
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIncorporation of antimicrobial peptides on electrospun nanofibres for biomedical applications†
Georgiana Amariei a,
Vanja Kokolb,
Karina Boltesa,
Pedro Letóna and
Roberto Rosal*a
a,
Vanja Kokolb,
Karina Boltesa,
Pedro Letóna and
Roberto Rosal*a
aDepartment of Chemical Engineering, University of Alcalá, E-28871 Alcalá de Henares, Madrid, Spain. E-mail: roberto.rosal@uah.es; Fax: +34 918855088; Tel: +34 918856395
bInstitute of Engineering Materials and Design, University of Maribor, SI-2000, Maribor, Slovenia
First published on 6th August 2018
Abstract
The aim of this work was to immobilize antimicrobial peptides onto a fibrous scaffold to create functional wound dressings. The scaffold was produced by electrospinning from a mixture of the water soluble polymers poly(acrylic acid) and poly(vinyl alcohol) and subsequently heat cured at 140 °C to produce a stable material with fibre diameter below micron size. The peptides were incorporated into the negatively charged scaffold by electrostatic interaction. The best results were obtained for lysozyme impregnated at pH 7, which rendered a loading of up to 3.0 × 10−4 mmol mg−1. The dressings were characterized using SEM, ATR-FTIR, elemental analysis, ζ-potential and confocal microscopy using fluorescamine as an amine-reactive probe. The dressings preserved their fibrous structure after impregnation and peptides were distributed homogeneously throughout the fibrous network. The antibacterial activity was assessed by solid agar diffusion tests and growth inhibition in liquid cultures using Staphylococcus aureus, a pathogenic strain generally found in infected wounds. The antibacterial activity caused clear halo inhibition zones for lysozyme-loaded dressings and a 4-fold decrease in S. aureus viable colonies after two weeks of contact of dressings with bacterial liquid cultures. The release profile in different media showed sustained release in acidic environments, and a rapid discharge at high pH values. The incorporation of lysozyme resulted in dressing surfaces essentially free of microbial growth after 14 days of contact with bacteria at pH 7.4 attributed to the peptide that remained attached to the dressing surface.
Introduction
Wound healing refers to the series of processes involved in the repair and restoration of tissues after suffering an external injury. During wound healing many early and late complications may arise, leading to significant morbidity and mortality.1 Among them, bacterial infections are of special concern. All wounds can be colonized by bacteria, which eventually form biofilm communities that protect them from host defences.2 Additionally, bacteria in wounds and ulcers are susceptible to developing multidrug resistance, which is a rapidly growing problem for healthcare systems.3 Normal skin is slightly acidic due to the characteristic compounds forming the stratum corneum, the outermost layer of the epidermis.4 It is also well-known that pH plays an important role in wound healing by influencing microbial activity, oxygen release or the creation of a new vasculature via angiogenesis.5 Specifically, alkaline pH is associated with higher infection and lower healing rates. In fact, pH control has emerged as a strategy in the treatment of wounds that can combine with traditional or new antiseptics into wound dressings.6Many synthetic and natural materials have been proposed to accelerate wound healing and to control the associated infections.7 In general, they are intended to mimic some of the characteristics of biological tissues and their choice is based on their biocompatibility and biodegradability.8 Wound healing materials comprise a wide range of natural and synthetic polymers either in a gel or fibrous form.9,10 In particular, positively charged polymers such as chitin and chitosan have been proposed due to their ability to interact with the negatively charged cell envelopes.11 However, the bioactivity of polymeric materials is limited to surface interaction and becomes deactivated by the nonspecific adsorption of proteins and undesired biomolecules.12 This drawback can be overcome by creating composite materials that not only modify interphase interaction, but also provide sustained and controlled release of therapeutic drugs.13
The loading of wound healing dressings with active substances is particularly suitable when systemic delivery may cause toxicity.8 Many biological or medicated dressings have been prepared that incorporate different agents intended to overcome the disadvantages of topical pharmaceuticals. Among them active dressings including antimicrobials and growth factors have been extensively studied.14,15 As antimicrobials, the use of conventional antibiotics, alone or in combination, is well documented.16 The inclusion of silver or silver nanoparticles has also been the subject topic of many studies because of their strong and broad-spectrum antimicrobial action.17 Antimicrobial peptides (AMP) are host-defence molecules expressed by multicellular organisms to control microbial proliferation and modulate the host's immune response. AMP play a key role as endogenous mediators of wound healing making their inclusion in active wound dressings an appealing strategy for the treatment of epithelial injuries.18 The interest in developing AMP as alternative antibacterial therapy has recently boosted due to the emergence of bacterial strains with increased resistance to conventional antibiotics.19,20
Electrospinning is a versatile method for the preparation of polymeric nanofibres in which a jet of charged fluid is ejected out of a capillary tube when the electric potential overcomes the surface tension of a molten or dissolved polymer.21 The thin jet of charged polymer draws out from the tip of a spinneret whipping in a fast moving spiral until it is recovered on a collector surface usually as a non-woven mesh.22 Electrospun fibres possess high pore interconnectivity, high surface area-to-volume ratio and easy surface functionalization.23 Due to these unique properties electrospun fibres have been proposed for uses in several biomedical applications including scaffolds for tissue engineering, medical implants, biosensors, and wound dressings.24,25
The functionalization of electrospun dressings with active substances has recently become an active research topic. Electrospun fibrous scaffolds benefit from their inherently small pores to inhibit microbial invasions and to control fluid drainage.26,27 Accordingly, a number of bioactive substances have been incorporated into electrospun scaffolds such as antibiotics, anti-inflammatory drugs or anticancer agents, among others.28–30 The incorporation of AMP into electrospun dressings by simple co-electrospinning or physical binding has been recently explored to improve wound healing with antimicrobial capacities.31,32 The functionalization of electrospun dressings with AMP is still a new formulation and further research should be conducted. Apart from operational questions such as dressing stability or releasing pattern, the possibility that AMP functionalized dressings may attract bacteria by electrostatic affinity or that regions with low charge density may allow live cells to adhere and create colonization points are issues that need to be addressed.33 Research is also needed on release ability and antimicrobial efficiency at the timescales required in real applications that are usually larger than those typically studied in laboratory tests.34 This work aims at assaying long term antimicrobial efficiency and release profiles of AMP-functionalized dressings prepared from poly(acrylic acid)–poly(vinyl alcohol) blends. Up to our best knowledge, poly(acrylic acid) has not yet been reported as base material for this purpose.
Poly(acrylic acid) (PAA) and polyvinyl alcohol (PVA) are water-soluble polymers that produce hydrogels with potential biomedical applications.35,36 PAA and PVA can be mutually crosslinked to produce water-stable materials with different swelling behaviour depending on their relative amounts and the annealing conditions.37 Both PAA and PVA have been used to prepare electrospun fibres in the nano- to few micrometre ranges. Electrospun PVA has been frequently proposed for wound dressings as it benefits from its excellent biocompatibility. Up to our knowledge PAA-containing nanofibrous materials have not been previously reported for that purpose. The relevance of this approach is that PAA-based polymers and blends impart strong antimicrobial activity to the resulting material. The effect is due to the sequestration of the divalent cations that stabilize prokaryotic membranes by carboxylic acid moieties.38
In this work, we prepared electrospun submicron fibres from PAA–PVA blends and functionalized their surface with two AMP to create antimicrobial wound dressings. The antibacterial activity assessment was performed using Staphylococcus aureus, which is the most commonly isolated bacterium in infected wounds also able to penetrate into bloodstream and internal organs.39,40 The antimicrobial assessment was performed using agar diffusion tests and liquid cultures in contact with AMP-loaded dressings. The release profile in different media was also studied during two-week periods.
Experimental
Materials
Poly(vinyl alcohol) (PVA, MW 89–98 kDa, 99+% hydrolysed) and poly(acrylic acid) (PAA, MW 450 kDa), and fluorescamine were purchased from Sigma-Aldrich. Lysozyme (L, from chicken egg white, MW 14.3 kDa, isoelectric point 10.9) and nisin (N, from Lactococcus lactis, MW 3.35 kDa, isoelectric point 8.8) were also acquired to Sigma-Aldrich. Nisin impairs bacteria by producing lesions in the cytoplasmic membrane, and lysozyme hydrolyses the peptidoglycan wall by breaking the β-1,4-glycosidic bonds between N-acetylmuramic acid and N-acetylglucosamine, both resulting in cell lysis.41,42 Ultrapure water (Millipore Milli-Q System) with a resistivity of at least 18 MΩ cm was used in all experiments. The components of culture media and buffers were purchased to Conda-Pronadisa (Spain).Preparation of electrospun fibres
The PAA–PVA electrospun fibres used for the immobilization of AMP were prepared as described elsewhere.38 Briefly, the spinning solution was a polymeric mixture of 8 wt% PAA and 15 wt% PVA in ultrapure water resulting in 35 wt% PAA and 65 wt% PVA in final dry fibres. Prior to electrospinning, the solution was stirred for 2 h at 25 °C and degassed. The solution was dispensed using a 5 mL syringe with a 23-gauge stainless steel blunt-tip needle and electrospun using the following parameters: working distance 23 cm, flow rate 0.8 mL h−1, voltage 23 kV, RH 40%, temperature 25 °C. A drum collector (PDrC-3000, Yflow Nanotechnology Solutions, Spain) rotating at 100 rpm was used to recover the nonwoven materials, which were subsequently dried at 50 °C, for 24 h.The electrospun fibres were crosslinked at 140 °C for 30 min, washed with distilled water, and dried under vacuum (10 kPa, 50 °C, 24 h). After the crosslinking process, specimens of the electrospun materials were kept in water for 24/48 h and the stability assessed by measuring the release of non-crosslinked materials in water as NPOC. The results showed that the amount of carbon released was <10 mg NPOC g−1 (equivalent to <1 wt%) after 24 h in water and <0.5 mg NPOC g−1 (equivalent to <0.05 wt%) during the following 24 h. Accordingly, the crosslinked electrospun fibres used for AMP impregnation were previously preconditioned in water for 48 h, to produce a water-insoluble material. The amount of carboxyl groups per unit mass of dry PAA/PVA fibres was 7.9 ± 0.3 mmol COOH per g of electrospun material determined by titration as explained below.
Immobilization of AMP onto electrospun fibres
The immobilization was performed by electrostatic adsorption. Pieces of about 50 mg of dry PAA–PVA fibres were immersed in phosphate (pH 7) or carbonate (pH 10) buffer solution containing AMP with molar ratio 0.5, 1 and 2 with respect to the carboxyl groups of PAA–PVA (7.9 mmol g−1). The choice of pH was based on the isoelectric points of the AMP as explained below. The mixture was kept under constant shaking (100 rpm) for 24 h at room temperature, after which the samples were removed and rinsed with ultrapure water to eliminate non-adsorbed AMP. Finally, the specimens were vacuum dried at room temperature for 24 h. The amount of AMP loaded per unit mass of dressing was calculated based on the total nitrogen content of the samples.Characterization of dressings
The morphology and diameter of the fibrous materials were determined using scanning electron microscopy (SEM, DSM-950 Zeiss, Oberkochen, Germany) operating at 15 kV on gold sputter-coated samples. Fibre diameters were determined using the ImageJ software (National Institute of Health, USA), with 50 measurements per sample. Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) spectra were obtained in the 4000–650 cm−1 range using a Thermo-Scientific Nicolet iS10 apparatus with a Smart iTR-Diamond ATR module. Total Organic Carbon (TOC) was measured as NPOC (Non-Purgeable Organic Carbon) using a Shimadzu, TOC-VCSH analyser.The amount of carboxyl groups per unit mass of fibres was measured by acid–base titration of carboxyl groups. Briefly, the samples were weighed and protonated using 0.1 M HCl for 24 h, washed with deionized water to remove excess HCl and immersed in 0.1 M NaOH for 24 h. The resulting NaOH solution was then titrated with 0.1 M HCl and the result expressed as moles of carboxyl groups per unit mass of dry sample. The experiments were carried out under nitrogen atmosphere. The nitrogen content of the samples was determined by elemental analysis using a LECO CHNS/O-932 equipment, which allowed calculating the amount of AMP per unit mass of dressing.
Surface ζ-potential measurements of neat and functionalized materials were performed using dynamic light scattering in a Zetasizer Nano-ZS apparatus equipped with the ZEN 1020 Surface Zeta Potential (Malvern Instruments Ltd., UK). A small section of the dressing was glued to the sample holder and inserted into a disposable 10 mm plastic cuvette containing 10 mM KCl, pH 7.0 with of 0.5 wt% PAA (450 kDa) as tracer. Measurements were conducted at 25 °C at six different distances the surface.
The presence of immobilized peptides on the surface of electrospun fibres was revealed using the fluorescamine assay, a coupling reaction of a reactive dye with primary amino groups.43 Briefly, 1 cm2 of each pre-wetted sample was put in contact with fluorescamine solution (100 μg mL−1 in acetone) in a Petri dish (10 μL fluorescamine solution per cm2) for 15 minutes at room temperature. The fluorescein isothiocyanate-labelled peptides were visualized by laser scanning confocal microscope (LSCM, Carl Zeiss LSM5100, Germany), at 365 nm excitation and 470 nm emission wavelengths.
Antibacterial and antibiofilm activity
The Gram-positive S. aureus (CETC 240) bacteria was used as reference strain for antibacterial testing. The microorganisms were preserved at −80 °C in glycerol (20% v/v) until use. Reactivation was performed in nutrient broth (NB, 10 g L−1 peptone, 5 g L−1 sodium chloride, 5 g L−1 meat extract and, for solid media, 15 g L−1 powder agar, pH 7.0 ± 0.1) at 30 °C under agitation (150 rpm) and routinely tracked by measuring optical density (OD) at 600 nm. The antimicrobial and antifouling effects of composite materials were evaluated according to the standardized ISO 22196 test, followed with minor modifications. Qualitative and quantitative assessments were employed for the bacterial inhibition studies using agar diffusion and liquid incubation methods, respectively.44For agar diffusion tests, neat and functionalized dressings (∼1 cm2) were placed onto the surface of soft agar plates previously inoculated with 0.6 mL S. aureus suspension with approximately 106 Colony Forming Units (CFU) per mL. Plates were incubated at 37 °C for 14 days. At pre-determined time intervals, the diameter of the inhibition zones was recorded, and the plates were digitally photographed as a measurement of antibacterial activity. All experiments were repeated three times.
The liquid incubation tests were carried out for neat and functionalized specimens. In brief, the samples (accurately weighed, ∼1 cm2 or 12.5 mg) were placed into the wells of sterile 6-well plates. Bacterial suspensions with a concentration of approximately 106 CFU mL−1 were pipetted into each well (0.4 mL mg−1) and incubated at 37 °C for 14 days. Control experiments were run in parallel without electrospun material and treated in the same way. After exposure, the fibrous material was transferred to clean 6-well plates containing each 2 mL phosphate buffer saline (PBS) and orbitally shaken at 5 °C for 15 min to remove non-adhered cells. The cells detached from the surface were recuperated using 2 mL per well SCDLP broth (soybean casein digest broth with lecithin and polyoxyethylene sorbitan monooleate) followed by 30 min shaking according to the ISO 22196. The cells from the supernatant liquid in contact with the surface and the cells removed from their surface were plate counted. Briefly, the aliquots were placed in sterile 96 well microtiter plates in 10-fold serial dilutions in PBS. Replicated 10 μL spots were plated on Petri dishes containing NB agar-medium as described above. After 24 h incubation at 37 °C, CFU were counted using a CL-1110 counting instrument (Acequilabs, Spain). For colony number estimations, at least three replicates of at least two serial dilutions were considered.
Confocal laser scanning microscopy (CLSM) was used to assess bacterial viability. The bacteria were stained with a Live/Dead kit (Live/Dead BacLight viability kit, Thermo Fisher, USA). This method differentiates viable and no-viability cells using Syto9, a fluorescent nucleic acid stain capable to penetrate cell membrane and bind DNA, and propidium iodide (PI), which is a fluorescent stain marking only membrane-damaged non-viable cells. The excitation/emission maxima were 480/500 nm for Syto9 and 490/635 nm for PI. The micrographs were obtained in a Leica Microsystems Confocal SP5 fluorescence microscope.
For SEM images, samples were washed twice with PBS, fixed in glutaraldehyde (2.5%) for 2 hours, and then dehydrated with gradient ethanol (30–100%) and acetone. Each sample was dried with hexamethyldisilazane for 15 minutes prior to sputter coating with gold for SEM observation.
In vitro release of lysozyme
The in vitro lysozyme release from functionalized materials was carried out in phosphate buffer saline (PBS, pH 7.4), acetate buffer (AB, pH 3.5) and carbonate buffer (CB, pH 10.0), as release media. Samples (approx. 50 mg) were placed into 125 mL flasks with 100 mL of the appropriate medium and incubated at room temperature under mild agitation. The medium was completely replaced with fresh one at predetermined time intervals. The concentration of lysozyme in solution was quantified by HPLC using an Agilent LC 1260 system, equipped with a 1260 Quaternary pump VL, 1260 DAD detector and automatic 1260 ALS injector. The automatization of analysis was done using the Agilent OpenLAB program. The column used was a Protein Green C4 5 μm, 150 × 4.6 mm purchased from Analisis Vínicos (Spain). A linear gradient of acetonitrile with 0.1% TFA in 0.1% TFA–water was used from 20% to 80% for 45 min with a flow rate of 1 mL min−1 at ambient temperature. Injection volume was 20 μL, and the detector wavelength was 210 nm. Under these conditions, lysozyme was eluted at 16.6 min and could be accurately measured in two concentration ranges: low range, below 20 mg L−1 with detection limit 1.66 mg L−1 and quantification limit 5.54 mg L−1, and high range, up to 800 mg L−1 with detection limit 20.6 mg L−1 and quantification limit 68.8 mg L−1. All measurements were performed in triplicate. The results were presented in terms of cumulative release as a function of release time.Results and discussion
Preparation and characterization of dressings
AMP immobilization was performed by electrostatic adsorption. The self-assembly of peptides is known to be mediated by weak intermolecular forces and depends on several factors such as peptide nature (amino-acid structure, molecular weight and size) and concentration as well as solvent properties (including pH) and the type of substrate.45 In this work, AMP concentration varied using molar ratios 0.5, 1 and 2 with respect to the carboxyl groups of PAA–PVA, while pH was adjusted using two different buffers: phosphate buffer at pH 7 and carbonate buffer at pH 10. The results are shown in Table 1 that indicates the amount of peptide loaded per unit mass of dressing together with surface charge.| Dressings | mmol AMP per mg | Surface ζ-potentiala (mV) | ||
|---|---|---|---|---|
| pH 7 | pH 10 | pH 7 | pH 10 | |
| a Measured at pH 7. The surface ζ-potential of neat PAA–PVA fibres was −36.5 ± 1.2 mV. | ||||
| N_1@PAA–PVA | 8.97 × 10−6 ± 5.3 × 10−7 | 7.41 × 10−6 ± 9 × 10−8 | −31.4 ± 1.3 | −34.1 ± 1.1 |
| N_2@PAA–PVA | 1.14 × 10−5 ± 2 × 10−7 | 6.5 × 10−6 ± 2.1 × 10−6 | −27.8 ± 0.8 | −31.6 ± 2.4 |
| N_3@PAA–PVA | 1.81 × 10−5 ± 6.8 × 10−6 | 8.17 × 10−6 ± 3.0 × 10−7 | −20.8 ± 2.7 | −31.0 ± 3.4 |
| L_1@PAA–PVA | 2.03 × 10−4 ± 5.6 × 10−5 | 1.56 × 10−5 ± 6 × 10−7 | −15.6 ± 0.6 | −28.1 ± 1.7 |
| L_2@PAA–PVA | 2.96 × 10−4 ± 8 × 10−6 | 2.54 × 10−5 ± 1.1 × 10−6 | −9.1 ± 1.8 | −25.1 ± 2.0 |
| L_3@PAA–PVA | 3.00 × 10−4 ± 2.5 × 10−5 | 3.9 × 10−5 ± 1.4 × 10−5 | −8.5 ± 1.4 | −22.7 ± 0.9 |
Table 1 shows that significant immobilization took place for both AMP, which can be explained in terms of the electrostatic interaction between the negatively charged PAA–PVA scaffold (ζ-potential −36.5 ± 1.2 mV as a consequence of dissociated carboxyl groups) and the positive charges of both peptides. Nisin consists of 34 amino acids including three lysine and two histidine residues. Lysozyme contains lysine, histidine, and cysteine residues. The protonation of their residual amino groups leads to an overall positive charge with 4 and 8 positively charged groups respectively at neutral conditions.46,47 The net positive charge of nisin and lysozyme allows their immobilization by adsorption onto negatively charged solid supports due to electrostatic interaction.48,49 In addition to electrostatic forces, the adsorption mechanisms between peptides and the backbone of PAA–PVA can be mediated by non-electrostatic interactions such as hydrogen bonding and dipole–dipole interactions with the polar groups in.44,50,51
For both peptides the amount loaded was considerably higher at neutral (pH 7) than at basic conditions (pH 10), and higher for lysozyme than for nisin. These results can be interpreted as a consequence of a decrease in the net charge of peptides, which is positive if pH is lower than the isoelectric point and negative if it is higher and supports the preferential role of electrostatic interactions in the adsorption of AMP on PAA–PVA fibres. Nisin has an isoelectric point of 8.8 and is negatively charged at pH 10.52 Lysozyme molecule is neutral at pH 10.9 and, therefore, no strong electrostatic interaction would be expected with PAA–PVA at pH 10.49 At neutral or slightly basic conditions, PAA–PVA was negatively charged leading to higher AMP loadings of the more molecules with hither isoelectric point, i.e.: those bearing higher positive charge, which is consistent with the fact that lysozyme was immobilized in considerably greater extension than nisin. The extent of loading increased with initial AMP concentration, and no further increase was observed for concentrations above 1 mol AMP per mol COOH, which can be interpreted as the result of surface saturation. Because of AMP adsorption, the ζ-potential of functionalized dressings became less negative, increasing up to 28 mV from the values obtained for PAA–PVA scaffold. Significantly, the amount of lysozyme loaded at pH 7 was directly related with the surface zeta potential of the resulting dressing. AMP bore a positive charge at acidic pH, but the large excess of carboxyl groups in PAA–PVA made the overall charge of the dressings negative even for the highest loadings.
Fig. 1 and S1 (ESI†) show the ATR-FTIR spectra of PAA–PVA fibres before and after AMP loading. The spectra of PAA–PVA revealed the characteristic peaks associated with PAA and PVA. The broad O–H stretching band (3200–3600 cm−1), the C–H alkyl stretching (2850–3000 cm−1), and the C–O stretching (1142 cm−1) are characteristic features of PVA. In addition, the typically carboxyl stretching frequency of PAA was observed at 1700 cm−1, while the symmetric and antisymmetric stretching frequencies of the carboxylate ion appeared at 1420 and 1560 cm−1.53 The commercial nisin preparation presented a broad band in the 3200–3600 cm−1 region resulting from N–H and O–H stretching vibrations that slightly increased from N_1@PAA–PVA to N_3@PAA–PVA. The bands in the 1720–1580 cm−1 region are attributed to the amide bands54 and increased with the amount of loaded nisin. The absorption band at 1620 cm−1 is associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching combined with N–H deformation of amide I group.55 Lysozyme exhibits characteristic symmetrical CO amide I peak at 1640 cm−1. The N–H amide II peak of the polypeptide chain is also clear at 1525 cm−1.56 These absorption bands typical of peptides were detected in all AMP functionalized dressings and, together with the bands corresponding to O–H and C–H stretching, increased with AMP loading as shown in Fig. 1 and S1.†
O stretching combined with N–H deformation of amide I group.55 Lysozyme exhibits characteristic symmetrical CO amide I peak at 1640 cm−1. The N–H amide II peak of the polypeptide chain is also clear at 1525 cm−1.56 These absorption bands typical of peptides were detected in all AMP functionalized dressings and, together with the bands corresponding to O–H and C–H stretching, increased with AMP loading as shown in Fig. 1 and S1.†
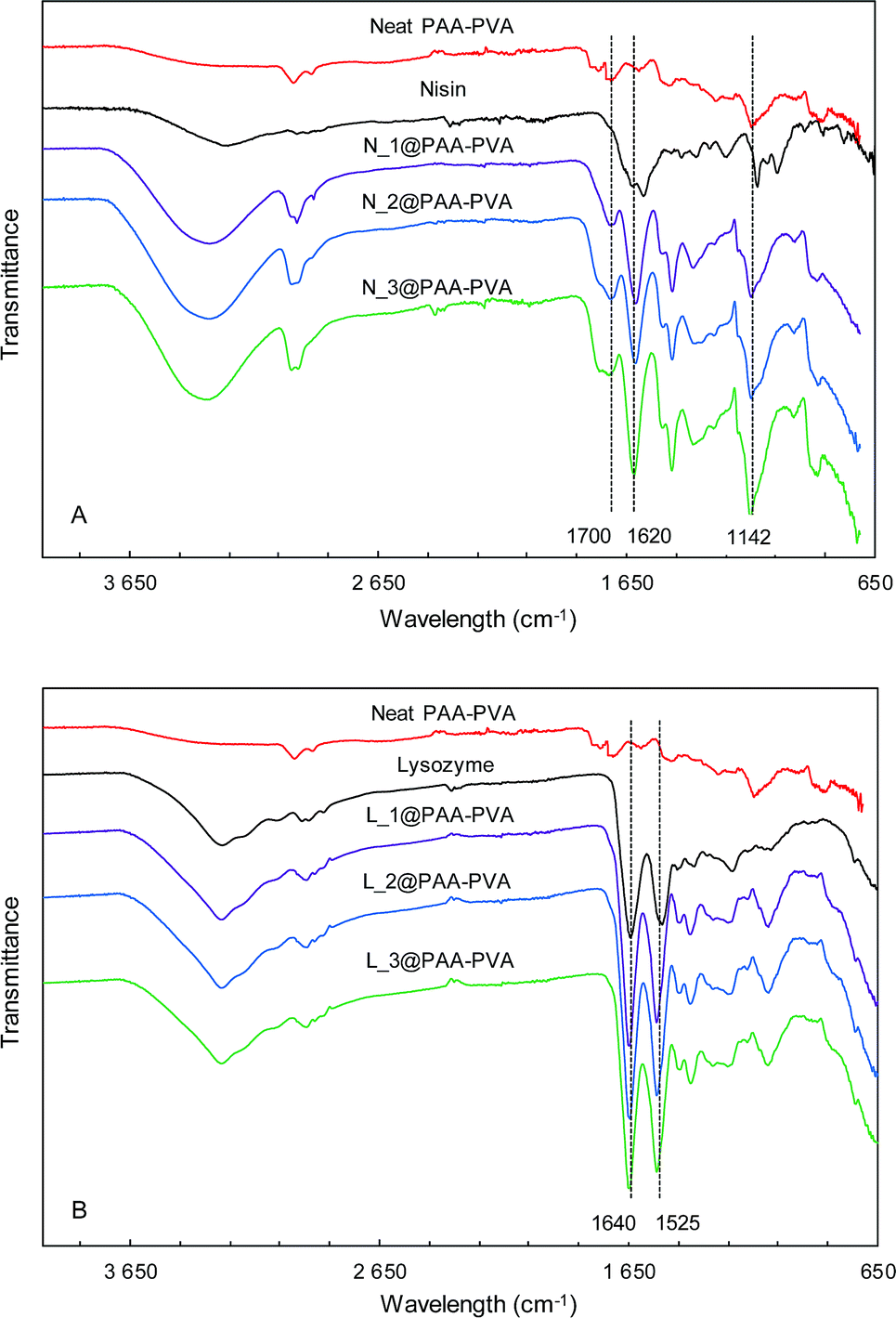 | ||
| Fig. 1 FTIR-ATR spectra of PAA–PVA functionalized dressings. Nisin (A) and lysozyme (B) functionalized at pH 7 (the spectra for dressings functionalized at pH 10, are shown in Fig. S1†). | ||
The morphology of neat and AMP loaded-dressings is shown in the SEM images of Fig. 2. All materials displayed well-preserved fibrous structure and little difference between neat and AMP functionalized dressings. The diameter of the fibres was 970 ± 170 (50 measurements). Fig. S2 (ESI†) presents a full set of SEM images at different magnifications.
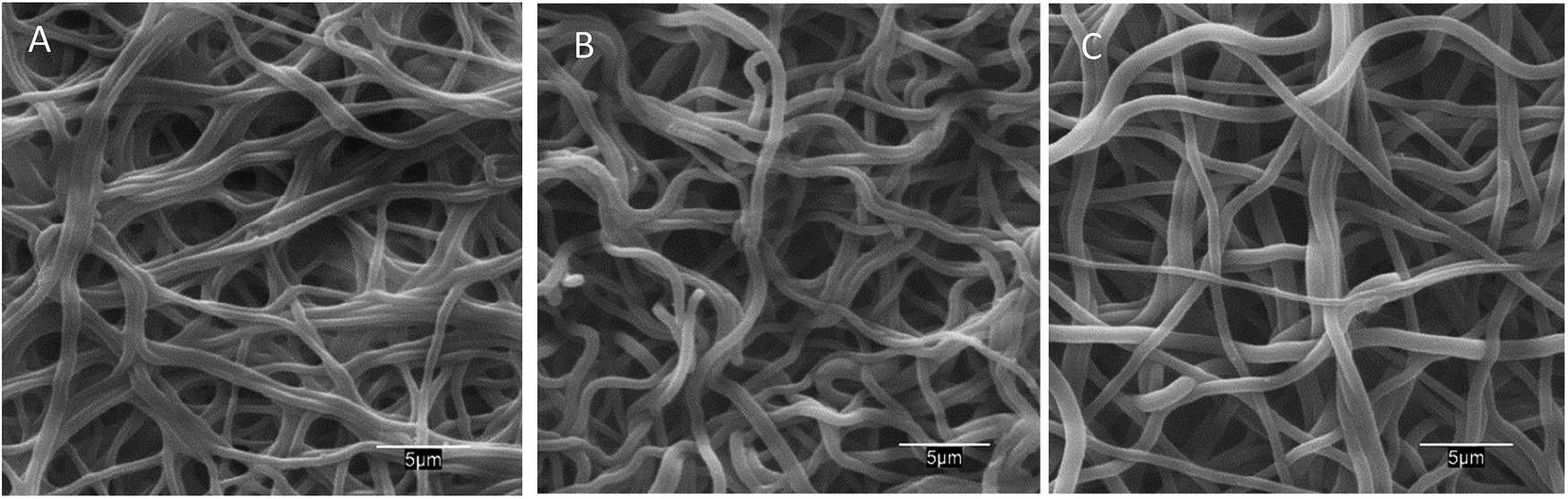 | ||
| Fig. 2 SEM images of PAA–PVA fibres before impregnation (A), and dressings after impregnation (pH 7) nisin (N_2@PAA–PVA, (B)) and lysozyme (L_2@PAA–PVA, (C)). | ||
Fig. 3 shows confocal laser scanning microscopy (CLSM) images of the fibrous materials stained with the amine-reactive dye fluorescamine. The fluorescence, absent in PAA–PVA, can be clearly observed in AMP loaded materials confirming the presence of amino groups with relatively homogeneous distribution. Fig. S3 (ESI†) shows a full set of CLSM images of the AMP loaded dressings prepared with different initial peptide concentrations and pH conditions. Nisin and lysozyme appeared well distributed through the fibrous dressing network, the fluorescence increasing with the AMP loading. The higher fluorescence emission was recorded for impregnation at pH 7 and was particularly high for lysozyme, which is consistent with the results shown in Table 1. Lower but significant fluorescamine emission was obtained for N_2/3@PAA–PVA at pH 7. Concerning lysozyme, a regular pattern of aggregates appeared for dressings prepared at pH 10 (Fig. S3†).
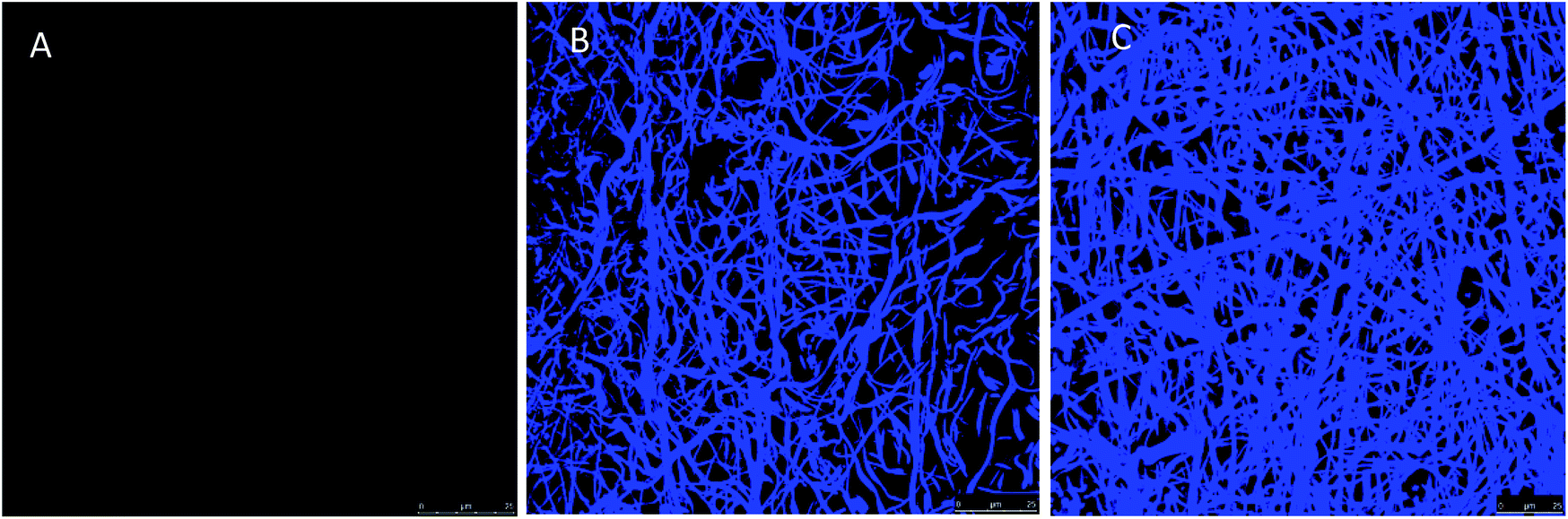 | ||
| Fig. 3 Confocal laser scanning microscopy images of PAA–PVA fibres (A), nisin-loaded PAA–PVA (N_3@PAA–PVA, pH 7, (B)) and lysozyme-loaded PAA–PVA (L_3@PAA–PVA, pH 7, (C)) after conjugation with fluorescamine. | ||
Antibacterial activity
The antimicrobial effect of AMP loaded dressings against the Gram-positive bacterium S. aureus is shown in Fig. 4 based on the inhibition zones recorded in agar diffusion tests. The figure shows quantitative results for lysozyme dressings impregnated at pH 7 and two representative images of inhibition zone experiments for nisin and lysozyme-loaded and neat PAA–PVA materials. A full set of images for all the materials listed in Table 1 is presented in Fig. S4 (ESI†). No significant inhibition zone could be recorded for neat PAA–PVA or for nisin-loaded dressings. Lysozyme-loaded composite materials exhibited significant antibacterial activity with inhibition zones that increased with incubation time. The surface free of bacterial growth more than doubled that of the dressings themselves after 14 days. Fig. 4A shows that the amount of lysozyme loaded onto the PAA–PVA fibrous material did not result in enhanced antimicrobial performance in the agar diffusion tests, which was attributed to mass transfer limitations and the fact that AMP molecules must first desorb from the PAA–PVA surface and then migrate through the agar surface before interacting the microorganisms.57 Nisin loaded dressings did not show appreciable halo inhibition in solid agar experiments in spite of its low minimum inhibitory concentration. The minimum inhibitory concentration of nisin to S. aureus was about 10 μg mL−1.58,59 If all nisin content of the wounds assayed in this work dispersed evenly in the agar plates, the concentration would be in the 6–12 μg mL−1, which roughly corresponds to the minimum inhibitory concentration. Therefore, the absence of inhibition halo in nisin-loaded dressings could be explained if the higher mobility of nisin as a result of its lower molecular weight resulted in a rapid diffusion throughout the agar plate. Lysozyme is a molecule larger than nisin and possess more amino groups that can dissociate and interact with the carboxylate moieties in the fibre backbone. Therefore, its mobility once attached to functionalized dressings should be lower. The amount of AMP available for antibacterial activity in halo inhibition tests is related to their capacity to desorb from functionalized surfaces. Consequently, the higher effect observed for lysozyme-loaded dressings can be interpreted as a result of the balance between adsorption strength and release capacity.44 The limited diffusion of lysozyme explains its antimicrobial effect because a rapid diffusion from the loaded dressings would quickly lead the concentration of lysozyme below its minimum inhibitory concentration. The higher antibacterial activity observed for the lysozyme-containing dressings prepared at pH 7 can be attributed to their higher AMP content, one order of magnitude higher than for dressings prepared at pH 10.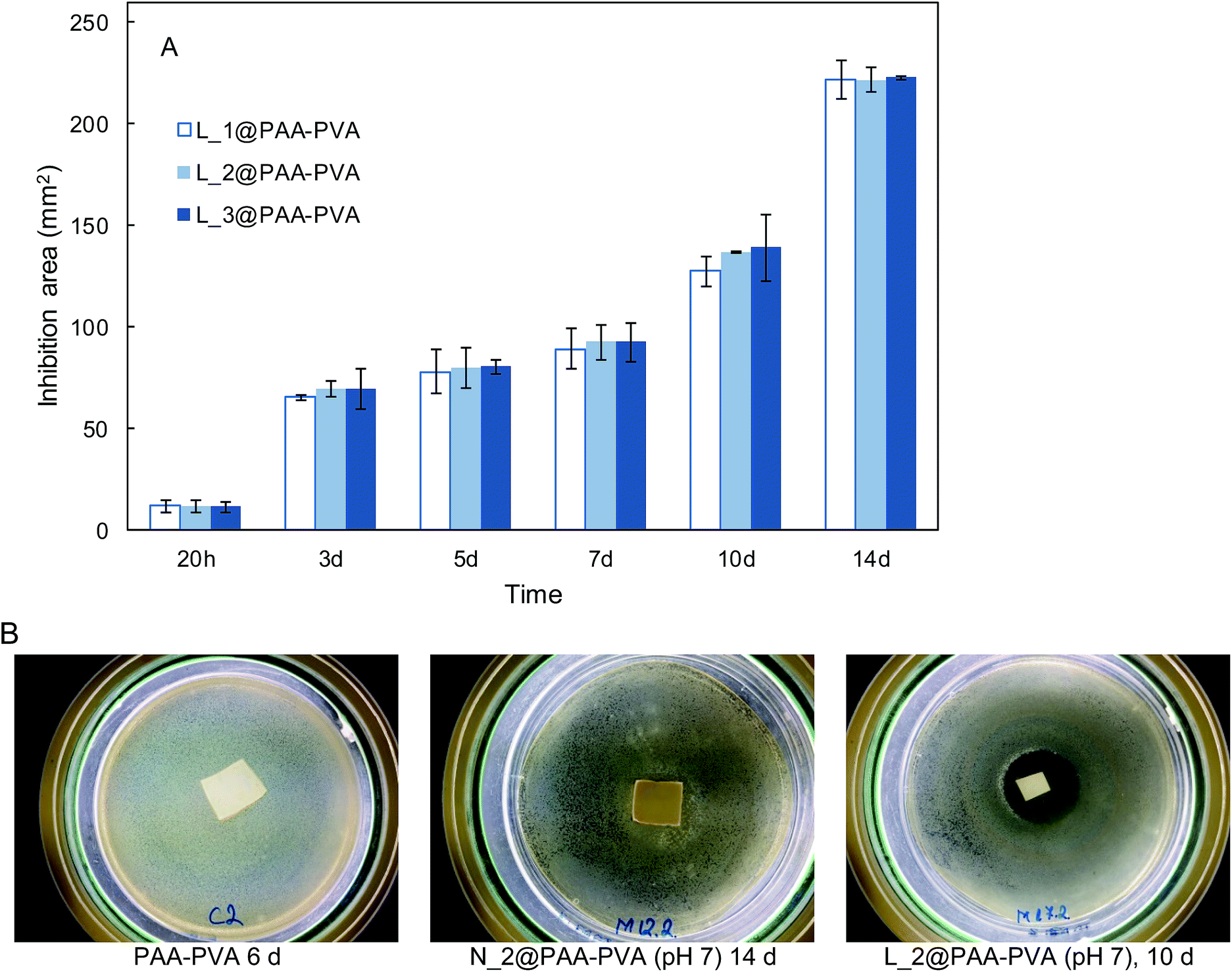 | ||
| Fig. 4 Halo inhibition zone expressed in mm2 for L_1@PAA–PVA, L_2@PAA–PVA and L_3@PAA–PVA (pH 7) along 14 day experiments with cultures of S. aureus in agar plates at 37 °C (A). Representative images of inhibition experiments corresponding to neat PAA–PAA, N_2@PAA–PVA at pH 7 and L_2@PAA–PVA at pH 7 (B). | ||
In view of the results from the agar diffusion tests, the formulation L_1@PAA–PVA was selected to perform liquid incubation tests with quantitative CFU measurements (Fig. 5). Lysozyme loaded fibres exhibited higher inhibition than the neat PAA–PVA scaffold with significant differences between 3 (72 h) and 5 days. The results are shown in Fig. 5 with single asterisks marking significant differences between loaded and non-loaded dressings and double asterisks indicating significant differences with respect to specimens in contact with cultures for 24 h. Fig. 5A corresponds to the liquid in contact with dressings incubated with bacterial suspensions (106 CFU mL−1, 0.4 mL mg−1 dressing). It was previously reported that PAA containing polymers exhibit considerable antimicrobial activity in contact with bacterial cultures.60 The effect was attributed to the chelation by carboxylate moieties of the divalent cations of bacteria in contact with dressings.38 Fig. 5B refers to cells detached from the surface using SCDLP broth as indicated in the ISO 22196. The antimicrobial effect of PAA-containing fibres was clear in liquid cultures, with CFU decreasing several orders of magnitude. However, as indicated in Fig. 5B, the dressings without lysozyme were still colonized by S. aureus: while PAA–PVA suffered significant surface colonization after 14 days, lysozyme-loaded dressings were much less colonized (>105 CFU g−1). The differences can be interpreted as a consequence of the tendency of S. aureus to form biofilms. When lysozyme is present, even after a prolonged exposure, the dressing kept its capacity to avoid bacterial colonization. The tendency to staphylococci to form biofilms is a well-known survival strategy because adopting a sessile mode of life, biofilm-embedded microorganisms benefit from a number of advantages over their planktonic counterparts.61
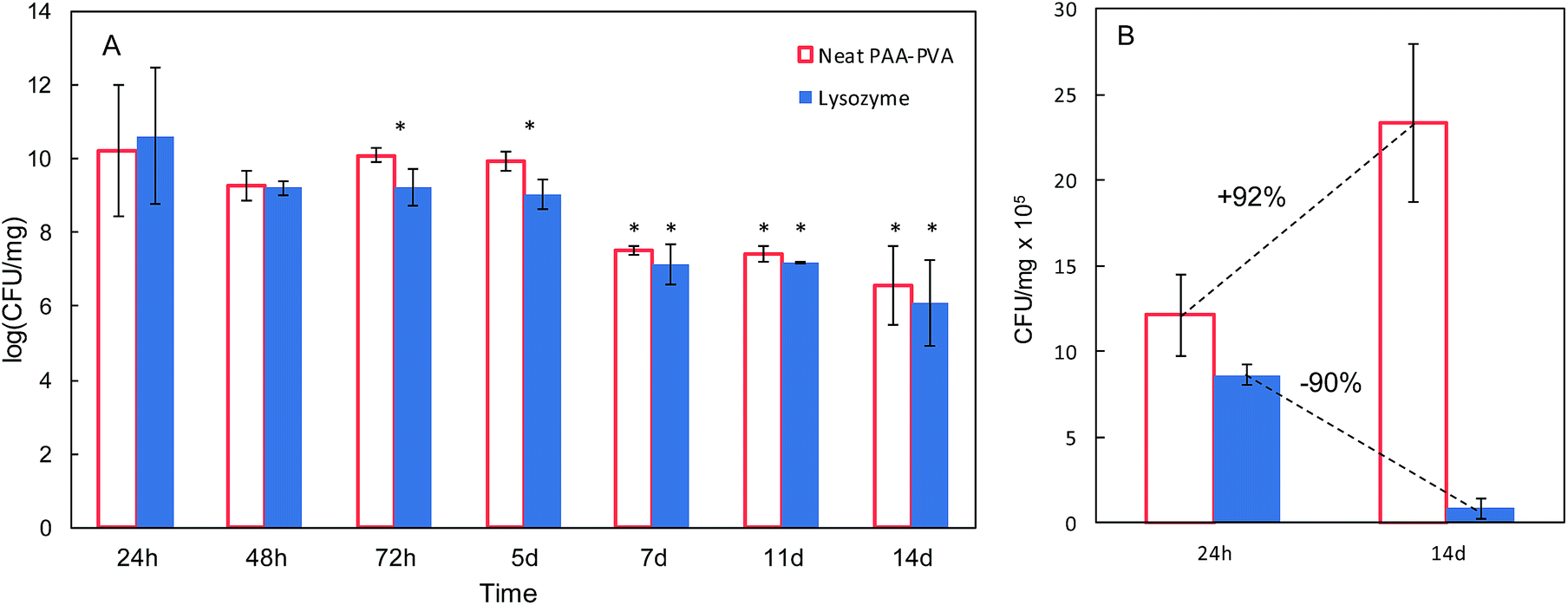 | ||
| Fig. 5 Colony-forming units (CFU) in liquid media in contact with PAA–PVA and L_1@PAA–PVA dressings (A) and for microorganisms detached from dressings (same dressings, after 24 h and 14 days, (B)). | ||
The antibiofilm effect against S. aureus colonization is apparent from the SEM images of dressings exposed to bacterial growth shown in Fig. 6. While S. aureus biofilms appeared clearly on the surface of PAA–PVA after 14 days exposed to cultures, lysozyme-loaded dressings were essentially free of colonization, with only a few S. aureus cells visible into the fibrous matrix but without signs of the extracellular structure of biofilms even after 2 weeks in contact with cells.
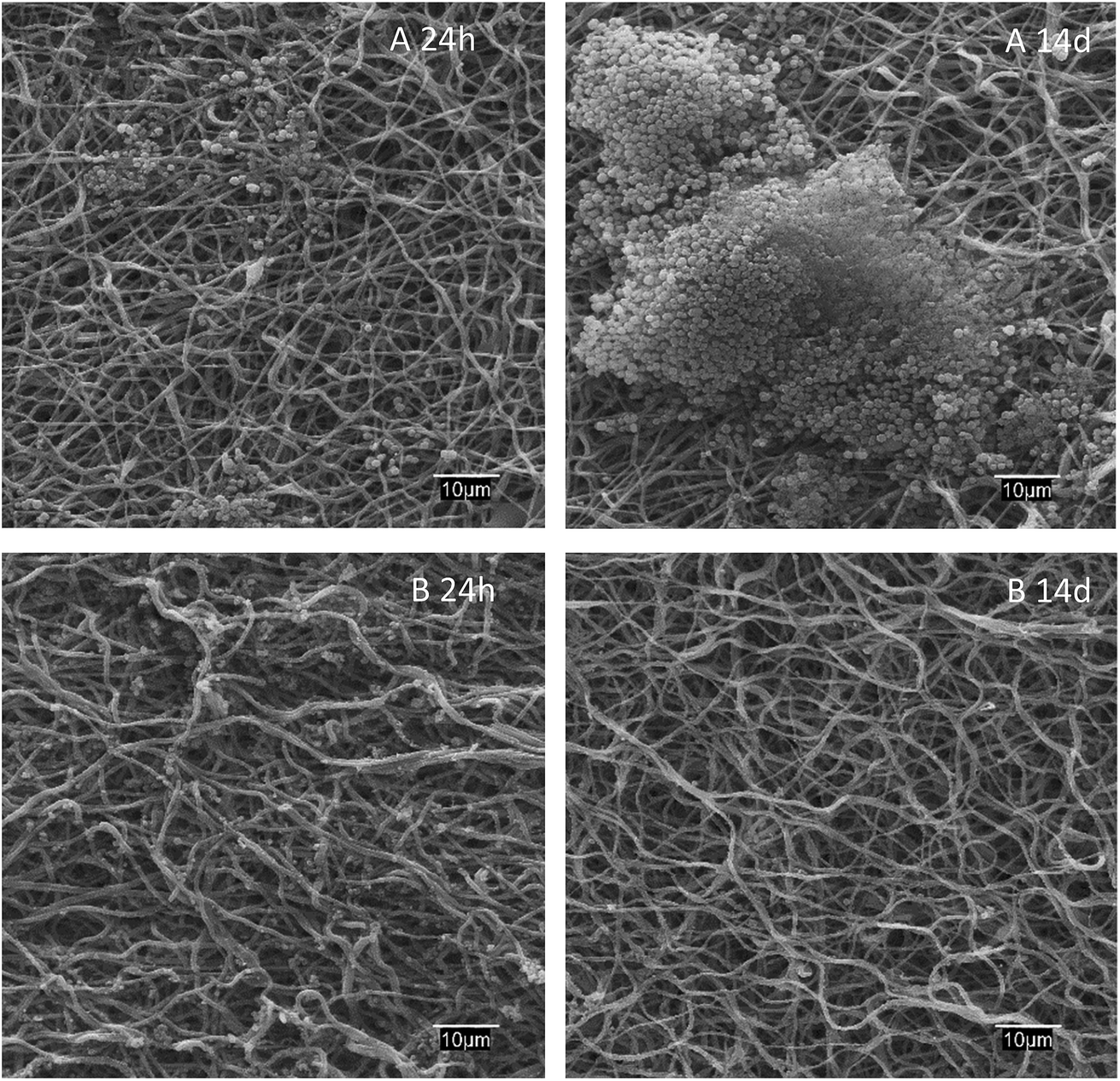 | ||
| Fig. 6 SEM micrographs of PAA–PVA (A) and lysozyme loaded L_1@PAA–PVA (B) dressings after 24 h and 14 days in contact with S. aureus cultures at 37 °C. | ||
Lysozyme release
Fig. 7 shows the lysozyme release profiles from L_1@PAA–PVA electrospun dressings in contact with different media during the same two-week period used for antimicrobial tests. The results showed that lysozyme rapidly released in basic media. During the first 2 h, the cumulative percentage of lysozyme released reached 20%, 71% and 88% for AB (pH 3.4), PBS (pH 7.4) and CB (pH 10.0), respectively. While dressings in CB and PBS displayed a high lysozyme discharge after 48 h, the release in AB at pH 3.5 was considerably slower, and about 40% of lysozyme remained in the dressing after 14 days. A similar profile consisting of an initial burst followed by a sustained release has been previously described for lysozyme-loaded chitosan-based nanofibrous dressings.62 It is to be noticed that this pattern is appropriate for the control of wound infections. While a rapid release of antimicrobial peptides during the first hours would kill most injury associated pathogens, a slow and constant release over the following few days would keep infection under control.63 As discussed above, PAA–PVA incorporated AMP via electrostatic interaction and the self-assembly onto the surface of electrospun fibres would weaken when pH approached the pKa value of lysozyme of about 10.9 at which the molecule becomes neutral.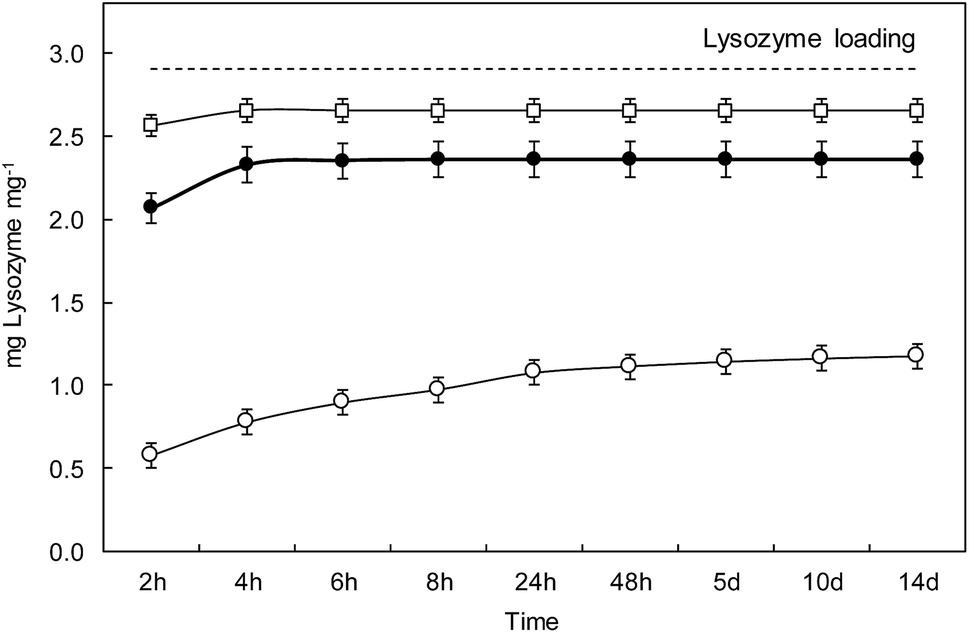 | ||
| Fig. 7 Release of lysozyme in different media: phosphate buffer saline pH 7.4 (●), carbonate buffer pH 10.0 (□) and acetate buffer pH 3.5 (○). The dashed line represents the lysozyme loading of specimens L_1@PAA–PVA. | ||
The choice of release media was based on wound pH arguments. The pH environment of open wounds varies from neutral to alkaline in a wide range from 6.5 to 8.9.64 This variability is representative of poorly healing wounds, that tend to be alkaline or close to neutral in those with better healing rates. Also, as wounds progress towards healing, the pH of non-infected wounds tends to become acidic, opposite to infected cases.65 Besides, wound surface pH is usually lowered using topical acidifying agents because acidic pH decreases infections, promotes epithelialization and increases healing rates.66 Acetic acid, lowering pH to values as low as 3, has been used to eradicate or prevent wound infections. Interestingly, polymer mixtures containing PAA also have the capability of reducing pH and this effect has been reported to cause stress to bacterial cell by disrupting cytoplasmic homeostasis.67 Our results showed that lysozyme release in AMP functionalized PAA–PVA dressings was greatly dependent on pH, which can in turn be modified by the acidic groups of PAA themselves as then can be easily protonated.
However, the amount of lysozyme still attached to the dressing after 14 days in contact with the release media was significant (∼20%) in PBS, pH 7.4, and high (∼60%) for the case of acetic acid, pH 3.5. The ATR-FTIR spectra of dressings after 14 days in contact with the different buffers are shown in Fig. S5 (ESI†). The bands at 1640 cm−1 and 1525 cm−1 were clear and could also be identified in the case of phosphate buffer, pH 7.4 for which the residual lysozyme content was 15%. The residual lysozyme in carbonate buffer, pH 10.0, was 5%, small amount but still visible in the FTIR-ATR spectra of dressings after 14 days in contact with it. The reason for the enhanced resistance to bacterial colonization of lysozyme-loaded dressings (as shown in Fig. 5B) can be attributed to the presence of lysozyme attached to the dressing even after 14 days. The presence of lysozyme firmly attached to the PAA–PVA surface can be explained by the fact that lysozyme has basic groups still charged at basic pH including 6 lysines (pKa 10.8) and 11 argines (pKa 12.5)68
Conclusions
This work shows the preparation of electrospun wound dressings from blends of PAA and PVA thermally stabilized by crosslinking and functionalized with AMP by electrostatic adsorption. The impregnation efficiency was higher at neutral pH, favoured by the electrostatic interaction of AMP with the negatively charged scaffold.The loading densities of AMP were in the 6.5 × 10−6 to 3.0 × 10−4 mmol mg−1 range. All functionalized dressings still displayed a negative surface charge due to the dissociation of carboxylic acid groups from PAA. The dressing kept fibrous structure after impregnation with fibre diameter near the micron size. Fluorescamine assay revealed peptides homogeneously distributed throughout the fibrous network.
The antimicrobial effect was tested using S. aureus in solid agar and in liquid cultures. Lysozyme-loaded dressings produced significant inhibition zones in solid agar diffusion tests and strongly inhibited bacterial growth in liquid cultures. The incorporation of lysozyme to the base scaffold resulted in surfaces essentially free of bacterial growth after 14 days.
The release of lysozyme was slower in acidic medium. Even after 14 days in contact with release media, significant amount of lysozyme was still attached to the dressing surface. The formulation containing lysozyme on PAA-based scaffolds is a promising candidate as antibacterial dressing with pH triggered release and capacity to retain AMP during extended periods.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the Spanish Ministry of Economy and Competitiveness, CTM2016-74927-C2-1-R and the Dirección General de Universidades e Investigación de la Comunidad de Madrid, Research Network S2013/MAE-2716. GA, thanks the University of Alcalá for the award of a pre-doctoral grant.References
- T. Velnar, T. Bailey and V. Smrkolj, J. Int. Med. Res., 2009, 37, 1528–1542 CrossRef PubMed.
- R. Edwards and K. G. Harding, Curr. Opin. Infect. Dis., 2004, 17, 91–96 CrossRef PubMed.
- I. C. Valencia, R. S. Kirsner and F. A. Kerdel, J. Am. Acad. Dermatol., 2004, 50, 845–849 CrossRef PubMed.
- H. Lambers, S. Piessens, A. Bloem, H. Pronk and P. Finkel, Int. J. Cosmet. Sci., 2006, 28, 359–370 CrossRef PubMed.
- L. A. Schneider, A. Korber, S. Grabbe and J. Dissemond, Arch. Dermatol. Res., 2007, 298, 413–420 CrossRef PubMed.
- S. L. Percival, S. Finnegan, G. Donelli, C. Vuotto, S. Rimmer and B. A. Lipsky, Crit. Rev. Microbiol., 2016, 42, 293–309 Search PubMed.
- M. Parani, G. Lokhande, A. Singh and A. K. Gaharwar, ACS Appl. Mater. Interfaces, 2016, 8, 10049–10069 CrossRef PubMed.
- J. S. Boateng, K. H. Matthews, H. N. E. Stevens and G. M. Eccleston, J. Pharm. Sci., 2008, 97, 2892–2923 CrossRef PubMed.
- G. D. Mogoşanu and A. M. Grumezescu, Int. J. Pharm., 2014, 463, 127–136 CrossRef PubMed.
- E. A. Kamoun, E. S. Kenawy and X. Chen, J. Adv. Res., 2017, 8, 217–233 CrossRef PubMed.
- K. Azuma, R. Izumi, T. Osaki, S. Ifuku, M. Morimoto, H. Saimoto, S. Minami and Y. Okamoto, J. Funct. Biomater., 2015, 6, 104 CrossRef PubMed.
- H. Kurniawan, Y. S. Ye, W. H. Kuo, J. T. Lai, M. J. Wang and H. S. Liu, Biochem. Eng. J., 2013, 78, 138–145 CrossRef.
- S. P. Zhong, Y. Z. Zhang and C. T. Lim, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 510–525 Search PubMed.
- Y. Wang, C. Fu, Z. Wu, L. Chen, X. Chen, Y. Wei and P. Zhang, Carbohydr. Polym., 2017, 174, 723–730 CrossRef PubMed.
- C. Dhand, M. Venkatesh, V. A. Barathi, S. Harini, S. Bairagi, E. Goh Tze Leng, N. Muruganandham, K. Z. W. Low, M. H. U. T. Fazil, X. J. Loh, D. K. Srinivasan, S. P. Liu, R. W. Beuerman, N. K. Verma, S. Ramakrishna and R. Lakshminarayanan, Biomaterials, 2017, 138, 153–168 CrossRef PubMed.
- J. Boateng and O. Catanzano, J. Pharm. Sci., 2015, 104, 3653–3680 CrossRef PubMed.
- J. Wu, Y. Zheng, W. Song, J. Luan, X. Wen, Z. Wu, X. Chen, Q. Wang and S. Guo, Carbohydr. Polym., 2014, 102, 762–771 CrossRef PubMed.
- M. L. Mangoni, A. M. McDermott and M. Zasloff, Exp. Dermatol., 2016, 25, 167–173 CrossRef PubMed.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- D. I. Andersson, D. Hughes and J. Z. Kubicek-Sutherland, Drug Resist. Updates, 2016, 26, 43–57 CrossRef PubMed.
- D. H. Reneker and A. L. Yarin, Polymer, 2008, 49, 2387–2425 CrossRef.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef PubMed.
- N. Bhardwaj and S. C. Kundu, Biotechnol. Adv., 2010, 28, 325–347 CrossRef PubMed.
- M. Abrigo, S. L. McArthur and P. Kingshott, Macromol. Biosci., 2014, 14, 772–792 CrossRef PubMed.
- J. Quirós, K. Boltes and R. Rosal, Polym. Rev., 2016, 56, 631–667 CrossRef.
- T. J. Sill and H. A. von Recum, Biomaterials, 2008, 29, 1989–2006 CrossRef PubMed.
- I. Jun, H. S. Han, J. R. Edwards and H. Jeon, Int. J. Mol. Sci., 2018, 19 Search PubMed.
- N. Yan, X. Zhang, Q. Cai, X. Yang, X. Zhou, B. Wang and X. Deng, J. Biomater. Sci., Polym. Ed., 2012, 23, 1005–1019 CrossRef PubMed.
- M. Ignatova, I. Rashkov and N. Manolova, Expert Opin. Drug Delivery, 2013, 10, 469–483 CrossRef PubMed.
- A. O. Basar, S. Castro, S. Torres-Giner, J. M. Lagaron and H. Turkoglu Sasmazel, Mater. Sci. Eng., C, 2017, 81, 459–468 CrossRef PubMed.
- T. H. B. Eriksen, E. Skovsen and P. Fojan, J. Biomed. Nanotechnol., 2013, 9, 492–498 CrossRef PubMed.
- D. W. Song, S. H. Kim, H. H. Kim, K. H. Lee, C. S. Ki and Y. H. Park, Acta Biomater., 2016, 39, 146–155 CrossRef PubMed.
- J. B. D. Green, T. Fulghum and M. A. Nordhaus, Chem. Rev., 2009, 109, 5437–5527 CrossRef PubMed.
- H. P. Felgueiras and M. T. P. Amorim, Colloids Surf., B, 2017, 156, 133–148 CrossRef PubMed.
- B. Kim, H. Park, S. H. Lee and W. M. Sigmund, Mater. Lett., 2005, 59, 829–832 CrossRef.
- C. Zhang, X. Yuan, L. Wu, Y. Han and J. Sheng, Eur. Polym. J., 2005, 41, 423–432 CrossRef.
- K. Kumeta, I. Nagashima, S. Matsui and K. Mizoguchi, J. Appl. Polym. Sci., 2003, 90, 2420–2427 CrossRef.
- J. Santiago-Morales, G. Amariei, P. Letón and R. Rosal, Colloids Surf., B, 2016, 146, 144–151 CrossRef PubMed.
- A. Chwalibog, E. Sawosz, A. Hotowy, J. Szeliga, S. Mitura, K. Mitura, M. Grodzik, P. Orlowski and A. Sokolowska, Int. J. Nanomed., 2010, 5, 1085–1094 CrossRef PubMed.
- L. J. Bessa, P. Fazii, M. Di Giulio and L. Cellini, Int. Wound J., 2015, 12, 47–52 CrossRef PubMed.
- W. Chung and R. E. W. Hancock, Int. J. Food Microbiol., 2000, 60, 25–32 CrossRef.
- L. Vanderkelen, J. M. Van Herreweghe, K. G. A. Vanoirbeek, G. Baggerman, B. Myrnes, P. J. Declerck, I. W. Nilsen, C. W. Michiels and L. Callewaert, Cell. Mol. Life Sci., 2011, 68, 1053–1064 CrossRef PubMed.
- X. Gao, X. Zhang, J. Song, X. Xu, A. Xu, M. Wang, B. Xie, E. Huang, F. Deng and S. Wei, Int. J. Nanomed., 2015, 10, 7109–7128 Search PubMed.
- L. Karam, C. Jama, A. S. Mamede, S. Boukla, P. Dhulster and N. E. Chihib, Appl. Microbiol. Biotechnol., 2013, 97, 10321–10328 CrossRef PubMed.
- P. Kumaraswamy, R. Lakshmanan, S. Sethuraman and U. M. Krishnan, Soft Matter, 2011, 7, 2744–2754 RSC.
- E. Breukink and B. de Kruijff, Biochim. Biophys. Acta, Biomembr., 1999, 1462, 223–234 CrossRef.
- S. M. M. Meira, A. I. Jardim and A. Brandelli, Food Chem., 2015, 188, 161–169 CrossRef PubMed.
- C. Ibarguren, P. M. Naranjo, C. Stötzel, M. C. Audisio, E. L. Sham, E. M. Farfán-Torres and F. A. Müller, Appl. Clay Sci., 2014, 90, 88–95 CrossRef.
- C. F. Narambuena, G. S. Longo and I. Szleifer, Soft Matter, 2015, 11, 6669–6679 RSC.
- S. P. Zustiak, Y. Wei and J. B. Leach, Tissue Eng., Part B, 2013, 19, 160–171 CrossRef PubMed.
- V. Tangpasuthadol, N. Pongchaisirikul and V. P. Hoven, Carbohydr. Res., 2003, 338, 937–942 CrossRef PubMed.
- D. Xiao, C. Gömmel, P. M. Davidson and Q. Zhong, J. Agric. Food Chem., 2011, 59, 9572–9580 CrossRef PubMed.
- G. Amariei, K. Boltes, P. Leton, I. Iriepa, I. Moraleda and R. Rosal, J. Mater. Chem. B, 2017, 5, 6776–6785 RSC.
- S. M. M. Meira, G. Zehetmeyer, J. M. Scheibel, J. O. Werner and A. Brandelli, LWT–Food Sci. Technol., 2016, 68, 226–234 CrossRef.
- M. Zohri, M. S. Alavidjeh, I. Haririan, M. S. Ardestani, S. E. S. Ebrahimi, H. T. Sani and S. K. Sadjadi, Probiotics Antimicrob. Proteins, 2010, 2, 258–266 CrossRef PubMed.
- A. Lončarević, M. Ivanković and A. Rogina, Journal of Tissue Repair and Regeneration, 2017, 1, 12–22 Search PubMed.
- C. K. Bower, J. McGuire and M. A. Daeschel, Appl. Environ. Microbiol., 1995, 61, 992–997 Search PubMed.
- S. Dosler and A. A. Gerceker, Chemotherapy, 2012, 57, 511–516 CrossRef PubMed.
- D. Field, R. O’ Connor, P. D. Cotter, R. P. Ross and C. Hill, Front. Microbiol., 2016, 7, 508 Search PubMed.
- G. Gratzl, S. Walkner, S. Hild, A. W. Hassel, H. K. Weber and C. Paulik, Colloids Surf., B, 2015, 126, 98–105 CrossRef PubMed.
- N. K. Archer, M. J. Mazaitis, J. W. Costerton, J. G. Leid, M. E. Powers and M. E. Shirtliff, Virulence, 2011, 2, 445–459 CrossRef PubMed.
- N. Charernsriwilaiwat, P. Opanasopit, T. Rojanarata and T. Ngawhirunpat, Int. J. Pharm., 2012, 427, 379–384 CrossRef PubMed.
- T. Heunis, O. Bshena, B. Klumperman and L. Dicks, Int. J. Mol. Sci., 2011, 12, 2158–2173 CrossRef PubMed.
- S. L. Percival, S. McCarty, J. A. Hunt and E. J. Woods, Wound Repair Regen., 2014, 22, 174–186 CrossRef PubMed.
- S. Ono, R. Imai, Y. Ida, D. Shibata, T. Komiya and H. Matsumura, Burns, 2015, 41, 820–824 CrossRef PubMed.
- V. K. Shukla, D. Shukla, S. K. Tiwary, S. Agrawal and A. Rastogi, J. Wound Care, 2007, 16, 291–294 CrossRef PubMed.
- G. Gratzl, C. Paulik, S. Hild, J. P. Guggenbichler and M. Lackner, Mater. Sci. Eng., C, 2014, 38, 94–100 CrossRef PubMed.
- M. Boström, D. R. M. Williams and B. W. Ninham, Biophys. J., 2003, 85, 686–694 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR-ATR spectra and SEM images of functionalized dressings; confocal laser scanning microscopy images of dressings after conjugation with fluorescamine and results of halo inhibition zone experiments. See DOI: 10.1039/c8ra03861a |
| This journal is © The Royal Society of Chemistry 2018 |