
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-delivery of doxorubicin and itraconazole by Pluronic® P123 coated liposomes to enhance the anticancer effect in breast cancers†
Yi Lin‡
a,
Xiaodan He‡b,
Dinglun Zhouc,
Li Li *a,
Jiawei Sunb and
Xuehua Jiangb
*a,
Jiawei Sunb and
Xuehua Jiangb
aNational Engineering Research Center for Biomaterials, Sichuan University, Chengdu 610064, China. E-mail: li_li@scu.edu.cn
bWest China School of Pharmacy, Sichuan University, Chengdu 610041, China
cWest China School of Public Health, Sichuan University, Chengdu 610041, China
First published on 29th June 2018
Abstract
To date, the combinational cancer therapy of anticancer and antiangiogenic agents represents a promising strategy to improve antitumor outcomes in clinics. However, combination therapy with drugs having distinct properties, such as solubility, limits the likelihood of simultaneous delivery. In our study, we aimed to develop a codelivery nanoparticulate system of hydrophilic doxorubicin (DOX) and hydrophobic itraconazole (ITZ) using liposomes coated with Pluronic® P123 (ITZ/DOX-PLip). The prepared ITZ/DOX-PLip exhibited a unimodal size distribution and high loading efficiency with sustained release profiles. Furthermore, cytotoxicity against 4T1 murine breast cancer cells and cellular uptake results revealed that the inhibitory effect of ITZ/DOX-Plip on tumor growth was superior to that of free DOX or DOX-loaded liposome (DOX-Lip), which was primarily attributed to the significantly higher intercellular DOX content. Cytotoxicity against HUVEC and wound healing tests confirmed that ITZ and ITZ formulations could inhibit the growth and migration of endothelial cells. In addition, in xenograft 4T1 bearing BALB/c mice, biodistribution experiments revealed that higher drug accumulation in tumors and decreased distribution in heart were observed for ITZ/DOX-PLip as compared to free DOX. Remarkably, ITZ/DOX-PLip significantly reduced tumor volume, tumor weight, liver metastasis and microvessel density in comparison with the same dose of ITZ injection or DOX-Lip. Overall, this Pluronic® P123 modified liposome-based codelivery system represents a promising nano-platform for combination therapy in cancers.
Introduction
The combination chemotherapy of anticancer drugs and antiangiogenic inhibitors, which not only suppresses tumor growth but also cuts off nutrient supply and oxygen delivery, represents a promising strategy to improve antitumor outcome in clinics.1 Clinical trials using combination chemotherapy of anti-cancer drugs and α-VEGF monoclonal antibody bevacizumab, a FDA approved anti-angiogenic agent, have been conducted around the world in the past two decades.1–4 However, the survival benefit associated with the combination therapy is marginal, and toxicities and cost are substantial.5,6 Furthermore, these two categories of drugs having distinct properties, such as solubility, generally require use of multiple carriers or solvents, which limits the likelihood of simultaneous delivery and action. Therefore, these results strongly support further development of novel anti-cancer and anti-angiogenic strategies for cancer therapy.Doxorubicin (DOX) is a DNA intercalation agent and inhibits DNA topoisomerase II. It was isolated from the pigment-producing Streptomyces peucetius early in the 1960s and remains the “evergreen” drug with broad clinical indications until now.7 However, the severe cardiotoxicity and acquired multi-drug resistance (MDR) reduced the anti-cancer outcomes in clinic.7 Itraconazole, an old antifungal drug (ITZ), has been found to have several attractive pharmacological activities. ITZ selectively inhibits endothelial cells over other cell types and the hedgehog signaling pathway, which results in blocking angiogenesis and tumor growth.8–10 Recent reports suggest that the possible mechanisms of inhibition of endothelial cell proliferation are activation of the simultaneous 5′ AMP-dependent protein kinase (AMPK) and perturbation of cholesterol trafficking pathway.11,12 Moreover, it is also a potent inhibitor of P-glycoprotein (P-gp), which plays an essential role in MDR.13 When it was co-administrated with free anticancer drugs such as vinblastine, daunorubicin, DOX and paclitaxel, the in vitro results demonstrated that the intracellular concentration of these anticancer drugs was increased via the inhibition of P-gp activity.13–16 ITZ has been repurposed as a treatment for different types of cancer in multiple clinical trials (https://www.clinicaltrials.gov). The early results have shown a promising efficacy against prostate cancer, lung cancer, pancreatic cancer and basal cell carcinoma.17–19 In the light of these results, the combination of DOX and ITZ is expected to improve the anticancer outcomes mainly due to the synergism of multiple functions including the enhanced intracellular DOX content induced by ITZ, inhibition of tumor growth and angiogenesis. Therefore, an effective and safe drugs co-delivery system is highly desirable for the combination administration of hydrophilic DOX·HCl and hydrophobic ITZ with enhanced anticancer activities and reduced adverse effects.
Liposomes are vesicles containing an aqueous core surrounded by a lipid bilayer membrane, which are excellent candidates for co-encapsulating hydrophilic and hydrophobic drugs.20 Liposomes have great benefits for protecting drugs from degradation, optimizing the pharmacokinetic profile and reducing toxicity in normal tissues.21,22 Polyethylene glycol (PEG) modification endows liposomes with “stealth” function which can avoid the capture by reticuloendothelial system (RES).23 However, traditional liposomes have none of capability to overcome MDR. Pluronic® block copolymers contain hydrophilic polyethylene oxide (PEO) (hydrophilic) and hydrophobic polypropylene oxide (PPO) (hydrophobic) moieties, which were reported to reduce the capture in vitro and in vivo by macrophages and extend their blood circulation time.24–26 This was primarily attributed to the steric barrier originated from PEO moieties.25 They are also potent inhibitors of the P-gp efflux system in MDR cells. The possible mechanism underlying the inhibition effect is related to the depletion in ATP level and membrane fluidization.27,28 Therefore, liposome modified by Pluronic® copolymers is expected to possess both stealth and anti-MDR capabilities.
In this study, Pluronic® P123 modified liposome for codelivery of DOX and ITX (ITZ/DOX-PLips) was fabricated. The size, size distribution, zeta potential, loading efficiency, and in vitro release profile were characterized. Cell experiments were conducted to assess the suppression efficiency of cell proliferation and migration both in 4T1 cells and HUVEC, and the cellular uptake in 4T1 cells. Moreover, the in vivo biodistribution, antitumor and angiogenesis activities against 4T1 murine breast cancer, and systemic toxicities were carefully evaluated.
Experimental
Materials, cells and animals
Soy phosphatidylcholine (SPC) was purchased from Lipoid GmbH (Ludwigshafen am Rhein, Germany). Cholesterol (Chol) and Pluronic® P123 (PEG20–PPO70–PEO20, molecular weight 5800) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Doxorubicin chloride (DOX·HCl) was purchased from Melun Biological Technology Co., Ltd. (Dalian, China). Itraconazole (ITZ) was a kind gift from Associate Professor Jing Ren (Chengdu University, China). ITZ Injection (Sporanox®) was purchased from Janssen Pharmaceutical Ltd. (Xi'an, China). 4′,6-Diamidino-2-phenylindole (DAPI) was purchased from Thermo-Fisher Scientific (Waltham, MA, USA). 2-(2-Methoxy-4-ni-trophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H tetrazolium, monosodium salt (CCK-8) assay kit was purchased from Dojindo Company (Kumamoto, Japan).Mouse mammary carcinoma cells 4T1 (a cell line derived from the BALB/c spontaneous mammary carcinoma) were purchased from Chinese Academy of Science Cell Bank for Type Culture Collection (Shanghai, China). RPMI 1640 culture media, fetal bovine serum (FBS), and streptomycin were purchased from Thermo-Fisher Scientific Inc. (Waltham, MA, USA). 4T1 cells were cultured in RPMI 1640 cell culture medium supplemented with 10% fetal bovine serum and 100 U mL−1 streptomycin in an incubator under 5% CO2 at 37 °C. Primary human umbilical vein endothelial cells (HUVEC) and endothelial cell medium (ECM) were purchased from Sciencell™ Research Laboratories (Carlsbad, California, USA). ECM consists of 500 mL of basal medium, 25 mL of FBS, 5 mL of endothelial cell growth supplement (ECGS) and 5 mL of penicillin/streptomycin solution. HUVEC were cultured in ECM in an incubator under 5% CO2 at 37 °C.
Female BALB/c mice and female BALB/c nude mice at 6 weeks old (weight 20 ± 2 g) were purchased from Dossy Biological Technology Co., Ltd. (Chengdu, China). The animal experiments were performed in agreement with the Experiment Animal Administrative committee of Sichuan University.
Preparation and characterization of liposomes
ITZ- and DOX-loaded liposomes coating with Pluronic® P123 (ITZ/DOX-PLip) were prepared by three steps.22,29 First, ITZ-loaded liposomes (ITZ-Lip) were prepared via thin film hydration method.22 Briefly, SPC, Chol and ITZ (mass ratio, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.6:1) were dissolved in methylene chloride: methanol (2:1, v/v) in a round-bottom flask. The organic solvent was removed by rotary vacuum evaporation and the formed lipid film was hydrated in ammonium-sulfate solution (300 mM) to achieve a final phospholipid concentration of 2 mg mL−1. The liposomes were sonicated in a sonication bath at 60 °C (180 W, 10 minutes) and then further sonicated by a probe sonication (Sonics & Materials Inc., Danbury, CT, USA) at 40 W for 2 min. External buffer was exchanged by dialyzing in 0.9% NaCl solution (MW cutoff: 6000–8000). The dialysis lasted for 6 h, with replacing the fresh dialysate every two hours. Secondly, DOX was loaded in liposomes via a pH gradient method.29 Briefly, DOX solution was added to liposomes at a SPC/drug mass ratio of 10:1 and incubated for 20 minutes at 60 °C with gentle stirring. After incubation, unencapsulated ITZ was removed by centrifugation at 3500 rpm for 5 min. Finally, the supernatant solution was separated and incubated with Pluronic® P123 solution at a SPC/Pluronic® P123 mass ratio of 1:1. Unabsorbed polymer and free DOX were removed by eluting through Sephadex G-50 column equilibrated with PBS (pH 7.4). The schematic diagram of preparation and structure of ITZ/DOX-PLip was shown in Scheme 1. The control groups including empty liposomes (Lip), empty liposomes coating with Pluronic® P123 (PLip), DOX-loaded liposomes (DOX-Lip), ITZ loaded liposomes (ITZ-Lip) and ITZ loaded liposomes coating with Pluronic® P123 (ITZ-PLip) were prepared in a similar procedure.
:0.6:1) were dissolved in methylene chloride: methanol (2:1, v/v) in a round-bottom flask. The organic solvent was removed by rotary vacuum evaporation and the formed lipid film was hydrated in ammonium-sulfate solution (300 mM) to achieve a final phospholipid concentration of 2 mg mL−1. The liposomes were sonicated in a sonication bath at 60 °C (180 W, 10 minutes) and then further sonicated by a probe sonication (Sonics & Materials Inc., Danbury, CT, USA) at 40 W for 2 min. External buffer was exchanged by dialyzing in 0.9% NaCl solution (MW cutoff: 6000–8000). The dialysis lasted for 6 h, with replacing the fresh dialysate every two hours. Secondly, DOX was loaded in liposomes via a pH gradient method.29 Briefly, DOX solution was added to liposomes at a SPC/drug mass ratio of 10:1 and incubated for 20 minutes at 60 °C with gentle stirring. After incubation, unencapsulated ITZ was removed by centrifugation at 3500 rpm for 5 min. Finally, the supernatant solution was separated and incubated with Pluronic® P123 solution at a SPC/Pluronic® P123 mass ratio of 1:1. Unabsorbed polymer and free DOX were removed by eluting through Sephadex G-50 column equilibrated with PBS (pH 7.4). The schematic diagram of preparation and structure of ITZ/DOX-PLip was shown in Scheme 1. The control groups including empty liposomes (Lip), empty liposomes coating with Pluronic® P123 (PLip), DOX-loaded liposomes (DOX-Lip), ITZ loaded liposomes (ITZ-Lip) and ITZ loaded liposomes coating with Pluronic® P123 (ITZ-PLip) were prepared in a similar procedure.
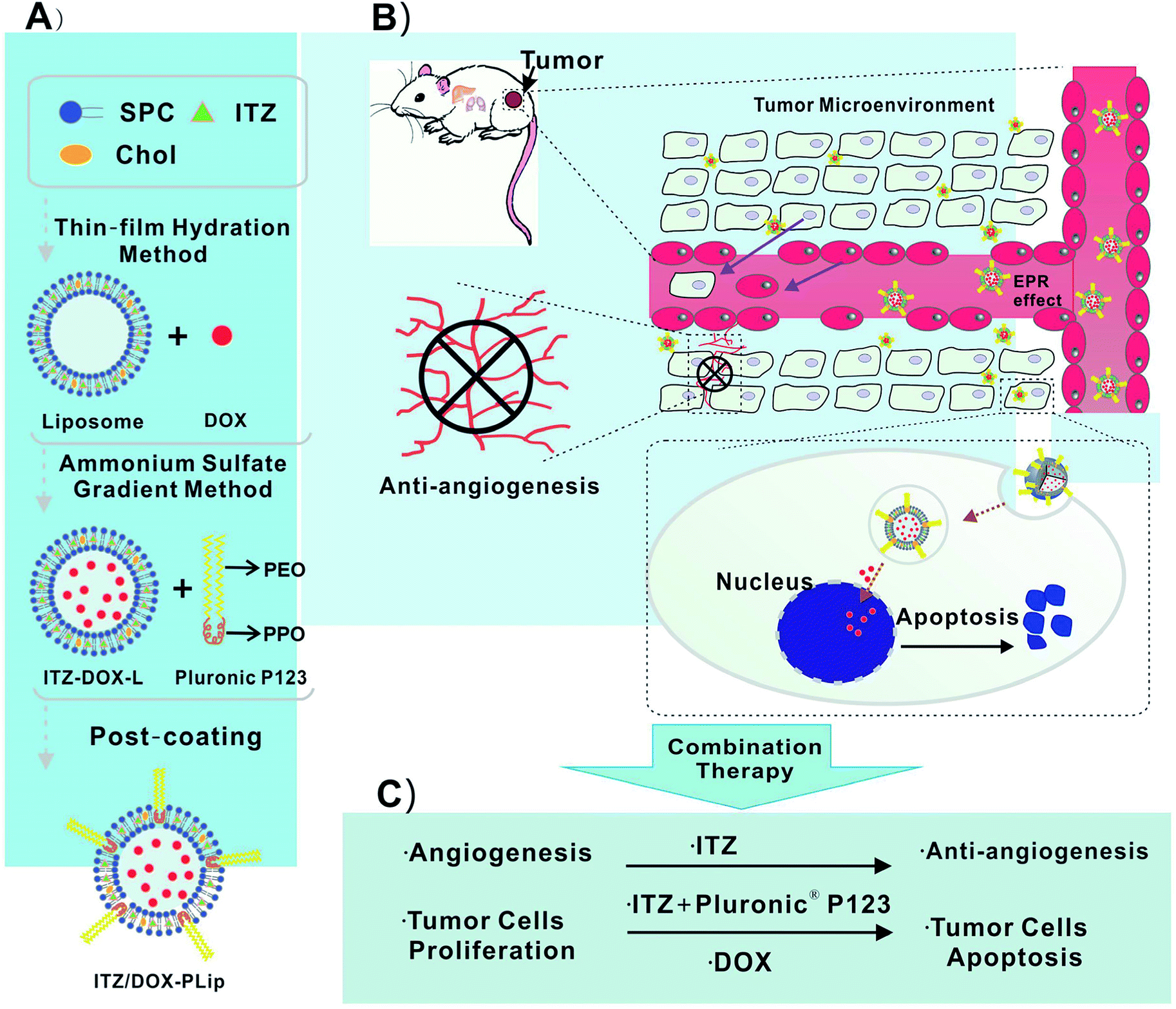 | ||
| Scheme 1 (A) Schematic diagram of the ITZ- and DOX-loaded liposomes coating with Pluronic® P123 (ITZ/DOX-PLip), (B and C) inhibition effects on tumor growth and tumor-associated angiogenesis of ITZ/DOX-PLip via combinatory therapy strategy. | ||
Liposome diameters and sample polydispersity index (PDI) were determined by means of dynamic light scattering using a Malvern Zetasizer Nano ZS90 (MalvernInstruments, Worcestershire, UK). Determinations were carried out at 25 °C at a fixed angle of 90° and a laser power of 40 mW. The thickness of the coating layers of Pluronic® P123 was measured by subtracting the size of the uncoated particles from the coated size and then dividing the figure by two.24
The zeta potential of the obtained liposomes was also measured by dynamic light scattering using a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Worcestershire, UK).
The drug contents were measured by high performance liquid chromatography (HPLC). The prepared liposomes were dissolved in methanol (liposome:methanol = 1:9, volume ratio). DOX analysis was performed using an ODS3 column (150 × 4.6 mm, 5 μm, Agilent, USA) and an Agilent HPLC system consisting of a 1260-pump and a 1260-ultraviolet detector (Agilent Technologies, Santa Clara, CA, USA). The mobile phase used water-acetonitrile (both containing 0.1% trifluoroacetic acid) (30:70, volume ratio). The UV detection wavelength was set to 480 nm, the column temperature was 25 °C, and the flow rate was 1.0 mL per minute. ITZ was determined to be in the same condition as DOX except a mobile phase consisting of water-acetonitrile (35:65, volume ratio), and the UV detection wavelength was 261 nm. DOX and ITZ loading efficiency and encapsulation efficiency were calculated as follows:
| Encapsulation efficiency (%) = (mass of drug found loaded/mass of drug input) × 100. | (1) |
| Loading efficiency (%) = (mass of drug found loaded/mass of drug-loaded liposomes) × 100. | (2) |
To demonstrate the stability of liposomes in storage and in blood, the particle sizes both in PBS buffer and in the presence of 50% fetal bovine serum (FBS) were measured using DLS.21 For the stability in storage, ITZ/DOX-PLips (phospholipid concentration 5 mg mL−1) were stored in a refrigerator at 4 °C. At predetermined time points (1, 3, 5 and 7 d), 100 μL of sample was diluted to 1 mL with PBS buffer (pH 7.4) for diameters measurement on a Malvern ZetasizerNano ZS90. For the stability in blood, ITZ/DOX-Plips (phospholipid concentration 5 mg mL−1) were gently mixed with equal volume of FBS and incubated at 37 °C. At predetermined time points (1, 4, 8 and 24 h), 100 μL of sample was diluted to 1 mL with PBS buffer (pH 7.4) for diameters measurement on a Malvern ZetasizerNano ZS90. The diameters of liposomes at time zero were determined in PBS buffer (pH 7.4).
Drug release
The in vitro release of DOX and ITZ from liposomes was measured by the modified dialysis method under sink conditions. Briefly, 0.5 mL ITZ/DOX-PLip and 0.5 mL fetal bovine serum (FBS) were added into one dialysis tube (Mw cutoff: 6000–8000), which was sealed tightly at both ends with clips. The dialysis tube was immersed into an EP tube with 50 mL of PBS (pH 7.4) containing 0.1% (mass/volume) Tween 80. This was then incubated at 37 °C with continuous shaking at a speed of 100 times per minute for 48 hours. At the predetermined time points (0.5, 1, 2, 4, 6, 8, 10, 12, 24, 48 hours), 0.5 mL of the release medium were withdrawn and replaced with an equal volume of fresh medium. The DOX content was measured by a Hitachi F-7000 spectrofluorimeter (Hitachi, Ltd., Tokyo, Japan) with the excitation wavelength 480 nm and emission wavelength 550 nm. ITZ content was assayed by HPLC as described in section “Preparation and characterization of liposomes”.Cellular location and uptake
Confocal laser scanning microscopy (CLSM) was employed to testify intracellular fate of DOX in 4T1 cells. 4T1 cells were seeded at a density of 3 × 103 cells in each glass-bottomed dish (35 × 12 mm, NEST Biotech Co. Ltd., Wuxi, China) and incubated for 12 h. ITZ/DOX-PLip, DOX-Lip and DOX·HCl solution with a 5 μg mL−1 DOX concentration in medium were introduced into each dish and incubated for 4 h. After that, the medium was removed, and the cells were washed with cold PBS for three times. The cells were fixed with 4% paraformaldehyde for 10 min and then stained by DAPI solution (100 ng mL−1) for 5 min. Finally, the cells were observed and imaged by CLSM (Leica TCP SP5, Mannheim, Switzerland). The cell-associated DOX was excited with an argon laser at 488 nm and the emitted fluorescence was detected at 575 nm. The excitation and emission wavelength of DAPI were 350 nm and 470 nm, respectively.Flow cytometry (FCM; Cytomics. FC 500; Beckman Coulter, Miami, FL, USA) was employed to quantify the internalization amount of DOX into 4T1 cells. 4T1 cells were seeded in 6-well culture plates at a density of 1 × 106 cells per well and allowed to attach to the plate overnight. Then the medium was replaced by 2 mL fresh medium containing ITZ/DOX-PLip, DOX-Lip and DOX·HCl (DOX equivalent, concentration of 5 μg mL−1) for 4 h, respectively. Control groups were performed by adding completed medium without DOX. After incubation for 4 h, the medium was removed, and 4T1 cells were rinsed thrice with cold PBS, followed by trypsinization and resuspension in 0.5 mL PBS. The fluorescent intensity of the DOX was measured by using FCM. The cell-associated DOX was excited with an argon laser at 488 nm and the emitted fluorescence was detected at 575 nm.
Cytotoxicity assay
4T1 cells and HUVEC cells were seeded in 96-well culture plates at a density of 3 × 103 cells per well. After cultured for 12 h, cells were exposed to the fresh medium containing various formulations. Cells treated with blank culture media were negative controls. After incubation for 48 h, the culture media were removed, and the cells were rinsed twice with PBS. 100 μL RPMI 1640 containing 10 μL CCK-8 was added into each well and further incubated at 37 °C for 2 h. The absorbance was measured by a microplate reader (Thermo Scientific, USA) at 450 nm. Cell viability was calculated as follows:| Cell viability (%) = (absorbance of cells treated with drugs − absorbance of blank culture medium containing CCK-8)/(absorbance of negative controls − absorbance of blank culture medium containing CCK-8) × 100. | (3) |
Half-maximal inhibitory concentration (IC50) of each group was calculated using Graph Pad Prism 5 software. The cytotoxicity of blank liposomes against 4T1 and HUVEC cells was evaluated by the same method as described above.
Migration assay
To investigate the effects of various formulations on migration of tumor and endothelial cells, the wound healing assay was performed similarly to Aftab B. T. et al.16 Briefly, 4T1 or HUVEC cells were plated in 24-well culture plates at a density of 1 × 105 cells per well and cultured overnight to form a confluent monolayer. The following day, a vertical scratch wound was generated using a 200 μL pipette tip with a ruler and then washed with PBS twice to remove exfoliated cells. Cells were incubated with the fresh medium, followed by adding ITZ injection, ITZ-PLip (ITZ equivalent, concentration of 1.5 μg mL−1) for 24 h, respectively. We captured the images using a microscope (Olympus, Japan) at the beginning and the ending to monitor the wound status. Migration was quantified as the percent decrease in mean migration zone area.In vivo imaging
Approximately 5 × 105 4T1 cells suspended in PBS (100 μL) were subcutaneously injected into the left backs of BALB/c nude mice. Tumor volume (V) was measured using the length (L) and width (W) and calculated as V = L × W2/2. The drug was administrated when the volume of tumor was about 50–100 mm3. The tumor-bearing nude mice were randomly assigned to two groups (nine animals per group). DOX·HCl and ITZ/DOX-PLip (DOX equivalent, dose of 5 mg kg−1) were administrated via tail vein with injection. At predetermined time intervals (6, 24 and 48 h), three mice were sacrificed at each time, and the organs were removed for fluorescence measurements by a Maestro in vivo imaging system (PerkinElmer Inc., Waltham, MA, USA). The images were analyzed and the average signals of DOX·HCl and ITZ/DOX-PLip distributed in different organs were quantified by Carestream Molecular Imaging software (v 5.0.7.23; Carestream Health Inc., Rochester, NY, USA).In vivo antitumor efficacy
The tumor-bearing mice were randomly assigned into four groups (n = 5). Saline, DOX·HCl (5 mg kg−1), ITZ injection (2.5 mg kg−1), ITZ/DOX-PLip (5 mg kg−1 DOX, 2.5 mg kg−1 ITZ) were administrated via tail vein injection, respectively. Formulations were injected at 10, 13 and 16 days after the tumor cells injection. The tumor sizes were measured with a digital caliper and the mice were weighted every two days until day 22. Toxicity was also evaluated by following the body weights of all mice throughout the entire experiment. After the experiment, the mice were sacrificed by cervical vertebra dislocation. TGI was calculated according to the equation as follows: TGI = (1 − (mean tumor weight of treatment group)/(mean tumor weight of control group)) × 100%. The tumor, heart, liver, spleen, lung and kidney of each mouse were collected and washed with saline, fixed in 10% formalin for a week. The tumors were embedded in paraffin and cut with a microtome into 5 μm-thick slices for hematoxylin and eosin (H&E) staining. The whole slide imaging (Leica Aperio® AT2, Leica Biosystems, Germany) was used for quantification the necrosis areas index. The necrosis areas index was calculated as the ratio of necrosis area to total area in each slide, using three parallel slides. For quantification of proliferative cells, Ki-67 was assessed with Anti-Ki67 antibody (Abcam, England). The Ki67 index was calculated as the ratio of proliferative cells to total cells in each field, using five random fields. For quantification of apoptotic cells, TUNEL assays were used with the in situ cell death detection kit-POD (Roche Group, Switzerland). The apoptotic index was calculated as the ratio of apoptotic cells to total cells in each field, using five random fields. For quantification of microvessel density in the tumor tissues, CD31 method was used with anti-CD31 antibody (Abcam, England). Five random fields were selected and the number of microvessel was counted and the mean was calculated.Statistical analysis
The results were presented as mean ± SD. Statistical comparisons were performed by the Student's t-test. Statistical difference was set at *p < 0.05, and statistically significant difference was set at **p < 0.01.Results and discussion
Characterization of liposomes
We designed a novel DOX and ITZ co-delivery liposome coated with Pluronic® P123. The liposomes were prepared by the thin film hydration and the ammonium sulfate gradient methods, followed by a post-coating method. As shown in Table 1, the ITZ and DOX co-loaded liposomes before coating (ITZ/DOX-Lip) were about 133 nm in size and PDI was 0.220. After incubation of ITZ/DOX-Lip with Pluronic® P123, there was an expected increase in average particle size which was mainly owing to the formation of an adsorbed coating layer of Pluronic® polymer on the liposome surface. Previous studied also presented that Pluronic® block copolymers could be absorbed onto the particle surface in an aqueous solution through hydrophobic interaction of the hydrophobic PPO moiety with particle surface.27,28 The thickness of the coating layers was about 6.5 nm, which was like those reported previously for poloxamer absorbed to PLGA particles (3–6 nm).27 Coated ITZ/DOX-Lip (ITZ/DOX-PLip) exhibited a slightly higher PDI value (0.258) than uncoated liposomes (0.220), which were also reflected the existence of Pluronic® P123 coating layer. Uncoated and coated liposomes had similar negative surface charges (−2.5 to −2.7 mV). In the light of the results, we assumed that the yielded liposome may contain three parts (as shown in Scheme 1A): the outmost part might be hydrophilic moieties PEO of Pluronic® P123. The middle part might be the bilayer of the liposomes and hydrophobic moiety PPO of Pluronic® P123 where the lipophilic drug ITZ was loaded in it.33 The inner part was the aqueous phase of liposomes where the hydrophilic drug DOX·HCl was loaded in it in the form of aggregated and gelatinous anthracycline sulfate salt.34 ITZ loading of the ITZ/DOX-PLip was slightly lower than that observed for ITZ-Lip at 3.1% (Table 2). This was attributed to the fact that a small part of ITZ at or near the surface of ITZ/DOX-PLip was readily released during liposome dialysis. DOX loading of ITZ/DOX-PLip was also lower than that the observed for DOX-Lip at 6.3%. It was likely because the incorporation of ITZ into the liposome bilayer changed the fluidity of liposome membrane.| Formulations | Particle size (nm) | PDI | Absorbed layer thickness (nm) | Zeta potential (mV) |
|---|---|---|---|---|
| ITZ/DOX-Lip | 133.3 ± 2.2 | 0.220 ± 0.009 | −2.5 ± 0.2 | |
| ITZ/DOX-PLip | 146.4 ± 1.6 | 0.258 ± 0.007 | 6.5 | −2.7 ± 0.3 |
| Formulations | ITZ loading (%, w/w) | ITZ encapsulating (%) | DOX loading (%, w/w) | DOX encapsulating |
|---|---|---|---|---|
| ITZ-Lip | 3.7 ± 0.5 | 38.8 ± 4.7 | ||
| DOX-Lip | — | — | 8.6 ± 0.2 | 87.7 ± 1.9 |
| ITZ/DOX-PLip | 3.1 ± 0.2 | 31.8 ± 1.6 | 6.3 ± 0.3 | 66.5 ± 3.8 |
Stability of ITZ/DOX-PLips both in storage and in serum was studied using DLS. As shown in Fig. 1B and C, little change was observed in the hydrodynamic diameter of ITZ/DOX-PLips neither in storage at 4 °C nor in 50% serum. The good stability is attributing to the hydrophilic PEO chain on the surface of the liposomes.35
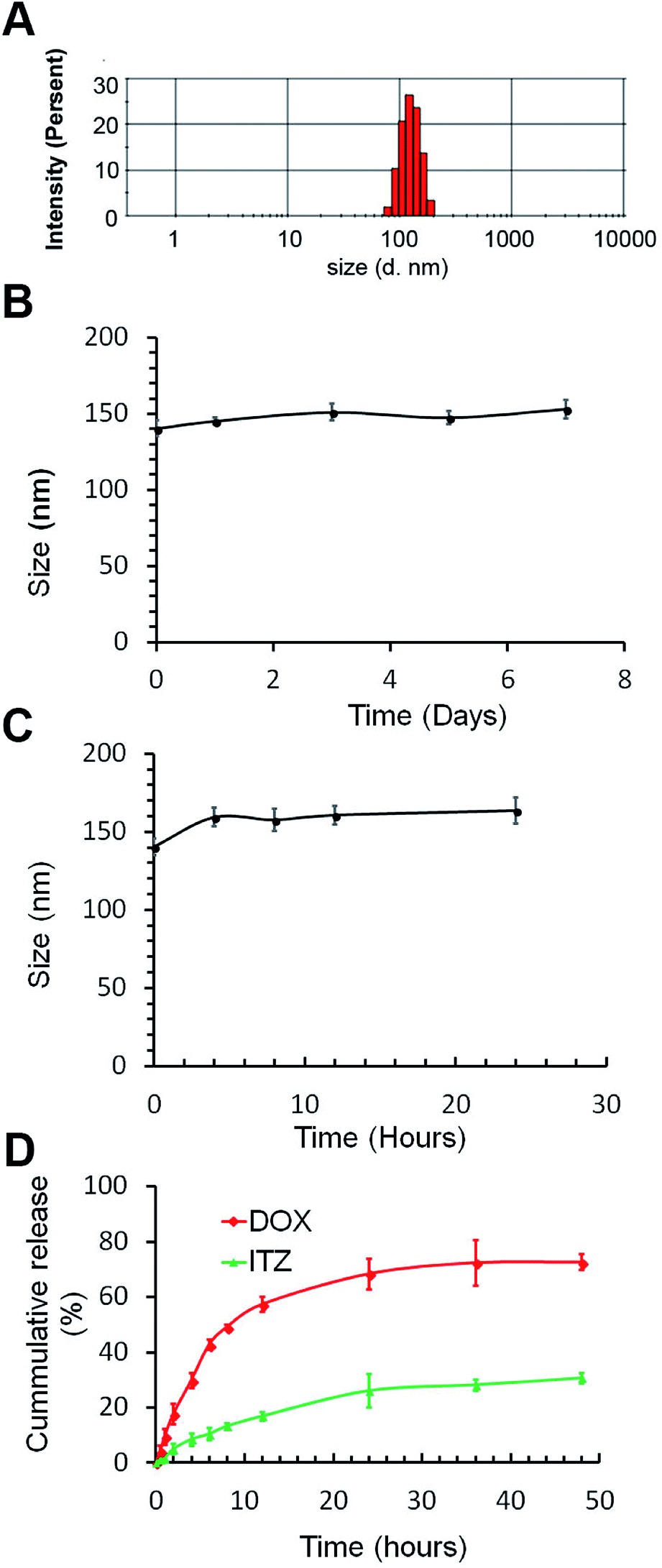 | ||
| Fig. 1 Characterization of ITZ/DOX-PLip. (A) Representative particle size distribution. (B) Particles size changes of ITZ/DOX-Lips in PBS buffer. (C) Particles size changes of ITZ/DOX-Lips in 50% FBS solution. (D) In vitro release profiles of both ITZ and DOX from ITZ/DOX-PLip in PBS (pH 7.4) with 0.1% (v/v) Tween 80 (n = 3, mean ± SD). | ||
Fig. 1D shows the in vitro release profiles of ITZ and DOX from ITZ/DOX-PLip. ITZ was continuously and slowly released from liposomes, without initial burst release effect, and the amount of cumulatively released ITZ over 48 hours was around 30%. In contrast, DOX released from ITZ/DOX-PLip (cumulative release of 72%) was faster than ITZ within 48 hours, but there was no significant burst release of DOX over the first 5 hours. This result proved that sustained release of ITZ and DOX from the liposomes.
