
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall molecule generators of biologically reactive sulfur species
Prerona Bora
,
Preeti Chauhan
,
Kundansingh A. Pardeshi
and
Harinath Chakrapani
 *
*
Department of Chemistry, Indian Institute of Science Education and Research Pune, Pune 411 008, Maharashtra, India. E-mail: harinath@iiserpune.ac.in
First published on 31st July 2018
Abstract
Sulfur metabolism is integral to cellular growth and survival. The presence of a wide range of oxidation states of sulfur in biology coupled with its unique reactivity are some key features of the biology of this element. In particular, nearly all oxidation states of sulfur not only occur but are also inter-convertible. In order to study the chemical biology of reactive sulfur species, tools to reliably detect as well as generate these species within cells are necessary. Herein, an overview of strategies to generate certain reactive sulfur species is presented. The donors of reactive sulfur species have been organized based on their oxidation states. These interesting small molecules have helped lay a strong foundation to study the biology of reactive sulfur species and some may have therapeutic applications in the future as well.
 Prerona Bora | Prerona Bora completed her Bachelor's (2010–2013) in chemistry from Hindu College, University of Delhi and her Master's (2013–2015) in chemistry from Tezpur University. She has been enrolled in the PhD programme at Indian Institute of Science Education and Research (IISER) Pune from 2016. She is currently working under the supervision of Dr Harinath Chakrapani and her research interest involves synthesis and evaluation of small molecule sulfur based signaling molecules. |
 Preeti Chauhan | Preeti Chauhan has completed her BSc (2009–2012) and MSc (2012–2014) in chemistry from Hindu College, University of Delhi. She has been enrolled in the PhD programme at Indian Institute of Science Education and Research (IISER) Pune from August 2014. She is currently working towards her PhD under the supervision of Dr Harinath Chakrapani. Her research interest is to design and synthesize small molecule based tools for the delivery of hydrogen sulfide (H2S) in a cellular system to explore the therapeutic potential of this gasotransmitter. |
 Kundansingh A. Pardeshi | Kundansingh Pardeshi has completed his Bachelors (2005–2008) and masters (2008–2010) in chemistry from Dr B. Ambedkar Marathwada University, Aurangabad, Maharashtra. He enrolled in PhD programme at Indian Institute of Science Education and Research (IISER) Pune in Aug 2011. He is currently working under the supervision of Dr Harinath Chakrapani. His research interest involves design, synthesis and evaluation of bioactivable donors of sulfur dioxide (SO2). |
 Harinath Chakrapani | Harinath Chakrapani completed his undergraduate and post-graduate studies in Chemistry from Loyola College (1994–97) and Indian Institute of Technology Madras (1997–99), respectively. In the fall of 1999, he moved to Duke University, USA to pursue his doctoral studies, which he completed under the supervision of Prof. Eric J. Toone in Dec. 2005. His post-doctoral research work was carried out at Wake Forest University, where he worked with Prof. S. Bruce King and the National Cancer Institute, under the mentorship of Dr Larry K. Keefer. He joined IISER Pune in July 2009 and is currently Associate Professor. |
Introduction
Sulfur is an essential element for life and plays critical roles in cellular functioning and growth. The naturally occurring amino acids cysteine and methionine are important components of numerous proteins. Since the emergence of life on earth, sulfur chemistry has been intricately involved in a multitude of metabolic pathways, the versatility of which can be attributed to the interconvertible oxidation states of sulfur (Fig. 1).1 Reactive sulfur species are sulfur-derived entities that when generated within cells can mediate biological processes and be cytoprotective in nature and are hence deployed by cells for defence.2,3 In contrast, reactive sulfur species can also damage cellular components, typically at elevated concentrations. The emergence of these species parallels the development of reactive oxygen species (ROS) that cause oxidative stress and reactive nitrogen species (RNS) that lead to nitrosative stress,4 from sulfate, which has an oxidation state of +6, to thiol/hydrogen sulfide, whose oxidation state is −2 with several intermediary states. Thus, nearly all these oxidation states occur in normal cells and may have critical roles to play in the functioning of cells.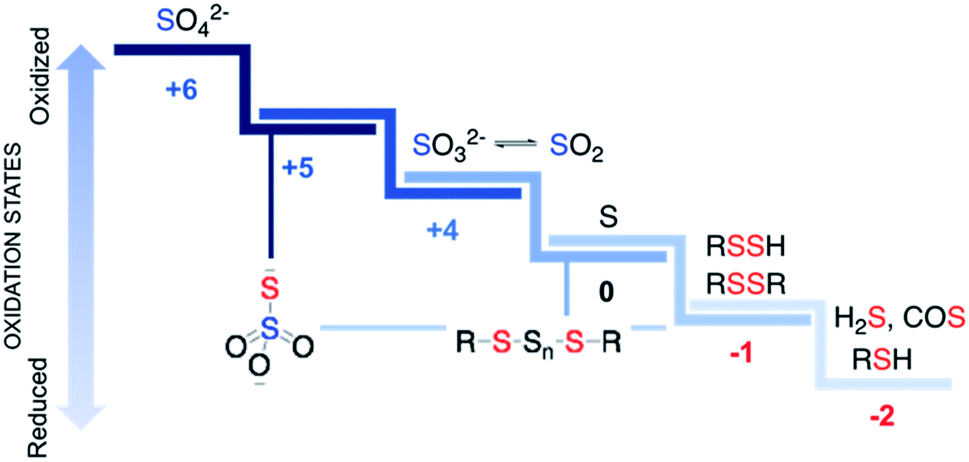 | ||
| Fig. 1 Some biologically relevant reactive sulfur species. Red and blue are used to designate sulfur in negative and positive oxidation states respectively. Thiosulfate (S2O32−) and polysulfides have sulfurs in different oxidation states. | ||
Reactive sulfur species
The lowest oxidation state of sulfur includes reactive species like thiols, hydrogen sulfide (H2S) and carbonyl sulfide (COS). H2S is obtained from L-cysteine via the transsulfuration pathway involving cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE) and 3-mercaptopyruvate sulfur transferase (3-MST).5–8 It can interact metal centres in haemoglobin9 and cytochrome c oxidase,10 act as an antioxidant or carry out downstream signaling by S-sulfhydration of proteins.11 H2S can be oxidized by the mitochondrial flavo-protein sulfide quinone oxidoreductase (SQR) forming a transient persulfide intermediate that can potentially S-sulfhydrate proteins or act as a substrate for the sulfurtransferase rhodanese to form thiosulfate.12,13Sulfite, which exists in equilibrium with SO2 can be generated by metabolism of L-cysteine by cysteine dioxygenase (CDO) and subsequently by aspartate aminotransferase (AAT).14 It can also be formed from thiosulfate in the presence of thiosulfate sulfurtransferase (TST). Interaction of sulfite with proteins via S-sulfonation is the most common pathway. However, the mechanisms by which SO2 elicits varied physiological roles are yet to be completely evaluated.15,16 Sulfite oxidation is catalysed by sulfite oxidase (SO) to form sulfate that is eventually excreted in the urine.17 Sulfate can be obtained from the diet as well, taken up by the cell through sulfate transporters. Sulfate assimilation in all living organisms is activated by the enzyme ATP sulfurylase to form adenosine-5′-phosphosulfate (APS) from ATP which subsequently gets converted to 3′-phosphoadenosine-5′-phosphosulfate (PAPS), leading to sulfonation and detoxification of xenobiotics (Fig. 2).18–20 Dissimilation of sulfate by reduction is quite common in microbes and plants, however in mammals this pathway is yet unknown. This manuscript has been arranged accordingly to address each of the oxidation states that occurs in cells and describe chemical approaches to generating these species within cells.
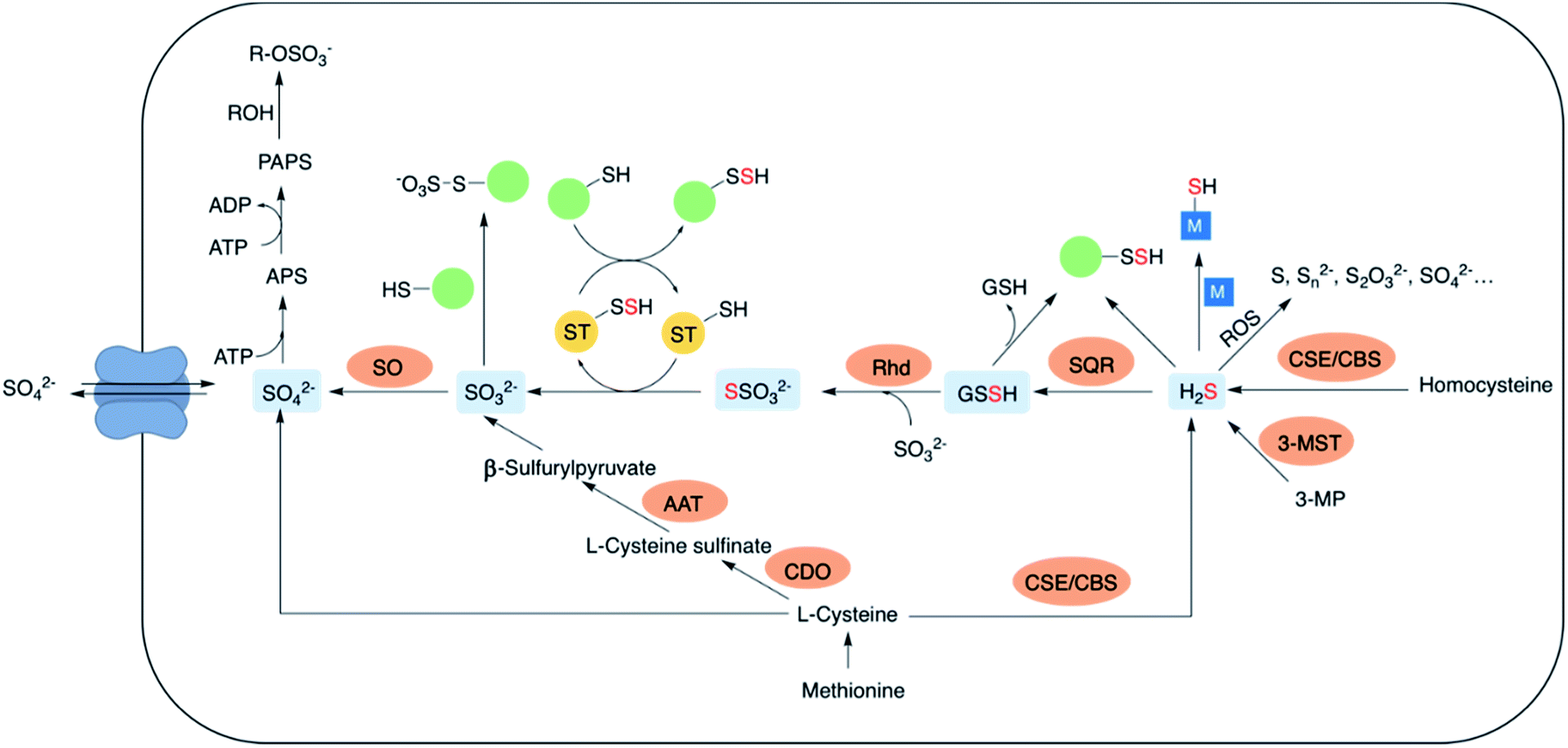 | ||
| Fig. 2 A partial ensemble of sulfur oxidation states and their distinct physiological roles. CBS: cystathionine-β-synthase; CSE: cystathionine-γ-lyase; 3-MST: 3-mercaptopyruvate sulfurtransferase; SQR: sulfide quinone oxidoreductase; Rhd: rhodanese; SO: sulfite oxidase; AAT: aspartate aminotransferase; CDO: cysteine dioxygenase; APS: adenosine-5′-phosphosulfate; PAPS: 3′-phosphoadenosine-5′-phosphosulfate. | ||
An important reactive sulfur species is hydrogen sulfide (H2S), which in the past two decades has become a major player in redox biology. Hydrogen sulfide (H2S) is an important gaseous form of sulfur known for its lethal nature and toxicity for centuries. However, over past 15 years it has emerged as an important signaling molecule along with nitric oxide (NO) and carbon monoxide (CO).21–31 Chemical sources of H2S is also proposed as an antioxidant and several small molecules are in various stages of pre-clinical evaluation for a number of indications. Being able to modulate levels of hydrogen sulfide may thus have tremendous potential in the treatment of certain pathophysiological conditions. A product of oxidation of hydrogen sulfide is sulfur dioxide (SO2), which is notorious for its environmental polluting activity, acid rain and allergic reactions in certain individuals.32,33 Its hydrated form, sulfite, is also used in the food industry as a preservative and an antioxidant.34–36 SO2 gas has also shown vasodilatory activity and may have regulatory roles as well.37
Thus, the fascinating chemical biology of the various redox forms of sulfur has triggered a lot of interest in systematically studying each of these forms in further detail. In order to do this, we need tools that can detect, generate as well as dissipate these species. Although most detection technologies have caveats associated with them, the use of multiple independent assays will help with enhanced reliability of detection of these species.
Specific dissipation (after generation) or inhibition of formation is another challenging problem. Most approaches have involved inhibiting a biosynthetic enzyme that catalyzes the formation of these species (in particular, hydrogen sulfide).38,39 Although a number of strategies are available, this remains an unsolved problem and merits further attention. In this review, we cover the various strategies to generate reactive sulfur species within cells. The major challenges are to develop carriers or caged compounds that are triggerable within cells with minimal byproduct toxicity.
Hydrogen sulfide
The production of H2S in cells is tightly regulated by the transsulfuration pathway enzymes. Therefore, abnormal changes in the level of H2S could lead to various pathological conditions, for example – neurological disorders, cardiovascular diseases, gastrointestinal disorders, inflammation etc. Exogenous administration of H2S under such conditions has proved beneficial which brings into picture the potential therapeutic effects of H2S. For example, exogenous administration of H2S attenuates myocardial ischemia reperfusion injury.40 Also, being a reducing agent, it readily reacts with reactive oxygen and nitrogen species (RONS) to reduce inflammation and therefore acts as an anti-inflammatory agent. H2S along with NSAIDs have been found to be more potent in treatment of gastrointestinal disorders as compared to NSAID alone.41,42 Therefore, to better explore the therapeutic potential of the gas and its mechanism of action, researchers from around the world have developed small molecule based H2S donors. Many of the donors are covered in some excellent reviews and will therefore be briefly discussed here.40,43,44 Hydrogen sulfide donors have been classified in three major categories as naturally occurring, hydrolysis based H2S donors, and triggerable H2S donors. In particular, we shall cover some recent developments in the use of carbonyl sulfide (COS) as a source of H2S and efforts to trigger the release of COS within cells.Naturally occurring polysulfides have been derived from allium plants like garlic and onion which have been known for their beneficial health effects for centuries.44 Consumption of garlic relates to the reduction in the risk of cardiovascular diseases such as high blood pressure, blood coagulation, and high cholesterol.45–47 These are rich source of organosulfur based polysulfides which are responsible for its pharmacological nature. Kraus and co-workers in 2007 showed that allicin which is produced upon crushing garlic rapidly decomposes to give diallyl based polysulfides – diallyl sulfide (DAS), diallyl disulfide (DADS) and diallyl trisulfide (DATS). Biological thiols like GSH and cysteine then act on these polysulfides to produce H2S (Fig. 3).48
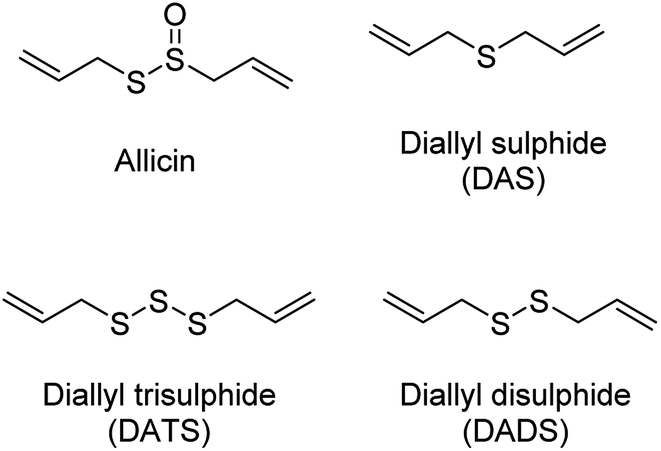 | ||
| Fig. 3 Naturally occurring H2S donors. | ||
Other sources of naturally occurring H2S donors include ovothiol from sea urchin eggs, leinamycin produced by streptomyces,49,50 and ergothioneine and lenthionine found in fungi.51 These natural products could act as potential antioxidant, antibacterial, antifungal and anti-cancer agents. The therapeutic potential of the naturally occurring H2S donors is yet to be completely evaluated.52,53
NaSH and Na2S are inorganic salts which upon dissolving in water release H2S.54 The salts are widely used as source of H2S but due to uncontrollable and burst release of sulfide, it is difficult to maintain a constant concentration in a cell and therefore are not considered ideal for studying the physiological and pharmacological effects of H2S. Lawesson's reagent is a chemical compound used for sulfurization reactions in organic synthesis.55,56 It is used as a source of sulfide by researchers due to slower rate of H2S release as compared to the inorganic salts.42,57 Thiophosphoryl chloride (PSCl3) is another inorganic salt that is known to produce H2S upon hydrolysis.58 However, the insoluble nature of these reagents limits its application.
Morpholin-4-ium 4 methoxyphenyl(morpholino) phosphinodithioate, GYY4137 a derivative of Lawesson's reagent, has better water solubility and produces H2S at a slower rate. GYY4137 has been widely and extensively used for studying (patho)physiological roles of H2S. Low level of H2S is maintained by GYY4137 even after 7 days.59,60 Although the donors exhibit H2S like activities but due to unavailability of appropriate negative controls the observed activities cannot be attributed to H2S production alone. Also, the rate of H2S release cannot be controlled due to lack of an appropriate handle. And site directed delivery is not possible with these donors. 1,2-Dithiole-3-thiones (DTTs) are yet another example of hydrolysis based H2S donors which are synthesized by sulfurization and dehydrogenation reaction of allylic methyl group. DTT coupled with NSAIDs have showed significant reduction in the gastrointestinal damage caused by the parent NSAID.61–64 5-(4-Hydroxyphenyl)-3H-1,2-dithiole-3-thione (ADT-OH) is a derivative of DTT and has also been used as a potent H2S donor. ADT-NSAID hybrids have also shown therapeutic potential (Fig. 4a)41,65,66 The mechanism of action of the donors remains to be completely elucidated.
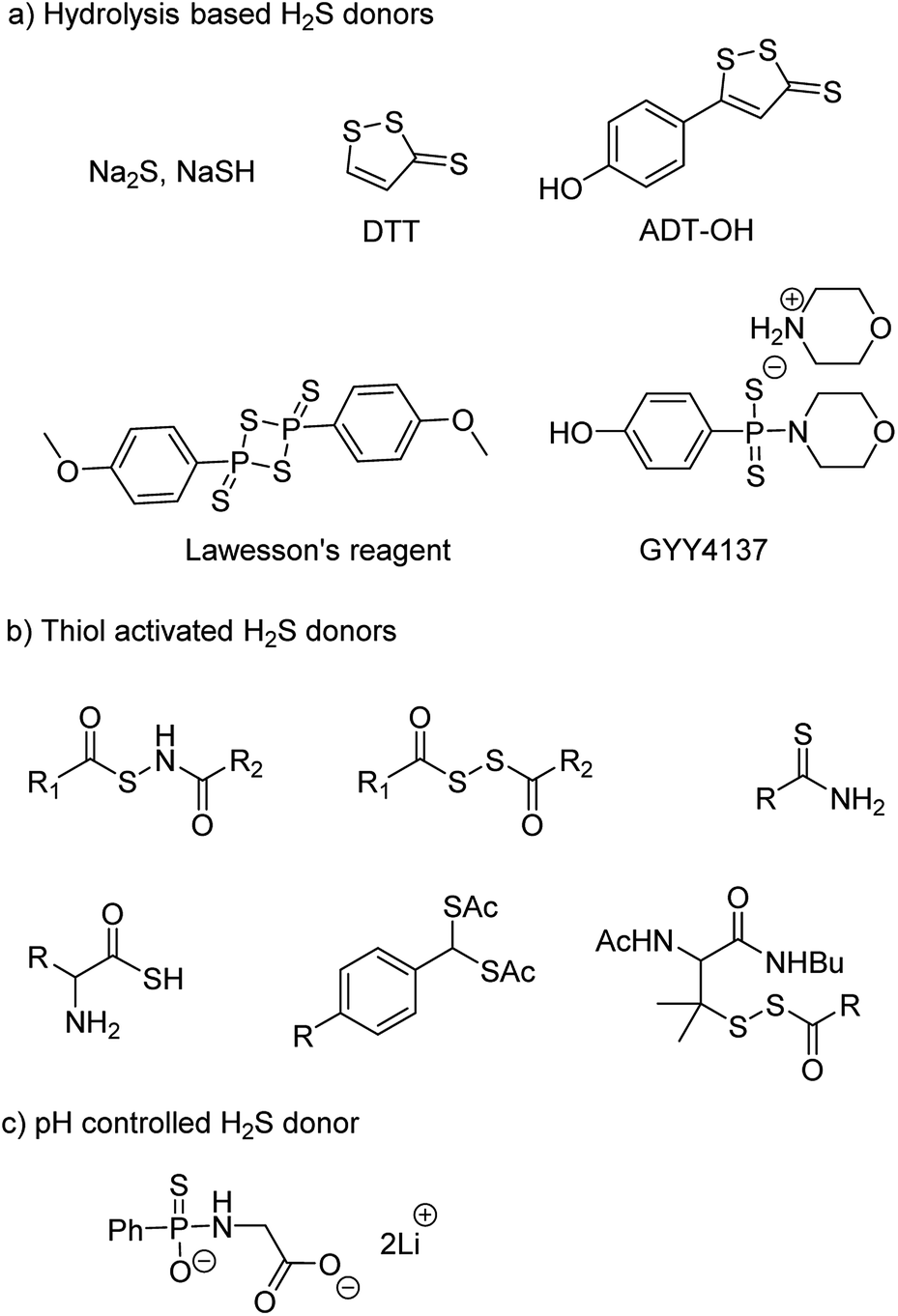 | ||
| Fig. 4 Representative examples of chemically activated H2S donors. | ||
The concept of controllable H2S donors was first introduced by Ming Xian and co-workers in the year 2011. They reported a series of N-(benzoylthio)benzamide derivatives which upon activation by cysteine form perthiols and further hydrolyse to produce H2S.67 Following a similar strategy, the same group reported cysteine perthiols protected with an acyl group. Cysteine perthiols are sensitive to glutathione (GSH) or cysteine and therefore get deprotected in the presence of thiols to yield H2S.68 Having studied N-SH and S-SH based donors, dithioperoxyanhydrides were introduced in 2013 by Galardon and co-workers as thiol activated H2S donors.69 Acylpersulfide was proposed as the key intermediate. Arylthioamides were also reported and categorized as thiol activated H2S donors.70 However, the yield of H2S produced from these donors was very low. Thiol activated gem dithiols were also reported as H2S donors (Fig. 4b).71 Due to large abundance of thiols in nearly all cells, the above-mentioned donors have limited potential for site-directed delivery of H2S.
In an effort to further improve upon the existing donors and overcome the limitations, Ming Xian and co-workers reported pH controlled H2S release based on GYY4137 scaffold (Fig. 4c).72 Donors were found to release H2S only at pH 5.0 and were stable at neutral and slightly alkaline pH. In vivo studies demonstrated cardioprotective nature of these donors at low doses of 50 and 100 μg kg−1.
With an aim to achieve localized delivery of H2S Ming Xian and co-workers reported light activated H2S donors. Upon irradiation with UV light, compounds decomposed to release H2S.73 Nakagawa and co-workers also contributed to the field by reporting thioether based scaffolds which are activated by UV light to produce H2S.74 However, the toxicity caused by UV light limits the use of these donors in a physiological system. Visible light driven H2S donors were reported in the year 2013 by You and co-workers where 1,3-diphenylisobenzothiophenone (DPBT) reacts with singlet oxygen to produce unstable endoperoxide which promptly undergoes fragmentation to give H2S.75 A very recent report of visible light activated H2S donor with real time monitoring has been reported by Singh and co-workers.76 Connal and co-workers also reported thiobenzaldehyde based H2S donors which are activated by light of 355 nm (Fig. 5a).77
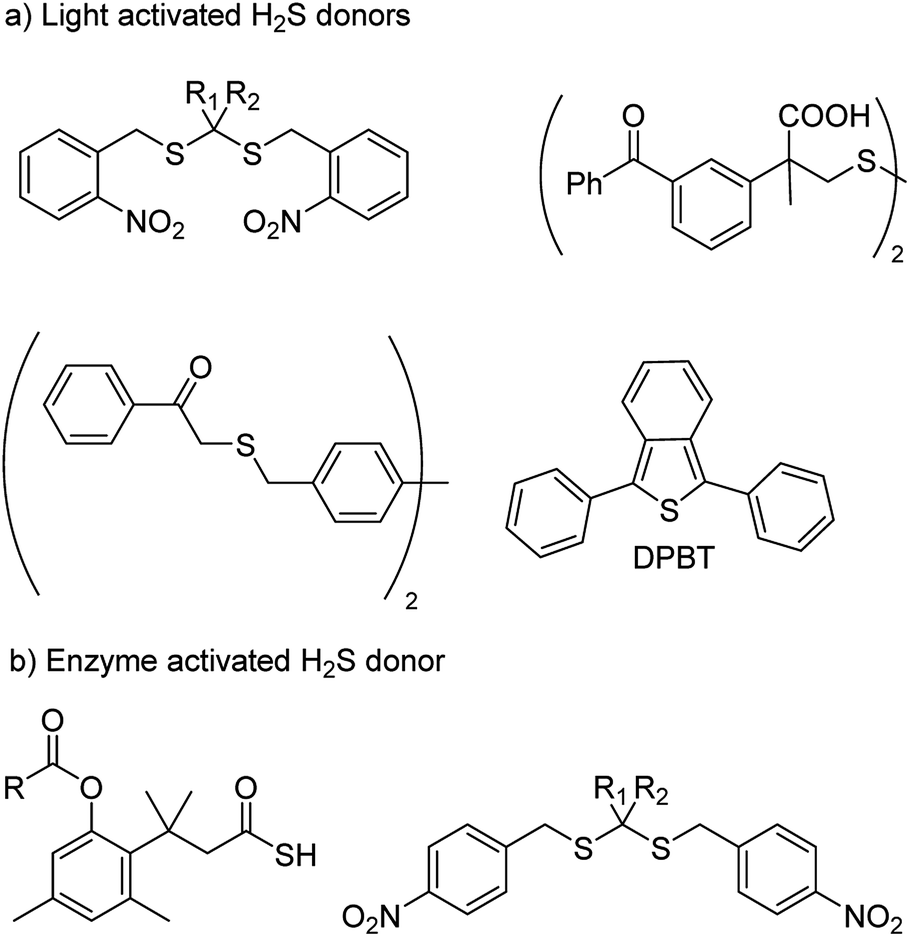 | ||
| Fig. 5 (a) Light activated H2S donors (b) H2S donors triggered by enzyme. | ||
In order to attain better and efficient release of H2S in a mammalian system Binghe Wang and co-workers in 2016 reported esterase sensitive H2S donors (Fig. 5b).78 The donors were found to attenuate the levels of pro-inflammatory cytokine TNF-α by releasing H2S. The first ever report of bacteria targeted delivery of H2S came in 2017 by Chakrapani and co-workers where they report nitroreductase (NTR) activated H2S donors for selectively generating H2S in bacteria (Fig. 5b).79 The donors were used to study the role of H2S in antibiotic resistance in patient-derived uropathogenic strains of Escherichia coli (E. coli). They find that modulating H2S levels has a tremendous impact on the bacterium's ability to counter stress inducted by an antibiotic. In particular, they find that multi-drug resistant strains of this pathogen could be sensitized to the antibiotic that they were previously resistant to by inhibiting biosynthesis of hydrogen sulfide. The resistance was restored by supplementing the bacterium with their donor. This study, with support from previous data, provided a significant advancement in our understanding of the role of hydrogen sulfide in antibiotic stress reduction and drug resistance.80
Although the rate of hydrolysis of COS in water is extremely slow 2.2 × 10−5 s−1, it readily hydrolyses in the presence of carbonic anhydrase (CA).81,82 The rate of COS hydrolysis by carbonyl sulfide hydrolase (COSase) obtained from T. thioparus strain TH1115 is reported as 58 s−1.83 CA obtained from bovine erythrocytes hydrolyses COS at the rate of 1.82 s−1 to give H2S and CO2.84 CA is a ubiquitous enzyme and this enzyme would therefore be available to hydrolyse COS within cells to produce hydrogen sulfide (Scheme 1). This strategy was employed to design a new class of H2S donors based on initial release of COS.
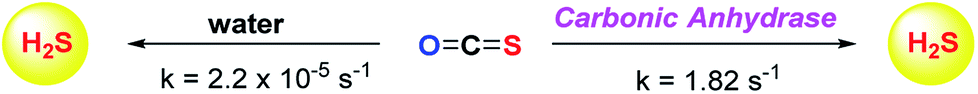 | ||
| Scheme 1 Hydrolysis of COS to H2S. | ||
Pluth and co-workers in 2016 reported O-alkyl based thiocarbamates with azide group as a trigger. The azide group was reduced in the presence of H2S resulting in analyte consumption which perturbed the homeostasis. Upon activation a self-immolative decomposition of thiocarbamates led to the formation of COS which was further hydrolysed by CA to give back H2S (Scheme 2a).84 Although it was the first report of triggerable COS donors, the activation required consumption of H2S and therefore this strategy is mainly useful for detection of hydrogen sulfide.
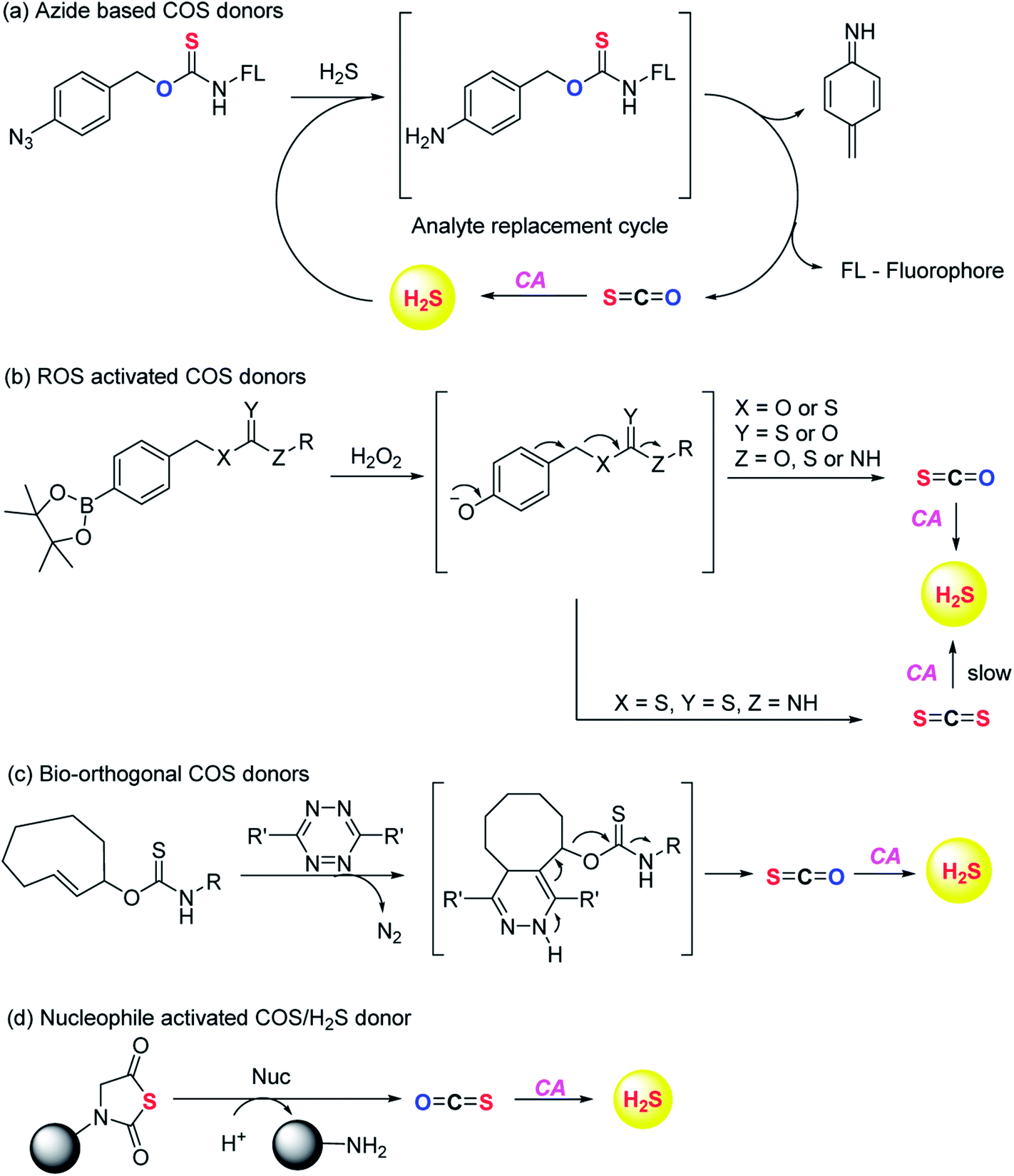 | ||
| Scheme 2 Chemically triggered COS donors. | ||
Later in the year, Zhao et al. from the same group reported COS/H2S donors which are activated by reactive oxygen and nitrogen species (RONS). Compounds were found to exhibit protective effects against H2O2 induced oxidative stress (Scheme 2b).85 A follow up on this report was published wherein the core structure of thiocarbamate scaffold was modified to attain different derivatives for achieving tunable release of H2S. However, in the case of electron withdrawing groups the donors were found to be unstable and resulted in isothiocyanate formation. Isothiocyanates being electron deficient can readily react with biological nucleophiles (thiols and amines) and therefore can be toxic.
One of the derivatives led to carbon disulfide formation which showed reduced yields of H2S due to slow rate of hydrolysis.86 Although its hydrolysis in cells is possible, carbon disulfide is toxic to the cells and prolonged exposure of cells to CS2 is not desirable. Hence, there is a need to have donors with broad range of tunability and less toxicity. Pluth and co-workers also reported COS donors using bio-orthogonal methods. The thiocarbamate moiety was attached with cyclooctene which upon reaction with tetrazine through inverse electron demand Diels–Alder click (IEDDA) reaction release COS (Scheme 2c). The donors were found to be incompatible with biological systems and therefore could not be used for further applications.87 Polymeric COS donors were reported by Matson and group in 2016 wherein they investigated N-thiocarboxyanhydride (NTA) based scaffolds which upon reaction with biological nucleophiles result in COS release (Fig. 6d). Both small molecule (NTA1) and polymer (pNTA1) were tested for their ability to produce H2S via COS release. NTA1 was found to promote cell proliferation.88
 | ||
| Fig. 6 Light activated COS donors. | ||
COS based strategy to release H2S was further explored by Chauhan and co-workers where they report esterase enzyme activated S-alkyl carbonothioates and carbamothioates as COS/H2S donors. The rate of H2S release from these donors was moderately dependent on the basicity of the leaving group amine. Being well tolerated by cells, the donors can be used for further applications (Scheme 3).89 A similar strategy was reported by Pluth and co-workers to release COS upon activation by esterase. The compounds were found to be toxic at 10 μM concentration and therefore could not be employed for biological studies. However, the toxicity was attributed to the reduced cellular respiration and ATP synthesis by inhibition of cytochrome c oxidase in the mitochondrial respiratory chain.90
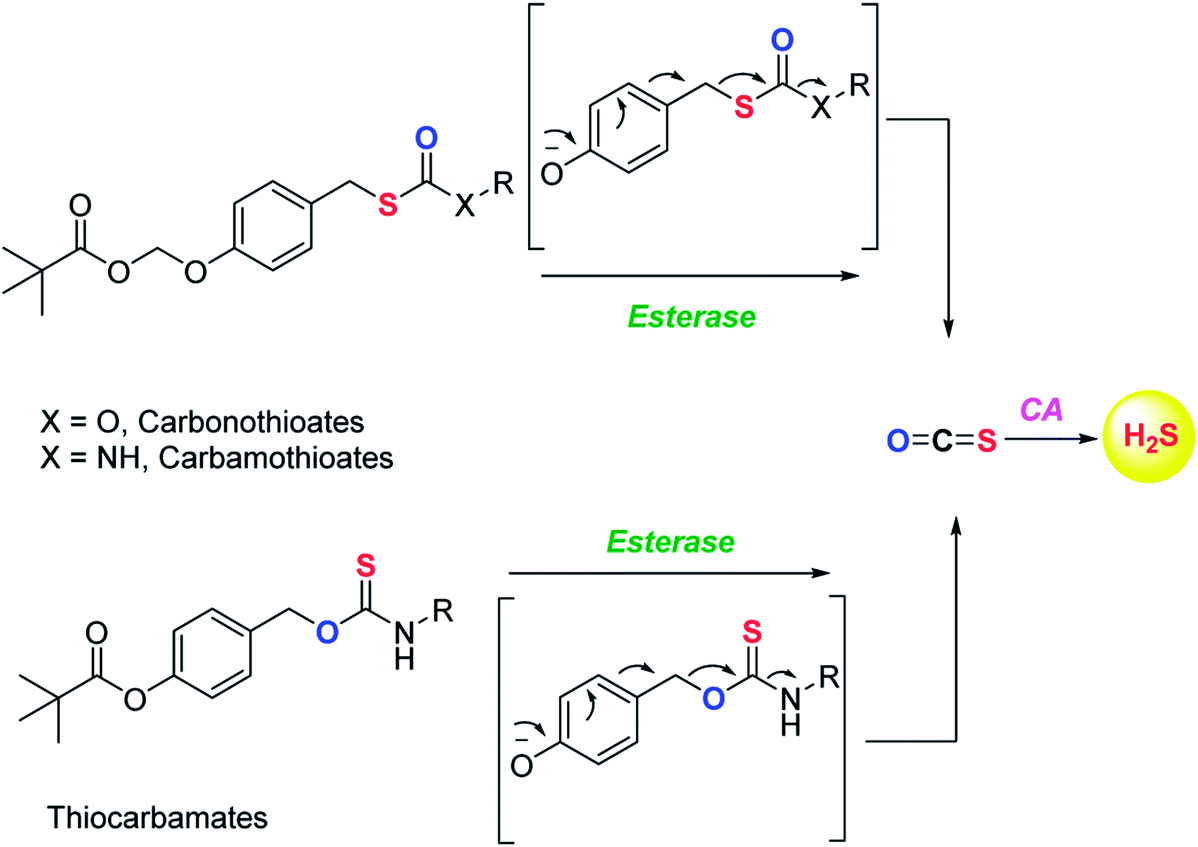 | ||
| Scheme 3 Esterase activated COS donors. | ||
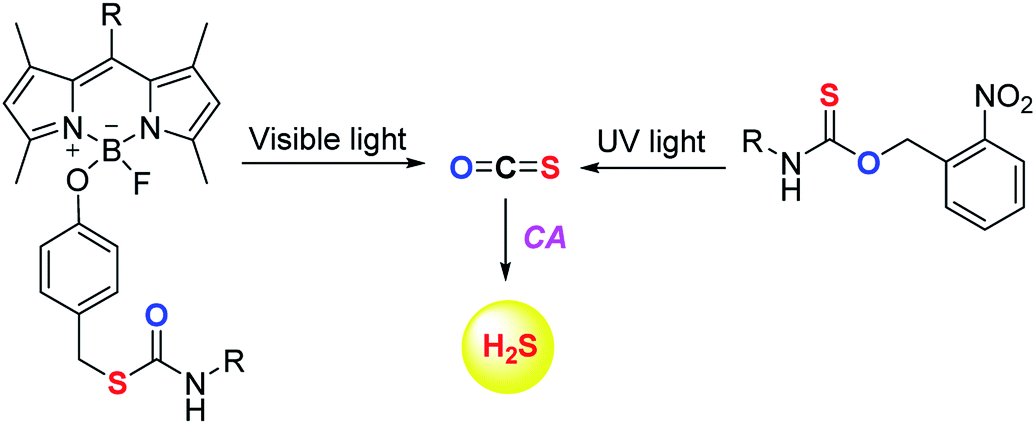
The scope of the COS/H2S donors was further expanded by the introduction of light activated COS donors. Pluth and co-workers reported UV light activated COS/H2S donors (Fig. 6).91 But due to the toxic effects of UV light these donors may not provide broad applicability.
Visible light activated COS/H2S donors reported by Sharma et al. overcame this limitation and the donor BDP-H2S reported by them works by activation by light at 480 nm (Fig. 6). The authors find that upon activation, an enhancement in fluorescence signal occurred. This serves as a convenient signal to infer activation. BDP-H2S was well tolerated by cells and may be useful for studying H2S related cell signaling.91 However, the wavelength of activation needs to be further increased as the light of ∼450–500 nm also induces oxidative stress, albeit at substantially lower levels when compared with UV light.92 Also, the aqueous solubility of these compounds was diminished and may be a major impediment to the wide use of this compound.
Although the field of H2S donors has made significant advancements over the past two decades, there is still a need to have better donors for studying precise biological effects of the gas. Also, the donors should be benign with minimal side-effects to explore the therapeutic potential of this gasotransmitter. Further work in this area should focus on greater efficiency of release with improved triggers. Also, the lack of reliable in vivo studies on most of these donor compounds makes evaluation of their therapeutic potential difficult.
Lastly, achieving tunability of H2S release from donors is a major challenge. Since the rate of generation of hydrogen sulfide has been previously shown to have major impact on the observed biological end-point, it is therefore important to develop donors with a good structural handle to modulate release rates. A number of polymer or materials-based methods for achieving this goal are in the literature.93–95 The rates of release can be typically varied from a few seconds to minutes to hours. However, the use of polymers is associated with issues such as toxicity, cell-permeability etc.
While these methods are useful, it is highly desirable to be able to modulate release rates using small molecules. Chauhan and co-workers have some preliminary evidence for the dependence of COS/H2S release rates on the amine. Thus, this problem needs to be addressed in future in order to better exploit the translational potential of this gas. A number of research groups including ours are working on solving this issue and it is anticipated that this problem will be adequately addressed in the near future.
Perthiols
Perthiols or persulfides (RSSH) form another class of reactive sulfur species with sulfur oxidation state −1 each. Protein S-sulfhydration is an oxidative post translational modification of cysteine residues wherein the cysteine thiol (RSH) gets modified to a persulfide intermediate (RSSH).11 This burgeoning area of research has gained considerable attention in the recent years because of its potential role as a mediator in H2S signaling pathways.2,96,97 Several protein targets have been identified to be S-sulfhydrated with various therapeutic outcomes but the role of H2S and the mechanism by which it carries out this modification remains largely elusive and warrants further attention.98,99 Since H2S cannot modify protein thiols directly due to redox constraints, it is considered to react with oxidized cysteine residues. However, recent studies indicate that other reactive sulfur species like polysulfides and persulfides are more potent S-sulfhydrating agents.100–104 The biogenesis of persulfides in mammals involve sulfurtransferases like 3-mercaptopyruvate sulfurtransferase (3-MST), human rhodanese and thiosulfate sulfurtransferase (TST), which form a transient persulfide intermediate in the active site cysteine, subsequently S-sulfhydrating a protein.2 Low molecular weight persulfides like CysSSH, on the other hand are biosynthesized by H2S producing transsulfuration pathway enzymes like CSE and CBS using cysteine as a substrate.100,105,106 Hence, as the biochemistry of persulfides begins to unravel, whether H2S is the actual signaling molecule or just a product of persulfide degradation is being debated. To better understand the nuances of these reactive sulfur species, there is a need to develop robust detection techniques and precursors or prodrugs that can efficiently generate these species in situ. In this regard, not much progress has been made in the field of persulfides, in contrast to H2S mainly due to the challenges associated with it. While there have been significant advances in developing methods for reliable detection of S-sulfhydration,11,107–112 there are limited number of donors available that can generate persulfides in situ in a controlled manner.The first report for the preparation of Low molecular weight persulfides or ‘hydrodisulfides’ date back to 1954 by Horst and Gerwalt.113 Following this there have been intermittent reports on the preparation of few isolated persulfides like ethyl, tert-butyl, benzyl, diphenylmethyl, trityl and adamantyl persulfides providing snippets of information about the chemical properties and reactivity of persulfides (Fig. 7).96,97 Trityl persulfides are relatively stable owing to its steric bulk, for it to be characterized by NMR, XRD, IR and Raman.114 Similarly, adamantyl persulfides can be stored indefinitely under inert conditions and at −20 °C.115,116 However, the chemical biology of these persulfides and their potency to S-sulfhydrate proteins were not reported. It is likely that this is due to their poor aqueous solubility and perhaps diminished rates of persulfidation due to steric hindrance.
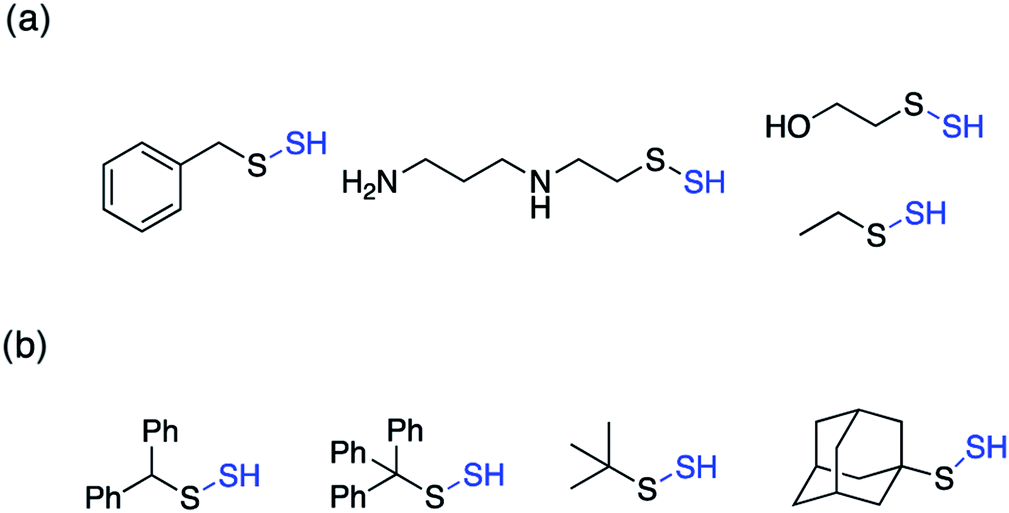 | ||
| Fig. 7 Low molecular weight (a) unhindered persulfides (b) hindered persulfides. | ||
In order to unravel the complex biochemistry of persulfides, small molecule perthiols like cysteine and glutathione persulfides were prepared in situ by the reaction of their disulfide counterpart with H2S.117–120 The reaction being reversible in nature suffer from obvious drawbacks. To address this, a small molecule persulfide precursor was developed by Erwan Galardon and Isabelle Artaud that spontaneously rearranges by an intramolecular acyl transfer in aqueous buffer (pH > 6) generating the persulfide in situ. The rate of persulfide formation was found to be pH dependant and maximum rate was observed under basic condition (Scheme 4).121
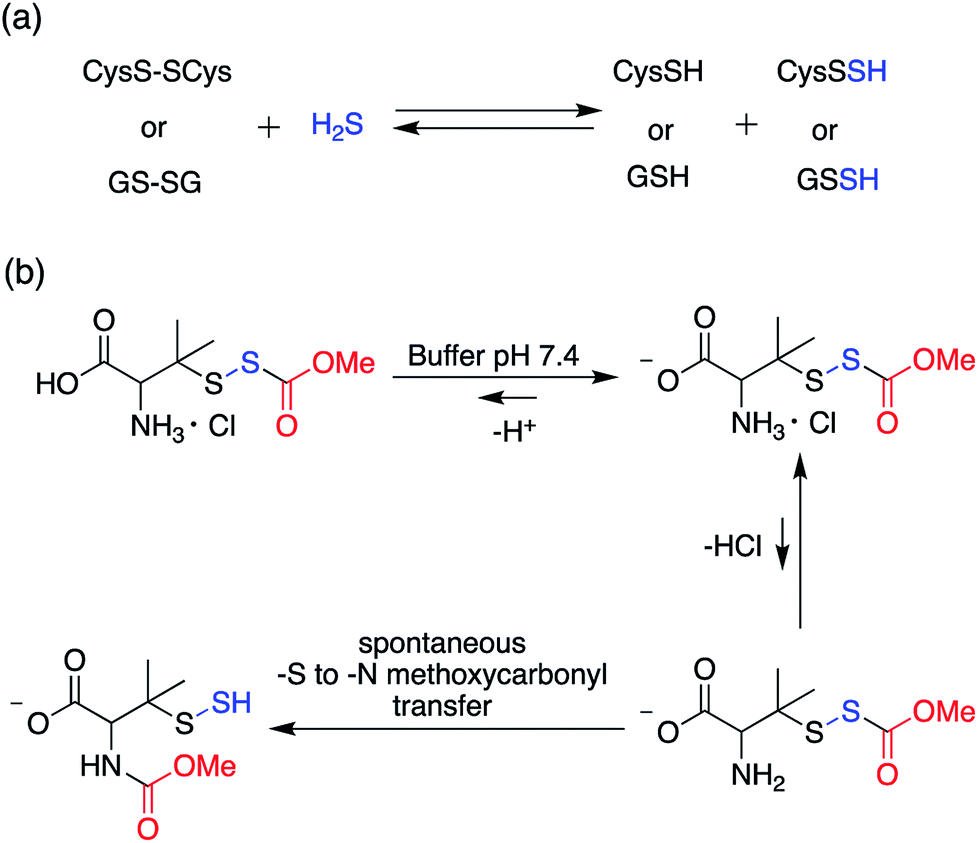 | ||
| Scheme 4 (a) Generation of small molecule persulfides by reaction of H2S with their corresponding disulfides (b) persulfide analogue of the nitrosothiol S-nitrosoacetylpenicillamine (SNAP). | ||
Ming Xian and co-workers have also developed a biomimetic persulfidation precursor, i.e. a functional 9-fluorenylmethyl disulfide (FmSSPy-A) for the formation of persulfides from the corresponding thiols. These disulfide precursors efficiently react with various small molecule thiols or protein thiols to form a base sensitive disulfide RSS-Fm adduct, which upon treatment with a suitable base generates the persulfide (Scheme 5a). The authors have used DBU as the base and have demonstrated the reactivity of persulfides towards various electrophiles. Using BSA as a model protein, DBU as a base and monobromobimane (mBBr)/iodoacetamide (IAM) as the persulfide blocking reagent, the authors have demonstrated the efficiency of their method to S-sulfhydrate protein thiols.122 Although this method provides an excellent platform to explore the chemical biology of persulfides, the use of chemical reagents like DBU limits its utility in biological systems.
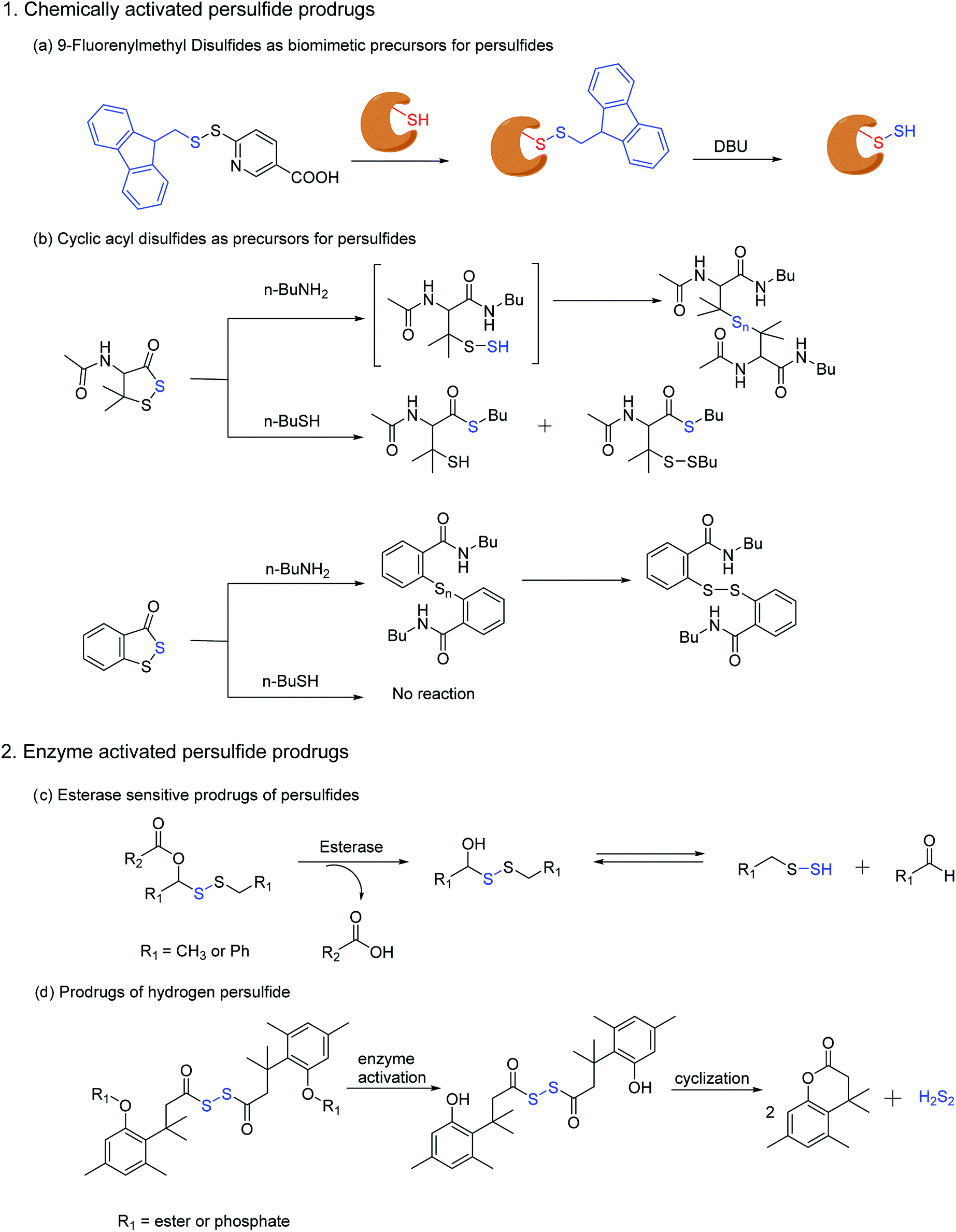 | ||
| Scheme 5 Prodrug strategies for the generation of persulfides (a) biomimetic prescussors for the generation of persulfides in presence of a base such as DBU (1,8-diazabicyclo(5.4.0)undec-7-ene). (b) Cyclic acyl disulfides as precursors for persulfides in presence of biological nucleophiles (c) prodrugs of persulfides activated by the widely prevalent esterase enzyme (d) trimethyl based prodrugs for hydrogen persulfides, activated by esterase and alkaline phosphatase. | ||
Ming Xian and co-workers have also recently reported another facile method for generation of persulfide species using cyclic acyl disulfides (Scheme 5b). Acyclic acyl disulfides have been frequently used as precursors for synthesis of low molecular weight persulfides. However, cyclic acyl disulfides as potential persulfide donors had not been considered. Two five membered ring substrates, dithiolane and benzodithiolane was reported which was made to react with physiologically significant nucleophiles like amines and thiols. Reaction of the dithiolane derivative with n-BuNH2 resulted in the formation of a polysulfide intermediate, presumably through the formation of an unstable persulfide intermediate. The intermediate was subsequently trapped using iodoacetamide, a commonly used thiol blocking agent. However, its reaction with thiols (n-BuSH) appeared to be complicated. The authors have proposed two plausible mechanisms for their observation, wherein the thiol can attack either the carbonyl or the sulfur next to it. On the other hand, the benzodithiolane proved to be more stable than the former with sluggish reactivity towards amines and no reactivity with thiols.123 Even though the persulfide-iodoacetamide adduct and H2S generation from these compounds in presence of cysteine or glutathione is a testament to the generation of a perthiol species, further validation under physiological conditions was not carried out.
Recently, Binghe Wang and co-workers have developed small molecule prodrugs for persulfide generation while demonstrating their therapeutic potential in murine model of myocardial infarction reperfusion (MI/R) injury. These esterase sensitive prodrugs would self-immolate in presence of the widely prevalent esterase enzyme to generate a perthiol species and the corresponding aldehyde as a by-product (Scheme 5c). The authors have systematically evaluated the formation of persulfide by trapping it with 2,4-dinitrofluorobenzene (DNFB) and S-methyl methanethiosulfonate (MMTS). The varying ester functionalities further provided a scope for tuning the release kinetics of persulfides.124 We anticipate that these prodrugs will serve as vital tools to decipher biological mechanisms of persulfides.
Hydrodisulfide or hydrogen persulfide (H2S2) has been considered as an important player in sulfur signaling. However, due to its rapid decomposition to produce H2S and elemental sulfur, it is difficult to differentiate its effects from H2S. Nevertheless, an enzyme activated prodrug for H2S2 has been developed by Binghe Wang and co-workers. The design employs a tri-methyl lock based system masked by ester and phosphate functionalities, such that upon activation by esterase or alkaline phosphatase H2S2 is generated after cyclization (Scheme 5d). Significant levels of persulfidation was observed in GAPDH when treated with the H2S2 prodrug. In contrast, conforming with earlier reports H2S alone failed to induce persulfidation but only in presence of H2O2. However, polysulfides formed from H2S2 degradation might be the actual persulfidating agent and not H2S2 itself.125 These handful of persulfide prodrugs offer distinct advantages to study the intricate biochemistry of persulfides and have initiated an area that will command further attention in future.
Sulfur dioxide
Sulfur dioxide has been extensively used in organic synthesis (Fig. 8) and needless to say, it is a versatile reagent.126,127 Sulfur dioxide is generated during burning of fossil fuels, volcanic eruptions and other industrial processes. Once released in the atmosphere, this gas can get further converted to acidic species of higher oxidation states including sulfuric acid. This, in part, leads to acidification of the atmosphere and contributes to acid rain. In addition, several respiratory illnesses can be traced back to excessive amounts of this gas in the environment.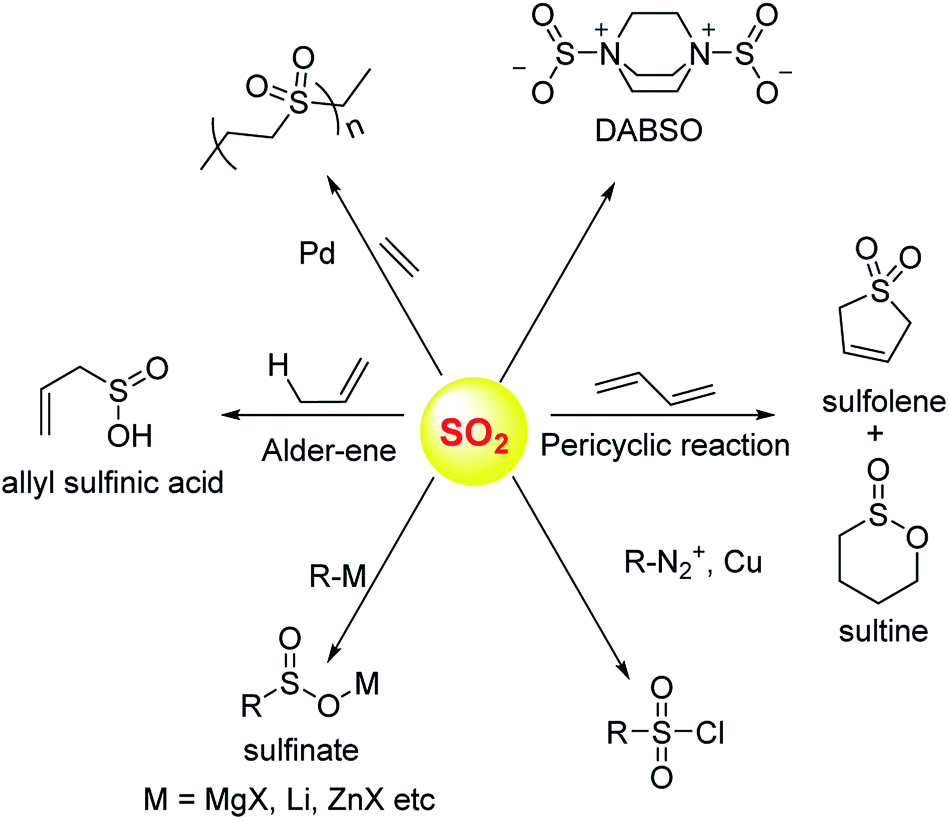 | ||
| Fig. 8 Selected synthetic transformations of sulfur dioxide. | ||
Once generated within cells, sulfur dioxide and its hydrated forms bisulfite and sulfite can interact with various biological macromolecules.33,128,129 Due to its propensity to get oxidized, sulfite can be classified as an antioxidant. The molybdopterin containing enzyme sulfite oxidase catalyzes this reaction. SO2 reacts with cysteine-containing proteins to produce S-sulfites (RS-SO3−), presumably by reaction of a disulfide with sulfite.130–132 Autoxidation of sulfite catalyzed by metal ions can generate highly reactive radical species which are known to damage DNA.
Donors of sulfur dioxide are envisaged as tools to study the precise biological effects of this gas as well as possibly exploiting its therapeutic potential. Although sulfur dioxide is used fairly well in organic synthesis, being a gas is somewhat cumbersome for use. Recent efforts to make caged sulfur dioxide donors for organic synthesis has been reviewed elsewhere.126,127,133 An example is DABSO,134 which is an adduct of DABCO and sulfur dioxide and is used to generate this gas in situ. The compounds discussed herein have been shown to produce SO2 under biologically relevant conditions.
In order to generate SO2, a sulfone or a sulfonate ester are good starting points (Scheme 6). The first report in the literature describing the anti-bacterial effects of sulfur dioxide donors was from our laboratory in 2012.135 The authors used a 2,4-dinitrophenylsulfonamide as the major functional group for generating sulfur dioxide.136 This functional group has been used as a protective group in organic synthesis previously and is deprotected by thiols.136 Using this observation as the cue, and since cellular thiols occur in large concentrations, Malwal and co-workers demonstrated the suitability of this series of compounds as sources of sulfur dioxide (Scheme 7). They find that the rate of sulfur dioxide generation was dependent on the nature of the amine.137 The more basic the amine was, the faster was the sulfur dioxide generation. Thus, simple structural modifications that affected the basicity of the amine formed the basis for modulating release rates.
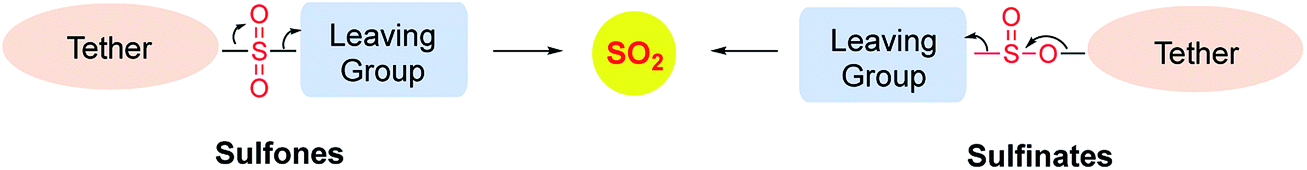 | ||
| Scheme 6 General strategies for SO2 generation. | ||
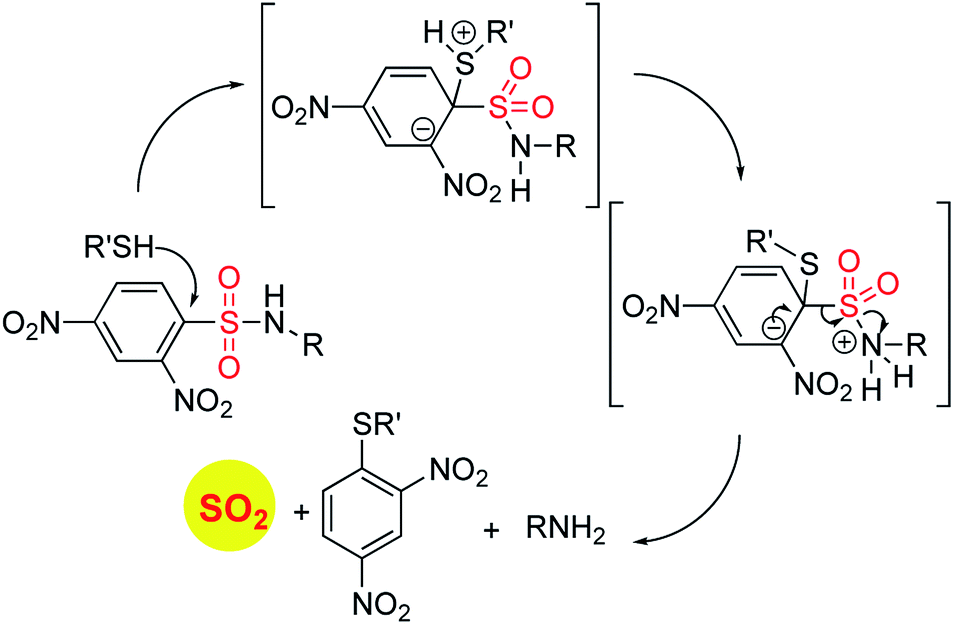 | ||
| Scheme 7 Thiol activated SO2 donors. | ||
The authors evaluated the potential for these compounds to inhibit bacterial growth and they found that, among closely related compounds, the propensity to inhibit mycobacterial growth was dependent on the rate of sulfur dioxide generation.135,137 The lead compound was found to have low micromolar inhibitory potency against Mycobacterium tuberculosis, the causative agent of tuberculosis, a disease that affects millions each year. This series of compounds was evaluated for the broad-spectrum antibacterial activity and it was found that the benzylamine derivative, which had potent anti-mycobacterial activity had no inhibitory activity against other Gram-positive as well as Gram-negative bacteria. A recent study on the effects of sulfur dioxide on tuberculosis occurrence in human populations was conducted.138 The authors found protective effects of this gas when patients were exposed to low-level ambient sulfur dioxide against TB.
Pardeshi and co-workers did a series of structural modifications that led to improved anti-bacterial properties, but only against Gram-positive pathogens.139 The structural modifications resulted in compounds with good inhibitory activity against drug-resistant strains of Staphylococcus aureus.
The benzylamine derivative has been used by several groups as a positive control for sulfur dioxide/sulfite generation.140 This compound thus reliably generates sulfur dioxide upon permeation into mammalian cells as well. Together, these compounds are useful as tools to generate sulfur dioxide and provide the scope for tuneable release of this gas. They display anti-bacterial properties and will need further evaluation for potential as therapeutic agents.
Sulfones have been reported to undergo cycloreversion to generation sulfur dioxide (Scheme 8).141,142 Typically elevated temperatures are required for this reaction. This strategy has been modified by Binghe Wang and co-workers to generate a highly reactive sulfone intermediate in situ (Scheme 9). The strategy followed was a “click and release” one, where a cycloaddition between and strained alkyne and a diene was used (Scheme 9).143 This reaction was carried out with two independent reactants and a follow-up on this work had an intramolecular reaction.144 The latter strategy has been used to tune release of sulfur dioxide from a few minutes to days.
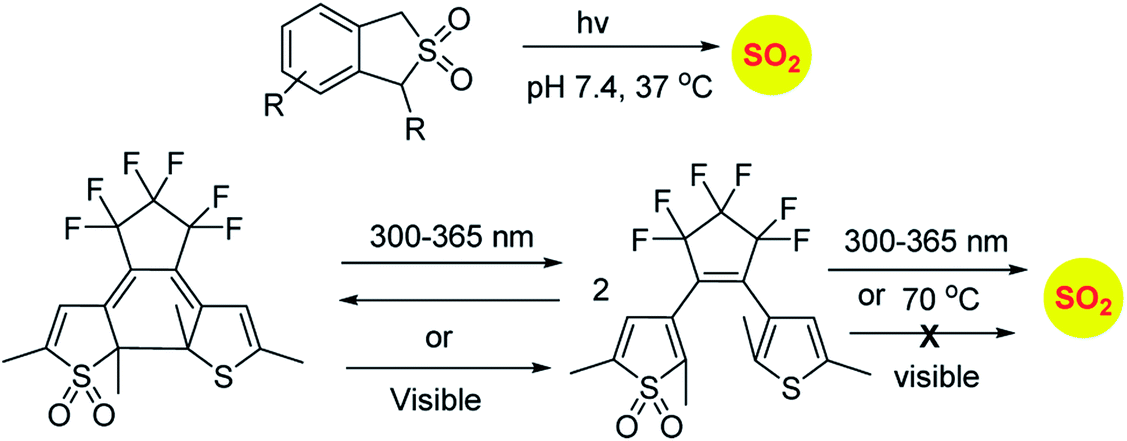 | ||
| Scheme 8 Photolabile SO2 donors. | ||
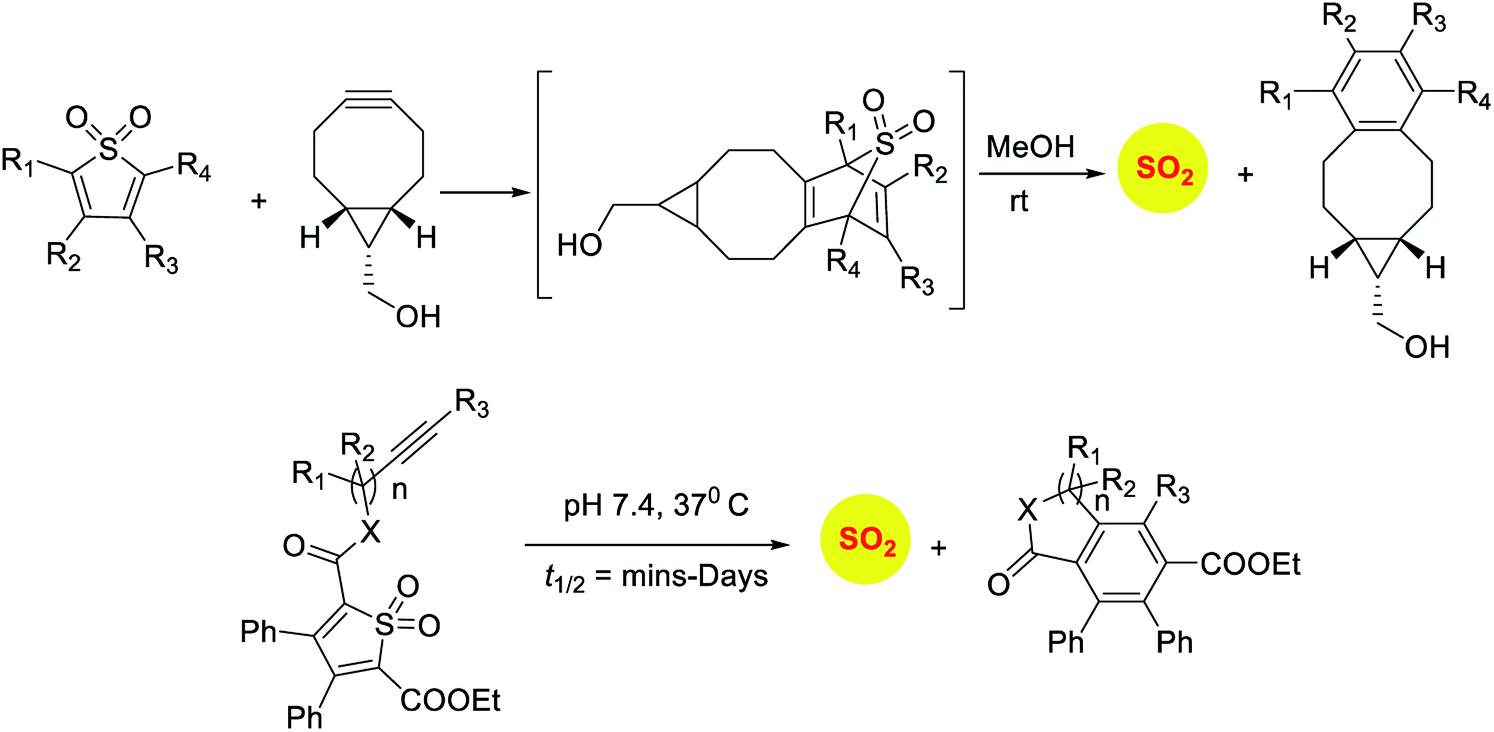 | ||
| Scheme 9 Click and release strategies for sulfur dioxide generation. | ||
Binghe Wang and co-workers recently reported a sulfone that was a candidate for Julia olefination reaction (Scheme 12).145 They modified this substrate to be triggered by esterase to generate and olefin and sulfur dioxide. Using a latent fluorophore that reacts with sulfite to become fluorescent, the donor was found to permeate cells to generate sulfur dioxide. The authors also show that the half-life of release of sulfur dioxide could be modulated by changing the substituent. Together, this donor may find wide use for cellular studies to interrogate the chemical biology of sulfur dioxide.
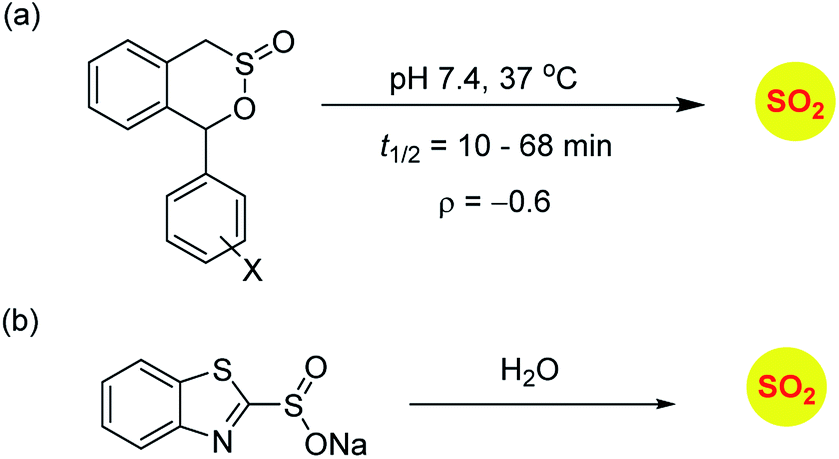 | ||
| Scheme 10 Sulfinate based SO2 donors. | ||
 | ||
| Scheme 11 Esterase activated SO2 donors. | ||
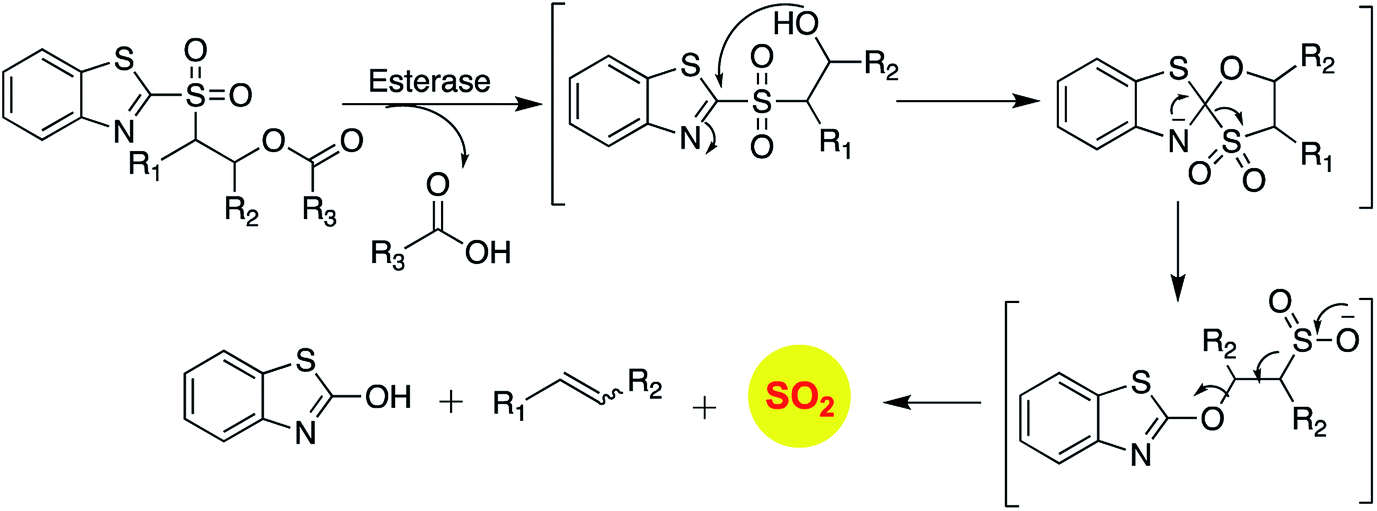 | ||
| Scheme 12 Esterase activated SO2 donors based on Julia olefination. | ||
Similarly, sulfones can also generate sulfur dioxide after exposure to light. Malwal and co-workers have demonstrated that benzosulfones can be irradiated with UV light to produce sulfur dioxide in pH 7.4 buffer.146 While this method allows for triggerable sulfur dioxide generation, the wavelength and intensity of light used is not entirely compatible for biological studies. Uchida and co-workers have reported a closed ring isomer of diaryl ethene bearing a sulfone group as a masked SO2 donor.147 In presence of visible light, the closed ring isomer gets converted into its open ring form but does not generate SO2 and also found to be stable at 70 °C. However, upon exposure to UV light, both the isomers exist in equilibrium and the open ring form efficiently generates SO2.
Sulfinate esters are also candidates for sulfur dioxide generation. A cycloreversion strategy was reported by Malwal and co-workers.148 Here, benzosultines were synthesized and these compounds were found to undergo retro Diels–Alder reaction to produce sulfur dioxide (Scheme 10a). A Hammett relationship study was carried out and the dependence of the rates of SO2 release on substituents on the carbon bearing the sulfinate ester was determined. A moderate electronic effect (ρ = −0.6) was found and the range of half-lives for SO2 generation was 10–68 min. A detailed computational investigation of the release mechanism was carried out and was found to be consistent with experimental data. This study showed that modulating the electronic environment around the sulfonate functional group may have an effect on release of sulfur dioxide.
Ming Xian and co-workers have developed benzothiazole sulfonate salt, a new sulfonate ester-based donor that undergoes hydrolysis in buffer to produce sulfur dioxide.149 This compound is water-soluble and release sulfur dioxide over several hours (Scheme 10b). They studied this donor's vasorelaxation properties as compared to the authentic SO2 gas solutions and found that the donor compared well with SO2 in this respect. This compound showed pH-dependent release and this study is a major advancement in the use of SO2 as a vasorelaxant.
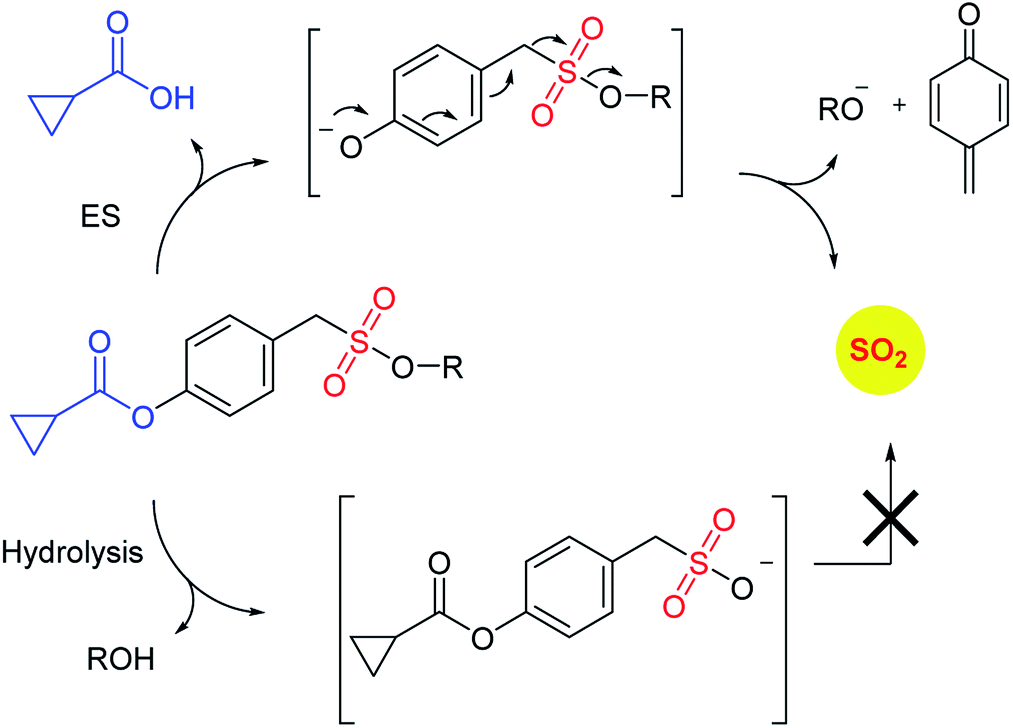
Recently, Pardeshi and co-workers have reported a sulfonate ester that undergoes self-immolation in the presence of esterase to produce sulfur dioxide and an alcohol (Scheme 11).150 The sulfonate esters are modelled on carbonates, which are frequently used as linkers in prodrugs. A large majority of the sulfonates evaluated were found to generate sulfur dioxide. Certain donors which are derived from aliphatic alcohols were found to hydrolyze even in the absence of esterase. The best donors were found to permeate cells to generate sulfite. Thus, this series of compounds are potentially useful for studying the chemical biology of sulfur dioxide.
Summary and outlook
A better understanding of the precise physiological roles and targets for reactive sulfur species is not only of academic interest but also may have numerous applications. A key aspect of gaining this knowledge is to generate in a controlled manner, reactive sulfur species. This has been an interest in our lab as well as in several laboratories globally. While numerous techniques are presently available, this is yet an emerging field of study. Some donors offer promise and may emerge as routine tools for redox biologists. The major challenge is to translate these interesting activities into therapy. Little work has been done in this regard and the next generation of studies will focus on this.In parallel, the development of tools to detect each of these reactive species has matured and robust methodologies are in place. However, no single methodology can unambiguously detect and quantify these reactive sulfur species, which remains an unsolved problem. Lastly, the work done towards dissipation or selective inhibition of biosynthesis is yet in its infancy. These technologies may hold the key for accurate interrogation of the physiological effects of these species. Tools that can reliably inhibit the biosynthesis can be used in conjunction with small molecules that can generate these species to study the effects of modulating the levels of reactive sulfur species within cells. In the past three decades, reactive nitrogen species such as nitric oxide (NO),151 nitroxyl5,142 and peroxynitrite152,153 have been systematically studied and this is, in part, due to the ready availability of small molecules that dissociate to generate these species.154–158
Reactive oxygen species (ROS) are another major redox regulator that have been found to be involved in many pathophysiological processes and regulating them has enormous implications in disease biology.159–162 While these reactive species have been looked at in isolation, they are often connected and can regulate each other both positively and negatively. Thus, the study of reactive species is often complicated by poor detection techniques, artefacts and uncharacterized cellular interactions. Nevertheless, the quest for efficient and benign methods for generating reactive species with minimal footprint will strengthen our understanding of the associated complex biology.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank the Department of Science and Technology (DST, Grant number EMR/2015/000668), Department of Biotechnology, India (BT/PR15848/MED/29/1025/2016) for financial support for our research. Council for Scientific and Industrial Research (CSIR) and the Department of Science and Technology – Innovation in Science Pursuit for Inspired Research (DST-INSPIRE) for fellowships.Notes and references
- M. Bouroushian, in Electrochemistry of Metal Chalcogenides, Monographs in Electrochemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 57–75 Search PubMed.
- T. V Mishanina, M. Libiad and R. Banerjee, Nat. Chem. Biol., 2015, 11, 457 CrossRef PubMed.
- O. Kabil, V. Vitvitsky and R. Banerjee, Annu. Rev. Nutr., 2014, 34, 171–205 CrossRef PubMed.
- C. Nathan and A. Cunningham-Bussel, Nat. Rev. Immunol., 2013, 13, 349–361 CrossRef PubMed.
- S. Bruce King, Free Radical Biol. Med., 2013, 55, 1–7 CrossRef PubMed.
- H. Kimura, Antioxid. Redox Signaling, 2015, 22, 347–349 CrossRef PubMed.
- F. Gallyas, Br. J. Pharmacol., 2012, 166, 2228–2230 CrossRef PubMed.
- A. Martelli, L. Testai, M. C. Breschi, C. Blandizzi, A. Virdis, S. Taddei and V. Calderone, Med. Res. Rev., 2012, 32, 1093–1130 CrossRef PubMed.
- R. Pietri, A. Lewis, R. G. León, G. Casabona, L. Kiger, S.-R. Yeh, S. Fernandez-Alberti, M. C. Marden, C. L. Cadilla and J. López-Garriga, Biochemistry, 2009, 48, 4881–4894 CrossRef PubMed.
- P. Nicholls and J.-K. Kim, Can. J. Biochem., 1982, 60, 613–623 CrossRef PubMed.
- A. K. Mustafa, M. M. Gadalla, N. Sen, S. Kim, W. Mu, S. K. Gazi, R. K. Barrow, G. Yang, R. Wang and S. H. Snyder, Sci. Signaling, 2009, 2, ra72 Search PubMed.
- M. H. Tatjana and K. G. Manfred, FEBS J., 2008, 275, 3352–3361 CrossRef PubMed.
- M. Libiad, P. K. Yadav, V. Vitvitsky, M. Martinov and R. Banerjee, J. Biol. Chem., 2014, 289, 30901–30910 CrossRef PubMed.
- T. P. Singer and E. B. Kearney, Arch. Biochem. Biophys., 1956, 61, 397–409 CrossRef PubMed.
- B. T. Hofstaetter, P. Gronski, E. J. Kanzy, H. U. Schorlemmer, F. R. Seller and A. G. Behringwerke, Vox Sang., 2009, 45, 144–154 Search PubMed.
- W. W. C. Chan, Biochemistry, 1968, 7, 4247–4254 CrossRef PubMed.
- M. H. Stipanuk, Annu. Rev. Nutr., 1986, 6, 179–209 CrossRef PubMed.
- W. J. Mueller and S. Naeem, FEBS J., 2013, 280, 3050–3057 CrossRef PubMed.
- N. B. Schwartz, S. Lyle, J. D. Ozeran, H. Li, A. Deyrup, K. Ng and J. Westley, Chem.-Biol. Interact., 1998, 109, 143–151 CrossRef PubMed.
- H. M. Logan, N. Cathala, C. Grignon and J.-C. Davidian, J. Biol. Chem., 1996, 271, 12227–12233 CrossRef PubMed.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef PubMed.
- H. Cheng, L. Wang, M. Mollica, A. T. Re, S. Wu and L. Zuo, Cancer Lett., 2014, 353, 1–7 CrossRef PubMed.
- A. Siegert, C. Rosenberg, W. D. Schmitt, C. Denkert and S. Hauptmann, Br. J. Cancer, 2002, 86, 1310–1315 CrossRef PubMed.
- D. D. Thomas, L. A. Ridnour, J. S. Isenberg, W. Flores-Santana, C. H. Switzer, S. Donzelli, P. Hussain, C. Vecoli, N. Paolocci, S. Ambs, C. A. Colton, C. C. Harris, D. D. Roberts and D. A. Wink, Free Radical Biol. Med., 2008, 45, 18–31 CrossRef PubMed.
- L. J. Ignarro, G. Cirino, A. Casini and C. Napoli, J. Cardiovasc. Pharmacol., 1999, 34, 879 CrossRef PubMed.
- D. A. Wink and J. B. Mitchell, Free Radical Biol. Med., 1998, 25, 434–456 CrossRef PubMed.
- R. M. J. Palmer, A. G. Ferrige and S. Moncada, Nature, 1987, 327, 524–526 CrossRef PubMed.
- D. A. Wink, H. B. Hines, R. Y. S. Cheng, C. H. Switzer, W. Flores-Santana, M. P. Vitek, L. A. Ridnour and C. A. Colton, J. Leukocyte Biol., 2011, 89, 873–891 CrossRef PubMed.
- B. Bonavida, S. Khineche, S. Huerta-Yepez and H. Garban, Drug Resist. Updates, 2006, 9, 157 CrossRef PubMed.
- K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066–1071 CrossRef PubMed.
- L. K. Wareham, R. K. Poole and M. Tinajero-Trejo, J. Biol. Chem., 2015, 290, 18999–19007 CrossRef PubMed.
- H. Niknahad and P. J. O'Brien, Chem.-Biol. Interact., 2008, 174, 147 CrossRef PubMed.
- A. F. Gunnison, Food Cosmet. Toxicol., 1981, 19, 667 CrossRef PubMed.
- C. S. Ough and E. A. Crowell, J. Food Sci., 1987, 52, 386 CrossRef.
- B. L. Wedzicha, Chemistry of Sulphur Dioxide in Foods, 1984 Search PubMed.
- M. A. Amerine, H. W. Berg, R. E. Kunkee, C. S. Ough, V. L. Singleton and A. D. Webb, Technology of Wine Making, 1980 Search PubMed.
- J. Li and Z. Meng, Nitric Oxide, 2009, 20, 166 CrossRef PubMed.
- K. Hanaoka, K. Sasakura, Y. Suwanai, S. Toma-Fukai, K. Shimamoto, Y. Takano, N. Shibuya, T. Terai, T. Komatsu, T. Ueno, Y. Ogasawara, Y. Tsuchiya, Y. Watanabe, H. Kimura, C. Wang, M. Uchiyama, H. Kojima, T. Okabe, Y. Urano, T. Shimizu and T. Nagano, Sci. Rep., 2017, 7, 40227 CrossRef PubMed.
- C. D. McCune, S. J. Chan, M. L. Beio, W. Shen, W. J. Chung, L. M. Szczesniak, C. Chai, S. Q. Koh, P. T.-H. Wong and D. B. Berkowitz, ACS Cent. Sci., 2016, 2, 242–252 CrossRef PubMed.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329–345 CrossRef PubMed.
- J. L. Wallace, Trends Pharmacol. Sci., 2007, 28, 501–505 CrossRef PubMed.
- J. L. Wallace, L. Vong, W. McKnight, M. Dicay and G. R. Martin, Gastroenterology, 2009, 137, 569–578.e1 CrossRef PubMed.
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef PubMed.
- Y. Zhao, T. D. Biggs and M. Xian, Chem. Commun., 2014, 50, 11788–11805 RSC.
- A. A. Powolny and S. V Singh, Cancer Lett., 2018, 269, 305–314 CrossRef PubMed.
- K. Rahman, J. Nutr., 2001, 131, 977S–979S CrossRef PubMed.
- J. A. Milner, in Adv. Exp. Med. Biol., Springer US, Boston, MA, 2001, pp. 69–81 Search PubMed.
- G. A. Benavides, G. L. Squadrito, R. W. Mills, H. D. Patel, T. S. Isbell, R. P. Patel, V. M. Darley-Usmar, J. E. Doeller and D. W. Kraus, Proc. Natl. Acad. Sci. U.S.A, 2007, 104, 17977–17982 CrossRef PubMed.
- Y.-Q. Cheng, G.-L. Tang and B. Shen, J. Bacteriol., 2002, 184, 7013–7024 CrossRef PubMed.
- S. Sivaramakrishnan and K. S. Gates, Bioorganic Med. Chem. Lett., 2008, 18, 3076–3080 CrossRef PubMed.
- C. Jacob, A. Anwar and T. Burkholz, Planta Med., 2008, 74, 1580–1592 CrossRef PubMed.
- E. G. Mueller, Nat. Chem. Biol., 2006, 2, 185 CrossRef PubMed.
- Z. J. Song, M. Y. Ng, Z.-W. Lee, W. Dai, T. Hagen, P. K. Moore, D. Huang, L.-W. Deng and C.-H. Tan, Medchemcomm, 2014, 5, 557–570 RSC.
- W. Giggenbach, Inorg. Chem., 1971, 10, 1333–1338 CrossRef.
- T. Ozturk, E. Ertas and O. Mert, Chem. Rev., 2007, 107, 5210–5278 CrossRef PubMed.
- H. Z. Lecher, R. A. Greenwood, K. C. Whitehouse and T. H. Chao, J. Am. Chem. Soc., 1956, 78, 5018–5022 CrossRef.
- F. Spiller, M. I. L. Orrico, D. C. Nascimento, P. G. Czaikoski, F. O. Souto, J. C. Alves-Filho, A. Freitas, D. Carlos, M. F. Montenegro, A. F. Neto, S. H. Ferreira, M. A. Rossi, J. S. Hothersall, J. Assreuy and F. Q. Cunha, Am. J. Respir. Crit. Care Med., 2010, 182, 360–368 CrossRef PubMed.
- C. Fee, D. C. Gard and D. R. Yang, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, New York, 2005 Search PubMed.
- Z. W. Lee, J. Zhou, C.-S. Chen, Y. Zhao, C.-H. Tan, L. Li, P. K. Moore and L.-W. Deng, PLoS One, 2011, 6, e21077 CrossRef PubMed.
- L. Li, M. Whiteman, Y. Y. Guan, K. L. Neo, Y. Cheng, S. W. Lee, Y. Zhao, R. Baskar, C.-H. Tan and P. K. Moore, Circulation, 2008, 117, 2351–2360 CrossRef PubMed.
- A. Sparatore, G. Santus, D. Giustarini, R. Rossi and P. Del Soldato, Expert Rev. Clin. Pharmacol., 2011, 4, 109–121 CrossRef PubMed.
- M. V. Chan and J. L. Wallace, Am. J. Physiol.: Gastrointest. Liver Physiol., 2013, 305, G467–G473 CrossRef PubMed.
- C. J. Gargallo and A. Lanas, J. Dig. Dis., 2012, 14, 55–61 CrossRef PubMed.
- G. Caliendo, G. Cirino, V. Santagada and J. L. Wallace, J. Med. Chem., 2010, 53, 6275–6286 CrossRef PubMed.
- L. Li, P. Rose and P. K. Moore, Annu. Rev. Pharmacol. Toxicol., 2011, 51, 169–187 CrossRef PubMed.
- L. Li, G. Rossoni, A. Sparatore, L. C. Lee, P. Del Soldato and P. K. Moore, Free Radical Biol. Med., 2007, 42, 706–719 CrossRef PubMed.
- Y. Zhao, H. Wang and M. Xian, J. Am. Chem. Soc., 2011, 133, 15–17 CrossRef PubMed.
- Y. Zhao, S. Bhushan, C. Yang, H. Otsuka, J. D. Stein, A. Pacheco, B. Peng, N. O. Devarie-Baez, H. C. Aguilar, D. J. Lefer and M. Xian, ACS Chem. Biol., 2013, 8, 1283–1290 CrossRef PubMed.
- T. Roger, F. Raynaud, F. Bouillaud, C. Ransy, S. Simonet, C. Crespo, M.-P. Bourguignon, N. Villeneuve, J.-P. Vilaine, I. Artaud and E. Galardon, ChemBioChem, 2013, 14, 2268–2271 CrossRef PubMed.
- A. Martelli, L. Testai, V. Citi, A. Marino, I. Pugliesi, E. Barresi, G. Nesi, S. Rapposelli, S. Taliani, F. Da Settimo, M. C. Breschi and V. Calderone, ACS Med. Chem. Lett., 2013, 4, 904–908 CrossRef PubMed.
- Y. Zhao, J. Kang, C.-M. Park, P. E. Bagdon, B. Peng and M. Xian, Org. Lett., 2014, 16, 4536–4539 CrossRef PubMed.
- J. Kang, Z. Li, C. L. Organ, C.-M. Park, C. Yang, A. Pacheco, D. Wang, D. J. Lefer and M. Xian, J. Am. Chem. Soc., 2016, 138, 6336–6339 CrossRef PubMed.
- N. O. Devarie-Baez, P. E. Bagdon, B. Peng, Y. Zhao, C.-M. Park and M. Xian, Org. Lett., 2013, 15, 2786–2789 CrossRef PubMed.
- N. Fukushima, N. Ieda, K. Sasakura, T. Nagano, K. Hanaoka, T. Suzuki, N. Miyata and H. Nakagawa, Chem. Commun., 2014, 50, 587–589 RSC.
- S. Y. Yi, Y. K. Moon, S. Kim, S. Kim, G. Park, J. J. Kim and Y. You, Chem. Commun., 2017, 53, 11830–11833 RSC.
- Y. Venkatesh, J. Das, A. Chaudhuri, A. Karmakar, T. K. Maiti and N. D. P. Singh, Chem. Commun., 2018, 54, 3106–3109 RSC.
- Z. Xiao, T. Bonnard, A. Shakouri-Motlagh, R. A. L. Wylie, J. Collins, J. White, D. E. Heath, C. E. Hagemeyer and L. A. Connal, Chem.–Eur. J., 2017, 23, 11294–11300 CrossRef PubMed.
- Y. Zheng, B. Yu, K. Ji, Z. Pan, V. Chittavong and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 4514–4518 CrossRef PubMed.
- P. Shukla, V. S. Khodade, M. SharathChandra, P. Chauhan, S. Mishra, S. Siddaramappa, B. E. Pradeep, A. Singh and H. Chakrapani, Chem. Sci., 2017, 8, 4967–4972 RSC.
- K. Shatalin, E. Shatalina, A. Mironov and E. Nudler, Science, 2011, 334, 986–990 CrossRef PubMed.
- S. Elliott, E. Lu and F. S. Rowland, Environ. Sci. Technol., 1989, 23, 458–461 CrossRef.
- H. W. Thompson, C. F. Kearton and S. A. Lamb, J. Chem. Soc., 1935, 1033–1037 RSC.
- T. Ogawa, K. Noguchi, M. Saito, Y. Nagahata, H. Kato, A. Ohtaki, H. Nakayama, N. Dohmae, Y. Matsushita, M. Odaka, M. Yohda, H. Nyunoya and Y. Katayama, J. Am. Chem. Soc., 2013, 135, 3818–3825 CrossRef PubMed.
- A. K. Steiger, S. Pardue, C. G. Kevil and M. D. Pluth, J. Am. Chem. Soc., 2016, 138, 7256–7259 CrossRef PubMed.
- Y. Zhao and M. D. Pluth, Angew. Chem., Int. Ed., 2016, 55, 14638–14642 CrossRef PubMed.
- Y. Zhao, H. A. Henthorn and M. D. Pluth, J. Am. Chem. Soc., 2017, 139, 16365–16376 CrossRef PubMed.
- A. K. Steiger, Y. Yang, M. Royzen and M. D. Pluth, Chem. Commun., 2017, 53, 1378–1380 RSC.
- C. R. Powell, J. C. Foster, B. Okyere, M. H. Theus and J. B. Matson, J. Am. Chem. Soc., 2016, 138, 13477–13480 CrossRef PubMed.
- P. Chauhan, P. Bora, G. Ravikumar, S. Jos and H. Chakrapani, Org. Lett., 2017, 19, 62–65 CrossRef PubMed.
- A. K. Steiger, M. Marcatti, C. Szabo, B. Szczesny and M. D. Pluth, ACS Chem. Biol., 2017, 12, 2117–2123 CrossRef PubMed.
- Y. Zhao, S. G. Bolton and M. D. Pluth, Org. Lett., 2017, 19, 2278–2281 CrossRef PubMed.
- Y. Nakashima, S. Ohta and A. M. Wolf, Free Radical Biol. Med., 2017, 108, 300–310 CrossRef PubMed.
- J. C. Foster, C. R. Powell, S. C. Radzinski and J. B. Matson, Org. Lett., 2014, 16, 1558–1561 CrossRef PubMed.
- J. C. Foster and J. B. Matson, Macromolecules, 2014, 47, 5089–5095 CrossRef.
- J. M. Carter, Y. Qian, J. C. Foster and J. B. Matson, Chem. Commun., 2015, 51, 13131–13134 RSC.
- M. R. Filipovic, J. Zivanovic, B. Alvarez and R. Banerjee, Chem. Rev., 2018, 118, 1253–1337 CrossRef PubMed.
- C.-M. Park, L. Weerasinghe, J. J. Day, J. M. Fukuto and M. Xian, Mol. BioSyst., 2015, 11, 1775–1785 RSC.
- M. S. Vandiver, B. D. Paul, R. Xu, S. Karuppagounder, F. Rao, A. M. Snowman, H. Seok Ko, Y. Il Lee, V. L. Dawson, T. M. Dawson, N. Sen and S. H. Snyder, Nat. Commun., 2013, 4, 1626 CrossRef PubMed.
- B. D. Paul and S. H. Snyder, Nat. Rev. Mol. Cell Biol., 2012, 13, 499 CrossRef PubMed.
- T. Ida, T. Sawa, H. Ihara, Y. Tsuchiya, Y. Watanabe, Y. Kumagai, M. Suematsu, H. Motohashi, S. Fujii, T. Matsunaga, M. Yamamoto, K. Ono, N. O. Devarie-Baez, M. Xian, J. M. Fukuto and T. Akaike, Proc. Natl. Acad. Sci. U.S.A, 2014, 111, 7606–7611 CrossRef PubMed.
- K. Ono, T. Akaike, T. Sawa, Y. Kumagai, D. A. Wink, D. J. Tantillo, A. J. Hobbs, P. Nagy, M. Xian, J. Lin and J. M. Fukuto, Free Radical Biol. Med., 2014, 77, 82–94 CrossRef PubMed.
- J. I. Toohey, Anal. Biochem., 2011, 413, 1–7 CrossRef PubMed.
- H. Kimura, Antioxid. Redox Signaling, 2014, 22, 347–349 CrossRef PubMed.
- Y. Kimura, Y. Mikami, K. Osumi, M. Tsugane, J. Oka and H. Kimura, FASEB J., 2013, 27, 2451–2457 CrossRef PubMed.
- T. Yamanishi and S. Tuboi, J. Biochem., 1981, 89, 1913–1921 CrossRef PubMed.
- P. K. Yadav, M. Martinov, V. Vitvitsky, J. Seravalli, R. Wedmann, M. R. Filipovic and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 289–299 CrossRef PubMed.
- D. Zhang, I. Macinkovic, N. O. Devarie-Baez, J. Pan, P. Chung-Min, K. S. Carroll, M. R. Filipovic and M. Xian, Angew. Chem., Int. Ed., 2013, 53, 575–581 CrossRef PubMed.
- N. Krishnan, C. Fu, D. J. Pappin and N. K. Tonks, Sci. Signaling, 2011, 4, ra86 CrossRef PubMed.
- N. Sen, B. D. Paul, M. M. Gadalla, A. K. Mustafa, T. Sen, R. Xu, S. Kim and S. H. Snyder, Mol. Cell, 2012, 45, 13–24 CrossRef PubMed.
- S. Longen, F. Richter, Y. Köhler, I. Wittig, K.-F. Beck and J. Pfeilschifter, Sci. Rep., 2016, 6, 29808 CrossRef PubMed.
- É. Dóka, I. Pader, A. Bíró, K. Johansson, Q. Cheng, K. Ballagó, J. R. Prigge, D. Pastor-Flores, T. P. Dick, E. E. Schmidt, E. S. J. Arnér and P. Nagy, Sci. Adv., 2016, 2, e1500968 Search PubMed.
- T. Sawahata and R. A. Neal, Anal. Biochem., 1982, 126, 360–364 CrossRef PubMed.
- B. Horst and Z. Gerwalt, Justus Liebigs Ann. Chem., 2006, 585, 142–149 Search PubMed.
- T. S. Bailey, L. N. Zakharov and M. D. Pluth, J. Am. Chem. Soc., 2014, 136, 10573–10576 CrossRef PubMed.
- T. S. Bailey and M. D. Pluth, Free Radical Biol. Med., 2015, 89, 662–667 CrossRef PubMed.
- N. E. Heimer, L. Field and J. A. Waites, J. Org. Chem., 1985, 50, 4164–4166 CrossRef.
- E. Cuevasanta, M. Lange, J. Bonanata, E. L. Coitiño, G. Ferrer-Sueta, M. R. Filipovic and B. Alvarez, J. Biol. Chem., 2015, 290, 26866–26880 CrossRef PubMed.
- D. K. Liu and S. G. Chang, Can. J. Chem., 1987, 65, 770–774 CrossRef.
- A. Vasas, É. Dóka, I. Fábián and P. Nagy, Nitric Oxide, 2015, 46, 93–101 CrossRef PubMed.
- G. S. Rao and G. Gorin, J. Org. Chem., 1959, 24, 749–753 CrossRef.
- A. Isabelle and G. Erwan, ChemBioChem, 2014, 15, 2361–2364 CrossRef PubMed.
- C.-M. Park, B. A. Johnson, J. Duan, J.-J. Park, J. J. Day, D. Gang, W.-J. Qian and M. Xian, Org. Lett., 2016, 18, 904–907 CrossRef PubMed.
- J. Kang, A. J. Ferrell, W. Chen, D. Wang and M. Xian, Org. Lett., 2018, 20, 852–855 CrossRef PubMed.
- Y. Zheng, B. Yu, Z. Li, Z. Yuan, C. L. Organ, R. K. Trivedi, S. Wang, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2017, 56, 11749–11753 CrossRef PubMed.
- B. Yu, Y. Zheng, Z. Yuan, S. Li, H. Zhu, L. K. De La Cruz, J. Zhang, K. Ji, S. Wang and B. Wang, J. Am. Chem. Soc., 2018, 140, 30–33 CrossRef PubMed.
- E. J. Emmett and M. C. Willis, Asian J. Org. Chem., 2015, 4, 602–611 CrossRef.
- I. Marco, A. L. Thompson, H. Woolven, C. Gonz and M. C. Willis, Org. Lett., 2011, 2005–2007 Search PubMed.
- D. Wu and Z. Meng, Arch. Environ. Contam. Toxicol., 2003, 45, 423 Search PubMed.
- G. Qin and Z. Meng, Toxicol. Lett., 2005, 160, 34 CrossRef PubMed.
- M. J. Pena-Egido, B. Garcia-Alonso and C. Garcia-Moreno, J. Agric. Food Chem., 2005, 53, 4198 CrossRef PubMed.
- G. Paino-Campa, M. J. Peña-Egido and C. García-Moreno, J. Sci. Food Agric., 1991, 56, 85 CrossRef.
- B. Garcia-Alonso, M. J. Pena-Egido and C. Garcia-Moreno, J. Agric. Food Chem., 2001, 49, 423 CrossRef PubMed.
- P. Bisseret and N. Blanchard, Org. Biomol. Chem., 2013, 11, 5393–5398 Search PubMed.
- H. Woolven, C. González-Rodríguez, I. Marco, A. L. Thompson and M. C. Willis, Org. Lett., 2011, 13, 4876–4878 CrossRef PubMed.
- S. R. Malwal, D. Sriram, P. Yogeeswari, V. B. Konkimalla and H. Chakrapani, J. Med. Chem., 2012, 55, 553–557 CrossRef PubMed.
- T. Fukuyama, M. Cheung, C.-K. Jow, Y. Hidai and T. Kan, Tetrahedron Lett., 1997, 38, 5831 CrossRef.
- S. R. Malwal, D. Sriram, P. Yogeeswari and H. Chakrapani, Bioorg. Med. Chem. Lett., 2012, 22, 3603–3606 CrossRef PubMed.
- E. Ge, M. Fan, H. Qiu, H. Hu, L. Tian, X. Wang, G. Xu and X. Wei, Environ. Pollut., 2017, 228, 408–415 CrossRef PubMed.
- K. A. Pardeshi, S. R. Malwal, A. Banerjee, S. Lahiri, R. Rangarajan and H. Chakrapani, Bioorg. Med. Chem. Lett., 2015, 25, 2694–2697 CrossRef PubMed.
- M.-Y. Wu, K. Li, C.-Y. Li, J.-T. Hou and X.-Q. Yu, Chem. Commun., 2014, 50, 183–185 RSC.
- P. Vogel, M. Turks, L. Bouchez, D. Marković, A. Varela-Álvarez and J. Á. Sordo, Acc. Chem. Res., 2007, 40, 931–942 CrossRef PubMed.
- F. Jung, M. Molin, R. Van der Elzen and T. Durst, J. Am. Chem. Soc., 1974, 96, 935–936 CrossRef.
- W. Wang, X. Ji, Z. Du and B. Wang, Chem. Commun., 2017, 53, 1370–1373 RSC.
- X. Ji, E. M. El-labbad, K. Ji, D. S. Lasheen, R. A. T. Serya, K. A. Abouzid and B. Wang, Org. Lett., 2017, 19, 818–821 CrossRef PubMed.
- W. Wang and B. Wang, Chem. Commun., 2017, 53, 10124–10127 RSC.
- S. R. Malwal and H. Chakrapani, Org. Biomol. Chem., 2015, 13, 2399–2406 RSC.
- R. Kodama, K. Sumaru, K. Morishita, T. Kanamori, K. Hyodo, T. Kamitanaka, M. Morimoto, S. Yokojima, S. Nakamura and K. Uchida, Chem. Commun., 2015, 51, 1736–1738 RSC.
- S. R. Malwal, M. Gudem, A. Hazra and H. Chakrapani, Org. Lett., 2013, 15, 1116–1119 CrossRef PubMed.
- J. J. Day, Z. Yang, W. Chen, A. Pacheco and M. Xian, ACS Chem. Biol., 2016, 11, 1647–1651 CrossRef PubMed.
- K. A. Pardeshi, G. Ravikumar and H. Chakrapani, Org. Lett., 2018, 20, 4–7 CrossRef PubMed.
- L. K. Keefer, ACS Chem. Biol., 2011, 6, 1147–1155 CrossRef PubMed.
- G. Ferrer-Sueta and R. Radi, ACS Chem. Biol., 2009, 4, 161–177 CrossRef PubMed.
- C. Szabo, H. Ischiropoulos and R. Radi, Nat. Rev. Drug Discovery, 2007, 6, 662–680 CrossRef PubMed.
- K. Sharma, A. Iyer, K. Sengupta and H. Chakrapani, Org. Lett., 2013, 15, 2636–2639 CrossRef PubMed.
- A. E. Maciag, J. E. Saavedra and H. Chakrapani, Anti-Cancer Agents Med. Chem., 2009, 9, 798 CrossRef PubMed.
- J. E. Saavedra, T. R. Billiar, D. L. Williams, Y.-M. Kim, S. C. Watkins and L. K. Keefer, J. Med. Chem., 1997, 40, 1947–1954 CrossRef PubMed.
- A. T. Dharmaraja, G. Ravikumar and H. Chakrapani, Org. Lett., 2014, 16, 2610–2613 CrossRef PubMed.
- K. Sharma, K. Sengupta and H. Chakrapani, Bioorg. Med. Chem. Lett., 2013, 23, 5964–5967 CrossRef PubMed.
- A. T. Dharmaraja, M. Alvala, D. Sriram, P. Yogeeswari and H. Chakrapani, Chem. Commun., 2012, 48, 10325–10327 RSC.
- A. T. Dharmaraja and H. Chakrapani, Org. Lett., 2014, 16, 398–401 CrossRef PubMed.
- A. T. Dharmaraja, C. Jain and H. Chakrapani, J. Org. Chem., 2014, 79, 9413–9417 CrossRef PubMed.
- R. H. Ritchie, G. R. Drummond, C. G. Sobey, T. M. De Silva and B. K. Kemp-Harper, Pharmacol. Res., 2017, 116, 57–69 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |