
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of the structure and characteristics of mesoporous TiO2 photocatalyst, and evaluation of its factors on gaseous formaldehyde removal by the analysis of ANOVA and S/N ratio
Shiuan-Shinn Leea,
Chi-Yuan Lu *ab and
Min-Chang Wua
*ab and
Min-Chang Wua
aDepartment of Public Health, Chung Shan Medical University, Taichung 40201, Taiwan, Republic of China. E-mail: cylu2280@csmu.edu.tw; Fax: +886-4-23248179; Tel: +886-4-24730022 ext. 12182
bDepartment of Family and Community Medicine, Chung Shan Medical University Hospital, Taichung 40201, Taiwan, Republic of China
First published on 18th June 2018
Abstract
This study differs from previous studies of TiO2/SiO2 in that 0.5–10 μm microsized TiO2-rutile based catalysts (TR catalysts) with varying proportions of titanium and silicon were synthesized using a one-step modified hydrothermal method. At Ti/Si = 1/9, a two-dimensional channel-structured catalyst with a morphology resembling that of SBA-15 was obtained. In contrast, at Ti/Si = 3/7 or 5/5, a three-dimensional porous structure was formed, and Ti–O–Si–C bonds appeared. The structure of the TR catalyst transformed due to the decrease in C–Si bond content and the increase in C–C bond content with increasing Ti/Si ratio. The results indicated that the rutile phase was the main crystal phase of the TR catalyst. The small crystal size and large rutile phase content of the mesoporous TR catalyst contributed to the low band gap energy below 3.0 eV. Under 2 × 10 W lamp irradiation with either UVA or visible light, the three TR catalysts showed better formaldehyde (HCHO) removal efficiency than P25. Furthermore, the Taguchi method was employed to evaluate the catalytic factors by analysis of variance (ANOVA) and S/N ratio. The results revealed the contributions of each of the three factors to HCHO removal efficiency over TR catalysts to be as follows: space velocity (62%), Ti ratio (32%), and time on stream (5%). The TR catalyst with Ti/Si = 1/9 showed good HCHO removal efficiency with a high SBET (787.1 m2 g−1) and large pore volume (0.95 cm3 g−1) for a residence time of over 2.29 × 10−1 s under visible light irradiation. Microwave-assisted EG reduction was successfully applied to dope a TR catalyst with nanosized Pt particles in a short synthesis time. After Pt doping, the removal efficiency in the stream improved and stabilized. The Pt particles were Pt0 and proved effective for improving the photocatalytic removal of HCHO over the TR catalyst by prolonging the separation time of the electron–hole pairs. Overall, the Pt/TR catalyst is a potential material for pollutant removal and can be easily separated from the pollutant removal system since the catalysts are microsized.
1. Introduction
With improvement in people's social living conditions, the pace of building renovations has increased. The pollution emitted by building-material manufacturing plants as well as furniture, decorations, and building materials used in homes all contain volatile organic compounds that indirectly release organic pollutant gases, deteriorating indoor air quality and damaging personal health. Considering that humans spend 80% of their time indoors,1 such as in offices, laboratories, or homes, improving the indoor air quality has become one of the most significant problems of this century. Of the various indoor pollutants, formaldehyde (HCHO) has attracted the most attention.2To efficiently remove hazardous organic gases, their deep oxidation and conversion to harmless CO2 and H2O is better than their adsorption since the absorbed organic gases may desorb due to some indoor factors such as temperature, moisture, volatilization of gases, and saturated absorption of the material. Among the various oxidation methods, photocatalytic oxidation has the potential to remove indoor organic gases with low energy consumption. Titanium dioxide (TiO2) is a classic photocatalyst; however, due to its extremely low activity under visible light, TiO2 reactions are performed only under UV light, which comprises less than 5% of the sun's total light output. If catalytic materials can be modified to function under visible light, photocatalysis can be extended to indoor applications and related uses.
Common modification methods include (1) addition of noble metals such as Pd, Au, Ag, and Pt;3–7 (2) combining TiO2 and other semiconductors such as CdS, CdSe, WO3, ZnO, SnO2, C3N4;8–11 (3) doping transition metals such as Co, Fe, Cu, and Cr;12–15 (4) adding nonmetallic molecules, e.g., C, N, S, and F;16–18 and (5) increasing the sensitization of TiO2 surface.19,20 Considering this, the current research directions involving surface modification/doping are as follows:3–20 (1) modification of absorption wavelength to shift the absorption range of photocatalyst from UV to visible wavelength; (2) reducing the band gap between the valence band and the conduction band; (3) effective electron–hole pair separation methods; (4) controlling reaction selectivity o reduce the production of intermediate products and increase the production of final product.
Besides the above studies, some investigations focused on the development of efficient photocatalysts with high specific surface areas (SBET) and large pore volumes, such as TiO2 hollow spheres,21 TiO2 nanofiber,22 Zr–TiO2 monolith,23 3D TiO2 foam,24 and mesoporous silica supported TiO2 catalyst.25
An ideal structure of a catalyst contributes to efficient catalysis by improving the four underlying catalytic mechanisms—absorption, migration, oxidation/reduction, and desorption.6,23–25 Santa Barbara Amorphous (SBA15), one of the silica family materials, has been modified with Ti atoms for broad-spectrum photocatalysis in recent years. Various mesoporous Ti-SBA15 catalysts have been synthesized by post-synthesis,25 sol–gel method,26 hydrothermal process,27,28 impregnation,29,30 grafting method,31 and gas deposition.32 These studies concluded that Ti-SBA15 synthesized with a well ratio of Si/Ti was advantageous for high photocatalytic activity such as high SBET (179–898 m2 g−1) and pore volume (0.34–1.45 cm3 g−1), large amount of OH group, good crystallinity of the anatase phase, and greater accessibility of photo-oxidative sites.25–32
However, most of the Ti-SBA15 catalysts were prepared by the grafting method after mixing the silica source and surfactant or post synthesis and then studied for liquid pollutant removal. Moreover, nanoscaled catalysts, Pt/TiO2,7 g-C3N4–TiO2,11 Pt/TiO2 nanofiber,22 and TiO2/diatomite,33 showed the good HCHO removal efficiencies. Few studies have focused on the removal of gaseous organic pollutants in a continuous flow reactor under low-power light irradiation over microscaled mesoporous photocatalyst since the indoor air quality is increasingly important nowadays. The aim of this study is to evaluate the potential of TiO2-rutile based (TR) catalyst for the removal of organic pollutant under visible light in a continuous flow reactor. Formaldehyde (HCHO) was chosen as the reactant in this study. Unlike the two-dimensional channel structure of Ti-SBA15 catalyst, two or three-dimensional structured TR catalysts were synthesized in this study by a one-step modified hydrothermal method via the premixing of silica and titanium precursors in three different Ti/Si ratios: 1/9, 3/7, and 5/5, respectively.
The catalytic efficiencies of one of the TR catalysts under UVA light were compared with that of a commercial TiO2–P25 catalyst. The photocatalytic activities of TR catalysts for HCHO removal for various Ti–Si proportions were studied under visible light irradiation. Then, the catalyst with the optimal reaction parameter was selected by Taguchi experimental design to evaluate the effects of factors such as ratio of Ti, space velocity, and time on stream. Finally, in order to tune the separation time of electron–hole pairs,34 Pt particles were doped on the TR catalyst by a microwave-assisted ethylene glycol (EG) reduction method to improve the removal efficiency of HCHO under low-power light irradiation. The catalysts were characterized by BET surface area analyzer, XRD, Raman spectroscopy, XPS, UV-visible DRS, PL spectroscopy, TEM, and FESEM.
2. Experimental
2.1. Synthesis of TR photocatalysts
TiO2-rutile based (TR) photocatalysts were synthesized using the modified hydrothermal method reported for SBA15 preparation. TR catalysts were obtained from the reaction of triblock copolymer Pluronic P123 (EO20PO70EO20, Aldrich), tetraethyl orthosilicate (TEOS, 98%, Aldrich), and titanium tetraisopropoxide (TTIP, 97%, Aldrich) under acidic conditions (HCl, 2 M) via the modified hydrothermal method. Ti/Si precursors, TEOS and TTIP, were pre-mixed in HCl solution, which was stirred at 20 °C. P123 was dissolved in HCl solution by agitation for 30 min. The solution of TEOS and TTIP was then added to the acidic solution of P123, and the mixture was heated at 100 °C for 24 h. The obtained material was filtered and washed with deionized water until pH = 7. Finally, the sample was calcined at 600 °C to remove the organic template and improve crystallinity. Three TR catalysts, TR(10), TR(30), and TR(50), were prepared with Ti/Si ratios of 1/9, 3/7, and 5/5, respectively.2.2. Synthesis of Pt/TR photocatalysts
Pt/TR photocatalysts were prepared by a microwave-assisted EG reduction method. The Pt precursor (H2PtCl6·6H2O) was added to EG and evenly stirred. This mixture was then added into the TR photocatalyst solution, prepared as mentioned above. After sufficient stirring, it was heated in a microwave at 500 W for 60 s. Subsequently, the Pt/TR photocatalyst was rinsed in alcohol and dried in an oven at 110 °C. Finally, the product was calcined to obtain the Pt/TR catalyst.2.3. Activity tests
Pre-prepared imitation gas was released into a catalytic reactor to initiate photocatalytic reaction. The catalytic reactor used in this study had an internal diameter of 20 mm, an external diameter of 22 mm, and a length of 310 mm. The reactor was placed upright to prevent bypass flow caused by catalyst sedimentation and to provide actual catalytic environment with minimal external interference from concentration and temperature gradients. A quartz filter plate was inserted into the quartz reaction tube to prepare the catalyst for photocatalytic reaction. Furthermore, the gaps in the filter plate allowed sufficient gas molecules to pass through. Following the reaction, portable gas analyzers (MIC-800) and Agilent gas chromatographs (Agilent GC-7890) were used to detect the change in gas concentration before and after the reaction. The data were entered online immediately using a personal computer. Finally, the reactor was irradiated with two low-power lamps from both the sides of reactor. The UVA light source was a T8 10 W black light lamp (365 nm), and the visible light source was a 10 W fluorescent lamp.The following activity tests were performed in this study. First, TR(10) catalyst was tested for HCHO removal and compared with P25 under UVA irradiation. A mixture of HCHO/oxygen in He + N2 was chosen to represent one of the organic pollutants. The HCHO was carried by He from a saturator filled with liquid HCHO. The concentration of HCHO was 40 ± 10 ppm, which was adjusted by the saturator temperature and mixed air flow rate. Second, three TR catalysts were tested under visible light irradiation. One of the catalysts with superior performances chosen for the study of optimum space velocity in a later experiment. Further, two TR catalysts with superior performances were chosen for Pt/TR catalyst preparation and HCHO removal. The HCHO removal efficiency was calculated from the pollutant consumption obtained by comparing the inlet and outlet concentrations of the pollutant.
2.4. Characterization of catalysts
Particle size, morphologies, and elemental composition of TR and Pt/TR catalysts were investigated by field emission scanning electron microscopy (FESEM; Model JSM-6700F, JEOL) operated at 5 kV accelerating voltage and equipped with X-ray energy-dispersive spectrometry (EDS). The dispersion of Pt particles in TR was observed using transmission electron microscopy (TEM, Philips 400T) operated at 120 keV. The samples were crushed in an agate mortar and suspended in ethanol. After ultrasonic dispersion, a droplet was deposited on a copper grid supporting a perforated carbon film.An X-ray powder diffractometer (XRD) (SIEMENS D5000) was used to identify the crystalline species of TR and Pt/TR catalysts. A Cu tube serving as the X-ray source was employed to estimate the active site phase at a working voltage and current of 40 kV and 30 mA, respectively. The powdered samples were pressed onto suitable holders and scanned in the 2θ range of 1 to 6° and 20° to 80°, respectively, at a scanning speed of 2° min−1. The diffraction patterns were manually analyzed with the Joint Committee of Powder Diffraction Standard (JCPDS) cards.
The specific surface areas of the TRcatalysts were measured at 77 K by gravimetric methods with an ASAP 2010 vacuum microbalance. The Brunauer–Emmett–Teller (BET) surface area was calculated from the adsorption isotherm, and the ratio of pore volume and average pore diameter was obtained from the pore size-volume distribution. UV-Vis diffuse reflectance (DRS) spectrum of the TR catalyst was measured from 190 to 1100 nm, and the photoluminescence (PL) spectrum of Pt/Ti-SBA15(I) catalyst was measured using a fluorescence spectrometer (Hitachi F-7000) with an excitation (EX) wavelength of 250 nm. X-ray photoelectron spectroscopy (XPS) was employed to analyze the chemical states of Ti, O, C, and Pt in the TR catalysts via Kratos Axis Ultra DLD.
2.5. Experimental design of Taguchi method
In order to study the effect of catalytic factors in this experiment, the Taguchi method35,36 was employed to efficiently design a representative experiment. Three factors were chosen as experimental parameters: the ratio of Ti, space velocity, and time on stream. According to the orthogonal arrays (OA) of Taguchi method, a L9 OA table with three levels was considered, as described in Table 1. The above three factors were labelled as A, B, and C, respectively, and the three levels were designed with different parameters. For factor A, 1, 2, and 3 were TR(10), TR(30), and TR(50); for factor B, 1, 2, and 3 were 3932 h−1, 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 727 h−1, and 31454 h−1; and for factor C, 1, 2, and 3 were 20, 40, and 60 min, respectively.
727 h−1, and 31454 h−1; and for factor C, 1, 2, and 3 were 20, 40, and 60 min, respectively.
| Level | Factor | ||
|---|---|---|---|
| A | B | C | |
| Ratio of Ti | Space velocity (h−1) | Time on stream (min) | |
| 1 | TR(10) | 3932 | 20 |
| 2 | TR(30) | 15727 | 40 |
| 3 | TR(50) | 31454 | 60 |
3. Results and discussion
3.1. Characterization of TR catalysts
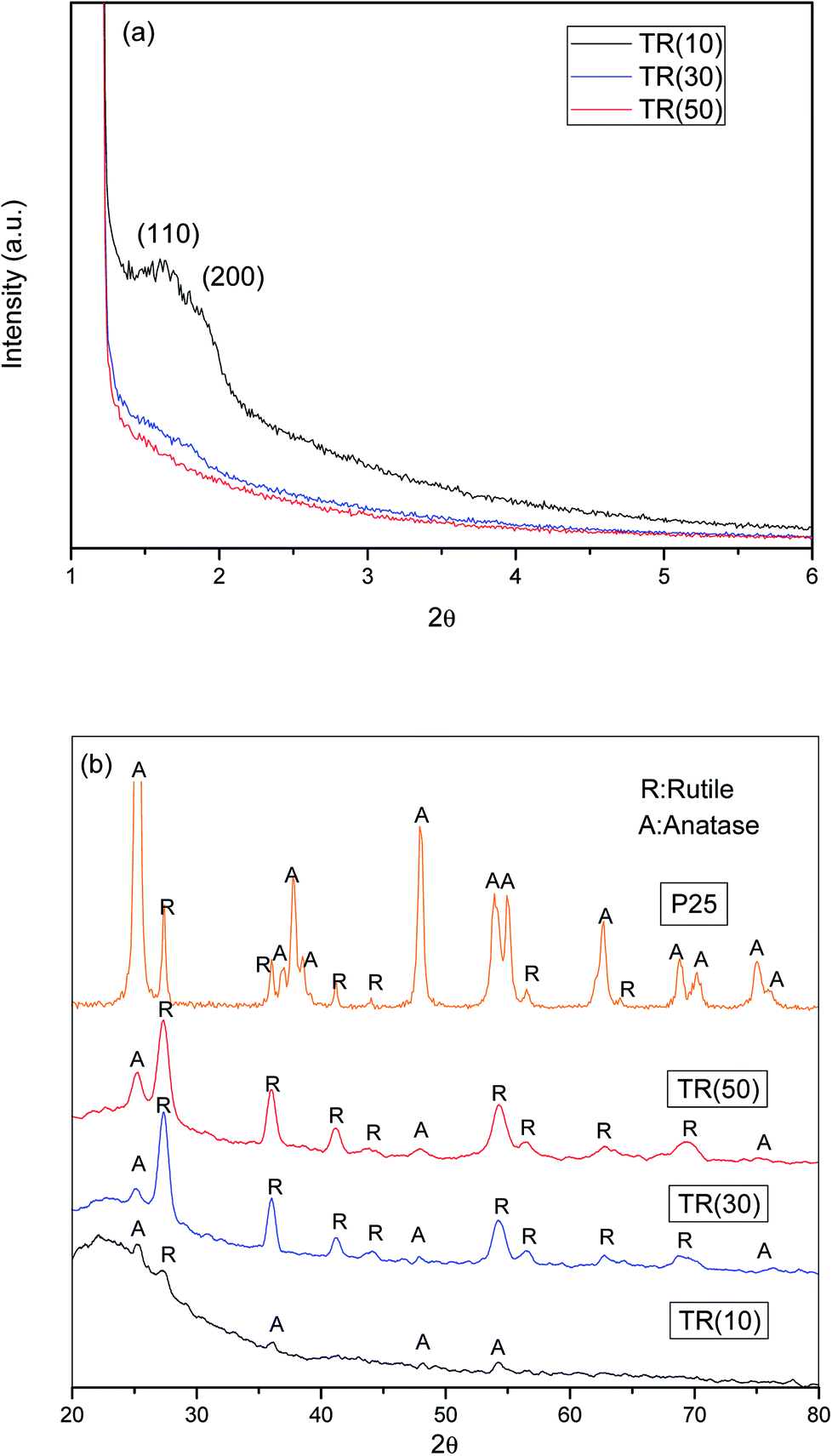 | ||
| Fig. 1 (a) Small angle XRD patterns TR catalysts; (b) XRD patterns of P25 and TR catalysts. | ||
(1) At pH < 1, P123–H+ formed from a neutral triblock copolymer surfactant, Pluronic 123, and H+ (HCl + H2O → H3O+ + Cl−).
(2) P123–H+ reacted with Cl− and H2O–TEOS+ to form P123–H+˙Cl−˙Si+.
(3) At longer reaction times, a hexagonal columnar structure (P6mm) formed through self-assembly due to condensation from the static electricity effect of 2{P123–H+˙Cl−·×H2O–Si+}m at a high temperature at longer reaction times.
(4) The obtained sample was calcined to remove the copolymer template.
The three TR catalysts were synthesized with different ratios of TTIP (Ti)/TEOS (Si). The hydrolysis rate and condensation of Ti atoms affected the framework of the TR catalyst. In Fig. 1(a), when the Ti/Si ratio was 1/9, only (110) and (200) peaks were observed in TR(10). When the Ti/Si ratio was increased to 5/5, all characteristic peaks disappeared in TR(50). As the Ti doping weight increased during the synthesis process, the results indicated that the structures of TR(30) and TR(50) changed from hexagonal straight pores (P6mm) to other structures, whereas TR(10) may have retained the main structure of SBA-15 since the (110) and (200) peaks remained.
Fig. 1(b) shows the XRD patterns of TiO2–P25 and the three TR catalysts measured from 20° to 80°. Comparison of the diffraction patterns shows that the crystalline species of Ti added in various proportions during the hydrothermal synthesis process differed slightly after calcination. The purpose of calcination was to remove the surfactant template efficiently and promote crystal formation by the constituent ions (Si4+ and Ti4+) after thermal treatment.37 However, at calcination temperatures higher than 600 °C, the unstable crystal phase of TiO2, anatase or brookite, transforms easily and completely into the rutile phase.38,39 In this study, when Ti/Si was 1/9, the main crystalline species in the TR(10) catalyst was anatase (2θ = 25.2°, 36.9°, 48.0°, and 53.8°). When the Ti/Si ratio was increased to 3/7, both the anatase and rutile crystalline species appeared in TR(30). Compared to that of P25, the crystalline diffraction peak of TR(30) was significantly higher for the rutile phase (2θ = 27.5°, 36.0°, 41.2°, 44.0°, 54.3°, 56.6°, 64.0°, and 69.0°) than for the anatase phase. However, the intensity of the anatase phase increased in TR(50) compared with that in TR(30) when the Ti/Si ratio increased to 5/5.
Acosta-Silva et al.25 reported that Si improves thermal stability and inhibits the transformation of anatase to rutile since the surrounding Si stabilizes the Ti–O bonds at the interface with SiO2. In addition, Resende et al.40 reported that a homogeneous silica layer was formed in contact with TiO2, which inhibited the appearance of the rutile phase because of insufficient atomic diffusion. The atomic mixing of Ti and Si retards phase separation and crystallization.41 Our results suggested that anatase phase transformation was inhibited in TR(10) when the ratio of Ti/Si was 1/9. However, when the Ti/Si ratio was 3/7 or 5/5, the two-dimensional hexagonal porous structure appeared to change, and the limited Si sites further decreased with Ti–O–Si bonding.29 Thus, at Ti/Si = 5/5 (TR(50)), the transformation of large amounts of anatase phase to the rutile phase was successfully inhibited.
000× magnification. Fig. 2(a) shows the classic pillar shape of SBA-15,37 with a particle size of approximately 1–3 μm, as shown in the inset of Fig. 2(a). SBA-15 has a two-dimensional channel structure with uniform pore channels, as shown in the 50000× image in Fig. 2(a). The pore channels could be clearly observed on the surface of SBA-15. Moreover, a small region of the samples were analyzed by EDS, as shown in Fig. (3). The EDS patterns of the three samples show that the elements in the particles are the same, including Ti, O, and Si.
 | ||
| Fig. 2 FESEM images of: (a) SBA15; (b) TR(10); (c) TR(30); (d) TR(50). | ||
 | ||
| Fig. 3 EDS analysis of: (a) TR(10); (b) TR(30); (c) TR(50). | ||
In contrast to traditional SBA-15, deformation occurred after the titanium precursor, TTIP, was premixed with the silica precursor, TEOS, during the hydrolysis/condensation process; the material no longer possessed the original structure of SBA-15. The FESEM images confirm the results of small-angle XRD analysis (Fig. 1(a)). As shown in Fig. 2(b), the TR(10) catalyst is irregularly shaped but has numerous holes distributed along its surface, similar to SBA-15. The structure of surface channels can be seen to have changed due to the embedding of Ti into the Si–O framework in TR(10). Moreover, the TR(10), with a diameter of approximately 10 μm, is larger than SBA-15, as shown in the inset of Fig. 2(b). The ionic radii of Ti and Si were 0.064 nm and 0.042 nm, respectively,42 which suggested that the bonding length of Ti–O–Si was longer than that of Si–O–Si. The Ti incorporated into the silica framework contributed to the large porous structure,28 as shown in the inset of Fig. 2(b).
With increasing Ti content, the structures of TR(30) and TR(50) transformed into a three-dimensional porous structure. Fig. 2(c) shows that the shape of TR(30) is a sphere formed by many long cylinders, resembling a cauliflower. The diameter of TR(30) is approximately 0.8 μm. Similar to TR(30), TR(50) (with Ti/Si = 5/5) is composed of large amounts of long cylinders and is shaped like an individual sphere. Although both TR(30) and TR(50) are spheres comprising innumerable long cylinders, TR(50) is slightly smaller at 0.5 μm, as shown in the inset of Fig. 2(d). Additionally, the cylinders in TR(50) are shorter than those in TR(30). The results suggest that the structure changed from two-dimensional to three-dimensional as the Ti/Si ratio increased to 3/7 or 5/5. The high concentration of Ti formed a different framework from that of the incorporated silica species.43 Thereafter, the decreased number of Si sites around the P123 surfactant and the shorter hydrolysis time of Ti resulted in a three-dimensional porous structure after the condensation process.27
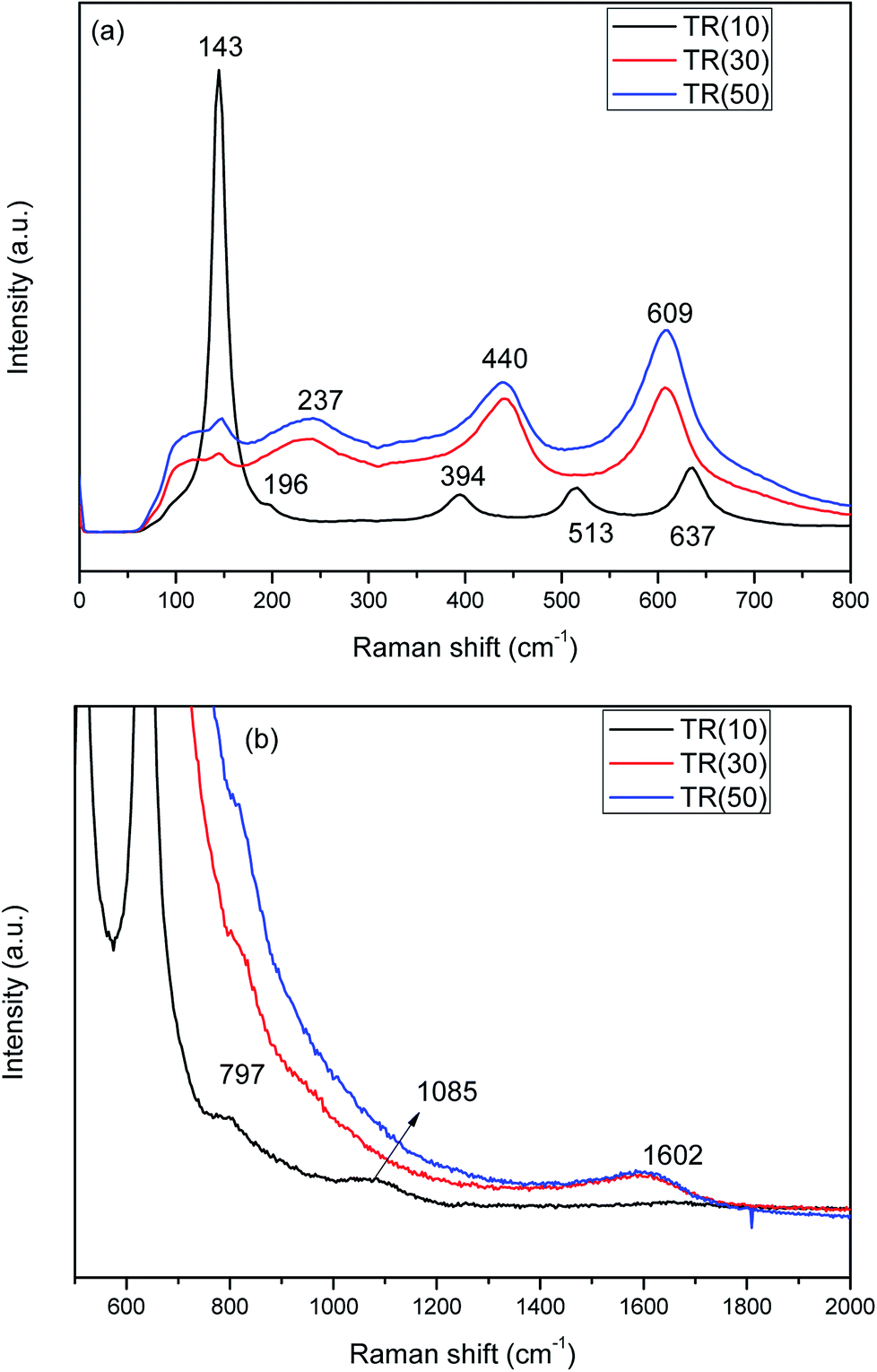 | ||
| Fig. 4 Raman spectra of three TR catalysts: (a) Raman shift: 0–800 cm−1; (b) Raman shift: 500–2000 cm−1. | ||
In contrast, the peaks at 237, 440, and 609 cm−1 are attributed to the rutile phase in TR(30) and TR(50). Furthermore, in TR(30) and TR(50), the anatase peaks are almost overlapped by the rutile peaks (237, 440, and 609 cm−1). In addition, the peak at 143 cm−1, which is characteristic of the anatase phase, shows a stronger intensity for TR(50) than for TR(30); this reveals that TR(50) possesses a greater amount of anatase phase than TR(30). The Raman results thus confirm the XRD analysis results.
Since the anatase and rutile phases coexisted in the TR catalysts, the ratio of the anatase/rutile phase was expected to affect the photocatalytic activity.45,46 Thus, the content fractions of anatase and rutile in the TR catalysts were calculated by the Spurr and Myers equation,47 as shown in Table 2. The fraction of rutile phase is in the order of TR(30) (81.5%) > TR(50) (77.9%) > TR(10) (56.9%).
| Catalyst | Crystal size of anatase | Crystal size of rutile | Average crystal size | Component of anatase | Component of rutile | Crystallinity of anatase | Crystallinity of rutile |
|---|---|---|---|---|---|---|---|
| (nm) | (%) | (%) | |||||
| P25 | 19.9 | 24.8 | 20.6 | 86.1 | 13.9 | 100 | 14.3 |
| TR(10) | 50.5 | 30.7 | 39.2 | 43.1 | 56.9 | 85.5 | 100 |
| TR(30) | 26.2 | 11.6 | 14.3 | 18.5 | 81.5 | 25.6 | 100 |
| TR(50) | 14.3 | 13.2 | 13.4 | 22.1 | 77.9 | 32.0 | 100 |
The rutile phase content varied with the Ti/Si ratios of the TR catalysts. According to earlier studies,25,29 Si enhances the thermal stability and crystallinity of TiO2 during calcination, as shown in Table 2. By comparing with the intensity of standard XRD patterns of TiO2, crystallinities of anatase and rutile phase were calculated. The higher degree of crystallinity would reduce the possibility of electron and hole recombination at a defective site and enhance the photocatalytic activity.39 As shown in Fig. 2(b), embedded Ti was observed on the surface of the two-dimensional channel structure over TR(10). At a low Ti content, Ti is incorporated into the silica framework, and Si inhibits the transformation of the anatase phase to the rutile phase. After calcination, the anatase phase content of TR(10) was 43.1%. However, the suppression of the TiO2 phase transformation in TR(10) was not the same as in TR(30) and TR(50). The transformation inhibition did not decrease with large amounts of added TiO2. In general, the low inhibition efficiency was mainly attributed to the reduced assistance from the silica framework. The three-dimensional porous structures of TR(30) and TR(50) also contributed to the different inhibition effects during calcination. The long cylinders of TR(30) led to a lower anatase content (18.5%) than the short cylinders of TR(50) (22.1%). The result suggests that the longer Si–O–Ti bonding length in a three-dimensional porous structure is one of the factors affecting the TiO2 crystal phase during calcination, leading to the lower inhibition of TiO2 phase transformation. Furthermore, the compositions of the TiO2 crystalline phase in the three TR catalysts were not the same as those in commercial TiO2–P25 (Table 2). The fraction of anatase phase in P25 (86.1%) is higher than that in the three TR catalysts. Moreover, the photocatalytic activity of the TR catalyst is different from that of traditional TiO2.
Fig. 4(b) shows the Raman spectra of TR catalysts measured from 500 to 2000 cm−1. The band at 797 cm−1 corresponds to the symmetric stretching mode of Si–O–Si in the three TR catalysts.48 With increasing Ti/Si ratio, the intensity of the Si–O–Si peak decreased in TR(30) and TR(50). The band located at 1085 cm−1 was assigned to the symmetric stretching vibration of the Ti–O–Si bond in TR(10).28 These results are in agreement with the results of FESEM and XRD. At a low Ti/Si ratio, the Ti ions were incorporated into the silica framework to form a two-dimensional channel structure (Fig. 2(b)). Thus, the thermal stability of the anatase phase improved and contributed to the inhibition of the phase transformation and sintering of TR(10).
In Fig. 4(b), the peak at 1602 cm−1 corresponds to the IG mode of carbon,49 whose sources are the precursors TEOS and TTIP and the surfactant P123. Graphite crystal was clearly observed in TR(30) and TR(50), whereas no defect carbon (ID mode of carbon) appeared at 1350 cm−1. Thus, the shift in the spectra was not affected by carbon. Without defect carbon, the excited electron would not scatter during analysis.50 This result indicates that C–C bonds existed in the structures of TR(30) and TR(50), while the decreased Si sites around the P123 surfactant and the shorter hydrolysis time of Ti contributed to the structure formed after the condensation process. These factors contributed to the transformation of a two-dimensional channel column to a three-dimensional porous spherical structure.
 | ||
| Fig. 5 Ti2p XPS spectra of TR(10), TR(30), and TR(50) catalysts. | ||
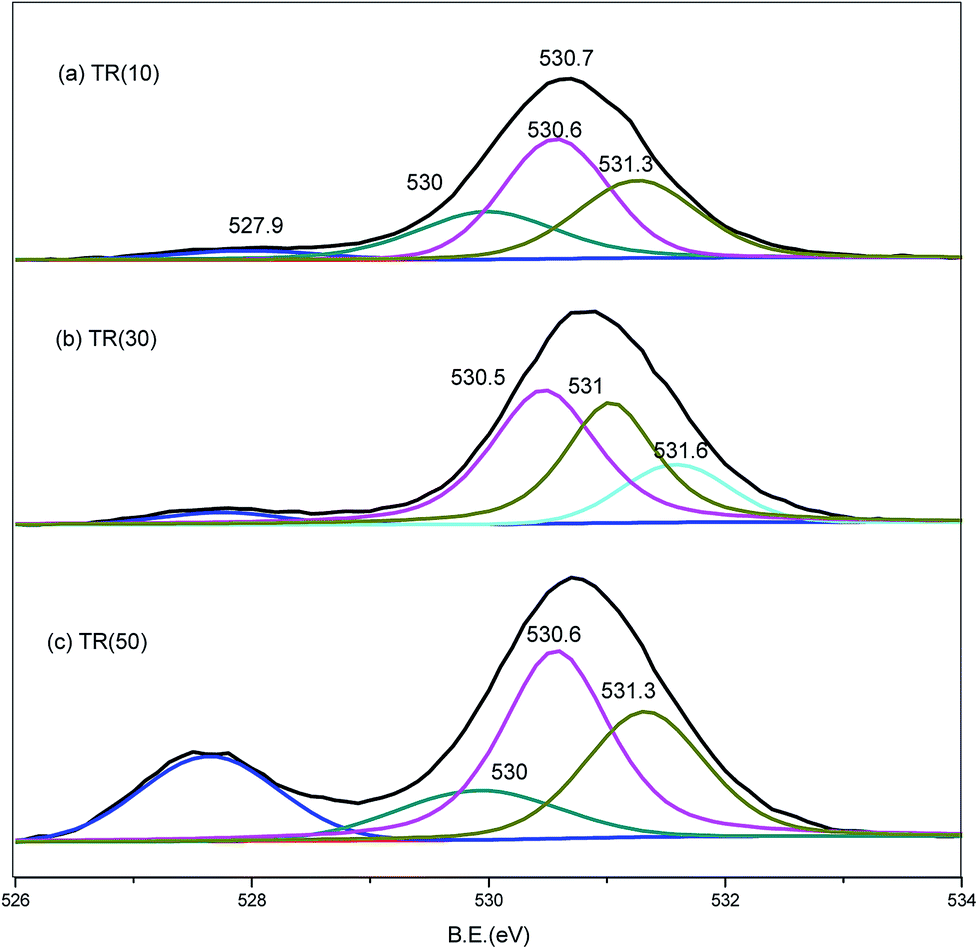 | ||
| Fig. 6 O1s XPS spectra of TR(10), TR(30), and TR(50) catalysts. | ||
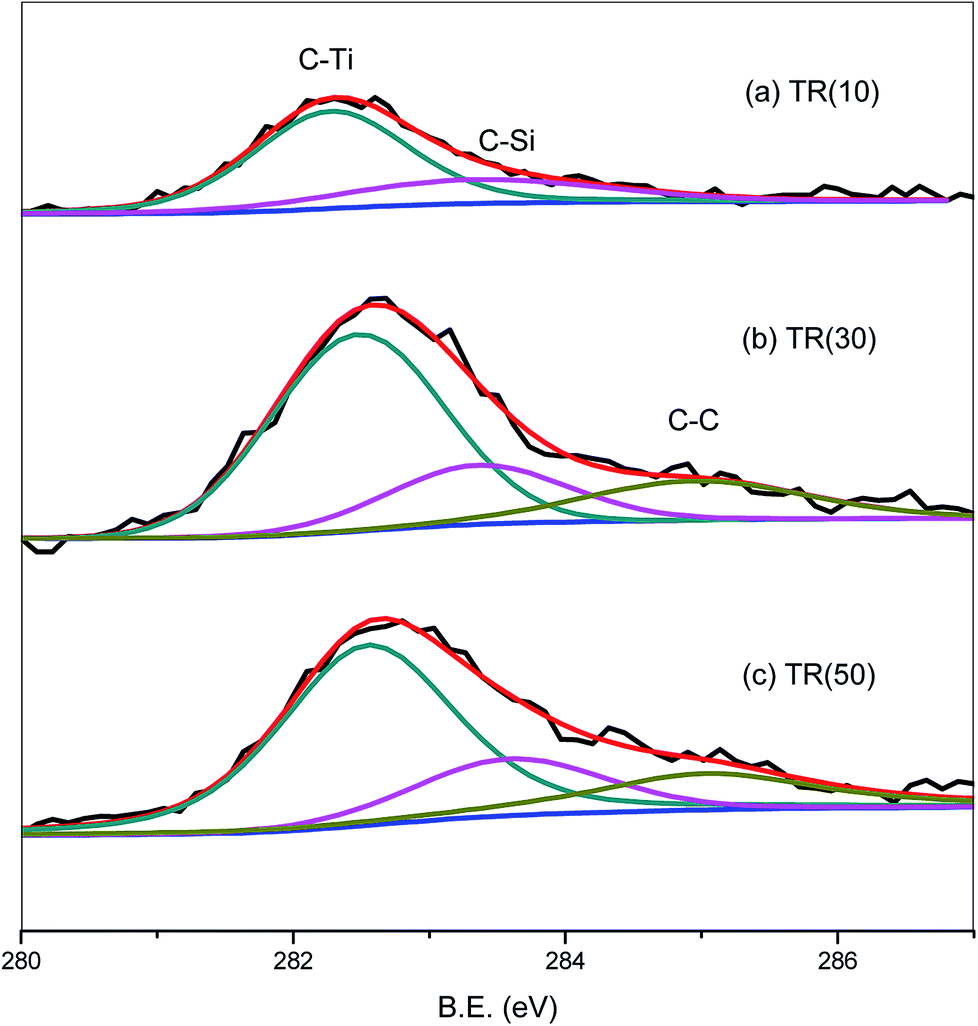 | ||
| Fig. 7 C1s XPS spectra of TR(10), TR(30), and TR(50) catalysts. | ||
Similar to the Ti2p spectra, the O1s spectra shifted downward to a binding energy of −2.0 eV for the three catalysts, as shown in Fig. 6. The band at 527.9 eV corresponded to the lattice oxygen of the Ti–O–Ti bond.25,28,43,51 As the Ti/Si ratio increased, the intensity of the Ti–O–Ti bond became stronger for TR(50). The O1s peak at 530.1 eV was deconvoluted into three peaks. The resolved peaks of TR(10) and TR(50) were similar, and the binding energies of the three peaks were 530.0, 530.6, and 531.3 eV, respectively. For TR(30), the O1s peak decomposed into three peaks with binding energies of 530.5, 531.0, and 531.6 eV.
The peak at 530.0 eV was related to the absorbed oxygen on the surface. Mazur et al.51 reported that the absorbed oxygen originated from H2O and hydroxyl radicals. No decomposed peak appeared at 530.0 eV in TR(30); however, there was a peak at 531.6 eV related to the Si–OH bond or absorbed H2O.53 The oxygen bond content arising from the H2O or hydroxyl groups was calculated in relation to the total oxygen bond content, and the order was TR(10) (23.9%) > TR(30) (15.6%) > TR(50) (13.0%). This result was in agreement with earlier studies,38,40 which suggested that Si facilitated the bonding of OH groups onto the TR catalyst. The peaks at 530.5/530.6 eV and 531.0./531.3 eV corresponded to the Ti–O–Si and Si–O–Si bonds, respectively. As mentioned above, the peak for defect oxygen arose from the Ti atoms incorporated into the silica framework of TR(10). In TR(30) and TR(50), the strong interaction of the Ti–O–Si bonds between TiO2 and SiO2 resulted in the formation of a three-dimensional porous structure.53
The C1s spectra of the three catalysts are shown in Fig. 7. The C1s peak was deconvoluted into three peaks related to the C–Ti, C–Si, and C–C bonds at 282.1, 283.1, and 284.3 eV, respectively.54 The C1s peak of TR(10) was weaker than that of the other two catalysts, possibly because the C–C bond, which appeared in samples with a Ti/Si ratio greater than 3/7, contributed to the stronger C1s peak. For TR(10), the carbon bond content is 71.8% for C–Ti and 28.2% for C–Si. With increasing Ti loading weight, the hydrolysis and condensation of Ti and Si surrounded by the P123 surfactant contributed to the production of C–C bonds after calcination. The C–C bond content was calculated in relation to the total carbon bond content, and the order was TR(50) (19.1%) > TR(30) (17.6%). The proportions of C–Ti and C–Si in TR(30) and TR(50) were lower than those in TR(10). The C–Si bond content was calculated in relation to the total carbon bonds, and the order was TR(10) (28.2%) > TR(30) (20.1%) > TR(50) (19.4%). Although the amount of carbon in the catalysts was very small, the analysis of the C–Si bonds revealed that the framework of the catalysts could be transformed by reducing the C–Si bond content and increasing the C–C bond content. This transformation might be the other factor responsible for the formation of a three-dimensional porous structure. Ti–O–Si–C bonds would appear in the three-dimensional structured catalyst. The results revealed that the observed G-band of the Raman spectra, which appeared only in TR(30) and TR(50), arose from the C–C bond, as shown in Fig. 4(b).
| Catalyst | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Avg. pore diameter (nm) | ||
|---|---|---|---|---|---|
| TR(10) | 787.1 | 0.95 | Macro | 0.01 | 4.82 |
| Meso | 0.6 | ||||
| Micro | 0.34 | ||||
| TR(30) | 463.5 | 0.88 | Macro | 0.03 | 7.61 |
| Meso | 0.66 | ||||
| Micro | 0.19 | ||||
| TR(50) | 419 | 0.54 | Macro | 0.01 | 5.18 |
| Meso | 0.37 | ||||
| Micro | 0.16 | ||||
| P25 | 53.4 | 0.13 | Macro | 0.02 | 9.88 |
| Meso | 0.09 | ||||
| Micro | 0.02 | ||||
The average pore diameter of the three samples decreased in the order TR(30) (7.61 nm) > TR(50) (5.18 nm) > TR(10) (4.82 nm). The FESEM images (Fig. 2) revealed that the pores of TR(30) and TR(50) mainly arose from the gaps between cylinders, whereas hexagonal channels formed the pores of TR(10). The proportion of mesoporous volume followed the order TR(30) (74.9%) > TR(50) (67.4%) > TR(10) (64.2%). The proportions of mesoporous structures for all TR catalysts were above 64%. This result suggested that the gaseous molecules could react efficiently with active sites over mesoporous TR catalysts during photocatalytic oxidation since the molecular size of HCHO is 0.27 nm.23
3.2. Formaldehyde removal efficiencies of TR catalysts
591 h−1 under 2 × 10 W UVA irradiation. The results of the experiment indicated that TR(10) possessed superior formaldehyde photocatalytic removal efficiency to that of P25. Within 20 min, TR(10) had removed 99% of the formaldehyde, whereas P25 had removed only 20% to 30%.
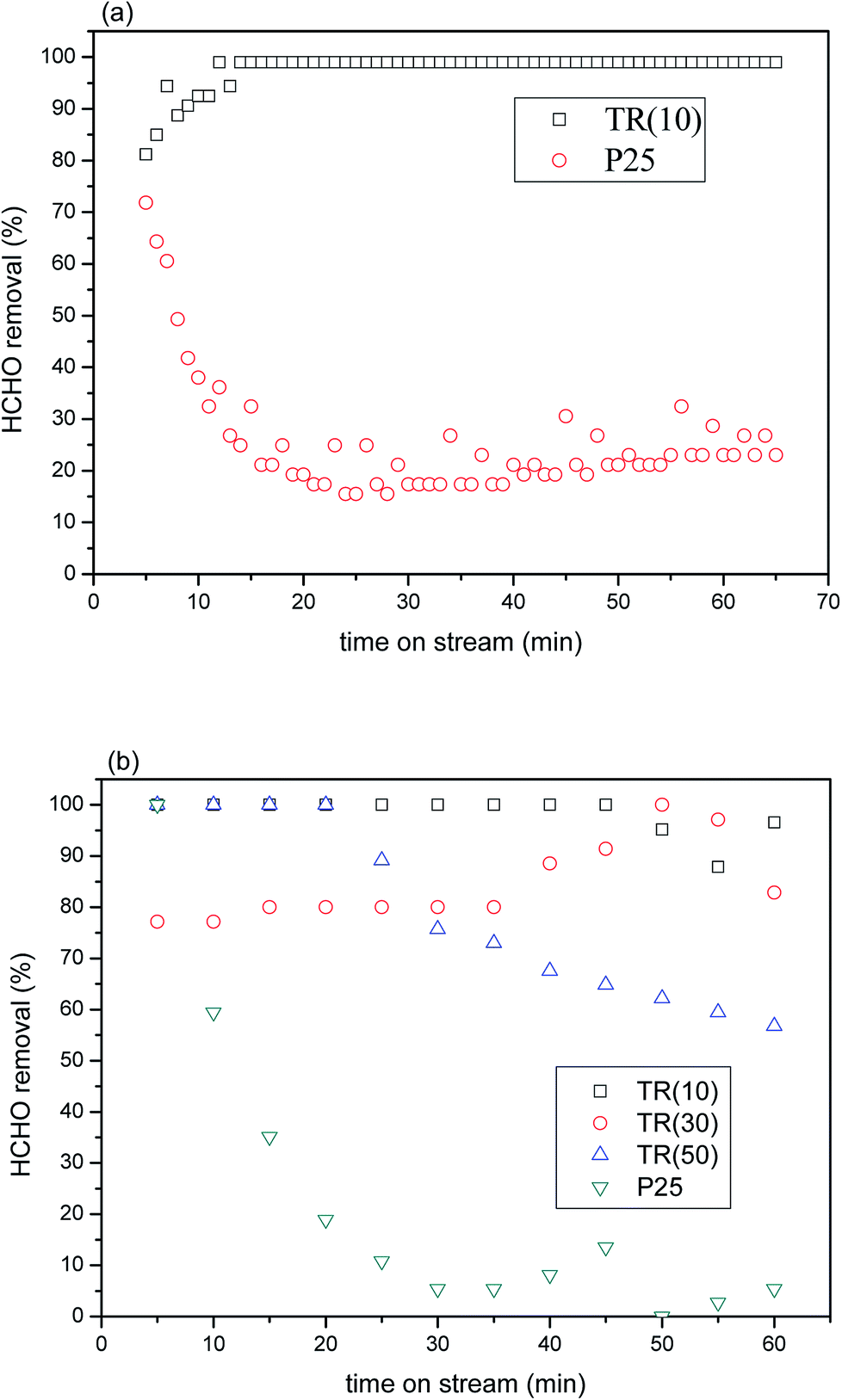 | ||
| Fig. 8 (a) HCHO removal over P25 and TR(10) catalysts as a function of reaction time under UVA irradiation (feed composition: 40 ppm HCHO, SV = 23591 h−1); (b) HCHO removal over P25 and three TR catalysts as a function of reaction time under visible light irradiation (feed composition: 40 ppm HCHO, SV = 3932 h−1). | ||
The results demonstrated that the shift in the light wavelength from the UVB to the UVA region contributed to the low photocatalytic activity of P25 with a retention time of 1.52 × 10−1 s. In general, the maximum absorption wavelength of light for commercial TiO2–P25 was 320 nm; thus, little energy was obtained from UVA (365 nm) irradiation. Therefore, few photons were excited for the separation of electron–hole pairs, which contributed to the low photocatalytic oxidation efficiency of HCHO.55 In contrast, the proportion of the rutile phase was above 56.9% in the three TR catalysts, whereas that in P25 was only 13.9%, as shown in Table 1. The results under near-visible light irradiation suggested that more superoxide radical anions would be produced from the photoexcitation process e− + O2ads → O2˙−, improving HCHO oxidation. Furthermore, the larger specific surface area and pore volume of TR(10) would provide stronger adsorption capacity during the photocatalytic reaction; then, the HCHO molecules would migrate and react with the active site efficiency via the mesoporous structure, possibly resulting in the superior photocatalytic capabilities of TR(10) to those of P25. The results indicated that a TR catalyst has potential for use as a photocatalytic material for HCHO removal.
The removal efficiencies of the TR catalysts varied after a period of reaction time in the HCHO stream. After 45 min of time on stream, TR(10) attained a removal efficiency of 87–96%. TR(30) attained a removal efficiency of approximately 82–97% after 35 min. TR(50) attained a removal efficiency of 56–89% after 20 min. Multiple factors were assumed to contribute to the differences in the removal efficiencies. First, the morphologies and textural structures of the three samples were different, as shown in Fig. 2 and Table 3. The SBET, pore volume, and average pore diameter directly affect the photocatalytic efficiency. All samples were mesoporous structures, and the HCHO molecule would be most easily absorbed on a catalyst with a high SBET and large pore volume, such as TR(10), which is why TR(10) showed the most stable HCHO removal for the longest time, as shown in Fig. 8(b).
The literature25,44,56 recommends adding an appropriate amount of SiO2 to improve the photocatalytic properties of TiO2 because increasing the SiO2 content may increase the surface acidity of TiO2. SiO2-modified TiO2 exhibited the greatest surface acidity improvement, not only increasing the adsorption of organic matter on the TiO2 surface but also forming strong OH groups in the hole capture sites on the surface, thereby reducing the probability of electron–hole pairing. This process generated OH groups with strong oxidation activity and improved the photocatalytic properties. According to the XPS analysis of O1s (Fig. 6), TR(10) showed 23.9% oxygen bonds arising from H2Oabs or hydroxyl groups, TR(30) showed 15.6%, and TR(50) showed 13.0%. The amount of absorbed oxygen corresponded to the removal efficiency of HCHO. Finally, comparing Fig. 8(b) and (a) could suggest that the photocatalytic activity of the TR catalyst might be affected by the different absorption of light energy under visible light irradiation since the proportion of the rutile phase was not the same in all TR catalysts, as shown in Table 2. Overall, the TR catalysts showed better photocatalytic efficiency than commercial P25 regardless of whether the light source was UVA or visible light, showing that TR catalysts have potential for use as photocatalytic materials.
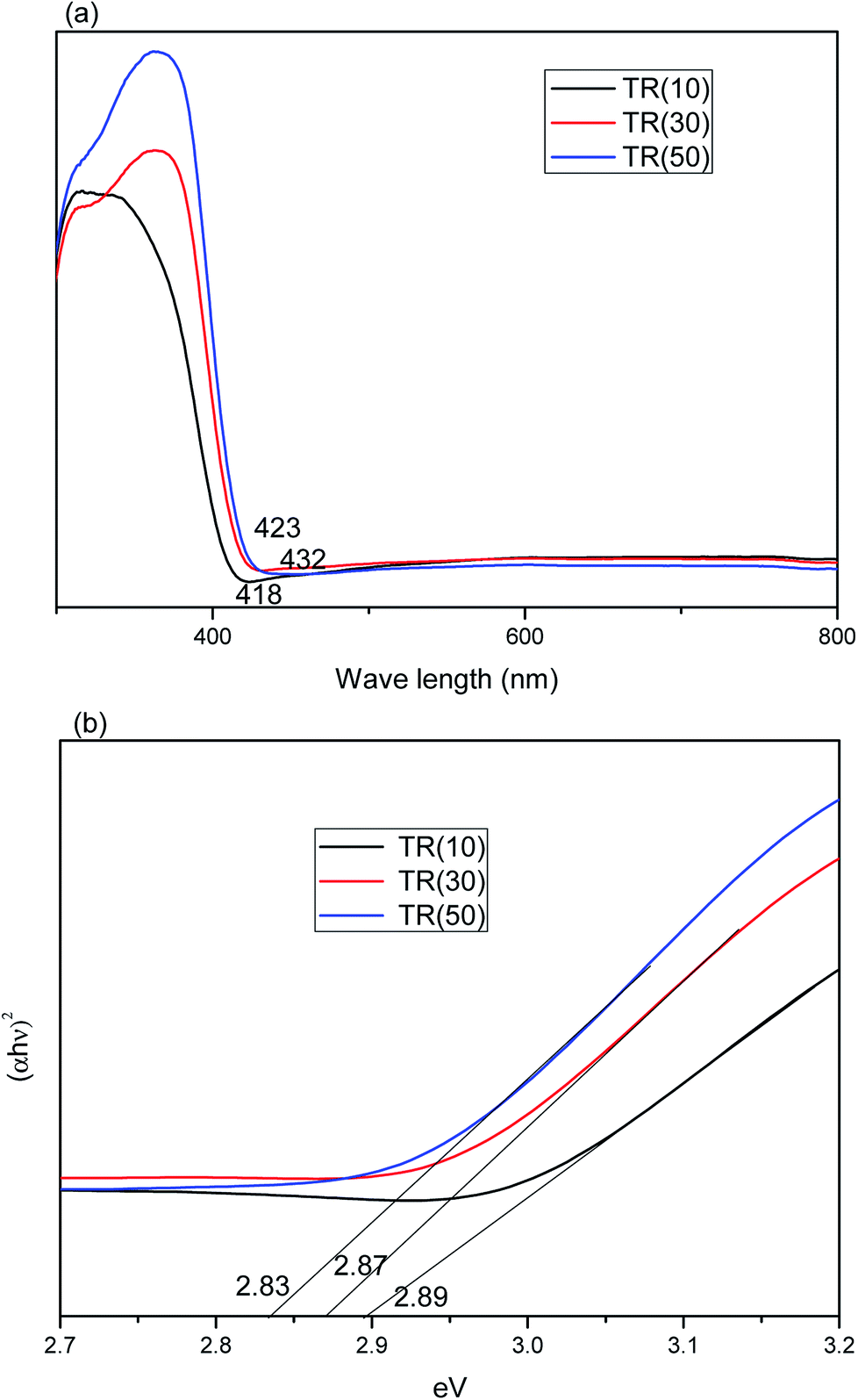 | ||
| Fig. 9 (a) Absorption spectra of TR(10), TR(30), and TR(50) catalysts; (b) band gap calculation of (αhν)2 vs. photon energy for TR catalysts. | ||
The total amount of rutile in the TR catalyst could be calculated by the formula Rtotal (%) = Ti weight (%) × XR (%). The Rtotal values for the photocatalysts followed the order TR(50) (38.9%) > TR(30) (24.4%) > TR(10) (5.6%). Moreover, the crystal size of the rutile phase in the TR catalysts was smaller than that of the anatase phase, as shown in Table 2. This result was not the same as that observed for commercial TiO2 (P25). The synergistic effect of the smaller rutile crystal size and higher rutile phase content would enhance the photocatalytic activity. Thus, the optical absorption edge of TR(50) was at a wavelength of 432 nm, longer than that of the others. This result indicated that TR(50) would separate electron–hole pairs under visible light irradiation more easily than the others.
Moreover, the UV-Vis DRS analysis results can be employed to calculate the band gap energy of the TR catalysts from the plot of [αhν]2 vs. the photo energy,55 as shown in Fig. 9(b). The band gap energy of the three TR catalysts was consistently below 3.2 eV (pure anatase phase) or 3.0 eV (pure rutile phase). The band gap energy of the TR catalyst followed the order TR(50) (2.83 eV) < TR(30) (2.87 eV) < TR(10) (2.89 eV). The results revealed that a higher rutile phase content contributed to a lower band gap. The band edge positions of the TR catalyst can be calculated by the empirical equation57 ECB = X − Ec − 0.5Eg, where ECB is the conduction band edge potential; X is the absolute electronegativity of the semiconductor (X value for TiO2 is 5.81 eV); Ec is the energy of free electrons on the hydrogen scale (approximately 4.5 eV); and Eg is the band gap of the TR catalyst. The predicted band positions at the point of zero charge of TR(10), TR(30), and TR(50) were −0.135 eV–2.755 eV, −0.125 eV–2.745 eV, and −0.105 eV–2.725 eV, respectively.
However, earlier studies45,46 indicated that the recombination of electron–hole pairs was faster for the rutile phase than for the anatase phase since photoexcitation would occur easily over the rutile phase with low band gap energy. This observation was corroborated by comparing Fig. 8(b) with Fig. 9(b): TR(50) showed the lowest band gap energy, but its HCHO removal efficiency decreased with reaction time due to the rapid recombination rate of the electron–hole pairs. Therefore, the drawback of the short recombination time of electron–hole pairs for the rutile phase needs to be resolved to prolong the reaction time of photocatalytic oxidation.
3.3. Taguchi method for optimizing the catalytic factor over TR catalysts
To tune the operational parameters for HCHO removal over TR catalysts, three catalytic factors were studied in nine tests designed by the orthogonal array L9 of the Taguchi method, as shown in Tables 1 and 4. The experimental results were analyzed by using the S/N ratio and one-way analysis of variance (ANOVA).| Run | Factor | Removal (%) | ||
|---|---|---|---|---|
| A | B | C | ||
| Ratio of Ti | Space velocity | Time on stream | ||
| 1 | Level 1 | Level 1 | Level 1 | 99.1 |
| 2 | Level 1 | Level 2 | Level 2 | 99.0 |
| 3 | Level 1 | Level 3 | Level 3 | 43.8 |
| 4 | Level 2 | Level 1 | Level 3 | 82.8 |
| 5 | Level 2 | Level 2 | Level 1 | 79.0 |
| 6 | Level 2 | Level 3 | Level 2 | 49.3 |
| 7 | Level 3 | Level 1 | Level 2 | 67.6 |
| 8 | Level 3 | Level 2 | Level 3 | 50.1 |
| 9 | Level 3 | Level 3 | Level 1 | 25.2 |
The S/N ratio is the ratio of signal to noise and was calculated according to the degradation efficiencies for the corresponding factors at each level tested. S/N serves as an indicator of the experimental quality, has the additive effect of an experimental factor and reduces interactions between factors. The larger the value of the S/N is, the smaller is the loss of experimental quality or the smaller is the variation in the quality. The corresponding chart of experimental results for the individual effects of various parameters at each level is shown in Fig. 10. For factor A (proportion of Ti), the S/N values followed the order TR(10) > TR(30) > TR(50). For factor B (space velocity), the S/N values were 3932 h−1 >15727 h−1 > 31454 h−1. For factor C (time on stream), the S/N values were 40 min > 20 min > 60 min. The more extreme the difference in the S/N values was, the more significant was the effect. The level effects of the three factors were in the order B (6.5) > A (4.6) > C (1.7). The ranks of the three factors varied in the order B > A > C. Factor B, space velocity, had a significant effect on the photocatalytic activity for HCHO removal, which indicated that mass transfer was the principal factor for TR catalysts, since the mesoporous structures of TR catalysts were variable. Factor A, the ratio of Ti, had a minor effect, which demonstrated that a low Ti loading wt% would result in better catalytic activity than a high Ti loading. Factor C, time on stream, had the least impact on the photocatalytic activity. The lack of an obvious decay effect on the TR catalysts during the reaction was suggested to be because appreciable coke deposition did not occur on the TR catalysts due to the amount of surface acidity on TR catalysts (discussed in Section 3.2.2). In this study, the optimum test parameter for the ratio of Ti was level 1 (TR(10)); that for the space velocity was level 1 (3932 h−1); and that for the time on stream was level 2 (40 min).
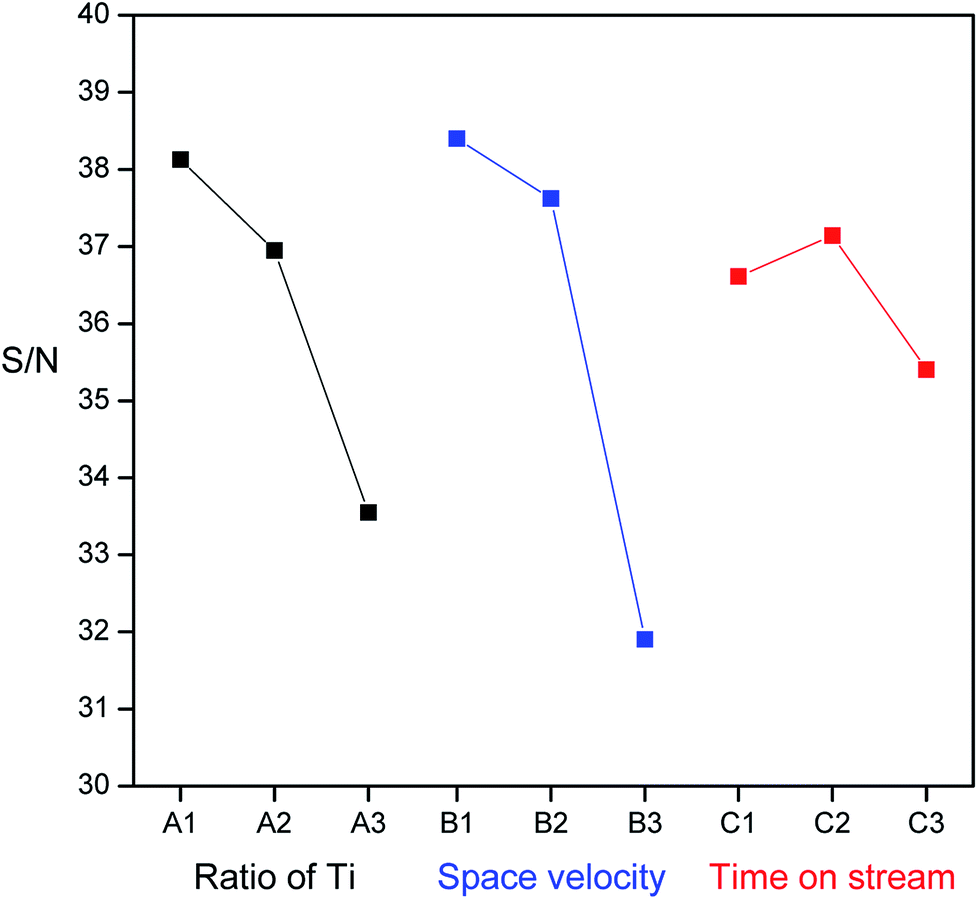 | ||
| Fig. 10 The effect of each factor (ratio of Ti, space velocity, and reaction time) on the values of S/N ratio. | ||
Then, ANOVA was used to analyze the influence of various test parameters on the HCHO removal efficiency in nine runs. Table 5 shows the ANOVA results for the photocatalytic removal of HCHO with a 95% confidence interval and two degrees of freedom. The degree of residual value in the ANOVA analysis was 6. The contributions of the three factors to HCHO removal efficiency over TR catalysts were 32%, 62%, and 5%, respectively. Space velocity had a significant influence on HCHO removal. The contributions of factors could be ranked as follows: space velocity > ratio of Ti > time on stream. The time on stream had very little effect on the TR catalyst and could be neglected for HCHO removal. Furthermore, the error contribution to the total variance was 1%, which is lower than 15%, and this result indicated that no significant parameters were ignored in the experimental design. The three factors chosen in this study were reasonable and credible.58 The effects of the studied factors on the response function were evaluated by the F value of the three factors, which varied in the order B (7.35) > A (2.13) > C (0.24). Only the F value of factor B was found to be larger than the critical value of F, where Fcritical (0.05, 2, 6) was 5.143. This finding rejected the null hypothesis that the standard deviations were equal and demonstrated differences in the variances in theory. The order of the effects of the three factors on HCHO removal over TR catalysts was factor B > factor A > factor C, which was the same as that obtained by investigation of the S/N ratio. Finally, the P value was studied. The F value of 7.35 and the P value of 0.02 for factor B (space velocity) demonstrated the real and high significance of factor B for HCHO removal over TR catalysts. In contrast, a P value > 0.05 meant that HCHO removal efficiency was not significantly affected by factor A and factor C, even though factor A showed a 32% contribution in the catalytic test.
| Factor | DF | SS | MS | Contribute | F value | P value |
|---|---|---|---|---|---|---|
| A | 2 | 1715.76 | 857.88 | 0.32 | 2.13 | 0.20 |
| B | 2 | 3310.38 | 1655.19 | 0.62 | 7.35 | 0.02 |
| C | 2 | 266.08 | 133.04 | 0.05 | 0.24 | 0.80 |
| Residual | 6 | 44.84 | 7.47 | 0.01 | ||
| Total | 12 | 5337.06 |
In the following study, the better-performing catalysts TR(10) and TR(30) were chosen for Pt/TR catalyst preparation to evaluate the assisting effect of metal-Pt particles on HCHO removal over TR catalysts. Before testing the activity of Pt/TR catalysts, the optimum space velocity should be considered to assess whether Pt/TR(10) or Pt/TR(30) could be a potential catalyst for HCHO removal.
3.4. Optimum space velocity for HCHO removal over TR catalysts
In this study, TR catalysts showed good photocatalytic activity for HCHO removal at a reaction space velocity of 3932 h−1 under visible light irradiation. To confirm the existence of a mass transfer gradient effect on the chemical reactions in the continuous reactor used in this study, we selected the better catalyst—TR(10)—to measure the actual effects of the catalytic reaction weight and gas flow velocity on the photocatalytic reactions. The primary factors controlling the heterogeneous photocatalytic reaction rates were the dispersion and surface reaction. When the size of the photoreactor and the light intensity were fixed, variations in the gas flow affected the photocatalytic reaction. A varying gas flow affects not only the residence time but also the reaction gas flow. Because low gas flows are slow, the flow in the reactor was similar to a laminar flow, hindering interaction between the reaction gas and the photocatalyst and limiting the ability of the reaction gas to diffuse onto the surface of the photocatalyst; thus, the reaction was deemed to have been caused by reaction gas diffusion. Conversely, as the gas flow was gradually increased, the flow created turbulence equivalent to a mixing process. Therefore, the probability of interaction between the reaction gas and the photocatalyst increased, accelerating diffusion. For low flows, the reaction rates are affected by mass transmission. Increasing the flow increases the reaction rate. For high flows, the reaction rates are not limited by mass transmission but are controlled by surface reactions.Fig. 11(a) shows the HCHO removal efficiency of TR(10) under visible light irradiation using various spatial velocities. The reaction conditions were as follows: 40 ppm HCHO and 20% O2 (balance He + N2) under 2 × 10 W fluorescent irradiation. The results of the experiment indicated that the photocatalytic reaction efficiency decreased with increasing the spatial velocity during the reaction. Spatial velocities of 3932 h−1 and 15727 h−1 provided the best reaction removal efficiency (greater than 99%). The HCHO removal efficiency began to decline when the spatial velocity exceeded 31454 h−1. The photocatalytic activity was clearly affected by the contact time between the HCHO and TR catalysts when the residence time was less than 1.14 × 10−1 s. A short residence time contributed to a low mass transfer efficiency and lowered the removal of HCHO. The minimum efficiency was obtained at 78635 h−1 (residence time of ∼0.46 × 10−1 s); however, the minimum efficiency was still 30%. The efficiency at a high spatial velocity still exceeded that of the commercial photocatalyst P25 (Fig. 8(b)). Fig. 11(b) shows the photocatalytic kinetic curves for the TR catalysts at different space velocities. A linear correlation was observed at high space velocities of 31454 h−1 and 78635 h−1; the values of the pseudo-first-order reaction rate constant for these two space velocities were 0.0127 m−1 and 0.0117 m−1, respectively. In contrast, no linear correlation was observed at low space velocities. To render subsequent tests comparable, this study set the spatial velocity for the following HCHO photocatalytic reaction tests at 31454 h−1.
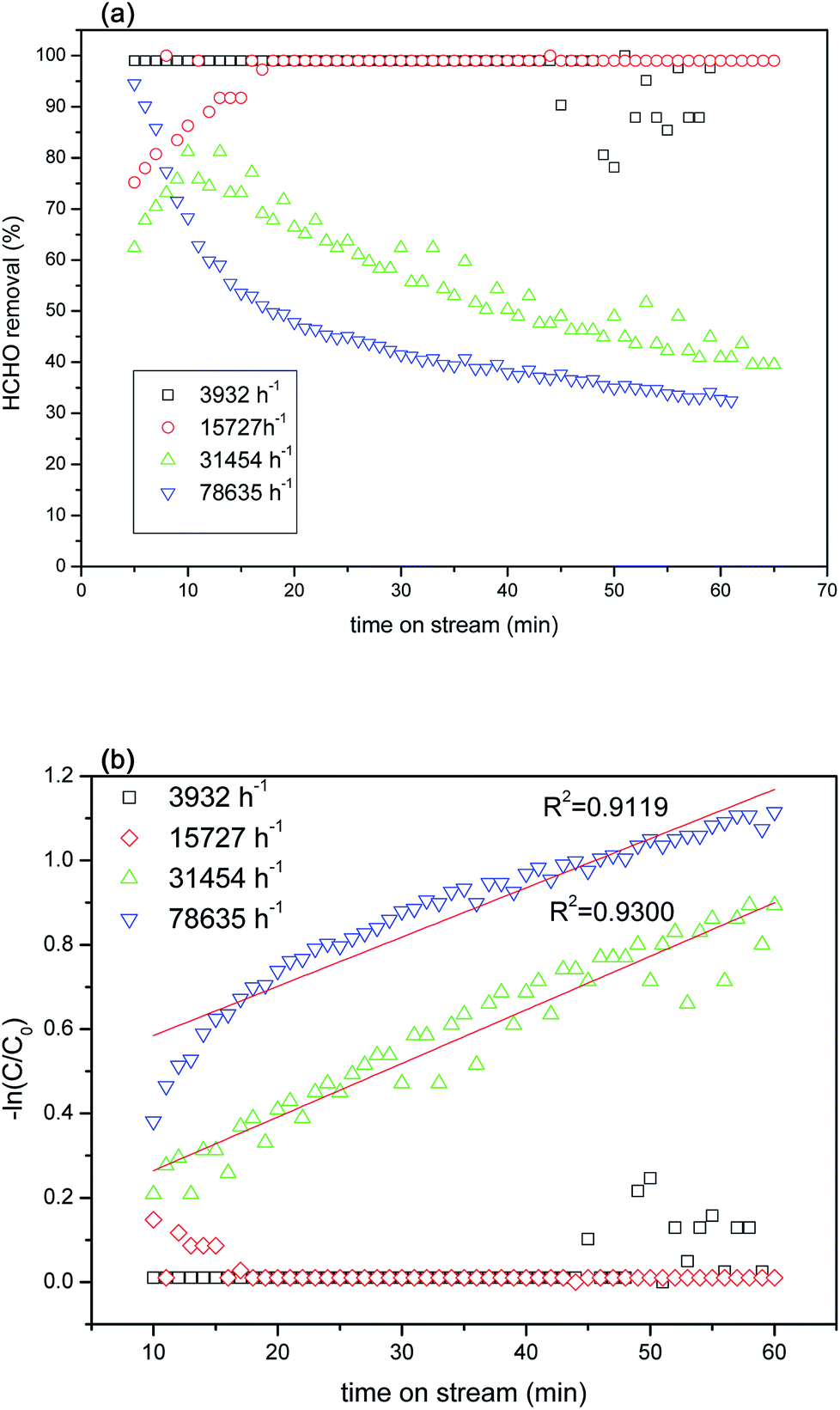 | ||
| Fig. 11 (a) HCHO removal over TR(10) catalyst at different space velocities as a function of reaction time under visible light irradiation; (b) photocatalytic kinetics of TR(10) catalyst at different space velocities as a function of reaction time on HCHO removal. | ||
3.5. Formaldehyde removal efficiency of Pt/TR catalysts
Fig. 12(a) shows TEM images of Pt/TR(10) at 200000× magnification, while the inset is at 30000× magnification. A comparison of the inset image of Fig. 12(a) with Fig. 2(b) showed that the morphology of TR(10) did not change after Pt doping. The hexagonal pore structure was retained, and the size of the Pt particles ranged from 4 to 30 nm (as indicated by red arrows) Moreover, X-rays were used to perform EDS, as shown in Fig. 12, which shows a good dispersion of Pt particles (light spots in Fig. 13(a)) and peak signals for Pt, Ti, O, and Si; therefore, the Pt particles on the surface of Pt/TR(10) were observable, and EDS analysis verified that Pt particles were indeed doped on Pt/TR(10). TEM images and EDS analysis showed that some Pt particles were dispersed on the surface of the Ti–O–Si structure, and some Pt particles were embedded in the two-dimensional channels of TR(10). The microwave-assisted ethylene glycol reduction method effectively dispersed Pt particles onto the catalysts.
 | ||
| Fig. 12 TEM images of Pt/TR catalysts: (a) Pt/TR(10); (b) Pt/TR(30). | ||
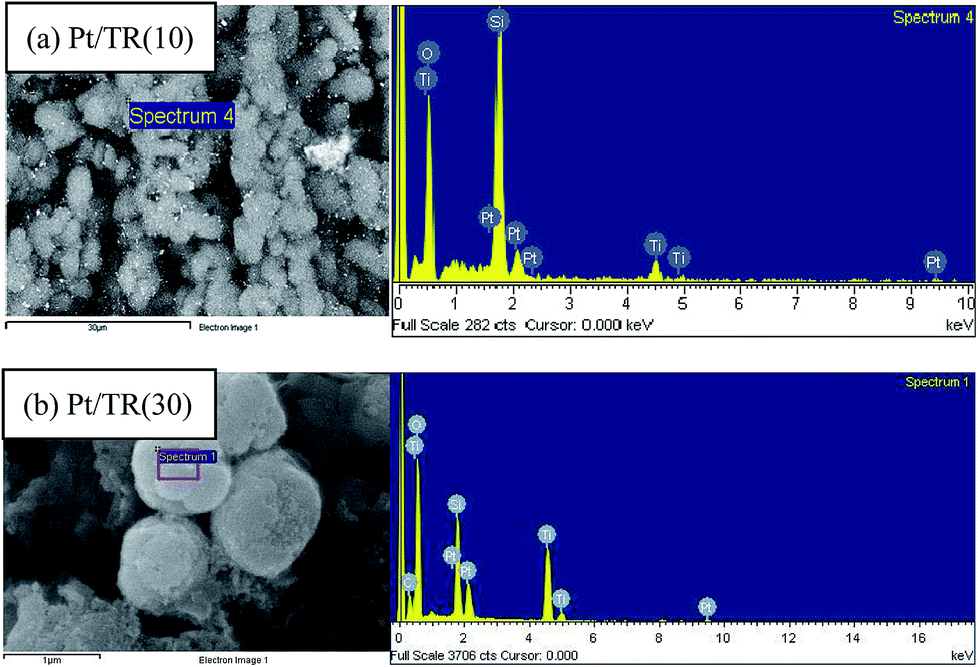 | ||
| Fig. 13 EDS analysis of: (a) Pt/TR(10); (b) Pt/TR(30). | ||
Fig. 12(b) shows TEM images of Pt/TR(30) at 100000× magnification, and the inset is at 400000× magnification. Similar to TR(30) (Fig. 2(c)), the shape of Pt/TR(30) was similar to that of a cauliflower. However, the internal texture was not observed clearly because the porous three-dimensional structure led to low electron beam penetration during TEM analysis. Also, EDS analysis shows that Pt, Ti, O, and Si were appeared on TR(30), as shown in Fig. 13(b). At high magnification (inset of Fig. 12(b)), the Pt particles were dispersed into the splits between cylinders, and the sizes of the Pt particles were approximately 5–10 nm. The results showed that when 1 wt% Pt was attached to TR photocatalysts with varying proportions of Ti/Si, the external shape of the photocatalysts remained relatively unchanged. The internal structure of the catalysts also remained unchanged after calcination, which indicated that TR catalysts possess good thermal stability.
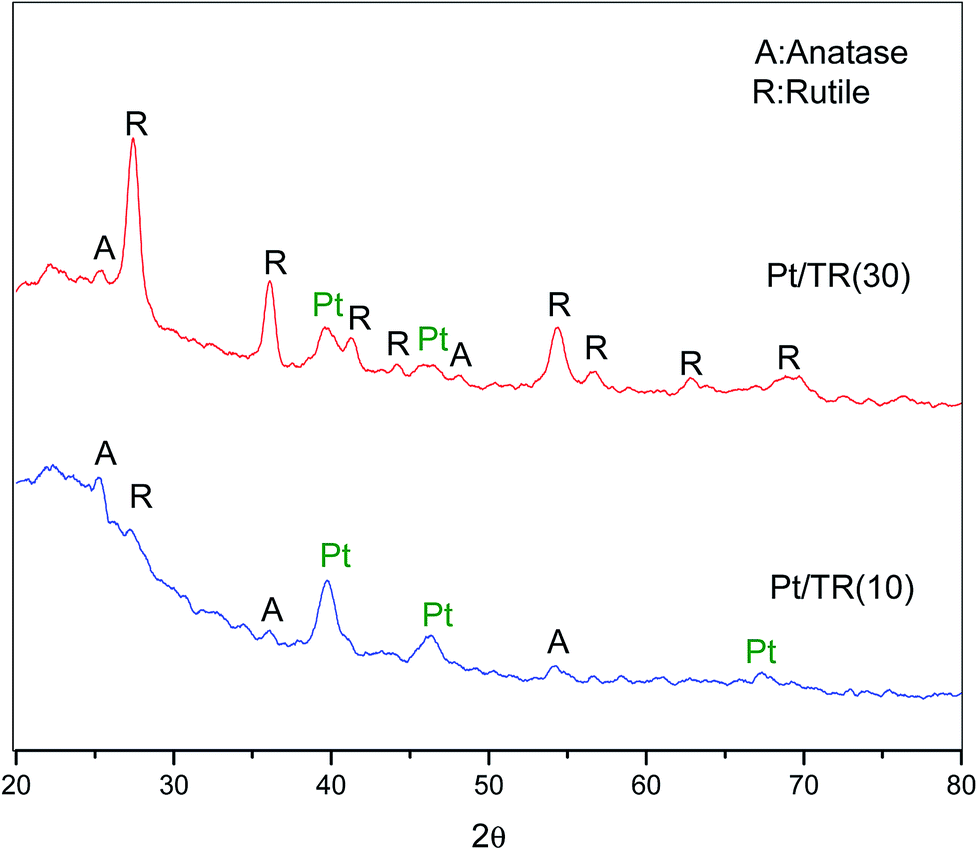 | ||
| Fig. 14 XRD patterns of Pt/TR(10) and Pt/TR(30) catalysts. | ||
The spectra for Pt4f of the Pt/TR catalysts are shown in Fig. 15. Similar to the Ti2p spectra, the Pt4f spectra showed a downward shift to a value of −2.0 eV in the binding energy for Pt/TR(10) and Pt/TR(30). For Pt/TR(10), the bands at 69.0 eV and 72.3 eV were attributed to Pt4f7/2 and Pt4f5/2.59 The gap between the two bands was 3.3 eV, and the peak area ratio was 1.26, which was assigned to zerovalent Pt (Pt0) formed in TR(10).60 A blueshift of the two bands of Pt/TR(30) was observed, possibly due to the quantum effect resulting from the smaller size of Pt particles, as seen in Fig. 12(b). This result revealed that the size of Pt particles calculated using the Scherrer equation would be affected by the diffraction of neighboring crystals. The gap and peak area ratio of the two bands were 3.3 eV and 1.30, respectively. Thus, Pt0 was also the main phase in TR(30). This result was confirmed by XRD.
 | ||
| Fig. 15 Pt4f XPS spectra of Pt/TR(10) and Pt/TR(30) catalysts. | ||
454 h−1 under 2 × 10 W fluorescent irradiation.
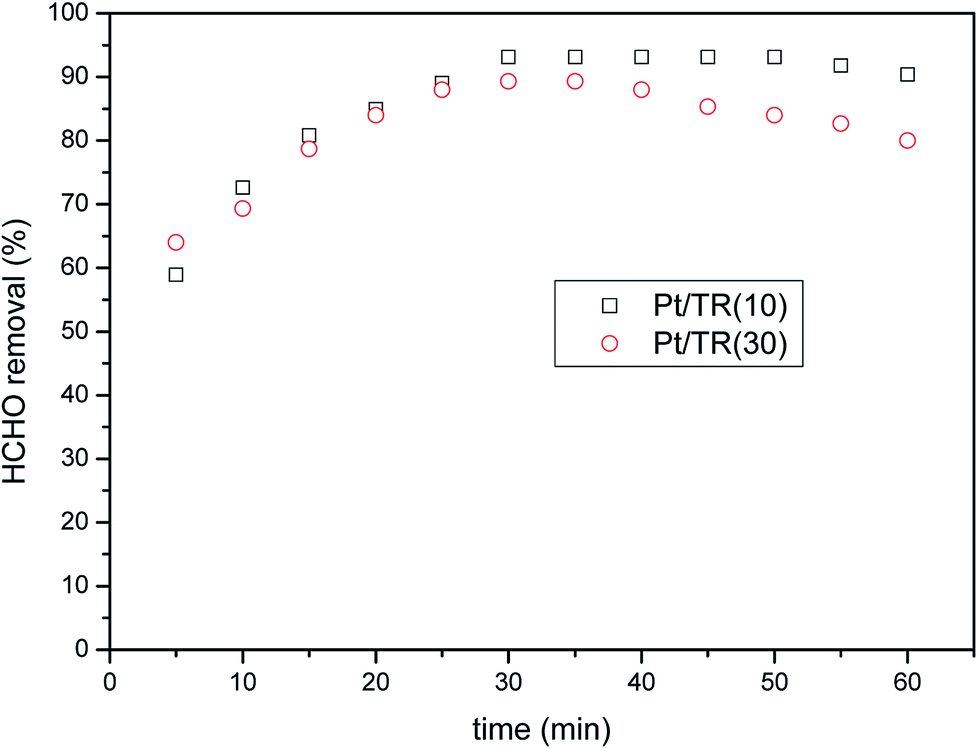 | ||
| Fig. 16 HCHO removal over Pt/TR(10) and Pt/TR(30) catalysts as a function of reaction time under visible light irradiation (feed composition: 40 ppm HCHO, SV = 31454 h−1). | ||
Comparison of Fig. 8(b) and 11 showed that Pt doping on the TR catalysts effectively increased the HCHO photocatalytic efficiency at a residence time of 1.14 × 10−1 s, increasing the removal efficiency of Pt/TR(10) and Pt/TR(30) to 90.4% and 80.0%, respectively, at a reaction time of 60 min. Furthermore, the physical and chemical advantages of TR catalysts led to a synergistic effect on HCHO removal. A possible reason for this increase in the photocatalytic efficiency was that the use of the microwave-assisted EG reduction method to synthesize Pt/TR catalysts can enhance Pt particle dispersion and control the particle size, as shown in Fig. 12. These uniformly dispersed and nanosized Pt particles can effectively capture electrons during electron–hole separation, thereby prolonging the electron–hole separation time and ultimately improving the photocatalytic efficiency of HCHO removal. Sun et al.61 reported that Pt particles would be photoactivated by visible light irradiation; then, the photoinduced electrons would be easily excited to the impurity band and conduction band. The result was also in agreement with reports made by Zhang et al.5 and suggested that nanoscale Pt0 particles would serve as electron sinks to reduce the recombination time of electron–hole pairs; then, an assisting effect would be created by the production of additional superoxide radical anions by electron transfer from TiO2 to the surface of the TR catalysts. To confirm the effect of the Pt particles, the PL spectra of Pt/TR(10) were analyzed, as shown in Fig. 17. The intensity of the Pt/TR(10) spectra was weaker than that of the TR(10) spectra. This result correlated with the longer separation time for electron–hole pairs noted for Pt/TR(10). The argument presented above was thus confirmed.
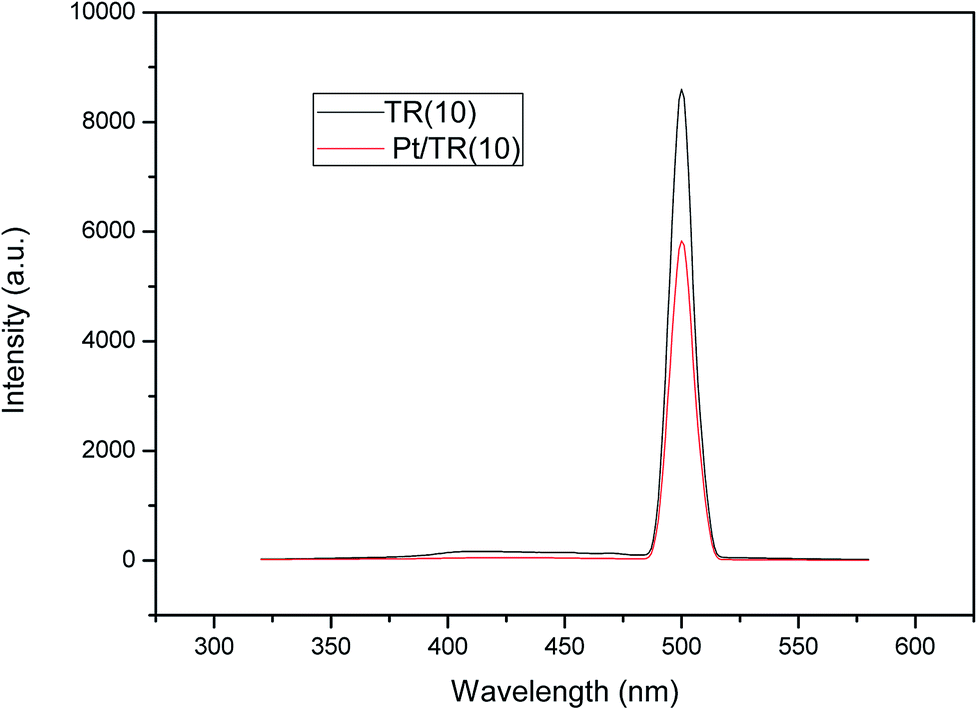 | ||
| Fig. 17 PL spectra of TR(10) and Pt/TR(10) catalysts. | ||
4. Conclusions
TiO2 is a common photocatalyst and is widely used in photocatalytic reactions performed under UV light with a wavelength of 325 nm. To expand the range of photocatalysis applications and to reduce their deleterious effects on health, in this study, TiO2-rutile-based catalysts were synthesized using the one-step modified hydrothermal method and then doped with metallic Pt particles. The resulting photocatalysts provided greater efficiency than that of traditional P25 photocatalysts, even under visible light. The following conclusions could be drawn based on the results obtained in this study:(1) The shape and structure of TR catalysts with various Ti proportions synthesized using the modified hydrothermal method differed from those of the porous material in traditional SBA-15. When the ratio of Ti/Si was 1/9, TR(10) maintained a two-dimensional hexagonal channel structure; however, Ti–O–Si bonds enlarged the TR(10) structure. When the ratio of Ti/Si was 3/7 or 5/5, a three-dimensional porous spherical structure was formed instead of a two-dimensional channel structure, with small particle sizes of less than 1 μm.
(2) XRD and Raman analyses showed that the rutile phase was the primary crystalline species in the TR catalysts. Si atoms inhibited the transformation of the anatase phase and improved the thermal stability of the TR catalysts during calcination. Raman and XPS analysis results demonstrated that the Ti–O–Si–C bond contributed to the transformation of the two-dimensional structure to the three-dimensional structure during TR preparation. The mesoporous structure of the TR catalysts improved their photocatalytic activity owing to its large pore volume and high SBET. DRS analysis showed that a higher rutile phase content and smaller crystal size of the rutile phase of the TR catalysts contributed to the lower band gap energy.
(3) The activity test results showed that when HCHO was used as the reactant, the TR catalysts were removed HCHO more effectively than the commercial photocatalyst P25 regardless of whether the reaction occurred under UV or visible light. The mesoporous structure and surface-absorbed oxygen of the TR catalyst might increase the adsorption of HCHO, improving its photocatalytic activity. Then, photoexcitation would occur easily because of the low band gap energy of the TR catalyst (<3.0 eV) under visible light irradiation.
(4) The microwave-assisted EG reduction method was shown to effectively disperse nanoscale Pt particles with low energy consumption and a short synthesis time. After the Pt/TR catalyst underwent calcination, the TR catalyst structure and the crystalline Ti remained unchanged and undamaged, demonstrating the excellent stability of TR, and the chemical state of platinum was Pt0. The Pt particles could increase the electron–hole pair separation time, thereby effectively improving the catalytic efficiency of the photocatalysts.
(5) The size of the TR catalysts followed the order TR(10) (10 μm) > TR(30) (0.8 μm) > TR(50) (0.5 μm). The Taguchi method was employed to study the effect factors for the microscale TR catalysts during the photocatalytic removal of HCHO. The results of ANOVA and the analysis of the S/N ratio were the same and showed that the level effect followed the order space velocity > ratio of Ti > time on stream.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed under the auspices of the Ministry of Science and Technology, ROC, under Contract MOST 98-2221-E-040-001-MY3 and MOST 102-2221-E-040-011-MY3, to which the authors wish to express their thanks.References
- T. Ohura, T. Amagai, X. Shen, S. Li, P. Zhang and L. Zhu, Comparative study on indoor air quality in Japan and China: characteristics of residential indoor and outdoor VOCs, Atmos. Environ., 2009, 43, 6352–6359 CrossRef.
- X. Tang, Y. Bai, A. Duong, M. T. Smith and L. Zhang, Formaldehyde in China: Production, consumption, exposure levels, and health effects, Environ. Int., 2009, 35, 1210–1224 CrossRef PubMed.
- W. Ho, J. C. Yu and S. Lee, Low-temperature hydrothermal synthesis of S-doped TiO2 with visible light photocatalytic activity, J. Solid State Chem., 2002, 179, 1171–1176 CrossRef.
- Y. Mizukoshi, Y. Makise, T. Shuto, J. Hu, A. Tominaga, S. Shironita and S. Tanabe, Immobilization of noble metal nanoparticles on the surface of TiO2 by the sonochemical method: Photocatalytic production of hydrogen from an aqueous solution of ethanol, Ultrason. Sonochem., 2007, 14, 387–392 CrossRef PubMed.
- X. J. Zheng, L. F. Wei, Z. H. Zhang, Q. J. Jiang, Y. J. Wei, B. Xie and M. B. Wei, Research on photocatalytic H2 production from acetic acid solution by Pt/TiO2 nanoparticles under UV irradiation, Int. J. Hydrogen Energy, 2009, 34, 9033–9041 CrossRef.
- S. Yurdakal, Ş. Ö. Yanar, S. Çetinkaya, O. Alagöz, P. Yalçın and L. Özcan, Green photocatalytic synthesis of vitamin B3 by Pt loaded TiO2 photocatalysts, Appl. Catal., B, 2017, 202, 500–508 CrossRef.
- G. J. Kim, S. M. Lee, S. C. Hong and S. S. Kim, Active oxygen species adsorbed on the catalyst surface and its effect on formaldehyde oxidation over Pt/TiO2 catalysts at room temperature; role of the Pt valence state on this reaction?, RSC Adv., 2018, 8, 3626–3636 RSC.
- N. Serpone, P. Maruthamuthu, P. Pichat, E. Pelizzetti and H. Hidaka, Exploiting the interparticle electron transfer process in the photocatalysed oxidation of phenol, 2-chlorophenol and pentachlorophenol: chemical evidence for electron and hole transfer between coupled semiconductors, J. Photochem. Photobiol., A, 1995, 85, 247–255 CrossRef.
- N. Kanai, T. Nuida, K. Ueta, K. Hashimoto, T. Watanabe and H. Ohsaki, Photocatalytic efficiency of TiO2/SnO2 thin film Stacks Prepared by DC magnetron sputtering, Vacuum, 2004, 74, 723–727 CrossRef.
- X. Chen and C. Burda, Photoelectron Spectroscopic Investigation of Nitrogen-Doped Titania Nanoparticles, J. Phys. Chem. B, 2004, 108, 15446–15449 CrossRef.
- J. Yu, S. Wang, J. Low and W. Xiao, Enhanced photocatalytic performance of direct Z-scheme-g-C3N4–TiO2 photocatalysts for the decomposition of formaldehyde in air, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- M. Iwasaki, M. Hara, H. Kawada, H. Tada and S. Ito, Cobalt Ion-Doped TiO2 Photocatalyst Response to Visible Light, J. Colloid Interface Sci., 2000, 224, 202–204 CrossRef PubMed.
- J. Zhu, F. Chen, J. Zhang, H. Chen and M. Aupo, Fe3+–TiO2 photocatalysts prepared by combining sol–gel method with hydrothermal treatment and their characterization, J. Photochem. Photobiol., A, 2006, 180, 196–204 CrossRef.
- I. H. Tseng, J. C. S. Wu and H. Y. Chou, Effect of sol–gel procedures on the photocatalysis of Cu/TiO2 in CO2 photoreduction, J. Catal., 2004, 221, 432–440 CrossRef.
- C. C. Pan and J. C. S. Wu, Visible-light response Cr-doped TiO2–XNX photocatalysts, Mater. Chem. Phys., 2006, 100, 102–107 CrossRef.
- V. Trevisan, A. Olivo, F. Pinna, M. Signoretto, F. Vindigni, G. Cerrato and C. L. Bianchi, C–N/TiO2 photocatalysts: effect of co-doping on the catalytic performance under visible light, Appl. Catal., B, 2014, 160–161, 152–160 CrossRef.
- S. Larumbe, M. Monge and C. Gómez-Polo, Comparative study of (N, Fe) doped TiO2 photocatalysts, Appl. Surf. Sci., 2015, 327, 490–497 CrossRef.
- J. H. Pan, Z. Y. Cai, Y. Yu and X. S. Zhao, Controllable synthesis of mesoporous F–TiO2 spheres for effective photocatalysis, J. Mater. Chem., 2011, 21, 11430–11438 RSC.
- D. Chatterjee and A. Mahata, Demineralization of organic pollutants on the dye modified TiO2 semiconductor particulate system using visible light, Appl. Catal., B, 2001, 33, 119–125 CrossRef.
- Y. Cho and W. Choi, Visible Light-induced Reactions of Humic Acids on TiO2, J. Photochem. Photobiol., A, 2002, 148, 129–135 CrossRef.
- X. Jiang, C. Li, S. Liu, F. Zhang, F. You and C. Yao, The synthesis and characterization of ytterbium doped TiO2 hollow spheres with enhanced visible-light photocatalytic activity,, RSC Adv., 2017, 7, 24598–24606 RSC.
- F. Xu, Y. Le, B. Cheng and C. Jiang, Effect of calcination temperature on formaldehyde oxidation performance of Pt/TiO2 nanofiber composite at room temperature, Appl. Surf. Sci., 2017, 426, 333–341 CrossRef.
- T. V. L. Thejaswini, D. Prabhakaran and M. Akhila Maheswari, Synthesis of mesoporous-like ZrO2–TiO2 monoliths and their photocatalytic applications towards organic dye degradation, J. Photochem. Photobiol., A, 2017, 344, 212–222 CrossRef.
- K. Zhang, W. Zhou, X. Zhang, B. Sun, L. Wang, K. Pan, B. Jiang, G. Tian and H. Fu, Self-floating amphiphilic black TiO2 foams with 3D macro-mesoporous architectures as efficient solar-driven photocatalysts, Appl. Catal., B, 2017, 206, 336–343 CrossRef.
- Y. J. Acosta-Silva, R. Nava, V. Hernández-Morales, S. A. Macías-Sánchez, M. L. Gómez-Herreera and B. Pawelec, Methylene blue photodegradation over titania-decorated SBA-15, Appl. Catal., B, 2011, 110, 108–117 CrossRef.
- M. J. López-Muñoz, R. van Grieken, J. Aguado and J. Marugán, Role of the support on the activity of silica-supported TiO2 photocatalyst: structure of the TiO2/SBA-15 photocatalyst, Catal. Today, 2005, 101, 307–314 CrossRef.
- K. De witte, A. M. Busuioc, V. Meyne, M. Mertens, N. Bilba, G. Van Tendeloo, P. Cool and E. F. Vansant, Influence of the synthesis parameters of TiO2-SBA-15 materials on the adsorption and photodegradation of rhodamine-6G, Microporous Mesoporous Mater., 2008, 110, 100–110 CrossRef.
- B. C. Ledesma, O. A. Anunziata and A. R. Beltramone, HDN of indole over Ir-modified Ti-SBA15, Appl. Catal., B, 2016, 192, 220–233 CrossRef.
- T. Ekou, C. Especel and S. Royer, Catalytic performances of large pore Ti-SBA15 supported Pt nanocomposites for the citral hydrogenation reaction, Catal. Today, 2011, 173, 44–52 CrossRef.
- M. G. Sorolla II, M. L. Dalida, P. Khemthong and N. Grisdanurak, Photocatalytic degradation of paraquat using nano-sized Cu–TiO2/SBA15 under UV and visible light, J. Environ. Sci., 2012, 24, 1125–1132 CrossRef.
- S. Perathoner, P. Lanzafame, R. Passalacqua, G. Centi, R. Schlögl and D. S. Su, Use of mesoporous SBA15 for nanostructuring titania for photocatalytic applications, Microporous Mesoporous Mater., 2006, 90, 347–361 CrossRef.
- F. Chiker, J. P. Nogier, F. Launay and J. L. Bonardet, Optimisation of gas phase deposition of titanium on mesoporous silica SBA15: active site counting and catalytic activity in cyclohexene epoxidation, Appl. Catal., A, 2004, 259, 153–162 CrossRef.
- G. Zhang, Z. Sun, Y. Duan, R. Ma and S. Zheng, Synthesis of nano-TiO2/diatomite composite and its photocatalytic degradation of gaseous formaldehyde, Appl. Surf. Sci., 2017, 412, 105–112 CrossRef.
- M. V. Dozzi, G. L. Chiarello, M. Pedroni, S. Livraghi, E. Giamello and E. Selli, Hight photocatalytic hydrogen production on Cu(II) pre-grafted Pt/TiO2, Appl. Catal., B, 2017, 209, 417–428 CrossRef.
- E. G. Villabona-Leal, J. P. Lóez-Neira, J. A. Pedraza-Avella, E. Pérez and O. Meza, Screening of factors influencing the photocatalytic activity of TiO2:Ln (Ln = La, Ce, Pr, Nd, Sm, Eu and Gd) in the degradation of dyes, Comput. Mater. Sci., 2015, 107, 48–53 CrossRef.
- R. Bashiri, N. M. Mohamed, C. F. Kait, S. Sufian, M. khatani and H. Hanaei, Effect of Preparation Parameters on Optical Properties of Cu and Ni Doped TiO2 Photocatalyst, Procedia Eng., 2016, 148, 151–157 CrossRef.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Triblock Copolymer Syntheses of Mesoporous Silica with Periodic 50 to 300 Angstrom Pores, Science, 1998, 273, 548–552 CrossRef.
- T. Armaroli, F. Milella, B. Notari, R. J. Willey and G. Busca, A spectroscopic study of amorphous and crystalline Ti-containing silicas and their surface acidity, Top. Catal., 2001, 15, 63–71 CrossRef.
- Y. Gao, H. Wang, J. Wu, R. Zhao, Y. Lu and B. Xin, Controlled facile synthesis and photocatalytic activity of ultrafine high crystallinity TiO2 nanocrystals with tunable anatase/rutile ratios, Appl. Surf. Sci., 2014, 294, 36–41 CrossRef.
- S. F. Resende, E. H. M. Nunes, M. Houmard and W. L. Vasconcelos, Simple sol–gel process to obtain silica-coated anatase particles with enhanced TiO2–SiO2 interfacial area, J. Colloid Interface Sci., 2014, 433, 211–217 CrossRef PubMed.
- Y. Zhao, L. Xu, Y. Wang, C. Gao and D. Liu, Preparation of Ti–Si mixed oxides by sol–gel one step hydrolysis, Catal. Today, 2004, 93–95, 583–588 CrossRef.
- H. C. Choi, Y. M. Jung and S. B. Kim, Size effects in the Raman spectra of TiO2 nanoparticles, Vib. Spectrosc., 2005, 37, 33–38 CrossRef.
- F. Chang, J. Wang, J. Luo, J. Sun, B. Deng and X. Hu, Enhanced visible -light-driven photocatalytic performance of mesoporous W–Ti-SBA-15 prepared through a facile hydrothermal route, Colloids Surf., A, 2016, 499, 69–78 CrossRef.
- D. Pang, Y. Wang, X. Ma and F. Ouyang, Fluorine promoted and silica supported TiO2 for photocatalytic decomposition of acrylonitrile under simulant solar light irradiation, Chem. Eng. J., 2014, 258, 43–50 CrossRef.
- S. S. Arbuj, R. R. Hawaldar, U. P. Mulik, B. N. Wani, D. P. Amalnerkar and S. B. Waghmode, Preparation, characterization and photocatalytic activity of TiO2 towards methylene blue degradation, Mater. Sci. Eng., B, 2010, 168, 90–94 CrossRef.
- N. Yuangpho, S. T. T. Le, T. Treerujiraphapong, W. Khanitchaidecha and A. Nakaruk, Enhanced photocatalytic performance of TiO2 particles via effect of anatase–rutile ratio, Phys. E, 2015, 67, 18–22 CrossRef.
- N. E. Quaranta, J. Soria, V. Cortés Corberán and J. L. G. Fierro, Selective Oxidation of Ethanol to Acetaldehyde on V2O5/TiO2/SiO2 Catalyst: Effect of TiO2 coating of the Silica Support, J. Catal., 1997, 171, 1–13 CrossRef.
- X. Gao, S. R. Bare, J. L. Fierro, M. A. Banares and I. E. Wachs, Preparation and In Situ Spectroscopic Characterization of Molecularly Dispersed Titanium Oxide on Silica, J. Phys. Chem. B, 1998, 102, 5653–5666 CrossRef.
- D. Goldbaum, P. Manimuda, G. Kamath, S. Descartes, J. E. Klemberg-Sapieha and R. R. Chromik, Tribological behavior of TiN and Ti(Si, C)N coatings on cold sprayed Ti substrates, Surf. Coat. Technol., 2016, 291, 264–275 CrossRef.
- H. Sharma, A. K. Shukla and V. D. Vankar, Structural modifications and enhanced Raman scattering from multiwalled carbon nanotubes grown on titanium coated silicon single crystals, Thin Solid Films, 2012, 520, 1902–1908 CrossRef.
- M. Mazur, D. Wojcieszak, D. Kaczmarek, J. Domaradzki, S. Song, D. Gibson, F. Placido, P. Mazur, M. Kalisz and A. Poniedzialek, Functional photocatalytically active and scratch resistant antireflective coating based on TiO2 and SiO2, Appl. Surf. Sci., 2016, 380, 165–171 CrossRef.
- L. Kernazhitsky, V. Shymanovska, T. Gavrilko, V. Naumov, L. Fedorenko, V. Kshnyakin and J. Baran, Photoluminescence of Cr-doped TiO2 induced by intense UV laser excitation, J. Lumin., 2015, 166, 253–258 CrossRef.
- S. P. Chenakin, G. Melaet, R. Szukiewicz and N. Kruse, XPS study of the surface chemical state of a Pd/(SiO2+TiO2) catalyst after methane oxidation and SO2 treatment, J. Catal., 2014, 312, 1–11 CrossRef.
- E. Thangavel, S. Lee, K. S. Nam, J. K. Kim and D. G. Kim, Synthesis and characterization of Ti–Si–C–N nanocomposite coatings prepared by a filtered vacuum arc method, Appl. Surf. Sci., 2013, 265, 60–65 CrossRef.
- A. Nakaruk, D. Ragazzon and C. C. Sorrell, Anatase–rutile transformation through high-temperature annealing of titania films produced by ultrasonic spray pyrolysis, Thin Solid Films, 2010, 518, 3735–3742 CrossRef.
- J. C. Kotz and P. Treichel, Chemistry & Chemical Reactivity, Harcourt Brace & Company, Orlando, Florida, 4th edn, 1999, pp. 828–831 Search PubMed.
- M. Han, T. Sun, P. Y. Tan, X. Chen, O. K. Tan and M. S. Tse, m-BiVO4@γ-Bi2O3 core–shell p–n heterogeneous nanostructure for enhanced visible-light photocatalytic performance, RSC Adv., 2013, 3, 24964–24970 RSC.
- Y. Ma, X. Nie, D. O. Northwood and H. Hu, Systematic study of the electrolytic plasma oxidation process on a Mg alloy for corrosion protection, Thin Solid Films, 2006, 494, 296–301 CrossRef.
- B. Hasa, E. Kalamaras, E. I. Papaioannou, L. Sygellou and A. Katsaounis, Electrochemical oxidation of alcohols on Pt–TiO2 binary electrodes, Int. J. Hydrogen Energy, 2013, 38, 15395–15404 CrossRef.
- E. A. Kozlova, T. P. Lyubina, M. A. Nasalevich, A. V. Vorontsov, A. V. Miller, V. V. Kaichev and V. N. Parmon, Influence of the method of platinum deposition on activity and stability of Pt/TiO2 photocatalysts in the photocatalytic oxidation of dimethylphosphonate, Catal. Commun., 2011, 12, 597–601 CrossRef.
- H. Sun, R. Ullah, S. Chong, H. M. Ang, M. O. Tadé and S. Wang, Room-light-induced indoor air purification using an efficient Pt/N–TiO2 photocatalyst, Appl. Catal., B, 2011, 108–109, 127–133 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |