DOI:
10.1039/C8RA03669A
(Review Article)
RSC Adv., 2018,
8, 21100-21132
Heterocorroles: corrole analogues containing heteroatom(s) in the core or at a meso-position
Received
28th April 2018
, Accepted 4th June 2018
First published on 8th June 2018
Abstract
Corroles are 18 π aromatic macrocyclic systems having one direct pyrrole–pyrrole linkage leading to a contracted cavity compared to porphyrins. Corroles exhibit contrasting coordination chemistry and properties compared to porphyrins. Structural modification of corroles by introducing a heteroatom in their aromatic conjugation circuit i.e., either in the core or at a meso position leads to a new class of corrinoids called heterocorroles. The core modification strategy includes replacing one or two core nitrogen atom(s) with O, S or C atoms and meso-modification involves replacing the meso-carbon atom at the 10-position with NH, NR, O, S, Se or Si atoms. This review article presents an overview of the progress in heterocorrole chemistry including their syntheses, key structural aspects and properties.
 Booruga Umasekhar | Booruga Umasekhar was born in the state of Andhra Pradesh, India in 1991. He received his BSc and MSc degrees from Sri Venkateswara University Tirupati in 2011 and 2013 respectively. He joined Professor M. Ravikanth's research group at Indian Institute of Technology Bombay in 2014 to pursue his PhD, where he is currently working on synthesis of meso-pyrrolyl dipyrromethane based fluorescent systems. |
 Vijayendra S. Shetti | Vijayendra S. Shetti was born in the state of Karnataka, India in 1981. He obtained his MSc degree from Karnatak University Dharwad and PhD from Indian Institute of Technology Bombay working under the supervision of Prof. M. Ravikanth. During his PhD, he worked on synthesis of meso-substituted 22-thiacorroles and core-modified porphyrin based molecular assemblies. After postdoctoral assignments at Taiwan and South Korea, he joined BMS College of Engineering, Bengaluru as an Assistant Professor of Chemistry in 2014. His current research is focused on synthesis of novel π conjugated molecules for organic electronic applications. |
 Mangalampalli Ravikanth | M. Ravikanth was born in India, in 1966. He received his BSc and MSc from Osmania University, Hyderabad and PhD from Indian Institute of Technology, Kanpur in 1994. After his postdoctoral stay in USA and Japan, he joined as a faculty at Indian Institute of Technology Bombay, where he is currently a full professor. His current research interest includes porphyrin and related macrocycles and boron dipyrromethenes. |
1. Introduction
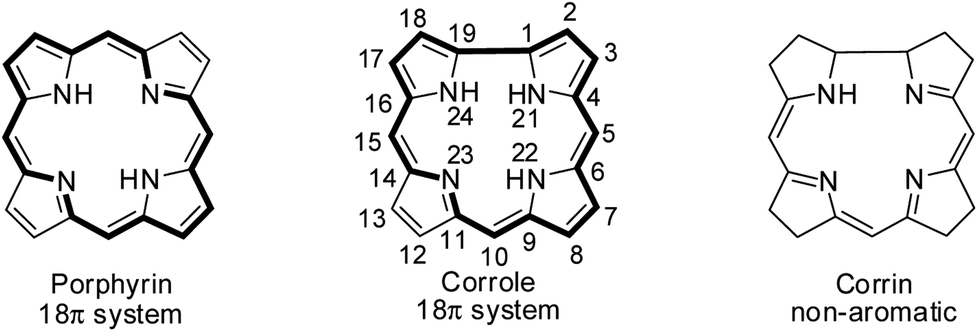
Hodgkin's work on the X-ray crystallography of β-substituted free-base corrole revealed for the first time that corroles resemble corrin, a cobalt coordinating ligand in vitamin B12.1 Corroles are tetrapyrrolic macrocycles which have a direct pyrrole–pyrrole linkage similar to corrin and preserve the aromaticity of porphyrins despite having one less meso-carbon atom (Fig. 1).2 Corroles differ from their congener porphyrin in having a contracted cavity and three ionisable core NH atoms. This leads to a contrasting coordination chemistry of corroles where they can stabilize metal ions in a higher oxidation state unlike porphyrins and also can exhibit unique reactivity for low valent metallocorroles.3 Corroles' ability to absorb light throughout the visible spectral range, their enhanced luminescence yield, their good photostability in most solvents and their relative ease of oxidation give them an edge over porphyrins in many applications.4–6
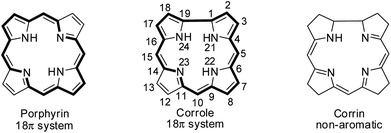 |
| | Fig. 1 Skeletal structure of porphyrin, corrole and corrin. | |
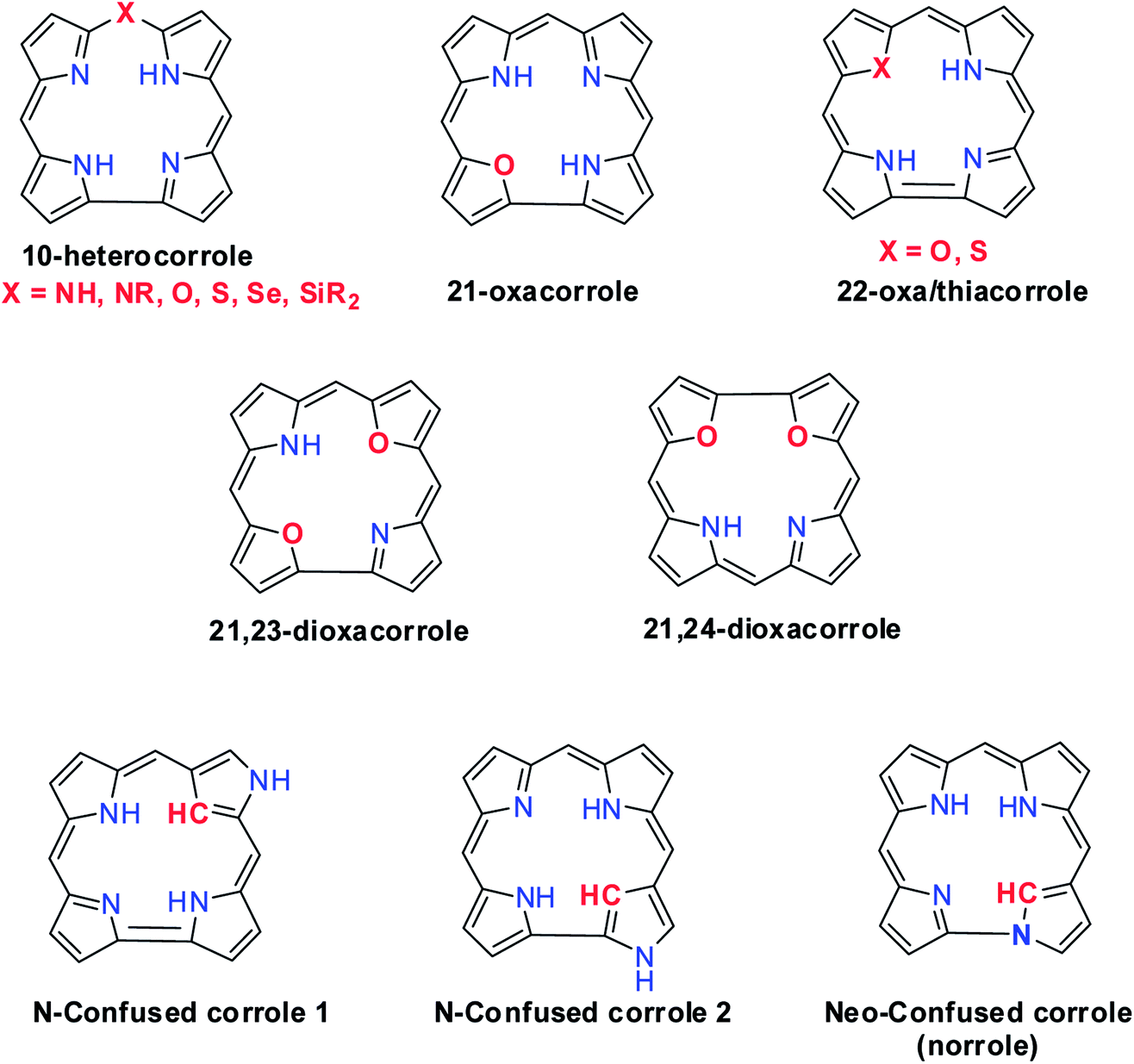
Structural modification of corroles by introducing a heteroatom on their aromatic conjugation circuit i.e., either at the core or at a meso position leads to a new class of corrinoids called heterocorroles.2,7,8 The core modification strategy includes replacing one or two core nitrogen atom(s) by oxygen, sulfur (21-, 22-, 21,23-heterocorroles) or carbon atoms (confused corrole) and meso-modification involves replacing meso-carbon atom at the 10-position by NH, NR, O, S, Se or even Si atoms (10-heterocorroles) (Fig. 2). Heterocorroles represent a hybrid class of molecules which have contracted cavity similar to corroles and are dianionic ligands like porphyrins. Because of this, they can serve as appropriate reference systems to study metallocorroles. In this review article, we wish to present an overview of the progress in the area of heterocorroles, especially in the area of core-modified corroles and mono meso-modified corroles. However, the discussion on corrolazines is not included in this article.
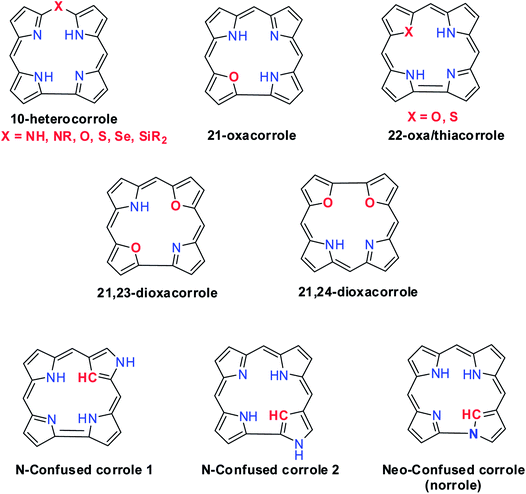 |
| | Fig. 2 Various examples of heterocorrole framework. | |
2. Corroles containing heteroatom at 10-position (meso-modified corroles)
2.1. 10-Azacorroles
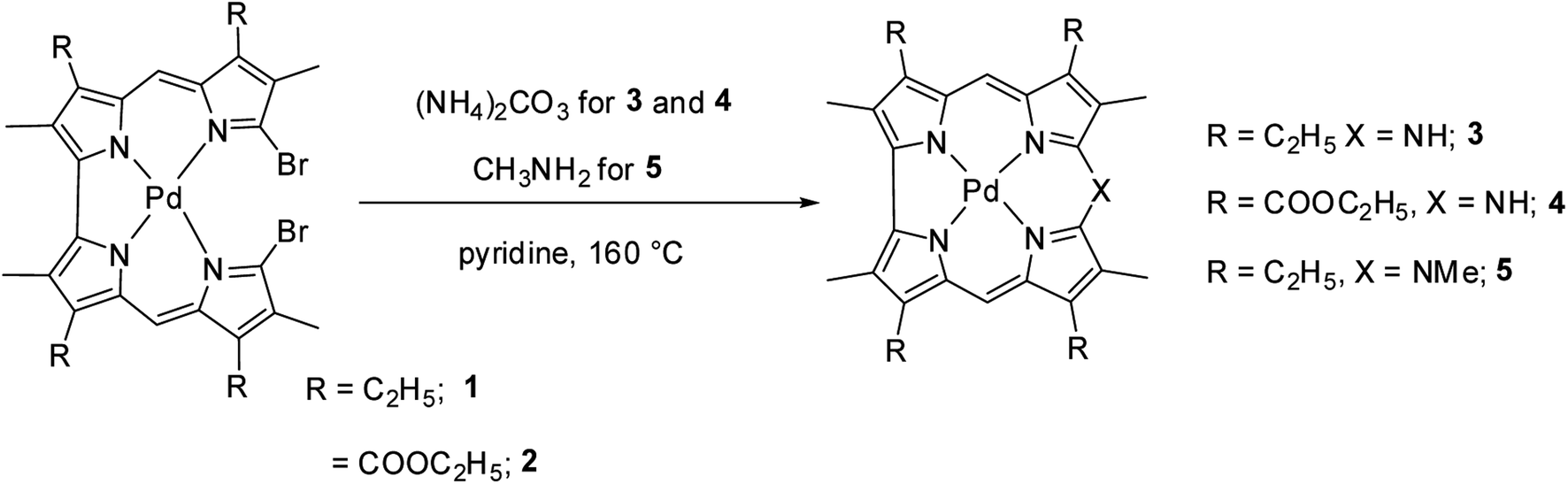
Johnson, Kay and Rodrigo in their pioneering work9 synthesized β-substituted palladium corroles 3–5 containing nitrogen atom at one of the meso positions starting from Pd(II) chelates of dibromo 5,5′-bi(dipyrromethene) 1 and 2 (Scheme 1). Authors' attempts to obtain free base corroles by demetallating the central metal atoms by acid treatment was not completely successful.
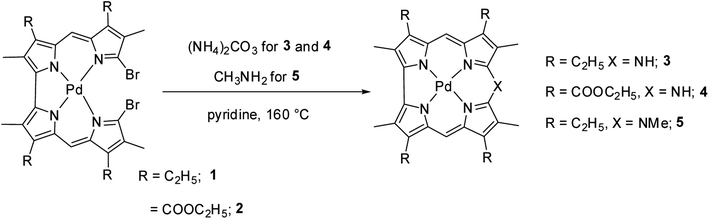 |
| | Scheme 1 Synthesis of β-substituted 10-azacorroles 3–5. | |
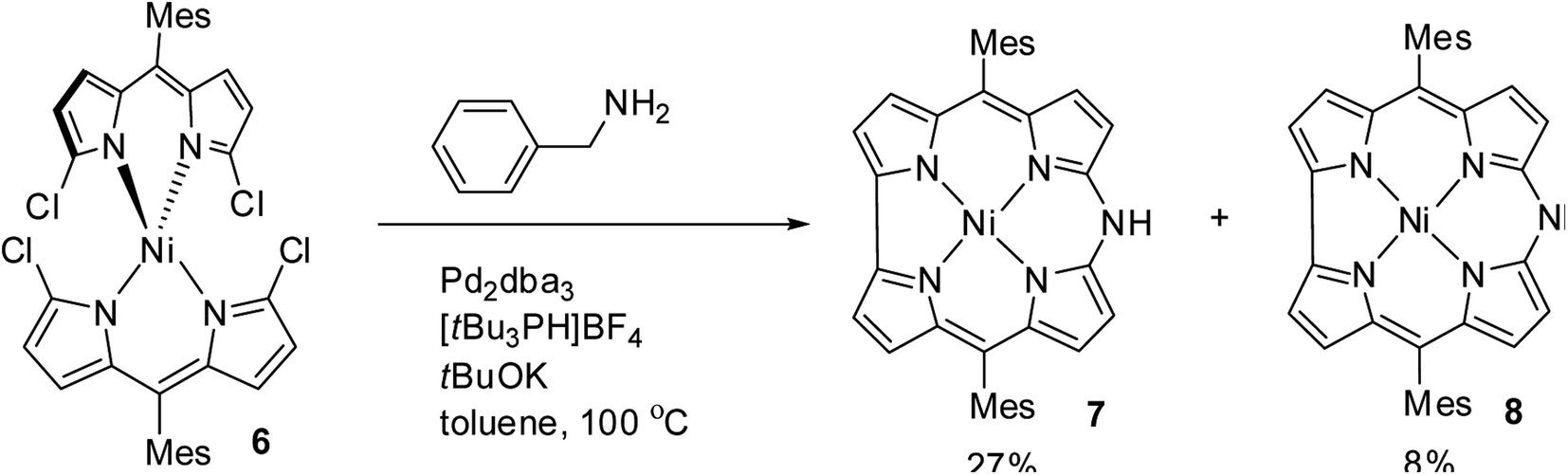
Shinokubo and co-workers10 reported the earliest synthesis of meso-aryl substituted azacorroles 7 & 8 by utilizing Buchwald–Hartwig coupling reaction between α,α′-dichlorodipyrrin Ni(II) complex 6 and benzylamine (Scheme 2). Though, this reaction resulted in a mixture of azacorroles 7 and 8, column chromatographic separation enabled authors to isolate 7 in 27% yield. 1H NMR spectral feature and nucleus independent chemical shift (NICS) studies revealed the aromatic character of azacorrole 7. To demonstrate regio-selective functionalization and the acidic nature of N–H bond at meso-position, authors performed acylation reaction of 7 with acetyl chloride (Scheme 3). The acylation reaction of 7 in the presence of a base resulted in the formation of N-acetylate product 9 in 62% yield. When the C-acylation was performed by using acetyl chloride and zinc, C8-acylated product 10 was obtained in 53% yield which is in contrast to the electrophilic substitution pattern noticed in normal corroles which generally yield C2 and C17 substituted products.
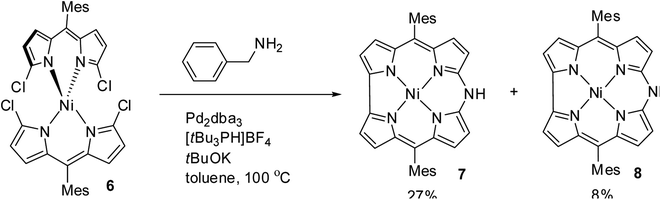 |
| | Scheme 2 Synthesis of 10-azacorroles 7 and 8 by Buchwald–Hartwig reaction. | |
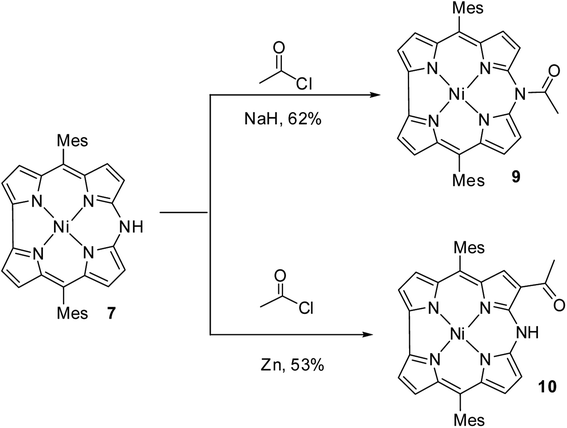 |
| | Scheme 3 N- and C-acylation reactions of 10-azacorrole 7. | |
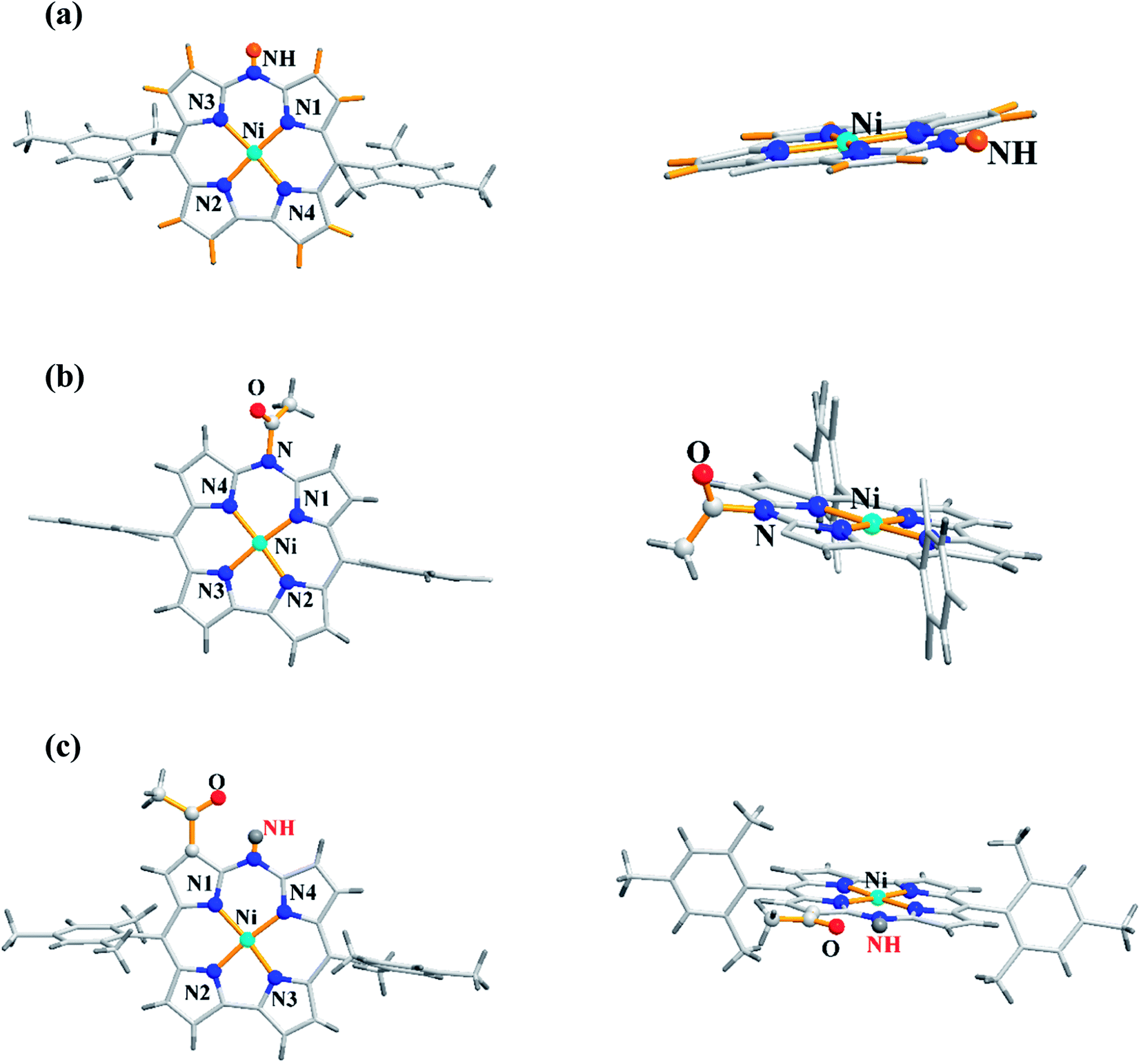
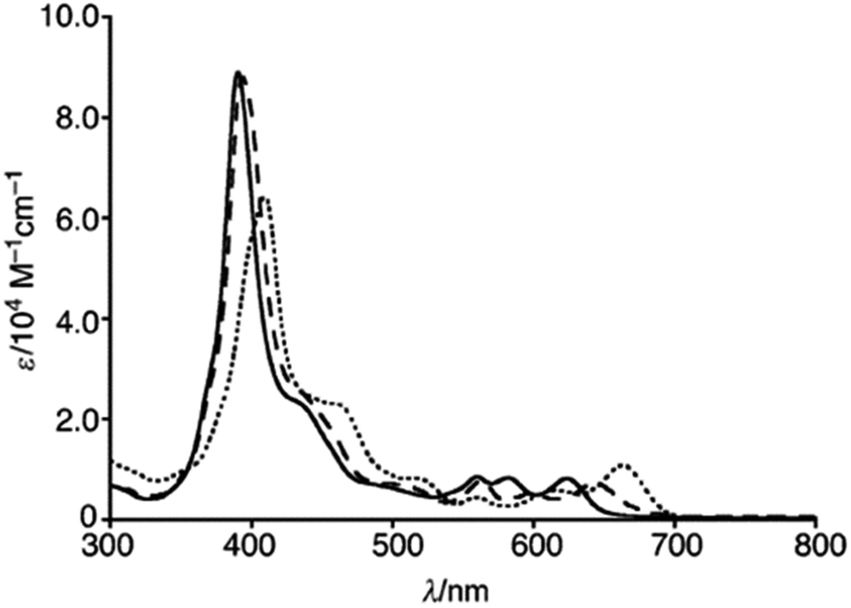
The single crystal X-ray analysis revealed highly planar structures of azacorroles 7, 9 and 10 (Fig. 3). The orientation of acetyl moiety in 9 was found to be perpendicular to the corrole plane, whereas in 10, the acetyl group was coplanar with corrole plane due to its intramolecular hydrogen bonding interaction with meso-NH proton. This resulted in an expanded conjugation leading to a substantial bathochromic shift of Q-like bands of 10 compared to 7 and 9 (Fig. 4). The electrochemical studies showed that the first oxidation of corrole 7 was rather easy compared to acylated corroles 9 and 10.
 |
| | Fig. 3 Top and side view of X-ray crystal structures of (a) 7 (CCDC 861382) (b) 9 (CCDC 861384) and (c) 10 (CCDC 861385). | |
 |
| | Fig. 4 UV-vis spectra of 7 (normal line), 9 (dashed line) and 10 (dotted line) recorded in dichloromethane. Reproduced from ref. 10 with permission. Copyright© 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
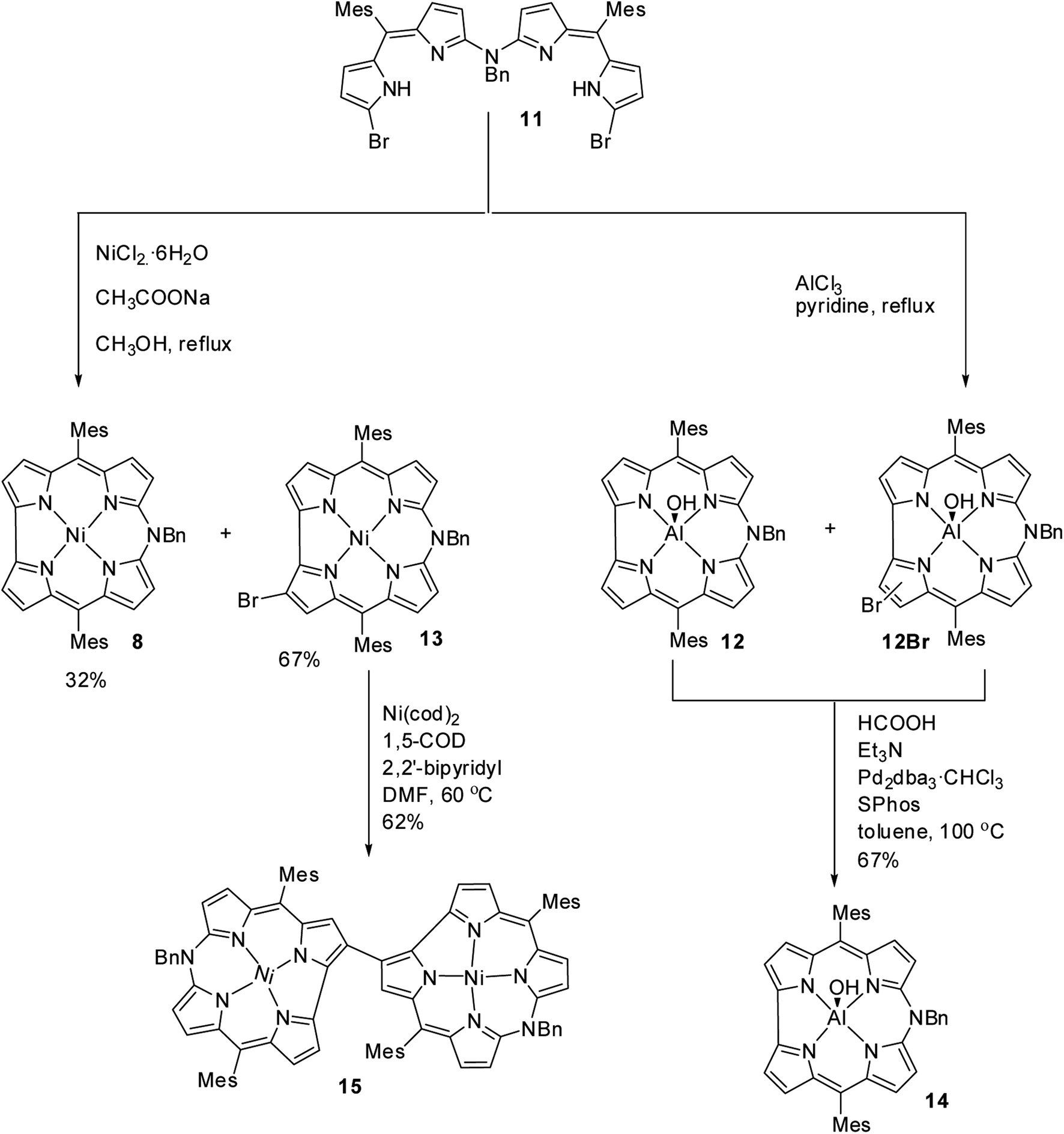
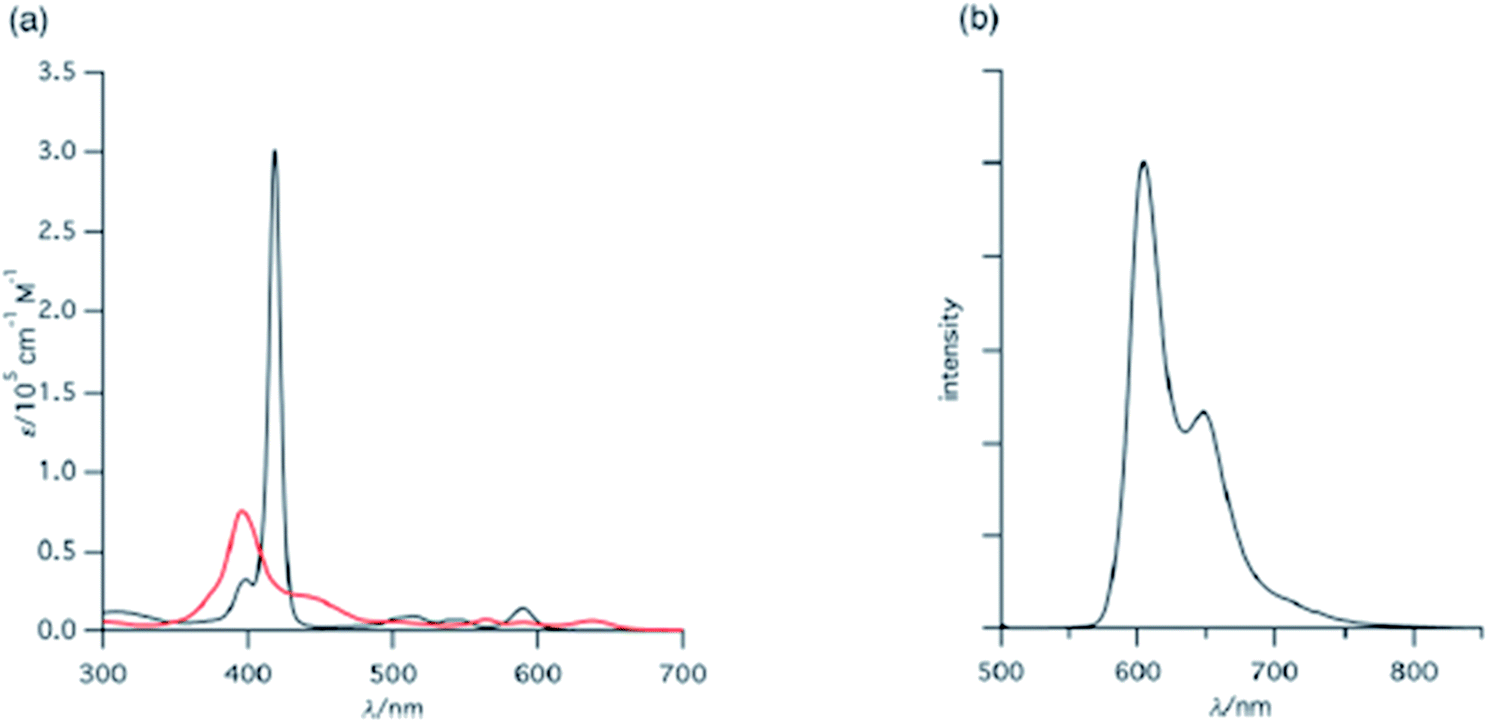
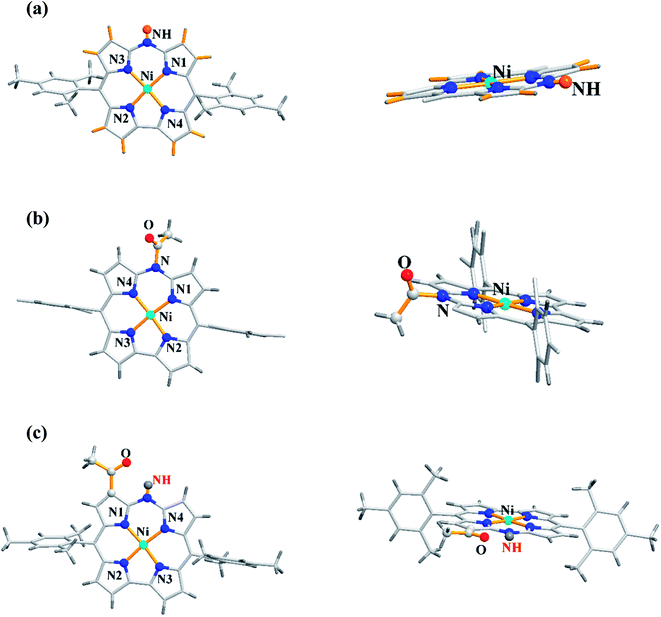
Shinokubo, Hiroto and co-workers synthesized Ni(II) and Al(III) complexes of 10-azacorrole by using coordination induced cyclization of nitrogen bridged bisdipyrrin.11 The bisdipyrrin containing two bromo groups at α-position 11 when reacted with excess NiCl2·6H2O yielded Ni(II) azacorrole 12 along with 2-bromo-10-azacorrole 13. The reaction of same bisdipyrrin with excess AlCl3 resulted in a mixture of Al(III) azacorroles including regioisomeric bromoazacorroles which upon subsequent reductive dehalogenation afforded Al(III) 10-azacorrole 14 in 67% yield (Scheme 4). The 2-bromo-10-azacorrole 13 was further subjected to coupling reaction in presence of Ni(cod)2/2,2′-bipyridyl (Yamamoto conditions) yielding to a 2,2′-linked corrole dimer 15 (Scheme 4). The Al and Ni corroles displayed contrasting absorption spectral features compared to their porphyrin counterparts. The Al corrole 14 exhibited a sharp Soret band and hypsochromically shifted lowest energy absorption band compared to the Ni corrole 12 (Fig. 5). The Al corrole 14 exhibited larger oxidation and reduction potential values compared to the Ni corrole due to electron withdrawing nature of Al(III). The Al(III) corrole 14 also represented the first examples of emissive 10-azacorroles with moderate quantum yield (Fig. 5).