Cell proliferation and cellular uptake
The cytotoxicity of blank liposomes including Lip and Plip against 4T1 cells and HUVEC cells was evaluated using CCK-8 assay. As shown in Fig. S1A and B,† cell viabilities after the treatment of blank liposomes were no less than 90%, suggesting that both Lip and PLip were almost non-toxic. In vitro anticancer efficiencies of DOX formulations including free DOX, DOX-Lip and ITZ/DOX-PLip were tested in 4T1 cells using CCK-8 assay. As shown in Table 3, DOX in ITZ/DOX-PLip exhibited the greatest cytotoxicity against 4T1 cells among all the tested formulations. IC50 of DOX in ITZ/DOX-PLip (0.015 ± 0.096 μg mL−1) was about 10-fold and 53-fold lower than that of DOX-Lip (0.073 ± 0.051 μg mL−1) and free DOX (0.797 ± 0.046 μg mL−1), respectively. ITZ, a commonly used antifungal drug, has been reported to have anti-angiogenesis activity.18 Considering DOX, a cytotoxicity anticancer drug, also can inhibit proliferation of endothelial cells and this kind of cells are very sensitivity to cytotoxic drug, in vitro anti-angiogenesis activities of ITZ formulations without DOX were tested in primary HUVEC. Table 4 exhibited IC50 values of ITZ in ITZ injection, ITZ-Lip and ITZ-PLip. The ITZ injection was a little more toxic than ITZ-Lip (P < 0.05), while the IC50 of ITZ-PLip lied between ITZ injection and ITZ-Lip with no statistic difference. The results suggested that ITZ-PLip and ITZ injection had the similar anti-angiogenesis activity. However, the ITZ-loaded liposomes in vitro accumulative release in 48 h was very low (∼30%). One possible explanation was that the liposome may trigger lipid fusion with plasm membranes in the process of internalization, which could greatly enhance cytosolic ITZ release.30 Conclusively, the results revealed that ITZ/DOX-PLip exhibited growth inhibition on both 4T1 and HUVEC.To understand the underling mechanism of the strong cytotoxicity of ITZ/DOX-PLip against 4T1 cells, cellular uptake assays were conducted. The cellular uptake efficiencies of various DOX formulations were detected by CLSM and FCM. First, we studied the effects of liposome, ITZ and Pluronic® P123 on the internalization and localization of DOX in 4T1 cells. As shown in Fig. 2A, DOX fluorescence were mainly located in nuclear, which is the action site of DOX. DOX fluorescence in DOX treated cells was much weaker than that in DOX-Lip group due to the nano-scaled liposome with ∼133 nm size facilitating the internalization of DOX. As reported in many studies, the liposomes with 100–200 nm size range could effectively be internalized by cells.21–23,31 And the uptake even increased with increased size over the range of 100–1000 nm.32 ITZ/DOX-PLip group presented the strongest red fluoresce, suggesting that ITZ/DOX-PLip could delivery much more DOX into cells as compared to DOX and DOX-Lip groups. The FCM result was in accordance with the finding of CLSM. As shown in Fig. 2B and C, DOX fluorescence in the DOX-Lip group was 52.4% higher than free DOX treated group (p < 0.01). Furthermore, DOX level in ITZ/DOX-PLip treated cells was 1.92-fold higher than that in DOX-Lip treated group. Two possible factors facilitated the internalization of ITZ/DOX-PLip by 4T1 cells. One factor was ITZ which could enhance the DOX internationalization by inhibiting P-gp activity.13–15 The other factor might be attributed to the presence of Pluronic® P123, which was able to efficiently improve the cell membrane permeability and inhibit P-gp activity, therefore promote the internalization of the drug.25,33
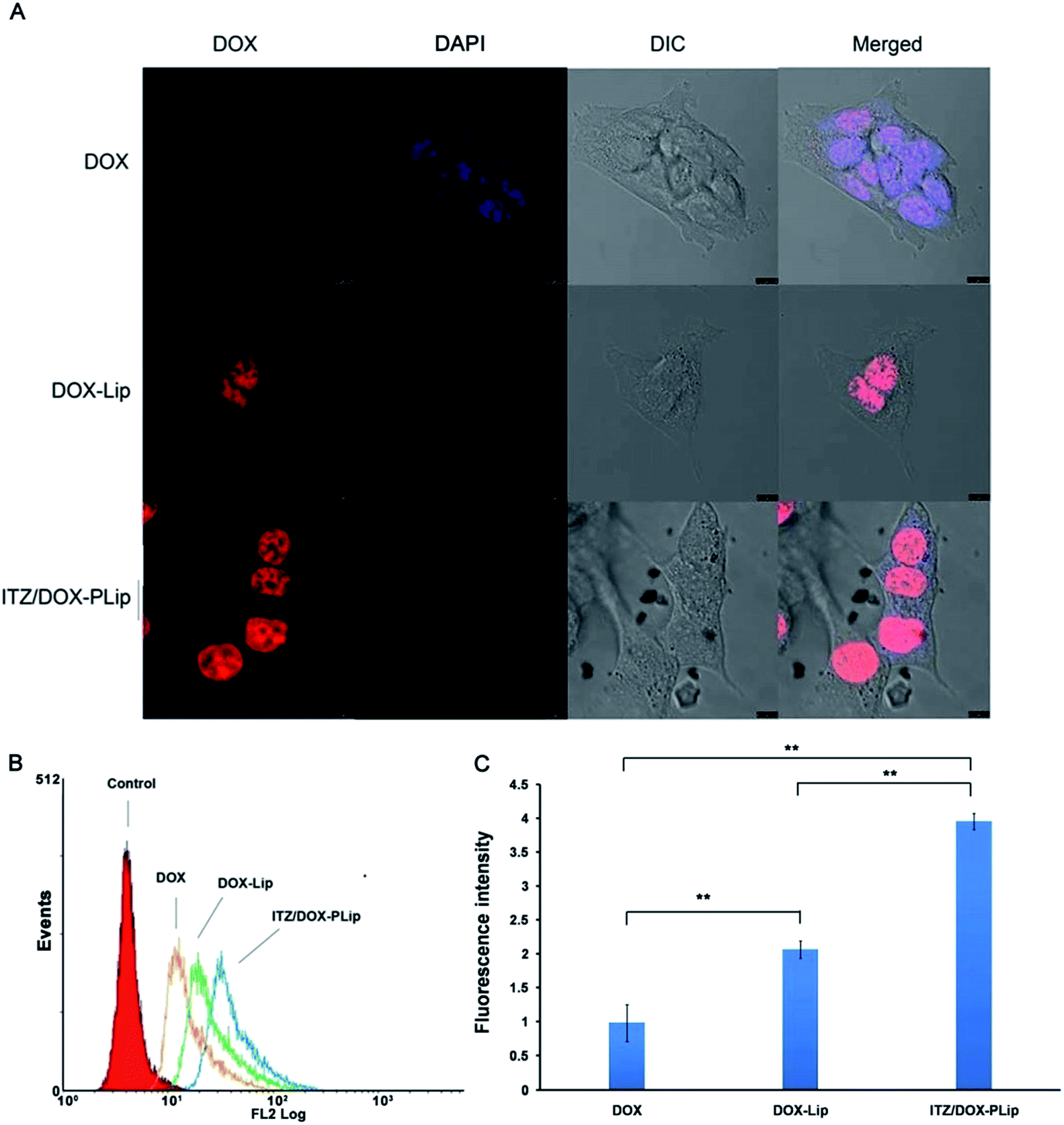 | ||
| Fig. 2 Cellular uptake studies of DOX formulations. (A) CLSM images of 4T1 cells incubated with DOX, DOX-Lip and ITZ/DOX-PLip for 4 hours at 37 °C, respectively. Red represents the fluorescence of DOX and blue represents the fluorescence of DAPI, scale bar represents 10 μm. (B) FCM curves of 4T1 cells and (C) quantification of intracellular DOX after incubation with DOX, DOX-Lip and ITZ/DOX-PLip for 4 hours at 37 °C, respectively. Each bar represents mean fluorescence intensity ± standard deviation (n = 3). **p < 0.01. | ||
Migration assay
Endothelial cell migration is an essential process in angiogenesis and tumor cell migration is important to tumor metastasis.36 ITZ has a potential to inhibit the migration of endothelia cells,16 while its effect on tumor cells was unclear. Here, the wound healing assay was used to investigate this effect on both 4T1 cells and HUVEC. To avoid the interference effect caused by DOX inducing apoptosis, the migration inhibition experiment was carried out using ITZ solution and ITZ-PLip. For 4T1 cells, as shown in Fig. 3A, the control group showed the strongest migration ability among the tested groups and the former scratch was scarcely seen. ITZ-PLip showed stronger inhibitory effect on the wound-healing as compared to free ITZ. It may be because that the liposomal ITZ could entry into cells more easily and result in higher intracellular concentration. For HUVEC cells, the result was like 4T1 cells. ITZ-PLip also demonstrated much stronger migration inhibitory capability with the relative migration rate of 25.59% than free ITZ with the relative migration rate of 73.40% (Fig. 3B). The result was in good agreement with what B. T. Aftab et al. reported before.16 These results suggested that ITZ-PLip had a superior inhibitory effect on migration of both tumor cells and endothelial cells, which played an essential role in metastasis and angiogenesis. | ||
| Fig. 3 Migration assay. (A) Wound healing images of 4T1cells and HUVEC incubated with ITZ and ITZ-PLip after 24 h at 37 °C, respectively. White lines represent the width of scratch. Scale bar represents 200 μm. (B) Percentage of migration areas calculated at 24 h compared with the scratch areas calculated at 0 h time point. Each bar represents mean percentage of scratch areas ± standard deviation (n = 3). **p < 0.01. | ||
In vivo biodistribution assay
The biodistribution of ITZ/DOX-PLip in nude mice bearing metastatic 4T1 breast cancer was investigated through determining the fluorescence intensity of DOX in each tissue after harvested at different time point. Free DOX was used as the control. The fluorescent images of each tissue and biodistribution profiles of DOX were shown in Fig. 4. In DOX group, DOX was widely distributed into most tissues and was rapid eliminated. The accumulation of DOX in tumor was neglectable after 24 h, and in heart it was detectable at 6 h, in contrast, in ITZ/DOX-PLip group, DOX was eliminated much more slowly compared with the DOX group. For this group, fluorescent signals of DOX in tumor was much higher than the DOX group (P < 0.01), while DOX signals in heart was much less (P < 0.01), which suggested that the ITZ/DOX-PLip group may show improved anticancer efficiency and less heart toxicity. Which was like the results of the work on PEGylated dendron-DOX conjugate nanoparticles done by Li et al.37 These phenomena could be mainly attributed to the EPR effect of nano-liposome and prolonged circulation half-life with the aid of hydrophilic PEO shell on the surface of the liposomes.23,27,37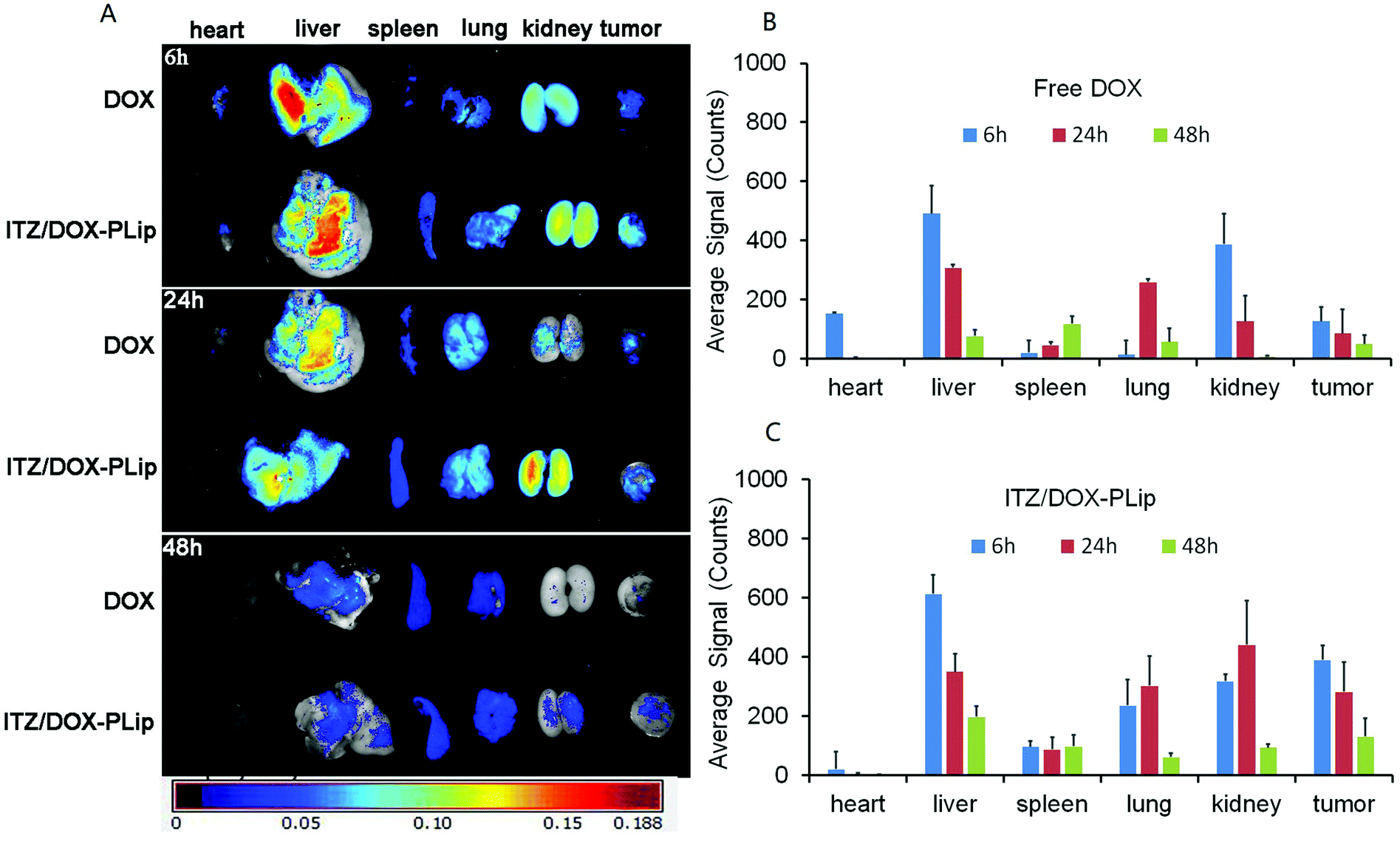 | ||
| Fig. 4 The ex vivo fluorescence images. The tumor-bearing nude mice were injected via tail vein with DOX·HCl and ITZ/DOX-PLip (DOX equivalent, dose of 5 mg kg−1). (A) The mice were sacrificed, and the organs were removed for ex-vivo fluorescence measurements at time intervals (6, 24 and 48 h). (B) Average fluorescent signals in different organs of free DOX·HCl. (C) Average fluorescent signals in different organs of ITZ/DOX-PLip. Each bar represents mean average signal ± standard deviation (n = 3). | ||
In vivo anticancer efficacy
The anticancer effect of DOX and/or ITZ was evaluated in 4T1 tumor-bearing BALB/c mice. The average tumor volume and body weight were measured during the experiment for the evaluation of the antitumor efficacy and the toxicity (Fig. 5A–C). The tumor grew rapidly in saline treated animals. In contrast, ITZ showed weak antitumor efficiency with TGI of 30.39% probably due to its anti-angiogenesis effect on tumor,38 which was consistent with the results of in vitro cytotoxicity experiments. Mice treated with DOX exhibited modest inhibitory effect on tumor growth with TGI of 57.65%. ITZ/DOX-PLip treatment showed the strongest inhibitory activity in tumor growth with TGI of 83.19%. The outstanding advantage of ITZ/DOX-PLip could be mainly caused by the synergy between DOX and ITZ, 15 and the enhanced accumulation of dual drugs in tumor with the aid of Pluronic® P123 modified liposome, which was confirmed by the cellular uptake and biodistribution results. DOX treated mice lost about 25% weight at the end of the experiment, while other mice did not lose weight, which suggested no significant systemic toxicity of ITZ and ITZ/DOX-PLip.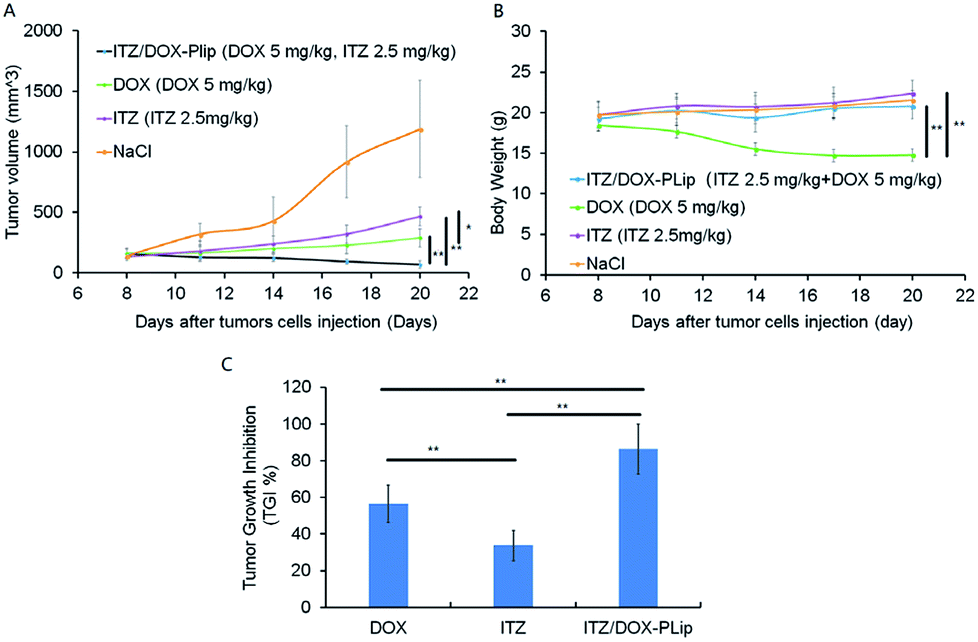 | ||
| Fig. 5 In vivo antitumor efficacy of free drugs and liposomes with different dosages in 4T1 cells tumor-bearing mice (n = 5). Mice were injected with normal saline as control, DOX (5 mg kg−1, every three days), ITZ (2.5 mg kg−1, every three days), and ITZ/DOX-PLip (DOX 5 mg kg−1, ITZ 2.5 mg kg−1, every three days). Throughout the study, mice were weighed and tumors were measured with calipers every two days. (A) The changes in tumor growth and (B) body weight were measured. At the end of the trial, tumor tissues were removed and weighted, and (C) the tumor growth inhibition (TGI%) were calculated. The results are shown as the mean ± standard deviation (n = 5). *p < 0.05, **p < 0.01. | ||
H&E staining (Fig. 6A) and whole slide digital images (Fig. SI2†) of tumor tissue demonstrated that apoptosis and necrosis areas were greatest in the ITZ/DOX-PLip group (81.85%) (Fig. 6B). H&E staining images of liver tissue showed that metastasis modules occupied a large part of the liver when mice were treated by saline, DOX, or ITZ, which indicated that the tumor metastasis suppressing of DOX or ITZ alone was quite poor to 4T1 metastatic mice. Mice treated with ITZ/DOX-PLip showed no tumor burden in the tested normal tissues. H&E staining images of heart tissue showed that negligible heart toxicity in the DOX group, which were in a good agreement with the body weight result. Totally, for ITZ/DOX-PLip group, the apoptosis and necrosis areas were significantly increased, the liver metastasis tumor nodule and the heart toxicity were reduced, which were mainly attributed to the effective EPR effect and the combination of apoptosis and anti-angiogenesis effects of DOX and ITZ. To further investigate the tumor suppression mechanism of liposomes, IHC assays of tumor tissue slices were performed at the end of the trial (Fig. 7). Ki-67 assay staining proliferation active cells was used to assess the inhibitory effect of treatments on tumor cells proliferation. ITZ/DOX-PLip with Ki-67 positive cell rate of 9.98% had the most significant suppression of tumor proliferation in vivo (P < 0.01), which was consistent well with the in vivo tumor volume and H&E results. TUNEL assay was used to detect programmed apoptotic cell death in situ. TUNEL assay revealed that treatments with ITZ/DOX-PLip resulted in much higher apoptosis levels of 61.95% than the other groups (P < 0.01).
 | ||
| Fig. 6 H&E staining images and analysis. (A) HE of different organs of mice treated with NaCl, DOX (5 mg kg−1), ITZ (2.5 mg kg−1) and ITZ/DOX-PLip DOX (5 mg kg−1), ITZ (2.5 mg kg−1) (all tissues: ×100). Black circles represent the necrosis zones in tumors and hearts. Black arrows represent metastases in livers. (B) HE whole slide digital analysis of necrosis area of tumors. Each bar represents mean necrosis area ± standard deviation (n = 3). **p < 0.01. | ||
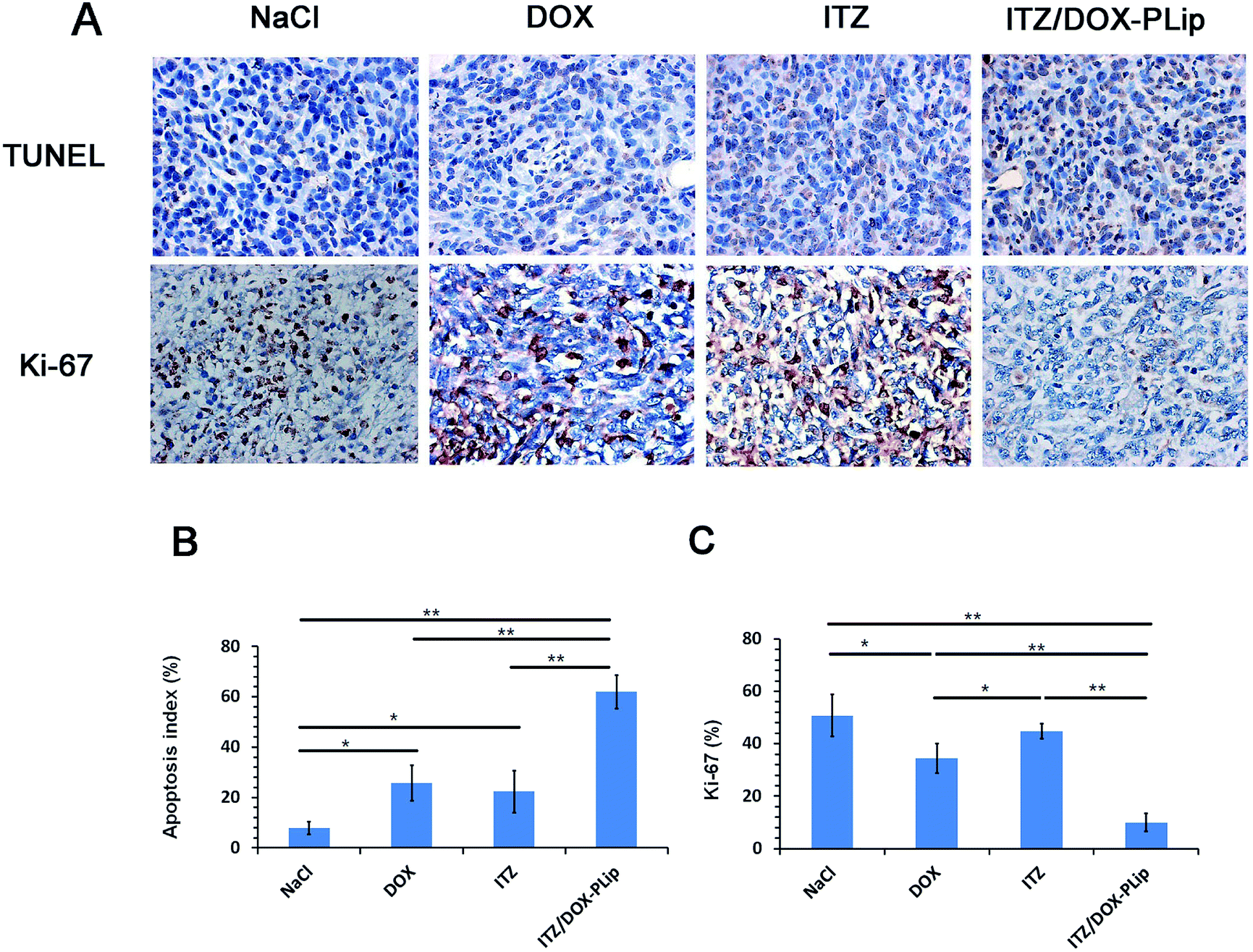 | ||
| Fig. 7 Immunohistochemical (IHC) staining of tumor tissues. (A) TUNEL and Ki-67 images of tumors in different groups. Positive staining was shown in brown. (all tissues: × 400). (B) Quantification of apoptotic cells by TUNEL assay. The apoptotic index was calculated as the ratio of apoptotic cells to total cells in each field, using five random fields. (C) Quantification of proliferative cells by Ki-67 assay. The Ki67 index was calculated as the ratio of proliferative cells to total cells in each field, using five random fields. The results are shown as the mean ± standard deviation (n = 5). *p < 0.05, **p < 0.01. | ||
In vivo angiogenesis
To evaluate the antiangiogenic activity of ITZ/DOX-PLip treatment in vivo, the microvessel density (MVD) was assessed by immunohistochemistry. As shown in Fig. 8A, microvessels were clearly observed by CD31 staining, and very few microvessels were observed in the ITZ/DOX-PLip treated group. For the angiogenesis treatment, the ITZ group was less effective than ITZ/DOX-PLip treated group, mainly due to the poor accumulation of ITZ in tumor site. In DOX and saline groups, MVD was much more than the other groups indicated negligible anti-angiogenic activity. | ||
| Fig. 8 CD31 immunohistochemical (IHC) staining of tumor tissues. (A) Representative micrographs of immunohistochemical detection of CD31-positive microvessel of 4T1 tumors from different groups: NaCl (A1), DOX (5 mg kg−1, every three days) (A2), ITZ (2.5 mg kg−1, every three days) (A3) and ITZ/DOX-PLip (DOX 5 mg kg−1, ITZ 2.5 mg kg−1, every three days) (A4). The brown areas indicate CD31 positive staining. (B) Quantification of microvessel density in the tumor tissues by CD31 assay. The microvessel counts were counted in five random fields. The results are shown as the mean ± standard deviation (n = 5). *p < 0.05, **p < 0.01. | ||
Conclusions
In this study, we designed a DOX and ITZ co-encapsulated liposome coating with Pluronic® P123 (ITZ/DOX-PLip) for anti-cancer and anti-angiogenic therapy. The ITZ/DOX-PLip was successfully prepared and exhibited a uniform size distribution, a high loading efficiency, and a sustained release profile. Compared with free DOX or DOX-Lip, ITZ/DOX-PLip could significantly enhance cytotoxicity and cellular uptake in 4T1 murine breast cancer cells. Furthermore, ITZ/DOX-PLip could effectively inhibit tumor growth in xenograft 4T1 bearing BALB/c mice with higher drug accumulation in tumors and decreased distribution in heart due to passive targeting and prolonged circulation. The results of endothelial cell proliferation, migration and MVD confirmed that the antiangiogenic activity of ITZ/DOX-Plip was superior to free ITZ or ITZ-Lip in vitro and in vivo. Overall, this DOX and ITZ co-delivery system may provide a promising strategy for cancer combinatory treatment by inhibiting tumour growth and tumour-associated angiogenesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Associate Professor Jing Ren, due to her presenting us Itraconazole as a gift. The work was supported by National Natural Science Foundation of China (81371666) and International Visiting Fund for Excellent Young Scholars of Sichuan University. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “Sichuan University” and Experiments were approved by the Animal Ethics Committee of “Sichuan University”.References
- A. Sandler, R. Gray, M. C. Perry, J. Brahmer, J. H. Schiller, A. Dowlati, R. Lilenbaum and D. H. Johnson, N. Engl. J. Med., 2006, 355, 2542–2550 CrossRef PubMed.
- R. R. McWilliams, J. B. Allred, J. A. Slostad, R. Katipamula, R. S. Dronca, K. M. Rumilla, L. A. Erickson, A. H. Bryce, R. W. Joseph, L. A. Kottschade, D. M. King, J. M. Leitch and S. N. Markovic, Cancer, 2018, 124, 537–545 CrossRef PubMed.
- B. Escudier, J. Bellmunt, S. Negrier, E. Bajetta, B. Melichar, S. Bracarda, A. Ravaud, S. Golding, S. Jethwa and V. Sneller, J. Clin. Oncol., 2010, 28, 2144–2150 CrossRef PubMed.
- N. J. Robert, V. Dieras, J. Glaspy, A. M. Brufsky, I. Bondarenko, O. N. Lipatov, E. A. Perez, D. A. Yardley, S. Y. T. Chan, X. Zhou, S.-C. Phan and J. O'Shaughnessy, J. Clin. Oncol., 2011, 29, 1252–1260 CrossRef PubMed.
- V. Ranpura, S. Hapani and S. H. Wu, JAMA, J. Am. Med. Assoc., 2011, 305, 487–494 CrossRef PubMed.
- T. Fojo and C. Grady, JNCI, J. Natl. Cancer Inst., 2009, 101, 1044–1048 CrossRef PubMed.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef PubMed.
- J. Kim, J. Y. Tang, R. Gong, J. Kim, J. J. Lee, K. V. Clemons, C. R. Chong, K. S. Chang, M. Fereshteh, D. Gardner, T. Reya, J. O. Liu, E. H. Epstein, D. A. Stevens and P. A. Beachy, Cancer Cell, 2010, 17, 388–399 CrossRef PubMed.
- C. R. Chong, J. Xu, J. Lu, S. Bhat, D. J. Sullivan Jr. and J. O. Liu, ACS Chem. Biol., 2007, 2, 263–270 CrossRef PubMed.
- R. Liu, J. Li, T. Zhang, L. Zou, Y. Chen, K. Wang, Y. Lei, K. Yuan, Y. Li, J. Lan, L. Cheng, N. Xie, R. Xiang, E. C. Nice, C. Huang and Y. Wei, Autophagy, 2014, 10, 1241–1255 CrossRef PubMed.
- S. A. Head, W. Q. Shi, E. J. Yang, B. A. Nacev, S. Y. Hong, K. K. Pasunooti, R. J. Li, J. S. Shim and J. O. Liu, ACS Chem. Biol., 2017, 12, 174–182 CrossRef PubMed.
- S. A. Head, W. Shi, L. Zhao, K. Gorshkov, K. Pasunooti, Y. Chen, Z. Y. Deng, R. J. Li, J. S. Shim, W. Z. Tang, T. Hartung, J. Zhang, Y. M. Zhao, M. Colombini and J. O. Liu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E7276–E7285 CrossRef PubMed.
- K. Takara, Y. Tanigawara, F. Komada, K. Nishiguchi, T. Sakaeda and K. Okumura, Biol. Pharm. Bull., 1999, 22, 1355–1359 CrossRef.
- N. Iida, K. Takara, N. Ohmoto, T. Nakamura, T. Kimura, A. Wada, M. Hirai, T. Sakaeda and K. Okumura, Biol. Pharm. Bull., 2001, 24, 1032–1036 CrossRef.
- M. Kurosawa, M. Okabe, N. Hara, K. Kawamura, S. Suzuki, K. Sakurada and M. Asaka, Ann. Hematol., 1996, 72, 17–21 CrossRef PubMed.
- B. T. Aftab, I. Dobromilskaya, J. O. Liu and C. M. Rudin, Cancer Res., 2011, 71, 6764–6772 CrossRef PubMed.
- H. Tsubamoto, T. Sonoda, S. Ikuta, S. Tani, K. Inoue and N. Yamanaka, Anticancer Res., 2015, 35, 4191–4196 Search PubMed.
- E. S. Antonarakis, E. I. Heath, D. C. Smith, D. Rathkopf, A. L. Blackford, D. C. Danila, S. King, A. Frost, A. S. Ajiboye, M. Zhao, J. Mendonca, S. K. Kachhap, M. A. Rudek and M. A. Carducci, Oncologist, 2013, 18, 163–173 CrossRef PubMed.
- D. J. Kim, J. Kim, K. Spaunhurst, J. Montoya, R. Khodosh, K. Chandra, T. Fu, A. Gilliam, M. Molgo, P. A. Beachy and J. Y. Tang, J. Clin. Oncol., 2014, 32, 745–751 CrossRef PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef PubMed.
- L. Jiang, L. Li, B. He, D. Pan, K. Luo, Q. Yi and Z. Gu, J. Biomed. Nanotechnol., 2016, 12, 79–90 CrossRef PubMed.
- L. Jiang, L. Li, X. D. He, Q. Y. Yi, B. He, J. Cao, W. S. Pan and Z. W. Gu, Biomaterials, 2015, 52, 126–139 CrossRef PubMed.
- J. Sun, L. Jiang, Y. Lin, E. M. Gerhard, X. Jiang, L. Li, J. Yang and Z. Gu, Int. J. Nanomed., 2017, 12, 1517–1537 CrossRef PubMed.
- H. M. Redhead, S. S. Davis and L. Illum, J. Controlled Release, 2001, 70, 353–363 CrossRef PubMed.
- L. Illum and S. S. Davis, FEBS Lett., 1984, 167, 79–82 CrossRef PubMed.
- Y. Z. Song, Q. J. Tian, Z. J. Huang, D. Fan, Z. N. She, X. R. Liu, X. B. Cheng, B. Yu and Y. H. Deng, Int. J. Nanomed., 2014, 9, 2307–2317 Search PubMed.
- A. V. Kabanov, E. V. Batrakova and D. W. Miller, Adv. Drug Delivery Rev., 2003, 55, 151–164 CrossRef PubMed.
- E. V. Batrakova, S. Li, S. V. Vinogradov, V. Y. Alakhov, D. W. Miller and A. V. Kabanov, J. Pharmacol. Exp. Ther., 2001, 299, 483–493 Search PubMed.
- G. Haran, R. Cohen, L. K. Bar and Y. Barenholz, Biochim. Biophys. Acta, 1993, 1151, 201–215 CrossRef.
- K. Raemdonck, K. Braeckmans, J. Demeester and S. C. De Smedt, Chem. Soc. Rev., 2014, 43, 444–472 RSC.
- T. H. Kim, H. H. Jiang, Y. S. Youn, C. W. Park, K. K. Tak, S. Lee, H. Kim, S. Jon, X. Chen and K. C. Lee, Int. J. Pharm., 2011, 403, 285–291 CrossRef PubMed.
- S. Chono, T. Tanino, T. Seki and K. Morimoto, J. Pharm. Pharmacol., 2007, 59, 75–80 CrossRef PubMed.
- W. Zhang, Y. A. Shi, Y. Z. Chen, J. A. Ye, X. Y. Sha and X. L. Fang, Biomaterials, 2011, 32, 2894–2906 CrossRef PubMed.
- W. Gao, Z. Q. Lin, M. W. Chen, X. C. Yang, Z. Cui, X. F. Zhang, L. Yuan and Q. Zhang, Int. J. Nanomed., 2014, 9, 3425–3437 CrossRef PubMed.
- D. Tretiakova, N. Onishchenko, I. Boldyrev, I. Mikhalyov, A. Tuzikov, N. Bovin, E. Evtushenko and E. Vodovozova, Colloids Surf., B, 2018, 166, 45–53 CrossRef PubMed.
- L. Lamalice, F. Le Boeuf and J. Huot, Circ. Res., 2007, 100, 782–794 CrossRef PubMed.
- N. Li, N. Li, Q. Yi, K. Luo, C. Guo, D. Pan and Z. Gu, Biomaterials, 2014, 35, 9529–9545 CrossRef PubMed.
- J. R. Pace, A. M. DeBerardinis, V. Sail, S. K. Tacheva-Grigorova, K. A. Chan, R. Tran, D. S. Raccuia, R. J. Wechsler-Reya and M. K. Hadden, J. Med. Chem., 2016, 59, 3635–3649 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03787f |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |