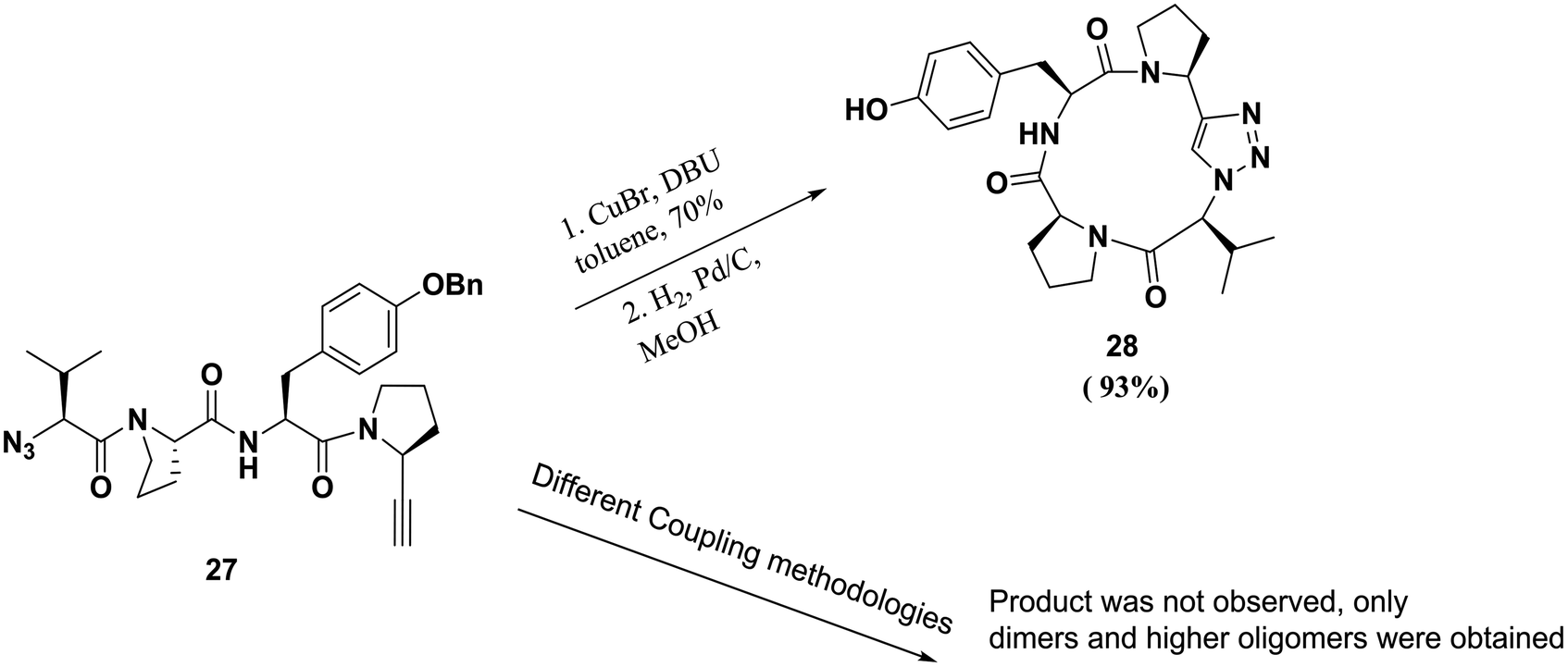 | ||
| Scheme 5 Application of click chemistry to the macrocyclization of peptidomimetics and the synthesis of cyclopeptides containing triazole skeletons. | ||
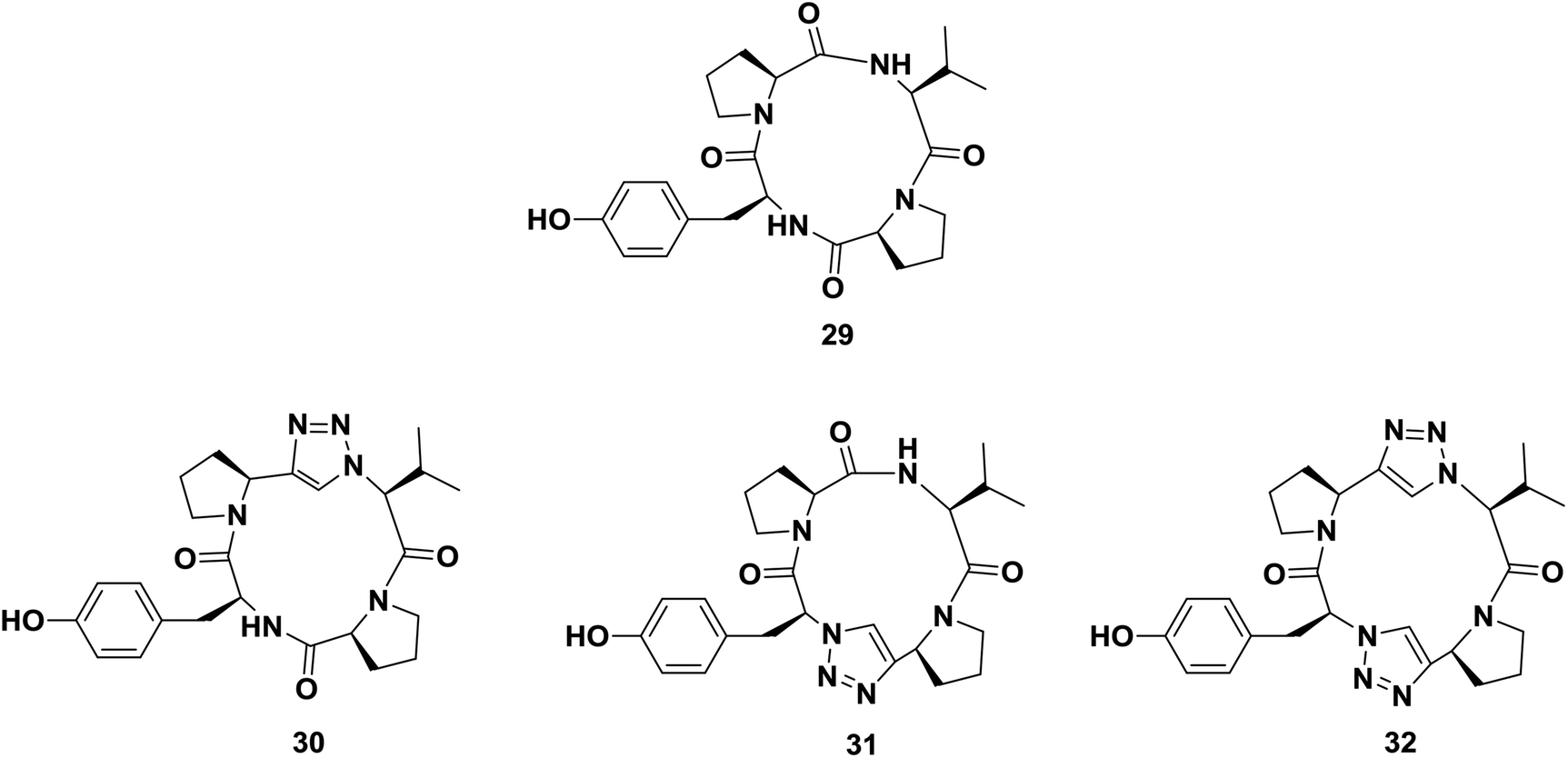 | ||
| Fig. 2 Examples of cyclopeptides and their triazolo-analogues synthesized by Bock. | ||
On the other hand, Davis and co-workers also reported triazole-peptide 34, an analogue of sansalvamide A 33, an inhibitor of heat shock protein 90 with cytotoxic effects against several cancer cell lines.64 In their work, they studied the utility of different moieties as amide bond surrogates (1,2,3-triazole, oxazole, thiazole, or pseudoprolines) in the same position. Evaluation of the compounds in vitro revealed that only triazole and thiazole heterocycles could be incorporated into the peptide without decreasing its cytotoxicity. In the case of triazole derivative 34, the potency of the reference compound 33 was preserved in terms of the inhibition of cell proliferation38 (Fig. 3).
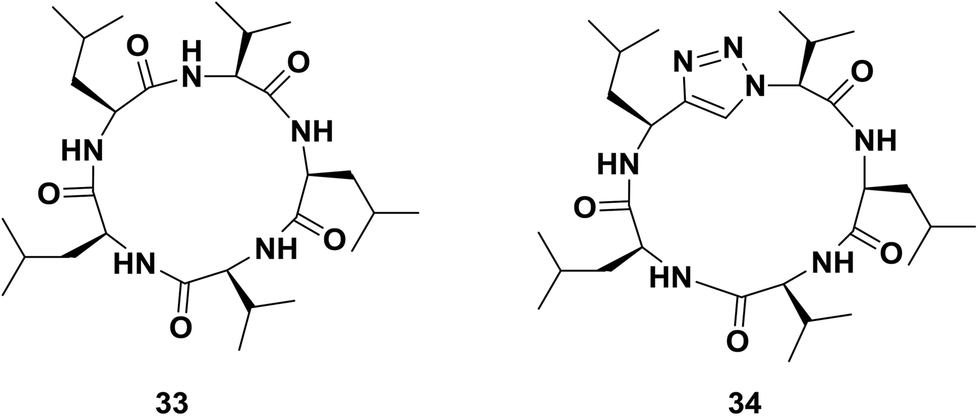 | ||
| Fig. 3 Examples of a cyclopeptide and its triazolo-analogue synthesized by Davis and co-workers. | ||
Horne and co-workers replaced an isoleucyl-pipecolyl residue with an isoleucyl-1,2,3-triazole-alanyl dipeptide mimic of apicidin 35, an inhibitor which binds with nanomolar affinity to several subtypes of histone deacetylases (HDACs; Fig. 4).60b Interestingly, the modification changed the subtype-specificity of the original compound. Compared to apicidin 35, triazole-peptide 36 showed an 8-fold decreased inhibitory effect against HDAC-subtype 1, while its effect against HDCA-subtype 3 was retained.
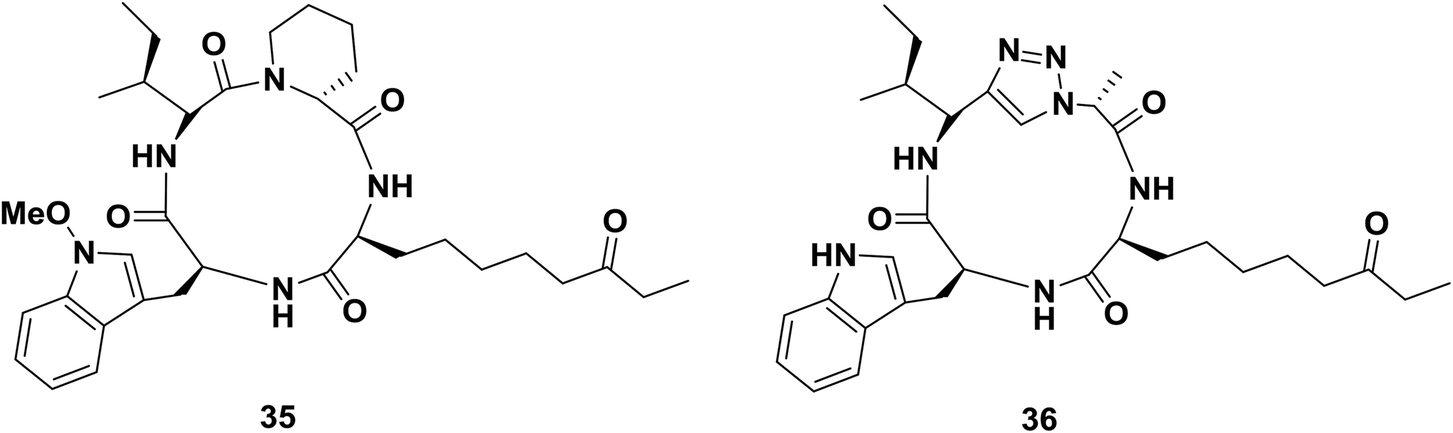 | ||
| Fig. 4 Examples of a cyclopeptide and a triazole-containing peptide synthesized by Horne and co-workers. | ||
Cyclic peptides containing the amino acid sequence Arg-Gly-Asp (cRGD) bind to αvβ3 integrins, which are involved in tumor angiogenesis and metastasis. Thus, several cRGD-based compounds have been developed to target tumor cells specifically. In 2008, Liu and co-workers reported the synthesis of a mimic of cyclopeptide 37 in which the D-phenylalanyl residue was replaced by a glycyl-1,2,3-triazole-glycyl dipeptide mimic (Fig. 5).65 In vitro studies performed with both compounds showed that triazole-peptide 38 had an affinity towards its receptor comparable to that of the parent compound 37.38
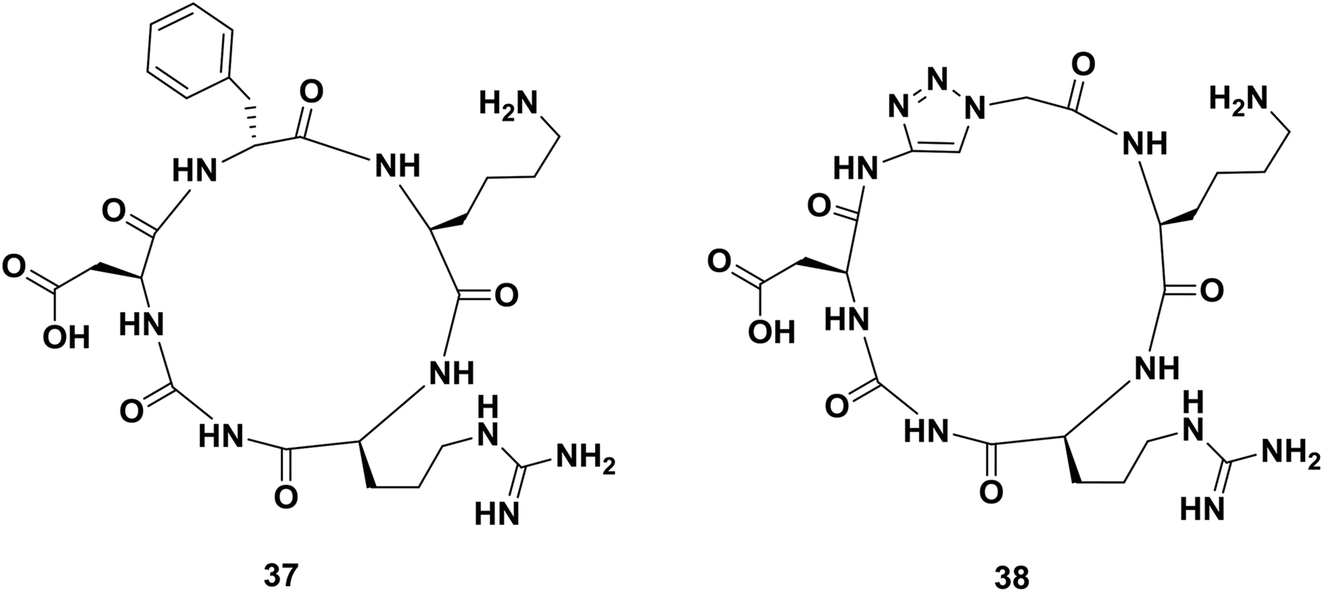 | ||
| Fig. 5 Examples of a cyclopeptide and a triazole-containing peptide synthesized by Liu and co-workers. | ||
Sunflower trypsin inhibitor 1 (SFTI-1) is a potent cyclic protease inhibitor with a turn conformation that is critical for its activity. Tischler and co-workers replaced a trans-amide bond located between the prolyl and the alanyl residue of SFTI-1 analog 39 with a prolyl-1,2,3-triazole-alanyl dipeptoid (Fig. 6).44 In their work, the authors reported the use of both 1,4- and 1,5-disubstituted 1,2,3-triazoles as amide-bond surrogates. As expected, only compound 40 derivatized with a 1,4-disubstituted triazole adopted a conformation close to that of the parent cyclopeptide and exhibited nanomolar affinity against bovine trypsin.
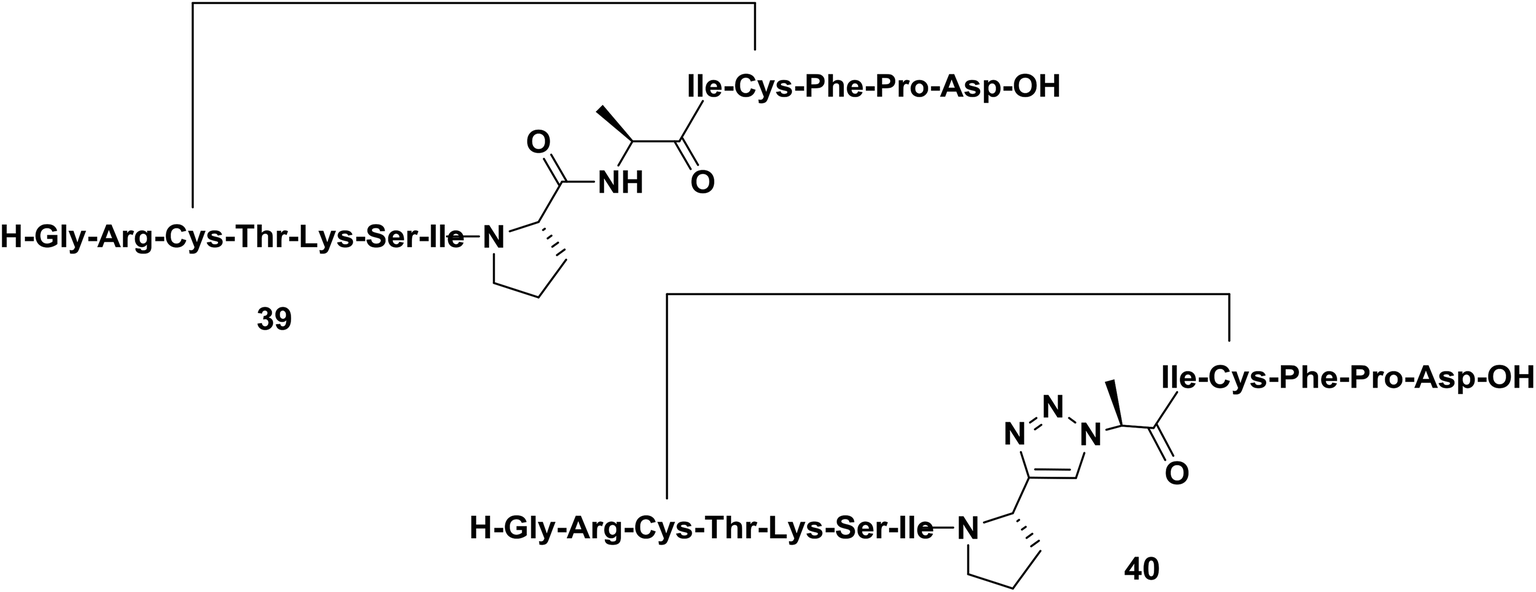 | ||
| Fig. 6 Examples of a cyclopeptide and a triazole-containing peptide synthesized by Tischler and co-workers. | ||
Later, Inguimbert and co-workers were able to establish a convenient method for the creation of a library of triazole peptidomimetics on a solid phase, as shown in Scheme 6.66 After the synthesis of the peptide, cyclization of the peptidyl-resin was performed by exposure to 0.5 equivalents of copper(I) iodide in the presence of sodium ascorbate and 2,6-lutidine in NMP/CH2Cl2. Then, the peptide was cleaved from the resin surface using TFA to afford the final cyclic triazole bridged peptidomimetic in good yield and high purity. Using this methodology, Inguimbert's group was able to successfully assemble and screen 18 conformationally constrained peptidomimetics; they discovered some promising lead compounds, such as compound 44, which exhibits potent bioactivity in inhibiting VEGFR 1.38
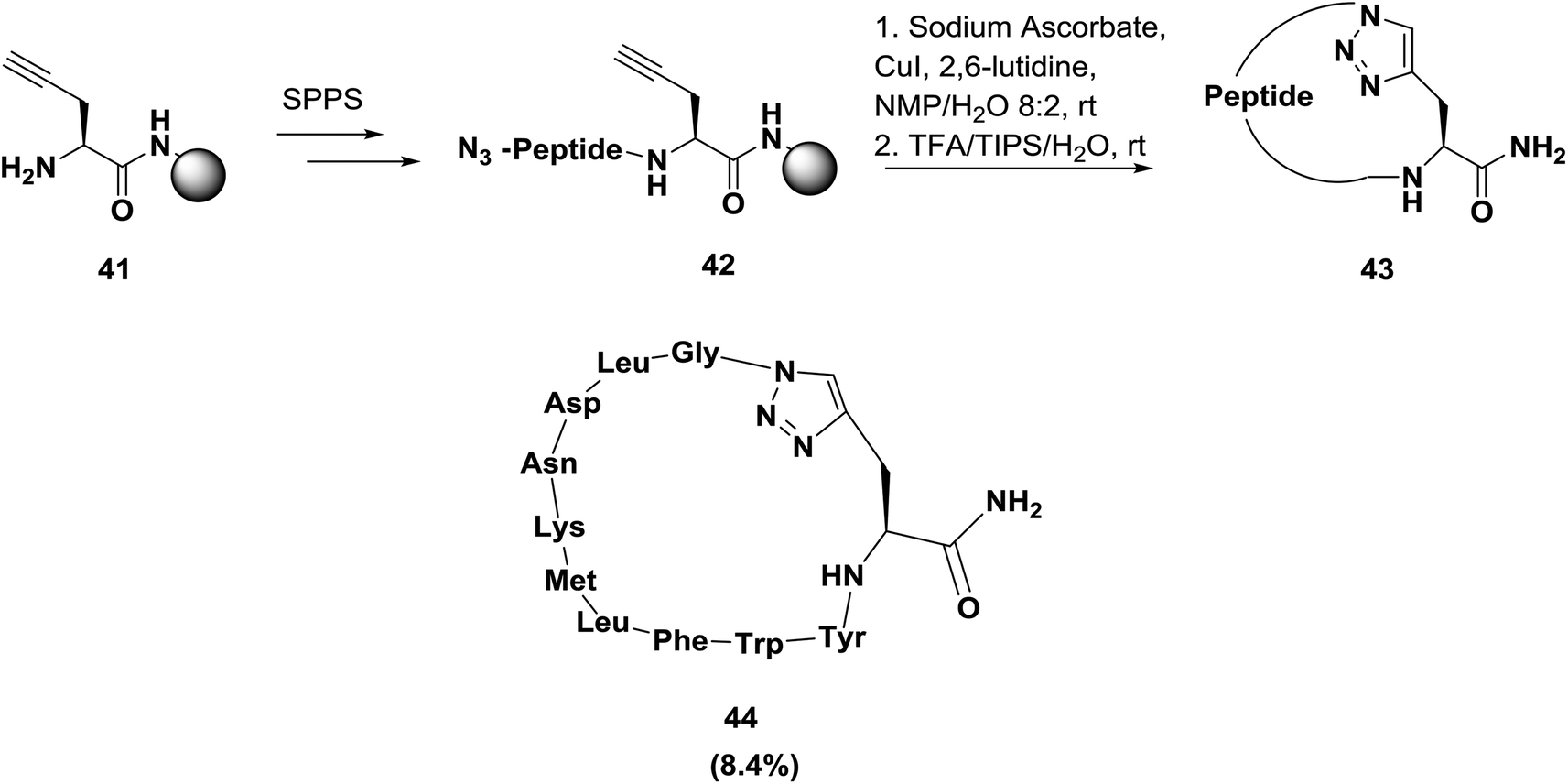 | ||
| Scheme 6 Application of click chemistry to the macrocyclization of peptidomimetics. | ||
Finn and co-workers successfully synthesized two cyclodimerized peptides, 22-residue cyclic peptide 46 and 38-residue cyclic peptide 48, as shown in Scheme 7.67 The cyclizations were performed by exposing each resin to 0.5 equivalents of CuI for 16 h at room temperature. The approximate yield of cyclodimerization was 60%. Meanwhile, this cyclization required a certain density on the solid support, and the dimeric pathway was favored even when the two chains were difficult to bring together. Based on their experimental observations, Finn and co-workers suggested that the reaction proceeds via two alkynes bound to a dicopper intermediate. The reason behind the formation of macrocyclodimers over the corresponding monomeric forms is that exo-like intermediates are favored over endo-like due to the geometric constraints of forming 1,4-disubstituted triazoles.67–69
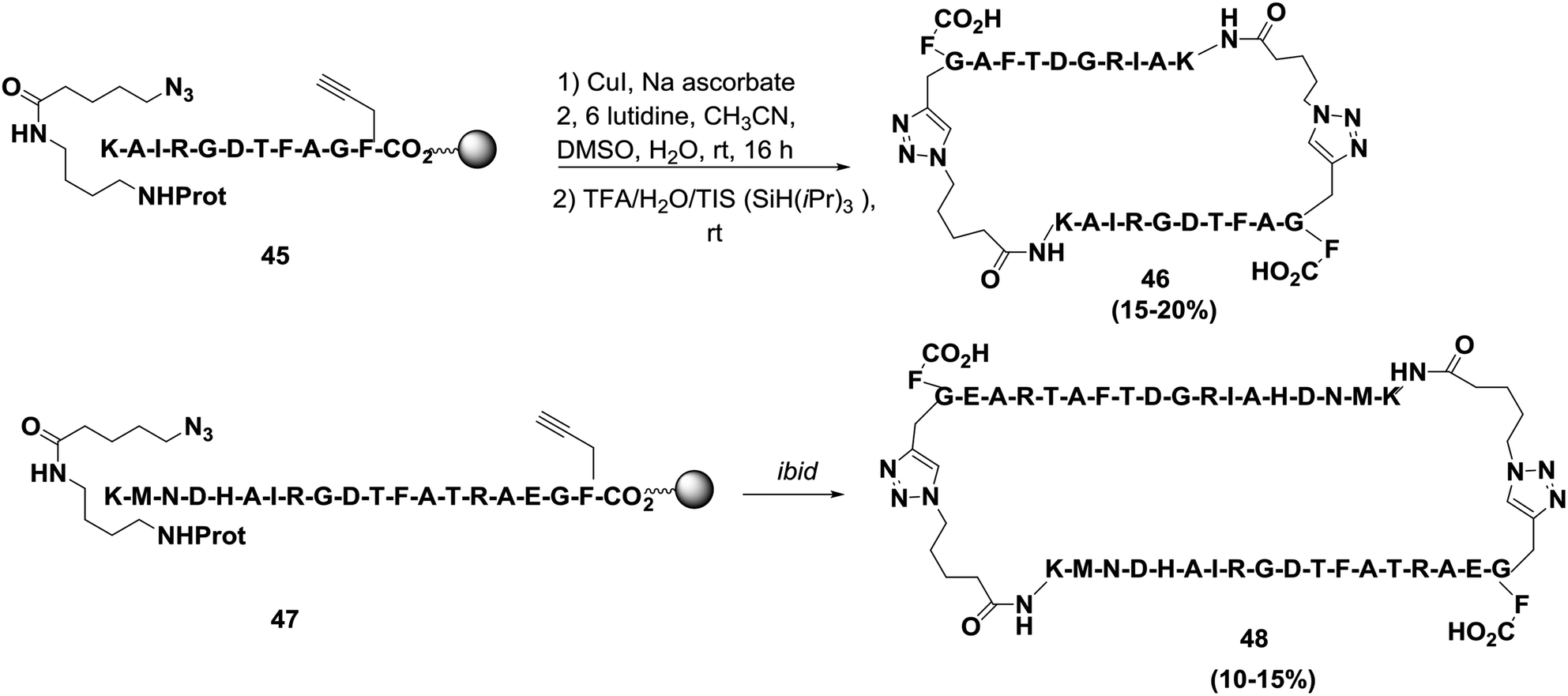 | ||
| Scheme 7 Preparation of cyclodimeric peptides using click chemistry. | ||
In another example of the development of tyrosine kinase-dependent signal transduction inhibitors, Burke's group synthesized triazole-containing macrocycles based on the Grb2-SH2 domain binding motif, as shown in Scheme 8.70 When the cycloaddition reaction was carried out at 2 mM substrate concentrations, the predominant products were dimeric macrocycles 50 (26% yield for 50a, 56% yield for 50b), and monomeric 52 was obtained as a minor product. Repeating the reaction with 1 mM substrate concentration reversed the product ratios and provided monomeric 52 (29% yield for 52a and 19% yield for 52b) as the predominant products, with dimeric 50 appearing as a minor product; thus, the dimerization reaction is highly concentration dependent. In Grb2-SH2 domain-binding assays, the corresponding dimeric macrocycle 51a was found to have greater than 50-fold higher affinity.
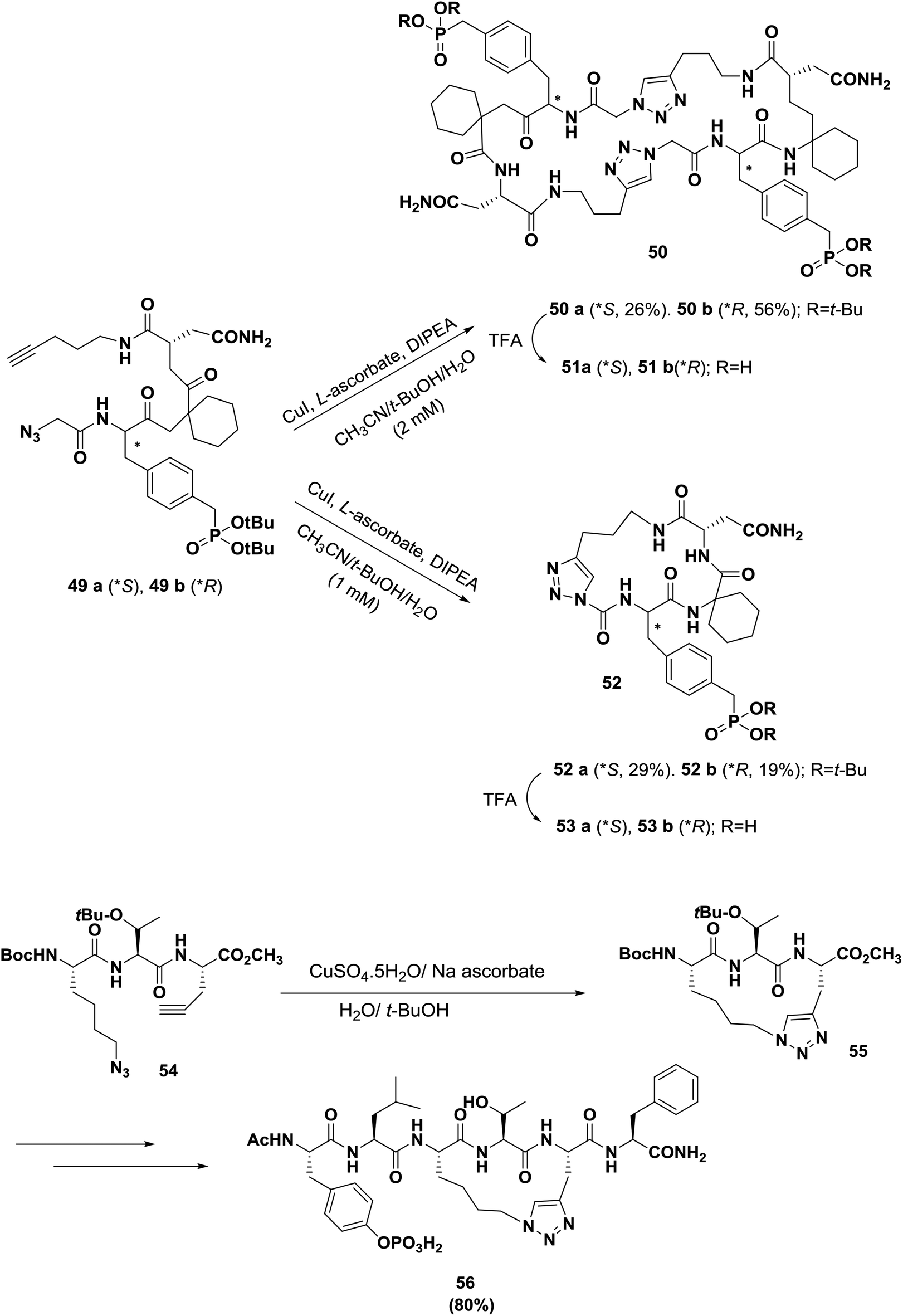 | ||
| Scheme 8 Application of click chemistry to construct triazole bridges in peptides. | ||
Wang's group also reported the design and synthesis of conformational constrained macrocyclic peptidomimetic 56 via click chemistry as an inhibitor of STAT3, as shown in Scheme 8.63,71 Compound 54 was converted to the key intermediate 55 in 80% yield in the presence of CuSO4·5H2O/sodium ascorbate. Macrocyclic compound 56 binding to STAT3 is a promising initial lead compound for further optimization for the design and development of potent small-molecule inhibitors of STAT3 as a new class of anticancer drugs.
Chorev and co-workers developed 1,2,3- triazole-containing cyclic peptides through solid-phase peptide side-chain-to-side-chain synthesis by assembling the azido and alkynyl side-chains within the peptide and cyclizing the resin-bound peptide through CuI-catalyzed azide-alkyne 1,3-dipolar cycloaddition.72 Two different approaches were used to insert the L-2-amino-6-azidohexanoic acid residue [Nle(ε-N3)] into the resin-bound peptide. In pathway A, the Nle (ε-N3) residue was produced via a diazo-transfer reaction on the selectively deprotected ε-amino moiety of the fully protected resin-bound peptide; meanwhile, in pathway B, Fmoc-Nle (ε-N3)–OH was incorporated as part of the standard stepwise on-resin peptide assembly strategy (Scheme 9). The assembly of the resin-bound peptides 58 and 59 by either DEPBT/DIEA or TBTU/HOBt/NMM-mediated coupling was accomplished in a straightforward manner by both pathways A and B. Deprotection of all side-chain protecting groups and simultaneous cleavage from the resin surface yielded the crude linear peptide 61. Complete conversion of the linear peptide 61 to the desired cyclic 1,2,3-triazolyl-containing peptide 62 was achieved after ON incubation at r.t. in t-BuOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v) in the presence of a 4.4-fold molar excess of CuSO4·5H2O and ascorbic acid.72 It is worth noting that all attempts to carry out on-resin intra-chain CuI-catalyzed side-chain-to-side-chain azide-alkyne 1,3-dipolar cycloaddition with different reported approaches failed.
:2 v/v) in the presence of a 4.4-fold molar excess of CuSO4·5H2O and ascorbic acid.72 It is worth noting that all attempts to carry out on-resin intra-chain CuI-catalyzed side-chain-to-side-chain azide-alkyne 1,3-dipolar cycloaddition with different reported approaches failed.
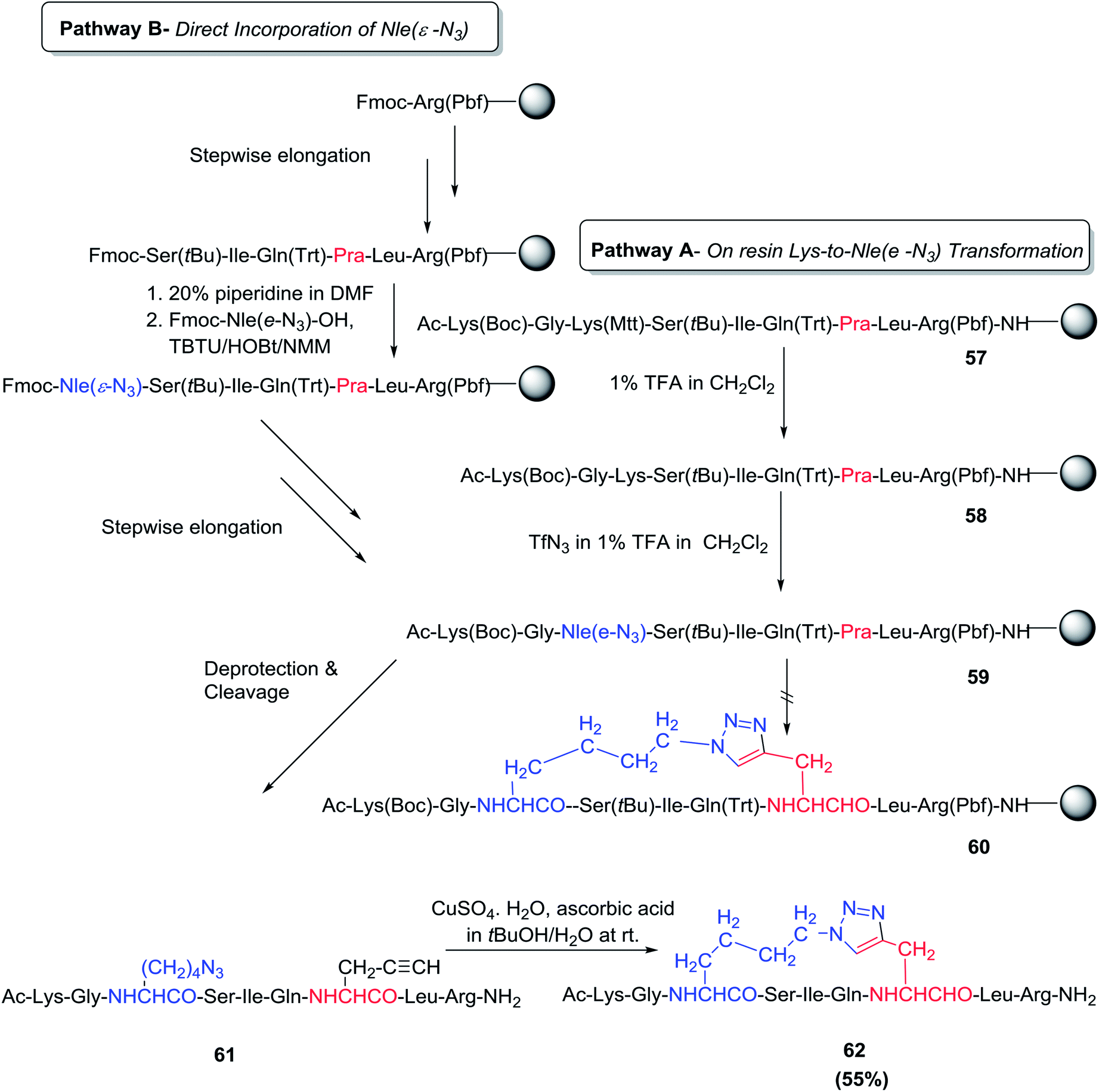 | ||
| Scheme 9 Strategies employed in the solid-phase synthesis of NαAc-[Xaa13(&1), Yaa17(&2)] hPTHrP (11–19)NH2]-[(&1(CH2)4-1,4-[1,2,3]triazolyl-CH2&2)]a. The & sign indicates the positions of the side-chain-to-side-chain cyclization.a Pathway A: stepwise assembly of the fully protected resin-bound peptide in which the ε-NH2 groups of the two lysine residues are differently protected to allow selective deprotection and subsequent diazo-transfer reaction in the on-resin transformation of Lys13 into Nle (ε-N3). Pathway B: stepwise on-resin assembly of the fully protected peptide 59 incorporating Fmoc-Nle (ε-N3)–OH as a building block. Cleavage and deprotection of 59 obtained by either pathway generated the linear precursor 61, which was then cyclized by CuI-catalyzed azide-alkyne 1,3-dipolar cycloaddition to yield the cyclic (1,2,3) triazolyl-containing peptide 62. | ||
A cyclopeptide-containing triazole moiety was reported by Balalaie and co-workers as a selective anti-lung cancer compound (A549, PC3, and C26 cells). To access this cyclopeptide, a heptapeptide was synthesized and was later used in a sequential Ugi/Huisgen 1,3-dipolar cyclization reaction.18 The detailed synthesis is shown in Schemes 10 and 11. This study clearly shows the importance of the triazole skeleton in the biological activities of the peptides. It may be possible to overcome the difficulties involved in synthesizing complex peptides by employing this elegant chemistry.
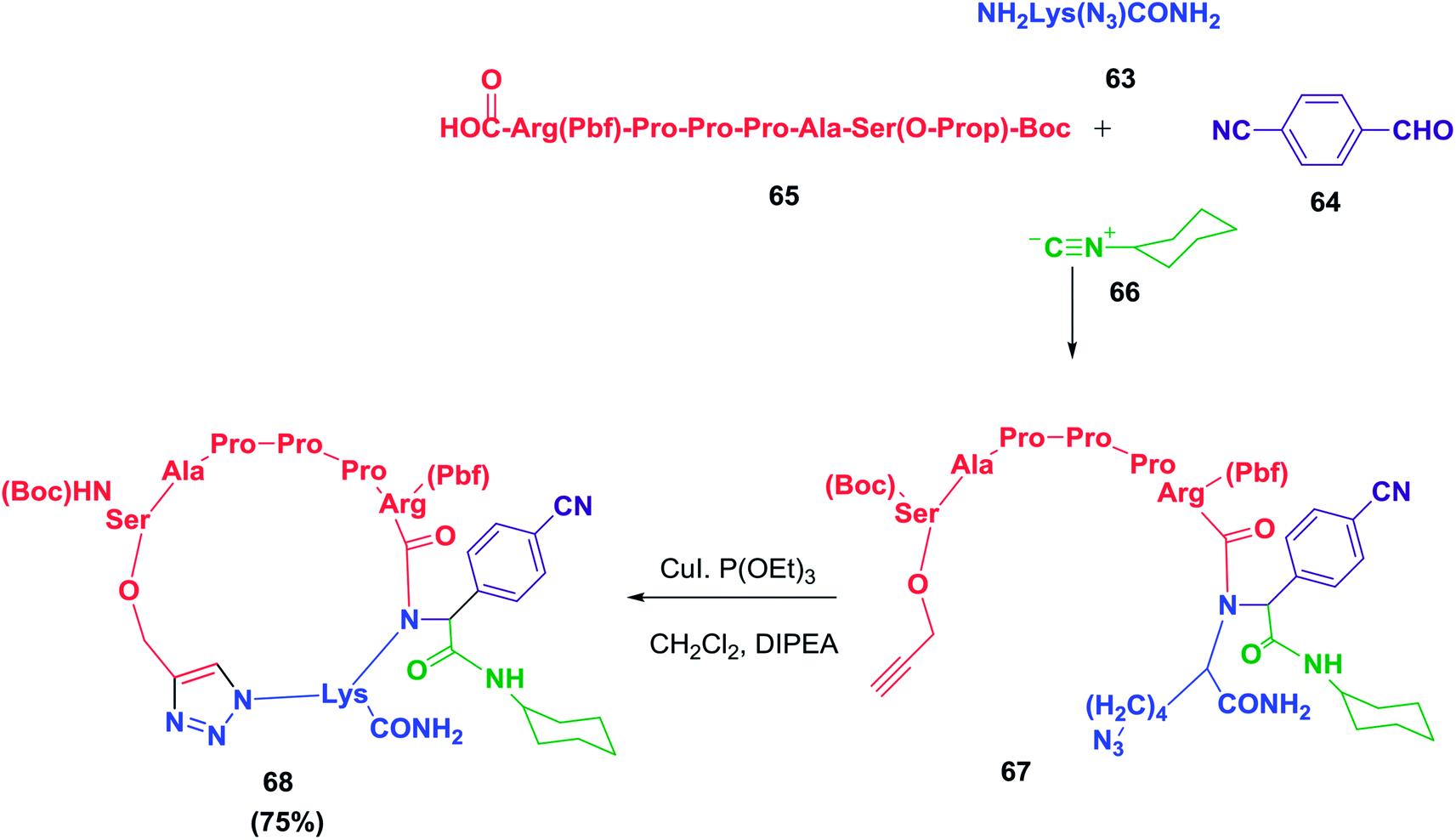 | ||
| Scheme 10 Sequential Ugi ligation/Huisgen 1,3-dipolar reactions to construct cyclopeptides containing triazole moieties. | ||
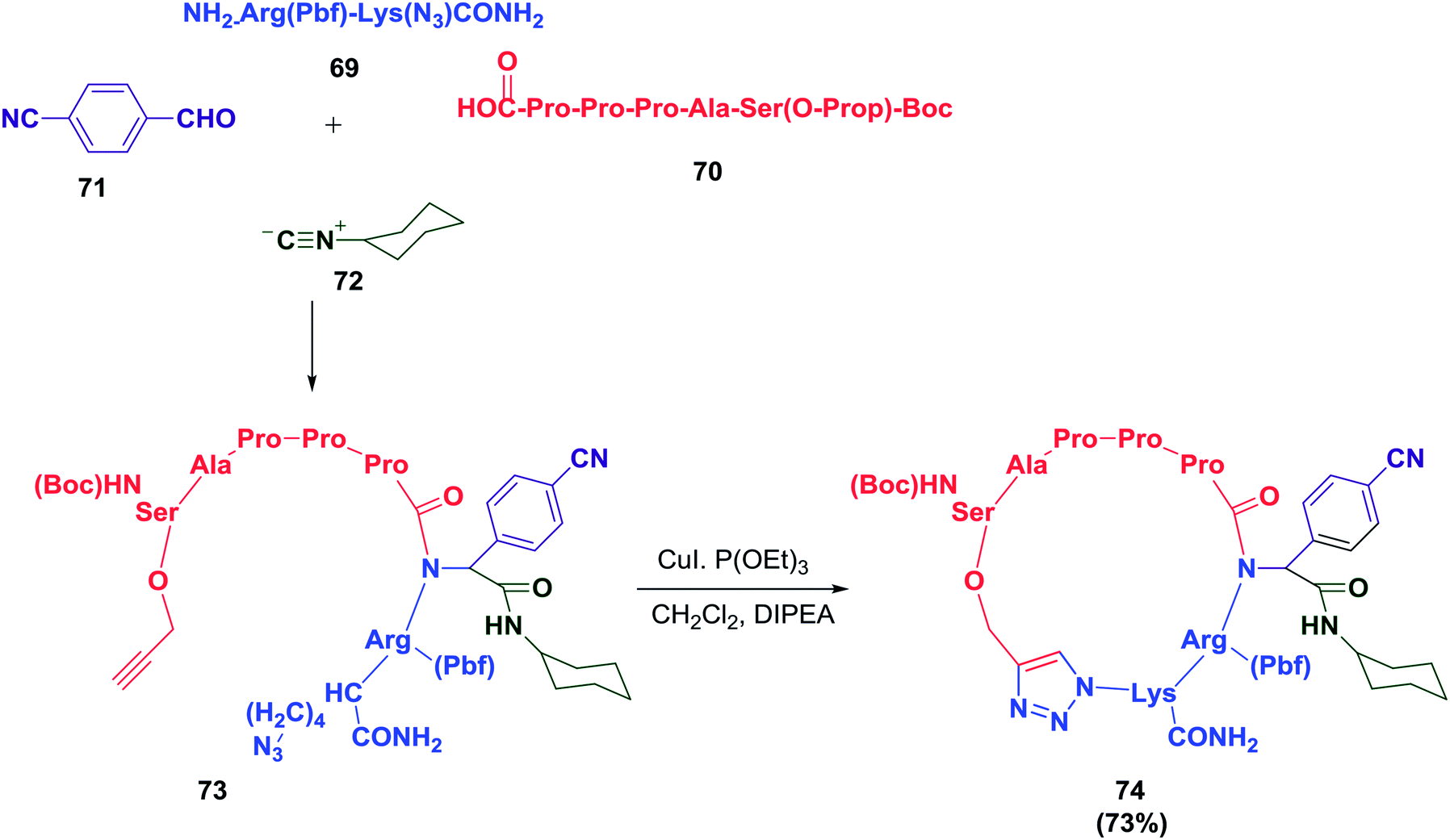 | ||
| Scheme 11 Sequential Ugi ligation/Huisgen 1,3-dipolar reactions to construct cyclopeptides containing triazole moieties. | ||
In addition to the above applications, click chemistry has been demonstrated to be a powerful tool in biomedical research, ranging from combinatorial chemistry and target-template in situ chemistry for lead discovery to providing an alternative approach for facile cyclization when regular approaches do not work. Moreover, this methodology will provide an effective strategy for the design and synthesis of peptide-based drugs with high bioavailability, metabolic stability, functional specificity, and potent activity.
3. Cyclopeptides with imidazole moieties
Imidazoles are well-known heterocyclic compounds that have important features in a variety of medicinal agents. Based on various literature studies, imidazole derivatives show different pharmacological activities, such as antibacterial, anticancer, analgesic, anti-inflammatory, cardiovascular, antineoplastic, anti-fungal, enzyme inhibition, anti-anthelmintic, anti-filarial, anti- viral, anti-HIV and anti-ulcer activities. Moreover, imidazole derivatives are structural isosteres of naturally occurring nucleotides, which permits them to interact simply with biopolymers of living systems; this accounts for their varied biological activities and functions. In addition to their pharmacological actions, they can also be used as dyestuff catalysts and polymerizing agents.73The imidazole moiety, which is present in the side chain of histidine, performs major roles in the biological functions of many peptides and proteins. Imidazoles, for example, are well known as ligands in many metalloenzymes (e.g. metalloproteases);74–76 also, due to their basicity, they have been proven to be main structural elements in the basic active sites of enzymes.77,78 However, cyclopeptides containing imidazoles have not yet been isolated from nature because the occurrence of the diaminopropanoic acid (Dap), which is an analogue of serine, is rare in natural sources.79,80
Haberhauer and co-workers described the synthesis of peptides 77–79 (Fig. 7) with imidazole units in their backbones; they resemble the naturally occurring marine cyclopeptides westiellamide 75 and ascidiacyclamide 76. In addition, they also investigated the structural modifications caused by the introduction of these imidazole moieties.81
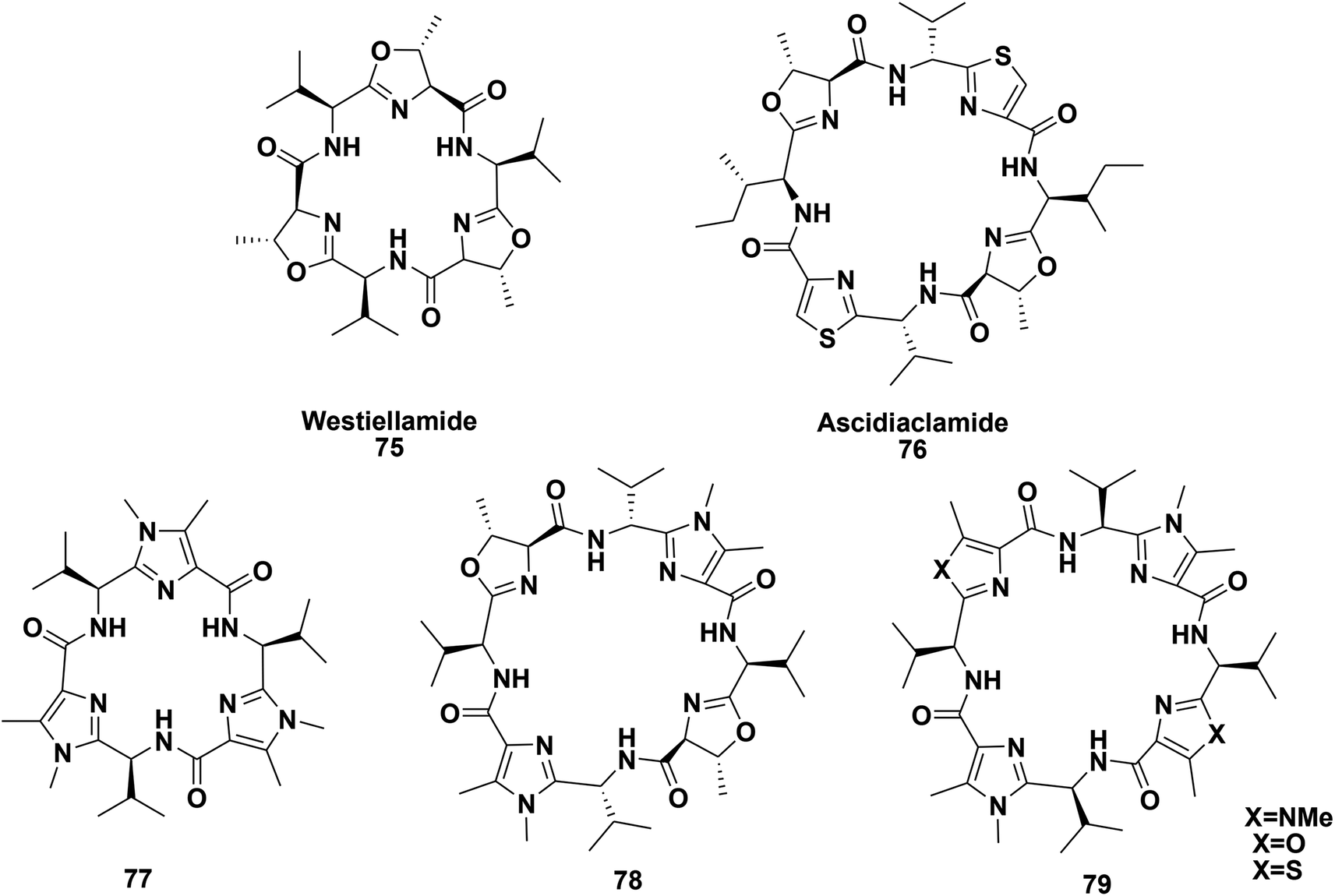 | ||
| Fig. 7 Lissoclinum cyclopeptides and their imidazole analogues. | ||
In continuation of their work, Haberhauer and co-workers also reported the synthesis of the cyclic peptides 86a–c and 87a–c based on the dipeptidyl imidazoles 83a–c (Scheme 12). Several approaches for one-pot macrocyclization of imidazoles 85a–c were tested. The most efficient route was to react the monomers (85a–c) with diphenyl phosphorazidate (DPPA) in the presence of an excess of Hünig's base in acetonitrile under highly diluted conditions (0.05 M) at room temperature. This method provided two products; the trimers 86a–c were obtained in relatively good yields (25% to 35%), and the tetrameric compounds 87a and 87b were obtained in lower yields (5% to 10%), whereas the yield of tetramer 87c was very low and it could not be isolated.81
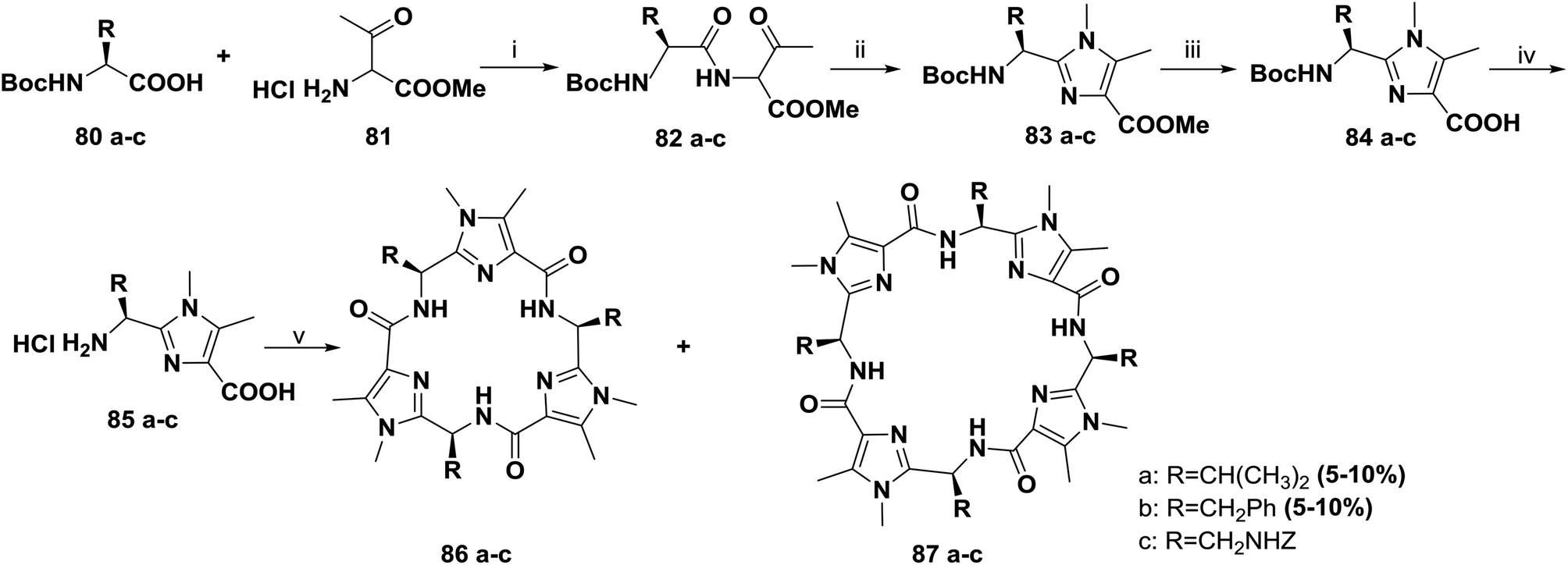 | ||
| Scheme 12 Synthesis of cyclopeptides 86a–c and 87a–c containing imidazole moieties. Reagents and conditions: (i) isobutyl chloroformate, NMM, THF, −20 °C, 85%; (ii) MeNH2, AcOH, xylenes, reflux, 70%; (iii) 2 M NaOH, MeOH/dioxane, rt, 95%; (iv) HCl/AcOEt, rt, quant.; (v) DPPA, iPr2NEt, CH3CN, rt, 25% to 35% for 86a–c, 5% to 10% for 87a, 87b. | ||
To improve the yield and the purity of the abovementioned reaction, Haberhauer and co-workers also developed an alternative synthetic route.82,83 The macrodimerization of the linear dimer 91 was carried out by pentafluorophenyl diphenylphosphinate (FDPP) activation and the addition of Hünig's base in acetonitrile; this afforded the tetramer 87a in 40% yield (Scheme 13).
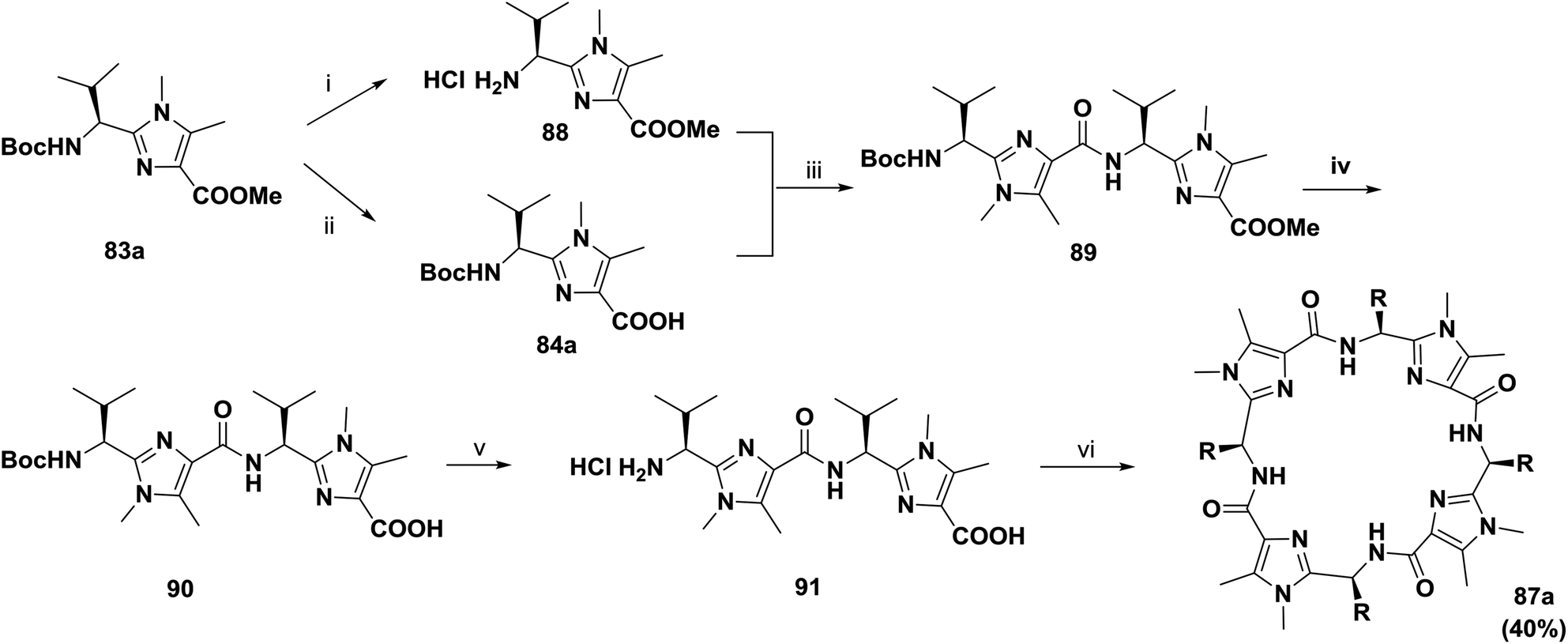 | ||
| Scheme 13 Synthesis of cyclopeptide 87a containing imidazole moieties. Reagents and conditions: (i) HCl/AcOEt, rt, quant.; (ii) 2 M NaOH, MeOH/dioxane, rt, 95%; (iii) DPPA, iPr2NEt, CH3CN, rt, 70%; (iv) 2 M NaOH, MeOH/dioxane, rt, 95%; (v) HCl/AcOEt, rt, quant.; (vi) FDPP, iPr2NEt, CH3CN, rt, 40%. | ||
Due to their synthetic challenges, macrocycles are not highly present in screening libraries.84,85 Meanwhile, many reports investigated the importance of the imidazole-4,5-dicarboxylic acid scaffold in drug discovery.86–89 To extend the library of peptide-macrocycles that contain imidazole-4,5-dicarboxylic acid, Baures and co-workers designed and synthesized a unique novel compound class inspired by macrocyclic natural products. The synthesized compounds are 14-membered macrocycles with differences in their stereochemistry and amino acid side chains. The synthesis started with commercially available N,N′-dialkylalkanediamines, 92, as shown in Scheme 14. To afford compound 93, compound 92 was treated with benzyloxycarbonyl chloride using an excess of the diamine. However, this method did not work well for 92 and resulted in significant amounts of deprotected diamine. However, a different synthetic approach was used by employing N-Boc protection first followed by benzyloxycarbonyl chloride protection to afford the orthogonally protected diamine. Finally, macrocyclic ring closure to form 99 was attained by dissolving intermediate I in CH2Cl2 and adding EDC with DMAP90 (Scheme 14).
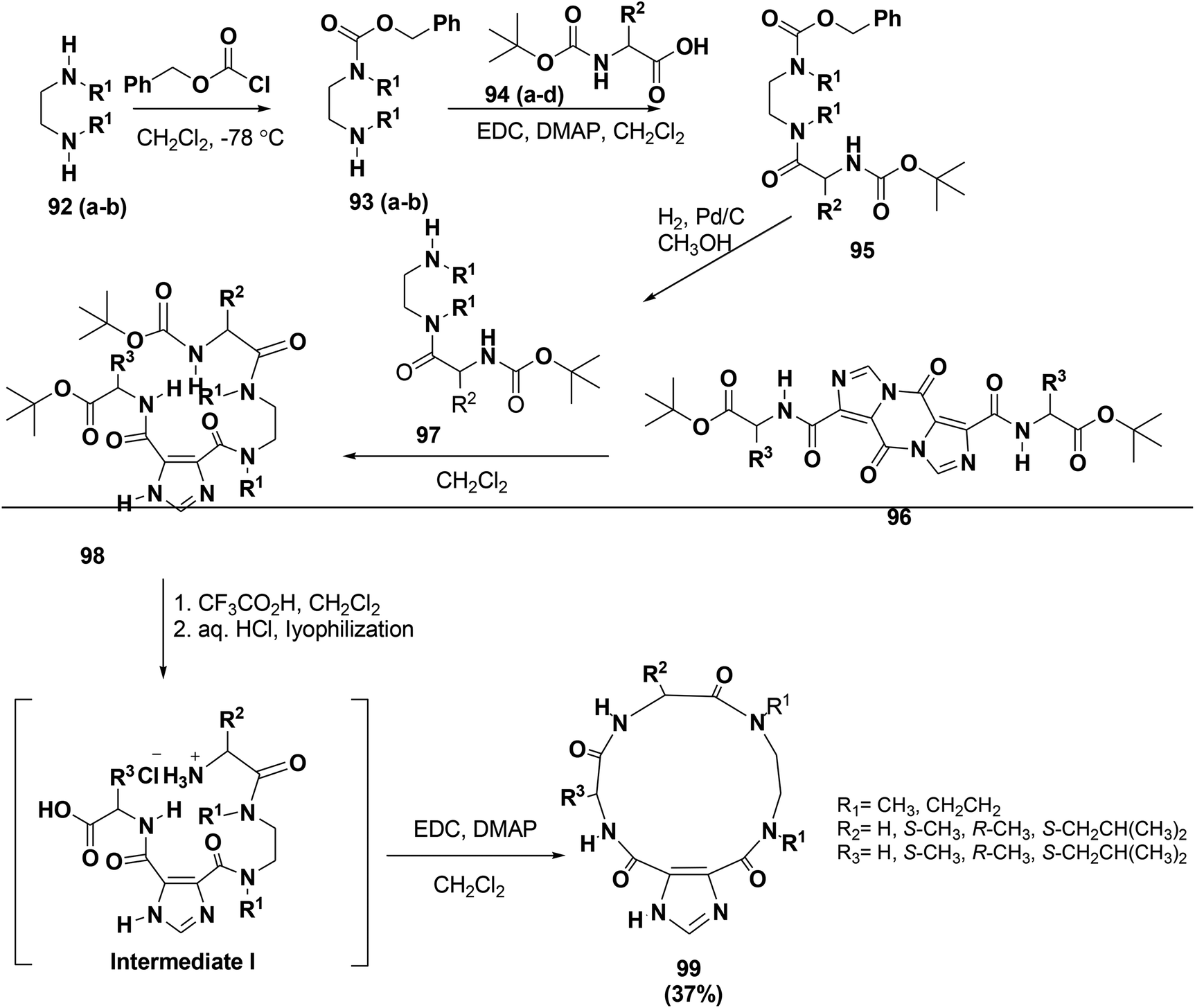 | ||
| Scheme 14 Synthetic strategy for the macrocycle synthesis. | ||
Papini and co-workers reported an efficient approach for the preparation of head-to-tail cyclic peptides on a solid phase which has wide application and is based on the principle of anchoring the peptide to the resin through a side chain.91,92 The reaction of Fmoc/tBu/allyl provided a highly flexible three-dimensional orthogonal compound that allowed the building of more complex peptides, such as cyclic peptides which have side chains that contain post-translational modifications or are conjugated to sugars and oligonucleotides.93,94 One approach to the synthesis of head-to-tail cyclic peptides was incorporating the imidazole ring within histidine residues into a trityl-resin.95 This approach was used to provide an efficient solid-phase Fmoc/tBu/allyl strategy.96 In order to establish this approach, Papini and co-workers synthesized a new building block, Fmoc-His-OAl 100. The trityl-chloride-resin 101a was treated with 100 under stirring. Around 0.4 to 0.6 mmol g−1 of the amino acid was chosen in order to obtain a final level of substitution. Firstly, deprotection at the N-terminal of the linear hexapeptide present on the resin surface was performed, followed by suspension in a solution of DIPEA and TBTU in DMF to allow the formation of the cyclopeptide. After cleavage from the resin with TFA–H2O (95:5) and purification by HPLC, cyclo (-His-Gly)3 103 was obtained97 (Scheme 15).
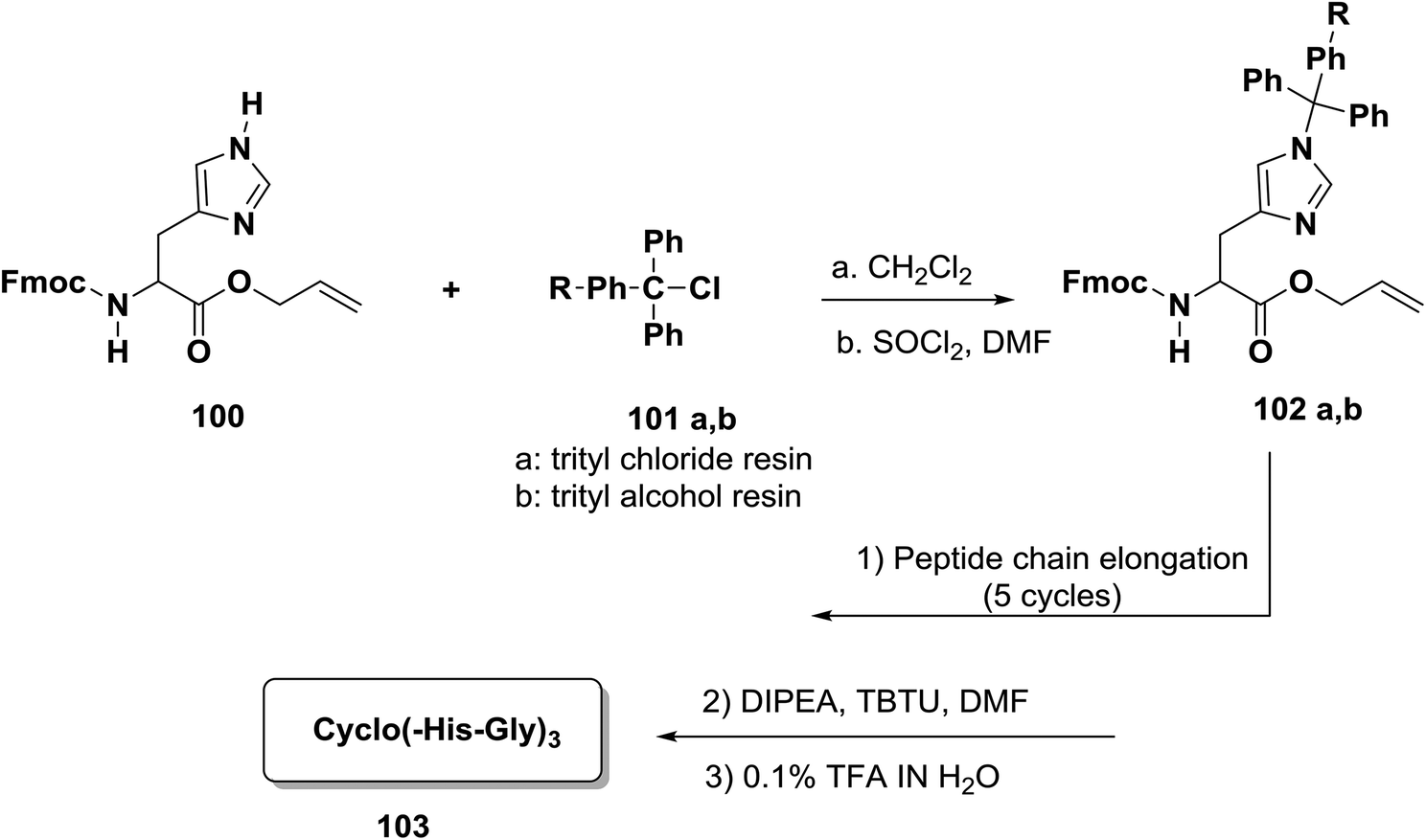 | ||
| Scheme 15 Cyclization of histidine-containing peptides. | ||
In another approach, Haberhauer and co-workers reported the synthesis of a larger receptor, 109, as shown in Scheme 16.98 Imidazole carboxylic acid 104 was coupled using FDPP in CH3CN to commercially available methyl 3-aminobenzoate 105 to afford building block 106. To acquire good yields of the products (41% for 106), the coupling required long reaction times (1 week at room temperature) due to the low nucleophilicity of aniline. Saponification allowed the deprotection of the carboxyl residue and afforded the acid 107. Later, the removal of the Boc group provided the free amino acid 108, which underwent cyclodimerization with FDPP to afford receptor 109 in a good yield of 52%.
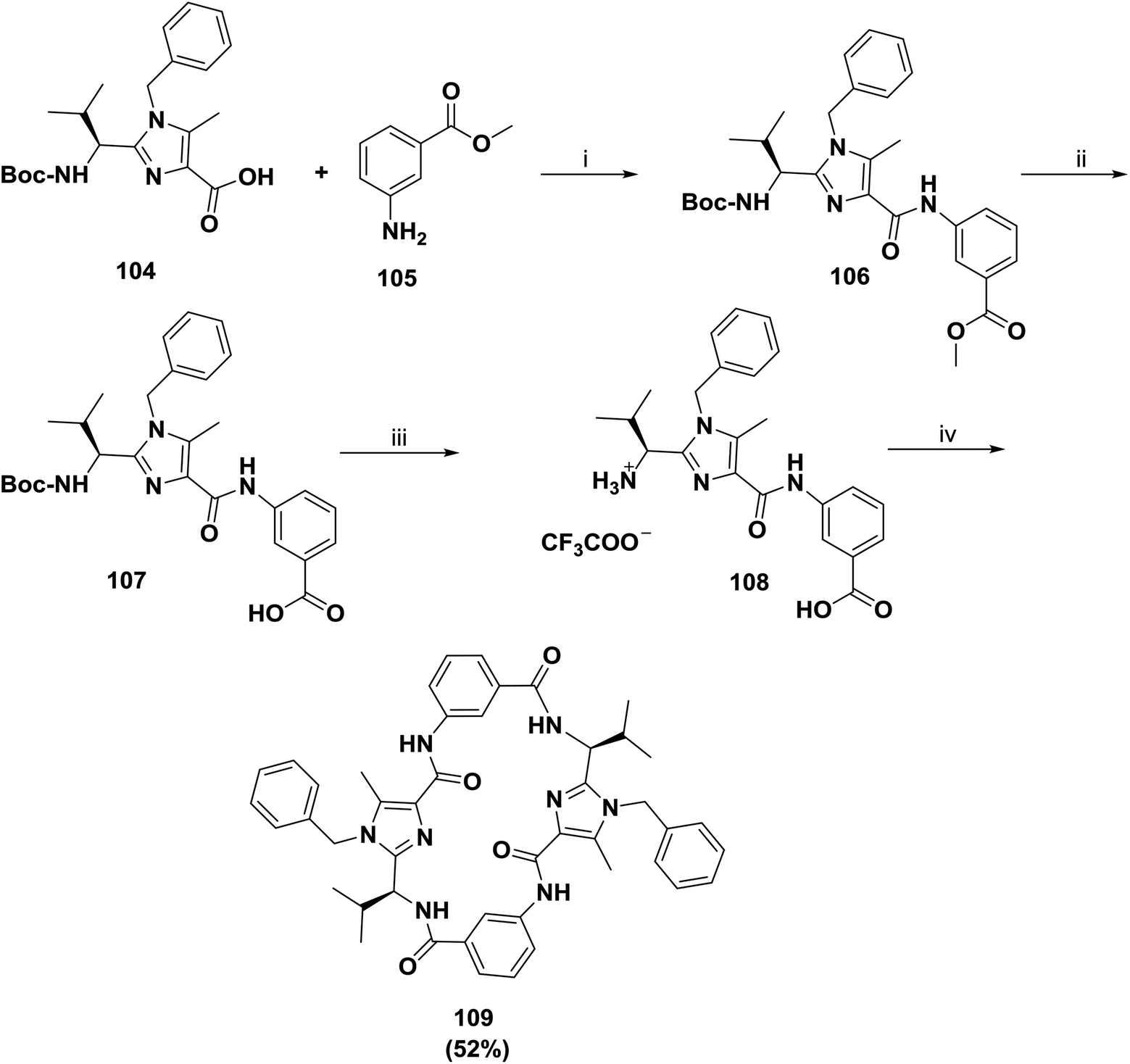 | ||
| Scheme 16 Synthesis of receptor 109. Reagents and conditions: (i) FDPP, iPr2NEt, CH3CN, r.t., 41%; (ii) 2N NaOH, MeOH/dioxane, 0 °C → rt, quant.; (iii) TFA, CH2Cl2, 0 °C → rt, quant.; (iv) FDPP, iPr2NEt, CH3CN, rt, 52%. | ||
Castle's group recently synthesized the indole-imidazole linkage through an oxidative coupling reaction during the synthesis of the antimitotic bicyclic peptide celogentin C, as shown in Scheme 17. The desired product 112 containing the required indole-imidazole linkage was obtained in 58% yield when the dipeptides 110 and 111 were subjected to NCS and 1,4-dimethylpiperazine in CH2Cl2 at room temperature. The resulting product was afterward cyclized to afford the peptidomimetic 113.99
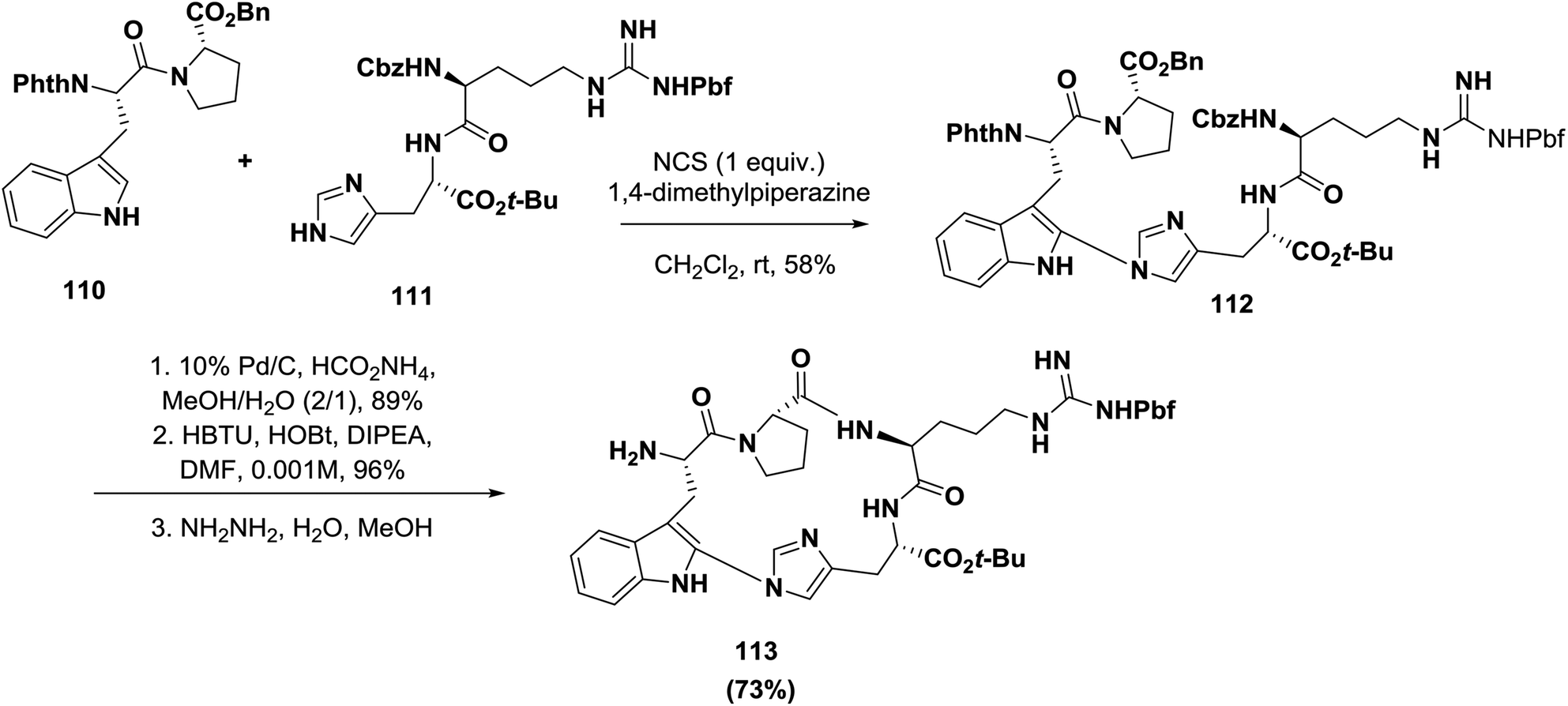 | ||
| Scheme 17 Synthesis of indole-imidazole linkage cyclic peptidomimetics by an oxidative coupling reaction. | ||
4. Cyclopeptides with tetrazole moieties
The synthesis and study of tetrazole compounds did not receive much early attention due to the late emergence of azoles in general. However, since the 1950s, tetrazoles have been widely applied in agriculture, pharmacology, medicine, biochemistry, explosives and other aspects; thus, tetrazole research has developed rapidly.100,101 The tetrazolyl functional group is considered to be a carboxylic acid isostere in drugs because its pKa is similar. In addition, it has almost the same required planar delocalized system space, and among the heterocyclic compounds, it provides the most nitrogen content.102 Due to their extended biological applications and unique structures, tetrazole and its derivatives have thus attracted the interest of scientists.103–106Kaczmarek and co-workers reported the synthesis of analogues of cyclolinopeptide A (CLA) in linear and cyclic forms. However, in their approach, two dipeptide segments (Val5-Pro6 and Pro6-Pro7) were replaced with tetrazole using the solid phase peptide synthesis (SPPS) technique and cyclized using TBTU (O-(benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate) as the coupling reagent. NMR and computational techniques were used to examine the conformational properties of the cyclic peptide containing the tetrazole moiety c(Leu1-Ile2-Ile3-Leu4-Val5-Pro6-Ψ[CN4]-Ala7-Phe8-Phe9).107 The overall solution structure of this cyclic peptide resembled that observed for CLA in the solid state. The synthesized cyclic tetrazole CLA analogue confirmed that the 1,5-disubstituted tetrazole ring functions as an effective, well-tolerated cis-amide bond mimic in solution.
On the other hand, somatostatin108 is a cyclic tetradecapeptide hormone (H-Ala1-Gly2-Cys3-Lys4-Asn5-Phe6-Phe7-Trp8-Lys9-Thr10-Phe11-Thr12-Ser13-Cys14-OH) whose role is to inhibit the release of a variety of other hormones, including gastrin, glucagon, insulin, and somatotropin (growth hormone).109 Moreover, analogues of somatostatin present promise in the treatment of hypersecretory disorders,110 as antineoplastic agents,111 and as tumor imaging reagents.112 Marshall and co-workers designed the synthesis of new analogues of somatostatin; in their approach, a 1,5-disubstituted tetrazole that is a cis-amide surrogate or mimic is incorporated into a cyclic hexapeptide analog of somatostatin in order to constrain the cis-amide bond by replacing the Xxx11-Xxx6 amide (Fig. 8). Both chemical and enzymatic methods were employed to accomplish the final cyclization. The product, cyclo (Ala6-Tyr7-D-Trp8-Lys9-Val9-Phe10-Ψ[CN]4), was found to have 83% of the activity of somatostatin.113
 | ||
| Fig. 8 Left: dipeptide with a cis amide bond. Right: Dipeptide with a cis amide bond replaced by the tetrazole surrogate (Ψ[CN4]). In this study, R′ = benzyl and R′′ = methyl. | ||
5. Cyclopeptides with oxazole/thiazole moieties
Oxazoles can be categorized as biologically active heterocyclic backbones that are found in extensive natural products and pharmaceuticals and are used as synthetic intermediates for further transformations. Oxazoles are synthesized in nature from serine and threonine. Oxazole derivatives have drawn considerable attention in medicinal research because they have extensive biological activities, such as hypoglycemic, anti-inflammatory, and antibacterial activities.114–116Meanwhile, thiazoles are essential scaffolds that exist in many natural sources, such as vitamin B1, thiamine, and other synthetic medicinally important compounds. The application of thiazoles in many drugs, such as penicillin, antimicrobials (sulfazole), antiretrovirals (ritonavir), antifungals (abafungin), and their antihistaminic and antithyroid activities demonstrate the importance of these heterocycles. In addition, thiazole derivatives have applications as anticancer drugs (tiazofurin), anthelmintics, vulcanizing accelerators (mercaptobenzothiazole) and photographic sensitizers.117
Since oxazoles and thiazoles were naturally discovered in the backbones of cyclopeptides, confirming that their presence can impose conformational restrictions on cyclic peptides, many studies have been performed on these heterocycles.118,119
Condensation between side chain cysteine thiols or threonine/serine hydroxy groups and neighboring amide bonds affords heterocyclic rings, as illustrated in Scheme 18, resulting in highly constrained pseudo-boat or saddle-shaped macrocycles. Wipf reviewed most of the syntheses investigated before 1995 33, and subsequent research was reviewed by Shioiri.120 Albericio and co-workers used both solid phase and solution phase approaches to synthesize trunkamide A 117121 (Scheme 18). In their approach, the linear peptide was constructed on a chlorotrityl resin; proline was the first amino acid linked to the resin, and the final two residues were D-Phe-[CSNH]Ser, which is the precursor of the thiazoline ring. Macrocyclization was carried out with PyAOP/DIEA. Post-cyclization using diethylamino sulfur trifluoride (DAST) afforded the thiazoline ring. Thiolysis of the oxazoline and the second cyclodehydration with DAST afforded 117.121
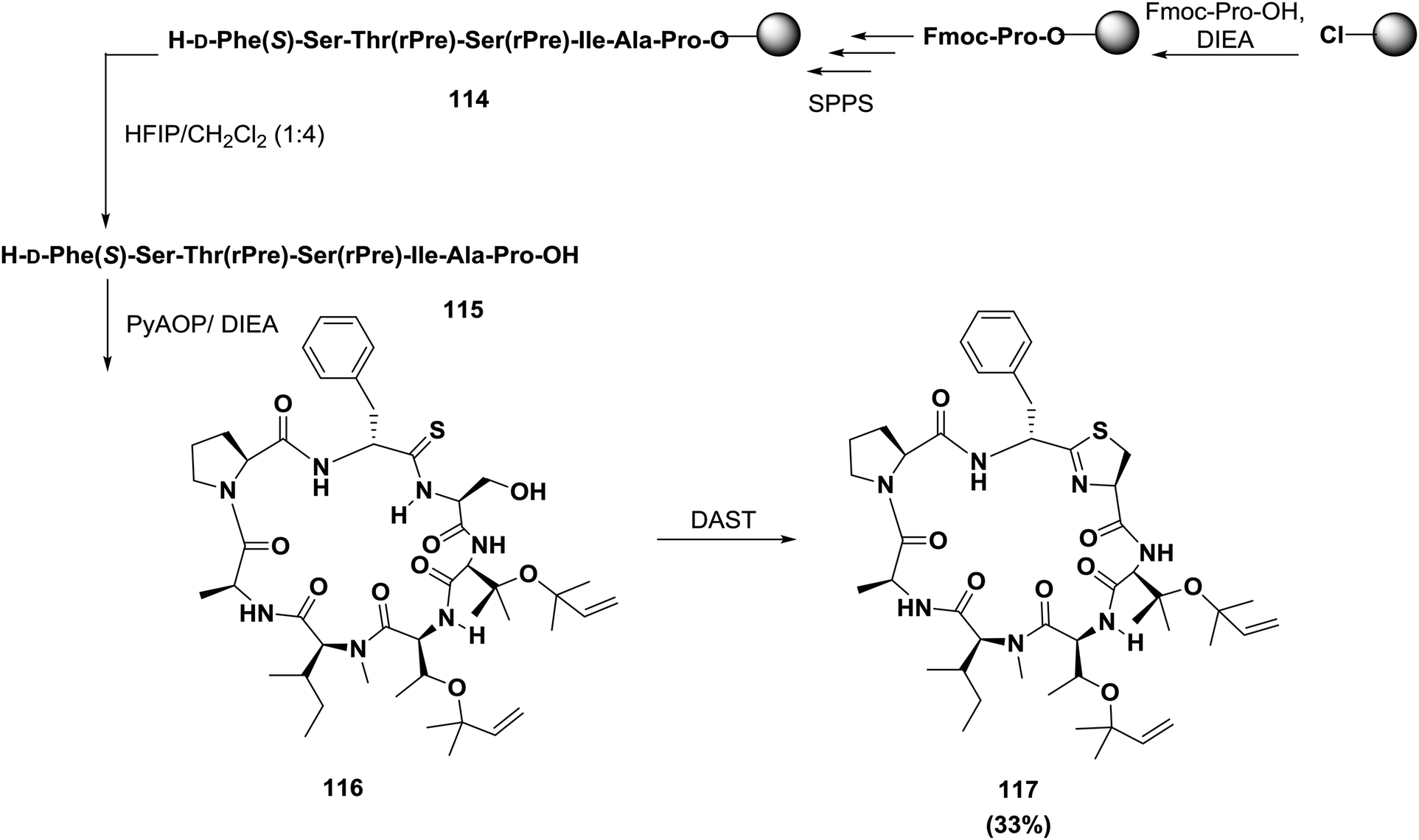 | ||
| Scheme 18 A synthetic strategy for Trunkamide A. | ||
On the other hand, McKeever and Pattenden initially failed to synthesize the cyclopeptide trunkamide A 117 in solution via cyclization of an acyclic thioamide precursor; however, when using a normal amide-Ser bond in the linear peptide and DPPA, they achieved successful cyclization, with the thiazoline ring 117 (35% yield) being formed later by treatment with DAST followed by H2S122 (Scheme 19).
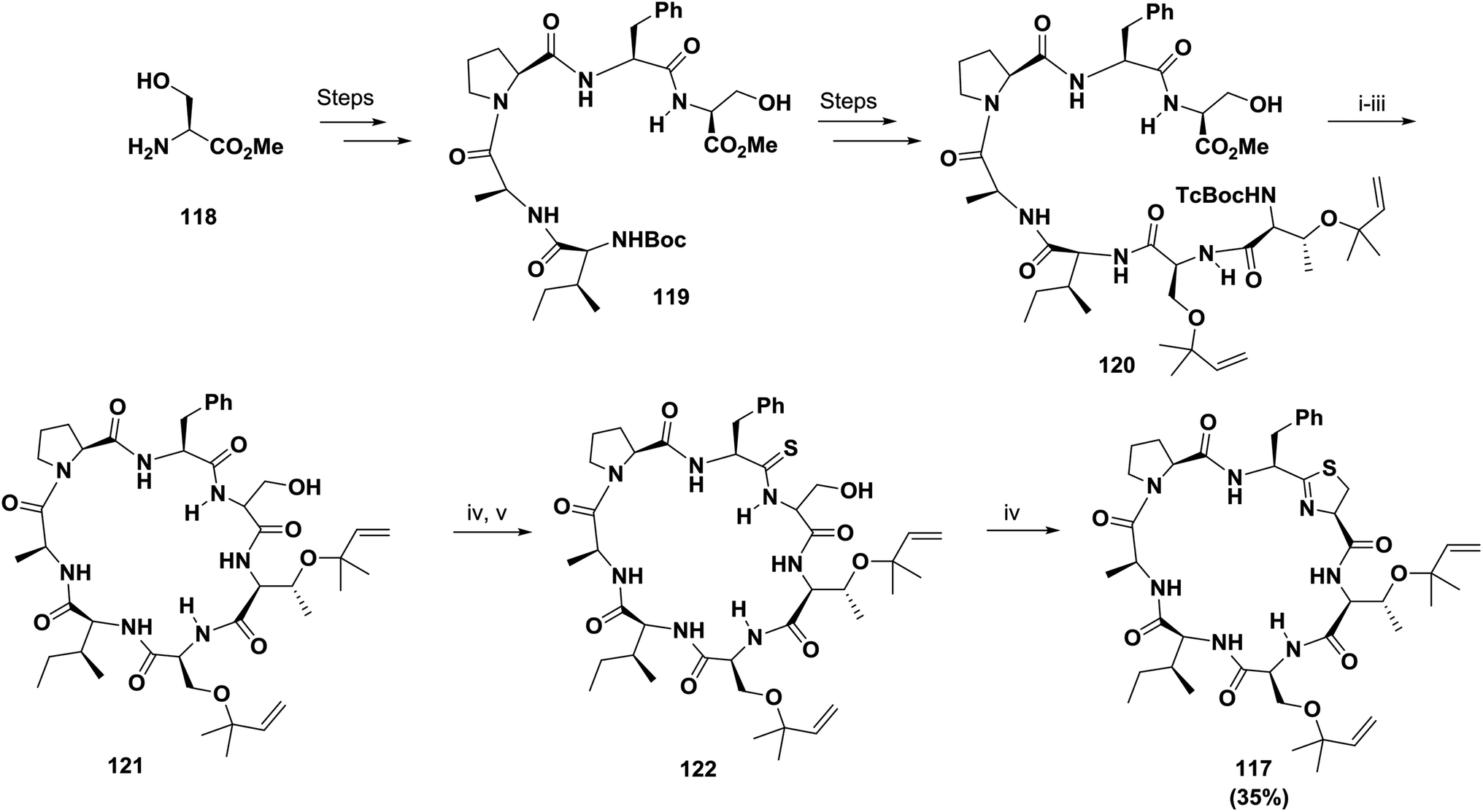 | ||
| Scheme 19 Synthesis of the cyclopeptide Trunkamide A. Reagents: (i) Cd:Pb, 1 M NH4OAc:THF (1:1), 98%; (ii) TBAH, THF, 0 °C; (iii) DPPA, DIPEA, DMF, 35% (two steps); (iv) DAST, CH2Cl2; (v) H2S, Et3N, MeOH. | ||
The synthesis of nostocyclamide 130 was also reported by Moody and co-workers; amino acids containing the thiazole moiety were successfully coupled to form a linear peptide which was further cyclized to 130 by activation using a pentafluorophenyl ester (74% yield)123 (Scheme 20).
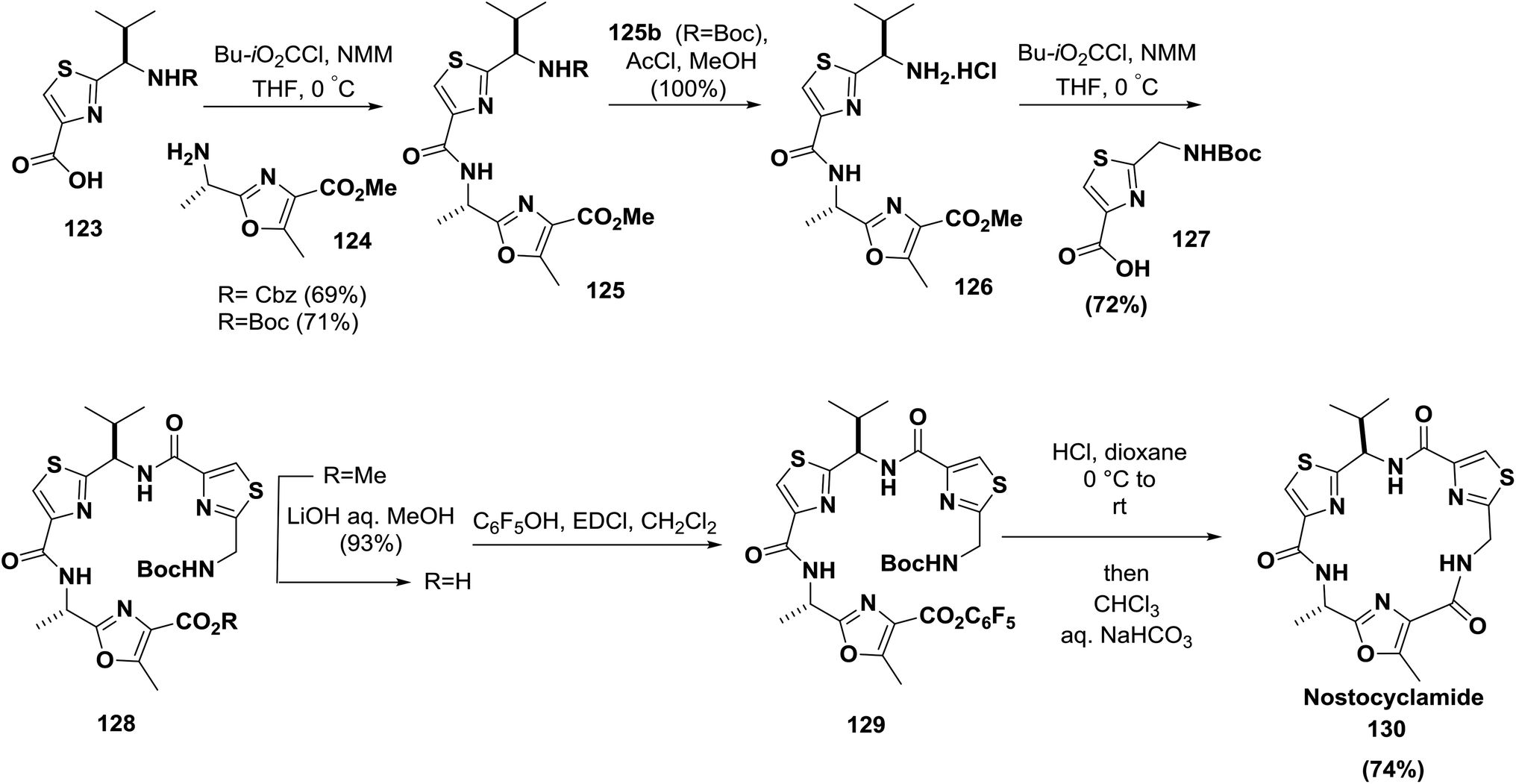 | ||
| Scheme 20 Synthesis of nostocyclamide. | ||
Later, Pattenden and co-workers reported another approach for the synthesis of nostocyclamide; they showed that amino acids containing oxazole and thiazole can undergo cyclization in the presence of FDPP to form nostocyclamide with yields controlled by the amount and type of the metal ions present in the reaction mixture124 (Scheme 21).
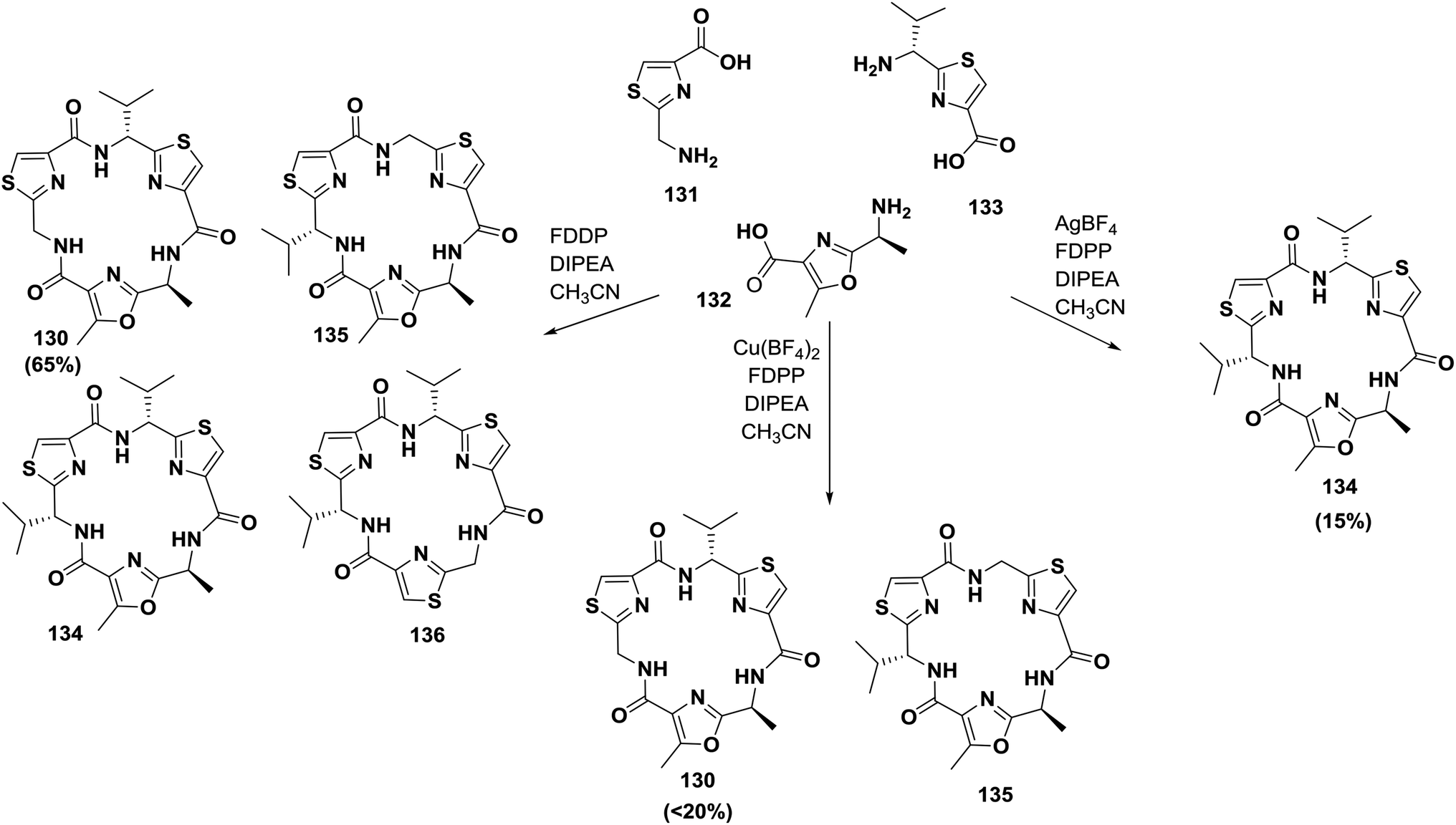 | ||
| Scheme 21 Synthesis of nostocyclamide and related cyclic peptides by metal-templated assembly. | ||
Smith and co-workers synthesized a related natural product, dendroamide A 143. The synthesis was performed using amino acids containing heterocycles. Cyclization of the linear peptide was performed using DPPA and afforded dendroamide A 143 (56% yield)125 (Scheme 22).
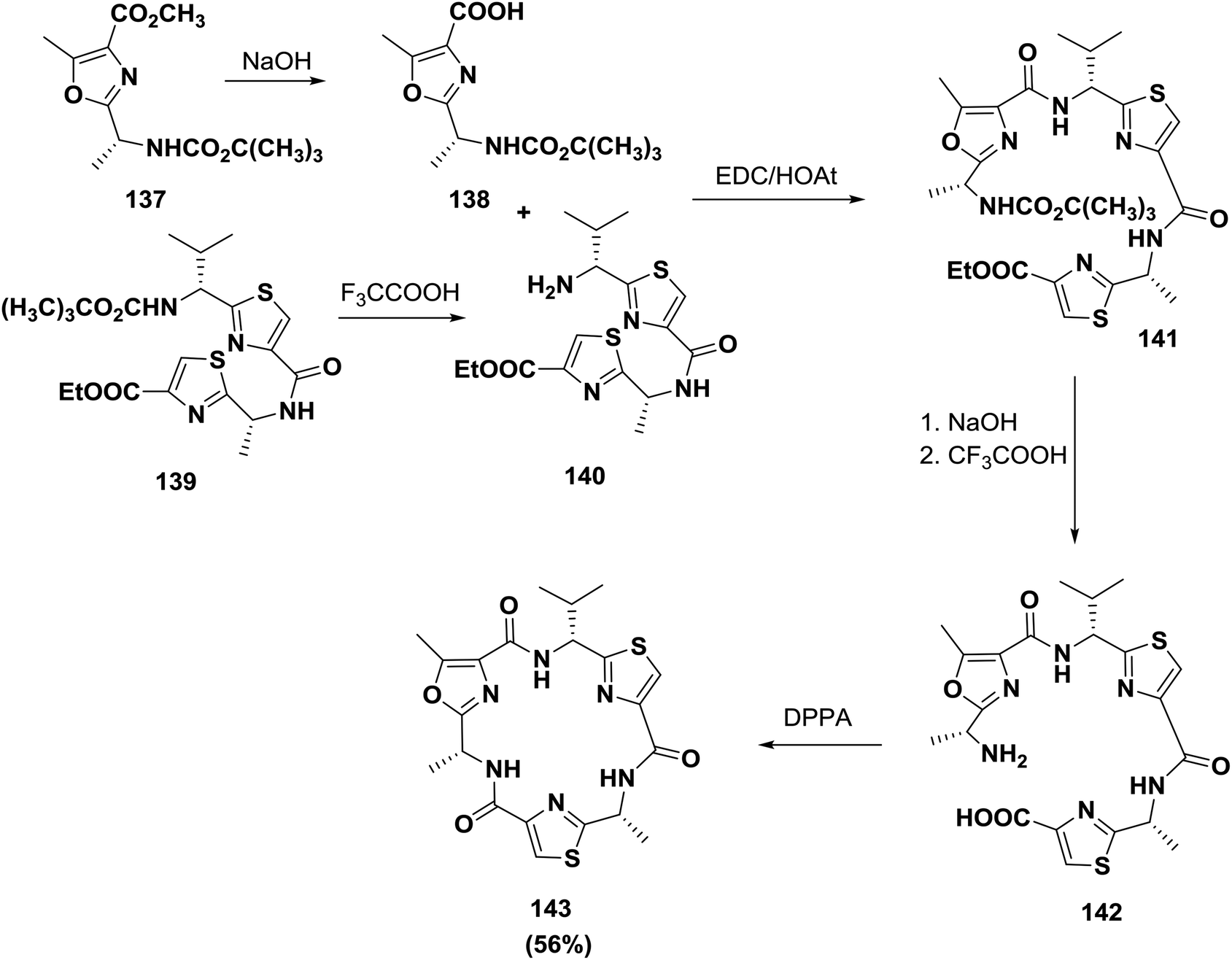 | ||
| Scheme 22 Synthesis of dendroamide A. | ||
Moody and co-workers, using a modified Bohlmann-Rahtz pyridine synthesis, described the total synthesis of the thiopeptide antibiotic promothiocin A 151 to establish the oxazolylthiazole-pyridine heterocycle of the antibiotic. The oxazole building blocks were obtained by a dirhodium(II)-catalyzed chemoselective carbenoid N–H insertion reaction followed by cyclodehydration and a Hantzsch reaction to afford the thiazoles. Two different macrocyclization strategies were successfully employed, and in the last steps of the synthesis, the dehydroalanine side chain of the natural product was introduced126 (Scheme 23).
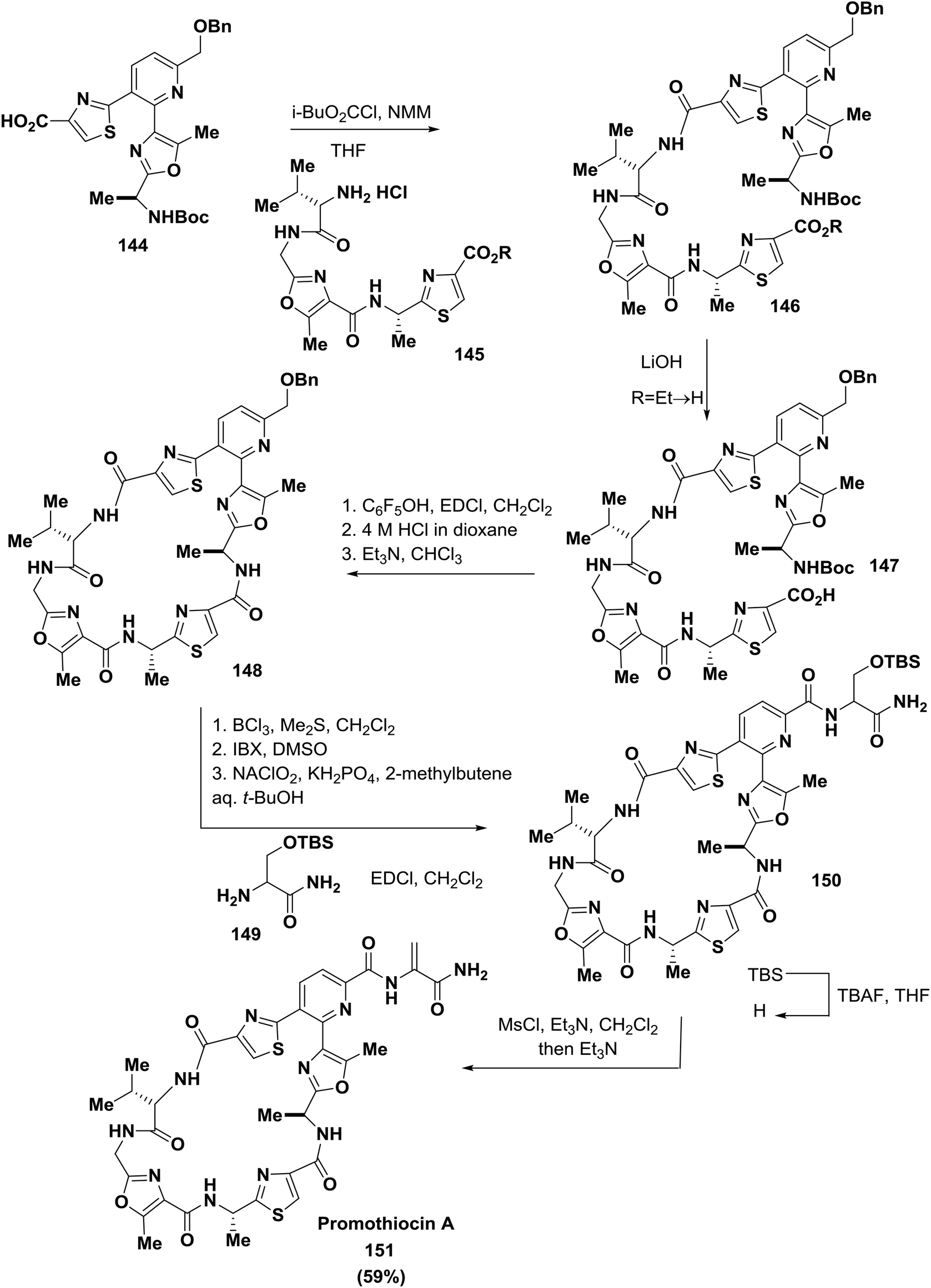 | ||
| Scheme 23 Synthesis of the cyclopeptide promothiocin A. | ||
Pattenden and co-workers also reported a convergent, complete synthetic approach of the directly linked tris-oxazole units in telomestatin 159 and YM-216391 167127 (Schemes 24 and 25).
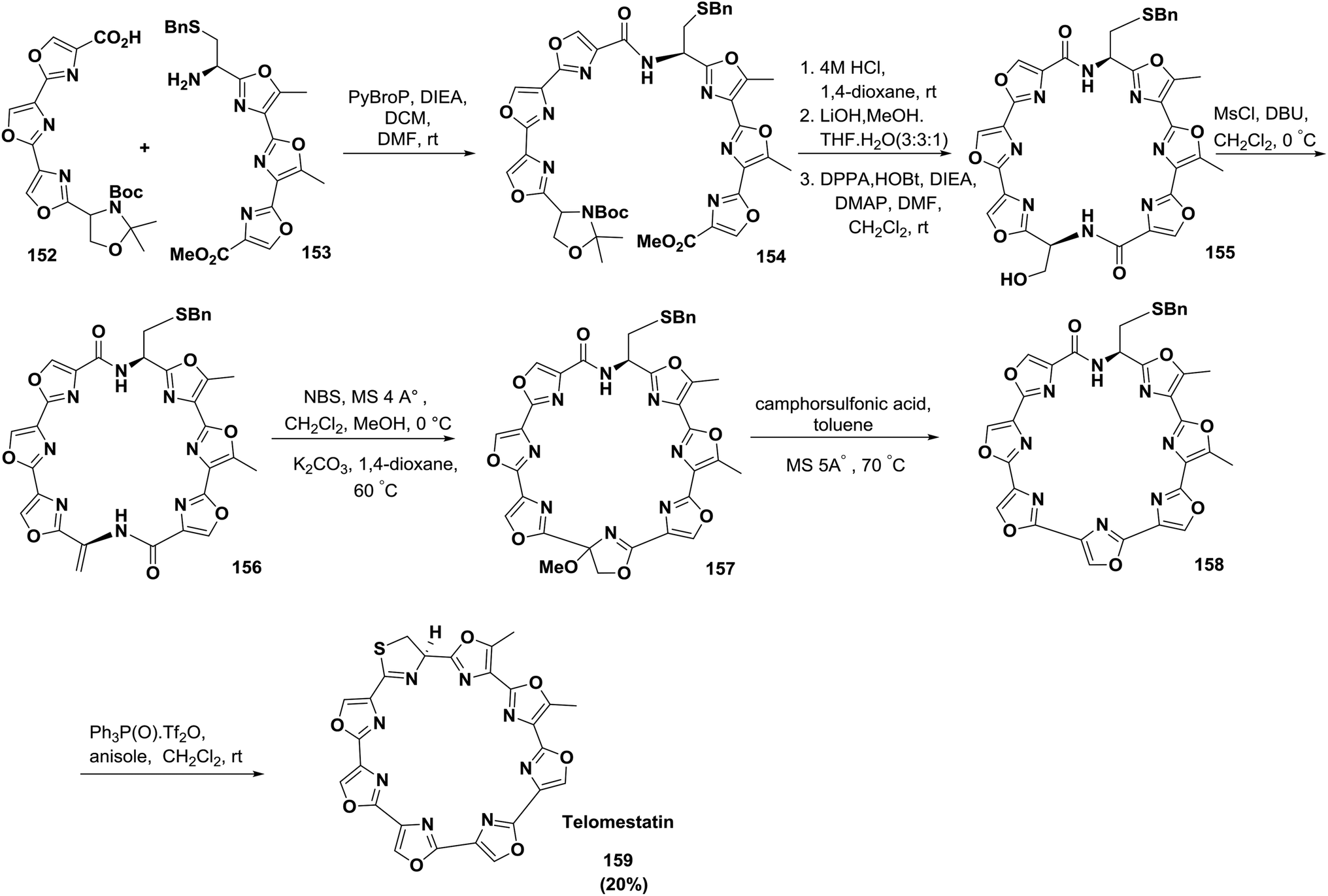 | ||
| Scheme 24 Synthesis of the cyclopeptide telomestatin. | ||
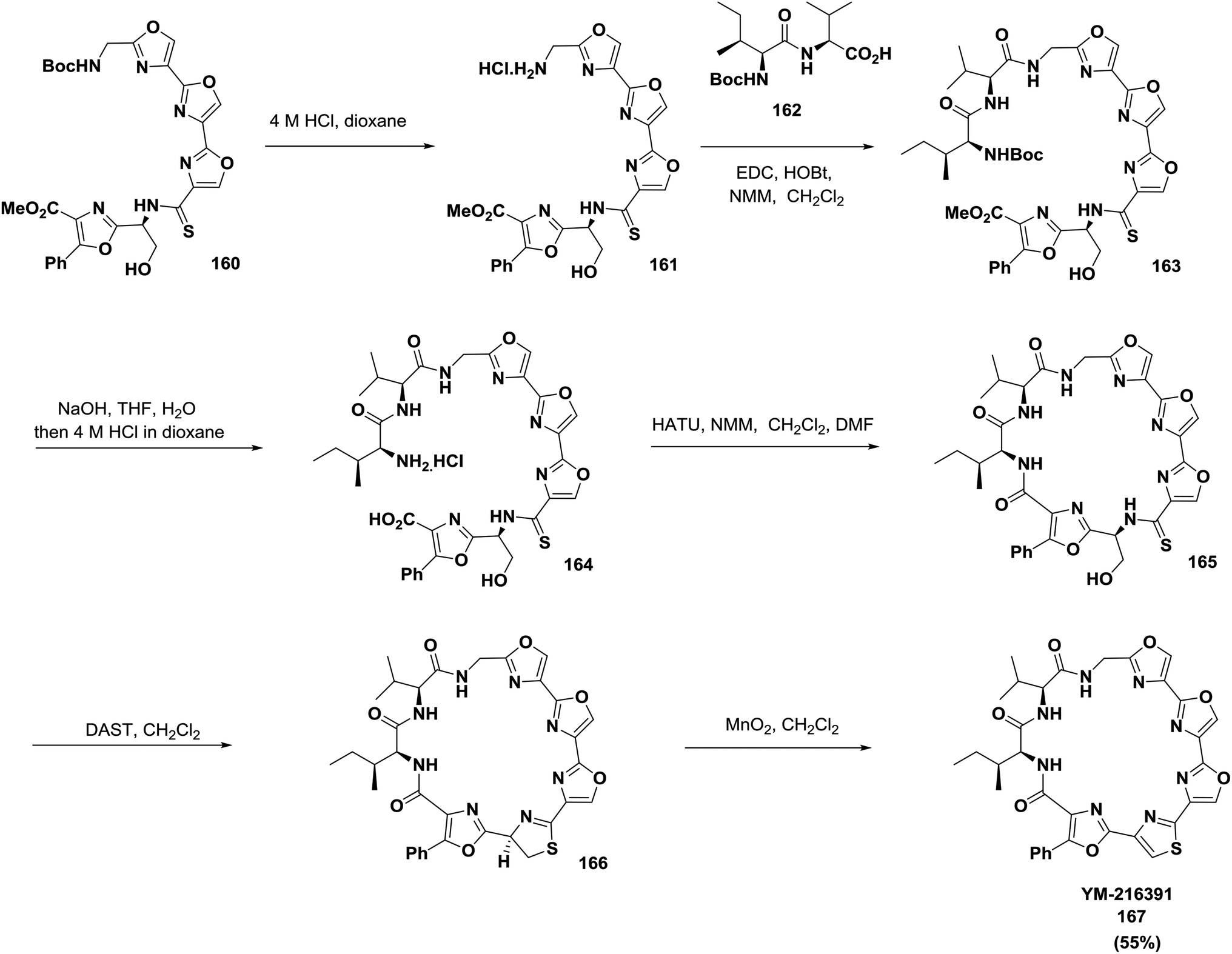 | ||
| Scheme 25 Synthesis of the cyclopeptide YM-216391. | ||
The synthesis of the marine-derived anti-cancer agent diazonamide A 168 is considered to be one of the most demanding synthetic challenges.13,128–131 Nicolaou and co-workers have established a new path to obtain diazonamide A. The approach described a SmI2-induced heteropinacol coupling cascade to assemble the 12-membered heterocyclic core of the target and an exceptional oxidation of an indoline to an oxindole using Pd(OH)2/C 168131 (Scheme 26).
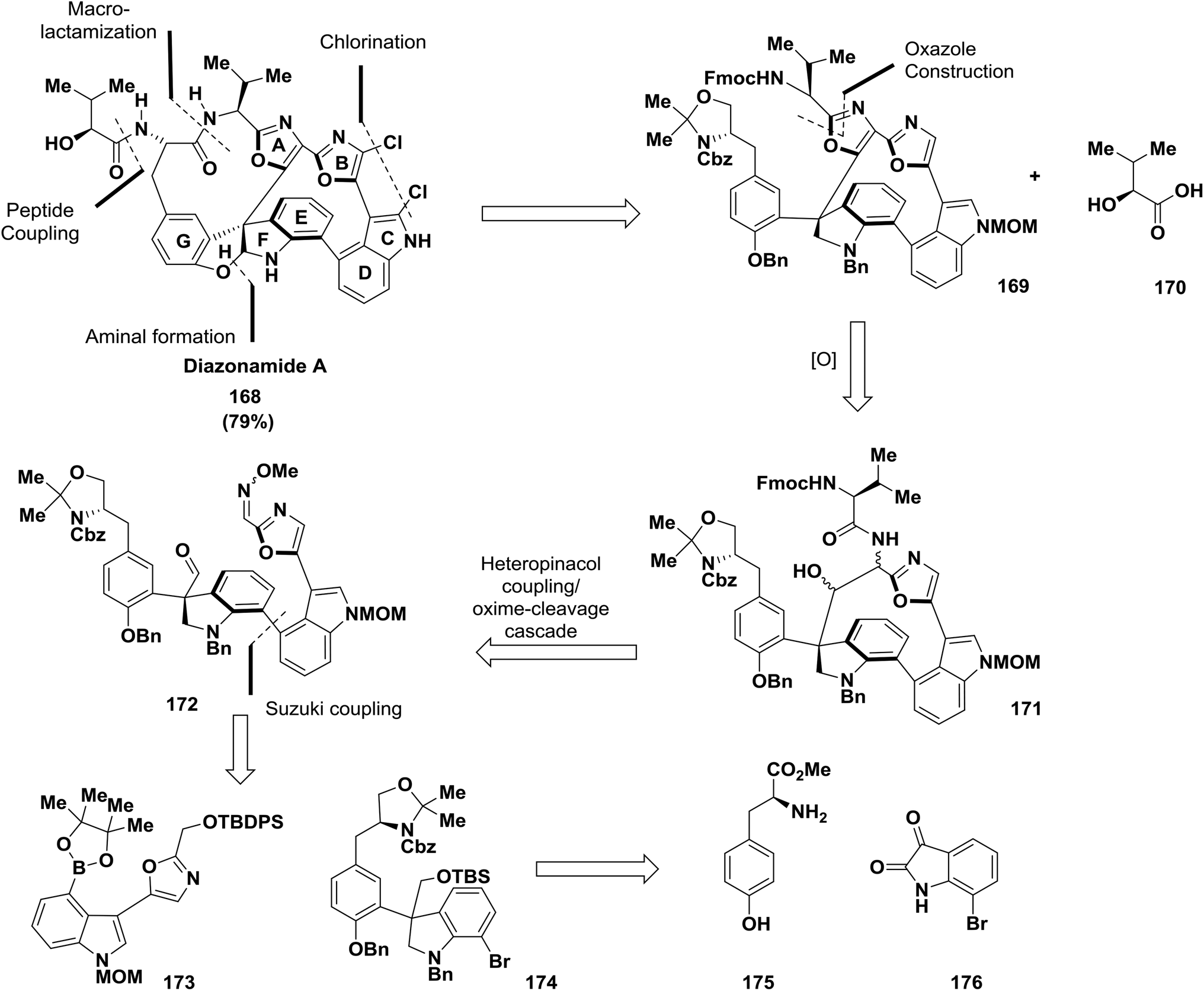 | ||
| Scheme 26 Structure of diazonamide A and general retrosynthetic analysis. | ||
In 2002, Palauan isolated the cyclic depsipeptide ulongamide A from collections of the marine cyanobacterium Lyngbya sp.; it possessed activity against some cancer types. Guzman and co-workers achieved the final macrocyclization of compound 177 using the coupling reagent BOP in 60% yield, as shown in Scheme 27.63,132,133
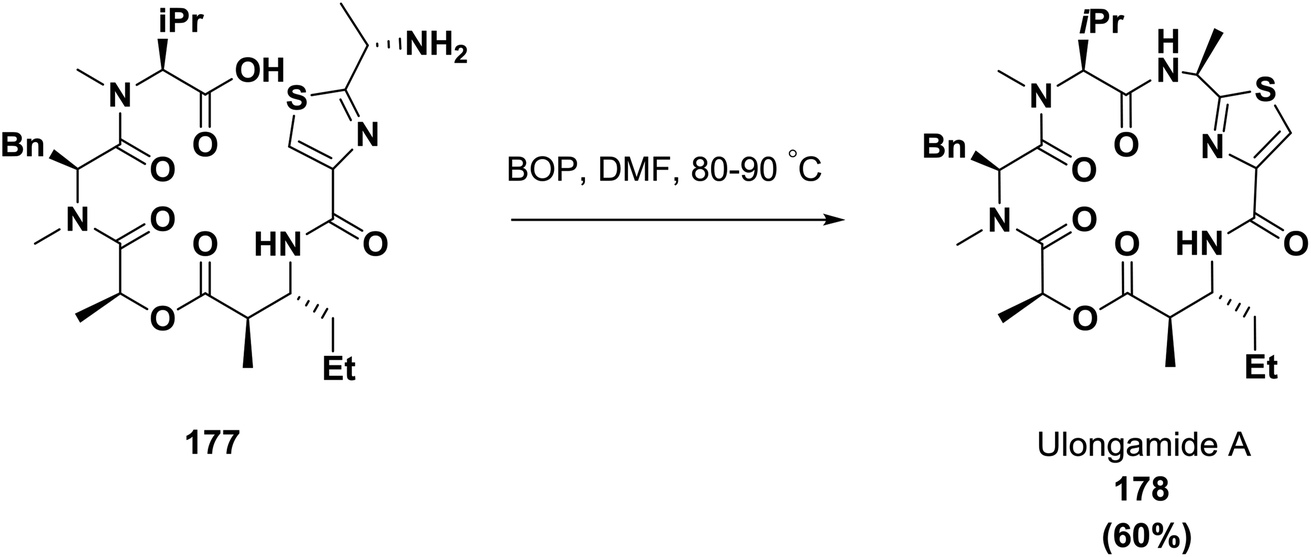 | ||
| Scheme 27 Synthesis of ulongamide A. | ||
In 2001, the cyclic depsipeptides halipeptin A and B were isolated from the marine sponge Haliclona sp. and appeared to have promising anti-inflammatory activities in vivo.134 Formation of the novel oxazetidine ring failed using the original approach, which was later revised to include thiazoline amino acid.135 The use of HOAt-derived uronium type reagents, such as HATU, proved to be efficient. For example, during the synthesis of halipeptin A, three approaches were used in different groups, including Ma's cyclization at the decanoic acid (HTMMD)/thiazoline-amino acid (alaThz) site (179); this macrocyclization was achieved in 27% yield using the highly efficient coupling reagent HATU136 (Scheme 28).
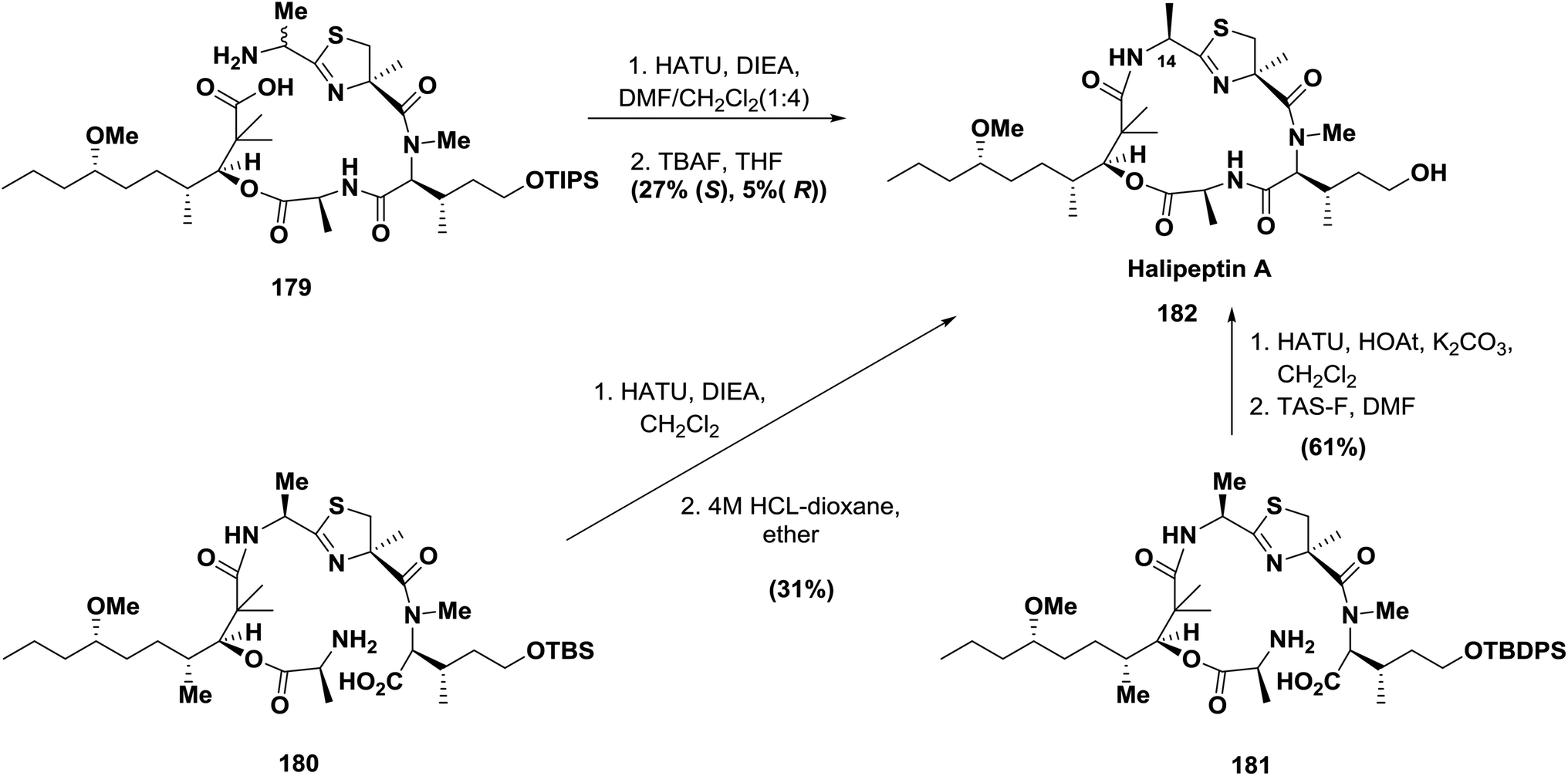 | ||
| Scheme 28 Different approaches for the synthesis of halipeptin A. | ||
On the other hand, Nicoloau's cyclization was performed between N-methylhydroxyisoleucine (N-MeOHIle) and L-alanine (Ala) (180) in 31% yield using the highly efficient coupling reagent HATU137 (Scheme 28).
Shortly after, Hamada and co-workers also succeeded in the synthesis of halipeptin A. Macrocyclization was achieved by using the coupling reagent HATU at the N-MeOHIle/Ala site (181), which provided halipeptin A 182 in 61% yield with 29% yield of the epimer, which was produced from racemization at the N-MeOHIle residue during the cyclization.138 (Scheme 28).
Hernandez and co-workers reported the synthesis of some analogs of the natural product IB-01211 183 from azole-based compounds via a biomimetic pathway based on cyclization–oxidation of serine-containing peptides combined with the Hantzsch synthesis. Macrocyclization of the linear peptides 188 and 189 was performed to afford IB-01211 (ref. 139) (Scheme 29).
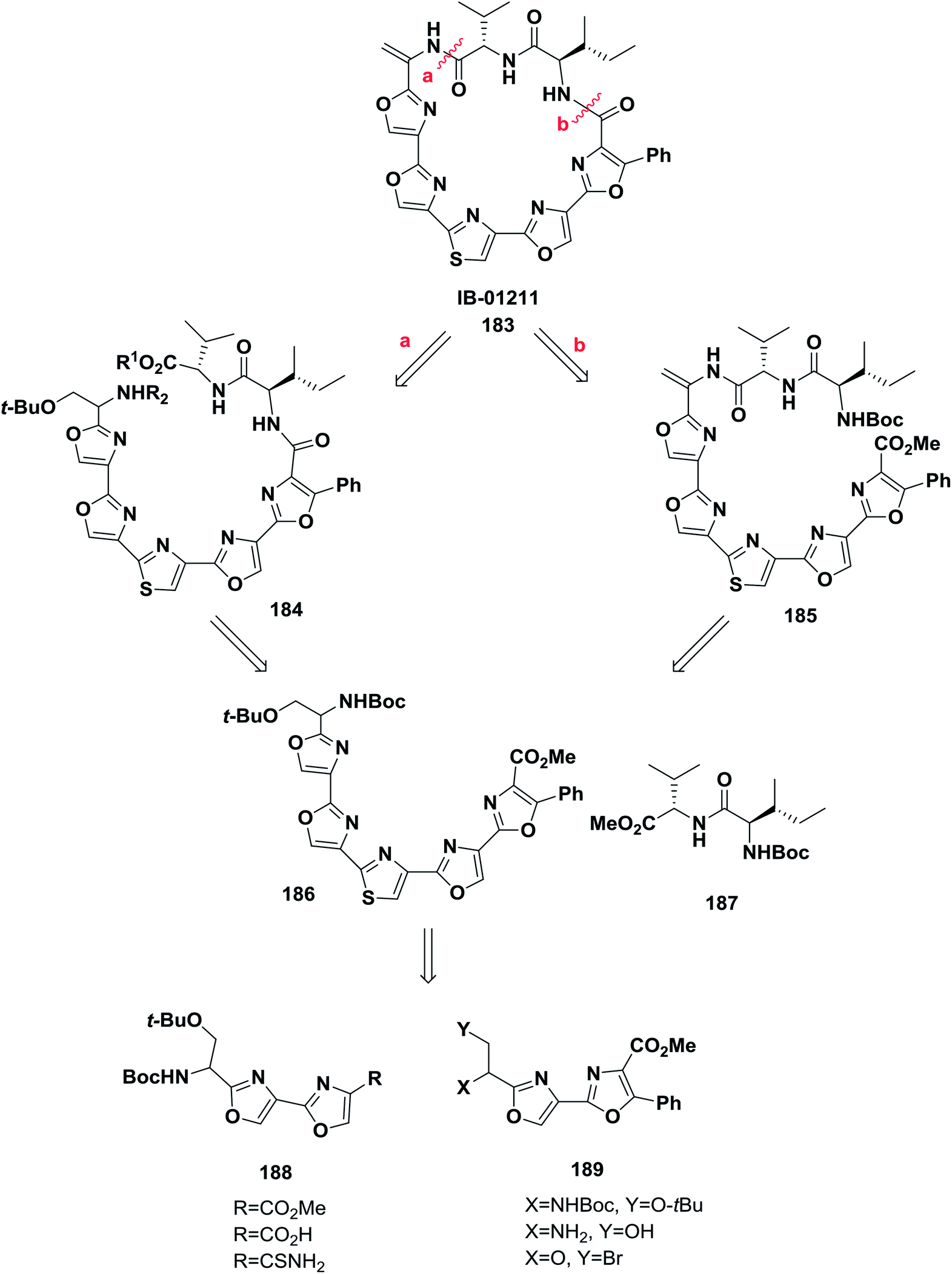 | ||
| Scheme 29 Retro-synthetic analysis of IB-01211. | ||
McAlpine and co-workers compared the solution phase route to the solid phase route for the synthesis of the cytotoxic natural product urukthapelstatin A (Ustat A) and ascertained that the solid phase method is superior. They reported the synthesis through two approaches: (A) the solution phase approach, which was difficult and involved cyclization of a ridged heterocyclic precursor; (B) the solid phase approach, which allowed the rapid and facile generation of a flexible linear peptide. Meanwhile, cyclization of the linear peptide was facile, and subsequent generation of three oxazoles located within the structure of Ustat A 190 was obtained in a relatively straightforward manner140 (Scheme 30).
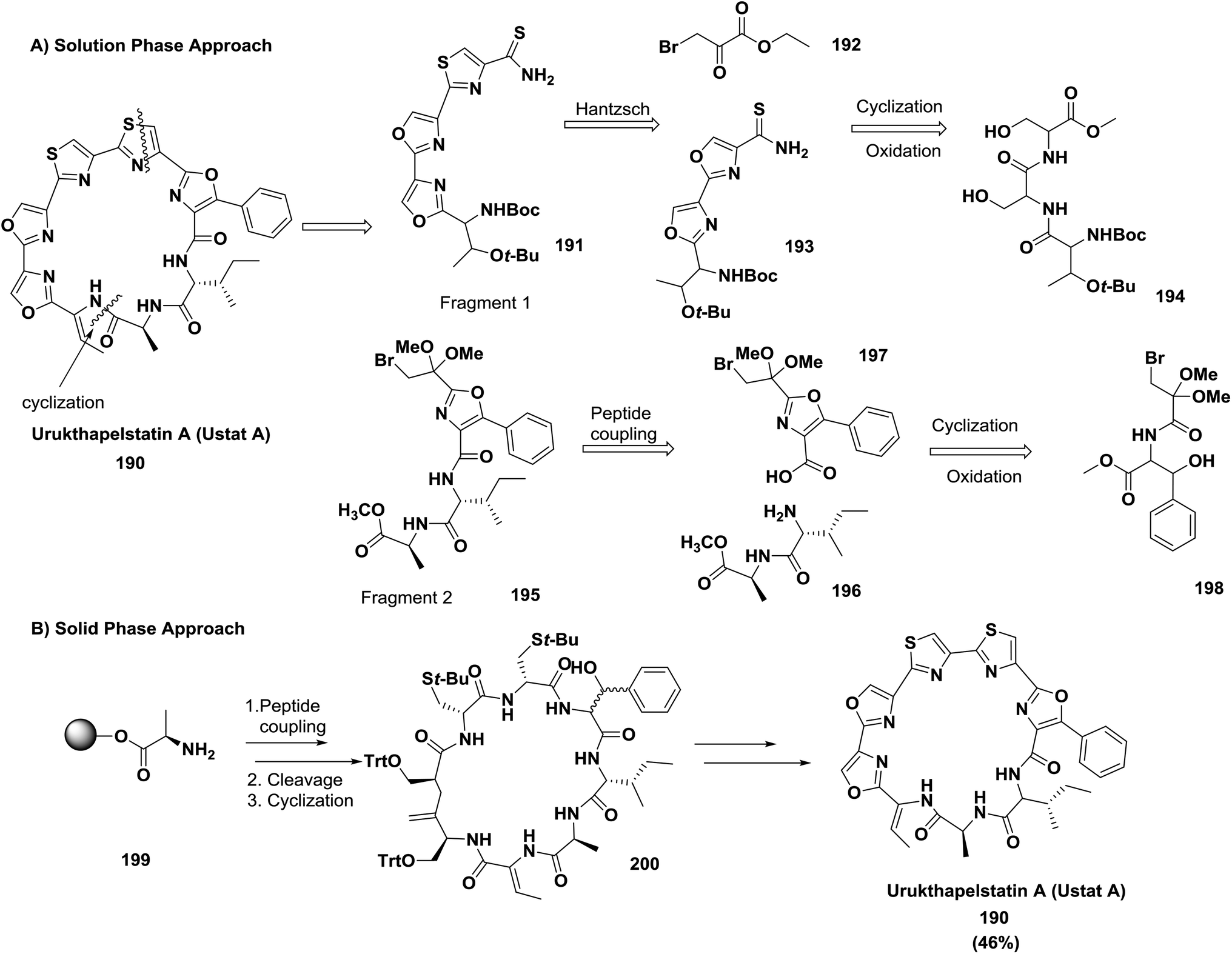 | ||
| Scheme 30 Two synthetic approaches: (A) solution phase retrosynthesis of Ustat A and (B) solid phase synthesis of Ustat A. | ||
Yokokawa and Shioiri and co-workers synthesized cis,cis-ceratospongamide 201 using a (5 + 2) convergent strategy to form a linear peptide which allowed cyclization via activation of the thiazole carboxyl group. The macrocyclization was examined with different coupling reagents, and FDPP enabled a yield of 63%. Finally, to obtain the oxazoline in the target compound, cyclodehydration was performed with bis(2-methoxyethyl) aminosulfur trifluoride (deoxo-fluor)141 (Scheme 31).
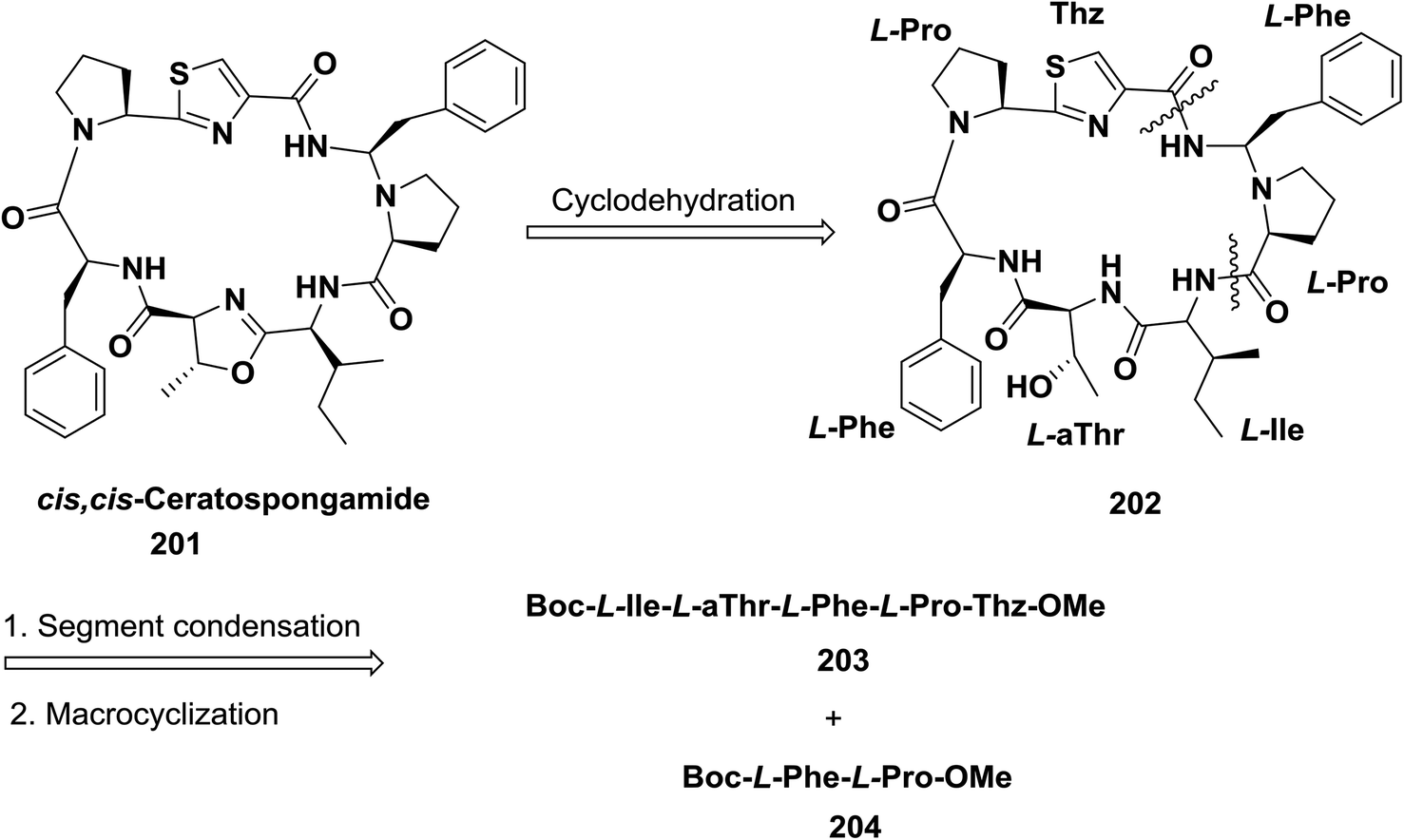 | ||
| Scheme 31 Retrosynthetic analysis of ceratospongamide. | ||
Yokokawa and co-workers also reported macrocyclization with amide formation at position (a) in cytotoxic metabolite lyngbyabellin A 205 using DPPA (58% yield). In this approach, the thiazole unit was built before cyclization142 (Scheme 32).
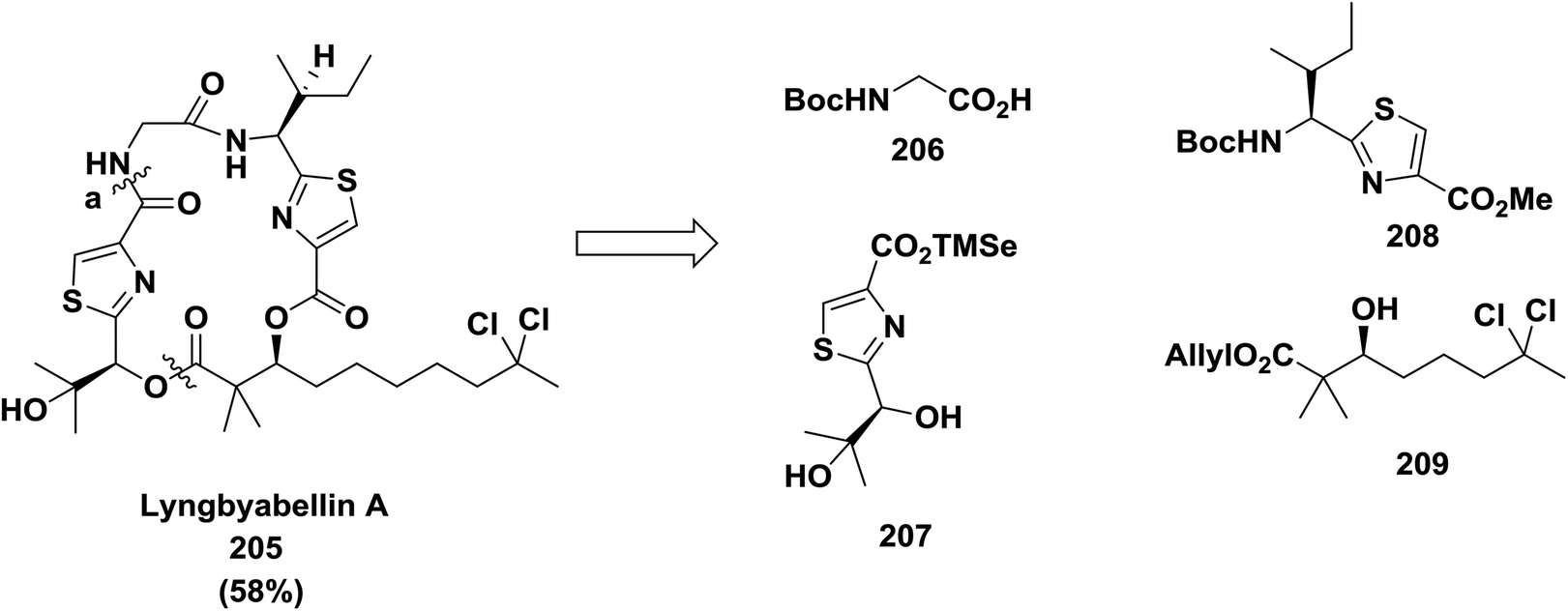 | ||
| Scheme 32 Retrosynthetic analysis of lyngbyabellin A. | ||
Meyers and co-workers reported the synthesis of griseoviridin 215, a member of the streptogramin family of antibiotics, in 68% yield. The oxazole carboxamide link was selected for macrolactamization using EDCI/HOBt; this was reported for the first time after many endeavors63,143 (Scheme 33).
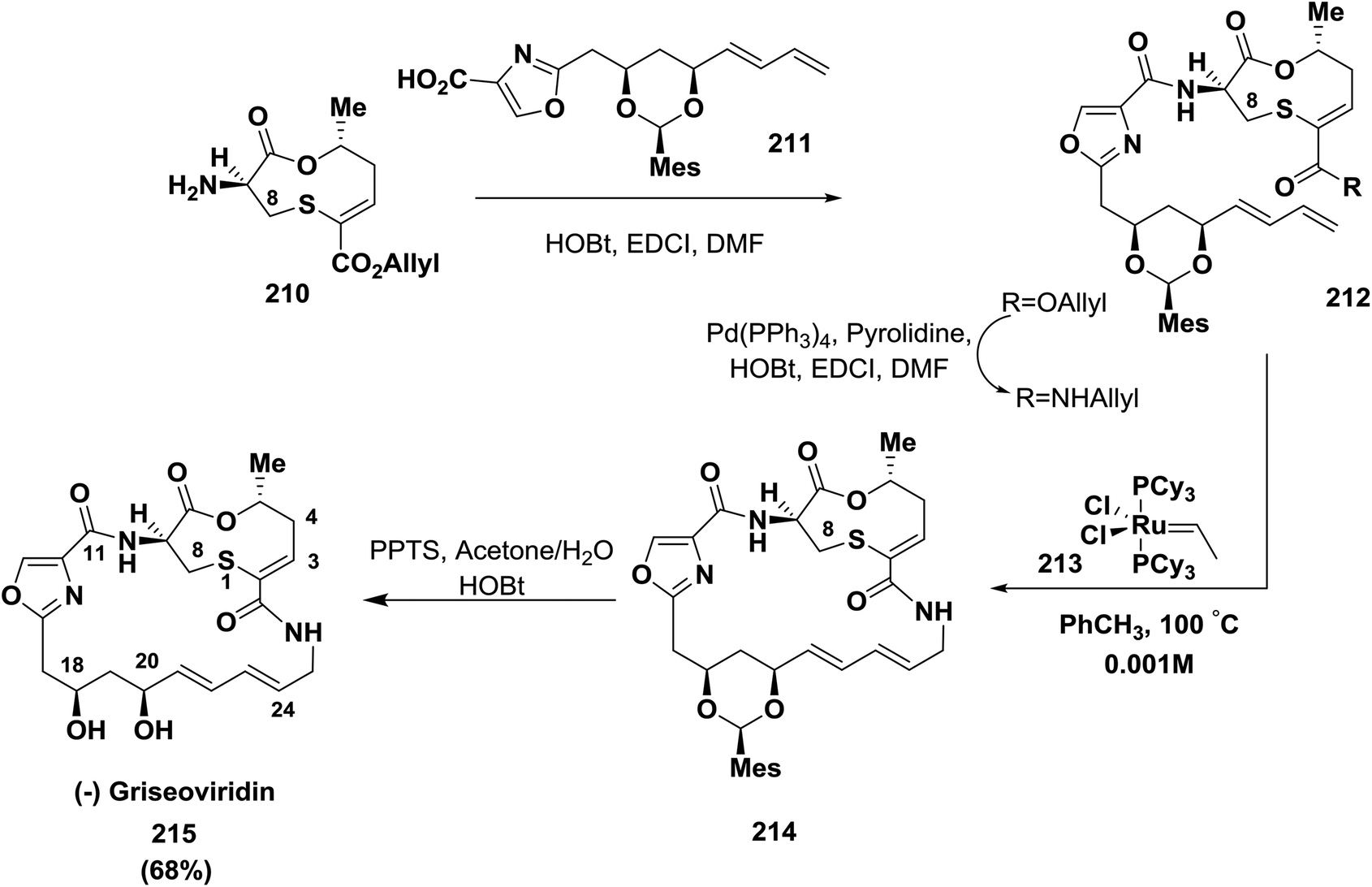 | ||
| Scheme 33 Synthesis of griseoviridin. | ||
Takahashi and co-workers described the synthesis of naturally occurring cyclic depsipeptide apratoxin A 217, which is isolated from Lyngbya majuscula and has potent cytotoxic activity, by adopting HATU as a coupling reagent to accomplish the macrolactamization of the linear precursor 216. HATU was able to activate the carboxylic group of the linear precursor 216, which resulted in selective formation of the amide bond63,144 (Scheme 34).
 | ||
| Scheme 34 Cyclization of apratoxin A. | ||
Ma and co-workers described an efficient approach for the synthesis of an oxazoline analogue of apratoxin A 218 (Fig. 9). A highly diastereoselective assembly of the Dtena moiety and consecutive installation of the oxazoline ring bearing an α,β-unsaturated ester side chain were performed. In addition, the success of macrocyclization at the N-Me-IIe-Pro site further indicates that this site is a suitable macrolactamization site to synthesize apratoxins and their analogues145 (Scheme 35).
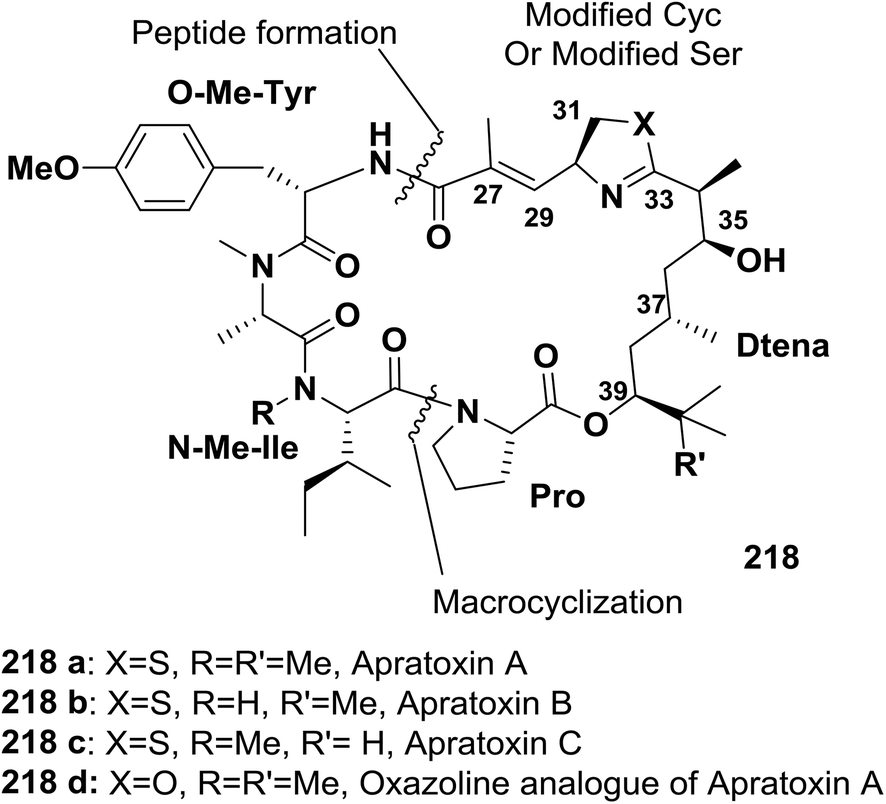 | ||
| Fig. 9 Structures of apratoxins A–C and an oxazoline analogue of apratoxin A. | ||
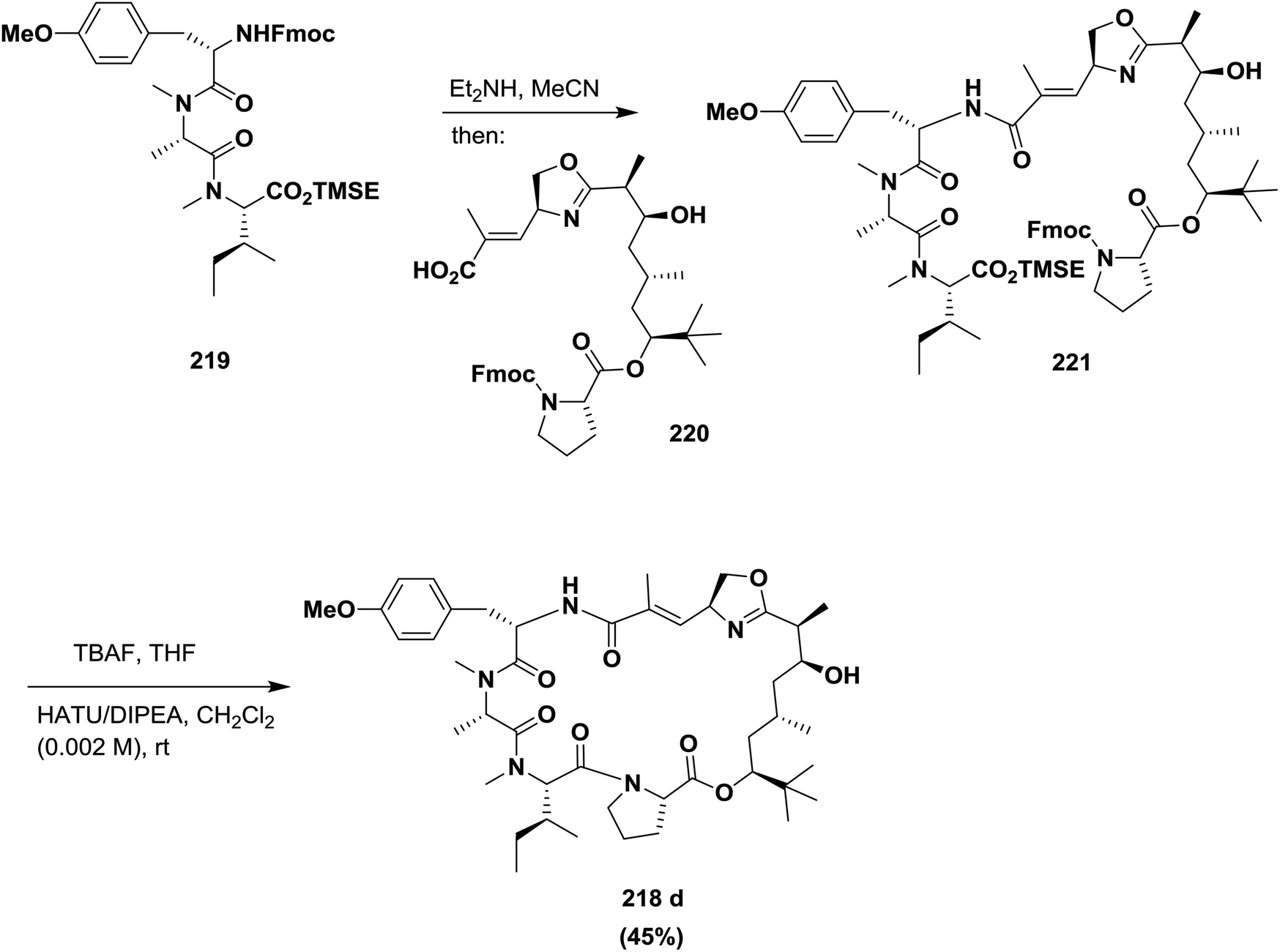 | ||
| Scheme 35 Synthesis of an oxazoline analogue of apratoxin A. | ||
Shin and co-workers synthesized micrococcin P 222 from protected fragments A–D, as shown in Fig. 10. The final macrocyclization using BOP occurred at position (a), affording total accordance between the physical and spectral properties of the synthetic and natural products. The same strategy has been applied to micrococcin P 223 with similar success146,147 (Fig. 10).
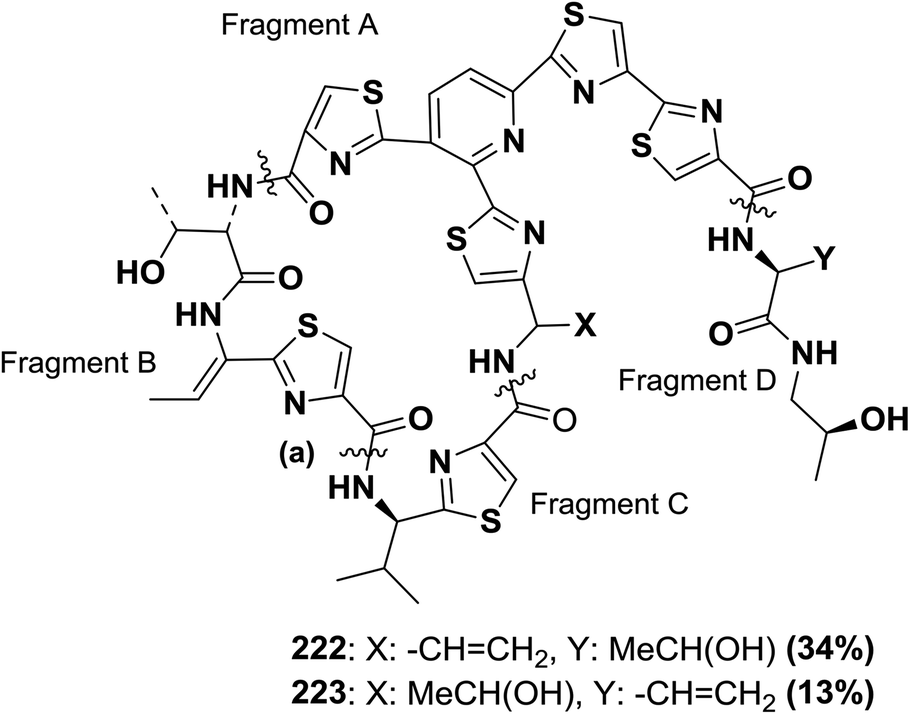 | ||
| Fig. 10 Structure of micrococcin P (222) and its analogue. | ||
6. Solid phase approach for the synthesis of azoles
Usually, azole-based amino acids are prepared in solution in advance and later used as building blocks in solution and solid-phase synthesis. There are some reported approaches for the synthesis of oxazoles, thiazoles, or imidazoles in solution, such as (1) a modified Hantzsch's procedure; (2) a condensation reaction between N-protected imino ethers and serine esters, cysteine esters, or 2,3-diaminopropionic acid esters; and (3) cyclodehydration of β-hydroxyamides or β-hydroxythioamides. The freshly synthesized 1,3-azolines are readily converted into 1,3-azoles by oxidation. Unfortunately, many of these methods have drawbacks, such as long synthetic sequences, harsh reaction conditions, or extensive purification procedures that lead to low yields.148Kessler and co-workers reported a solid-phase approach for the synthesis of oxazole, thiazole, and imidazole-based peptides. The reported procedures are compatible with Fmoc-solid phase peptide synthesis. This reported method opens the way for the synthesis of natural product libraries of peptidomimetics containing 1,3-azoles148 (Schemes 36–40).
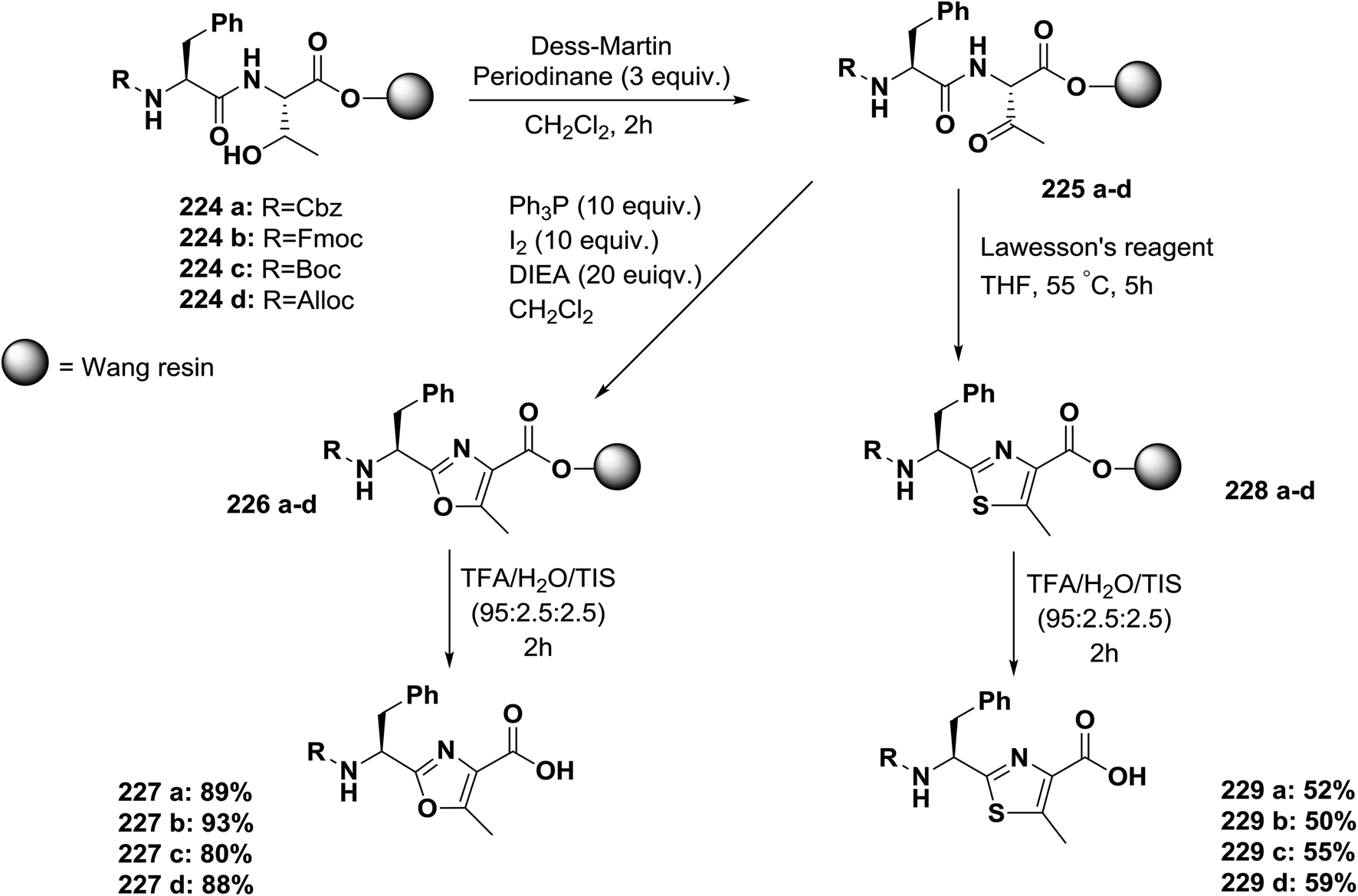 | ||
| Scheme 36 Solid-phase synthesis of oxazole- and thiazole-based peptides from threonine-containing dipeptides. | ||
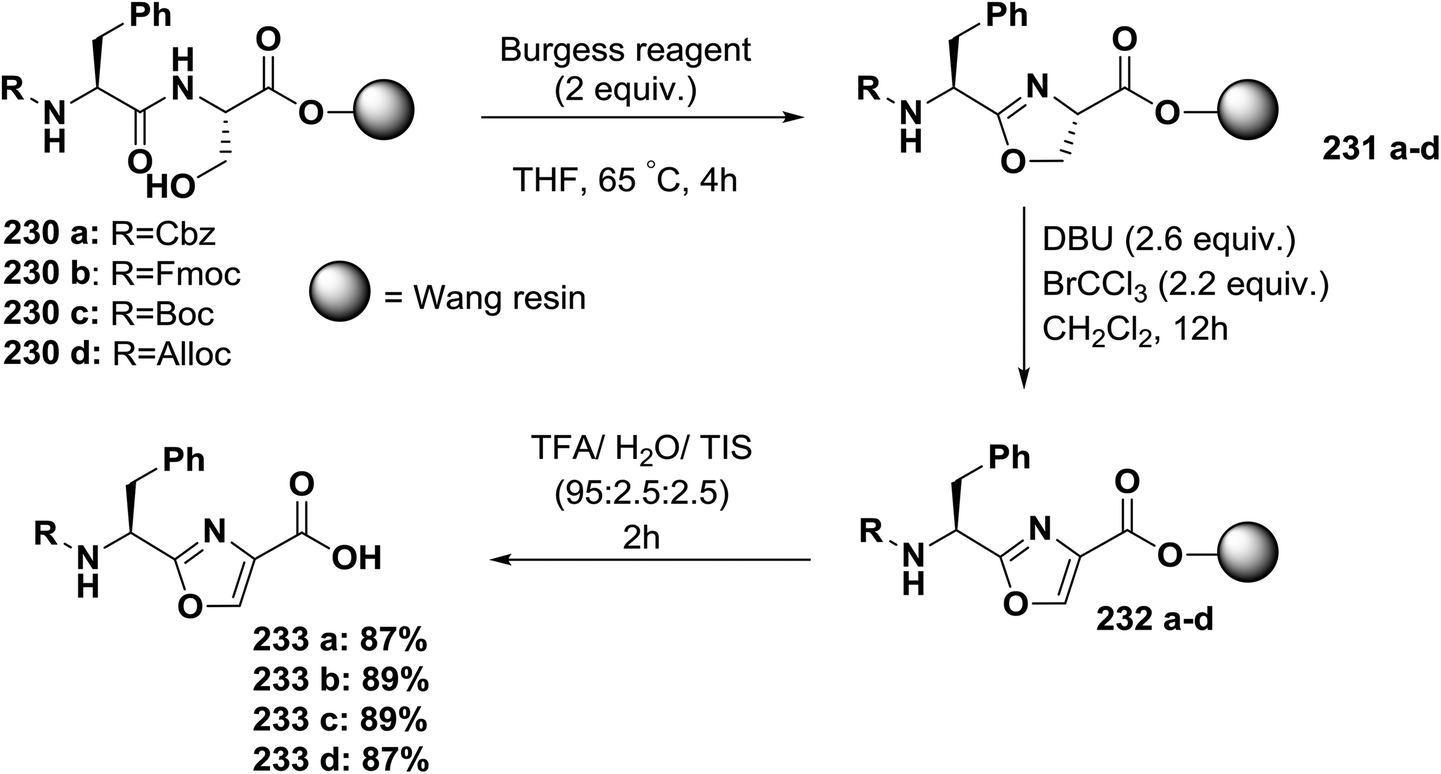 | ||
| Scheme 37 Solid-phase synthesis of 1,3-oxazole-based peptides from serine-containing dipeptides. | ||
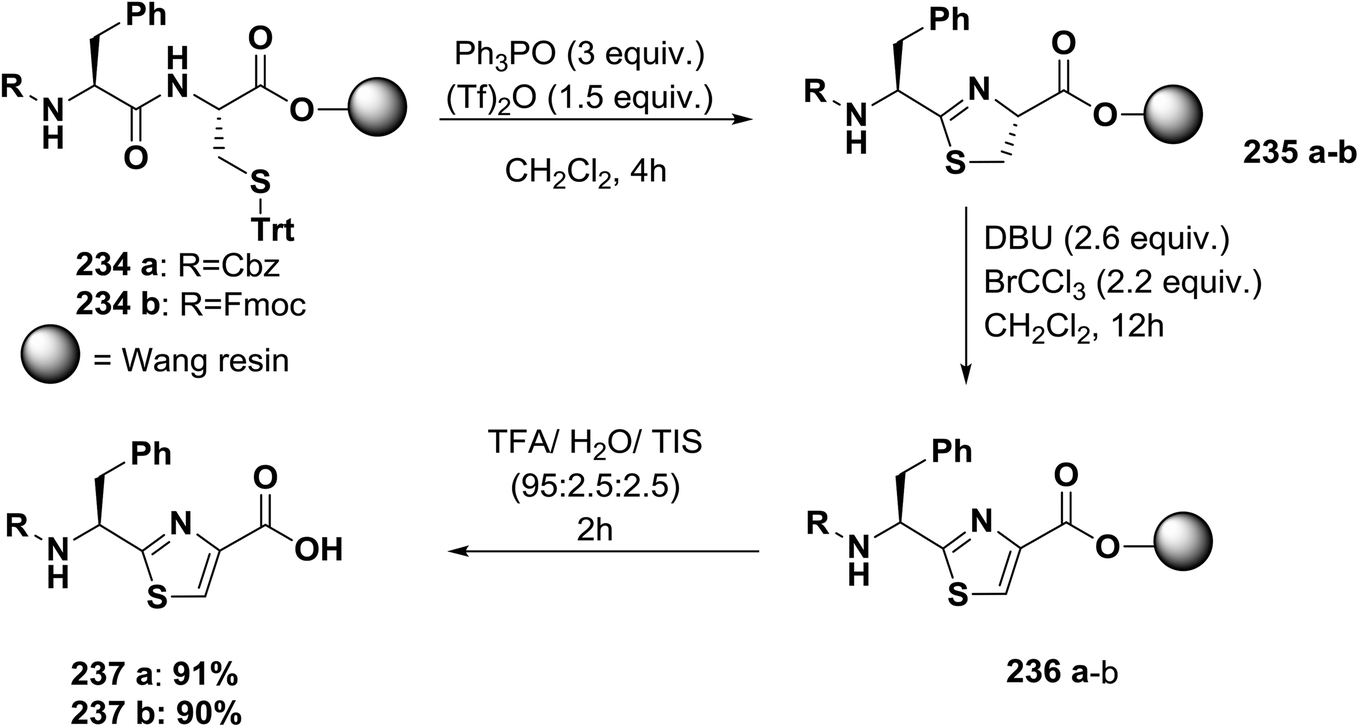 | ||
| Scheme 38 Solid-phase synthesis of thiazole-based peptides. | ||
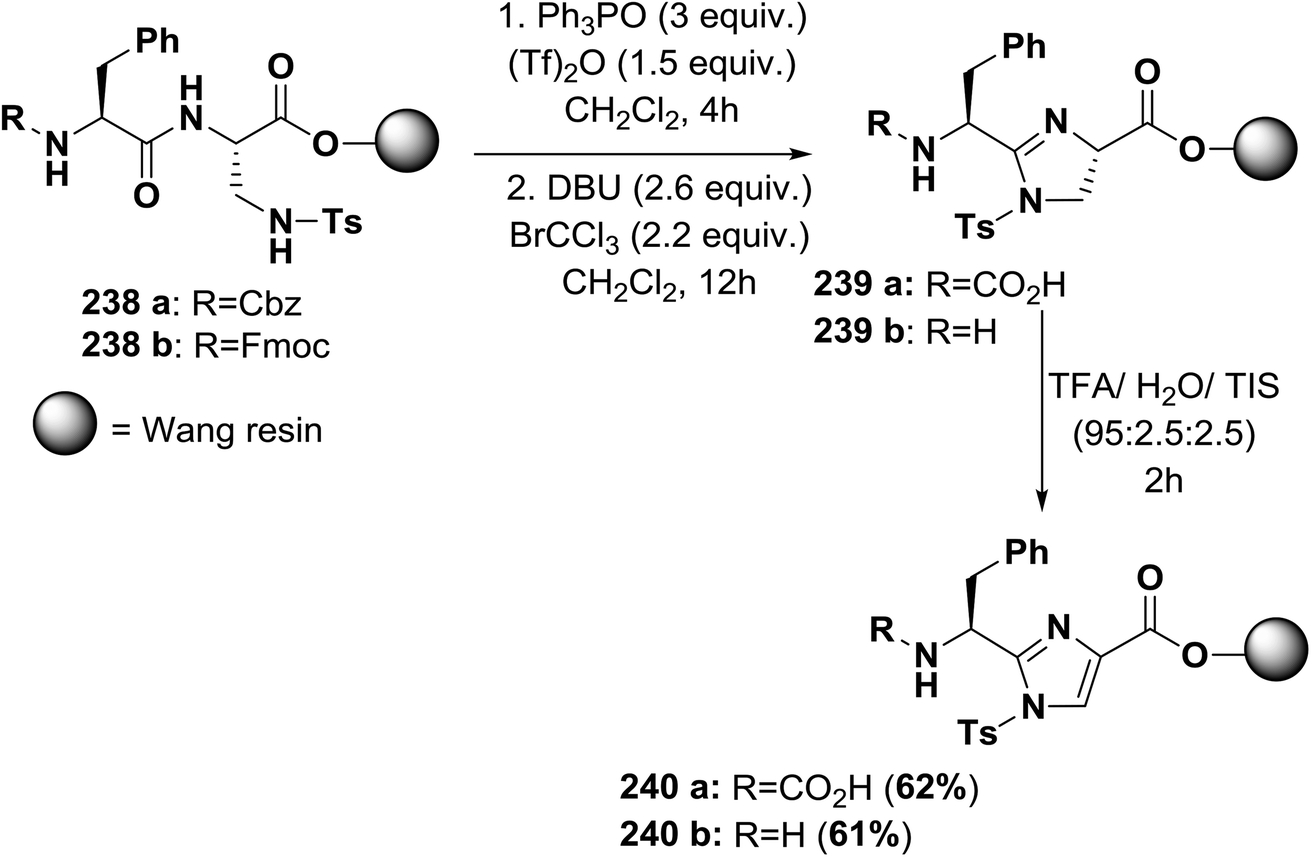 | ||
| Scheme 39 Solid-phase synthesis of imidazole-based peptides. | ||
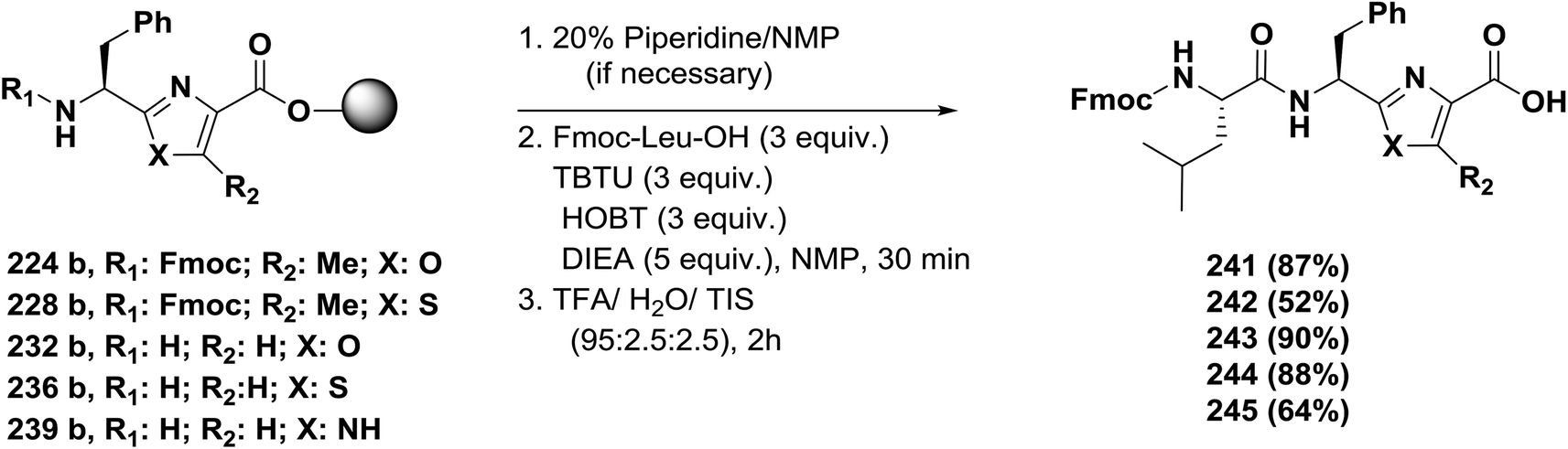 | ||
| Scheme 40 Solid-phase synthesis of azole-containing tripeptides. | ||
7. Conclusion
In conclusion, cyclopeptides have attracted the attention of synthetic chemists not only because of their unique, promising properties and biological significance but also because of the high synthetic complexity of these endeavors. In future, we will witness many approaches involving various sophisticated synthetic methods. Moreover, naturally occurring cyclopeptides containing heterocyclic skeletons in various sources, such as plant cyclopeptides, marine cyclopeptides, and fungi, have confirmed the importance of these skeletons in the extensive biological activities of these cyclopeptides.There are many resources of biologically active structures isolated from marine organisms, plants, and fungi that consist of cyclic peptides embedded in heterocyclic skeletons. In many examples, the existence of thiazole and oxazole rings establishes conformational restrictions in the corresponding cyclopeptides. Consequently, the incorporation of heterocycles into the backbones of cyclopeptides is eliciting growing interest, especially for medicinal chemistry prospects.
Moreover, studies have demonstrated that the incorporation of heterocyclic skeletons into cyclopeptides has a good influence on the conformations of the cyclopeptides. In addition, it allows the formation of noncovalent interactions that are important in molecular recognition, which is a key process in chemistry and biology. These interactions are illustrated in four groups: (1) hydrogen bonds, (2) cation-π interactions, (3) ion-pair interactions and (4) London dispersion forces. Variation of the azole units within the heterocycle skeleton enables scientists to design receptors that are either selective or have affinities for specific anions, allowing stronger interactions between the cyclopeptide and its target.
In summary, it is hoped that this review has successfully highlighted different approaches for peptide cyclizations containing heterocyclic skeletons. The examples discussed in this review reflect the complexity of nature's methods of adding constraints to peptides and illustrate the persisting challenges in their syntheses, such as control of stereochemistry, incompatibility of chemical functionalities, and difficulties with solid-phase approaches; these constraints can currently be mimicked utilizing a vast range of organic chemical techniques in the laboratory, but further research is still required to establish more widely applicable synthetic methodologies. These demanding synthetic challenges will afford necessary knowledge that will help to create mimics and design molecules which will definitely be useful in the development of more effective pharmaceuticals.
Conflicts of interest
There are no conflicts to declare.Abbreviations
| CuAAC | Copper-catalyzed azide-alkyne cycloaddition |
| RuAAC | Ruthenium-catalyzed azide-alkyne cycloaddition |
| THF | Tetrahydrofuran |
| DMS | Dimethyl sulfide |
| SPPS | Solid phase peptide synthesis |
| Tyr: Y | Tyrosine |
| Pro: P | Proline |
| Val: V | Valine |
| Arg: R | Arginine |
| Gly: G | Glycine |
| Asp: D | Aspartic acid |
| Phe: F | Phenylalanine |
| Cys: C | Cysteine |
| Thr: T | Threonine |
| Ser: S | Serine |
| Lys: K | Lysine |
| Ile: I | Isoleucine |
| Glu: E | Glutamic acid |
| Met: M | Methionine |
| Asn: N | Asparagine |
| Leu: L | Leucine |
| Trp: W | Tryptophan |
| NMP | N-Methyl-2-pyrrolidone |
| TBTA | Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate |
| TFA | Trifluoroacetic acid |
| TIPS | Triisopropylsilyl ether |
| HDAC | Histone deacetylases |
| Dap | Diaminopropanoic acid |
| NIe | Norleucine |
| DEPBT | 3-(Diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one |
| DIEA: DIPEA | N,N-Diisopropylethylamine or Hünig's base |
| NMM | N-Methylmorpholine |
| TBTU | 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate |
| FDPP | Pentafluorophenyl diphenylphosphinate |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| DMAP | 4-Dimethylaminopyridine |
| DMF | Dimethylformamide |
| HPLC | High performance liquid chromatography |
| NCS | N-Chlorosuccinimide |
| PyAOP | (7-Azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate |
| DAST | Diethylaminosulfur trifluoride |
| DPPA | Diphenylphosphoryl azide |
| IBX | 2-Iodoxybenzoic acid |
| DBU | 1,8-Diazabicyclo [5.4.0] undec-7-ene |
| NBS | N-Bromosuccinimide |
| Bop | (Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate |
| Thz | Thiazole |
| aThr | Alloc-L-threonine |
| Dtena | 3,7-Dihydroxy-2,5,8,8- tetramethylnonanoic acid |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| PPTS | Pyridinium p-toluenesulfonate |
| TIS | Triisopropylsilane |
Acknowledgements
We thank the National Institute for Medical Research Development (NIMAD) (Grant No. 943185) for financial support.References
- V. J. Hruby, Nat. Rev. Drug Discovery, 2002, 1, 847–858 CrossRef CAS PubMed.
- J. Vagner, H. Qu and V. J. Hruby, Curr. Opin. Chem. Biol., 2008, 12, 292–296 CrossRef CAS PubMed.
- P. Li, P. P. Roller and J. Xu, Curr. Org. Chem., 2002, 6, 411–440 CrossRef CAS.
- S. Hess, Y. Linde, O. Ovadia, E. Safrai, D. E. Shalev, A. Swed, E. Halbfinger, T. Lapidot, I. Winkler, Y. Gabinet, A. Faier, D. Yarden, Z. Xiang, P. F. Portillo, C. Haskell-Luevano, C. Gilon and A. Hoffman, J. Med. Chem., 2008, 51, 1026–1034 CrossRef CAS PubMed.
- M. Hurevich, Y. Tal-Gan, S. Klein, Y. Barda, A. Levitzki and C. Gilon, J. Pept. Sci., 2010, 16, 178–185 CAS.
- M. Siedlecka, G. Goch, A. Ejchart, H. Sticht and A. Bierzynski, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 903–908 CrossRef CAS.
- J. X. Yang, K. Zhao, Y.-X. Gong, A. Vologodskii and N. R. Kallenbach, J. Am. Chem. Soc., 1998, 120, 10646–10652 CrossRef CAS.
- N. H. Tan and J. Zhou, Chem. Rev., 2006, 106, 840–895 CrossRef CAS PubMed.
- A. Wele, Y. Zhang, I. Ndoye, J. P. Brouard, J. L. Pousset and B. Bodo, J. Nat. Prod., 2004, 67, 1577–1579 CrossRef CAS PubMed.
- P. W. Hsieh, F. R. Chang, C. C. Wu, K. Y. Wu, C. M. Li, S. L. Chen and Y. C. Wu, J. Nat. Prod., 2004, 67, 1522–1527 CrossRef CAS PubMed.
- D. J. Craik, N. L. Daly, J. Mulvenna, M. R. Plan and M. Trabi, Curr. Protein Pept. Sci., 2004, 5, 297–315 CrossRef CAS PubMed.
- N. Fusetani and S. Matsunaga, Bioactive sponge peptides, Chem. Rev., 1993, 93, 1793–1806 CrossRef CAS.
- J. S. Davies, J. Pept. Sci., 2003, 9, 471–501 CrossRef CAS PubMed.
- J. P. Michael and G. Pattenden, Angew. Chem., Int. Ed., 1993, 32, 1–23 CrossRef.
- T. H. Wieland, Peptides of Poisonous Amanita Mushrooms, Springer-Verlag, Berlin, Heidelberg, 1986 Search PubMed.
- K. Yamada, M. Unno, K. Kobayashi, H. Oku, H. Yamamura, S. Araki, H. Matsumoto, R. Katakai and M. Kawai, J. Am. Chem. Soc., 2002, 124, 12684–12688 CrossRef CAS PubMed.
- C. M. Harris and T. M. Harris, J. Am. Chem. Soc., 1982, 104, 4293–4295 CrossRef CAS.
- F. Tahoori, S. Balalaie, R. Sheikhnejad, M. Sadjadi and P. Boloori, Amino Acids, 2014, 46, 1033–1046 CrossRef CAS PubMed.
- Q. Mu, R. W. Teng, C. M. Li, D. Z. Wang, Y. Wu, H. D. Sun and C. Q. Hu, Chin. Chem. Lett., 2001, 12, 607–610 CAS.
- M. Goodman, C. Zapf and Y. Rew, Biopolymers, 2001, 60, 229–245 CrossRef CAS PubMed.
- A. Loffet, J. Pept. Sci., 2002, 8, 1–7 CrossRef CAS PubMed.
- S. Cappelletti, P. Annoni, G. DiGregori, O. Storace and M. Pinori, Chim. Oggi, 2002, 20, 47–50 CAS.
- M. Verlander, Chim. Oggi, 2002, 20, 62–66 CAS.
- M. Mizhiritskii and Y. Shpernat, Chim. Oggi, 2002, 20, 43–45 CAS.
- J. N. Lambert, J. P. Mitchell and K. D. Roberts, J. Chem. Soc., Perkin Trans. 1, 2001, 0, 471–484 RSC.
- J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS PubMed.
- A. Spatola and P. Romanovskis, J. Pept. Res., 1998, 51, 356–374 Search PubMed.
- D. A. Horton, G. T. Bourne and M. L. Smythe, J. Comput.-Aided Mol. Des., 2002, 16, 415–430 CrossRef CAS PubMed.
- W. C. Chan, Fmoc Solid Phase Peptide Synthesis, Oxford University Press Inc., New York, 2000 Search PubMed.
- W.-Y. Xu, S.-M. Zhao, G.-Z. Zeng, W.-J. He, H.-M. Xu, N.-H. Tan, S.-M. Zhao and H.-M. Xu, Acta Pharm. Sin., 2012, 47, 271–279 CAS.
- (a) M. Kaiser and S. Stolze, Synthesis, 2012, 44, 1755–1777 CrossRef; (b) K. Kobayashi, H. Suzuki, K. Shimbo, K. Takeya and H. Morita, J. Org. Chem., 2001, 66, 6626–6633 CrossRef PubMed.
- Y. Liang, X.-Q. Wu and Y. Zhang, China J. Chin. Mater. Med., 2006, 31, 709–714 CAS.
- P. Wipf, Chem. Rev., 1995, 95, 2115–2134 CrossRef CAS.
- J. Peng, J. Li and M. T. Hamann, in The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Elsevier Inc, 2005 Search PubMed.
- X.-Y. Wang, Q. Wang, X.-Y. Huang, T. Wang and X.-Q. Yu, ARKIVOC, 2006, 2006, 148–154 Search PubMed.
- S. Enck, A. Geyer, F. Kopp and M. A. Marahiel, Org. Biomol. Chem., 2010, 8, 559–563 RSC.
- X.-Y. Huang, T. Wang, Ch-Q. Xia, X.-Q. Yu and R.-G. Xie, Chin. J. Org. Chem., 2004, 24, 1629–1632 CAS.
- I. E. Valverde and T. L. Mindt, Chimia, 2013, 67, 262–266 CrossRef CAS PubMed.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., 2001, 113, 2056–2075 CrossRef; (b) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (c) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef; (d) J. Totobenazara and A. J. Burke, Tetrahedron Lett., 2015, 56, 2853–2859 CrossRef CAS.
- L. D. Quin and J. A. Tyrell, Fundamentals of Heterocyclic Chemistry: Importance in Nature and in the Synthesis of Pharmaceuticals, Wiley-VCH, Weinheim, 2010 Search PubMed.
- J. T. Lundquist and J. C. Pelletier, Org. Lett., 2001, 3, 781–783 CrossRef CAS PubMed.
- J. Alsina and F. Albericio, Pept. Sci., 2003, 71, 454–477 CrossRef CAS PubMed.
- A. Tam, U. Arnold, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2007, 129, 12670–12671 CrossRef CAS PubMed.
- M. Tischler, D. Nasu, M. Empting, S. Schmelz, D. W. Heinz, P. Rottmann, H. Kolmar, G. Buntkowsky, D. Tietze and O. Avrutina, Angew. Chem., Int. Ed., 2012, 51, 3708–3712 CrossRef CAS PubMed.
- I. E. Valverde, F. Lecaille, G. Lalmanach, V. Aucagne and A. F. Delmas, Angew. Chem., Int. Ed., 2012, 51, 718–722 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- S. I. Presolski, V. Hong, S.-H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570–14576 CrossRef CAS PubMed.
- V. O. Rodionov, S. I. Presolski, D. D. Diaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705 CrossRef CAS PubMed.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed.
- Z. Zhou and C. J. Fahrney, J. Am. Chem. Soc., 2004, 126, 8862–8863 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- V. D. Bock, R. Perciaccante, T. P. Jansen, H. Hiemstra and J. H. van Maarseveen, Org. Lett., 2006, 8, 919–922 CrossRef CAS PubMed.
- V. D. Bock, D. Speijer, H. Hiemstra and J. H. van Maarseveen, Org. Biomol. Chem., 2007, 5, 971–975 RSC.
- R. A. Turner, A. G. Oliver and R. S. Lokey, Org. Lett., 2007, 9, 5011–5014 CrossRef CAS PubMed.
- J. H. van Maarseveen, W. S. Horne and M. R. Ghadiri, Org. Lett., 2005, 7, 4503–4506 CrossRef CAS PubMed.
- S. W. Horne, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2003, 125, 9372–9376 CrossRef PubMed.
- (a) J. M. Beierle, S. W. Horne, J. H. van Maarseveen, B. Waser, J. C. Reubi and M. R. Ghadiri, Angew. Chem., Int. Ed., 2009, 48, 4725–4729 CrossRef CAS PubMed; (b) S. W. Horne, C. A. Olsen, J. M. Beierle, A. Montero and M. R. Ghadiri, Angew. Chem., Int. Ed., 2009, 48, 4718–4724 CrossRef PubMed.
- M. Ahsanullah and J. Rademann, Angew. Chem., Int. Ed., 2010, 49, 5378–5382 CrossRef PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- S. Jiang, Z. Li, K. Ding and P. P. Roller, Curr. Org. Chem., 2008, 12, 1502–1542 CrossRef CAS.
- M. R. Davis, E. K. Singh, H. Wahyudi, L. D. Alexander, J. B. Kunicki, L. A. Nazarova, K. A. Fairweather, A. M. Giltrap, K. A. Jolliffe and S. R. McAlpine, Tetrahedron, 2012, 68, 1029–1051 CrossRef CAS PubMed.
- Y. Q. Liu, L. H. Zhang, J. P. Wan, Y. S. Li, Y. H. Xu and Y. J. Pan, Tetrahedron, 2008, 64, 10728–10734 CrossRef CAS.
- V. Goncalves, B. Gautier, A. Regazzetti, P. Coric, S. Bouaziz, C. Garbay, M. Vidal and N. Inguimbert, Bioorg. Med. Chem. Lett., 2007, 17, 5590–5594 CrossRef CAS PubMed.
- S. Punna, J. Kuzelka, Q. Wang and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2215–2220 CrossRef CAS PubMed.
- J. F. Billing and U. J. Nilsson, J. Org. Chem., 2005, 70, 4847–4850 CrossRef CAS PubMed.
- Y. Angell and K. Burgess, J. Org. Chem., 2005, 70, 9595–9598 CrossRef CAS PubMed.
- W. J. Choi, Z. Shi, K. M. Worthy, L. Bindu, R. G. Karki, M. C. Nicklaus, R. J. Fisher and T. R. Burke Jr, Bioorg. Med. Chem. Lett., 2006, 16, 5265–5269 CrossRef CAS PubMed.
- J. Chen, Z. Nikolovska-Coleska, C.-Y. Yang, C. Gomez, W. Gao, K. Krajewski, S. Jiang, P. P. Roller and S. Wang, Bioorg. Med. Chem. Lett., 2007, 17, 3939–3942 CrossRef CAS PubMed.
- S. Cantel, A. L. Chevalier Isaad, M. Scrima, J. J. Levy, R. D. DiMarchi, P. Rovero, J. A. Halperin, A. M. D’ Ursi, A. M. Papini and M. Chorev, J. Org. Chem., 2008, 73, 5663–5674 CrossRef CAS PubMed.
- B. Narasimhan, D. Sharma and P. Kumar, Med. Chem. Res., 2011, 20, 1119–1140 CrossRef CAS.
- B. L. Vallee and D. S. Auld, Biochemistry, 1990, 29, 5647–5659 CrossRef CAS PubMed.
- B. L. Vallee and D. S. Auld, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 220–224 CrossRef CAS.
- B. W. Matthews, Acc. Chem. Res., 1988, 21, 333–340 CrossRef CAS.
- J. Rebek Jr, Struct. Chem., 1990, 1, 129–131 CrossRef.
- F. K. Winkler, A. D'Arcy and W. Hunziker, Nature, 1990, 343, 771–774 CrossRef CAS PubMed.
- F. Ikegami and I. Murakoshi, Phytochemistry, 1994, 35, 1089–1104 CrossRef CAS.
- M. Wang and S. J. Gould, J. Org. Chem., 1993, 58, 5176–5180 CrossRef CAS.
- G. Haberhauer and F. Rominger, Eur. J. Org. Chem., 2003, 2003, 3209–3218 CrossRef.
- G. Haberhauer and F. Rominger, Tetrahedron Lett., 2002, 43, 6335–6338 CrossRef CAS.
- Á. Pintér and G. Haberhauer, Tetrahedron, 2009, 65, 2217–2225 CrossRef.
- N. K. Terrett, Drug Discovery Today: Technol., 2010, 7, e97–e104 CrossRef CAS PubMed.
- A. Isidro-Llobet, T. Murillo, P. Bello, A. Cilibrizzi, J. T. Hodgkinson, W. R. J. D. Galloway, A. Bender, M. Welch and D. R. Spring, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6793–6798 CrossRef CAS PubMed.
- P. W. Baures, Trends Heterocycl. Chem., 2006, 11, 1–22 CAS.
- Z. Xu, J. C. DiCesare and P. W. Baures, J. Comb. Chem., 2010, 12, 248–254 CrossRef CAS PubMed.
- R. Solinas, J. C. DiCesare and P. W. Baures, Molecules, 2009, 14, 352–363 CrossRef CAS PubMed.
- R. Solinas, J. C. DiCesare and P. W. Baures, Molecules, 2008, 13, 3149–3170 CrossRef CAS PubMed.
- Z. Xu, K. A. Wheeler and P. W. Baures, Molecules, 2012, 17, 5346–5362 CrossRef CAS PubMed.
- G. Sabatino, M. Chelli, S. Mazzucco, M. Ginanneschi and A. M. Papini, Tetrahedron Lett., 1999, 40, 809–812 CrossRef CAS.
- M. C. Alcaro, G. Sabatino, J. Uziel, M. Chelli, M. Ginanneschi, P. Rovero and A. M. Papini, J. Pept. Sci., 2004, 10, 218–228 CrossRef CAS PubMed.
- F. Albericio, G. Barany, G. B. Fields, D. Hudson, S. A. Kates, M. H. Lyttle, N. A. Solé, Proceedings of the Twenty-Second European Peptide Symposium, Interlaken, Switzerland, 1992, vol. 22, pp. 191–192 Search PubMed.
- A. Trzeciak and W. Bannwarth, Tetrahedron Lett., 1992, 33, 4557–4560 CrossRef CAS.
- K. Barlos, D. Gatos, J. Kallitsis, G. Papaphotiu, P. Sotiriu, Y. Wenglng and W. Schiller, Tetrahedron Lett., 1989, 30, 3943–3946 CrossRef CAS.
- S. A. Kates, N. A. Solé, C. R. Johnson, D. Hudson, G. Barany and F. Albericio, Tetrahedron Lett., 1993, 34, 1549–1552 CrossRef CAS.
- G. Sabatino, M. Chelli, S. Mazzucco, M. Ginanneschi and A. M. Papini, Tetrahedron Lett., 1999, 40, 809–812 CrossRef CAS.
- M. Schnopp, S. Ernst and G. Haberhauer, Eur. J. Org. Chem., 2009, 2009, 213–222 CrossRef.
- L. He, L. Yang and S. L. Castle, Org. Lett., 2006, 8, 1165–1168 CrossRef CAS PubMed.
- W. K. Su, Z. Hong, W. G. Shan and X. X. Zhang, Eur. J. Org. Chem., 2006, 37, 2723–2726 CrossRef.
- (a) F. R. Benson, Theoretical principles of the chemistry of heterocycles, in Heterocyclic Compounds, John Wiley & Sons, Inc., New York, 1967, vol. 1 Search PubMed; (b) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles: Structures, Reactions, Synthesis, and Applications, John Wiley & Sons, Inc., New York, 2010 Search PubMed.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945–7950 CrossRef CAS PubMed.
- S. Berghmans, J. Hunt, A. Roach and P. Goldsmith, Epilepsy Res., 2007, 75, 18–28 CrossRef CAS PubMed.
- J. H. Toney, P. M. Fitzgerald, N. Grover-Sharma, S. H. Olson, W. J. May, J. G. Sundelof, D. E. Vanderwall, K. A. Cleary, S. K. Grant and J. K. Wu, Chem. Biol., 1998, 5, 185–196 CrossRef CAS PubMed.
- Y. Tamura, F. Watanabe, T. Nakatani, K. Yasui, M. Fuji, T. Komurasaki, H. Tsuzuki, R. Maekawa, T. Yoshioka and K. Kawada, J. Med. Chem., 1998, 41, 640–649 CrossRef CAS PubMed.
- C.-X. Wei, M. Bian and G.-H. Gong, Molecules, 2015, 20, 5528–5553 CrossRef CAS PubMed.
- K. Kaczmarek, S. Jankowski, I. Z. Siemion, Z. Wieczorek, E. Benedetti, P. Di Lello, C. Isernia, M. Saviano and J. Zabrocki, Biopolymers, 2002, 63, 343–357 CrossRef CAS PubMed.
- W. Vale, P. Brazeau, C. Rivier, M. Brown, B. Boss, J. Rivier, R. Burgus, N. Ling and R. Guillermin, Recent Prog. Horm. Res., 1975, 31, 365–397 CAS.
- S. Reichlin, N. Engl. J. Med., 1983, 309, 1495–1501 CrossRef CAS PubMed.
- V. M. Macaulay and D. N. Carney, Cancer Invest., 1991, 9, 659–673 CrossRef CAS PubMed.
- B. M. Evers, D. Parekh, C. M. J. Townsend and J. C. Thompson, Ann. Surg., 1991, 213, 190–198 CrossRef CAS PubMed.
- S. W. J. Lamberts, W. H. Bakker, J. C. Reubi and E. P. Krenning, N. Engl. J. Med., 1990, 323, 1246–1249 CrossRef CAS PubMed.
- (a) D. D. Beusen, J. Zabrocki, U. Slomczynska, R. D. Head, J. L. F. Kao and G. R. Marshall, Biopolymers, 1995, 36, 181–200 CrossRef CAS PubMed; (b) J. Zabrocki, U. Slomczynska and G. R. Marshall, in Proceedings of the 11th American Peptide Symposium: Peptides: Chemistry, Structure and Biology, ed. J. Rivier and G. R. Marshall, ESCOM, Leiden, 1990, pp. 195–197 Search PubMed.
- D. C. Palmer and S. Venkatraman, in The Chemistry of Heterocyclic compounds, Oxazoles: Synthesis, Reactions, and Spectroscopy: Part A, ed. D. C. Palmer DC, John Wiley & Sons, Inc.; Hoboken, New Jersey, 2003, vol. 60, pp. 138–149 Search PubMed.
- A. Rauf and N. N. Farshori, Springerbriefs in Molecular Science, 2011, vol. 3, pp. 9–14 Search PubMed.
- L. Swellmeen, Der Pharma Chemica., 2016, 8, 269–286 CAS.
- M. T. Chhabria, S. Patel, P. Modi and P. S. Brahmkshatriya, Curr. Top. Med. Chem., 2016, 16, 2841–2862 CrossRef CAS PubMed.
- D. Davyt and G. Serra, Mar. Drugs, 2010, 8, 2755–2780 CrossRef CAS PubMed.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munroa and M. R. Prinsep, Nat. Prod. Rep., 2014, 31, 160–258 RSC.
- Y. Hamada and T. Shioiri, Chem. Rev., 2005, 105, 4441–4482 CrossRef CAS PubMed.
- J. M. Caba, I. M. Rodriguez, I. Manzanares, E. Giralt and F. Albericio, J. Org. Chem., 2001, 66, 7568–7574 CrossRef CAS PubMed.
- B. McKeever and G. Pattenden, Tetrahedron Lett., 2001, 42, 2573–2577 CrossRef CAS.
- C. J. Moody and M. C. Bagley, J. Chem. Soc., Perkin Trans. 1, 1998, 601–607 RSC.
- A. Bertram and G. Pattenden, Synlett, 2001, 12, 1873–1874 CrossRef.
- Z. Xia and C. D. Smith, J. Org. Chem., 2001, 66, 3459–3466 CrossRef CAS PubMed.
- M. C. Bagley, K. E. Bashford, C. L. Hesketh and C. J. Moody, J. Am. Chem. Soc., 2000, 122, 3301–3313 CrossRef CAS.
- J. Deeley, A. Bertram and G. Pattenden, Org. Biomol. Chem., 2008, 6, 1994–2010 RSC.
- J. Li, S. Jeong, L. Esser and P. G. Harran, Angew. Chem., Int. Ed., 2001, 40, 4765–4770 CrossRef CAS PubMed.
- J. Li, A. N. G. Burgett, L. Esser, C. Amezcua and P. G. Harran, Angew. Chem., Int. Ed., 2001, 40, 4770–4773 CrossRef CAS PubMed.
- K. C. Nicolaou, M. Bella, D. Y. K. Chen, X. H. Huang, T. T. Ling and S. A. Snyder, Angew. Chem., Int. Ed., 2002, 41, 3495–3499 CrossRef CAS PubMed.
- K. C. Nicolaou, P. B. Rao, J. Hao, M. V. Reddy, G. Rassias, X. Huang, D. Y. K. Chen and S. A. Snyder, Angew. Chem., Int. Ed., 2003, 42, 1753–1758 CrossRef CAS PubMed.
- C. Alvarado, E. Díaz and A. Guzmán, Tetrahedron Lett., 2007, 48, 603–607 CrossRef CAS.
- S. Yu, X. Pa, X. Lin and D. Ma, Angew. Chem., Int. Ed., 2005, 44, 135–138 CrossRef CAS PubMed.
- A. Randazzo, G. Bifulco, C. Giannini, M. Bucci, C. Debitus, G. Cirino and L. Gomez-Paloma, J. Am. Chem. Soc., 2001, 123, 10870–10876 CrossRef CAS PubMed.
- C. D. Monica, A. Randazzo, G. Bifulco, P. Cimino, M. Aquino, I. Izzo, F. De Riccardis and L. Gomez-Paloma, Tetrahedron Lett., 2002, 43, 5707–5710 CrossRef.
- W. Li, A. Schlecker and D. Ma, Chem. Commun., 2010, 46, 5403–5420 RSC.
- K. C. Nicolau, D. W. Kim, D. Schlawe, D. E. Lizos, R. G. de Noronha and D. A. Longbottom, Angew. Chem., Int. Ed., 2005, 44, 4925–4929 CrossRef PubMed.
- S. Hara, K. Makino and Y. Hamada, Tetrahedron Lett., 2006, 47, 1081–1085 CrossRef CAS.
- D. Hernandez, E. Riego, A. Francesch, C. Cuevas, F. Albericio and M. Alvarez, Tetrahedron, 2007, 63, 9862–9870 CrossRef CAS.
- S. J. Kim and S. R. McAlpine, Molecules, 2013, 18, 1111–1121 CrossRef CAS PubMed.
- F. Yokokawa, H. Sameshima and T. Shioiri, Synlett, 2001, 42, 986–988 CrossRef.
- F. Yokokawa, H. Sameshima and T. Shioiri, Tetrahedron Lett., 2001, 42, 4171–4174 CrossRef CAS.
- C. A. Dvorak, W. D. Schmitz, D. J. Poon, D. C. Pryde, J. P. Lawson, R. A. Amos and A. I. Meyers, Angew. Chem., Int. Ed., 2000, 3, 1664 CrossRef.
- Y. Numajiri, T. Takahashi and T. Doi, Chem. - Asian J., 2009, 4, 111–125 CrossRef CAS PubMed.
- B. Zou, J. Wei, G. Cai and D. Ma, Org. Lett., 2003, 5, 3503–3506 CrossRef CAS PubMed.
- J. S. Davies, in Amino Acids, Peptides and Proteins, 1999, vol. 32, pp. 303–306 Search PubMed.
- K. Okumura, Y. Nakamura and C. g. Shin, Bull. Chem. Soc. Jpn., 1999, 72, 1561–1569 CrossRef CAS.
- E. Biron, J. Chatterjee and H. Kessler, Org. Lett., 2006, 8, 2417–2420 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Prof. Lutz Tietze on the occasion of his 75th birthday. |
| This journal is © The Royal Society of Chemistry 2018 |