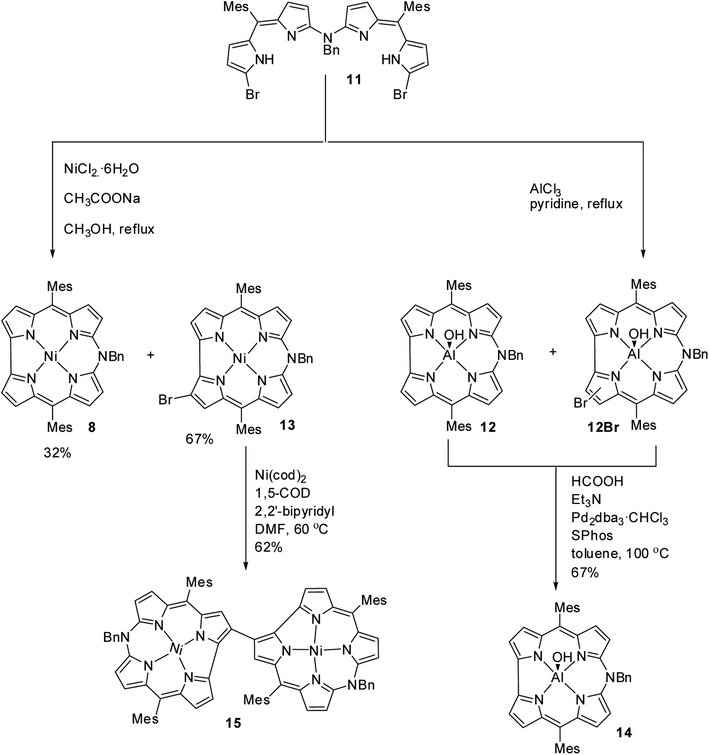 |
| | Scheme 4 Synthesis of 2-bromo-10-azacorrole 13, Al(III) azacorrole 14 and corrole dyad 15. | |
 |
| | Fig. 5 (a) UV-vis spectra of 12 (red), 14 (black) (b) emission spectrum of 14 (λex = 420 nm) recorded in dichloromethane. Reproduced from ref. 11 with permission. Copyright© 2016 Royal Society of Chemistry. | |
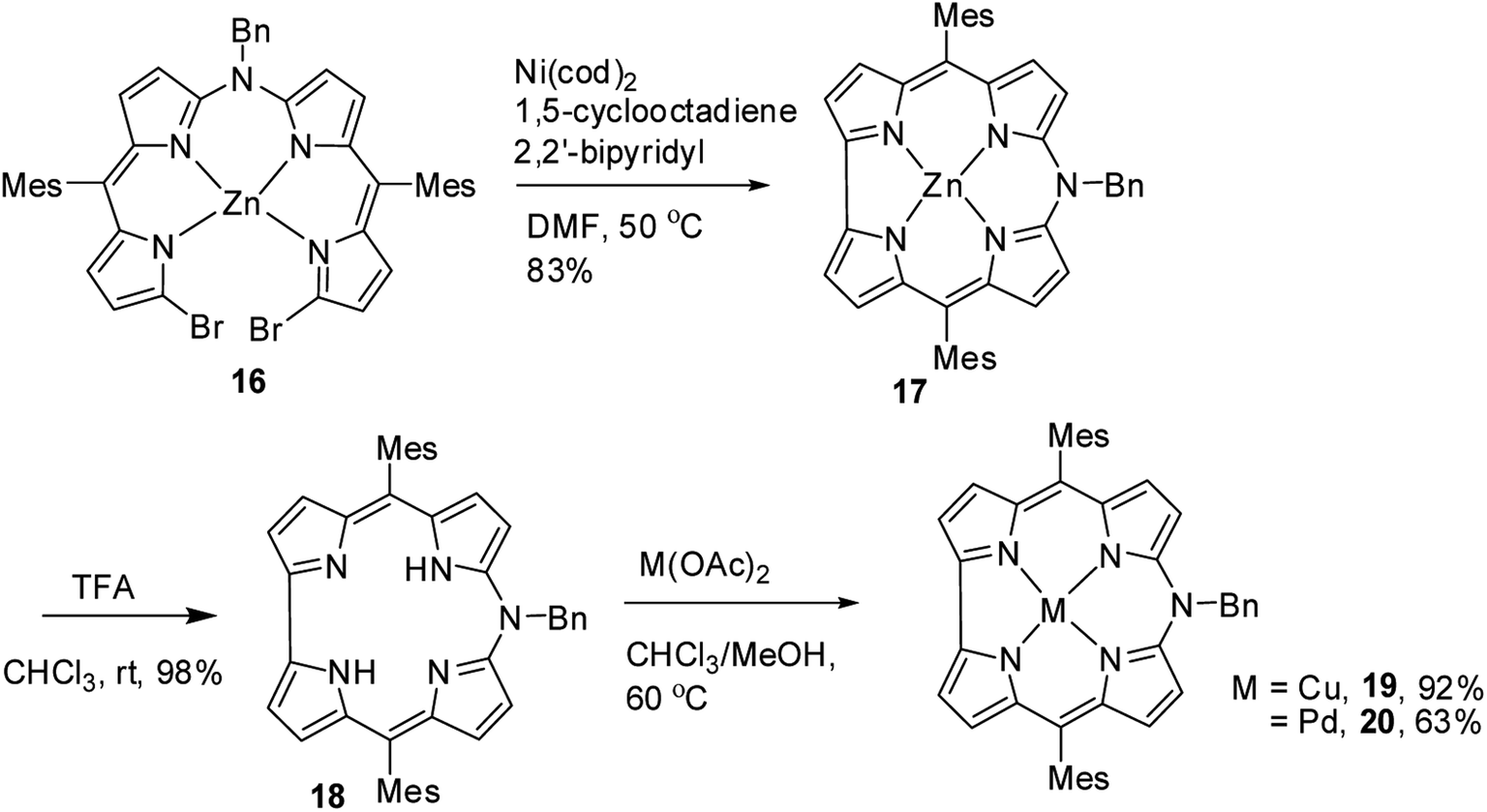
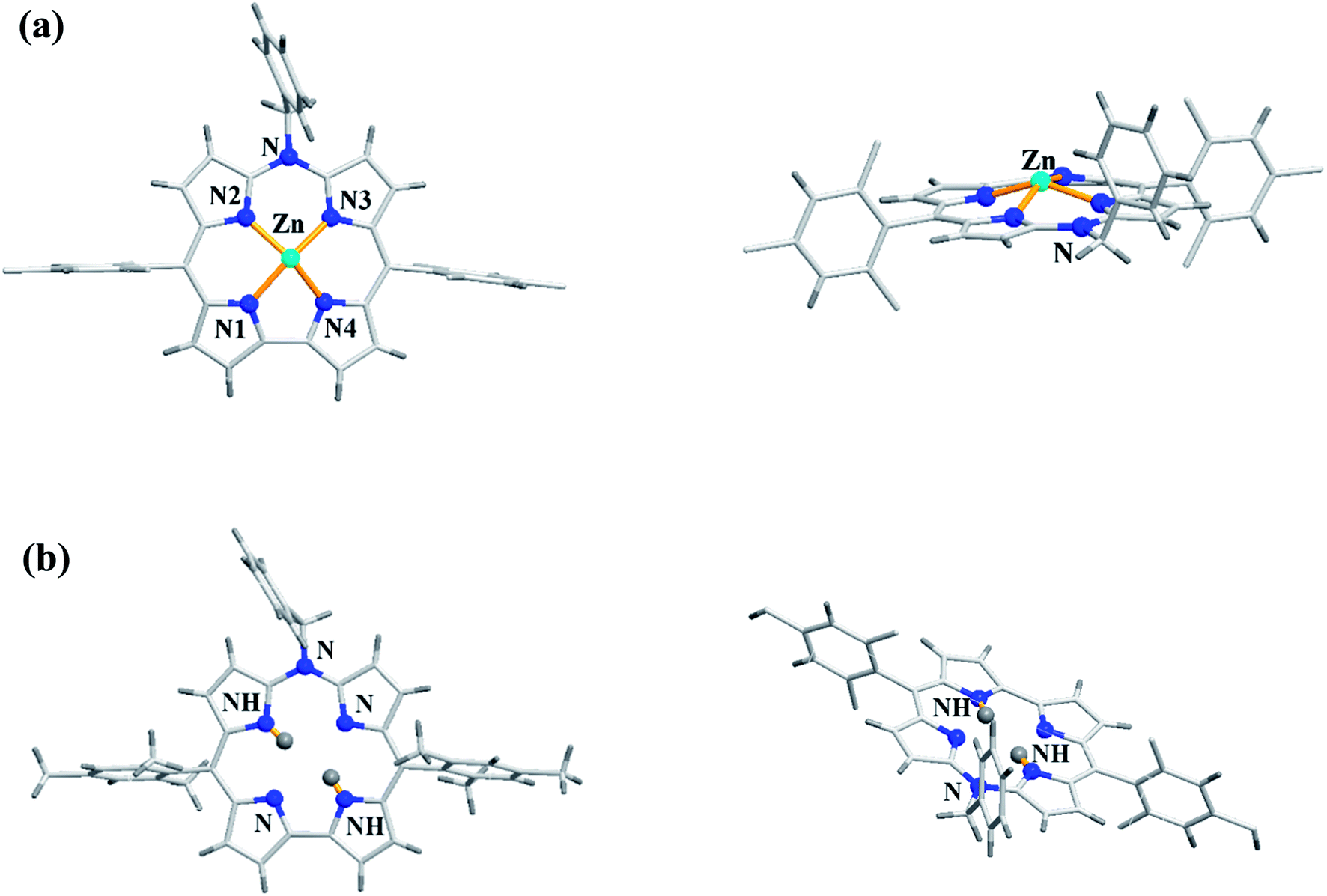
As demetallation of Ni(II) and Al(III) from above corroles was challenging and unsuccessful, the same authors, in their pursuit to find a synthetic protocol to make free base meso-azacorroles, devised a method involving Zn(II) complex of bisdipyrrin 16.12 The Zn(II) bisdipyrrin 16 was subjected to Yamamoto reaction conditions to facilitate cyclization leading to Zn(II) 10-azacorrole (Scheme 5). The TFA induced demetallation yielded corresponding free base 10-azacorrole 18 in quantitative yield. The single crystal X-ray structures of compound 17 and 18 are shown in Fig. 6. The single crystal X-ray analysis of zinc corrole 17 showed a pentacoordinated Zn(II) in the core and the mean plane deviation of zinc (0.088 Å) was found to be greater than the corresponding Ni complex 8 (0.020 Å). The X-ray structure of free base corrole 18 showed a highly planar structure with two core-NH atoms. The chemical shift value of the core NH resonance in 1H NMR spectrum (5.23 ppm) reflected on its lower aromatic character. The Cu(II) 19 and Pd(II) 20 azacorroles were synthesized by metallating free base azacorrole 18 with acetates of copper and palladium respectively. The Zn(II) 17 and Cu(II) 19 azacorroles showed red shifted absorption bands compared to free base azacorrole 18. The electrochemical studies revealed marginally reduced HOMO–LUMO gap for Zn(II) 17 and Pd(II) 20 corroles compared to their free base analogue. The fluorescence quantum yield of zinc corrole 17 was comparable to earlier reported Al(III) azacorrole 14.
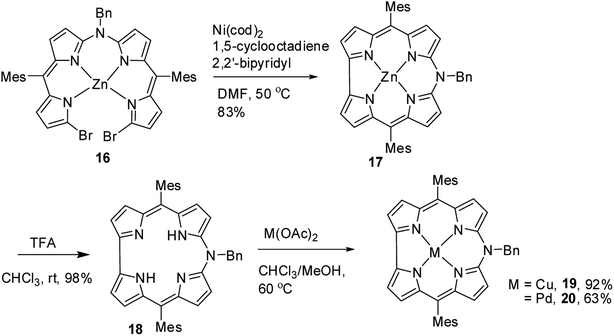 |
| | Scheme 5 Synthesis of free base 10-azacorrole 18 and its metal complexes 17, 19 & 20. | |
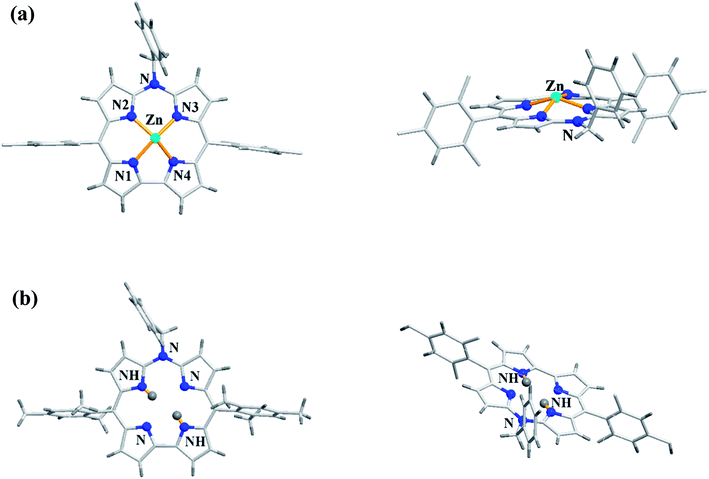 |
| | Fig. 6 Top and side view of X-ray crystal structures of (a) 17 (CCDC 1477606) (b) 18 (CCDC 1477608). | |
2.2. Corroles containing chalcogen atom at 10-position
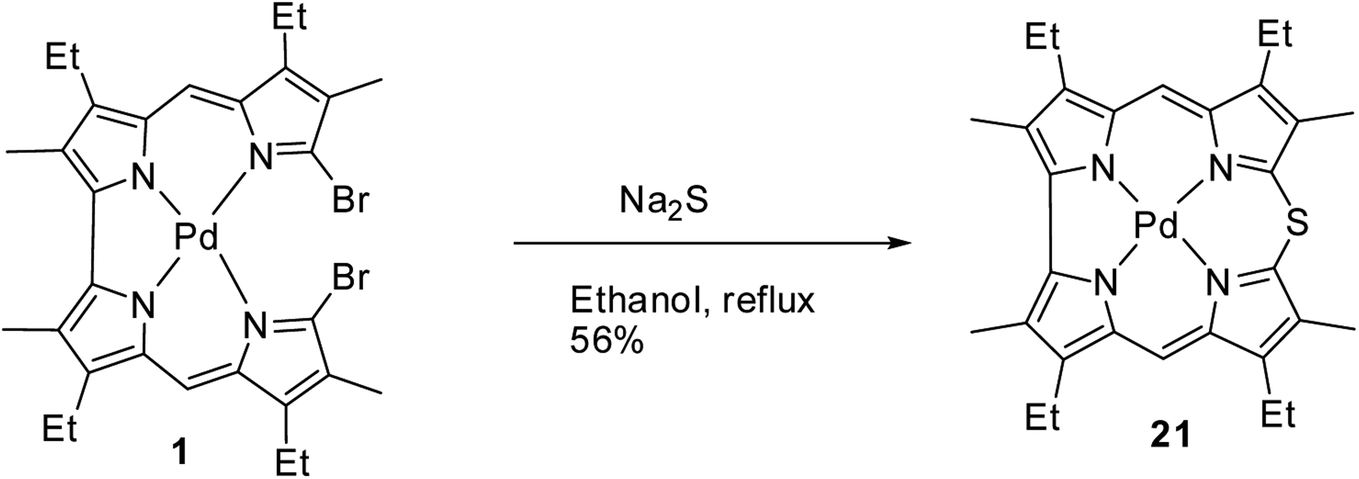
Johnson, Kay and Rodrigo in their 1963 paper,9 also reported the synthesis of palladium derivative of β-substituted meso-thiacorrole 21 by treating palladium chelate of 5,5′-bi(dipyrromethene) 1 with sodium sulphide in ethanol at refluxing temperature (Scheme 6).
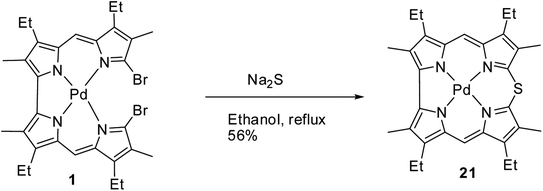 |
| | Scheme 6 Synthesis of β-substituted Pd(II) 10-thiacorrole 21. | |
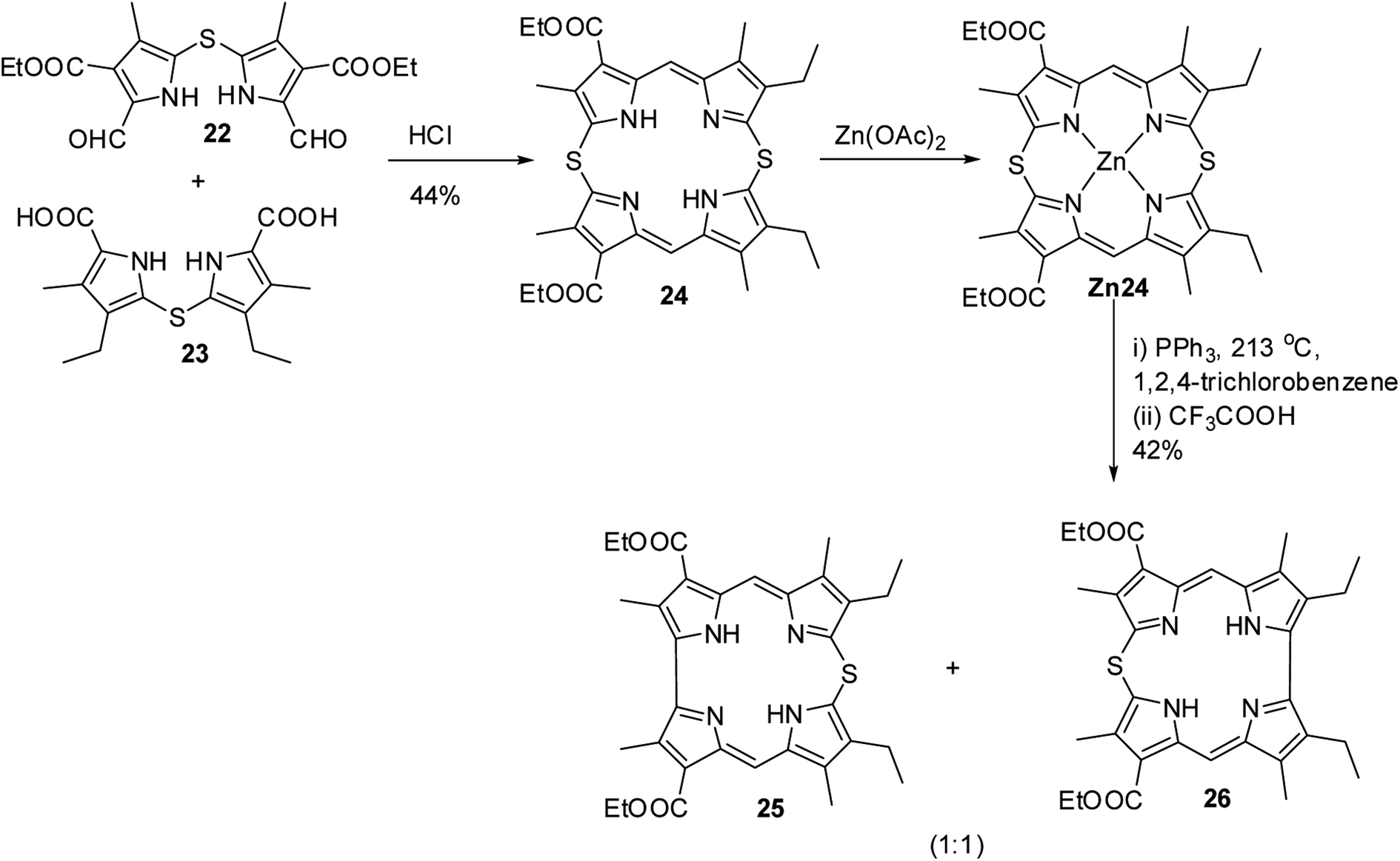
Further, in 1972, Grigg, Broadhurst and Johnson13 reported the synthesis of β-substituted meso-thiacorroles 25 & 26 by thermally driven sulfur extrusion reaction of Zn(II) complex of meso-dithiamacrocycle Zn24 in presence of triphenylphosphine (PPh3) followed by TFA assisted demetallation (Scheme 7). However, after these two initial reports on meso-thiacorroles, the further developments in this area were halted for almost four decades presumably owing to the difficulties involved in the synthesis of such macrocycles.
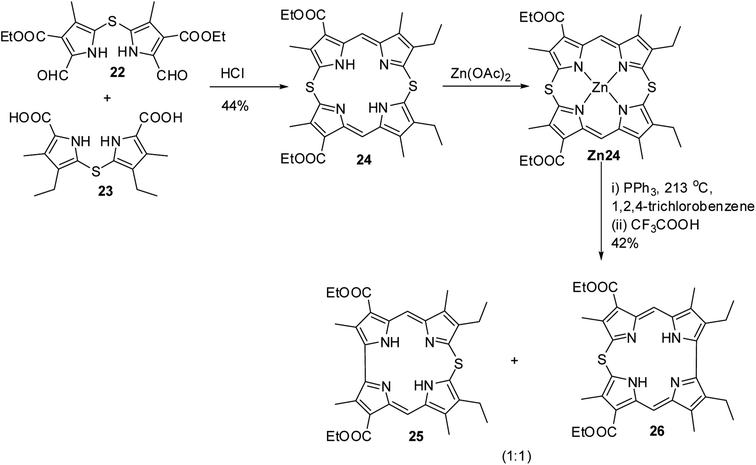 |
| | Scheme 7 Synthesis of β-substituted free base 10-thiacorroles 25 and 26. | |
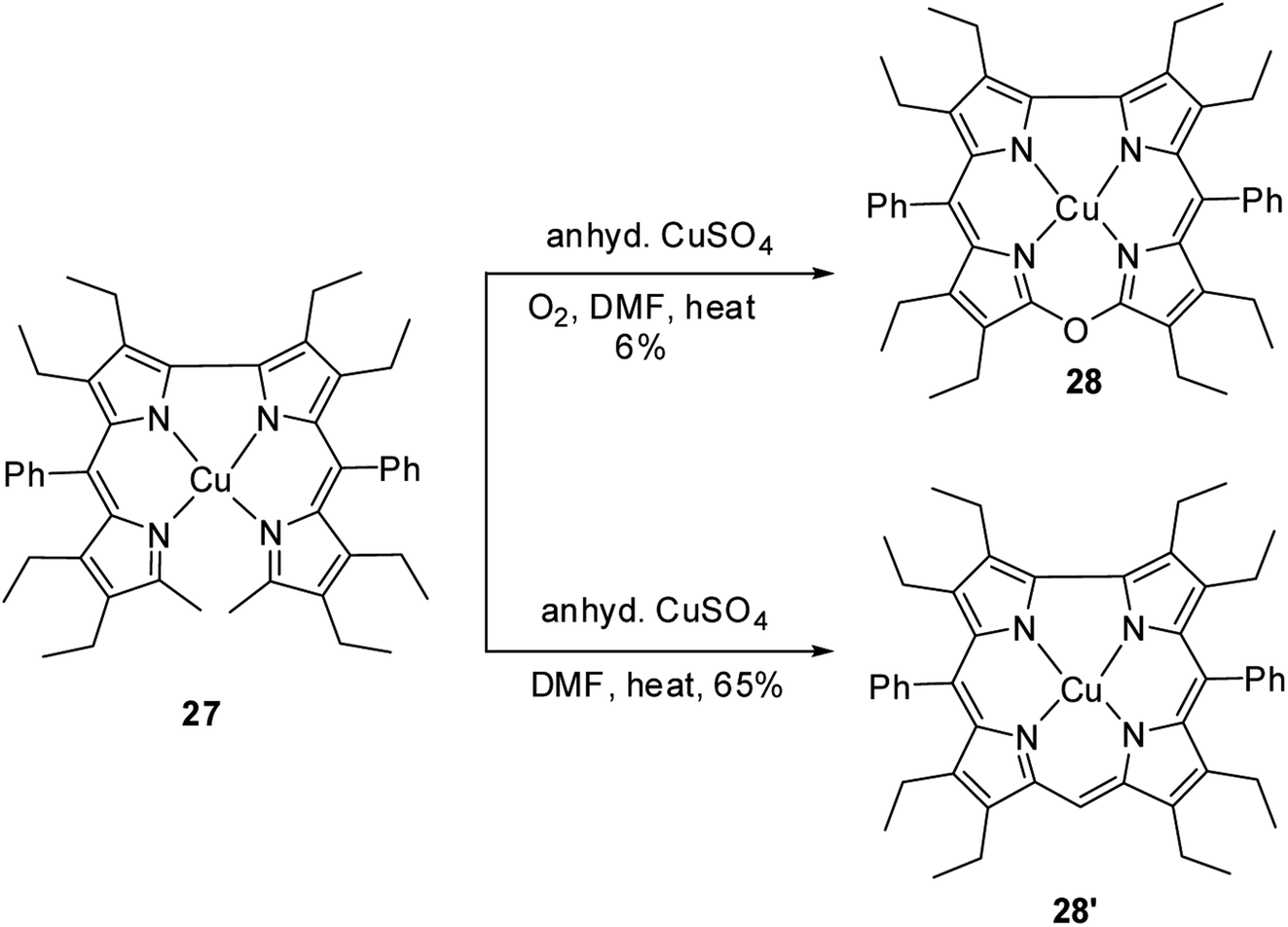
Bröring, Holthausen and co-workers14 reported the synthesis of copper derivative of 10-oxacorrole 28 by anhydrous copper(II) sulphate assisted oxidative macrocyclization of 2,2′-bidipyrrins 27 in the presence of oxygen (Scheme 8). The oxacorrole 28 was used as an appropriate reference compound to study the electronic ground state of copper in normal corrole 28′ as it has similarity in cavity size to 28′ and its resemblance to porphyrins in having double negative charge arising from its core. Absorption spectroscopy of both 28 and 28′ confirmed their typical porphyrinoid macrocyclic structure. The normal corrole 28′ was EPR silent whereas the oxacorrole 28 exhibited a EPR pattern typical of Cu(II) porphyrinoid. However, the comparison of X-ray structures of copper corrole 28′ with isostructural 28 followed by a detailed computational study on 28′ highlighted the possible electronic interaction between central Cu ion and macrocyclic π-orbital and hence led authors to conclude that copper's oxidation state in corrole 28′ can be better addressed as Cu(II) rather than Cu(III).
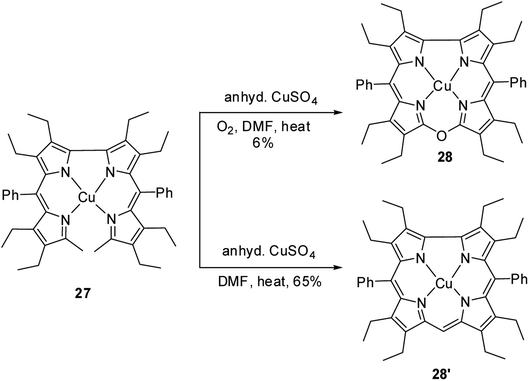 |
| | Scheme 8 Synthesis of copper derivatives of 10-oxacorrole 28 and normal corrole 28′. | |
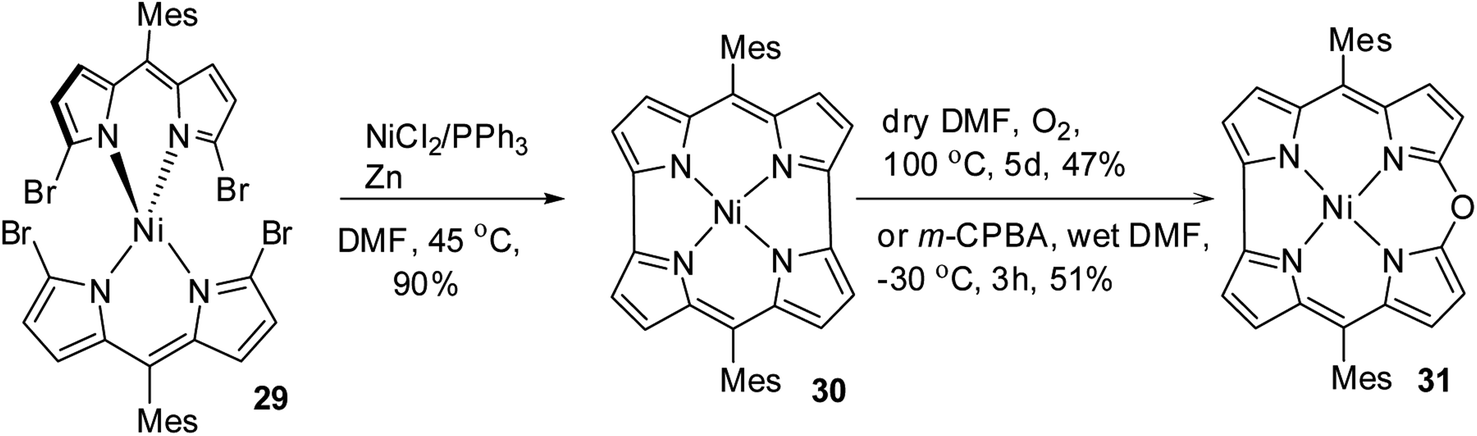
Shinokubo, Kobayashi and co-workers15 reported a remarkable gram-scale synthesis of strained macrocycle called Ni(II) norcorrole 30, which lacks two meso-carbon atoms compared to porphyrins.16 This Ni(II) norcorrole 30 showed a very good solid and solution state stability for an antiaromatic compound. However, authors observed that norcorrole was slowly getting oxidized in the presence of air at elevated temperatures to form an aromatic Ni(II) 10-oxacorrole 31 (Scheme 9), where the lone pair of electron on the meso-oxygen contributes to aromaticity.
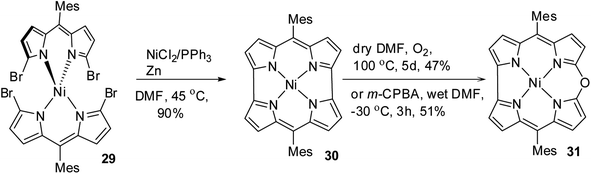 |
| | Scheme 9 Synthesis of meso-substituted Ni(II) 10-oxacorrole 31. | |
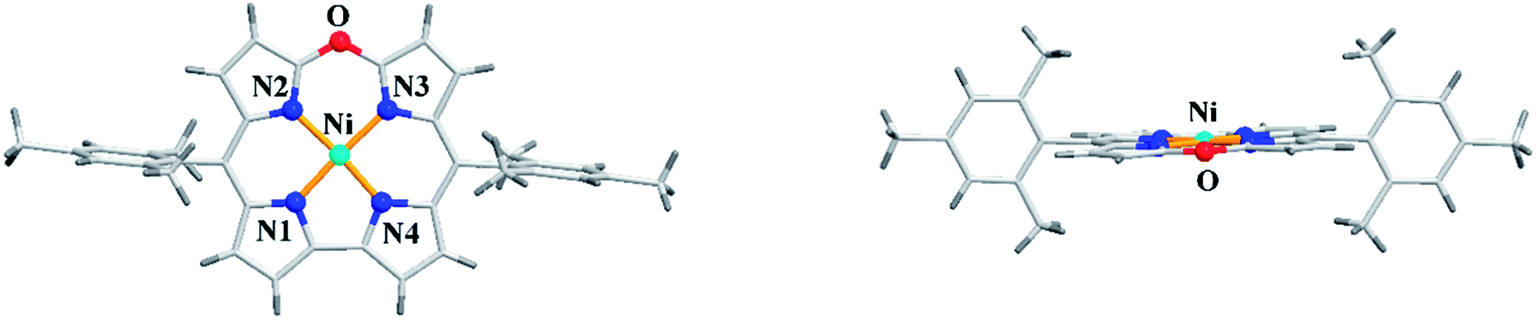
Alternatively, the treatment of norcorrole 30 with m-chloroperbenzoic acid (m-CPBA) led to formation of Ni(II) 10-oxacorrole 31 in 51% yield (Scheme 9). The single crystal X-ray analysis showed a planar conformation for oxacorrole 31 (Fig. 7) and the absorption and Magnetic Circular Dichroism (MCD) studies revealed features typical of aromatic porphyrinoid with lower molecular symmetry.
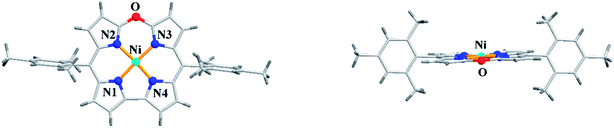 |
| | Fig. 7 Top and side view of X-ray crystal structure of 31 (CCDC 883625). | |
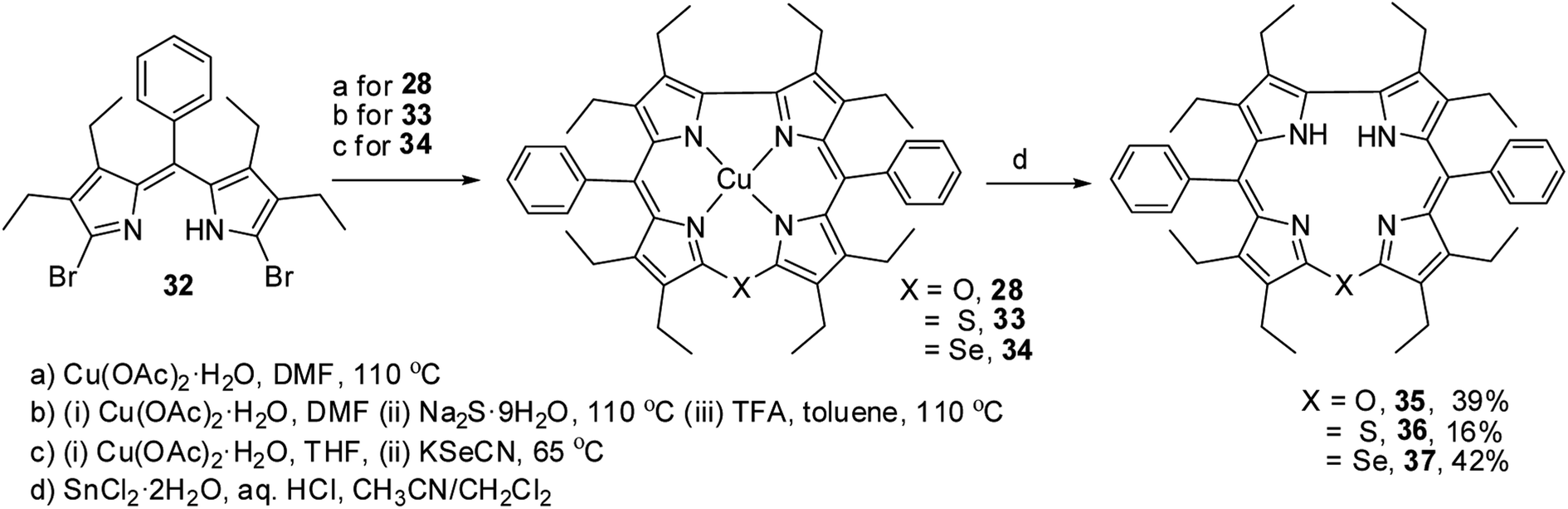
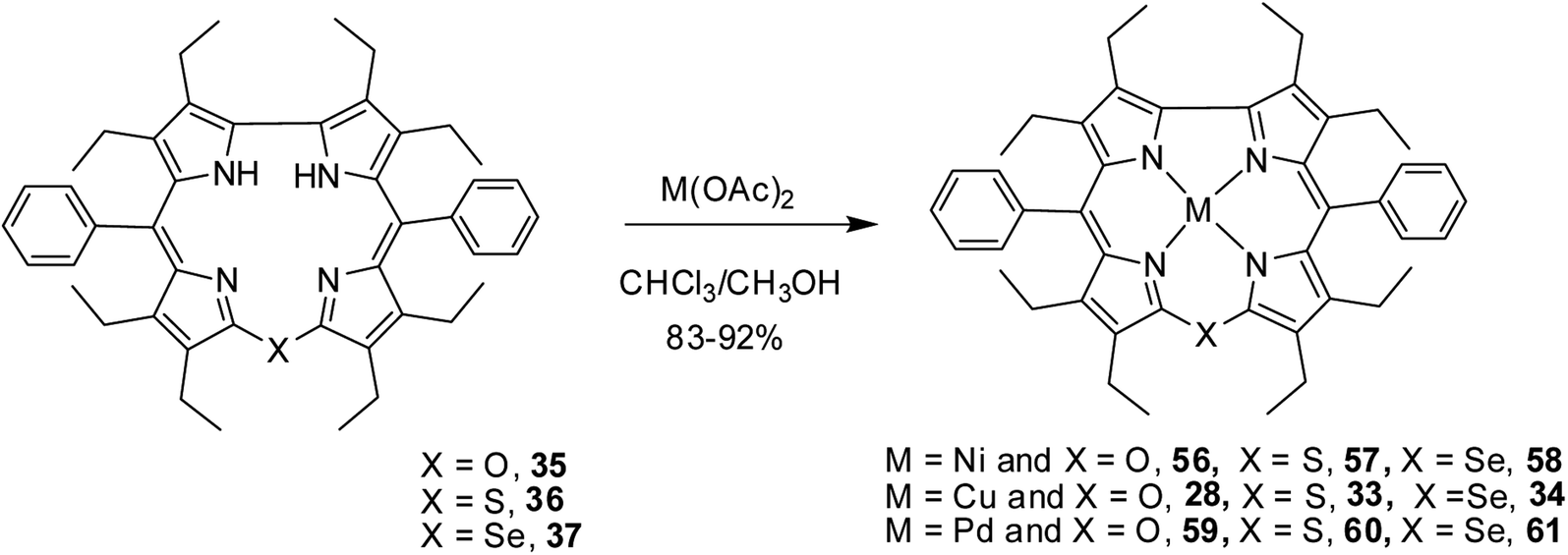
Bröring and co-workers17 synthesised a series of octaethyl derivatives of 10-heterocorroles 35–37 containing O, S, Se atoms at the 10-position by a metal-promoted two-step macrocyclization of dipyrrin 32 as depicted in Scheme 10.
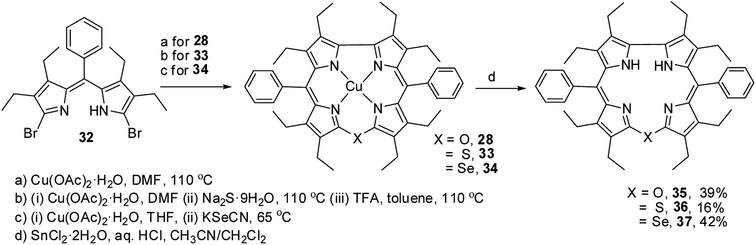 |
| | Scheme 10 Synthesis of octaethyl Cu(II) 10-heterocorroles 28, 33, 34 and their free base analogues 35–37. | |
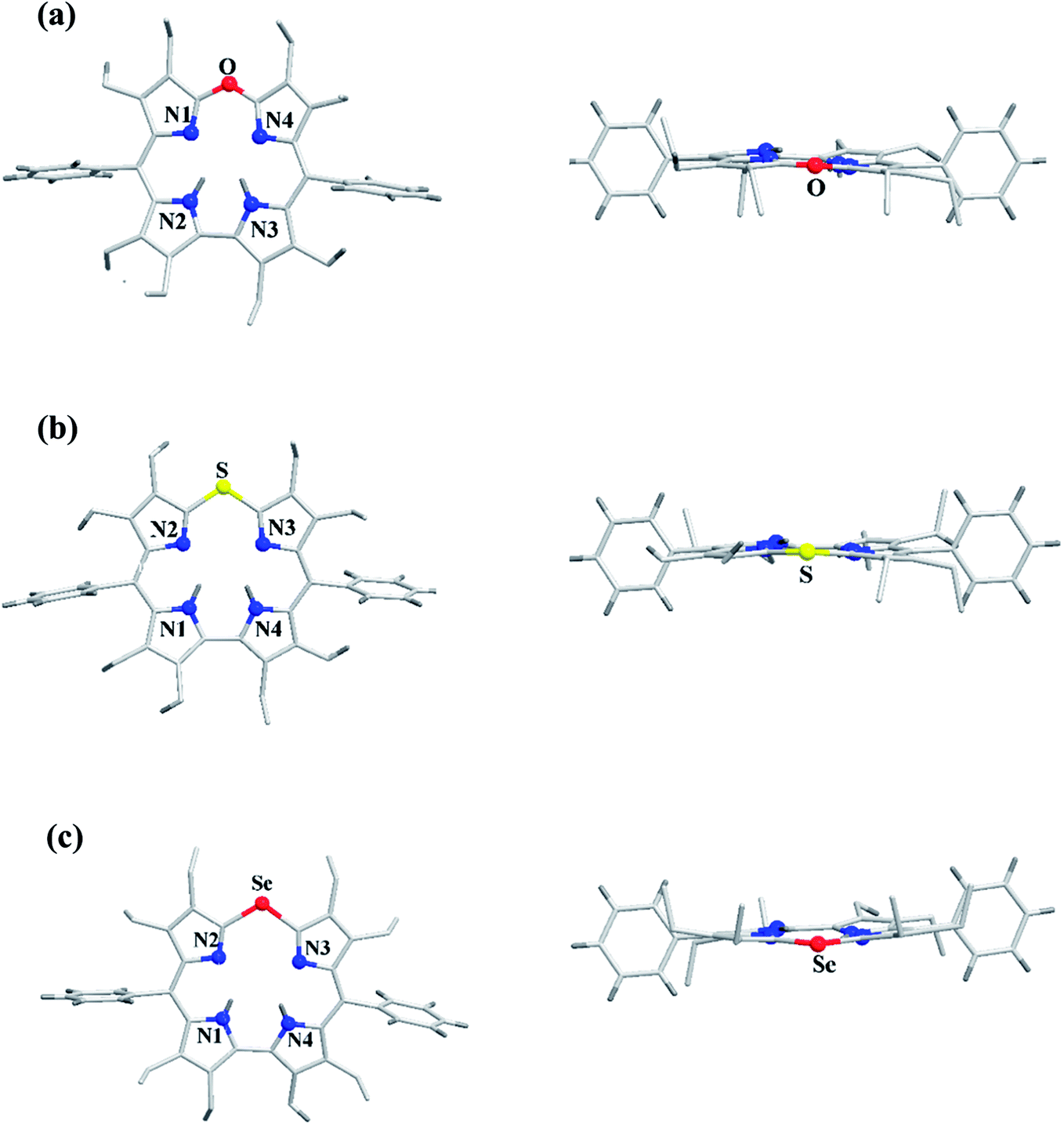
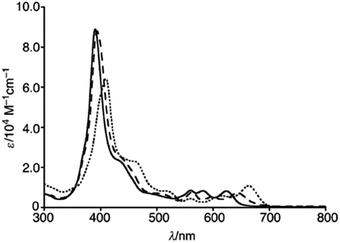
The chemical shift position of core NH resonance for free base corroles 35–37 and meso-H resonance for meso-unsubstituted analogues of 35–37 (not shown in the scheme) revealed that the aromaticity of these macrocycles follow the order: 10-thiacorrole 36 > 10-selenacorrole 37 > 10-oxacorrole 35. The X-ray structure of 10-heterocorroles 35–37 showed that they are almost planar with little saddle-shaped distortions and both the core-NH protons were located on the N-atoms of bipyrrolic unit (Fig. 8). The macrocyclic cavity size was found to increase from oxa 35 (7.05 Å2) to thia 36 (7.57 Å2) to selenacorroles 37 (7.77 Å2). The thia 36 and selena 37 corroles displayed absorption spectral bands typical of porphyrinoids whereas the oxacorrole 35 showed broadened and hypsochromically shifted absorption bands expected for a macrocycle with diminished aromatic nature (Fig. 9).
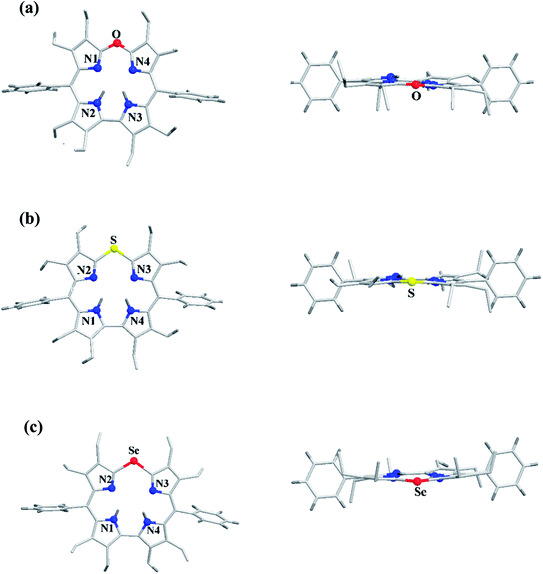 |
| | Fig. 8 Top and side view of X-ray crystal structures of (a) 35 (CCDC 921878) (b) 36 (CCDC 921879) and (c) 37 (CCDC 921880). | |
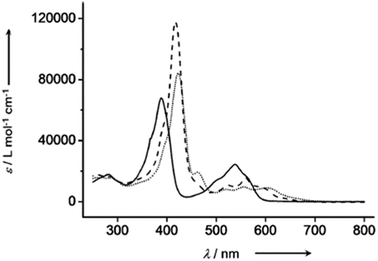 |
| | Fig. 9 UV-vis spectra of 35 (normal line), 36 (dashed line) and 37 (dotted line) recorded in dichloromethane. Reproduced from ref. 17 with permission. Copyright© 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
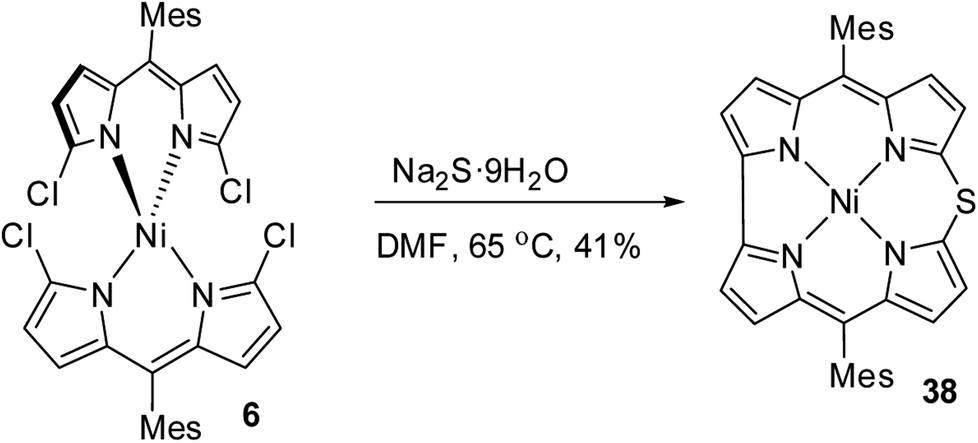
Shinokubo and co-workers18 achieved the synthesis of meso-substituted Ni(II) 10-thiacorrole 38 by sulfidation of α,α′-dichlorodipyrrin Ni(II) complex 6 as shown in Scheme 11.
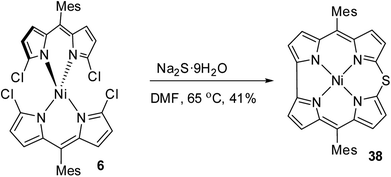 |
| | Scheme 11 Synthesis of meso-substituted Ni(II) 10-thiacorrole 38. | |
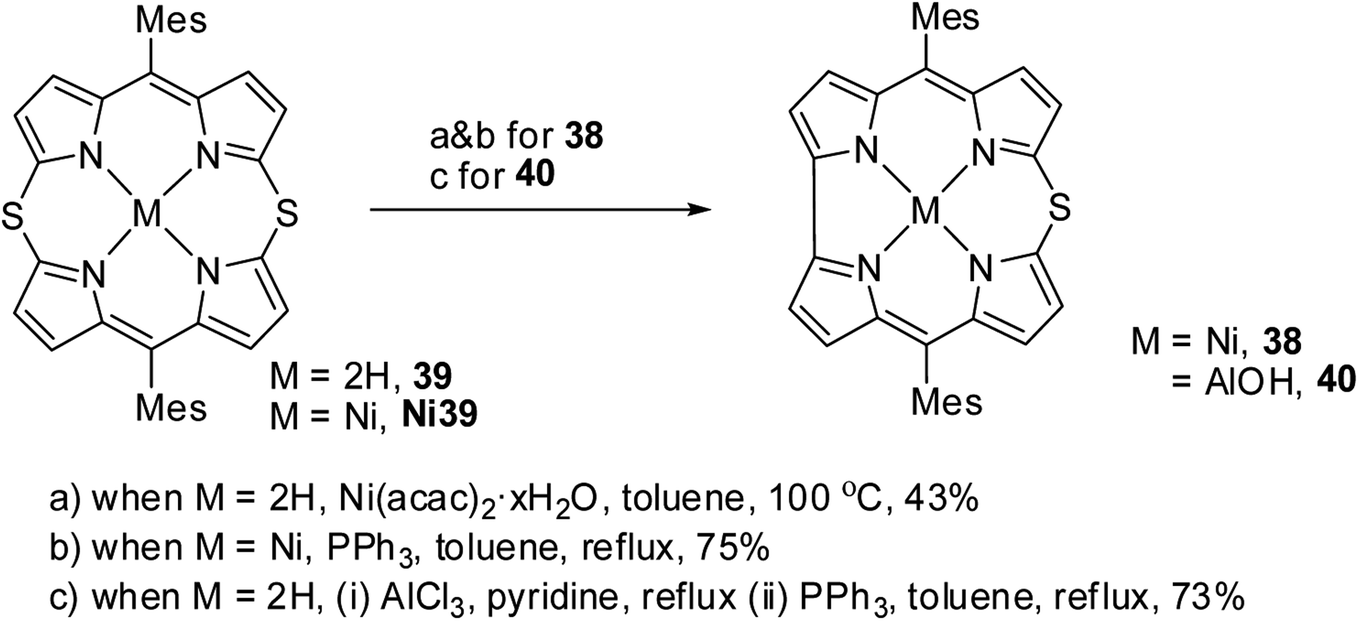
Alternatively, the insertion reaction on 5,15-dithiaporphyrin 39 in refluxing toluene and sulfur extrusion reaction of nickel dithiaporphyrin Ni39 upon treatment with triphenylphosphine resulted in the formation of Ni thiacorrole 38. Also, Al(III) thiacorrole 40 was synthesized by refluxing dithiaporphyrin 39 with AlCl3 in pyridine (Scheme 12). The same research group also observed similar sulfur extrusion reaction when Cu(II) 5,15-dithiaporphyrin was treated with PPh3 to yield Cu(II) 10-thiacorrole (not shown in the scheme).19 The 1H NMR spectral studies and absorption spectral patterns revealed that Al corrole 40 possessed strong aromaticity compared to Ni corrole 38.
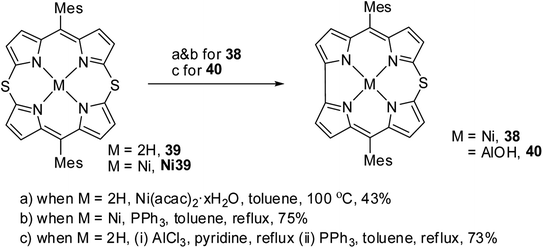 |
| | Scheme 12 Synthesis of Ni(II) 10-thiacorrole 38 and Al(III) thiacorrole 40 from 5,15-dithiaporphyrin. | |
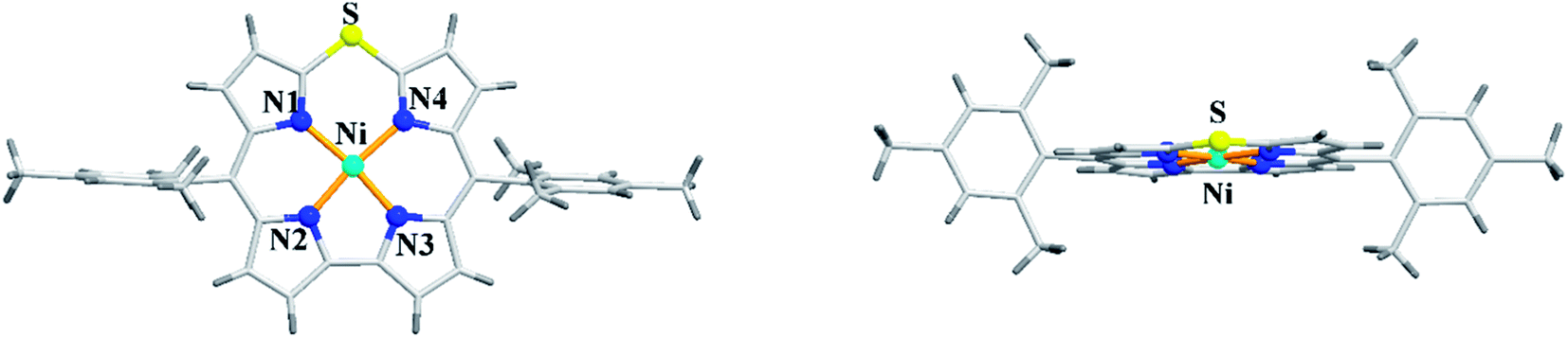
The X-ray structure of Ni(II) 10-thiacorrole 38 showed a planar conformation and the bond length of C–S (1.698(8) Å and 1.680(8) Å) was decreased compared to typical C–S bond length (1.82 Å) (Fig. 10) highlighting the partial double bond character of C–S bond and hence an indication of effective delocalization of sulfur's lone pair into the macrocyclic π system.
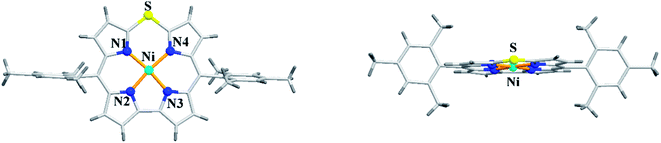 |
| | Fig. 10 Top and side view of X-ray crystal structure of 38 (CCDC 893774). | |
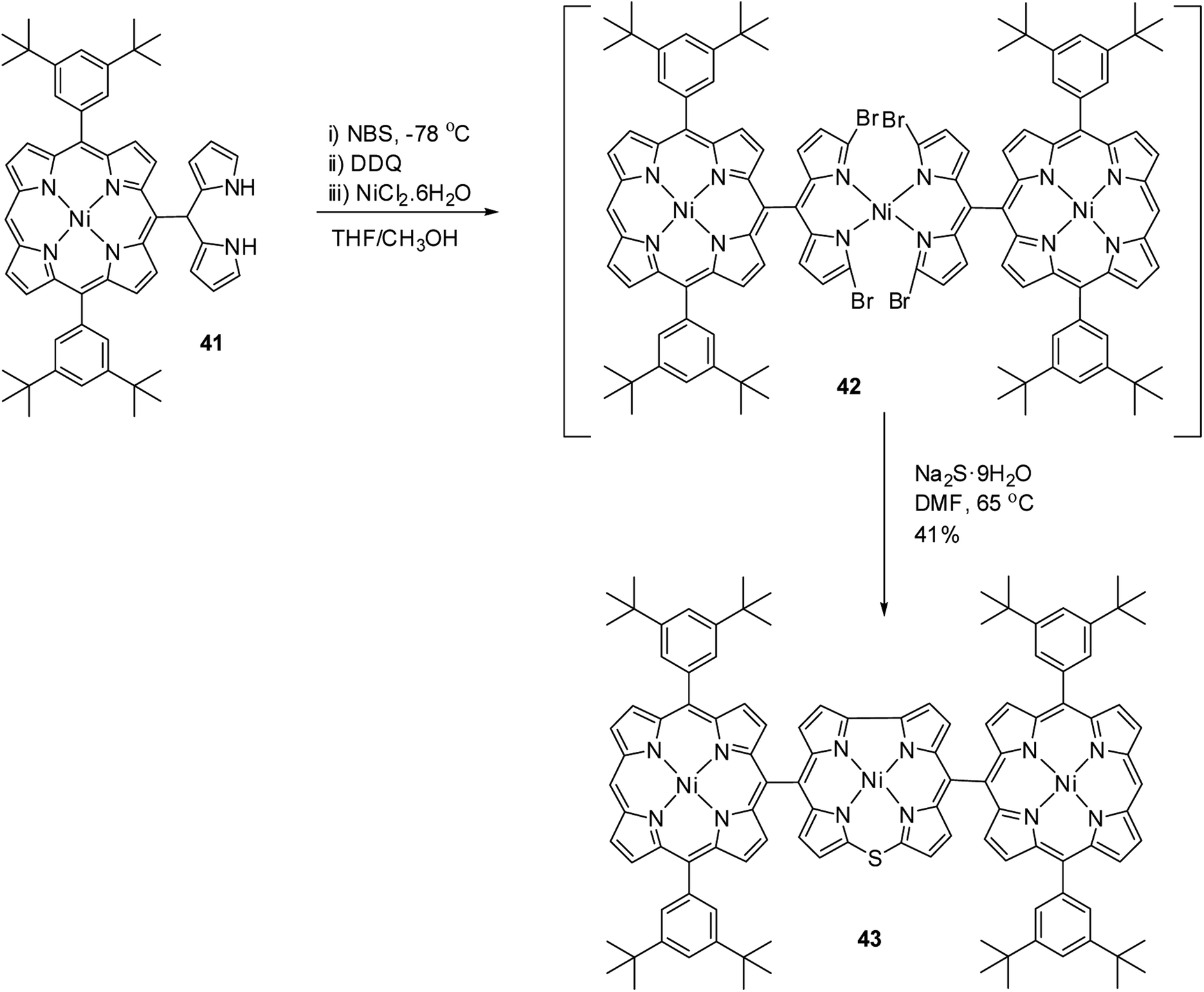
Kong and co-workers20 reported the synthesis of a meso–meso linked porphyrin–corrole hybrid (triad) in which 10-thiacorrole was flanked by two porphyrin units (Scheme 13). Porphyrinyl dipyrromethane 41 upon consecutive bromination, 2,3-dichloro-5,6-dicyano-benzoquinone (DDQ) oxidation and treatment with Ni(II) salt resulted in an unstable intermediate 42, which on treatment with Na2S·9H2O yielded triad 43 in 41% yield. In triad 43, the β-CH resonances of porphyrin subunits were slightly downfield shifted compared to thiacorrole unit and 43 displayed a broad absorption spectral band.
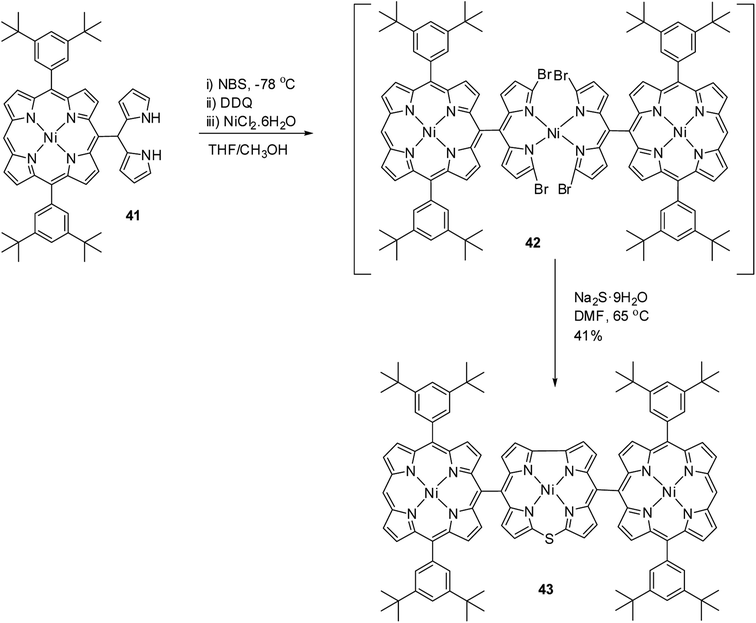 |
| | Scheme 13 Synthesis of meso–meso linked porphyrin-corrole hybrid 43. | |
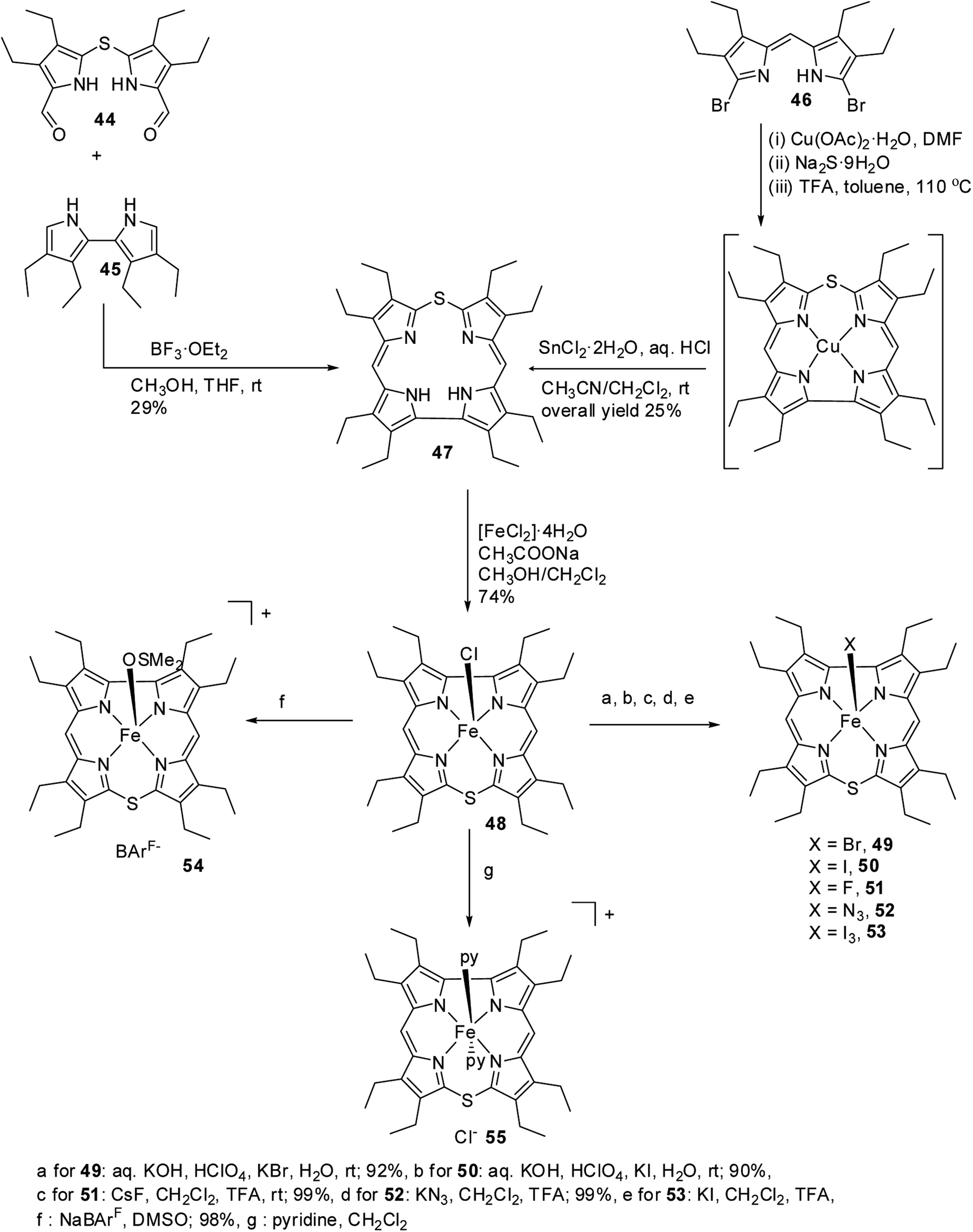
Bröring and co-workers21,22 carried out a systematic study on a series of iron 10-thiacorroles 48–55 containing halogenido, pseudo-halogenido and solvent-derived axial ligands to study the ground state spin state of Fe(III) ion in such complexes. The free base octaethyl-10-thiacorrole 47 was synthesized by either MacDonald-type [2 + 2] condensation of bis(3,4-diethyl-5-formylpyrrole)sulphide 44 and tetraethyl-2,2′-bipyrrole 45 or Cu(II) templated macrocyclization of dibromodipyrrin 46 (Scheme 14).
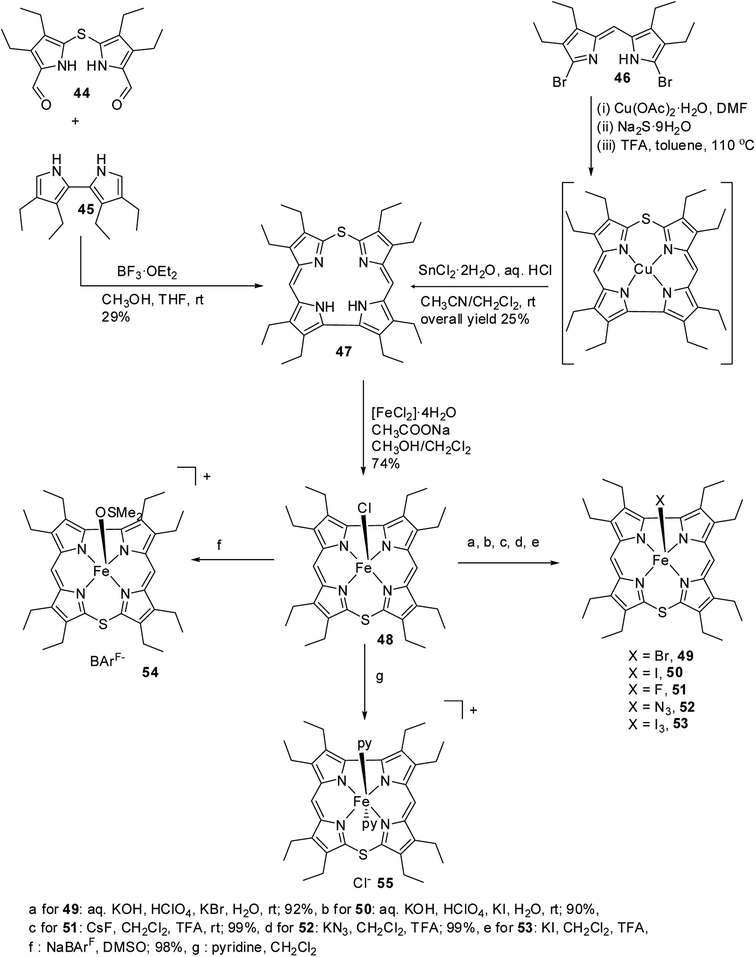 |
| | Scheme 14 Synthesis of octaethyl-10-thiacorrole 47 and its Fe(III) complexes 48–55 with various axial ligands. | |
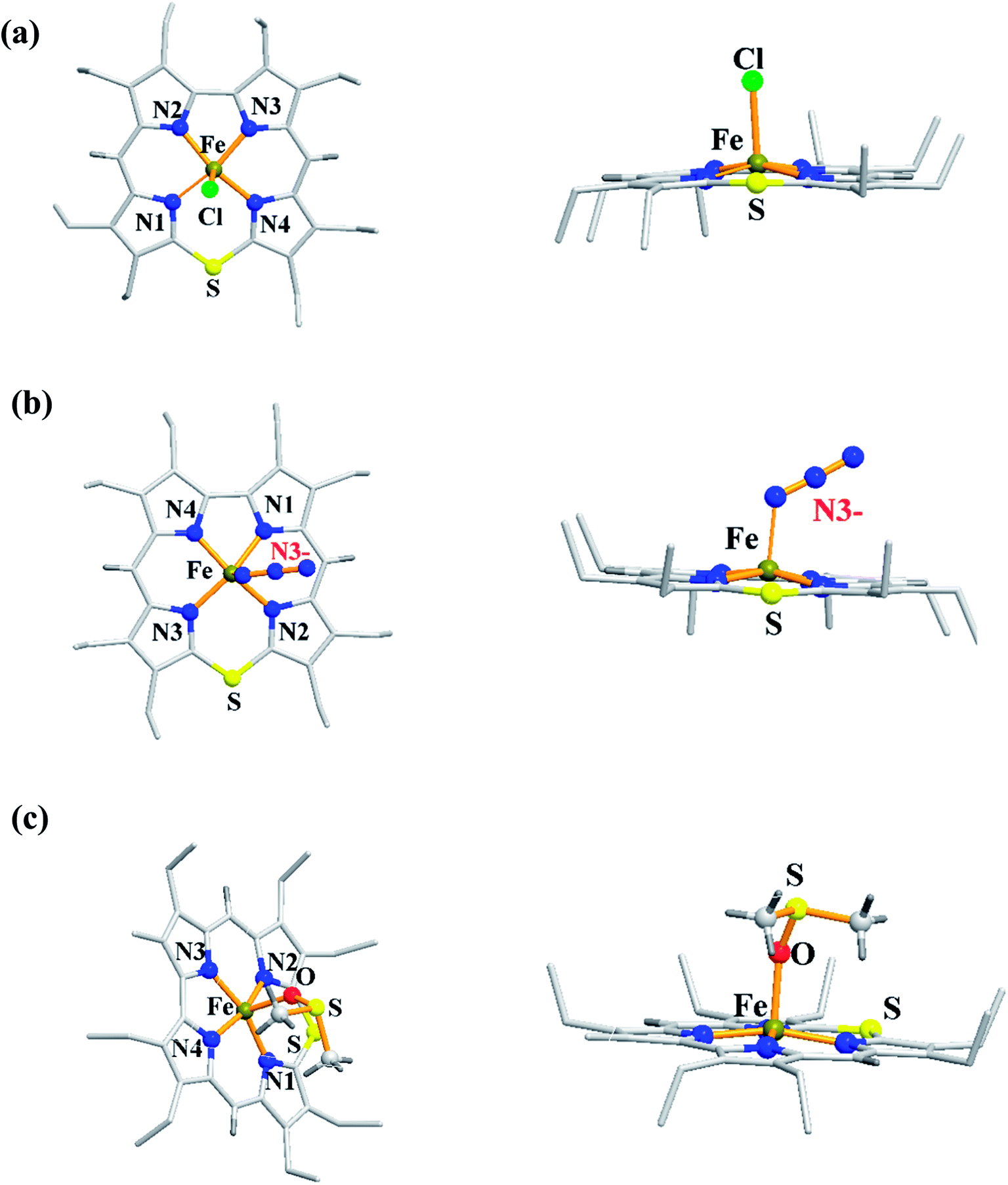
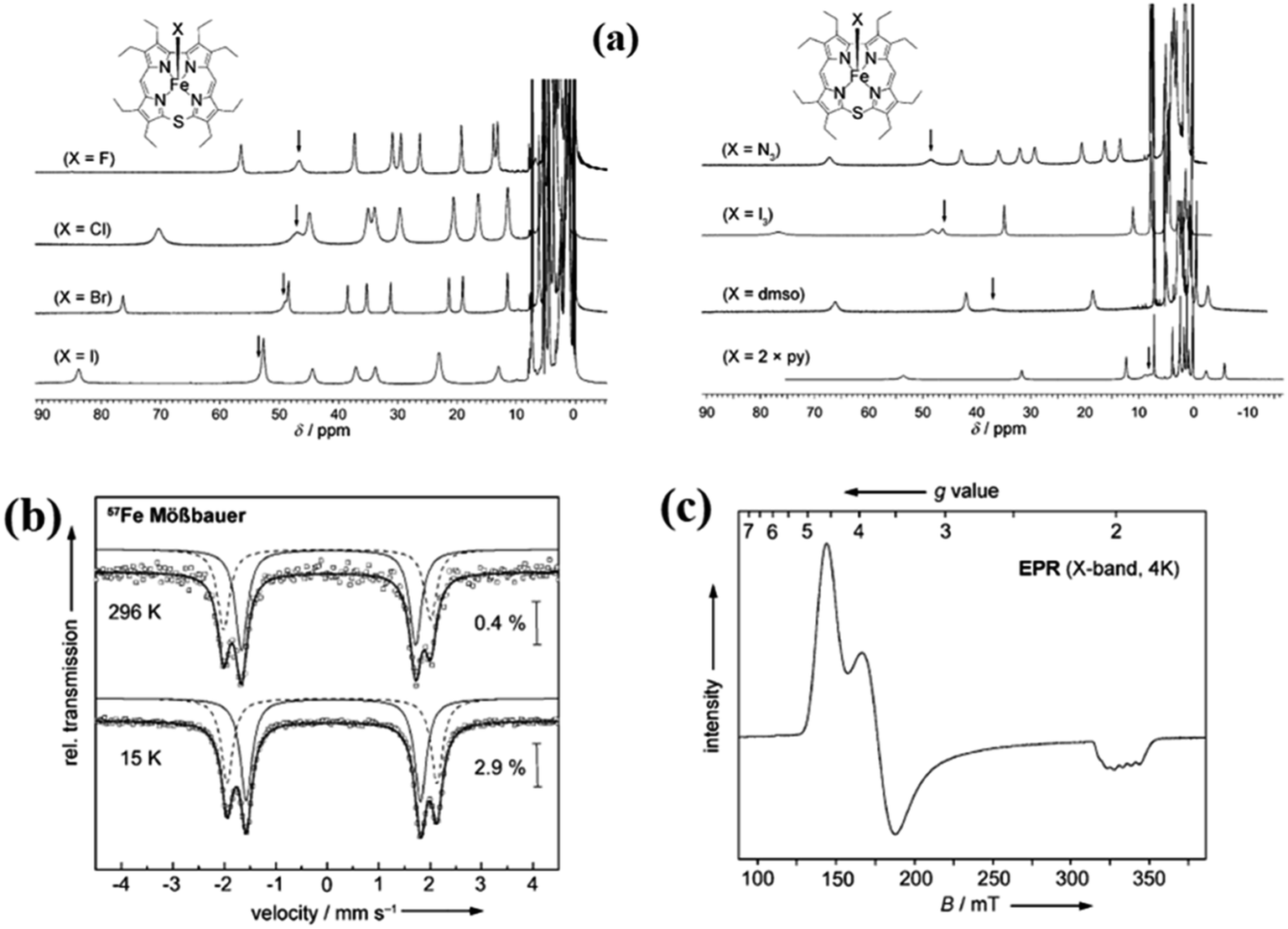
The synthesis of Fe(III) complexes of 47 with halogenido 48–51, pseudo-halogenido 52, 53 and solvent-derived axial ligands 54, 55 are also shown in the Scheme 14. The X-ray structures of compounds 48, 52, and 54 are shown in Fig. 11. The iron corroles containing halogen axial ligand 48–51 showed porphyrinoid like absorption features such as intense Soret band and several minor Q-bands. Most of these iron corroles (48–50, 52–54) displayed elongated Fe-axial ligand and short equatorial Fe–N bonds compared to iron porphyrins. The chemical shift values for meso-CH protons were in the down field region (δ = 35–50 ppm). They also displayed rhombic EPR signals; gz strongly split by superhyperfine coupling with axial donor's nuclear spin. The Mossbauer spectra revealed their large quadrupole splitting values (ΔEQ of >2.5 mm s−1) (Fig. 12). The redox potential values of these corroles exhibited cathodic shift in comparison to iron porphyrins. All these observations served as a corroborative evidence to support the fact that smaller cavity size of these iron meso-thiacorroles resulted in an intermediate spin (i.s., S = 3/2) state for ground state of Fe(III) ion. This intermediate spin state was found to be unperturbed and was temperature independent. This observation is in contrast to Fe(III) porphyrins which typically display high spin (S = 5/2) nature.
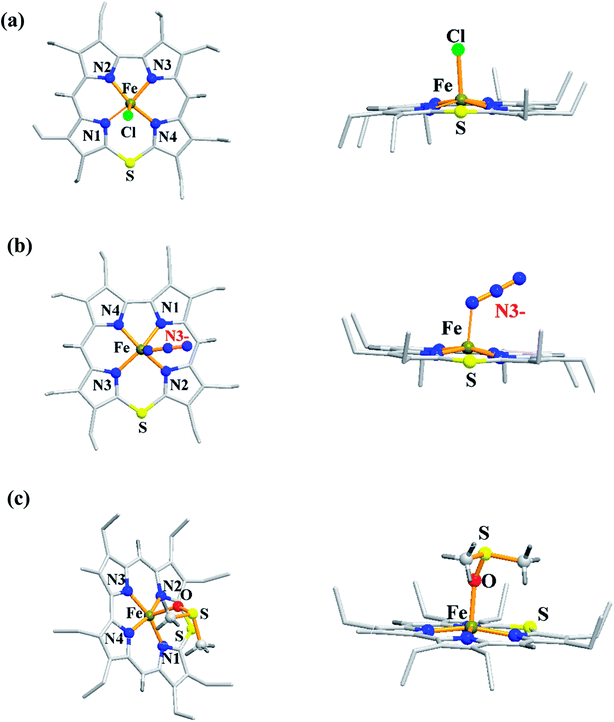 |
| | Fig. 11 Top and side view of X-ray crystal structures of (a) 48 (CCDC 962051) (b) 52 (CCDC 962050) and (c) 54 (CCDC 962046). | |
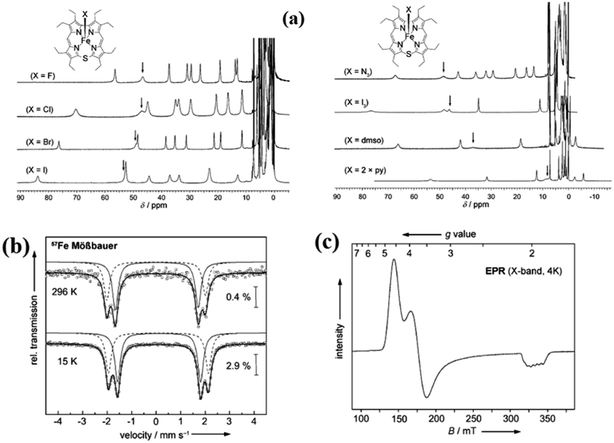 |
| | Fig. 12 (a) 1H NMR spectra of iron(III) thiacorroles 48–55 in CDCl3 (b) 57Fe Mössbauer spectra of 50 (powder) (c) EPR spectrum of 50 (solution, CHCl3/CH2Cl2 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 4 K). Reproduced from ref. 21 with permission. Copyright© 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. :1, 4 K). Reproduced from ref. 21 with permission. Copyright© 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
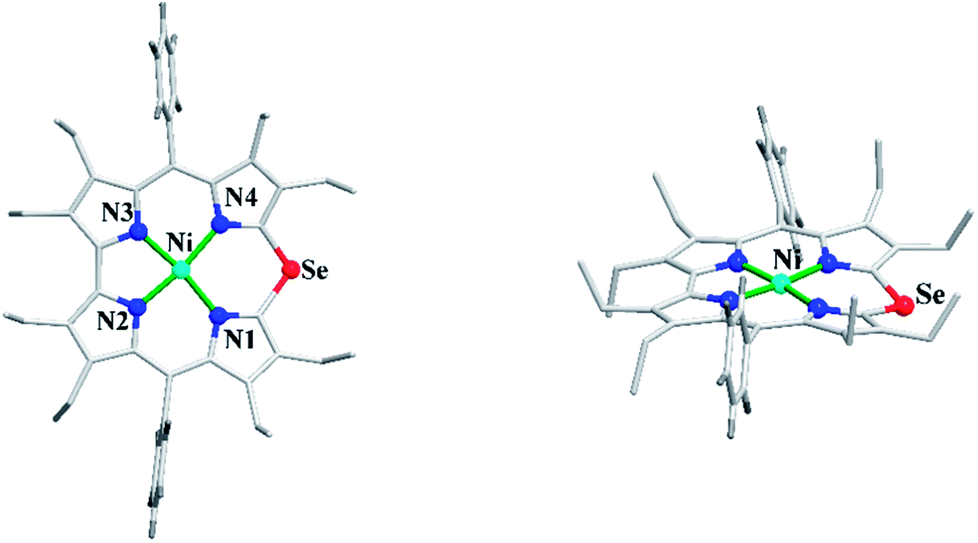
Bröring and co-workers23 further studied Ni(II), Cu(II) and Pd(II) complexes of 10-heterocorrole ligands containing O, S, Se at 10-position (35–37) to understand macrocycle's adaptability towards metal ions of different sizes while forming complexes. The synthesis of Ni, Cu, and Pd derivatives of these corroles is shown in Scheme 15. Their findings, mainly based on the X-ray structural data, revealed that ruffling dominates as out-of-plane distortion mode in complexes where a mismatch between the ligand cavity size and the radius of metal ion exists. The maximum ruffling was observed in the case of nickel 10-selenacorrole 58 (Fig. 13). This paper, for the first time, revealed that corroles can indeed exhibit ruffling conformations while forming complexes despite the presence of a direct pyrrole–pyrrole connection.
 |
| | Scheme 15 Synthesis of Cu(II) (28, 33, 34) Ni(II) (56–58), and Pd(II) (59–61) complexes of 10-heterocorrole 35–37. | |
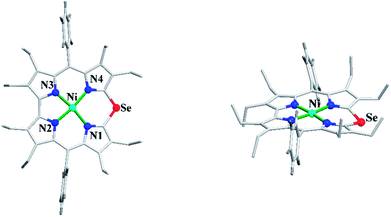 |
| | Fig. 13 Top and side view of X-ray crystal structure of 58 (CCDC 1544170). | |
2.3 10-Silacorroles
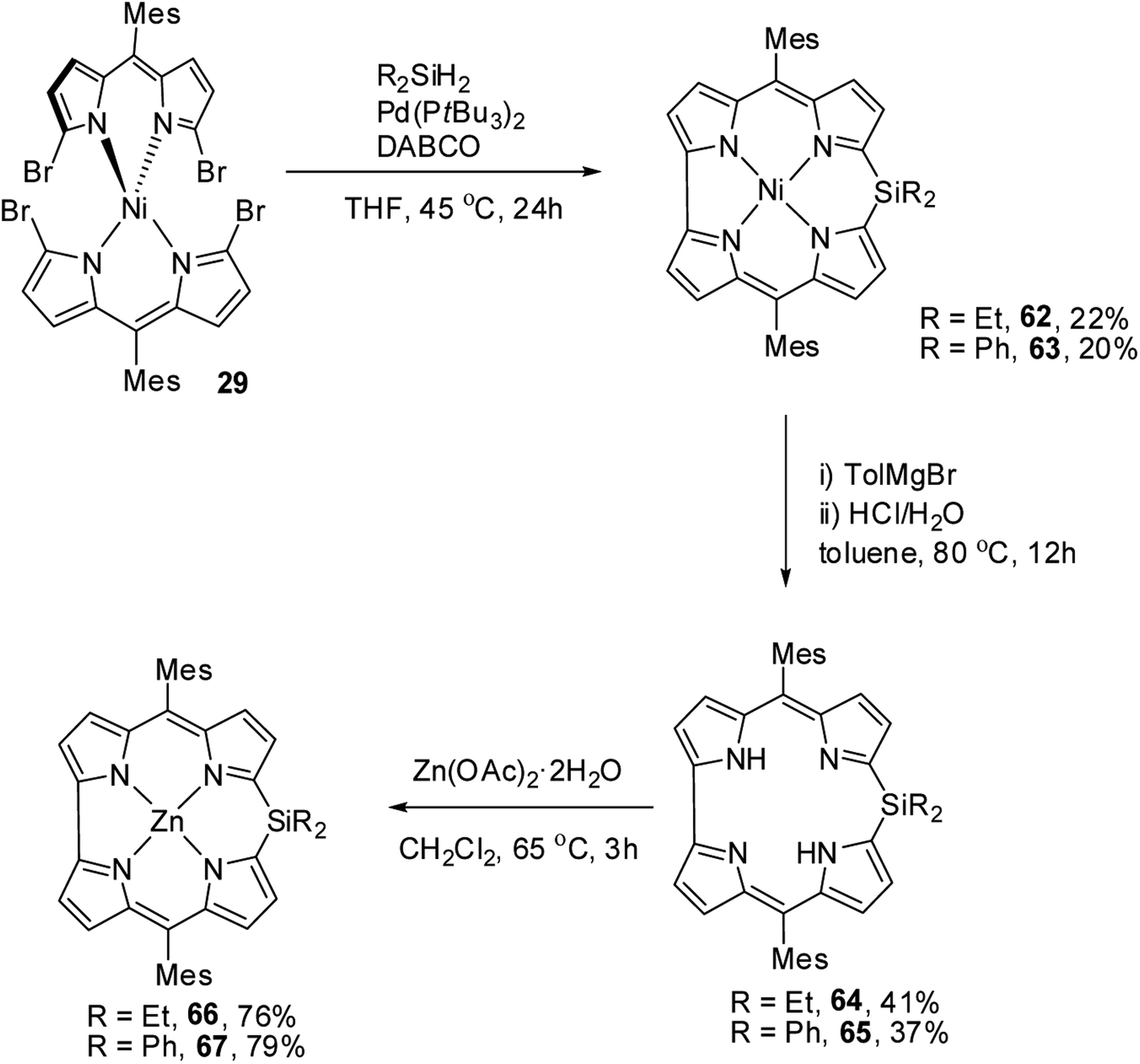
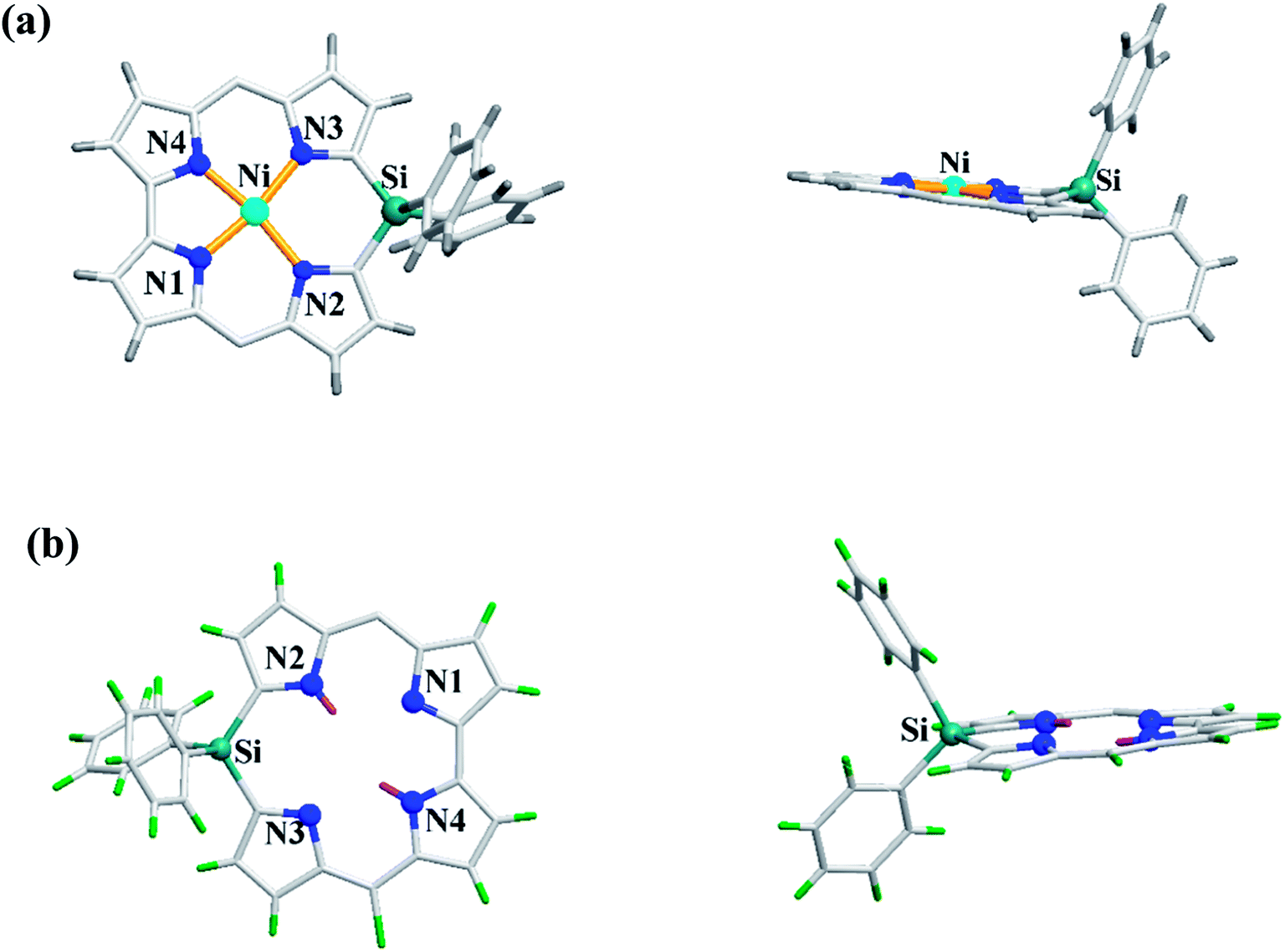
Recently, Shinokubo, Hiroto and co-workers24 reported the synthesis of Ni(II) (62, 63) free base (64, 65), and Zn(II) (66, 67) complexes of 10-silacorroles as shown in the Scheme 16. The Pd catalyzed silylation reaction of bis(α,α′-dibromodipyrrin) Ni(II) complex 29 resulted Ni(II) silacorrole 62 and 63, which upon demetallation and subsequent zinc insertion reaction yielded free base (64, 65) and Zn(II) silacorrole (66, 67) respectively. Although the Ni(II) and free base silacorroles adopt highly planar conformation as per their X-ray structures (Fig. 14), the 1H NMR spectral features such as β-pyrrolic proton resonances (δ = 5.99 to 6.31 for Ni silacorrole 62) and core NH resonance (δ = 15.1 ppm for free base silacorrole 64) were indicative of their non-aromatic nature. The absorption spectral band of Ni corrole was far red-shifted than zinc and free base corroles and reaches into NIR region (Fig. 15). The zinc silacorrole 66 also exhibited emission in NIR region (Fig. 15) with fluorescence quantum yield = 0.014 and emission lifetime = 1.78 ns. The electrochemical and theoretical studies carried out on these silacorroles showed the presence of σ*–π* conjugation between the silylene group and the tetrapyrrole system. Also, their electronic structures were susceptible to the central metal ions and the substituents present on silicon at meso-position.
 |
| | Scheme 16 Synthesis of Ni(II) (62, 63) free base (64, 65), and Zn(II) (66, 67) complexes of meso-substituted 10-silacorroles. | |
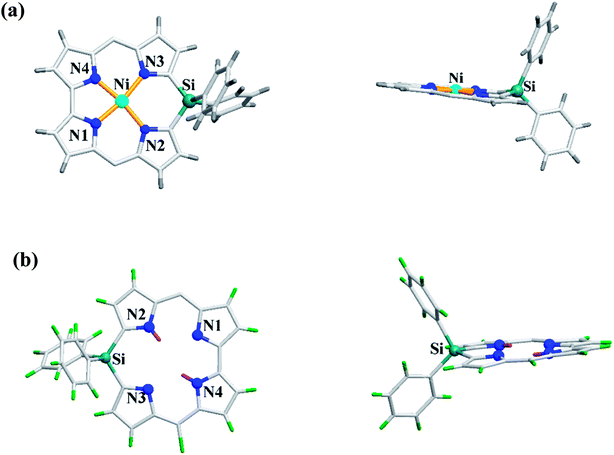 |
| | Fig. 14 Top and side view of X-ray crystal structures of (a) 63 (CCDC 1520052) (b) 65 (CCDC 1520055) (meso-mesityl groups are omitted for clarity). | |
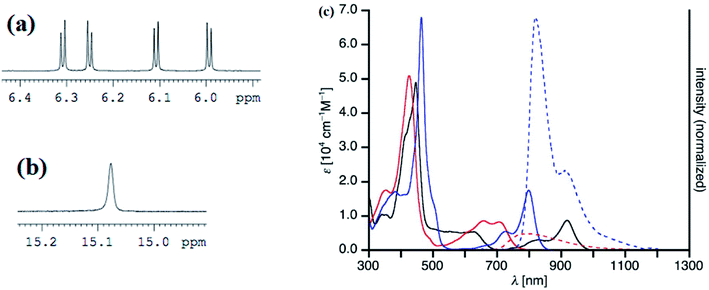 |
| | Fig. 15 Selected regions of 1H NMR spectra of (a) 62 (b) 64 and (c) UV-vis/NIR absorption spectra (normal line) of 62 (black), 64 (red), 66 (blue) and emission spectra (dashed line) of 64 (red, λmax = 426 nm), 66 (blue, λmax = 463 nm) recorded in dichloromethane. Reproduced from ref. 24 with permission. Copyright© 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
3. Corroles containing heteroatom(s) at their core (core modified corroles)
3.1. 22-Thiacorroles
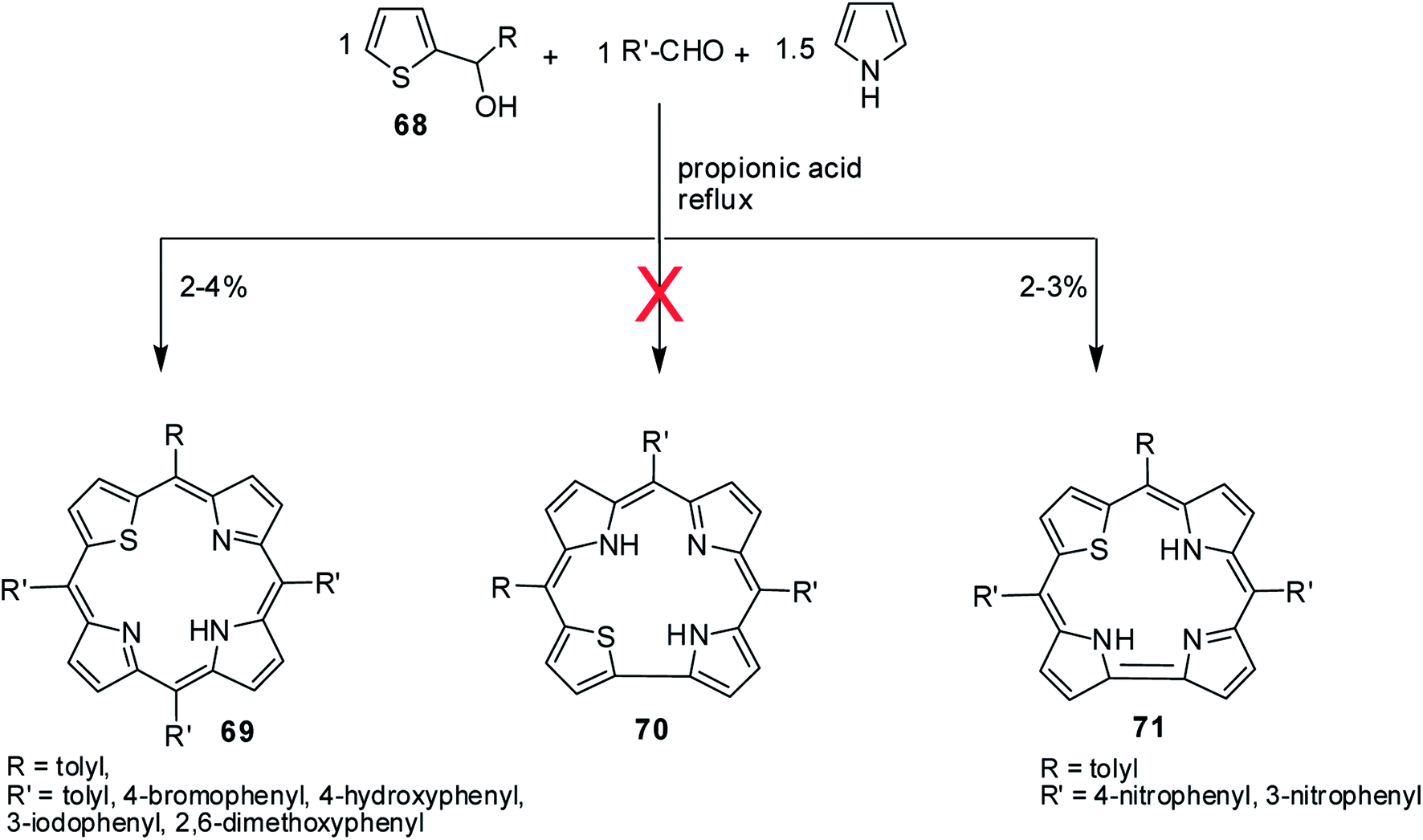
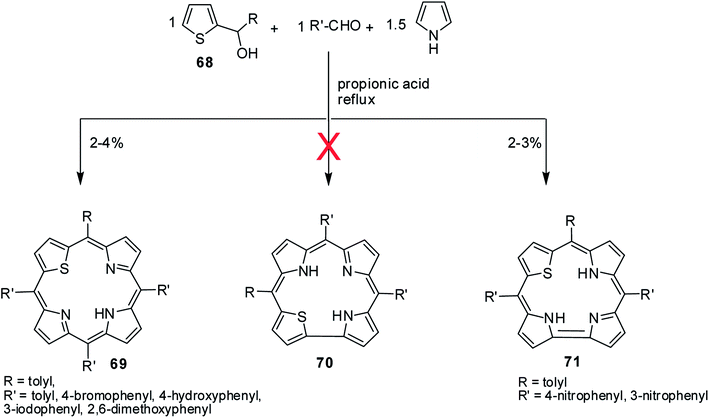
A perusal of literature reveals that there is only one report available on 22-thiacorroles that has direct pyrrole–pyrrole bond.25 The other possible isomer, 21-thiacorrole having direct pyrrole-thiophene link and dithiacorroles containing two core sulfur atoms in place of two pyrrole nitrogens are yet to be synthesized. Ravikanth and co-workers' attempts to make thiacorroles by condensing thiophene mono-carbinol 68 (1 eq.), aromatic aldehyde (1 eq.) and pyrrole (1.5 eq.) in refluxing propionic acid predominantly resulted in the formation of 21-thiaporphyrin 69 (ref. 26) (Scheme 17). Although authors observed the formation of thiacorrole in few cases, their isolation was not possible due to the decomposition of corrole during column chromatography. However, authors were successful in isolating stable thiacorroles 71 in ∼3% yield when 4-/3-nitrobenzaldehyde was used in the above condensation in otherwise similar conditions. Interestingly, no formation of 21-thiaporphyrin was noticed in this reaction. Authors found that changing the reaction conditions to mild Lindsey's protocol27 did not result in formation of thiacorroles. The thiacorrole formed in this condensation was expected to have direct α-α thiophene-pyrrole link 70 (21-thiacorroles) but the structural elucidation by detailed NMR studies revealed that the corrole was having direct α-α pyrrole–pyrrole link 71 (22-thiacorroles). Unfortunately, no crystal structure was available for 22-thiacorrole, but the combined NMR and Density Functional Theory (DFT) studies [B3LYP/6-31G(d) level] revealed their diminished aromatic character and non-planar conformation of 22-thiacorrole macrocycle.
 |
| | Scheme 17 Synthesis of meso-substituted 22-thiacorrole 71. | |
The 22-thiacorroles 71 exhibited one strong Soret-like band at ∼435 nm and one broad Q-band like transition at ∼620 nm due to their less symmetric nature and were also weakly fluorescent with one broad ill-defined fluorescence band at ∼660 nm. The electrochemical studies revealed that 22-thiacorroles 71 were easier to reduce compared to 21-thiaporphyrins supporting their electron deficient nature.
3.2. 21-Oxacorroles
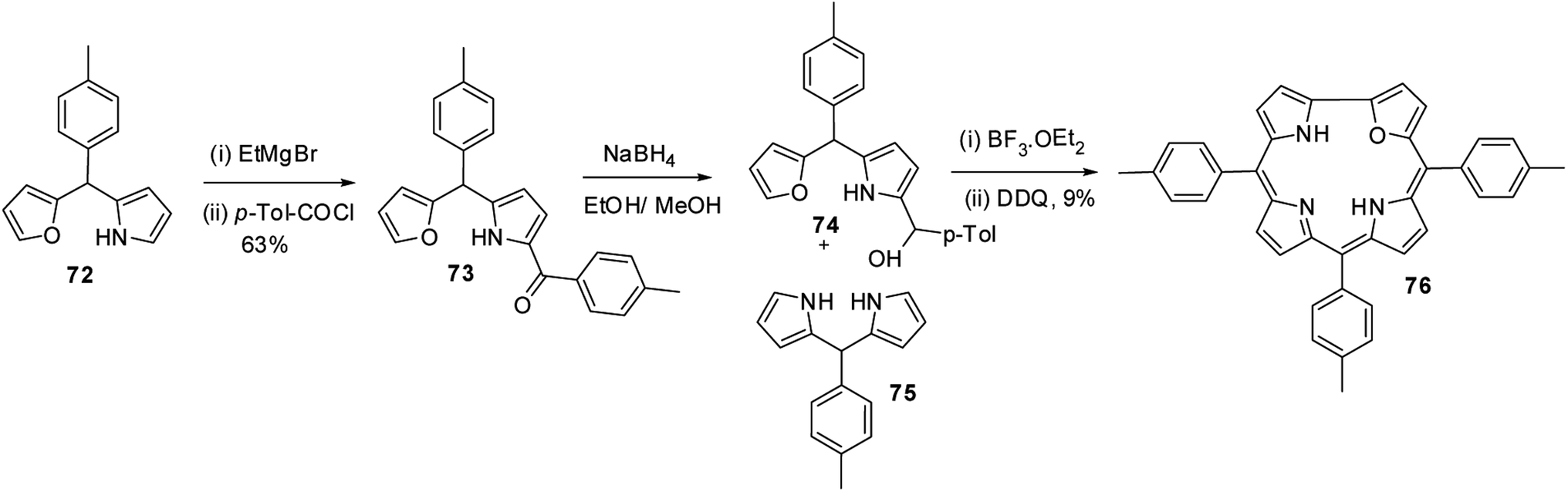
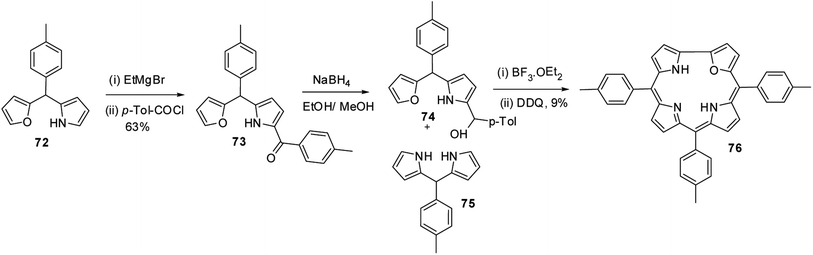
Lee and co-workers28,29 developed a rational method to synthesize the first examples of 21-oxacorroles containing direct furan-pyrrole bond 76 as shown in Scheme 18.
 |
| | Scheme 18 Synthesis of meso-substituted 21-oxacorrole 76. | |
The p-tolyl-(furan-2-yl)-(pyrrol-2-yl)methane 72 was treated with EtMgBr in THF followed by p-toluoyl chloride resulted in an exclusive formation of pyrrole acylated product 73 in 63% yield. The pyrrole acylated compound 73 was then reduced to the corresponding alcohol 74 with excess of NaBH4. The 21-oxacorrole 76 was synthesized in 9% yield by acid-catalyzed [2 + 2] condensation of furyl-pyrrolyl mono-ol 74 with meso-aryl dipyrromethane 75 followed by the column chromatographic purification.
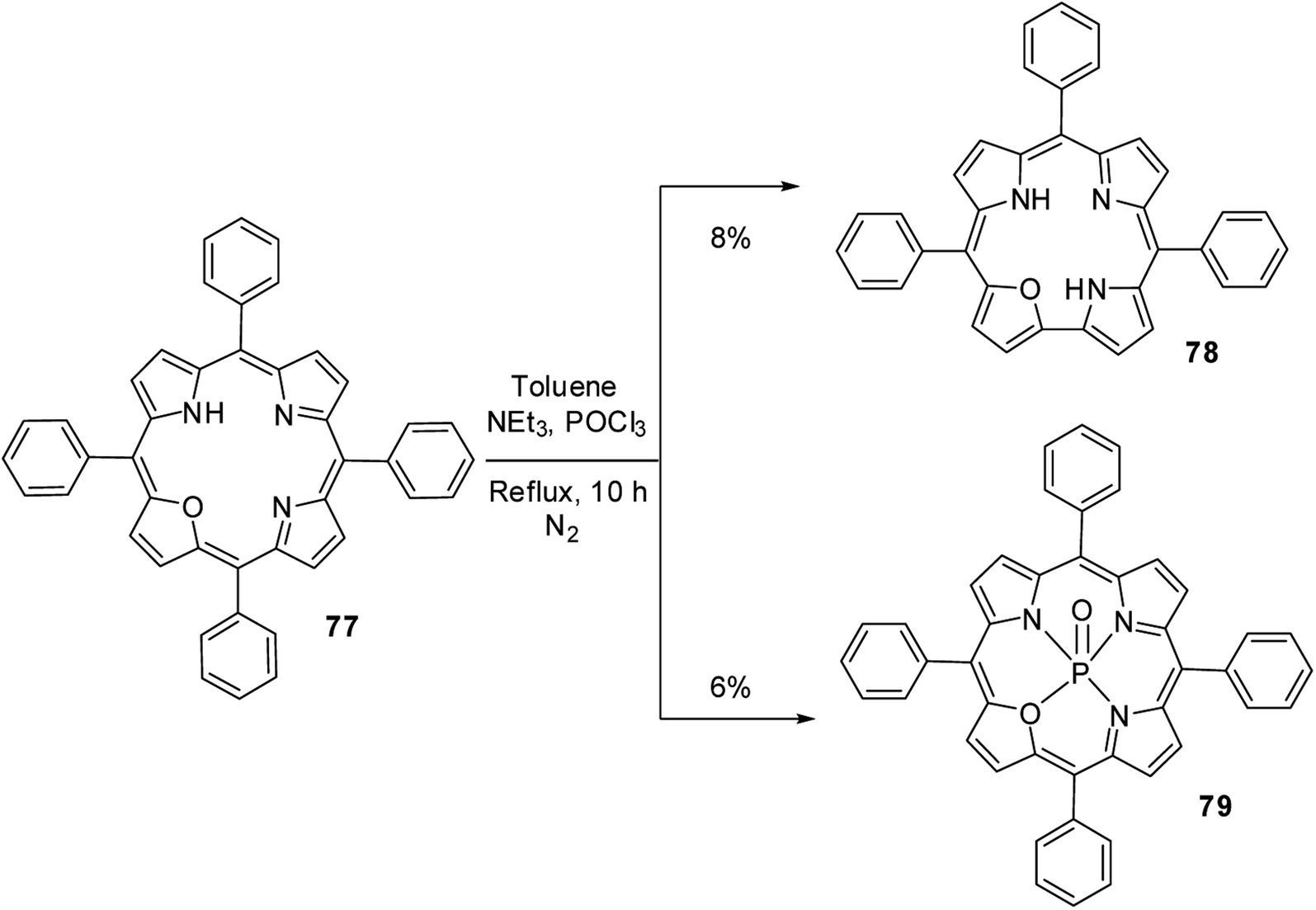
Ravikanth and co-workers30 serendipitously noticed the formation of 21-oxacorrole 78 while preparing the phosphoryl complex of 21-oxaporphyrin 77. The meso-tetraaryl 21-oxaporphyrin 77 was treated with excess POCl3 in toluene in the presence of triethylamine at reflux followed by column chromatographic purification afforded meso-triaryl-21-oxacorrole 78 in 8% yield along with phosphoryl complex of meso-tetraaryl 21-oxaporphyrin 79 in 6% yield (Scheme 19). The structure of 21-oxacorrole 78 was also confirmed by X-ray crystallography. As per the mechanism proposed by authors, the meso-tetrarayl 21-oxaporphyrin 77 underwent ring contraction upon treatment with POCl3 via dearylation of meso-aryl group present between furan and pyrrole moieties to form meso-triaryl-21-oxacorrole 78.
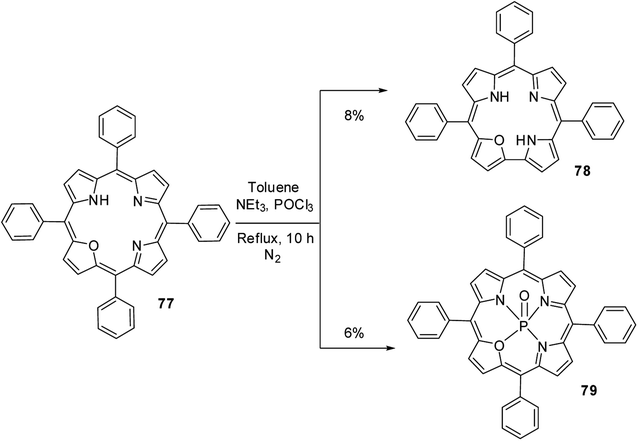 |
| | Scheme 19 Synthesis of meso-substituted 21-oxacorrole 78 from 21-oxaporphyrin 77. | |
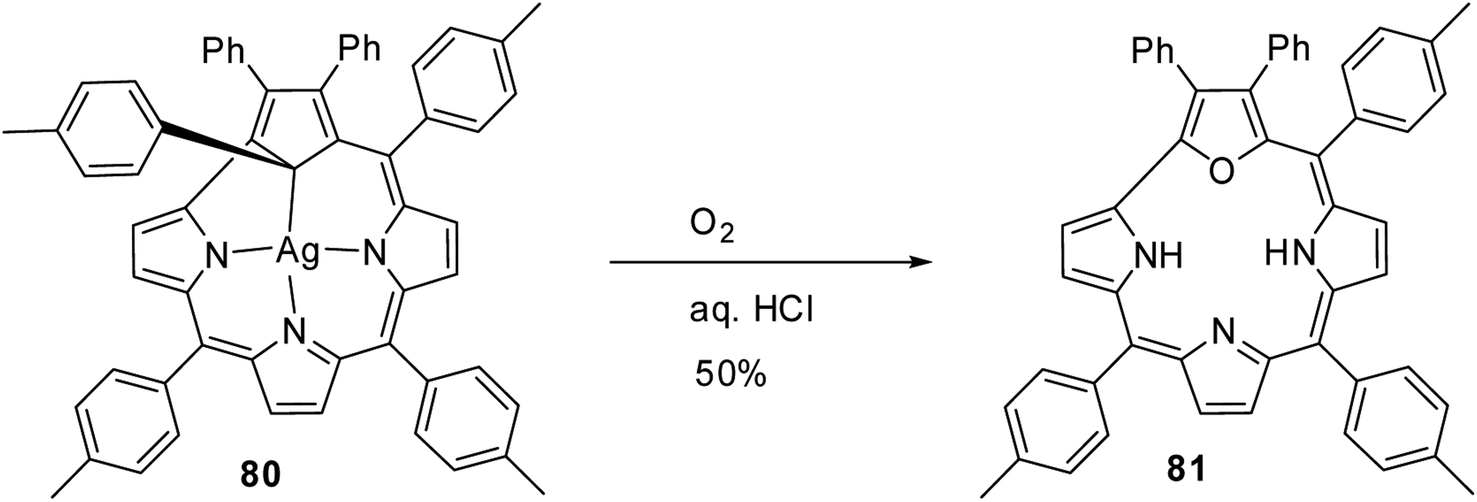
In an interesting report, Grażyński and co-workers noticed the formation of 21-oxacorrole 81, when the silver(III) carbacorrole 80 was reacted with dioxygen in presence of aqueous HCl (Scheme 20).31
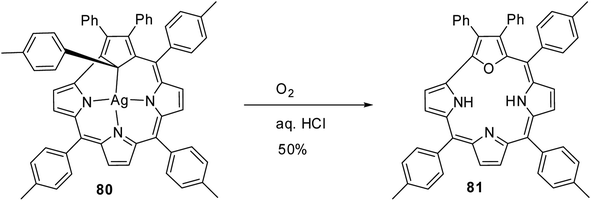 |
| | Scheme 20 Synthesis of 21-oxacorrole 81 from Ag(III) carbacorrole 80. | |
3.3. 22-Oxacorroles
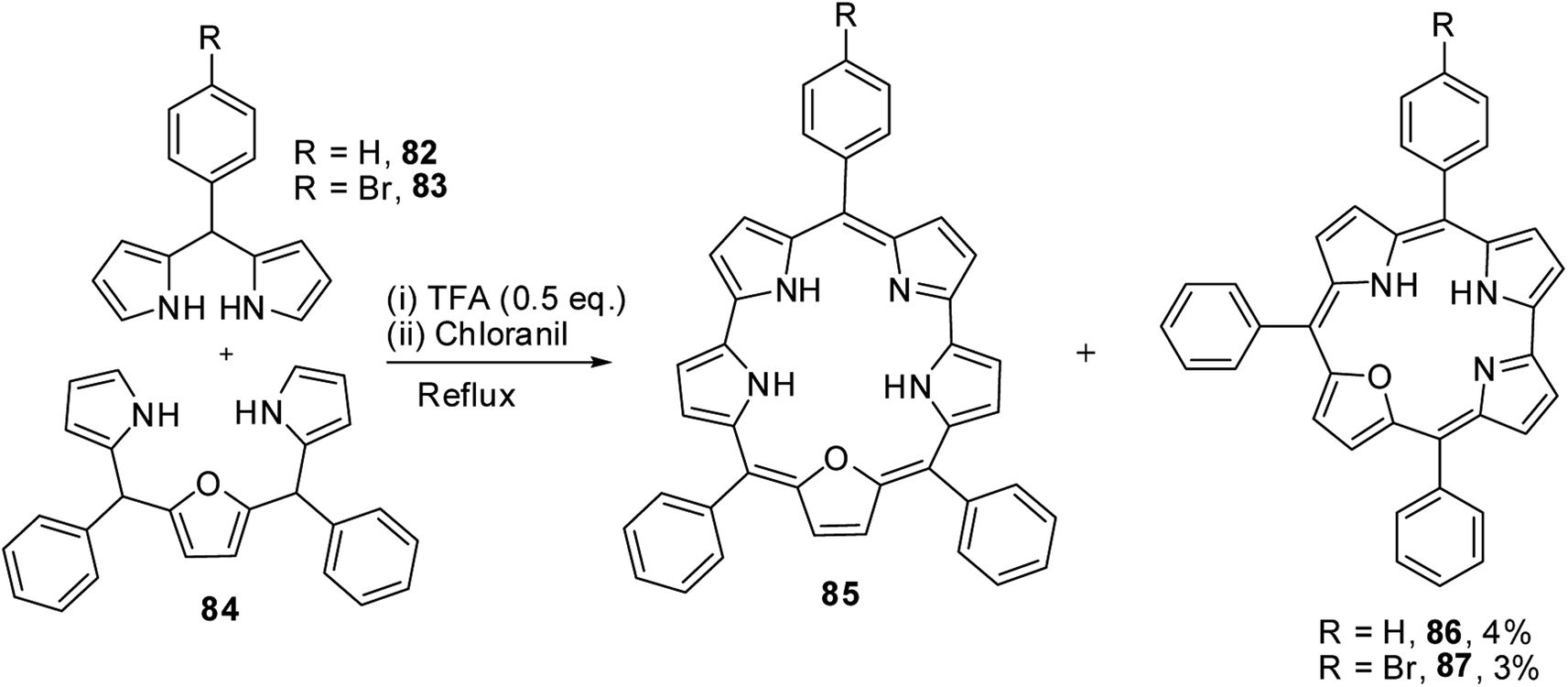
Chandrashekar and co-workers32 observed the formation of 22-oxacorrole containing direct pyrrole–pyrrole bond 86, 87 as a byproduct during their synthesis of expanded porphyrin, 25-oxasmaragdyrin 85. The TFA-catalyzed oxidative coupling between 16-oxatripyrrane 84 and meso-aryl dipyrromethanes 82, 83 followed by oxidation with chloranil resulted in the formation of 25-oxasmaragdyrin 85 as a major product (∼50% yield) and 22-oxacorroles 86, 87 in 3–4% yield (Scheme 21). Although this method yielded 21-oxacorroles in low yields, it was later proved to be an easy and versatile method to make meso-substituted 22-oxacorroles.
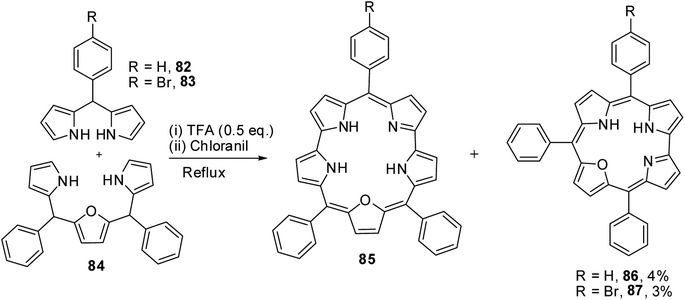 |
| | Scheme 21 Synthesis of meso-substituted 22-oxacorroles 86 and 87. | |
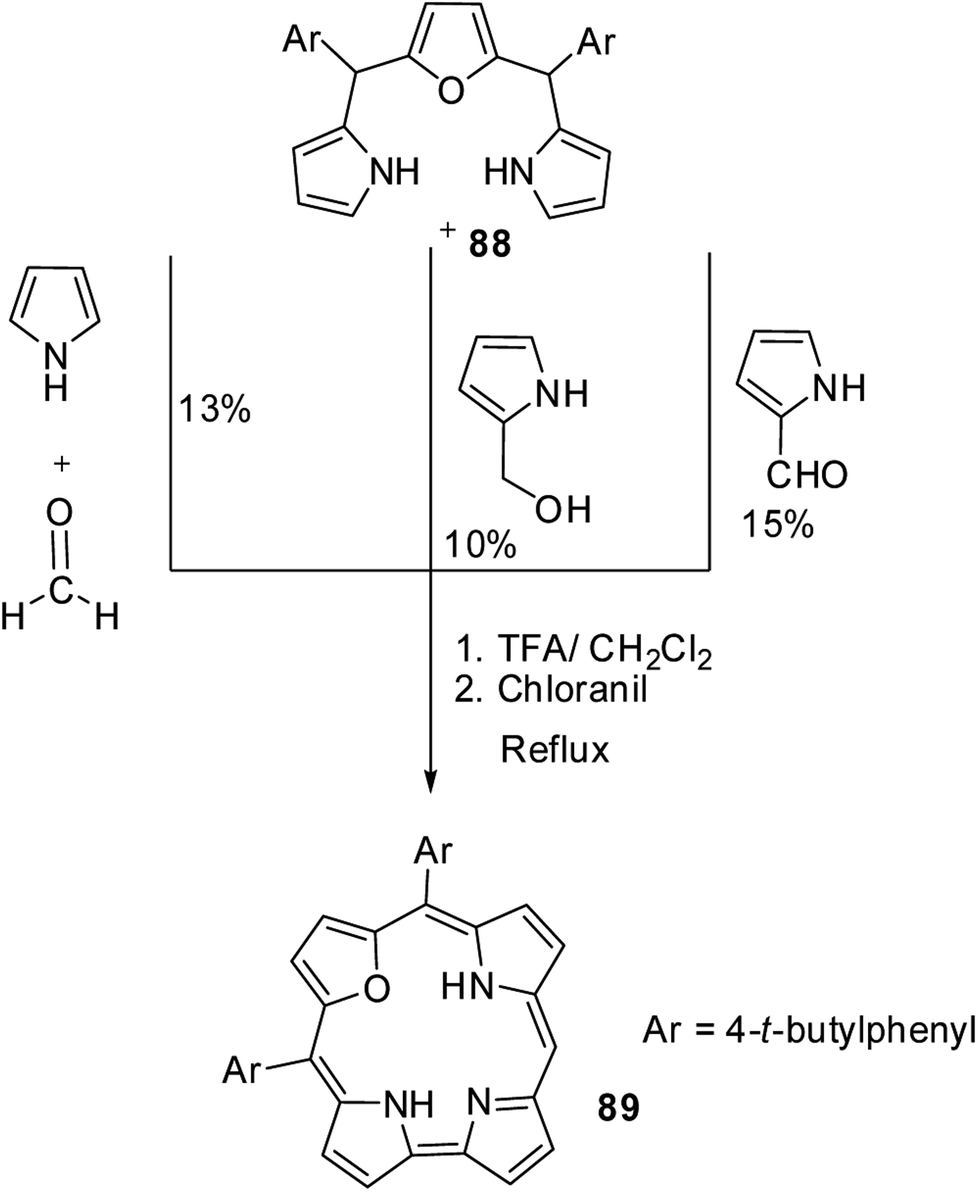
Chandrashekar and co-workers33 subsequently developed three different [3 + 1] synthetic routes to prepare 22-oxacorroles containing one meso-free carbon 89 as shown in Scheme 22. The acid catalyzed condensation of meso-substituted 16-oxatripyrrane 88 was done in three ways: in the first method, the 16-oxatripyrrane 88 was condensed with pyrrole and paraformaldehyde; in second method, 88 was condensed with 2-(hydroxymethyl)pyrrole and in the third method, 88 was condensed with pyrrole-2-carboxaldehyde. In all three methods, the condensation was followed by a chloranil oxidation resulting in mono meso-free 22-oxacorrole 89 in 10–15% yields.
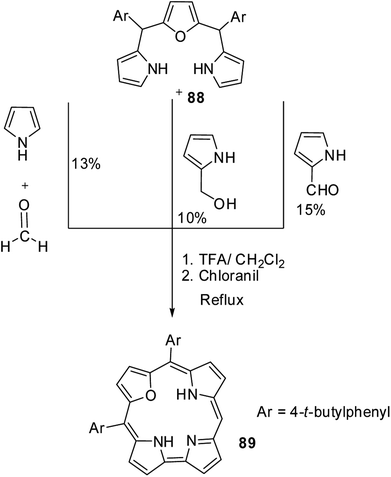 |
| | Scheme 22 Three different synthetic routes to prepare meso-substituted 22-oxacorrole 89. | |
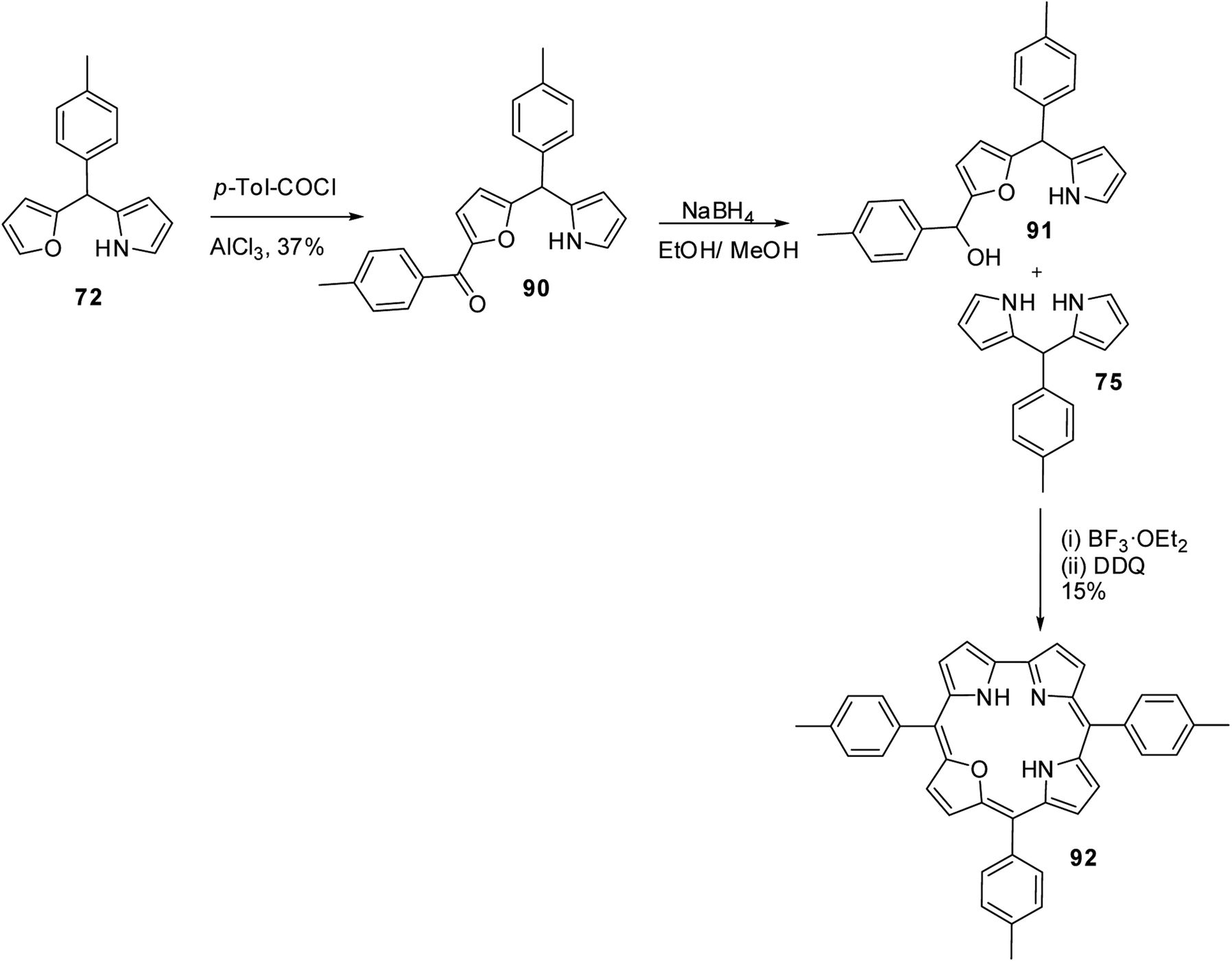
Lee and co-workers also successfully synthesized 22-oxacorroles by [2 + 2] condensation strategy as shown in Scheme 23.28,29 The AlCl3 catalysed acylation of p-tolyl-(furan-2-yl)-(pyrrol-2-yl)methane 72 followed by NaBH4 reduction yielded corresponding alcohol 91, which upon Lewis acid catalyzed condensation with meso-aryl dipyrromethane 75 followed by column chromatographic purification afforded 22-oxacorrole 92 in 15% yield.
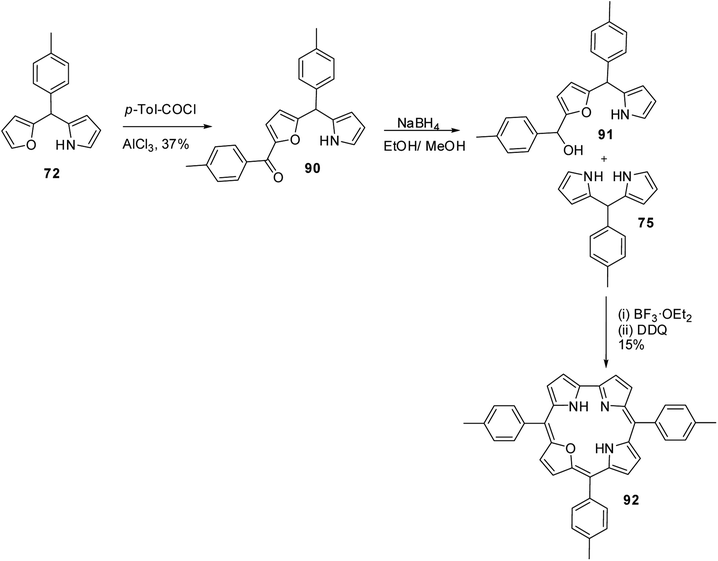 |
| | Scheme 23 Synthesis of meso-substituted 22-oxacorroles 92. | |
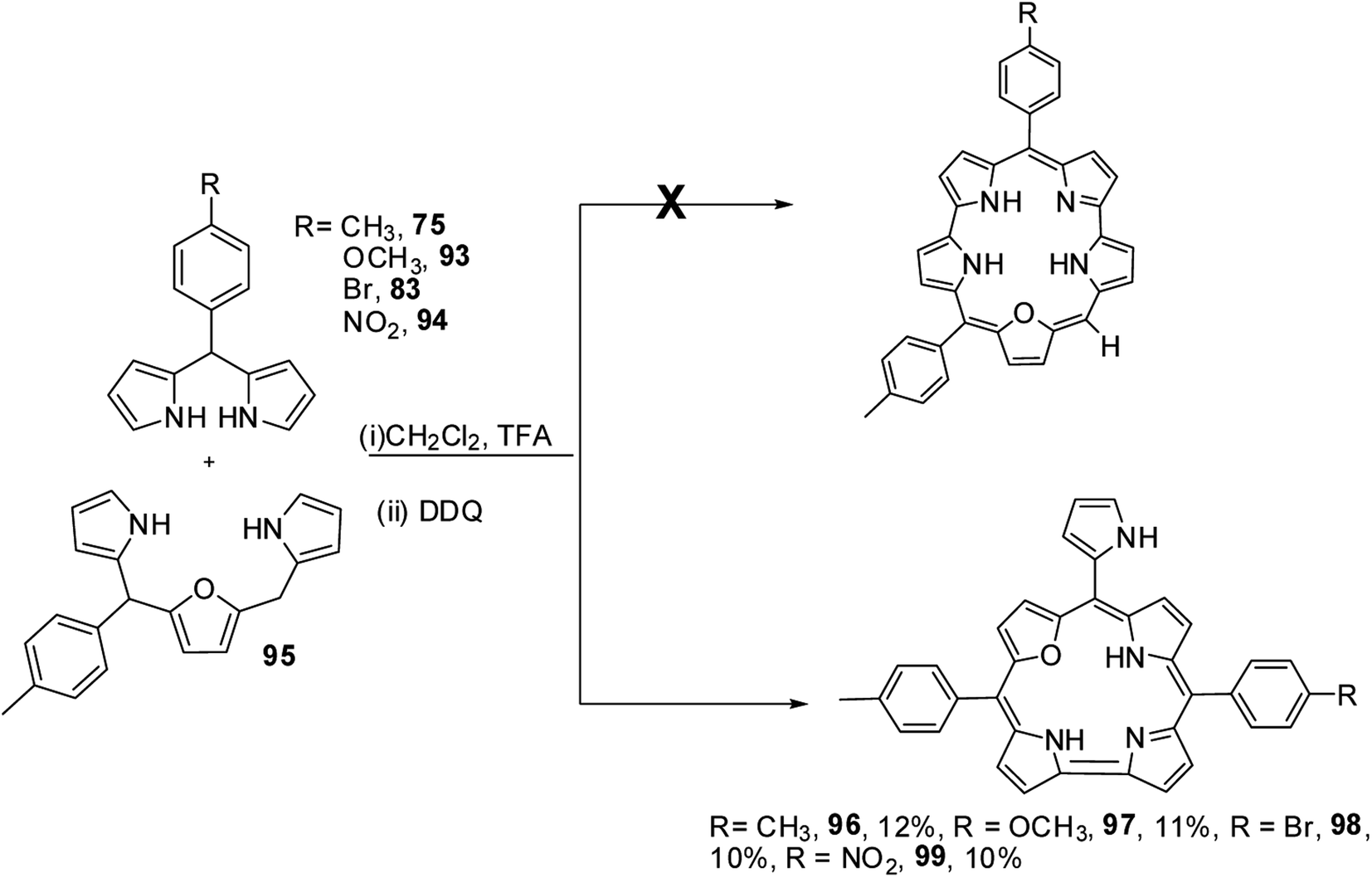
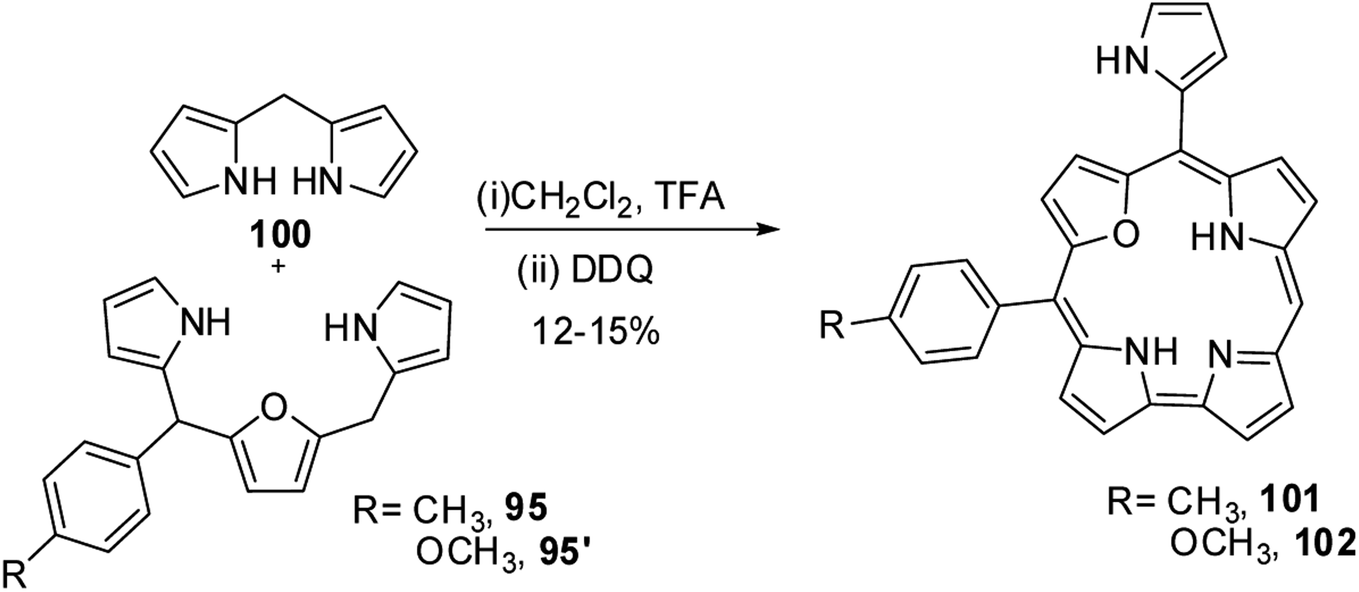
Ravikanth and co-workers adopted the same [3 + 2] synthetic strategy to prepare mono meso-pyrrolyl 22-oxacorroles 96–99.34 The 22-oxacorroles containing two meso-aryl and one meso-pyrrolyl groups 96–99 were synthesized by condensing 16-oxatripyrrane 95 with meso-aryl substituted dipyrromethanes 75, 83, 93 and 94 under mild acid catalyzed conditions followed by DDQ oxidation (Scheme 24). Kalita and Ravikanth further extended this synthetic strategy to prepare meso-free mono-meso-pyrrolyl 22-oxacorroles 101, 102 as shown in Scheme 25.35
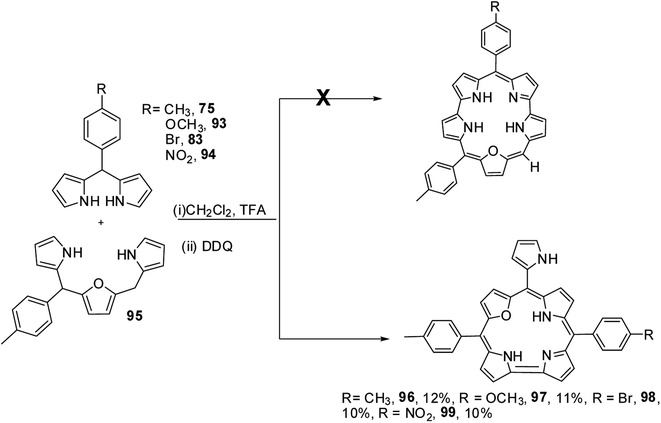 |
| | Scheme 24 Synthesis of meso-pyrrolyl 22-oxacorrole 96–99. | |
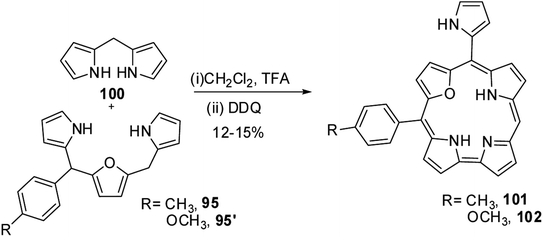 |
| | Scheme 25 Synthesis of mono meso-free 22-oxacorrole 101 and 102. | |
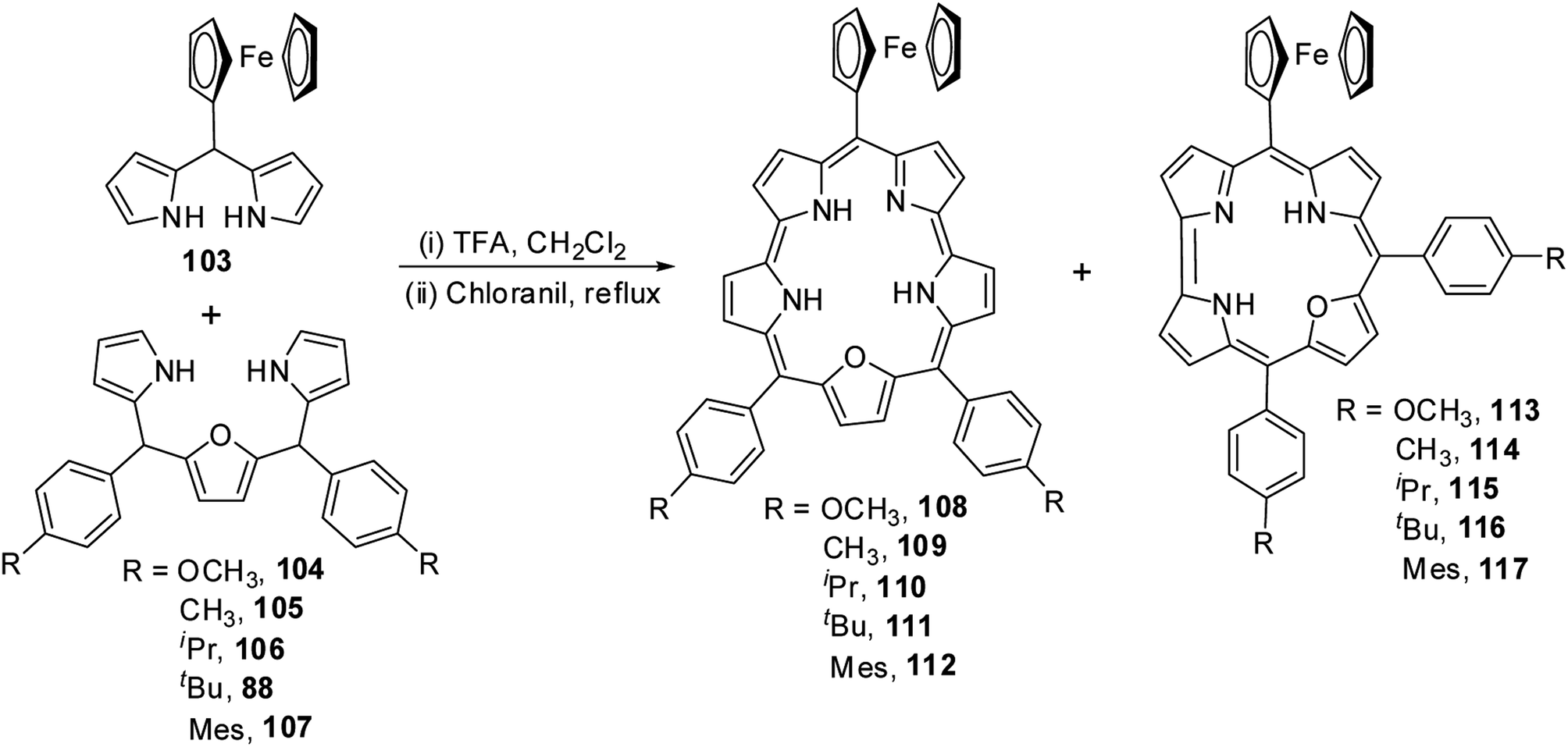
Chandrashekar and co-workers36 obtained meso-ferrocenyl 22-oxacorroles 113–117 as minor products (∼3% yield) along with meso-ferrocenyl 25-oxasmaragdyrins 108–112 (∼20% yield) by condensing appropriate 16-oxatripyrranes 88, 104–107 with meso-ferrocenyl dipyrromethane 103 under TFA-catalyzed conditions (Scheme 26).
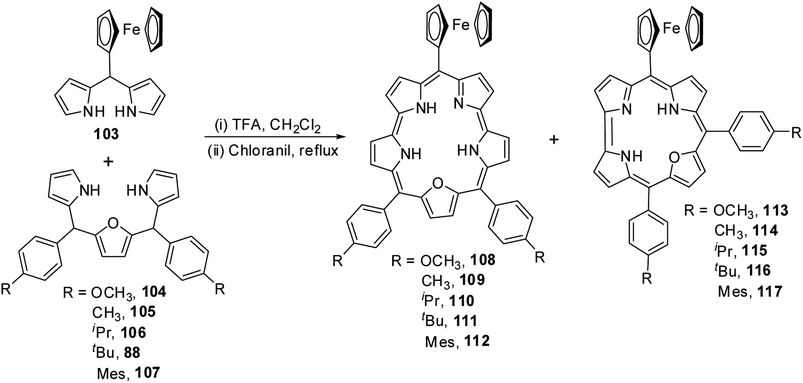 |
| | Scheme 26 Synthesis of mono meso-ferrocenyl 22-oxacorroles 113–117. | |
3.4. X-ray structures of 21-oxacorrole and 22-oxacorrole
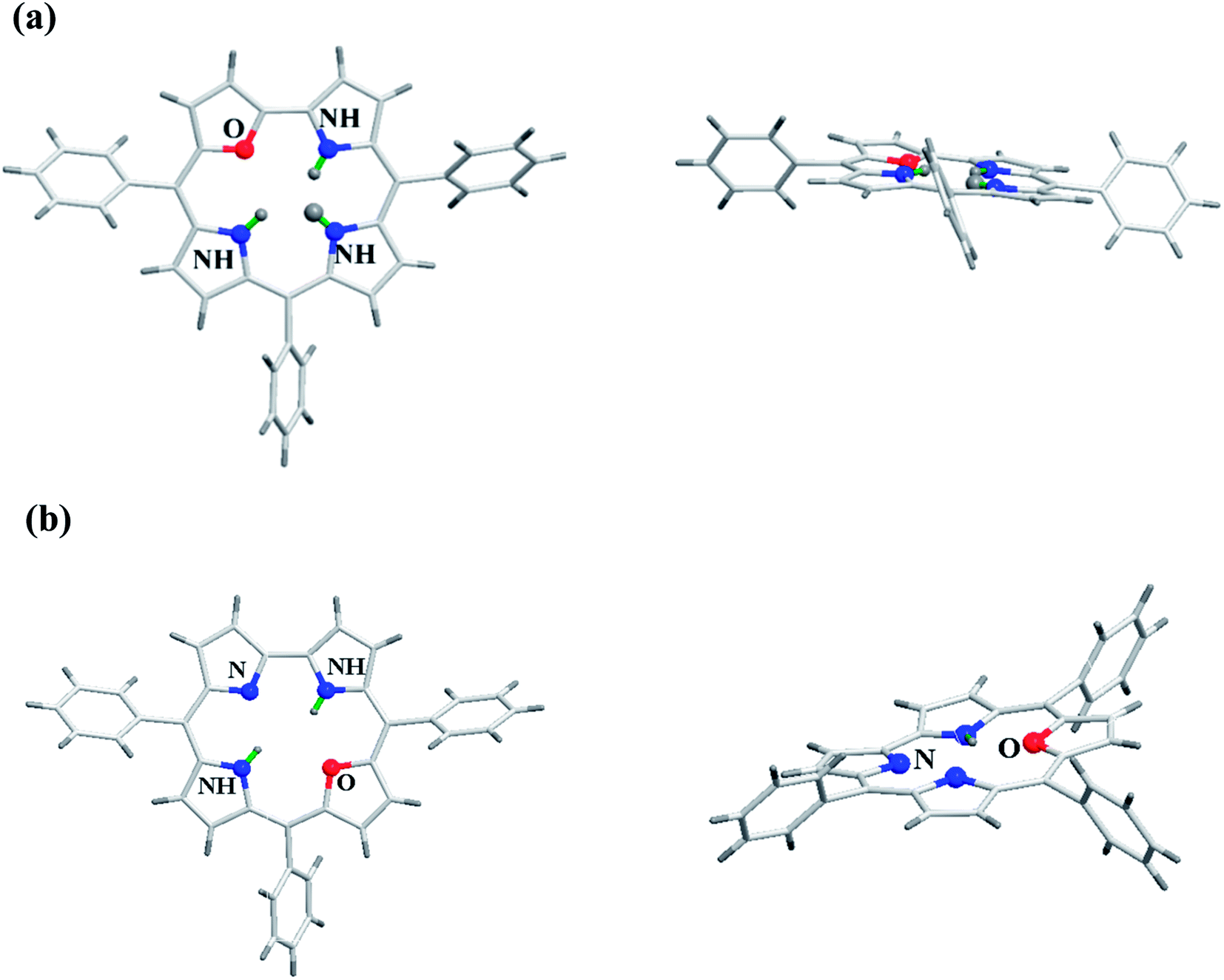
Gratifyingly, the crystal structures for both meso-triaryl 21-oxacorrole 78 and meso-triaryl 22-oxacorrole 86 were available by two different research groups and are presented in Fig. 16.30,32 Compared to meso-tetraaryl 21-oxaporphyrin, the 21-oxacorrole and 22-oxacorroles 78 and 86 adopted a near-planar conformation owing to the presence of direct furan-pyrrole bond and direct pyrrole–pyrrole bond respectively. The deviation of the furan ring from the mean plane of the macrocycle was much less in 21-oxacorrole 78 (5.69°) and 22-oxacorrole 86 (5.50°) compared to 21-oxaporphyrin (11.35°). Similarly, the deviation of three pyrrole rings from the mean plane of the macrocycle was more pronounced in 21-oxaporphyrin compared to 21-oxacorrole and 22-oxacorrole. Furthermore, the direct distance between the pyrrole and furan ring in 21-oxacorrole (1.40 Å) was little shorter than the direct pyrrole–pyrrole distance in 22-oxacorrole (1.43 Å). The meso-phenyl groups were more coplanar with the macrocycle in 21-oxacorrole and 22-oxacorrole compared to 21-oxaporphyrin.
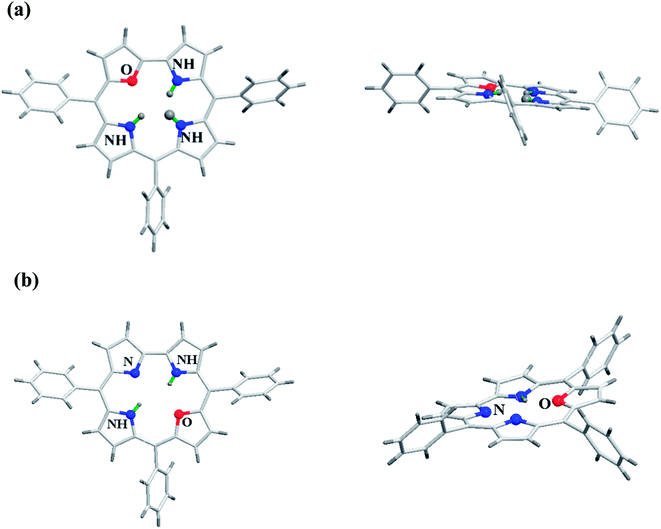 |
| | Fig. 16 Top and side view of X-ray crystal structures of (a) meso-substituted 21-oxacorrole 78 (CCDC 913385) (b) meso-substituted 22-oxacorrole 86 (CCDC 112074). | |
3.5. Spectral and electrochemical properties of 21-oxacorroles and 22-oxacorroles
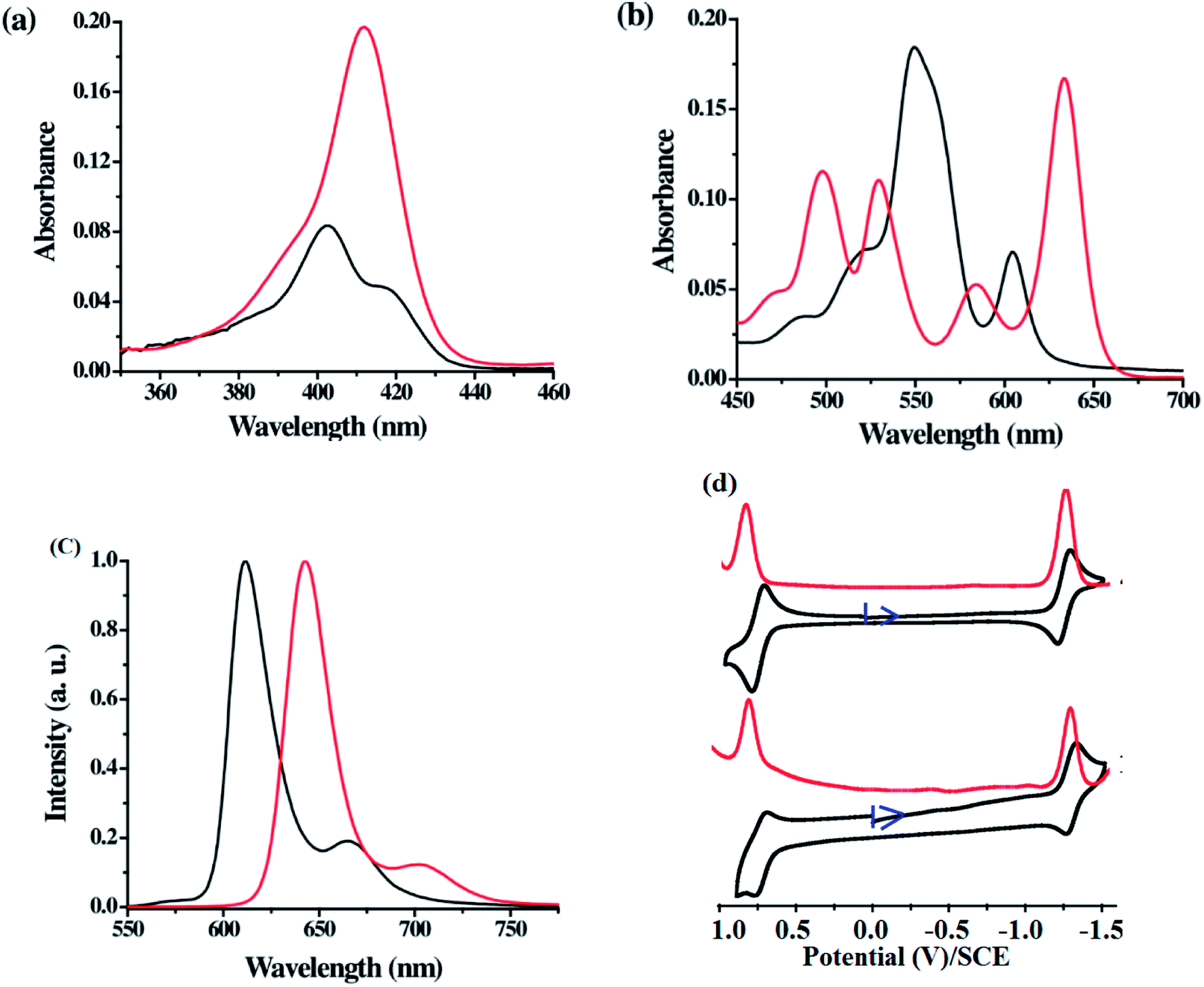
The comparison of Soret and Q-band absorption spectra, fluorescence spectra and cyclic voltammograms of 21-oxacorrole 78 and 22-oxacorrole 86 are presented in Fig. 17. The 21-oxacorrole displayed Q-type bands in the region 480–610 nm and a split Soret band in 400–420 nm, whereas the 22-oxacorroles showed four well defined Q-type bands in the region 490–650 nm and a strong Soret band at 411 nm. The blue shifted absorption bands of 21-oxacorrole compared to 22-oxacorrole indicate that the 21-oxacorrole is less flexible and more resonance stabilized than 22-oxacorrole. The nature and presence of meso-substituent on the absorption spectral pattern was also evident as meso-pyrrolyl 22-oxacorroles and meso-ferrocenyl 22-oxacorroles showed slight bathochromic shifts in their peak maxima whereas the meso-free 22-oxacorroles showed slight hypsochromic shifts compared to meso-triaryl 22-oxacorroles.
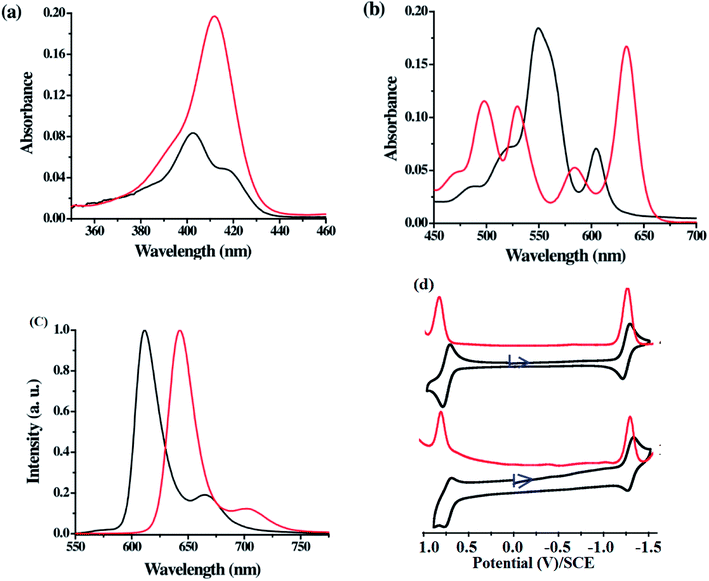 |
| | Fig. 17 (a) Soret band (b) Q-bands of meso-substituted 21-oxacorrole 78 (black), 22-oxacorrole 86 (red), (c) emission profiles of 21-oxacorrole 78 (black, λex = 402 nm), 22-oxacorrole 86 (red, λex = 412 nm) recorded in dichloromethane (d) cyclic voltammogram (black) and DPV plots (red) of 22-oxacorrole 78 (top) and 21-oxacorrole 86 (bottom) recorded in dichloromethane with TBAP as supporting electrolyte. Reproduced from ref. 30 with permission. Copyright© 2013 American Chemical Society. | |
Both 21-oxacorrole 78 and 22-oxacorrole 86 were brightly fluorescent with emission maxima in 610–650 nm range and the quantum yields were in the range of 0.3 to 0.5. In general, the oxacorroles exhibit one quasi-reversible and one irreversible peaks for both oxidation and reduction processes. Although both 21-oxacorrole 78 and 22-oxacorrole 86 were stable under redox conditions and exhibited similar redox behaviour, their oxidation/reduction was found to be difficult compared to 21-oxaporphyrins. The presence of different meso-substituents such as pyrrole and ferrocenyl groups slightly altered their redox behaviour. For example, the meso-pyrrolyl 22-oxacorroles were easier to reduce compared to meso-triaryl 22-oxacorroles.
3.6. Metal complexes of 22-oxacorroles
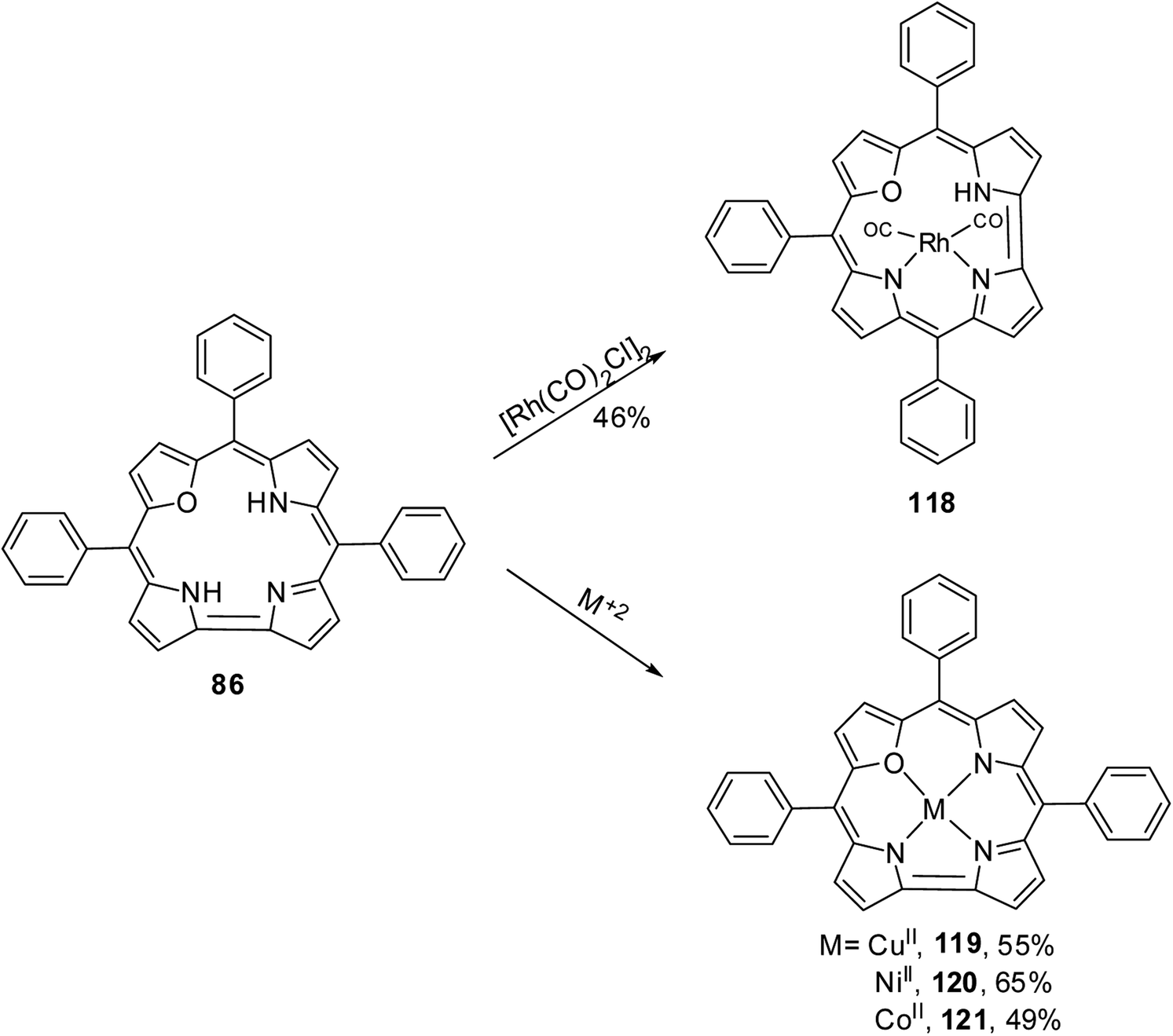
The 22-oxacorroles with two ionizable protons are expected to form complexes readily with divalent metals unlike normal corroles which are known to stabilise metals in higher oxidation states due to three ionizable protons. Chandrashekar and co-workers37 reported one monovalent Rh(I) complex 118 and three divalent metal complexes such as Cu(II) 119, Ni(II) 120 and Co(II) 121 by treating the 22-oxacorrole 86 with appropriate metal precursors under standard metallation reaction conditions (Scheme 27). The X-ray structures of Rh(I) 118 and Ni(II) 120 complexes are presented in Fig. 18. The crystal structure of Rh(I) complex 118 revealed that Rh(I) was located above the macrocyclic plane in a near square planar geometry coordinated by one imino and one amino nitrogen of the 22-oxacorrole macrocycle and remaining two coordination sites were occupied by carbonyl ligand (Fig. 18). Since Rh(I) was situated out of plane, the pyrrole rings of dipyrromethene moiety twisted above the mean corrole plane by an angle of 23.51° and 19.69° respectively. The angle between the plane containing the Rh(I) with its coordinated atoms and the mean corrole plane was 62.64°.
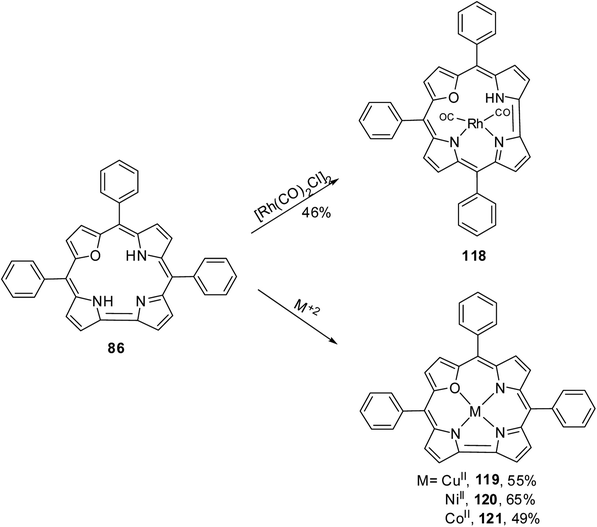 |
| | Scheme 27 Synthesis of metal derivatives of 22-oxacorrole 118–121. | |
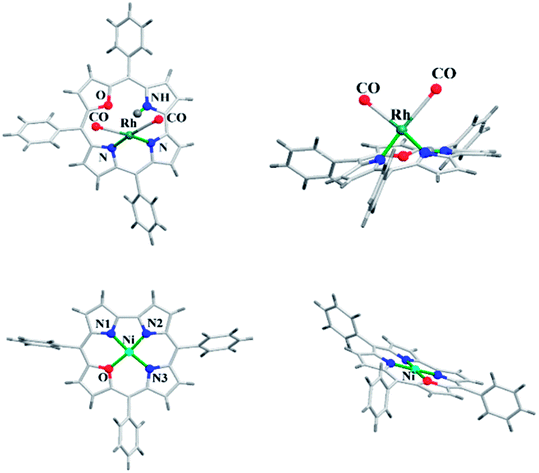 |
| | Fig. 18 Top and side view of X-ray crystal structures of Rh(I) 118 (CCDC 135916) (top) and Ni(II) 120 (CCDC 135917) (bottom) complexes of meso-substituted 22-oxacorrole. | |
The Ni(II) insertion into the 22-oxacorrole core flattens the macrocycle to almost planar structure and Ni(II) was coordinated in distorted square planar conformation. The Ni(II) ion lies above the mean plane of the macrocycle by only 0.008 Å. The Ni–N and Ni–O distances were shorter compared to the corresponding distances in Ni(II) derivative of 21-oxaporphyrin due to the reduced core size of 22-oxacorrole.
The Cu(II) 119 and Co(II) 121 complexes of 22-oxacorrole were paramagnetic and EPR spectra of these complexes displayed significant decrease in the metal hyperfine couplings compared to the corresponding porphyrin complexes. The absorption spectra of the metallated derivatives of 22-oxacorrole showed characteristic Soret-type and Q-type bands in the region 400–700 nm like their free base counterparts. The Cu(II) and Co(II) complexes showed broad Soret type band whereas the Rh(I) and Ni(II) showed split Soret band due to their lower symmetry in solution. The extinction coefficient values of all the metallated derivatives were smaller than the free base 22-oxacorrole due to decreased π-electron conjugation in metal derivatives. The electrochemical studies of these metal complexes revealed that the redox process was only macrocycle centered.
3.7. Functionalized 22-oxacorroles and covalently linked 22-oxacorrole dyads
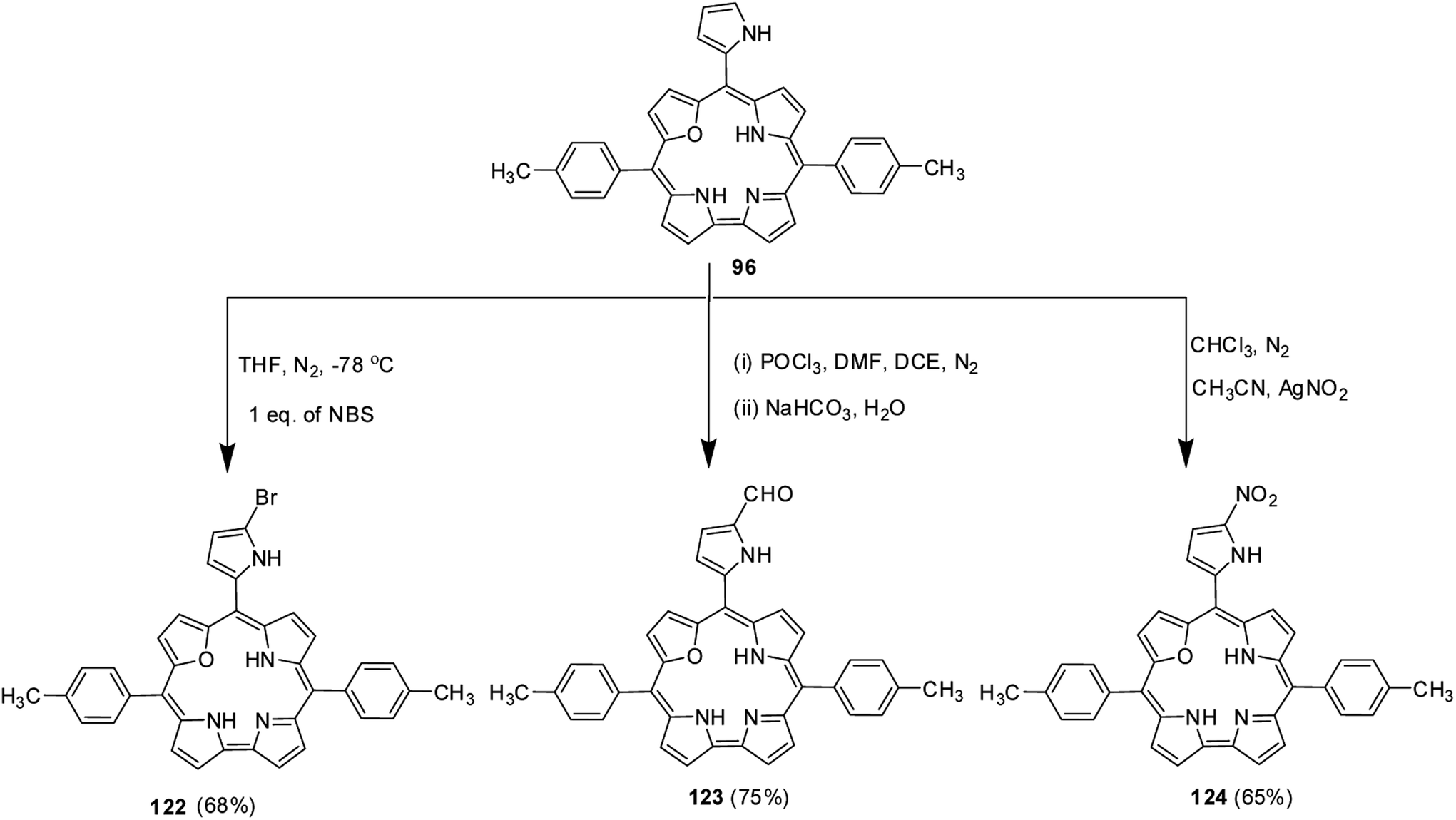
Ravikanth and co-workers explored the functionalization reactions of meso-pyrrole substituted 22-oxacorroles by taking advantage of the reactivity of the α-position of pyrrole ring present at the meso-position.34 Bromination, formylation and nitration reactions were carried out on meso-pyrrolyl substituted 22-oxacorrole 96 by using N-bromosuccinimide (NBS), Vilsmeier reagent and AgNO2 respectively, as shown in Scheme 28. The crystal structure of meso-pyrrole nitrated 22-oxacorrole 124 showed that (Fig. 19) the corrole macrocycle was almost planar with slight deviations ranging from 2.98 to 11.52° with respect to the mean plane defined by three meso-carbon atoms, three pyrrole rings and one furan ring. The nitro group and meso-pyrrole groups were perpendicular to the plane of the macrocycle.
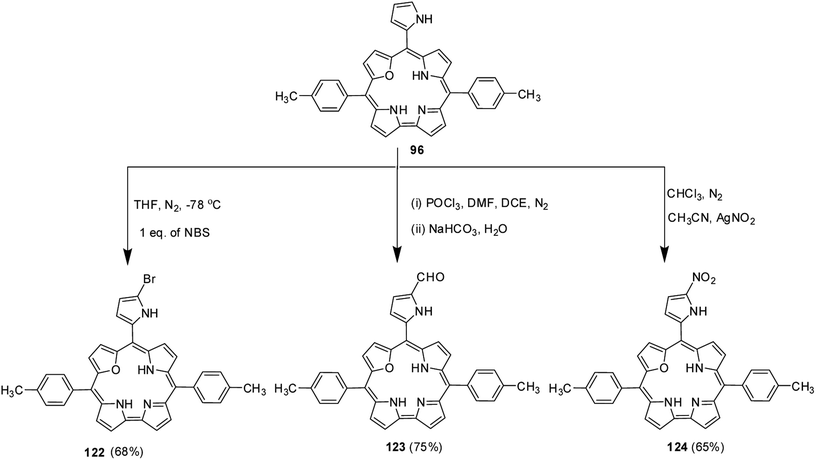 |
| | Scheme 28 Functionalization reactions of meso-pyrrolyl 22-oxacorrole 96. | |
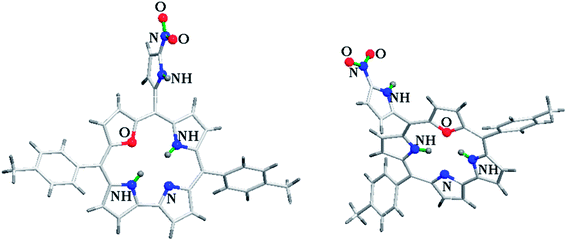 |
| | Fig. 19 Top and side view of X-ray crystal structures of 124 (CCDC 973515). | |
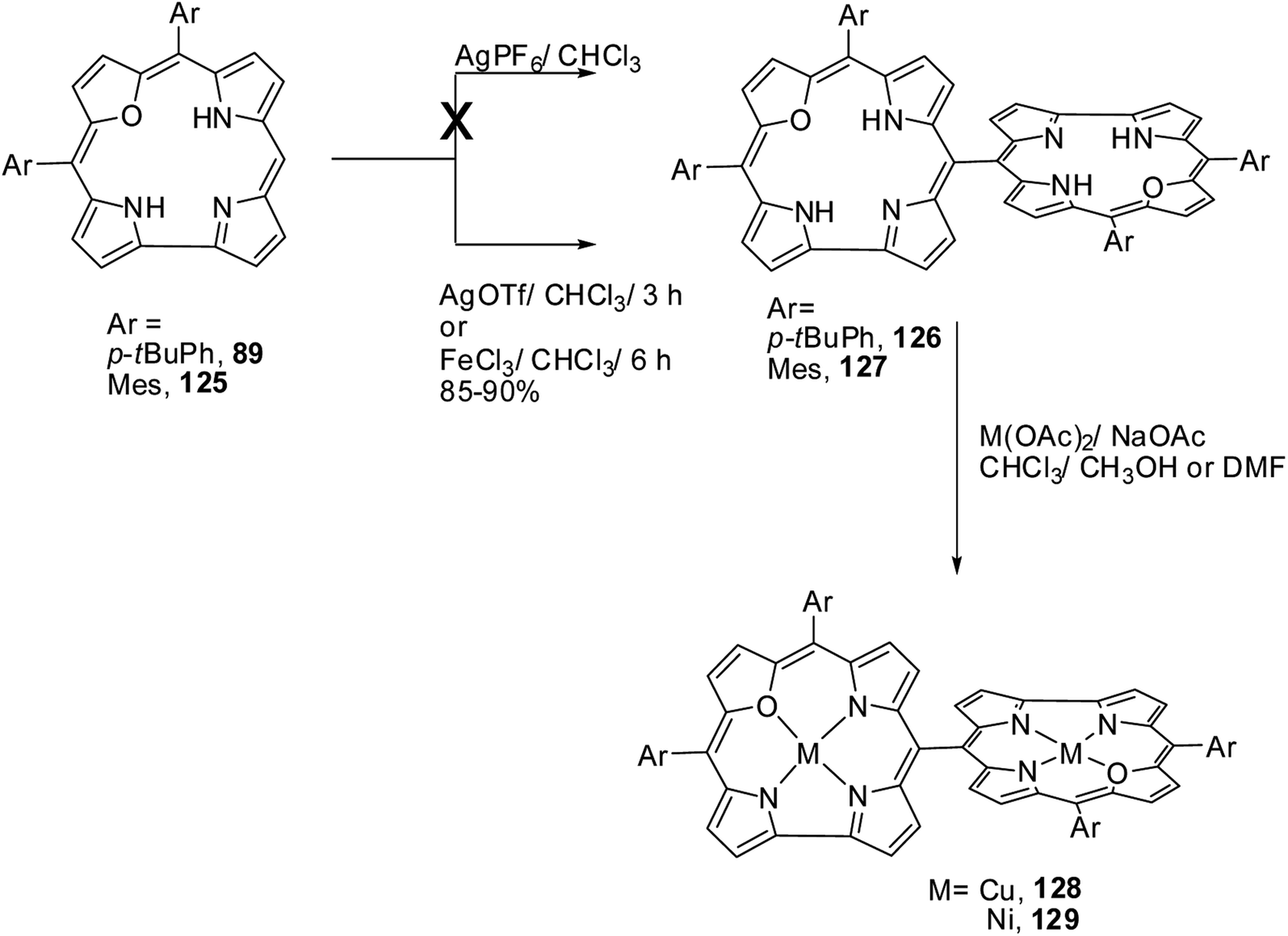
Chandrashekar and co-workers38 successfully prepared the first examples of meso–meso linked 22-oxacorrole dimers 126, 127 in near quantitative yield by AgOTf or FeCl3 catalyzed coupling reaction of meso-free 22-oxacorroles 89 and 125 as shown in Scheme 29. The absence of meso-CH resonance signal in 1H NMR spectrum provided a strong evidence for the formation of corrole dimer. The copper and nickel derivatives of meso–meso linked 22-oxacorrole dimers 128, 129 respectively, were obtained by metallating dimers with appropriate metal salts (Scheme 29) However, authors did not succeed in obtaining hetero bis-metal derivatives of 22-oxacorrole dimer.
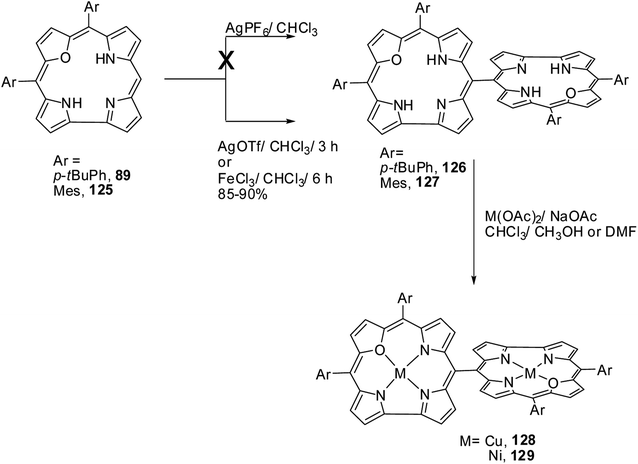 |
| | Scheme 29 Synthesis of meso–meso linked oxacorrole dimer 126, 127 and their bis metal derivatives 128, 129. | |
The absorption spectral studies revealed weak interactions between the two sub-units in dimers 126, 127 and the exciton coupling observed for the free base 22-oxacorrole dimer was lower than the corresponding protonated derivatives. The NMR and DFT (B3LYP-631G-level) studies indicated a noncoplanar arrangement of two corrole units in dimers. The electron paramagnetic resonance and magnetic studies on bis-Cu(II) complex of 22-oxacorrole dimer 128 indicated that both copper ions behave as independent spins without any noticeable interaction. The fluorescence maxima of 22-oxacorrole dimers experienced substantial bathochromic shift (60 nm) compared to monomeric 22-oxacorrole. The authors also measured hyperpolarizability (β) by hyper Rayleigh scattering (HRS) method which revealed that the β-values were doubled in 22-oxacorrole dimers compared to the corresponding 22-oxacorrole monomers due to enhanced π-conjugation in the dimers. The bis-copper complex was explored for the photocleavage of DNA and the studies revealed that the bis-copper complex selectively cleaves the nucleic acids without affecting the proteins. This observation suggests possible application of this bis copper complex in the removal of nucleic acid contamination from protein extracts through a simple photolytic pathway.
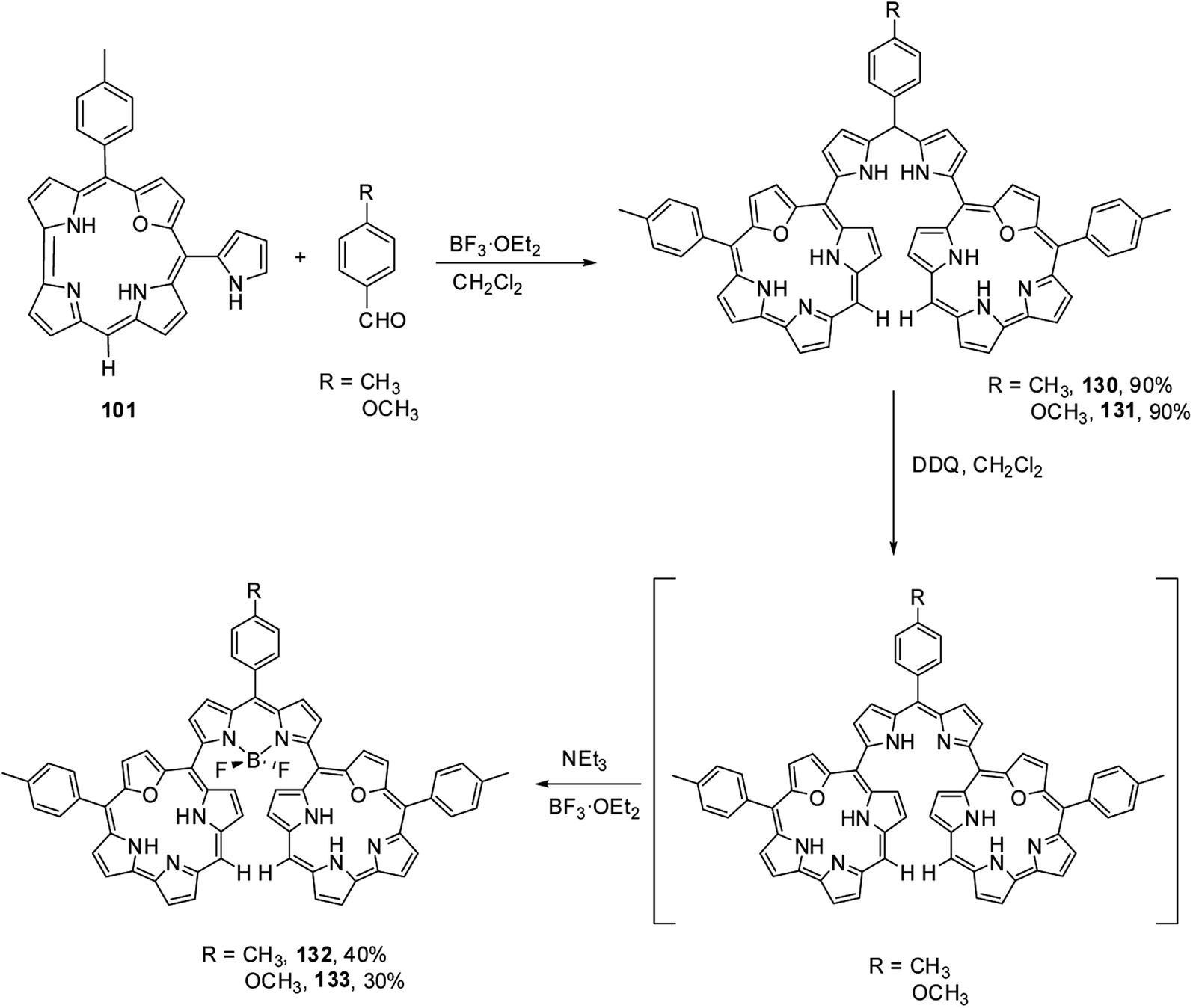
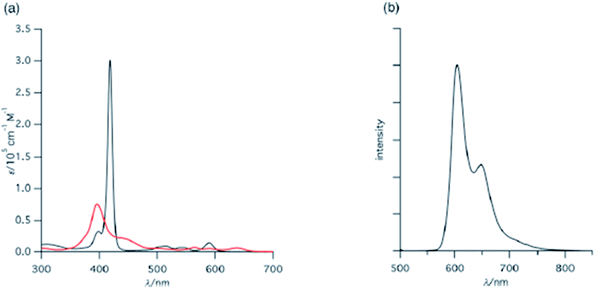
Kalita and Ravikanth35 reported the synthesis of novel boron-dipyrromethene (BODIPY)-bridged 22-oxacorrole dyads 132, 133 by using meso-pyrrolyl 22-oxacorrole 101 as a key synthon as shown in Scheme 30. The BODIPY-bridged corrole dyads 132, 133 showed an absorption band at 520 nm due to the BODIPY moiety and three bands at 399, 572 and 618 nm due to corrole units (Fig. 20). The absorption bands of BODIPY-bridged 22-oxacorrole dyads were slightly blueshifted with an increase in their extinction coefficients compared to monomeric meso-pyrrolyl 22-oxacorrole. The BODIPY-bridged 22-oxacorrole dyads 132, 133 were weakly fluorescent and showed one broad emission band at 640 nm with a quantum yield of 0.03–0.05.
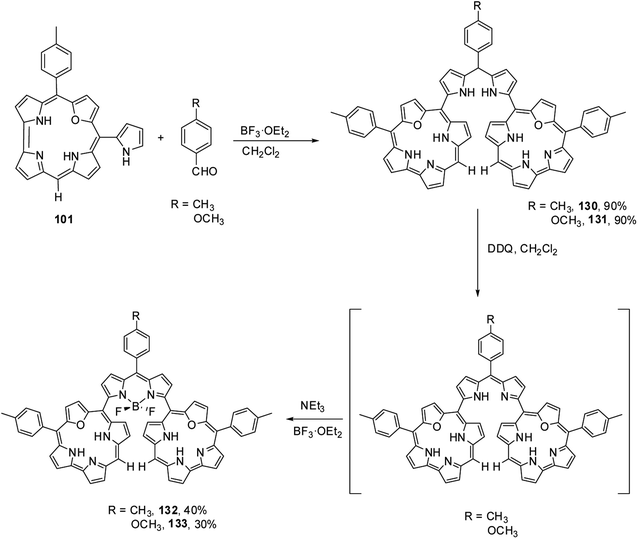 |
| | Scheme 30 Synthesis of BODIPY bridged 22-oxacorrole dyad 132, 133. | |
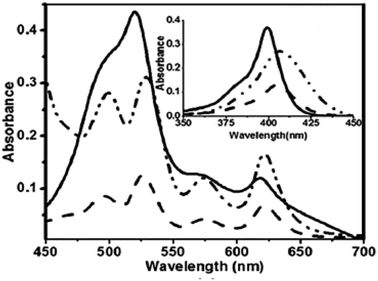 |
| | Fig. 20 Absorption spectra of BODIPY bridged corrole dyad 132 (normal line), corrole dyad (dotted line) 130 and monomer oxacorrole 101 (dashed line) recorded in dichloromethane. Reproduced from ref. 35 with permission. Copyright© 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
3.8. Dioxacorroles
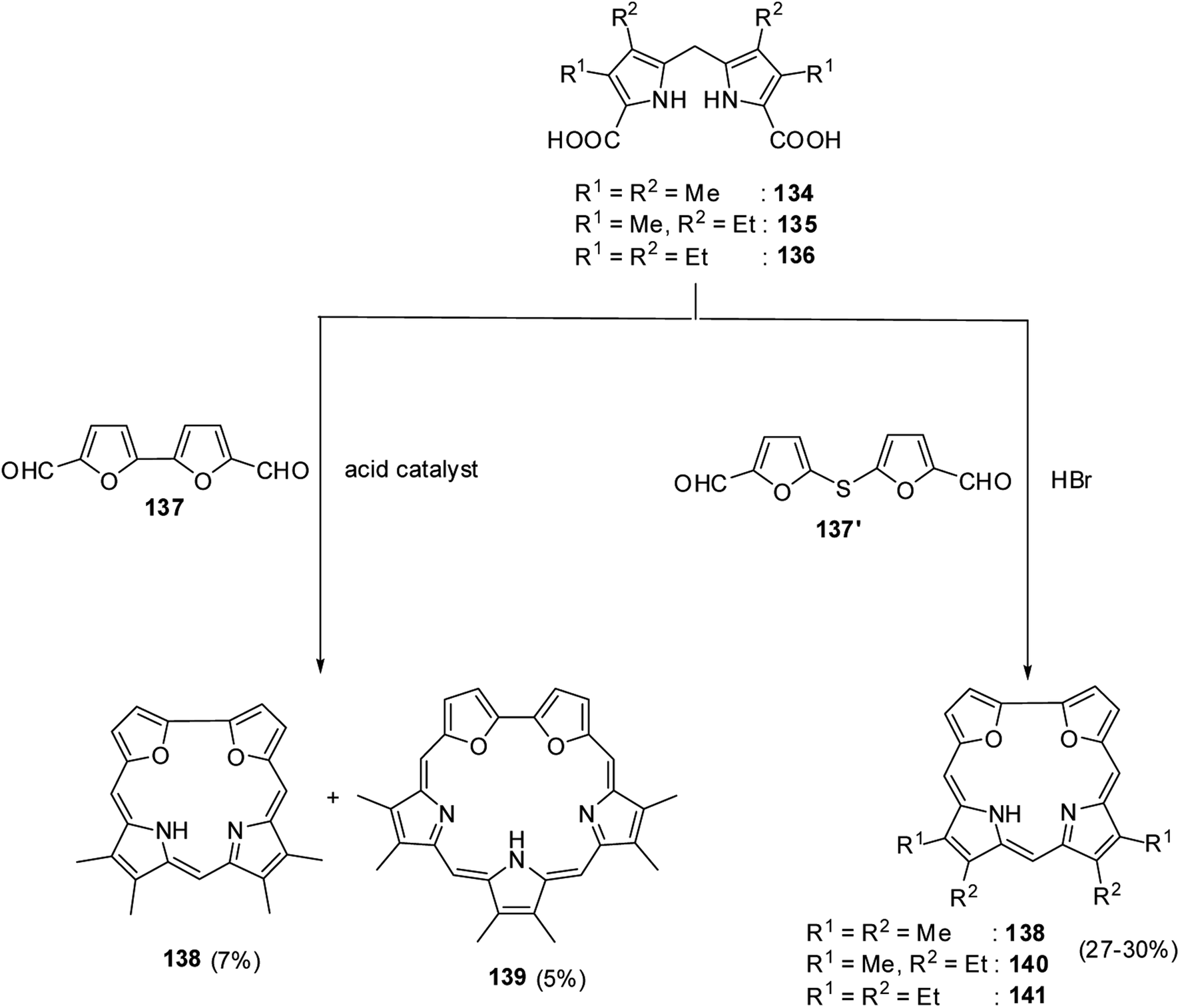
Broadhurst, Grigg and Johnson39 reported the synthesis of meso-free 21,24-dioxacorroles 138, 140 and 141 by utilizing the acid catalyzed, MacDonald type condensation of β-substituted dipyrrolylmethane diacids 134–136 with either bifuran dialdehyde 137 or diformyl-difuryl sulphide 137′ (Scheme 31). The condensation of β-substituted dipyrrolylmethane diacids 134–136 with bifuran dialdehyde 137 yielded dioxacorrole 138 along with an expanded porphyrin, heterosapphyrin 139 which were eventually separated by column chromatography. Alternatively, the acid-catalyzed condensation of diformyl-difuryl sulphide 137′ with dipyrrolylmethane diacids 134–136 yielded dioxacorroles 138, 140 and 141 in 27–30% yields. Authors' attempts to make metal derivatives of 21,24-dioxacorroles were unsuccessful.
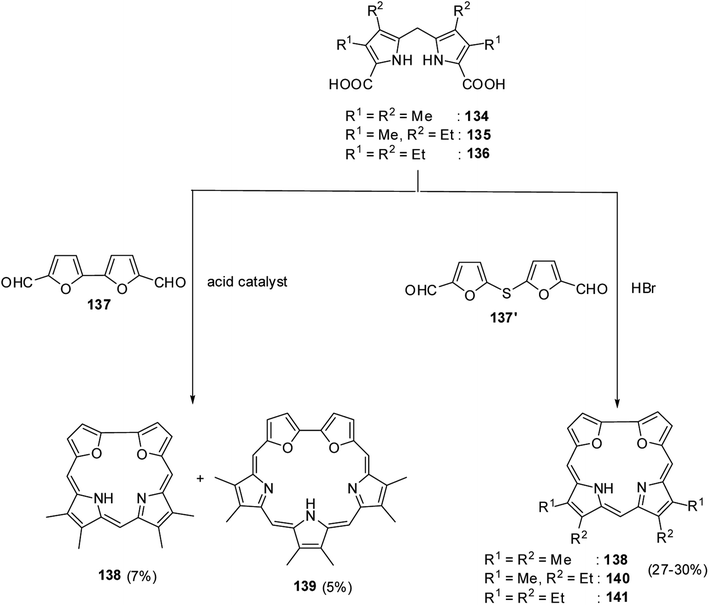 |
| | Scheme 31 Synthesis of meso-free 21,24-dioxacorroles 138–141. | |
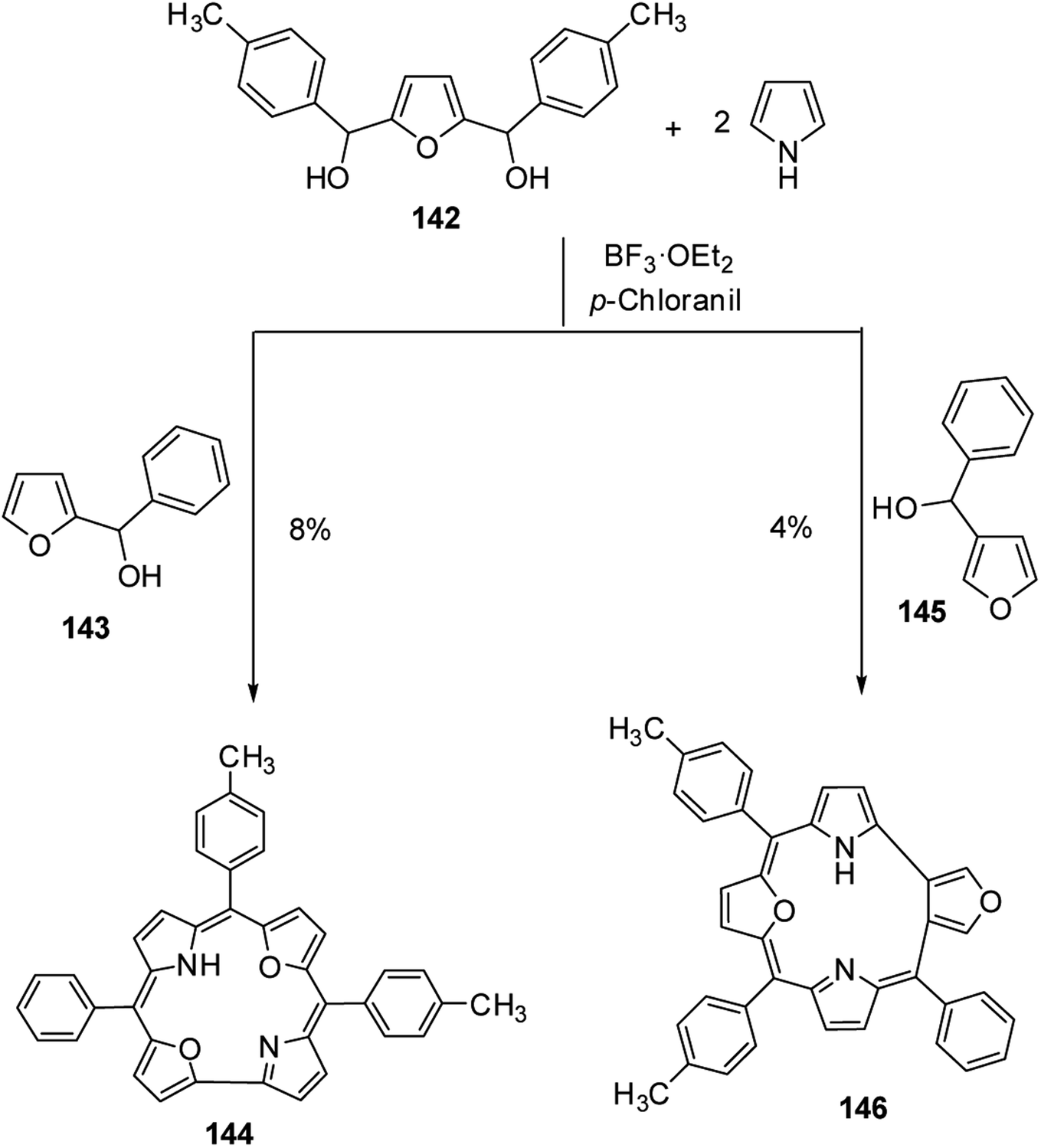
Latos-Grażyński and co-workers40 reported the synthesis of 5-phenyl-10,15-bis(p-tolyl)-21,23-dioxacorrole 144 by condensing furan diol 142 with 2-phenylhydroxymethylfuran 143 in a synthetic route depicted in Scheme 32. The aromatic character of dioxacorrole 144 was evident from its NMR spectroscopic features such as downfield and upfield shifted resonance signals for β-CH of furan and core NH protons respectively.
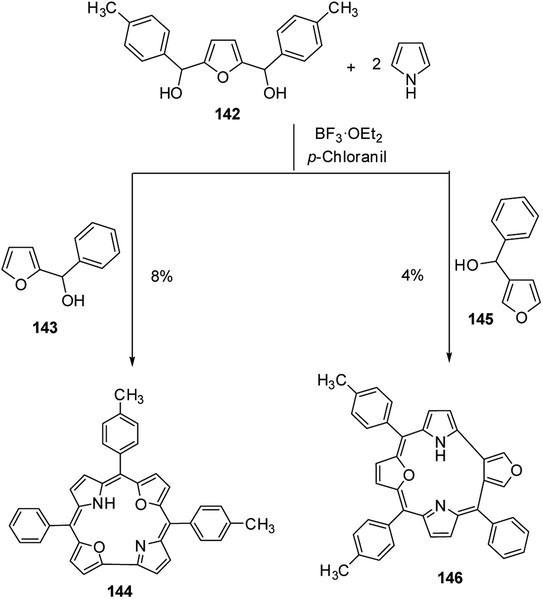 |
| | Scheme 32 Synthesis of meso-substituted 21,23-dioxacorrole 144 and its isomer 146. | |
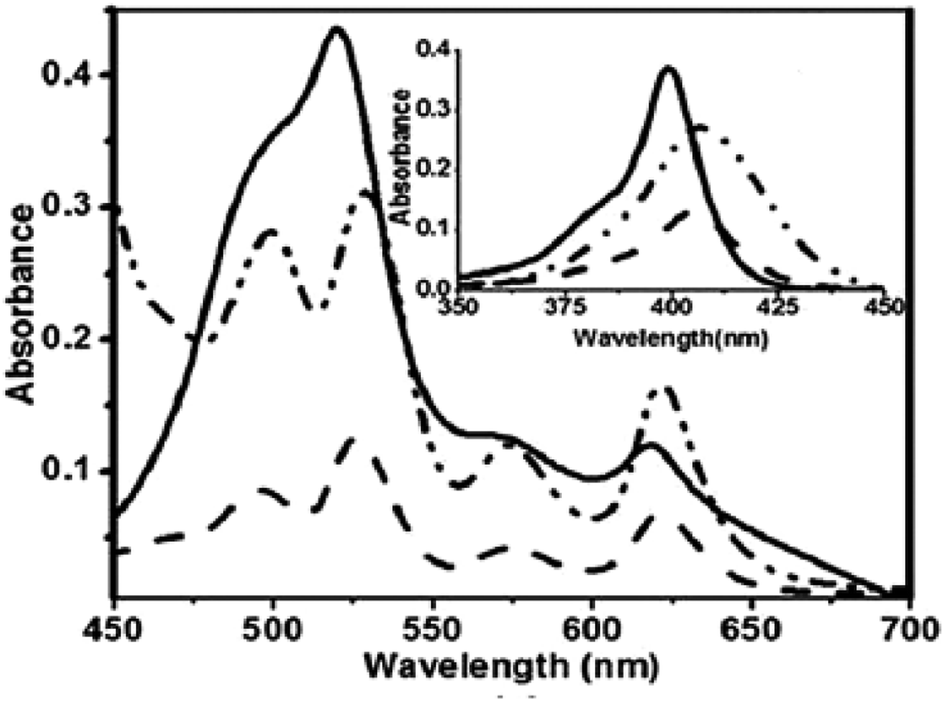
The core NH protons exhibited rapid tautomerism by exchanging sites between two structurally inequivalent nitrogen atoms even at 188 K. Protonation by TFA arrested the tautomerism and resonance signals for inner core NH protons were observed at −1.46 and −1.98 ppm. Also, dioxacorrole 144 exhibited acid dependent chemical shift of NH resonance emphasizing the role of counter-anion's interaction with monocation of dioxacorrole. The single crystal X-ray structure of dioxacorrole 144 is shown in Fig. 21. The furan moieties displayed longer and shorter bond lengths for Cα–Cβ and Cβ–Cβ respectively when compared to free furan indicating an alteration in π delocalization of furan rings in dioxacorrole. The dioxacorrole 144 exhibited markedly split Soret bands in 395–425 nm region reflecting reduced symmetry due to presence of two oxygen atoms in trans position along with series of Q-bands in 480–640 nm range.
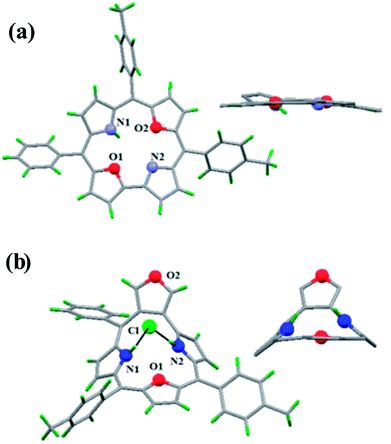 |
| | Fig. 21 Top and side view of X-ray crystal structures of (a) 144 (b) 146H+. | |
Further, when 3-phenylhydroxymethylfuran 145 was condensed with 142 and pyrrole under identical reaction conditions, the formation of an unusual isomer of dioxacorrole containing protruding furan ring 146 was formed (Scheme 32). Relatively upfield shifted β-H resonances with respect to 142 and markedly down field shifted resonance of NH (17.71 ppm) convincingly demonstrated non aromatic character of 146. The X-ray crystal structure of protonated corrole 146H+ showed a puckered structure arising due to the contraction of internal ring by one carbon atom compared to regular corrole core (Fig. 21). The protonated species 146H+ behaved as an anion receptor by exhibiting N–H⋯Cl hydrogen bond by two NH groups. The crystallographic data also revealed that the protruding furan ring preserved all the features typical of isolated furan whereas the second furan moiety in the macrocycle underwent perturbation in π-delocalization. The absorption spectroscopic features of 146 and its protonated form 146H+ were different from 144 and 144H+. The electronic absorption spectrum of 146 exhibited a split Soret band at 375 and 409 nm along with a broad band at 753 nm which was remarkably red shifted to 962 nm in compound 146H+.
The five-coordinate high-spin (21,23-O2Cor)NiIICl complex Ni144 was prepared by treating 21,23-dioxacorrole with nickel(II) chloride in boiling DMF. The 1H NMR spectral pattern displayed considerably downfield shifted resonances for β-H of pyrrole and furan ring resonances of Ni144 indicating its paramagnetic behavior. Ni144 displayed an absorption pattern typical of corroles. However, synthesis of Ni complex of dioxacorrole 146 under similar reaction conditions was unsuccessful.
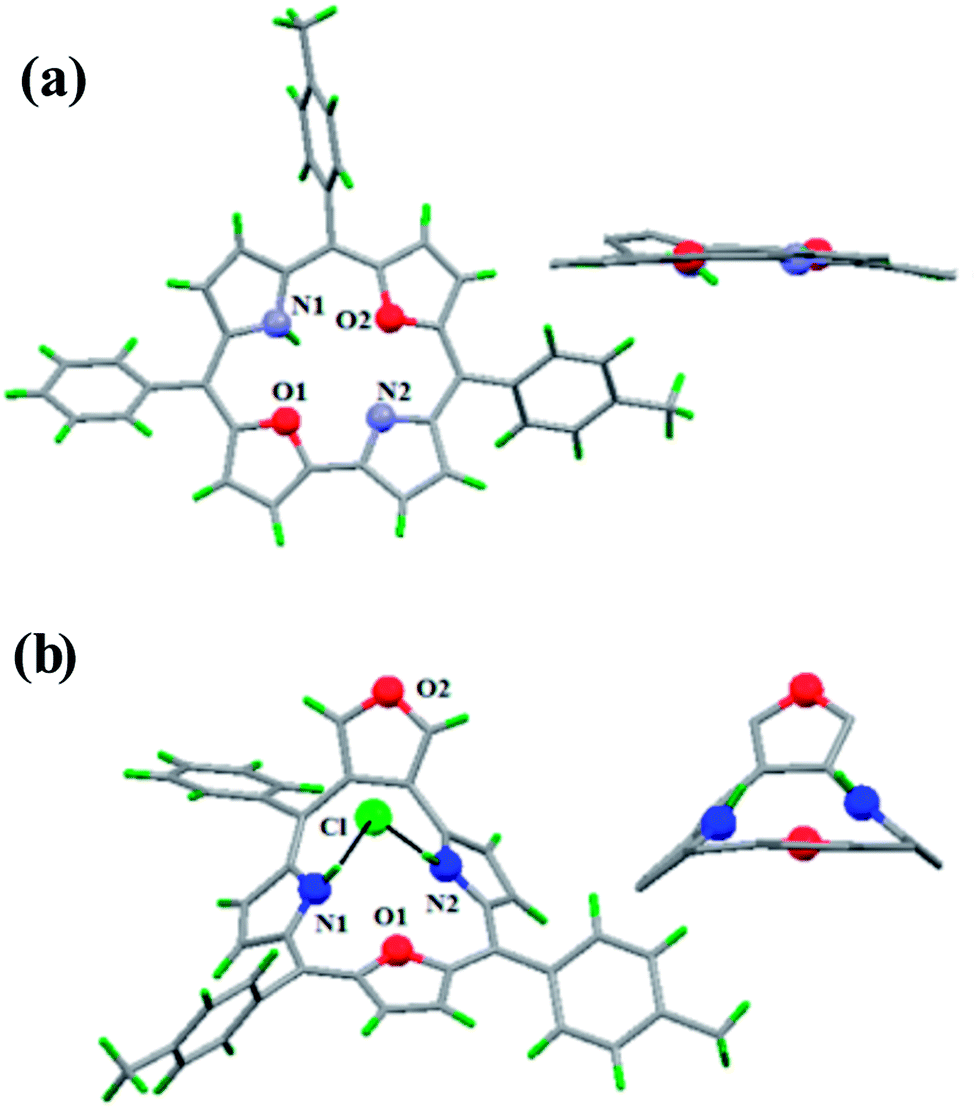
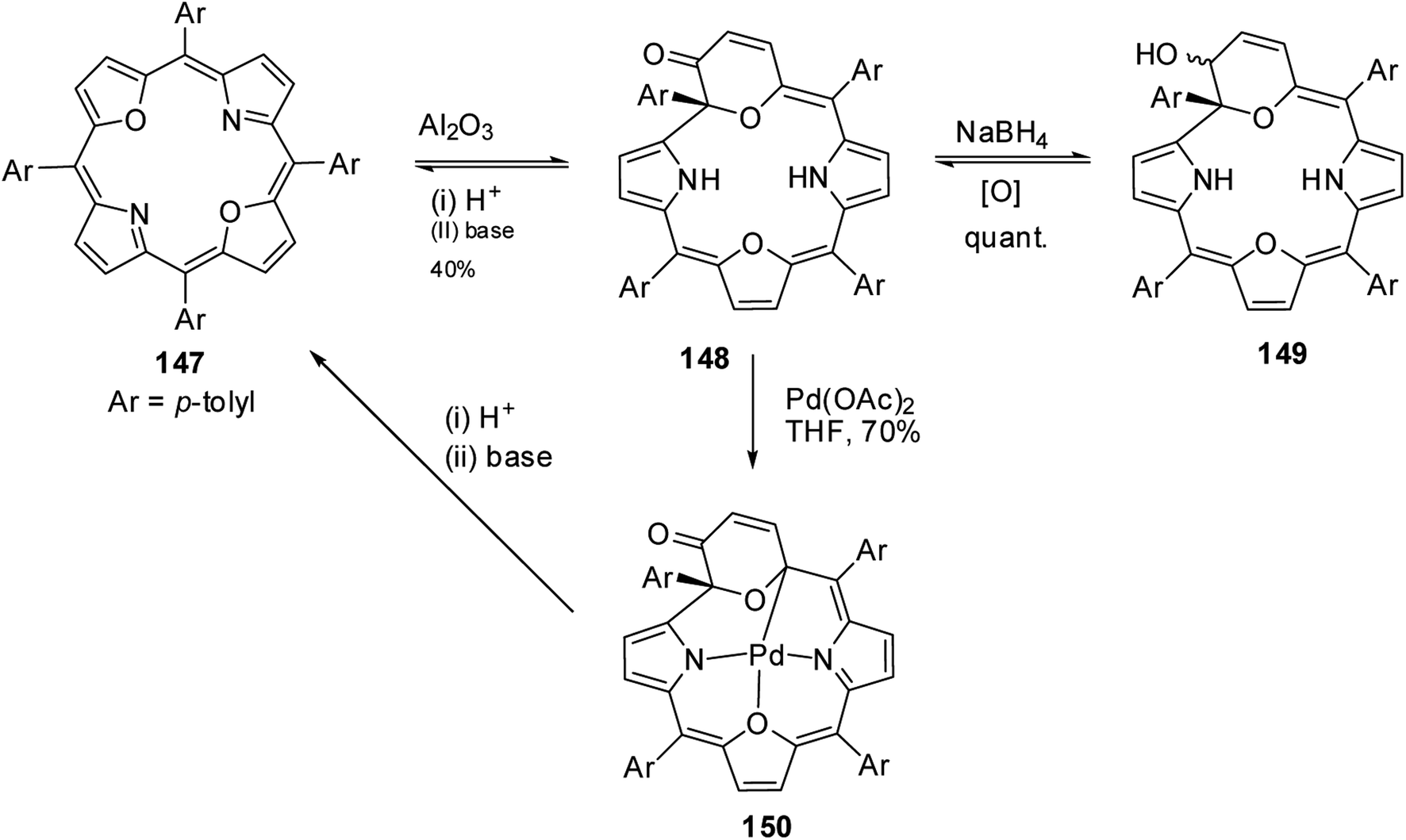
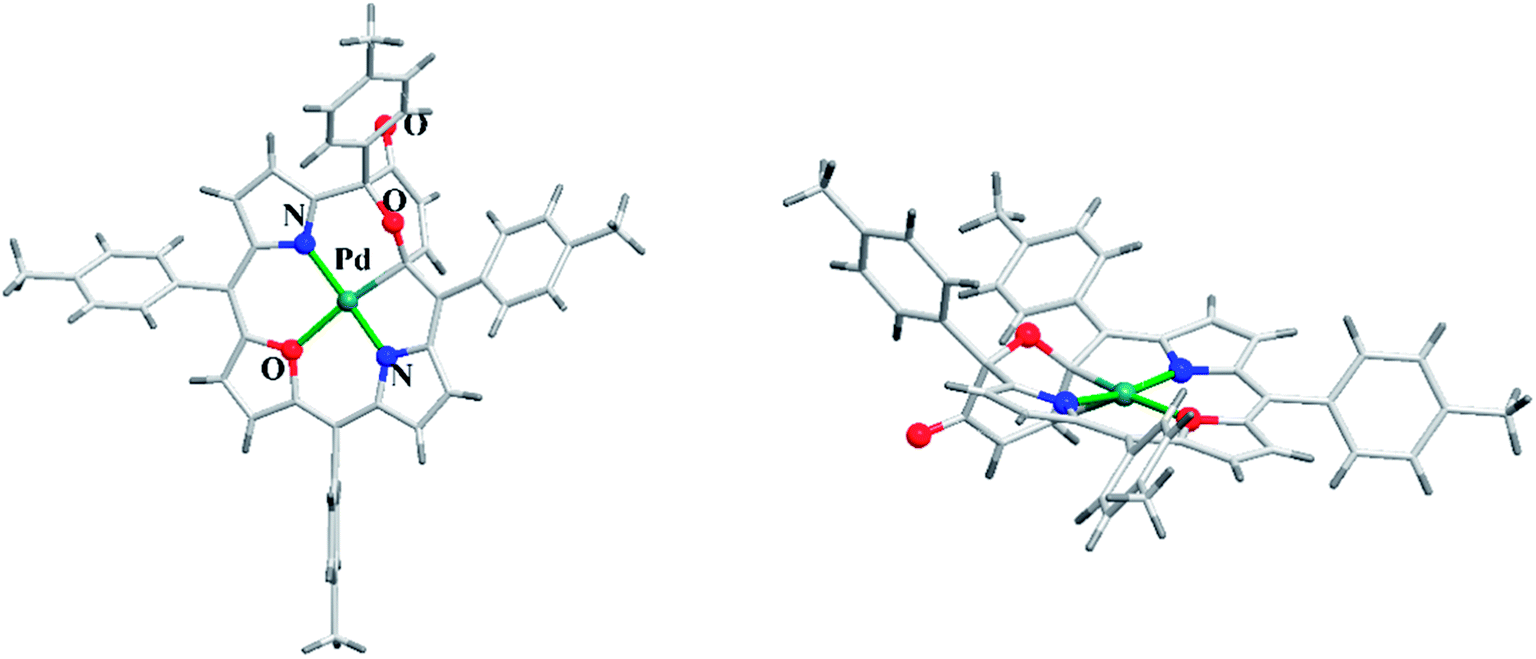
Latos-Grażyński and co-workers41 reported a unique example of 3-pyranone dioxacorrole 148 by adopting an unprecedented route of Achmatowicz rearrangement in the tetrarayl-21,23-dioxaporphyrin 147 (Scheme 33). The aromatic, symmetrical tetratolyl-21,23-dioxaporphyrin 147 when placed on the basic alumina G II converts to a green compound which was eluted with dichloromethane and identified as 3-pyranone dioxacorrole 148 by detailed 1D & 2D NMR spectroscopy and mass spectral analysis. The formation of 148 was reversible and the acid treatment of 148, regenerated tetraaryl-21,23-dioxacorrole 147 without the need of any reducing agent. The 3-pyranone dioxacorrole 148 readily forms Pd(II) complex upon treatment with Pd(OAc)2. The X-ray structure obtained for Pd(II) complex of 3-pyranone dioxacorrole 150 showed that Pd(II) ion was in [CNON] coordination environment and forms an organometallic complex (Fig. 22). The coordination of Pd(II) ion forces the 3-pyranone dioxacorrole ligand to acquire an extraordinary conformation in which the pyranone ring was almost perpendicular to the plane of the rest of the macrocycle.
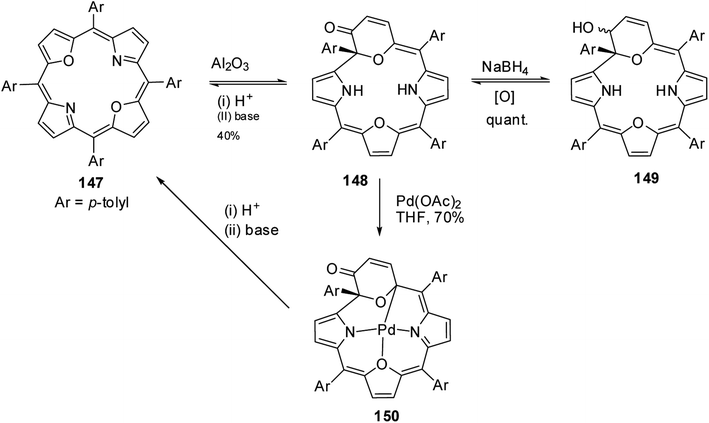 |
| | Scheme 33 Synthesis of 3-pyranone dioxacorrole 148 and its Pd(II) derivative 150. | |
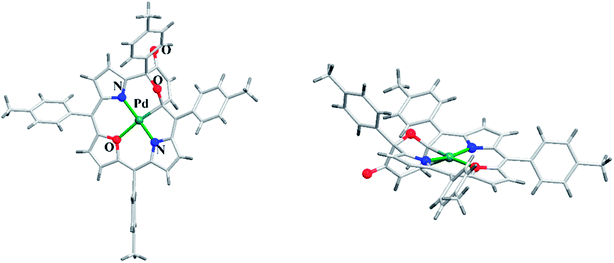 |
| | Fig. 22 Top and side view of X-ray crystal structures of 150 (CCDC 859245). | |
3.9. N-confused and neo-confused corroles
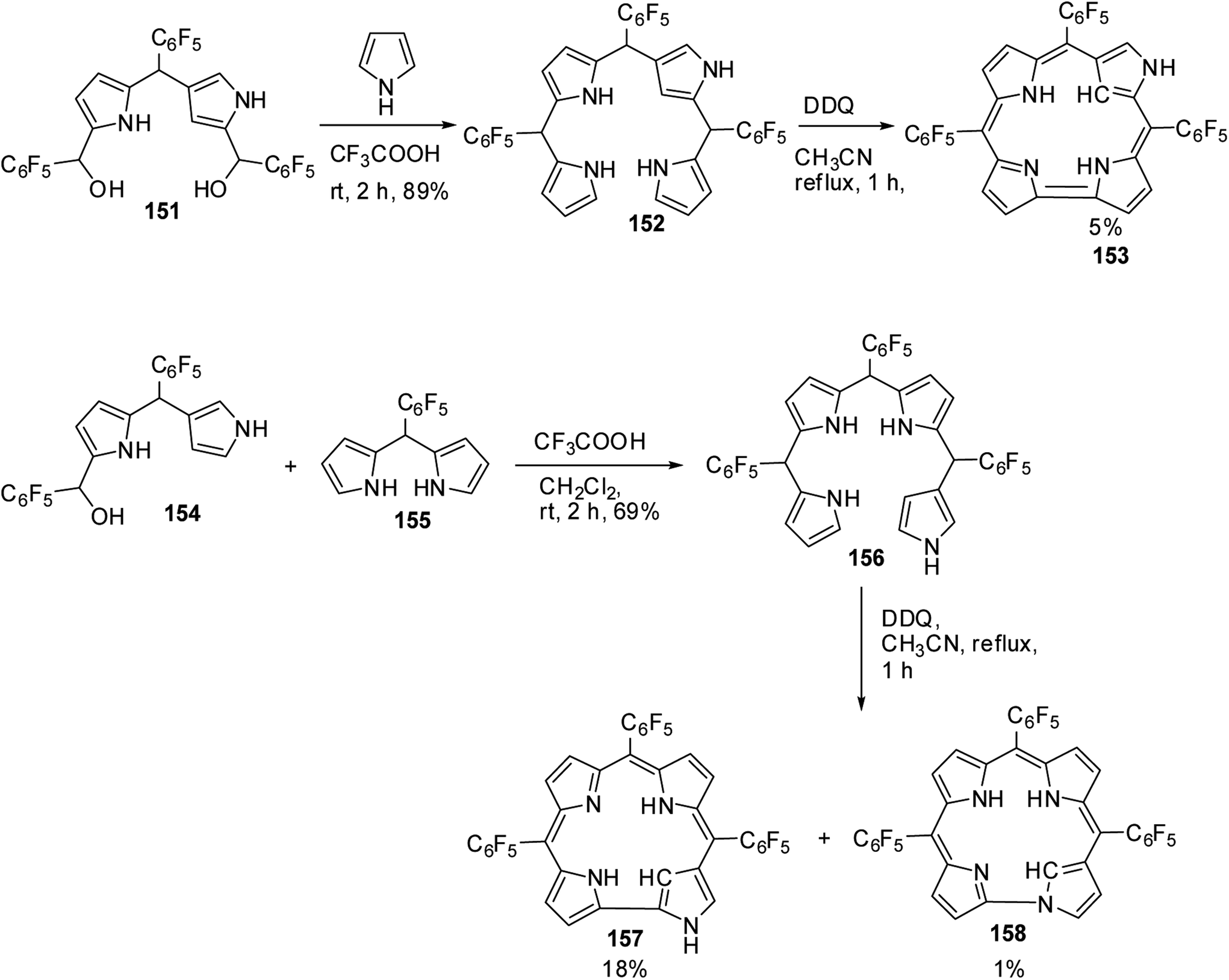
Furuta and co-workers42 reported the synthesis of corrole isomers 153, 157 and 158 where one of the pyrrole rings adopted a confused conformation (Scheme 34). The N-confused corroles 153 and 157 displayed a relatively sharp singlet corresponding to the interior CH at −0.91 and 1.84 ppm respectively in their 1H NMR spectra. The inner and peripheral NH protons of 153 and 157 displayed resonance signals in upfield and downfield regions respectively throwing light on their aromatic nature. However, the corrole 157 was relatively less aromatic than 153.
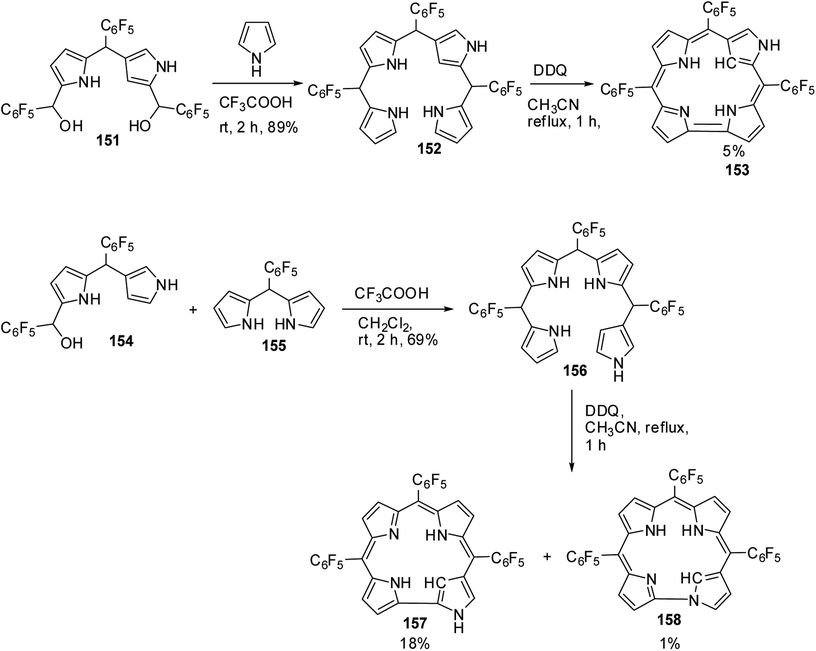 |
| | Scheme 34 Synthesis of N-confused corroles 153, 157 and norrole 158. | |
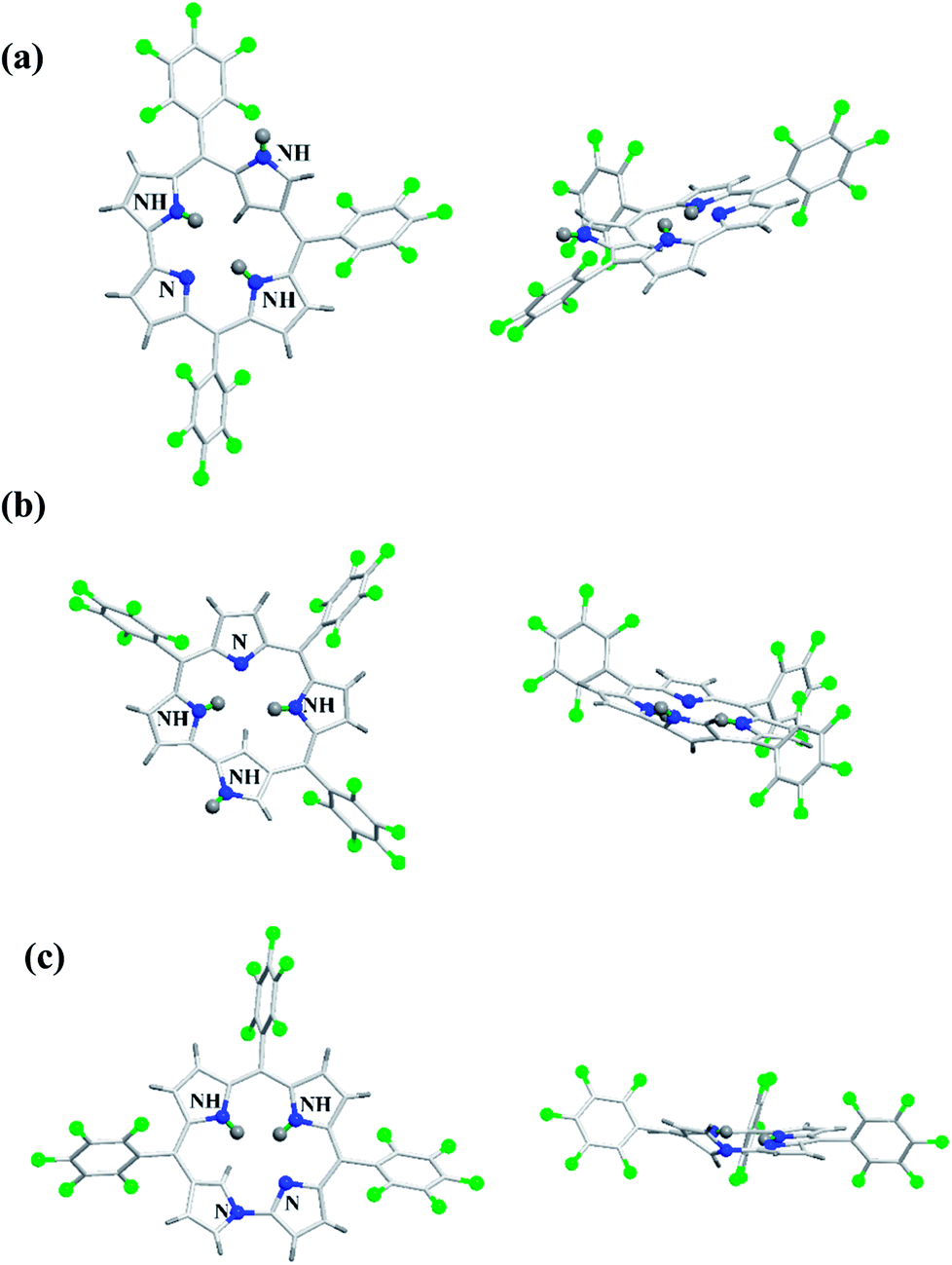
The neo-confused corrole (norrole) 158 displayed a unique resonance signal at δ = 1.21 ppm for β-pyrrole CH in its 1H NMR spectrum. The X-ray structures for N-confused corroles 153, 157 and norrole 158 is shown in Fig. 23. The confused pyrrole moiety in 153 and 157 was found to be of amino type and was significantly tilted from the corrole mean plane. The confused corroles 153, 157 and 158 displayed dramatic bathochromic shifts in their absorption bands compared to parent corrole. The neo-confused corrole 158 showed highest quantum yield compared to N-confused corroles 153 and 157 in dichloromethane (Fig. 25).
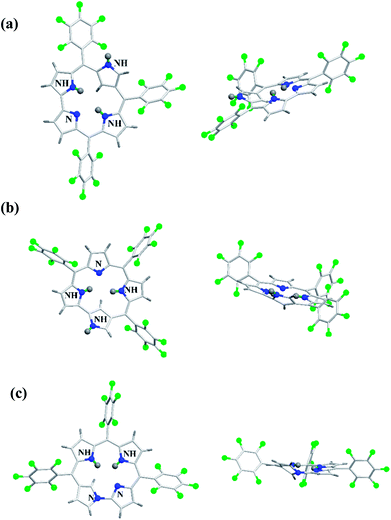 |
| | Fig. 23 Top and side view of X-ray crystal structures of (a) 153 (CCDC 719319) (b) 157 (CCDC 719320) (c) 158 (CCDC 719321). | |
The other striking feature of these confused corroles was their large Stokes shift values (774–1445 cm−1) compared to the corresponding regular corrole (221 cm−1). The N-confused corroles 153, 157 and 158, by virtue of presence of NH group on confused pyrrole, displayed anion binding properties through hydrogen bonding.
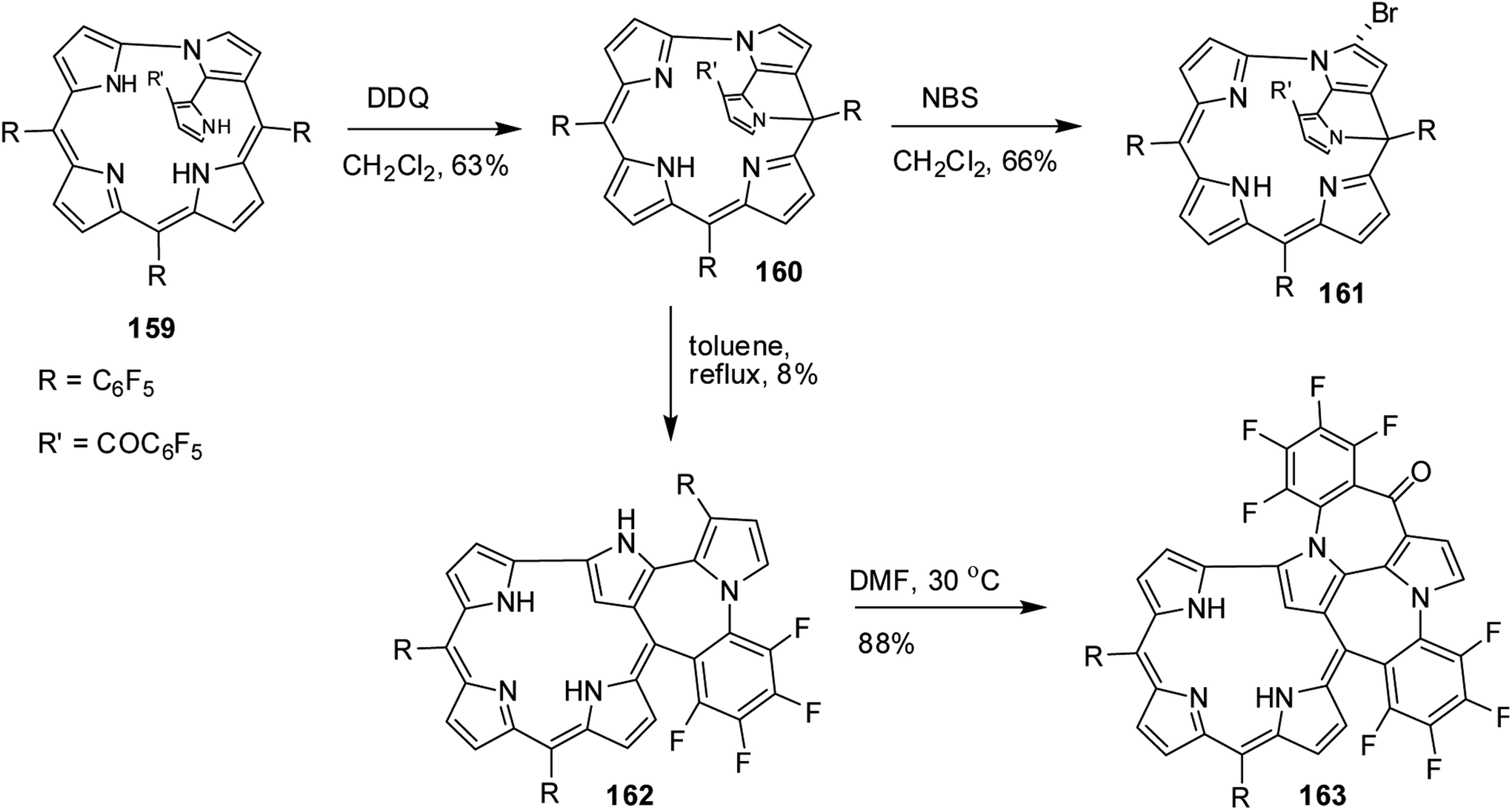
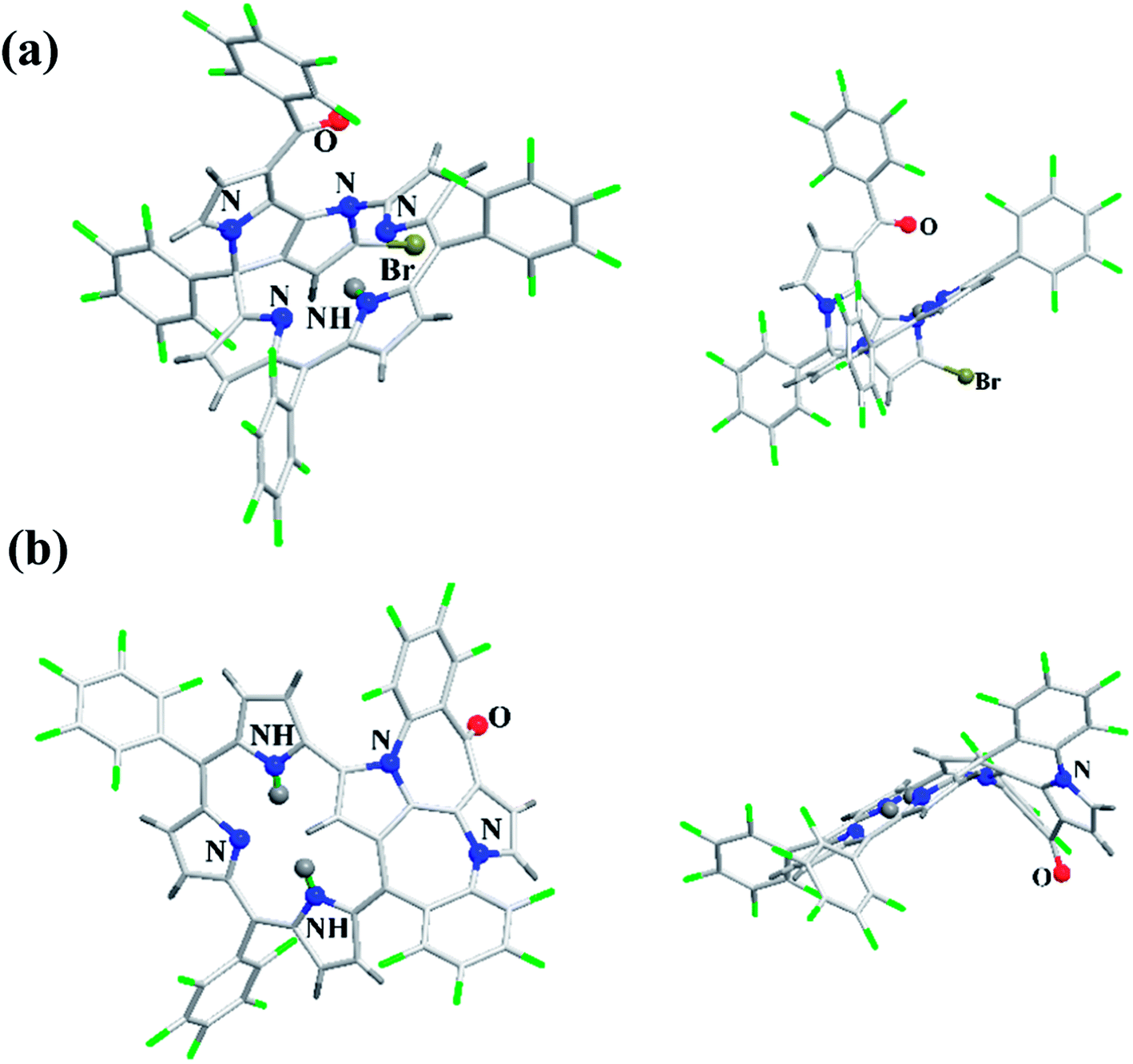
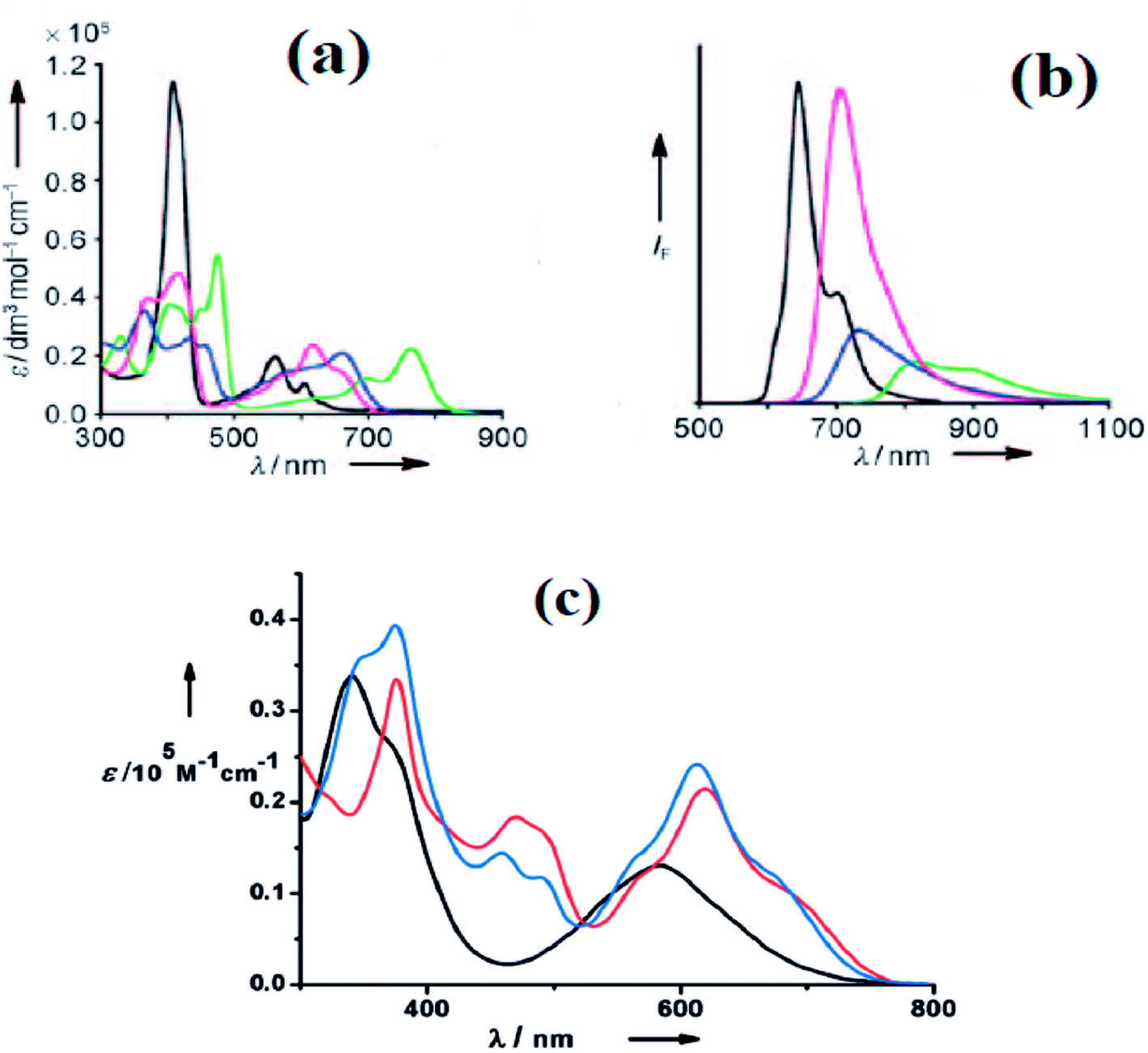
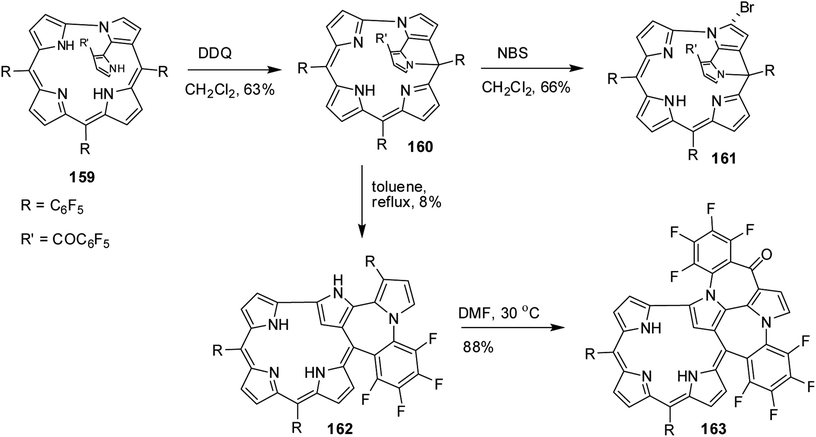
Furuta, Xie and co-workers43 recently disclosed a skeletal transformation reactions of norrole 159 (ref. 44) to its non-aromatic isomer isonorrole 160 and subsequently to N-confused corroles containing one and two N-CAr-fused ring 162 and 163 respectively (Scheme 35). The authors explored the reactivity of neo-confused corrole 159 containing an appended pyrrole moiety to generate these interesting molecules. The 1H NMR spectrum of 160 revealed decrease in number of resonance signals for NH protons confirming the oxidative dehydrogenation. The pyrrolic β-H signals in 160 were upfield shifted compared to 159 as an indication of its nonaromatic character. The bromination of 160 was regioselective and yielded brominated macrocycle 161 (Scheme 35). The compounds 162 and 163 were obtained respectively by refluxing 160 in toluene for ten hours and by treating 162 in DMF at 30 °C. The compounds 162 and 163 displayed 1H NMR spectral patterns typical for aromatic N-confused corroles. The X-ray structures of 161 and 163 are shown in Fig. 24. The absorption spectra of 160, 162 and 163 revealed two sets of broad bands in the range 340–380 nm and 460–750 nm, respectively (Fig. 25). The red shifted absorption bands of 162 and 163 compared to 160 is consistent with their respective aromatic nature. Surprisingly, corrole 160, despite being non-aromatic, showed smallest HOMO–LUMO gap compared to 162 and 163.
 |
| | Scheme 35 Synthesis of singly and doubly N-CAr-fused N-Confused corroles 162 and 163. | |
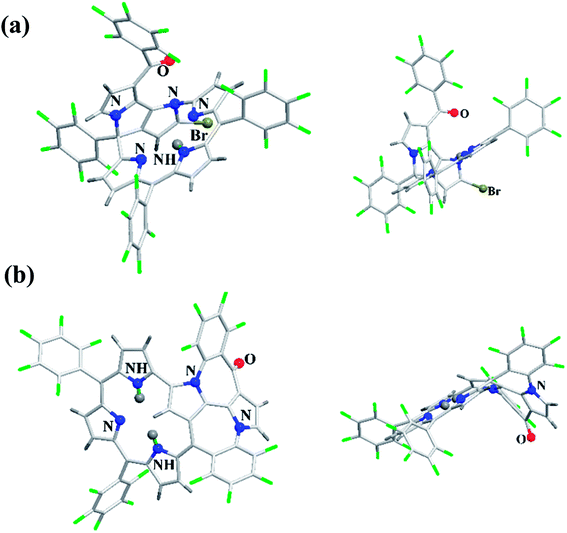 |
| | Fig. 24 Top and side view of X-ray crystal structures of (a) 161 (CCDC 1434096) (b) 163 (CCDC 1434097). | |
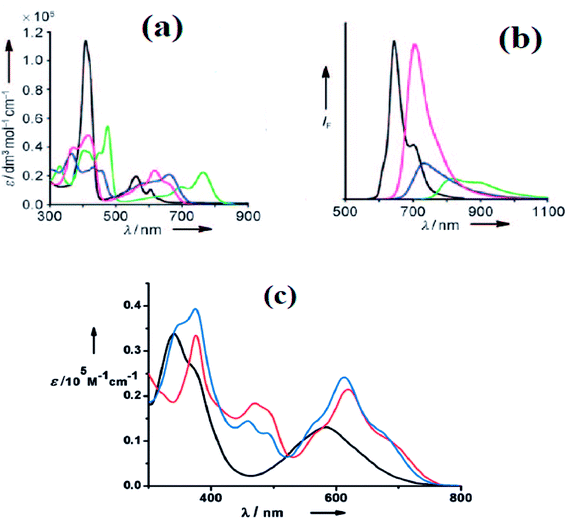 |
| | Fig. 25 (a) Absorption spectra of normal corrole (black), 153 (green), 157 (blue) and 158 (red) (b) their emission spectra (λex = 634, 764, 662, 659 nm for normal corrole, 153, 157 and 158 respectively) (c) absorption spectra of ring fused norrole 160 (black), 162 (red) and 163 (blue) recorded in dichloromethane. Reproduced from ref. 42 and 43 with permission. Copyright© 2011 and 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
4. Conclusions and outlook
In this article, the synthesis, structure and properties of heterocorroles containing heteroatom(s) at the core and also at a meso-position are described. It is evident from the discussion that, although the first examples of heterocorroles were reported long ago, the measurable growth in this area has happened only since 1999. The incorporation of heteroatom on macrocycle's aromatic conjugation pathway has imparted several new properties to heterocorroles. Owing to their contracted cavity size and presence of two inner ionisable NH atoms, heterocorroles have behaved as appropriate ligands to study coordination chemistry of porphyrins. The N-confused corroles and their isomer such as norrole have potential to exhibit novel coordination chemistry owing to the presence of carbon atom in the macrocycle's core. The near-infrared absorption and emission features of 10-silacorroles make them potential candidates for light harvesting applications. However, compared to heteroporphyrins, the progress in heterocorrole chemistry is rather slow presumably due to the difficulty involved in their synthesis and their inherent instability. The design of newer chemical synthesis of heterocorrole derivatives, their conjugates and multichromophoric systems containing heterocorroles would be an interesting proposition. The synthesis of heterocorroles containing heteroatom at a meso-position and also at the core without disrupting the aromaticity of the macrocycle will be interesting from their physico-chemical properties and coordination chemistry perspective. We are optimistic that such efforts will be undertaken in near future and the true potential of heterocorroles for various applications will be unveiled.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Authors wish to thank all the researchers who have contributed to the growth of heterocorrole chemistry. B. U. thanks UGC for the fellowship. V. S. S. thanks Science and Engineering Research Board (SERB), Govt. of India for a research grant (File no. YSS/2015/001557/CS). M. R. acknowledges financial support from Science and Engineering Research Board (SERB), Govt. of India (Project No. EMR/2015/002196).
References
- H. R. Harrison, O. J. R. Hodder and D. C. Hodgkin, J. Chem. Soc. B, 1971, 640–645 RSC.
- R. Orłowski, D. Gryko and D. T. Gryko, Chem. Rev., 2017, 117, 3102–3137 CrossRef PubMed , and references cited therein.
- I. Aviv-Harel and Z. Gross, Coord. Chem. Rev., 2011, 255, 717–736 CrossRef , and references cited therein.
- R. D. Teo, J. Y. Hwang, J. Termini, Z. Gross and H. B. Gray, Chem. Rev., 2017, 117, 2711–2729 CrossRef PubMed , and references cited therein.
- L. Flamigni and D. T. Gryko, Chem. Soc. Rev., 2009, 38, 1635–1646 RSC , and references cited therein.
- I. Aviv-Harel and Z. Gross, Chem.–Eur. J., 2009, 15, 8382–8394 CrossRef PubMed , and references cited therein.
- T. Chatterjee, V. S. Shetti, R. Sharma and M. Ravikanth, Chem. Rev., 2017, 117, 3254–3328 CrossRef PubMed , and references cited therein.
- D. T. Gryko, J. P. Fox and D. P. Goldberg, J. Porphyrins Phthalocyanines, 2004, 08, 1091–1105 CrossRef , and references cited therein.
- A. W. Johnson, I. T. Kay and R. Rodrigo, J. Chem. Soc., 1963, 2336–2342 RSC.
- M. Horie, Y. Hayashi, S. Yamaguchi and H. Shinokubo, Chem.–Eur. J., 2012, 18, 5919–5923 CrossRef PubMed.
- H. Omori, S. Hiroto and H. Shinokubo, Chem. Commun., 2016, 52, 3540–3543 RSC.
- H. Omori, S. Hiroto and H. Shinokubo, Org. Lett., 2016, 18, 2978–2981 CrossRef PubMed.
- M. J. Broadhurst, R. Grigg and A. W. Johnson, J. Chem. Soc., Perkin Trans. 1, 1972, 1124–1135 RSC.
- M. Bröring, F. Brégier, E. C. Tejero, C. Hell and M. C. Holthausen, Angew. Chem., Int. Ed., 2007, 46, 445–448 CrossRef PubMed.
- T. Ito, Y. Hayashi, S. Shimizu, J.-Y. Shin, N. Kobayashi and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 8542–8545 CrossRef PubMed.
- M. Bröring, S. Köhler and C. Kleeberg, Angew. Chem., Int. Ed., 2008, 47, 5658–5660 CrossRef PubMed.
- D. Sakow, B. Böker, K. Brandhorst, O. Burghaus and M. Bröring, Angew. Chem., Int. Ed., 2013, 52, 4912–4915 CrossRef PubMed.
- H. Kamiya, T. Kondo, T. Sakida, S. Yamaguchi and H. Shinokubo, Chem.–Eur. J., 2012, 18, 16129–16135 CrossRef PubMed.
- N. Wachi, T. Kondo, S. Ito, S. Hiroto, J.-Y. Shin and H. Shinokubo, J. Porphyrins Phthalocyanines, 2014, 18, 675–678 CrossRef.
- R. He, H. Yue and J. Kong, Molecules, 2017, 22, 1400 CrossRef PubMed.
- D. Sakow, D. Baabe, B. Böker, O. Burghaus, M. Funk, C. Kleeberg, D. Menzel, C. Pietzonka and M. Bröring, Chem.–Eur. J., 2014, 20, 2913–2924 CrossRef PubMed.
- M. Bröring, S. Köhler and C. Pietzonka, J. Porphyrins Phthalocyanines, 2012, 16, 641–650 CrossRef.
- J. Rösner, B. Cordes, S. Bahnmüller, G. Homolya, D. Sakow, P. Schweyen, R. Wicht and M. Bröring, Angew. Chem., Int. Ed., 2017, 56, 9967–9970 CrossRef PubMed.
- H. Omori, S. Hiroto and H. Shinokubo, Chem.–Eur. J., 2017, 23, 7866–7870 CrossRef PubMed.
- V. S. Shetti, U. R. Prabhu and M. Ravikanth, J. Org. Chem., 2010, 75, 4172–4182 CrossRef PubMed.
- I. Gupta and M. Ravikanth, J. Org. Chem., 2004, 69, 6796–6811 CrossRef PubMed.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827–836 CrossRef.
- C.-H. C. Lee, W.-S. Cho, J.-W. Ka, H.-J. Kim and P. H. Lee, Bull. Korean Chem. Soc., 2000, 21, 429–433 Search PubMed.
- W.-S. Cho and C.-H. Lee, Tetrahedron Lett., 2000, 41, 697–701 CrossRef.
- A. Ghosh, T. Chatterjee, W.-Z. Lee and M. Ravikanth, Org. Lett., 2013, 15, 1040–1043 CrossRef PubMed.
- S. Janusz, L. Latos-Grażyński and L. Szterenberg, Chem.–Eur. J., 2008, 14, 4861–4874 CrossRef PubMed.
- S. J. Narayanan, B. Sridevi, T. K. Chandrashekar, U. Englich and K. Ruhlandt-Senge, Org. Lett., 1999, 1, 587–590 CrossRef.
- J. Sankar, H. Rath, V. PrabhuRaja, T. K. Chandrashekar and J. J. Vittal, J. Org. Chem., 2004, 69, 5135–5138 CrossRef PubMed.
- H. Kalita, D. Kalita, W.-Z. Lee, J. Bellare and M. Ravikanth, Chem.–Eur. J., 2014, 20, 10404–10413 CrossRef PubMed.
- H. Kalita and M. Ravikanth, Chem.–Eur. J., 2015, 21, 7399–7402 CrossRef PubMed.
- S. Venkatraman, R. Kumar, J. Sankar, T. K. Chandrashekar, K. Sendhil, C. Vijayan, A. Kelling and M. O. Senge, Chem.–Eur. J., 2004, 10, 1423–1432 CrossRef PubMed.
- B. Sridevi, S. Jeyaprakash Narayanan, T. K. Chandrashekar, U. Englich and K. Ruhlandt-Senge, Chem.–Eur. J., 2000, 6, 2554–2563 CrossRef.
- J. Sankar, H. Rath, V. Prabhuraja, S. Gokulnath, T. K. Chandrashekar, C. S. Purohit and S. Verma, Chem.–Eur. J., 2007, 13, 105–114 CrossRef PubMed.
- M. J. Broadhurst, R. Grigg and A. W. Johnson, J. Chem. Soc. D, 1969, 23–24 RSC.
- M. Pawlicki, L. Latos-Grażyński and L. Szterenberg, J. Org. Chem., 2002, 67, 5644–5653 CrossRef PubMed.
- M. Pawlicki, D. Bykowski, L. Szterenberg and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2012, 51, 2500–2504 CrossRef PubMed.
- K. Fujino, Y. Hirata, Y. Kawabe, T. Morimoto, A. Srinivasan, M. Toganoh, Y. Miseki, A. Kudo and H. Furuta, Angew. Chem., Int. Ed., 2011, 50, 6855–6859 CrossRef PubMed.
- M. Li, P. Wei, M. Ishida, X. Li, M. Savage, R. Guo, Z. Ou, S. Yang, H. Furuta and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 3063–3067 CrossRef PubMed.
- Y. Xie, P. Wei, X. Li, T. Hong, K. Zhang and H. Furuta, J. Am. Chem. Soc., 2013, 135, 19119–19122 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Mangalampalli Ravikanth
*b and
Mangalampalli Ravikanth