DOI:
10.1039/C8RA03550D
(Paper)
RSC Adv., 2018,
8, 22764-22776
Injectable alendronate-functionalized GelMA hydrogels for mineralization and osteogenesis†
Received
25th April 2018
, Accepted 11th June 2018
First published on 20th June 2018
Abstract
In a minimally invasive procedure, hydrogels can be injected into an affected area for drug loading and tissue repair. Here, gelatin methacryloyl (GelMA) was modified with alendronate (ALN), and three different alendronate-functionalized GelMA (GelMA-ALN) hydrogels were prepared. The modification greatly improved the swelling ratio, protein adsorption and mineralization. The GelMA-ALN hydrogels significantly promoted the in vitro osteogenic differentiation of hFOB cells as indicated by their higher alkaline phosphatase (ALP) activity, denser mineralization and up-regulated osteogenesis-related genes at both the mRNA and protein levels. Meanwhile, the cells maintained their activity and differentiated into osteoblasts when encapsulated in the GelMA-ALN hydrogels. Alendronate-modified hydrogels have potential for use in the minimally invasive treatment of irregular bone defects.
1 Introduction
Bone defects, especially skull defects with complex shapes, require tissue engineering scaffolds that match the shape of the defect. Hydrogels are naturally promising materials because of their injectability and in situ formability. Gelatin methacryloyl (GelMA), a photopolymerizable gelatin material that is modified by adding methacrylate groups to its amine-containing side groups, contains arginine-glycine-aspartic acid (RGD) sequences1 and is similar to the native extracellular matrix (ECM).2 Due to their collagen-like backbone and mild polymerization conditions, GelMA hydrogels exhibit cell-responsive characteristics, such as promoting cell adhesion, proliferation and differentiation.3,4 GelMA hydrogels are suitable for encapsulating cells and undergo in situ crosslinking after in vivo injection.5–7
Although hydrogels can match the shape of natural bone, they still have different compositions. Phosphate groups are thought to play an important role in assisting and modulating calcification in the early stages of bone formation.8–13 Benoit14 used phosphate-functionalized polyethylene glycol to promote the osteogenesis of human mesenchymal stem cells (hMSCs) in normal growth media, demonstrating that osteogenesis was caused by the presence of small molecule-phosphate groups. This method is a simple means for preparing biomimetic bone biopolymers. Subsequent studies confirmed that phosphate groups can promote mineralization15–18 and adsorb proteins to improve biological function.19–22 Furthermore, phosphate groups have been identified as a signalling unit that regulates the cell cycle, proliferation rate and signal transduction pathways.23–26 Bisphosphonates (BPs), which can inhibit bone resorption, are widely used in the therapy of osteoporosis.27,28 Furthermore, BP groups strongly and reversibly bind calcium ions29,30 and can also bind to hydroxyapatite, calcium phosphate and other calcium crystals.31,32 BP groups were shown to have a high affinity for bone minerals, which leads to their selective uptake by bone.33 Although the activity34–39 and osteogenesis40–45 of cells encapsulated in 3D GelMA hydrogels have been widely studied, fewer studies have focused on the osteogenesis of cells cultured in 3D BP-functionalized hydrogels.
In this study, alendronate-functionalized GelMA (GelMA-ALN) hydrogels with three different alendronate concentrations were prepared. The hydrogel-based bone repair material can be easily injected in vivo by syringe. Based on our previous findings, it was hypothesized that this chemical modification could enhance osteogenesis in a dose-dependent manner. First, the protein adsorption and mineralization of the GelMA-ALN hydrogels were evaluated. Second, osteogenic differentiation was demonstrated by the higher alkaline phosphatase (ALP) activity, denser mineralization and up-regulation of osteogenesis-related genes, including runt-related transcription factor 2 (Runx 2), ALP, osteocalcin (OCN), collagen I (COL I) and osteopontin (OPN), at both the mRNA and protein levels. Finally, the live/dead staining and immunofluorescence staining of cells encapsulated in the hydrogels were performed to determine the activity and osteogenic differentiation, respectively, in a three-dimensional environment. The results showed that the alendronate modification markedly increased osteogenesis in both the 2D and 3D environments.
2 Materials and methods
2.1 Materials
Alendronate (ALN) was purchased from Aladdin (China). Gelatin, methacrylic anhydride, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), acetic acid, tris(hydroxymethyl)aminomethane hydrochloride, alizarin red S, sodium dodecyl sulfate (SDS), and 2-(N-morpholino) ethanesulfonic acid (4-morpholineethanesulfonic acid) (MES) were purchased from Sigma (USA). Dulbecco's modified Eagle's medium (DMEM)/F12 (DMEM-F12), phosphate-buffered saline (PBS), trypsin–EDTA, and foetal bovine serum (FBS) were obtained from Gibco (USA).
2.2 Synthesis of GelMA
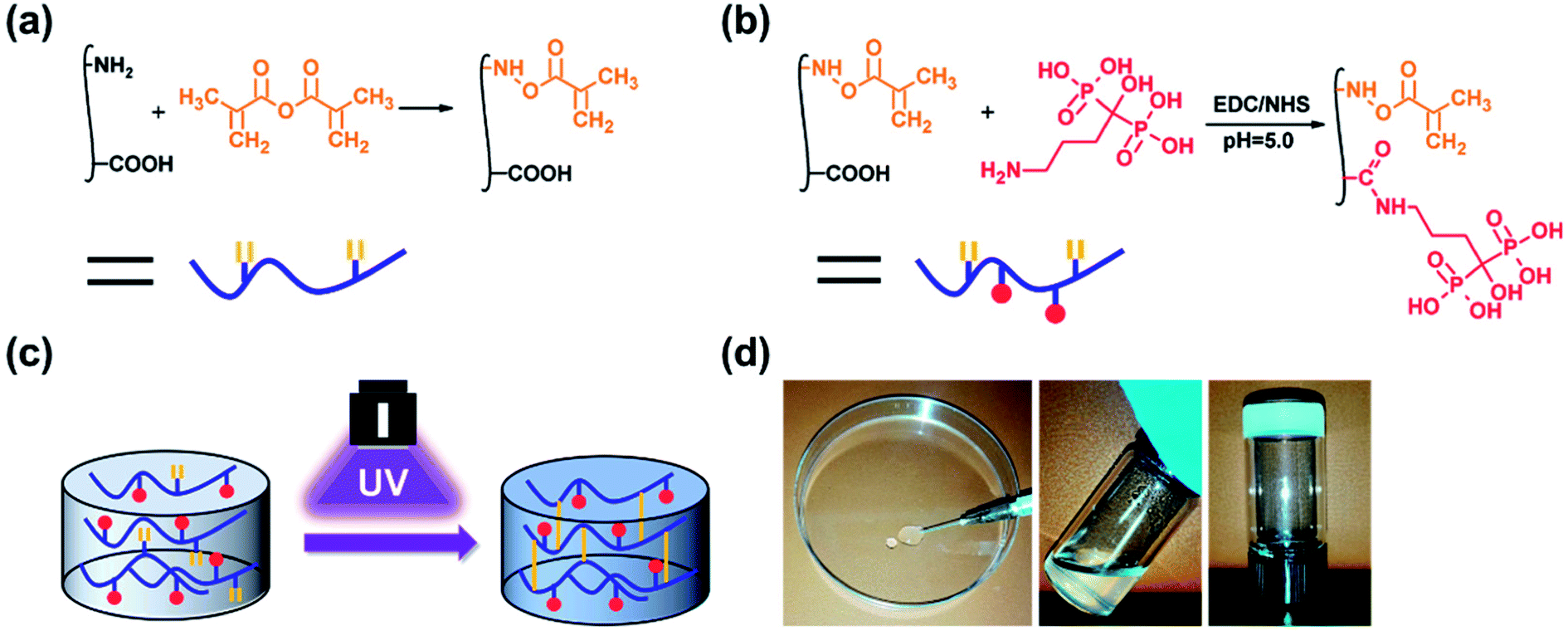
GelMA was prepared according to our previously reported procedure (Fig. 1a).46 Briefly, 10 g of gelatin type A was dissolved in 100 mL 1× PBS at 50 °C. Then, 20 mL of methacrylic anhydride was added to the mixture, and the acylation reaction was performed under stirring for 1 h. The mixture was dialysed for 5 days against warm deionized water through a dialysis membrane (Fisherbrand code, 12–14 kDa cut-off) and lyophilized for 2 days.
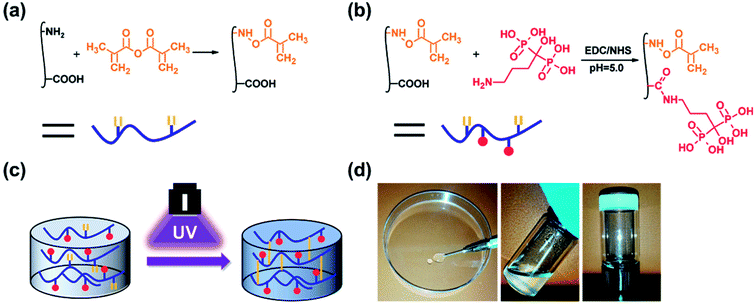 |
| | Fig. 1 Schematic illustrations of the (a) synthesis of GelMA, (b) synthesis of GelMA-ALN and (c) preparation of the hydrogel. (d) Injection of the hydrogel. | |
2.3 Synthesis of GelMA-ALN
Alendronate-functionalized GelMA (GelMA-ALN) was synthesized according to previously reported methods (Fig. 1b). Briefly, 1 g of GelMA was dissolved in 20 mL of a 0.5 M MES buffer (pH = 5) at 60 °C, and the mixture was then cooled to 37 °C. Three mmol of MES and 3 mmol of NHS were added to the solution, which was vigorously stirred for 20 min to activate the carboxyl groups on GelMA. Three mmol of alendronate was dissolved in 10 mL of a 0.5 M MES buffer at 60 °C, and the mixture was added to the reaction solution, which was subsequently reacted for another 24 h. The mixture was then dialysed for 5 days against warm deionized water through a dialysis membrane (Fisherbrand code, 12–14 kDa cut-off) and lyophilized for 2 days. By controlling the feed ratio, GelMA-ALN was prepared with three different alendronate contents (G-LALN, G-MALN and G-HALN represent low, medium and high alendronate contents, respectively) (Table S1†). GelMA and GelMA-ALN hydrogels were mixed in PBS at concentration of 10 w/v% with 0.5 w/v% photoinitiator and dissolved at 60 °C. The solution was transferred to mould and crosslinked under UV light.
2.4 Characterization of GelMA-ALN
The FTIR spectra (CCR-1, Thermo-Nicolet, USA) were collected between 400 cm−1 and 4000 cm−1 with a resolution of 2 cm−1. 1H and 31P NMR spectra were obtained using a 400 MHz spectrometer (AVANCE Digital 400, Bruker, Germany). The samples were dissolved in D2O with 1% acetic acid-d4 to a concentration of 10 mg mL−1. The 31P NMR spectra represent 1000 scans. The surface elemental compositions of GelMA and GelMA-ALN were characterized using an X-ray photoelectron spectrometer (XPS, Thermo Escalab 250Xi) operated at 150 W using Al Kα radiation as the excitation source (15 kV). The diameter of the analysed spot was approximately 500 μm. To calibrate the broadening of the spectral lines and the drift towards higher binding energies, a binding energy of 285.0 eV was assigned to the aliphatic carbon (–CH2) C 1s peak to correct the other binding energies.
2.5 Topography of the hydrogels
The morphologies and microstructures of the hydrogels were observed by a tungsten filament scanning electron microscope (Thermo Fisher Scientific, USA). The hydrogels were lyophilized under vacuum at −20 °C and then mounted on aluminium stubs. After sputter-coating the samples with gold for 60 s, the morphologies of the hydrogel surfaces were observed at an accelerating voltage of 10 kV.
2.6 Swelling ratio and equilibrium water absorption
To study the influence of ALN on the swelling ratio, the GelMA and GelMA-ALN hydrogels (3 mm high, 5 mm in diameter) were immersed in a PBS solution. The mass of each wet sample (Ww) was measured at different times. Finally, the samples were lyophilized to obtain the dry weights (Wd). The swelling ratio (%) was calculated using the formula:
| Swelling ratio = (Ww − Wd)/Wd × 100% |
The equilibrium swelling ratio of the hydrogels was determined in a PBS solution at 37 °C. The hydrogels were lyophilized and weighed to obtain the initial weight (Wi). Then, they were immersed in PBS for 48 h and reweighed to obtain the hydrated weight (WH). The swelling ability of the hydrogels was calculated using the following equation:
| Equilibrium swelling ratio = (WH − Wi)/Wi × 100% |
where
Wi and
WH are the initial dried and hydrated weights, respectively.
2.7 Mechanical testing
The mechanical properties of the GelMA and GelMA-ALN hydrogels were examined with a universal material testing machine (Intron5967, Instron, USA). Cylindrical samples (10 mm in diameter, 5 mm in thickness) were constructed and subjected to a constant compression speed of 3 mm min−1. At least 5 samples were measured for each group of hydrogels to obtain the compression curves and average Young's modulus, which was calculated using the slope of the stress–strain curves between 0% and 10%.
2.8 Rheological measurements
The rheological properties of GelMA and GelMA-ALN hydrogels were performed using a stress-controlled rheometer (Anton Paar MCR302) in plate–plate geometry (plate diameter of 25 mm, distance of 0.3 mm). The UV-initiated polymerization was investigated through UV light irradiation at 7.0 mW cm−2. The rheological properties of GelMA and GelMA-ALN hydrogels were recorded by oscillatory frequency sweeps (0.01–100 rad s−1, 1% strain) at 37 °C.
2.9 Mineralization
2× SBF was prepared by sequentially dissolving NaCl, NaHCO3, KCl, K2HPO4·3H2O, MgCl2·6H2O, CaCl2, and Na2SO4 in distilled deionized water at 37 °C. The GelMA and GelMA-ALN hydrogels were incubated in 2× SBF for 3 days, washed three times with deionized water, and then lyophilized. After coating the hydrogels with gold, they were examined by a tungsten filament scanning electron microscope (Thermo Fisher Scientific, USA) at an accelerating voltage of 10 kV.
2.10 Protein adsorption
The protein adsorption on the hydrogels was examined by measuring the total amount of proteins.22,47 Before protein adsorption, the hydrogels were extensively washed with phosphate buffer saline (PBS). They were allowed to swell in DMEM-F12 media without serum in a humidified incubator at 37 °C for 48 h. Then, the hydrogels were placed in a 24-well plate with 1 mL DMEM-F12 supplemented with 10% serum. After 6, 12 and 24 h in a humidified incubator at 37 °C, the hydrogels were transferred to a clean 24-well plate and gently rinsed with PBS for 10 s to remove loosely attached proteins. Then, 0.5 mL of a 1 w/v% sodium dodecyl sulfate (SDS) solution were pipetted into each well, and the well plate was placed on a shaker for 30 min for protein desorption. The concentration of proteins in the SDS solution was measured using a BCA protein assay kit (P0012S, Beyotime Company, China) according to the manufacturer's instructions.
2.11 Enzymatic degradation
To characterize the enzymatic degradation properties of GelMA hydrogel, we placed the hydrogel samples in 2.0 mL centrifuge tubes with 1.5 mL PBS containing 2.5 U mL−1 collagenase type II at 37 °C. At a pre-defined time, the GelMA and GelMA-ALN hydrogels were removed, frozen, and lyophilized. Mass loss was determined as the ratio of the final weight to the original dry weight. All experiments were repeated 4 times.
2.12 Cell culture
The hFOB cell line (CRL-11372, ATCC, USA) was maintained at 34 °C in DMEM-F12 containing 10% foetal bovine serum. The cells were cultured in a humidified atmosphere of 5% CO2 until confluency was achieved.
2.13 Cells cultured on the 2D hydrogel surfaces
2.13.1 Cell proliferation. Before cell seeding, the hydrogels were disinfected with 70% ethanol for 30 min and then washed with PBS, which was changed five times, for 5 min. The hydrogels were further incubated in DMEM-F12 overnight. Then, 400 μL of a cell suspension containing 5 × 104 cells were seeded on the scaffolds and incubated for 45 min to allow the cells to attach. The medium was changed every 2 days. The cell numbers were determined using a CCK-8 assay kit and by live/dead staining. The live/dead staining was observed by laser confocal microscopy (TCS SP8, Leica, Germany).
2.13.2 Alizarin red S staining and quantification. The mineralization of the hFOB cells cultured on the hydrogels was determined by alizarin red S staining.48 After incubation for 7 and 14 days, the cells with adherent mineralized nodules were removed from the hydrogels with trypsin–EDTA and washed with physiological saline. These cells were then stained with alizarin red S for 2 min. After rinsing with ultrapure water 5 times to remove the non-specific stain, the stained nodules were placed in a 96-well plate and observed by microscopy. The alizarin red S stain was extracted by sonication in 10 mM HCl in 70% ethanol for 10 min. The extracts were then diluted with 5 volumes of PBS, and the absorbance was measured at 550 nm using a multifunctional microplate reader (Thermo Scientific 3001, USA) for the determination of the mineralization.
2.13.3 ALP activity. The ALP activity was assayed using an alkaline phosphatase assay kit (P0012S, Beyotime Company, China) according to the manufacturer's instructions. Briefly, after culturing for 3, 7 and 14 days, the cells were washed three times with PBS and incubated in RIPA lysis buffer (P0013K, Beyotime Company, China) for 2 h at 4 °C. Then, 50 μL of the lysate of each sample was incubated in alkaline phosphatase buffer for 30 min at 37 °C. The reaction was stopped by adding 100 μL of a reaction stop solution, and the absorbance was measured at 405 nm using a multifunctional microplate reader (Thermo Scientific 3001, USA). A calibration curve was prepared using different concentrations of p-nitrophenyl, and the ALP activity was normalized to the total protein content of each scaffold using a commercially available protein assay kit (P0012S, Beyotime Company, China).
2.13.4 RNA isolation and quantitative real-time PCR. The marker genes of osteogenic differentiation (Runx 2, COL I, ALP, OPN and OCN) in the hFOB cells were detected by quantitative real-time PCR. The hFOB cells were seeded on the scaffolds at a density of 2 × 105 cm−2 and subsequently cultured for 3, 7 and 14 days at 37 °C. The total RNA was isolated using a HiPure Total RNA kit (Magentec, China) at the specified time intervals. The RNA was then reverse-transcribed into cDNA using a PrimeScript RT reagent kit with gDNA Eraser (TaKaRa Biotechnology, Japan). Finally, qRT-PCR analysis was performed using a SYBR Green system (GeneCopoeia) on a RT-PCR instrument (QuantStudio 6 Flex, Life Technologies). The relative quantification of the target genes was normalized to that of beta actin, and the 2−ΔΔCt method was used to calculate the gene expression.
2.13.5 Immunofluorescence staining. After culturing for 3, 7 and 14 days, the cells were fixed in a 4% neutral formaldehyde solution for 30 min, permeabilized with 0.1% Triton X-100 for 20 min and then incubated in 10% normal goat serum for 1 h to block non-specific proteins. The cells were incubated in the primary antibody (Runx 2 ((27 K): sc-101145, Santa Cruz), ALP ((C-8): sc-373737, Santa Cruz), OCN ((ABOC-5021): sc-73464, Santa Cruz) or OPN ((LFMb-14): sc-73631, Santa Cruz)) overnight at 4 °C and then incubated in the secondary antibody (donkey anti-mouse IgG H&L (Alexa Fluor® 647, ab150107) for Runx 2, OCN and OPN; donkey anti-mouse IgG H&L (Alexa Fluor® 594, ab150108) for ALP) for 1 h at room temperature. The Cell Navigator F-Actin labelling kit (F-actin, AAT Bioquest, Inc.) was used to stain the cell skeleton (green) for 1 h at room temperature. After gentle washing with PBS, the cell nuclei (blue) were stained with 4′,6′-diamidino-2-phenylindole (DAPI, Beyotime, China) for 5 min at room temperature. The cells were then observed by laser confocal microscopy (TCS SP8, Leica, Germany). To estimate the Runx 2, ALP, OCN and OPN expression, the ImageJ software was used to determine the ratio between the nuclear and cytoplasmic fluorescence intensities of the hFOB cells stained with the relevant antibody.49
2.14 Cells encapsulated in the 3D environment
2.14.1 Cell activity in the 3D environment. GelMA and GelMA-ALN were dissolved in a PBS solution at concentration of 10 w/v% with 0.5 w/v% Irgacure 2959 and then sterilized. Cells were digested and added to the solution to prepare a cell suspension of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 cells per mL. The cell suspension was transferred to well plates and crosslinked by UV light irradiation (5 mW) for 5 min. After adding the medium, the well plates were transferred to the incubator. For the cell activity studies, the cells encapsulated in the hydrogels were cultured at 34 °C for 1 and 5 days. Then, they were stained by live/dead staining and observed by laser confocal microscopy (TCS SP8, Leica, Germany).
000000 cells per mL. The cell suspension was transferred to well plates and crosslinked by UV light irradiation (5 mW) for 5 min. After adding the medium, the well plates were transferred to the incubator. For the cell activity studies, the cells encapsulated in the hydrogels were cultured at 34 °C for 1 and 5 days. Then, they were stained by live/dead staining and observed by laser confocal microscopy (TCS SP8, Leica, Germany).
2.14.2 Immunofluorescence staining in the 3D environment. The immunofluorescence staining method for the cells encapsulated in the 3D hydrogels was similar to that used for the cells on the 2D surface. After encapsulation in the hydrogels and culturing for 7 and 14 days, the cells were fixed in a 4% neutral formaldehyde solution for 1 h, permeabilized with 0.1% Triton X-100 for 20 min and then incubated in 10% normal goat serum for 1 h to block non-specific proteins. The cells were incubated in the primary antibody (ALP ((C-8): sc-373737, Santa Cruz), OCN ((ABOC-5021): sc-73464, Santa Cruz) or COL I (pan CEA (COL-1): sc-47756, Santa Cruz)) overnight at 4 °C and then incubated in the secondary antibody (donkey anti-mouse IgG H&L (Alexa Fluor® 647, ab150107) for COL I and OCN and donkey anti-mouse IgG H&L (Alexa Fluor® 594, ab150108) for ALP) for 1 h at room temperature. F-actin was used to stain the cell skeleton (green) for 1 h at room temperature. DAPI was used to stain the cell nuclei (blue) for 5 min at room temperature. The cells were then observed by laser confocal microscopy (TCS SP8, Leica, Germany). To estimate the COL I, ALP, and OCN expression, the ImageJ software was used to determine the ratio between the nuclear and cytoplasmic fluorescence intensities of the hFOB cells stained with the relevant antibody.49
2.15 Statistical analyses
Statistical analyses were performed using the SPSS software, version 12.0 (SPSS Inc, USA). The data were presented as the mean ± SD, and the levels were compared by the Student's t-test. P values of less than 0.05 were considered to be significant.
3 Results and discussion
As shown in Fig. 1, double bonds were first introduced into gelatin to enable UV crosslinking. Then, using zero grafting technology, which prevents the introduction of unrelated groups or links, alendronate was grafted onto GelMA.
3.1 Characterization of GelMA and GelMA-ALN
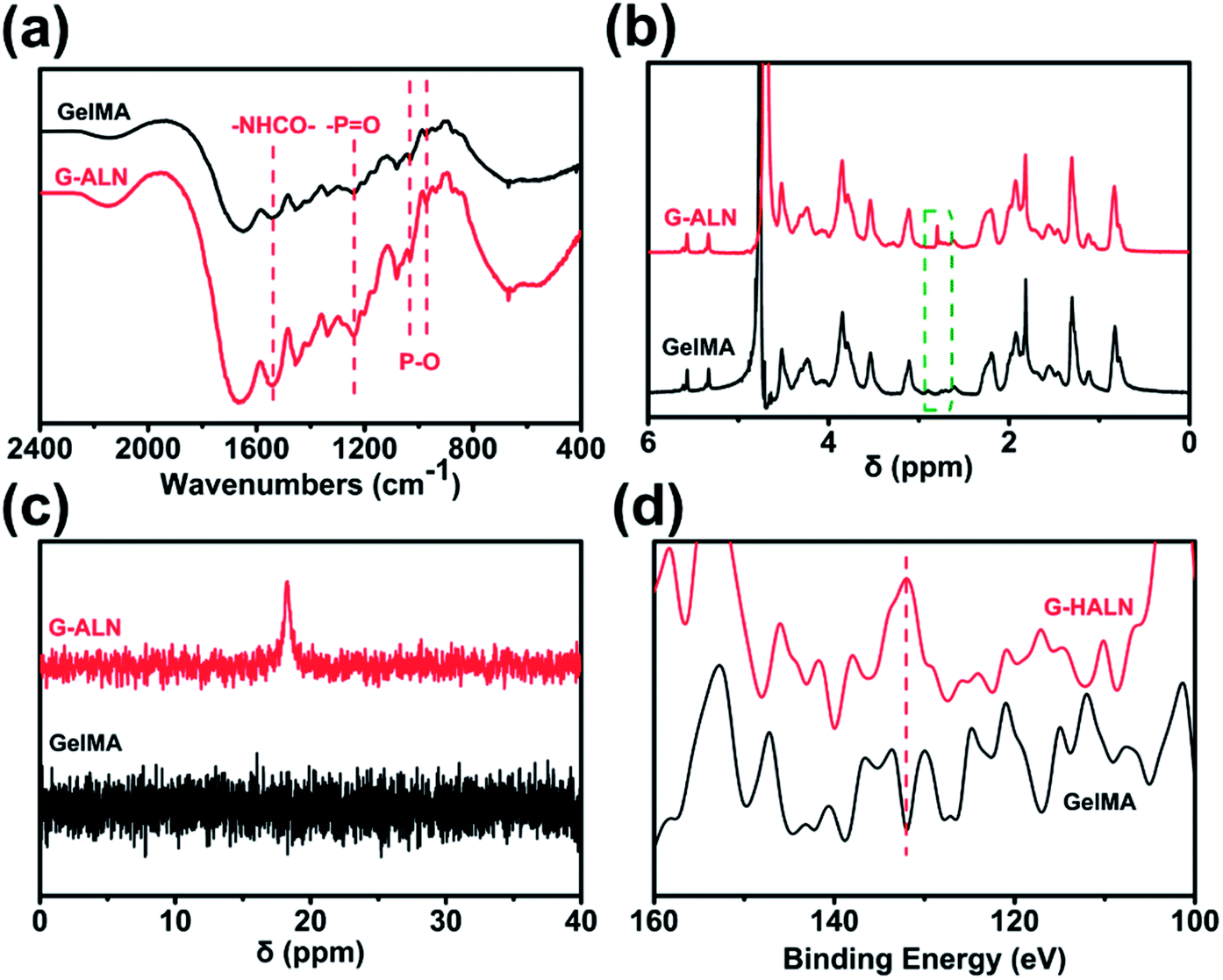
As shown in Fig. S1,† the proton peaks of the methyl groups (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) appeared at 5.3 and 5.6 ppm, demonstrating the successful introduction of double bonds into gelatin and the synthesis of GelMA. GelMA-ALN was characterized by FTIR, 1H NMR, 31P NMR and XPS. As shown in Fig. 2a, increases in the absorbances of the peaks at 1080, 975 and 920 cm−1, which overlapped with those of GelMA, indicated the presence of P–O. Furthermore, an increase in the absorbance of the amino II stretch at 1550 cm−1 was also observed, demonstrating that alendronate was grafted onto gelatin by an amide reaction. In the 1H NMR spectrum of GelMA-ALN (Fig. 2b), the peak at 2.8 ppm was attributed to the protons of the phosphate group in alendronate. The 31P NMR spectra (Fig. 2c) revealed an obvious resonance peak at 17.6 ppm for GelMA-ALN.50 The surface chemical composition was determined by XPS. As shown in Fig. 2d, the presence of the P 2p signals at 134 eV further demonstrated that bisphosphonate groups were successfully introduced into GelMA. Thus, the FTIR, 1H NMR, 31P NMR and XPS data demonstrated that alendronate was successfully grafted onto GelMA.
CH2) appeared at 5.3 and 5.6 ppm, demonstrating the successful introduction of double bonds into gelatin and the synthesis of GelMA. GelMA-ALN was characterized by FTIR, 1H NMR, 31P NMR and XPS. As shown in Fig. 2a, increases in the absorbances of the peaks at 1080, 975 and 920 cm−1, which overlapped with those of GelMA, indicated the presence of P–O. Furthermore, an increase in the absorbance of the amino II stretch at 1550 cm−1 was also observed, demonstrating that alendronate was grafted onto gelatin by an amide reaction. In the 1H NMR spectrum of GelMA-ALN (Fig. 2b), the peak at 2.8 ppm was attributed to the protons of the phosphate group in alendronate. The 31P NMR spectra (Fig. 2c) revealed an obvious resonance peak at 17.6 ppm for GelMA-ALN.50 The surface chemical composition was determined by XPS. As shown in Fig. 2d, the presence of the P 2p signals at 134 eV further demonstrated that bisphosphonate groups were successfully introduced into GelMA. Thus, the FTIR, 1H NMR, 31P NMR and XPS data demonstrated that alendronate was successfully grafted onto GelMA.
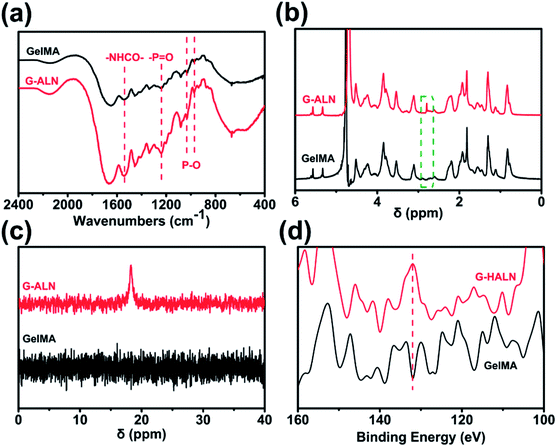 |
| | Fig. 2 (a) FTIR, (b) 1H NMR, (c) 31P NMR and (d) XPS spectra of the GelMA, G-LALN, G-MALN and G-HALN materials. | |
3.2 Topography and properties of hydrogels
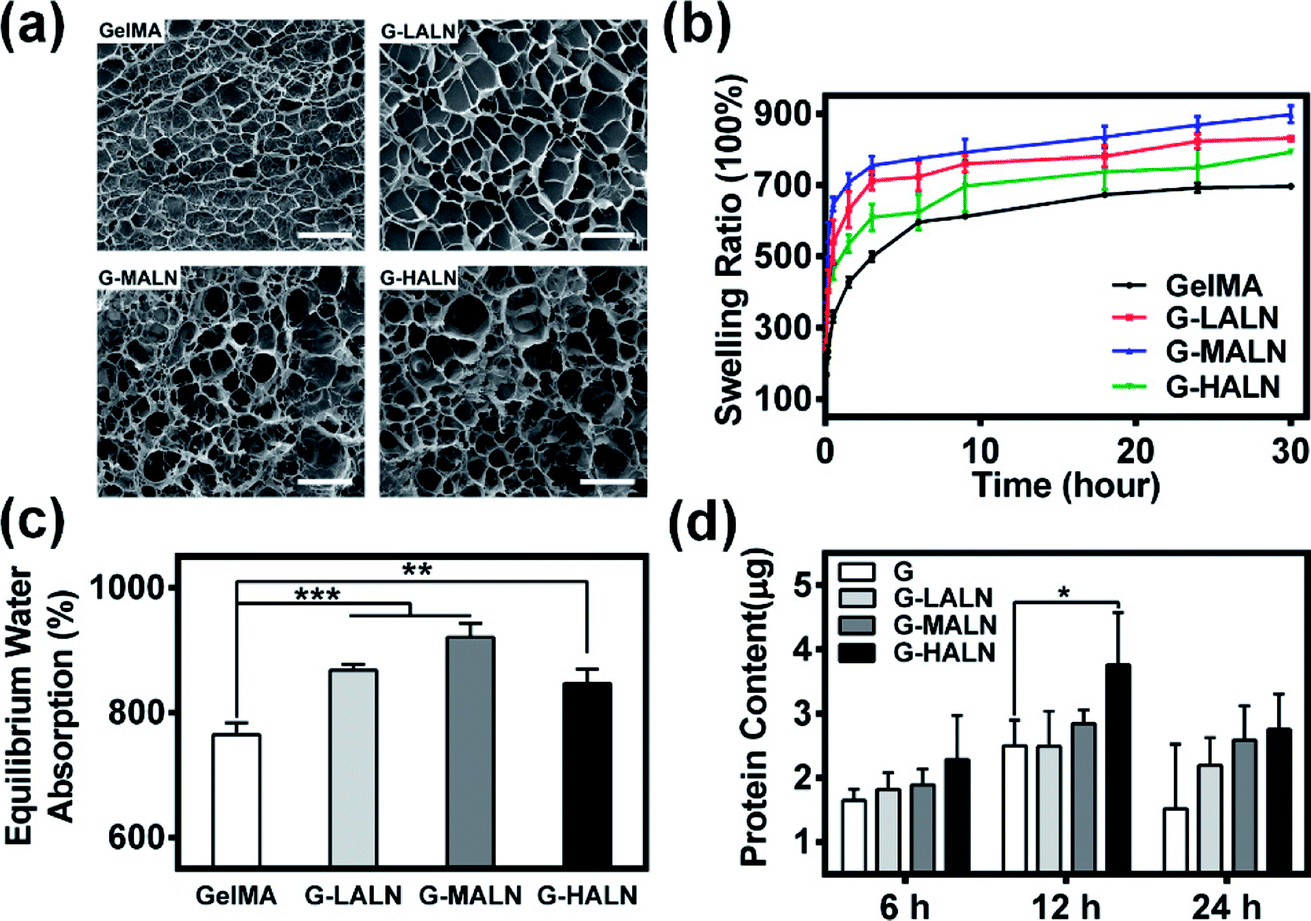
The microstructure of the hydrogels is shown in Fig. 3a. All hydrogels typically have porous microstructures with pore sizes on the order of tens of microns. The GelMA and G-LALN hydrogels had a less dense 3D structure with larger interconnected pores. The G-MALN and G-HALN hydrogels had an interconnected microstructure with a fibrillar mesh, indicating that the network structure of the hydrogel was maintained after in situ processing. The connected porous microstructure of the hydrogels enables nutrient transport and waste disposal. The swelling behaviour of GelMA and the alendronate-modified GelMA hydrogels was investigated, as shown in Fig. 3b and c. The swelling ratios of the hydrogels first increased and then decreased as the alendronate content increased. The equilibrium swelling ratio (Fig. 3c) exhibited the same trend as the swelling ratio. All the alendronate-modified GelMA hydrogels had higher equilibrium swelling ratios than the GelMA hydrogel. The equilibrium swelling ratio increased from 765% to 921% and then decreased to 846% as the alendronate content increased. The substitution of the amino groups by BP groups led to an increase in the hydrophilicity of the hydrogels. The dramatic decrease in the swelling of the G-HALN hydrogels might have been due to intramolecular hydrogen bonding.
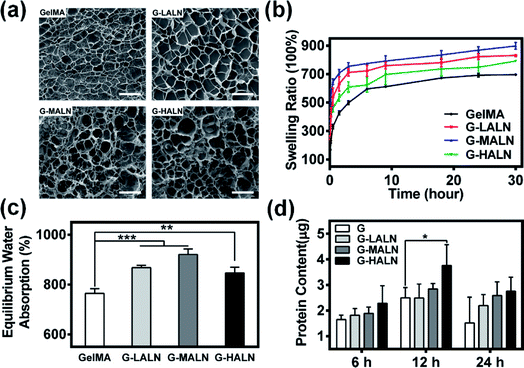 |
| | Fig. 3 (a) SEM images of the microstructures of the hydrogels. The scale bar is 100 μm. (b) Swelling ratio, (c) equilibrium water absorption and (d) protein adsorption of the GelMA, G-LALN, G-MALN and G-HALN hydrogels. | |
The mechanical properties of the hydrogels are shown in Fig. S2.† The modification of GelMA with ALN showed a significant decrease in compressive modulus. The compressive modulus was determined to be 0.29 ± 0.01, 0.24 ± 0.02, 0.14 ± 0.02 and 0.11 ± 0.01 kPa for GelMA, G-LALN, G-MALN and G-HALN, respectively. To further investigate the effect of modification on the mechanical properties of hydrogels, rheological measurements were performed before and after the polymerization during the formation of the hydrogels. Oscillatory frequency sweeps during the polymerization process showed that the polymerization started within 15 s (Fig. S3a†), which provides the possibility of rapid photocross-linking after injection. As shown in Fig. S3b,† each hydrogel showed significantly higher G′ compared to G′′ and the G-HALN hydrogel showed lowest values of G′ at all shear stress amplitudes tested. Moreover, the frequency sweep curves illustrated that the G′ remained the same irrespective of the frequency applied which is a typical behaviour of solid-like materials.
Previous research showed that adsorbed serum components can mediate cell attachment to and spreading on hydrogels.47 The protein adsorption capacity was determined by immersing the hydrogels in a medium with 10% foetal bovine serum. As demonstrated in Fig. 3d, alendronate modification could promote protein adsorption. After soaking for 6 h, the protein adsorption of the hydrogels increased but was not significantly different. The protein adsorption on the G-HALN hydrogel was significantly enhanced compared to that on the GelMA hydrogel at 12 h, suggesting that alendronate modification resulted in a remarkable increase in the protein adsorption capacity. This result could be explained by the fact that the zeta potential of GelMA was altered by incorporating charged –PO4 groups into it through the grafting of alendronate.47,51 This explanation was further supported by the observed increase in the protein adsorption on the GelMA-ALN hydrogels with increasing alendronate content.
To study the effect of alendronate modification on degradation of hybrid hydrogels, we studied enzymatic degradation of GelMA and GelMA-ALN hydrogels in collagenase solution by monitoring the percentage of hydrogel residual mass (Fig. S4†). Due to the highly interconnected and porous network, the collagenase solution can easily permeate the hydrogel network and degrade the entire network uniformly and rapidly. Furthermore, the GelMA and G-LALN hydrogels underwent complete degradation within 36 h, whereas the G-MALN and G-HALN hydrogels lasted for 48 h. This illustrates the feasibility of cell-mediated enzymatic degradation of the hydrogels, a critical feature for enabling encapsulated cells to remodel their microenvironment to form a continuous tissue.
3.3 Mineralization
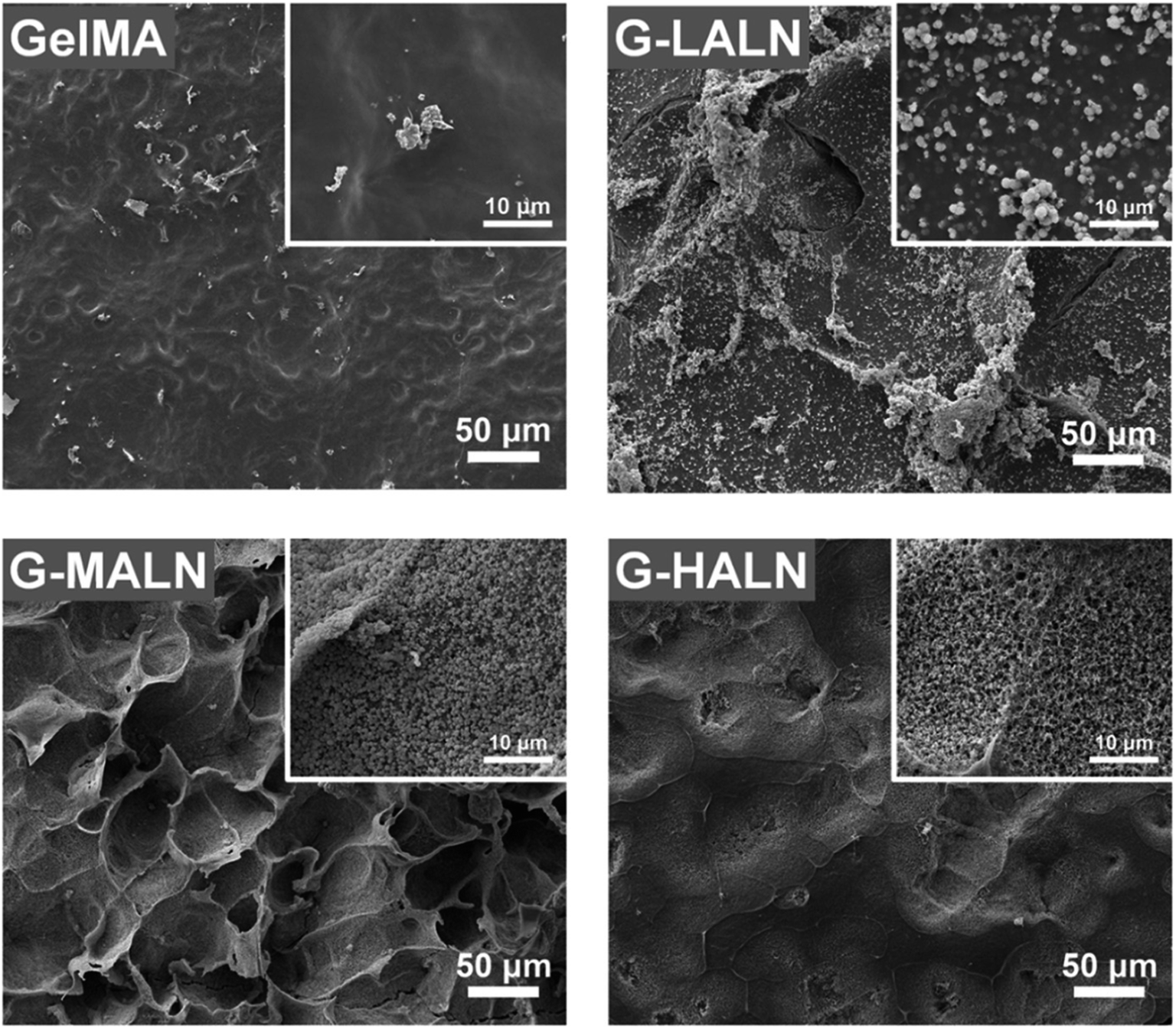
The in vitro osteogenic bioactivity of the hydrogel network could be evaluated by the in vitro biomineralization. The formation of apatite-like deposits on a biomaterial surface upon submersion in SBF indicates its bone-bonding capability. In this study, mineralization was characterized by immersing the hydrogels in 2× SBF for 3 days. As shown in Fig. 4, very few irregular apatite particles formed on the surface of the GelMA hydrogel, whereas many spherical apatite clusters, which might be an amorphous precursor of crystalline hydroxyapatite, formed on the surfaces of the alendronate-modified hydrogels.52 The formation of the apatite particles correlated with the BP content; as the content of the BP groups increased, more and smaller apatite particles were formed. These results suggested that the BP groups provided additional sites for hydroxyapatite nucleation and growth.
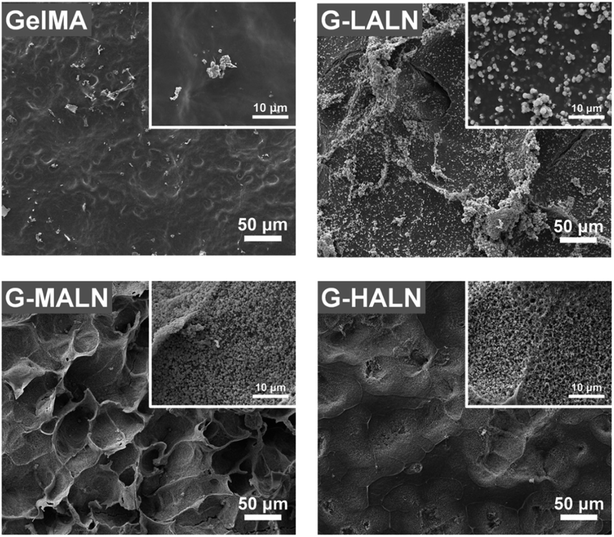 |
| | Fig. 4 SEM images of the GelMA, G-LALN, G-MALN and G-HALN hydrogels after mineralization in a 2× SBF solution for 3 days. | |
3.4 Cell proliferation
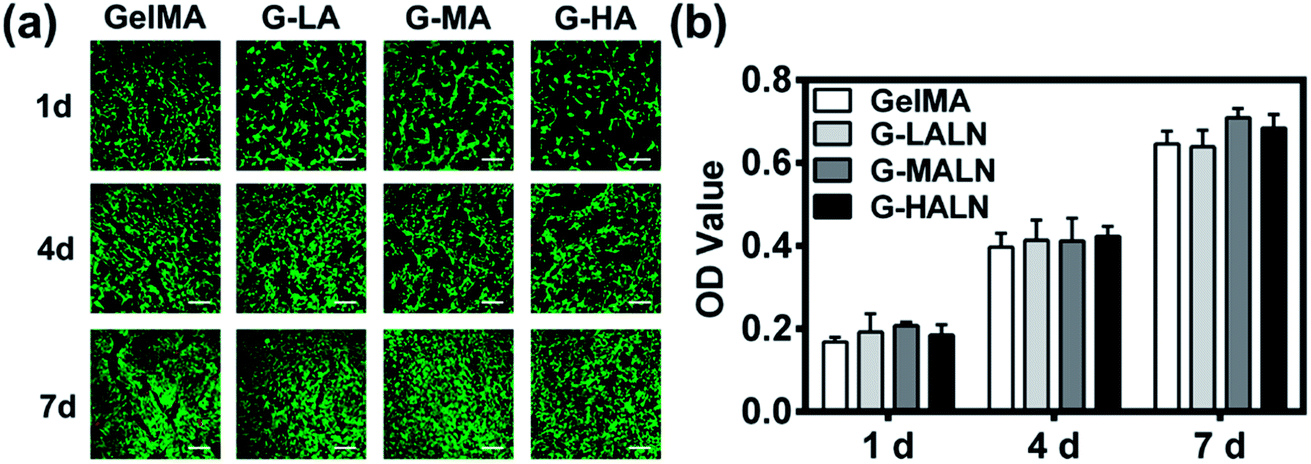
The ability to support cell proliferation is an important requirement for tissue engineering scaffolds. To evaluate the cytocompatibility of the hydrogels, CCK-8 tests and live/dead staining were performed. The live/dead staining of hFOB cells cultured on the hydrogels is shown in Fig. 5a. Most of the cells in the four different groups were green, and few dead cells (red) were observed because of cell metabolism and apoptosis. The cells proliferated well on all the hydrogels, and the cell numbers on all the hydrogels increased obviously from day 1 to day 7. The cells nearly covered the hydrogel surfaces at day 7. The quantitative data (Fig. 5b) of cell proliferation were further supported by CCK-8 tests. The proliferation rates of the hFOB cells on the GelMA and GelMA-ALN hydrogels were not significantly different. These results indicated that the introduction of BP groups did not affect the biological activity of the GelMA hydrogel.
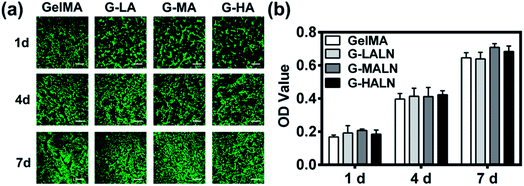 |
| | Fig. 5 Proliferation and spread of hFOB cells cultured on the GelMA, G-LALN, G-MALN and G-HALN hydrogels. (a) Confocal fluorescence microscopy images of the hFOB cells stained with a live/dead reagent on days 1, 4 and 7. The live and dead cells were stained green and red, respectively. The scale bar is 500 μm. (b) CCK-8 assay of the hFOB cells on the CS and CS-P scaffolds after 1, 4, and 7 days of culture. | |
3.5 Alizarin red S staining and ALP activity
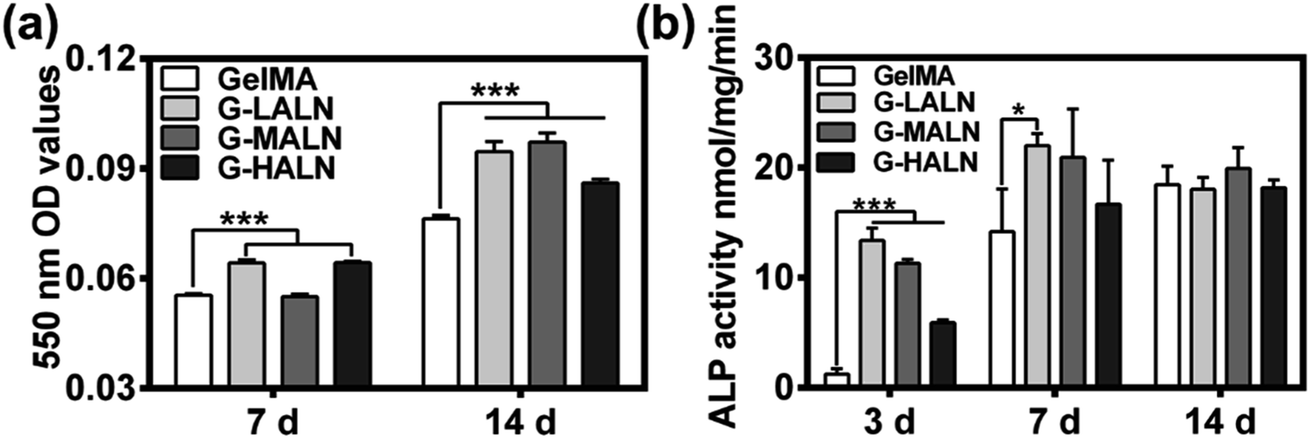
To investigate the effects of the alendronate modification on osteogenesis, hFOB cells were cultured on the hydrogels for 7 days (early stage) or up to 14 days (late stage) after initiation of induction. Alizarin red S staining allows the comparison of bone-like calcium deposits.53 Both quantitative and qualitative evaluations of the alizarin red S staining were performed. The mineralization level visualized by alizarin red S staining revealed the deposition of calcium. As shown in Fig. S2,† after 14 days of induction at 37 °C, mineralized nodules were observed in all the groups. The alizarin red S staining of the GelMA-ALN hydrogels was more distinct than that of the GelMA hydrogel. This result indicated that the alendronate-modified GelMA hydrogels facilitated more mineralization than the GelMA hydrogel, which was further confirmed by dissolving the stained calcium nodules and measuring the OD value of the solution at 550 nm. As shown in Fig. 6b, the OD values of the G-LALN and G-HALN groups were significantly higher than that of the GelMA group on day 7. Moreover, the OD values of all the alendronate-modified GelMA groups were significantly higher than that of the GelMA group (P < 0.001) on day 14, which demonstrated the greater capacity of the alendronate-modified GelMA groups for supporting mineralization. The ALP activities of hFOB cells cultured on the hydrogels for 3, 7 and 14 days was presented in Fig. 6c. The hFOB cells cultured on the alendronate-modified GelMA hydrogels had significantly higher ALP activities than those cultured on the GelMA hydrogel. The ALP activity of the cells cultured on the G-LALN hydrogel for 7 days was significantly higher than that of the cells cultured on the GelMA hydrogel. The higher ALP activity and alizarin red S staining of the GelMA-ALN groups indicated their greater ability to support cell osteogenic differentiation.
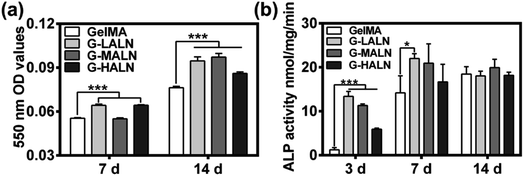 |
| | Fig. 6 (a) Quantitative mineralization results, that is, the absorbance of the extracted alizarin red S dye measured at 7 and 14 days. (b) ALP activity of hFOB cells cultured for 3, 7 and 14 days (*P < 0.05, **P < 0.01, ***P < 0.001). | |
3.6 Gene expression related to osteogenesis
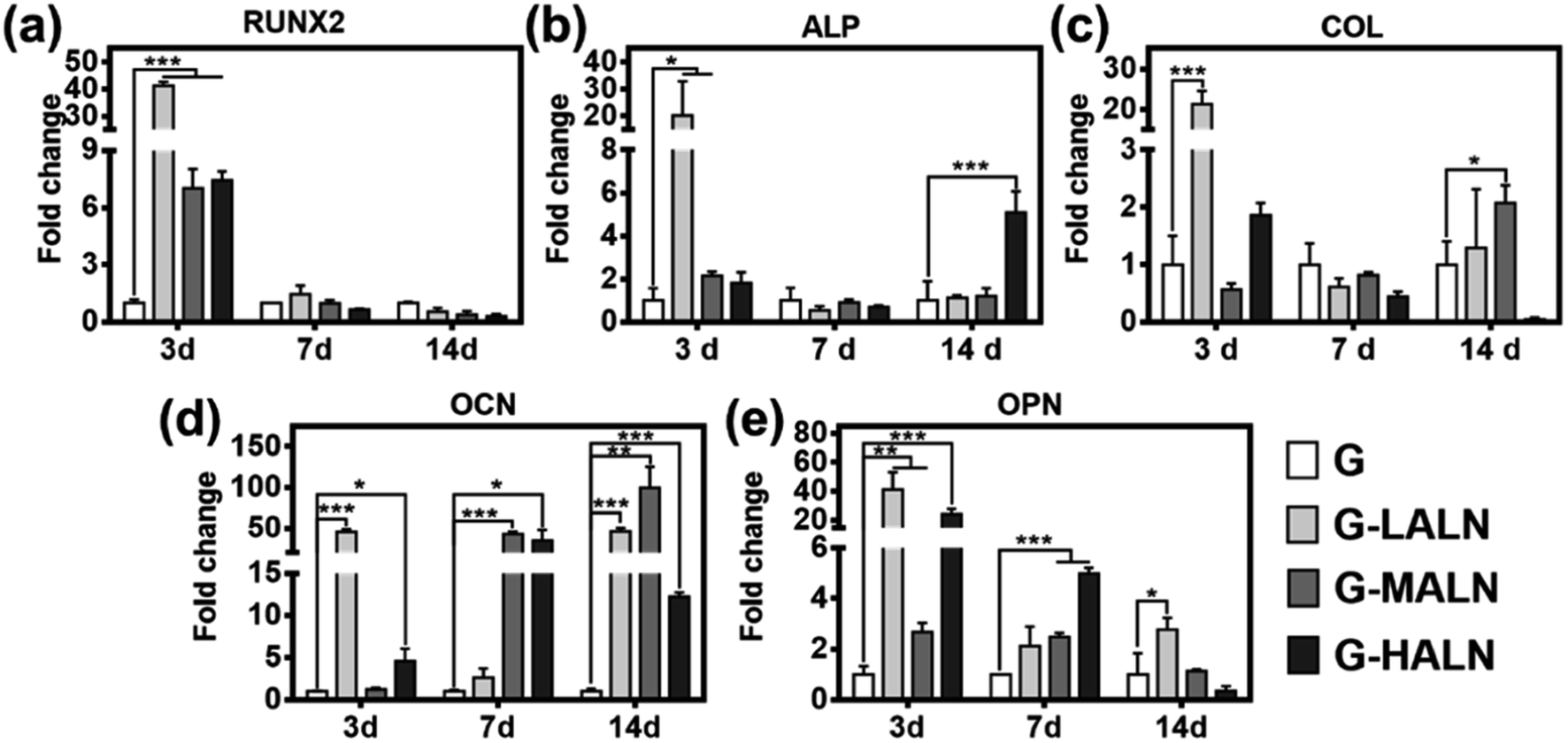
To assess the functional effects of the alendronate modification on osteogenic differentiation in the hFOB cells, the expression of key marker genes associated with osteogenesis, namely, Runx 2, ALP, COL I, OCN and OPN, are shown in Fig. 7. At day 3, all the alendronate-modified GelMA groups exhibited significant differences in the expression of Runx 2. Specifically, the five key osteogenic markers in the G-LALN group were significantly up-regulated compared to those in the control group. At day 7, the expression of OCN and OPN in the G-MALN group and of OPN in the G-HALN group was significantly higher than that in the control group. At day 14, the levels of OCN in the cells cultured on the alendronate-modified GelMA groups were significantly higher than that in the control group. The cells on the alendronate-modified GelMA hydrogels exhibited a significantly higher expression of the well-established early (Runx 2) and late (OCN) bone markers. The marked increases in the levels of Runx 2, ALP, OCN and OPN expression further indicated the osteogenic differentiation of the hFOB cells on the alendronate-modified GelMA hydrogels.
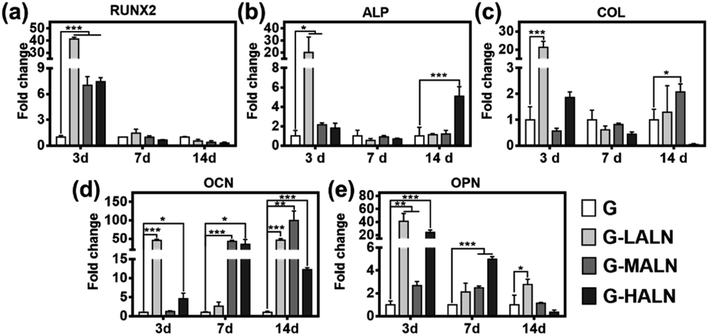 |
| | Fig. 7 qRT-PCR analysis of the mRNA levels of osteogenic genes. Relative gene expression of (a) Runx 2, (b) ALP, (c) COL I, (d) OCN and (e) OPN of the hFOB cells after 7 and 14 days of culture. Statistically significant differences between the different materials at the same time point (*P < 0.05, **P < 0.01, ***P < 0.001). | |
3.7 Immunofluorescence staining
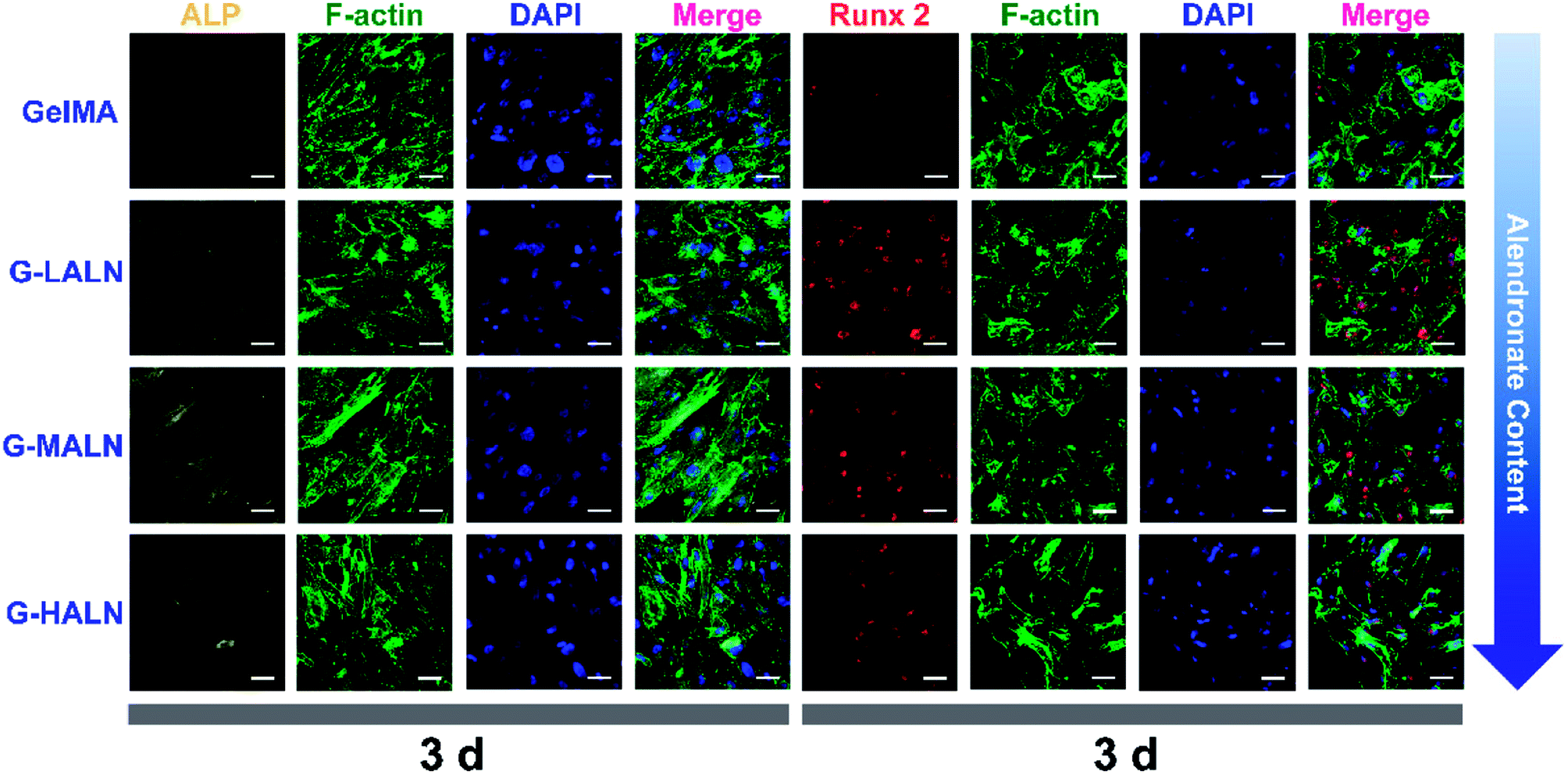
This finding was further confirmed by analysing the protein markers Runx 2, ALP, OPN and OCN, which are osteogenesis markers in the early and late stages of osteogenic differentiation, by the immunofluorescence staining of hFOB cells on the GelMA and alendronate-modified GelMA hydrogels. As shown in Fig. 8, the early osteogenesis proteins, ALP and Runx 2, were evaluated at day 3. At this early time in the osteogenic differentiation process, the G-LALN and G-MALN groups exhibited higher fluorescence intensities for Runx 2 and ALP. The Runx 2 stain was mainly concentrated in the nuclei in the GelMA group, whereas it was concentrated in the nuclei and their surroundings in the G-LALN and G-MALN groups. The ALP stain was distributed throughout the cells. Some irregular staining was observed in the GelMA group. In contrast, the cells in the G-LALN and G-MALN groups were almost entirely stained.
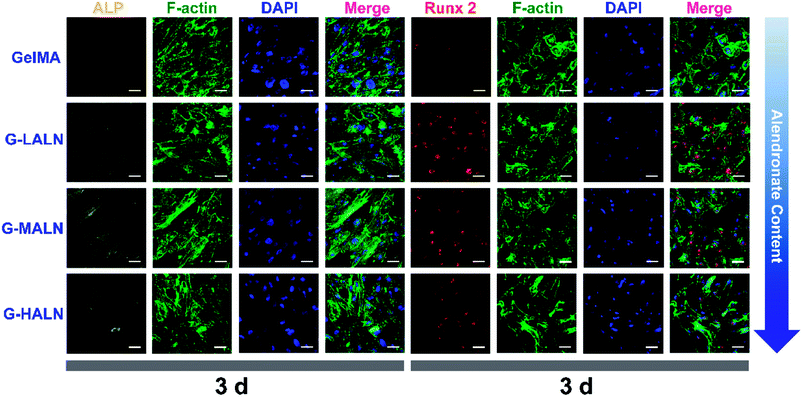 |
| | Fig. 8 Immunofluorescence staining of hFOB cells cultured on the GelMA, G-LALN, G-MALN and G-HALN hydrogels. ALP and Runx 2 expression of hFOB cells cultured for 3 days. The scale bar is 50 μm. | |
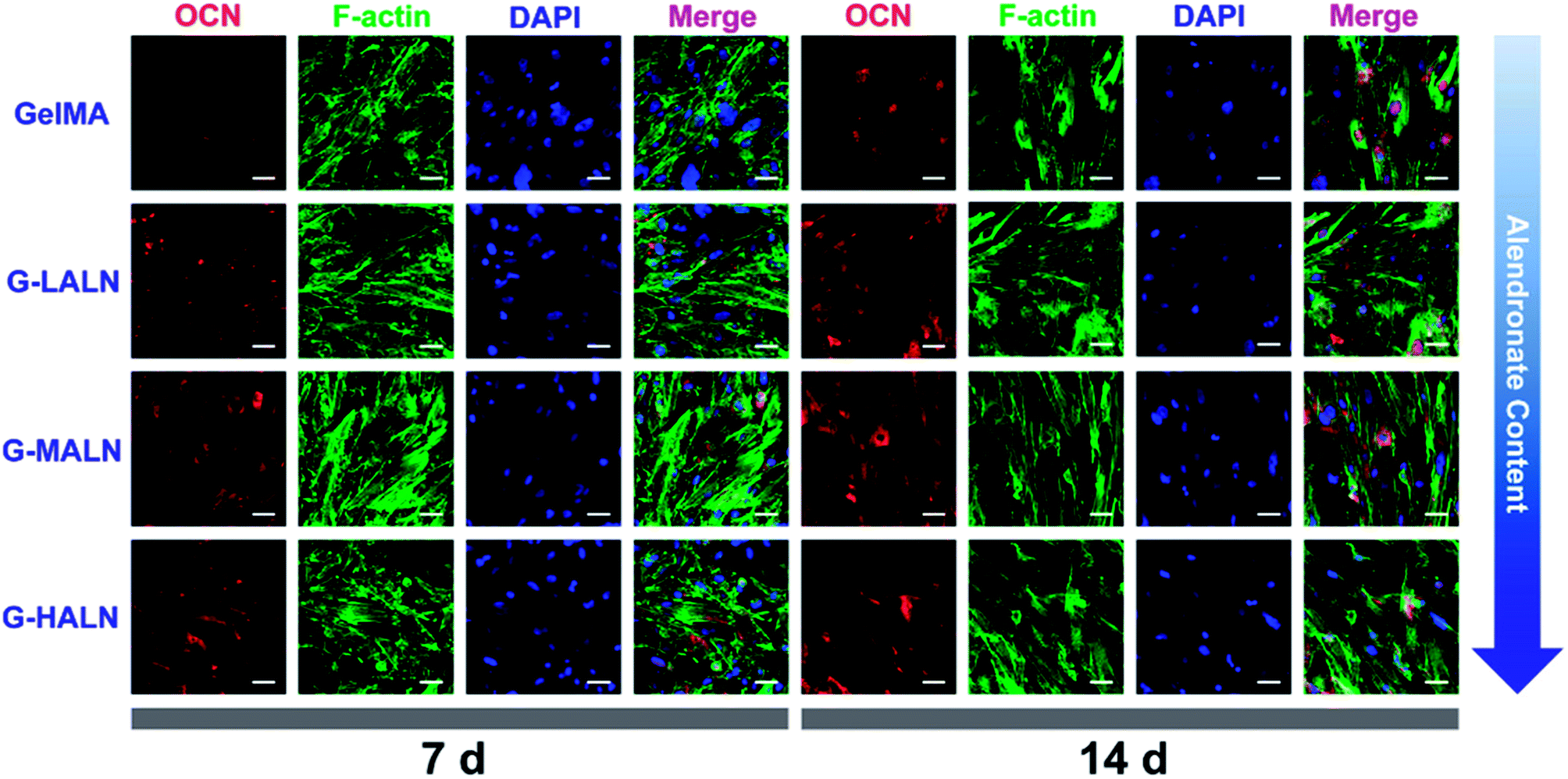
OCN, which is the most abundant protein in the bone matrix, participates in both endocrine and mineralization processes and plays an essential role in bone formation. As shown in Fig. 9, as the culture time increased from 7 days to 14 days, the OCN staining of all the groups became brighter, because the amount of the OCN protein secreted by the hFOB cells increased with increasing culture time. Some irregular staining of the cells was observed at 7 days; however, the cells were almost entirely stained at 14 days. More OCN staining was observed in the G-LALN, G-MALN and G-HALN groups than in the GelMA group at both 7 and 14 days.
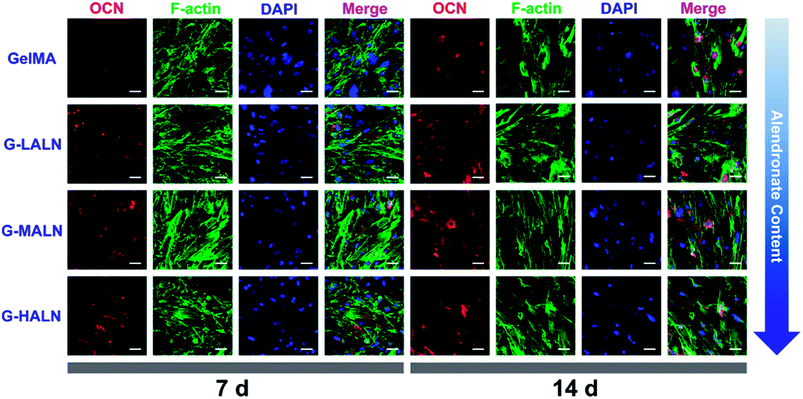 |
| | Fig. 9 Immunofluorescence staining of hFOB cells cultured on the GelMA, G-LALN, G-MALN and G-HALN hydrogels. OCN expression of hFOB cells cultured for 7 and 14 days. The scale bar is 50 μm. | |
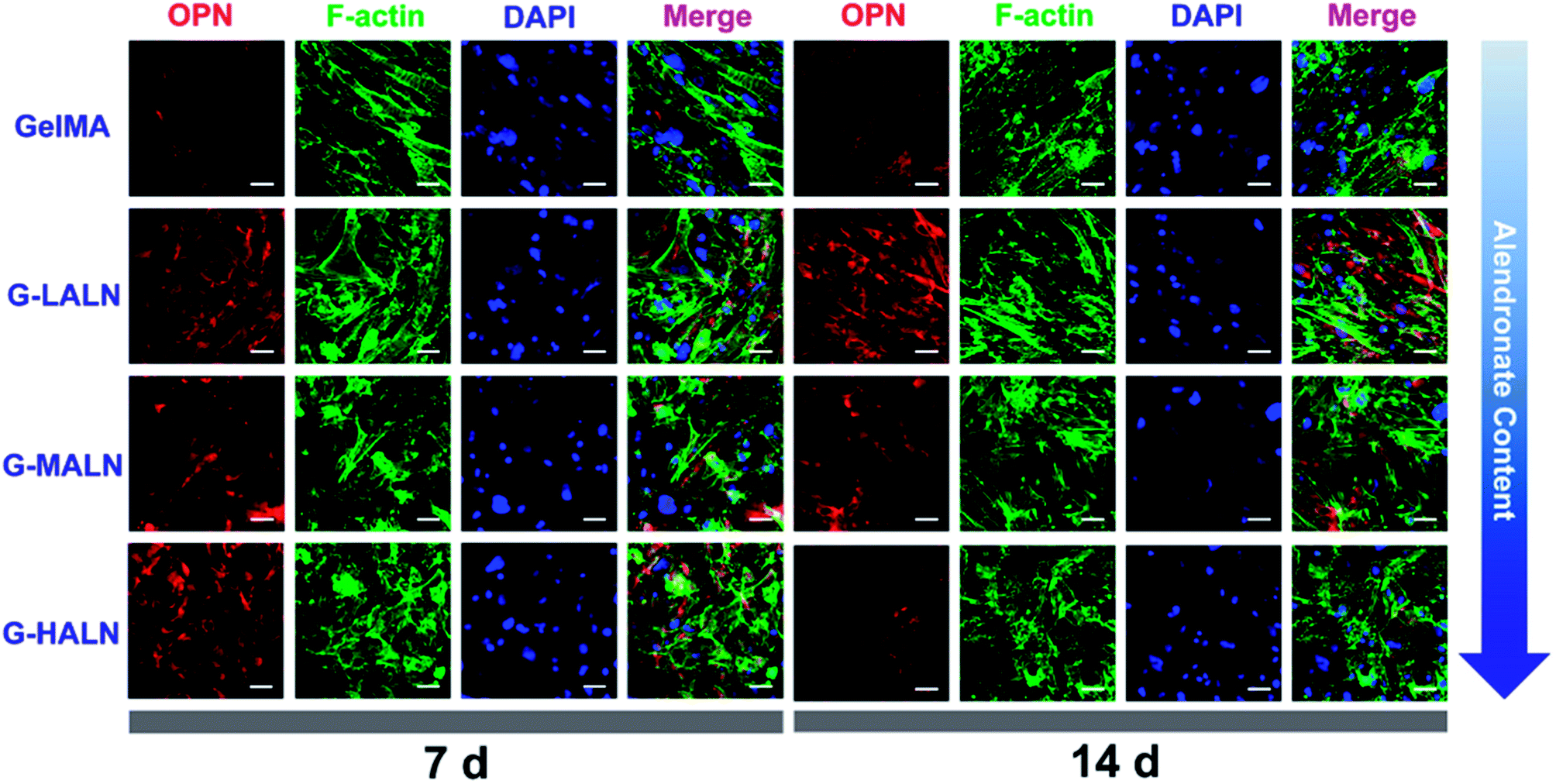
Similar phenomena were observed for the OPN staining (Fig. 10). As the culture time increased from 7 days to 14 days, the OPN staining of all the groups became brighter. More OPN staining was observed in the G-LALN and G-MALN groups than in the GelMA group at both 7 and 14 days. Semi-quantitative analysis of the immunofluorescence staining was performed using the ImageJ software to quantify the ability of the hydrogels to promote osteogenic differentiation. As shown in Fig. S3,† the observed trend was similar to those in the visual immunofluorescence staining observations and the quantitative results of qPCR. At a culture time of 3 days (Fig. S3a†), all the alendronate-modified GelMA hydrogels exhibited significant differences in the expression of Runx 2 and ALP. The semi-quantitative analysis of the OCN immunofluorescence staining (Fig. S3b†) showed that the OCN expression of all the alendronate-modified GelMA groups was significantly higher than that of the control group at both 7 and 14 days. The semi-quantitative analysis of the OPN immunofluorescence staining (Fig. S3c†) showed that the OPN expression of the G-LALN and G-MALN groups was significantly higher than that of the control group at both 7 and 14 days. The results of the immunofluorescence staining and semi-quantitative analyses demonstrated that the G-LALN and G-MALN groups significantly promoted the secretion of osteogenesis proteins in the 2D environment. This is consistent with previous research that low-dose of alendronate can serve as an osteo-inductive factor to promote osteogenesis in vitro and accelerate bone regeneration in vivo.54–57 Overall, these results showed that the alendronate modification significantly accelerated the osteoblastic differentiation of hFOB cells without the need for osteogenesis-inducing supplements.
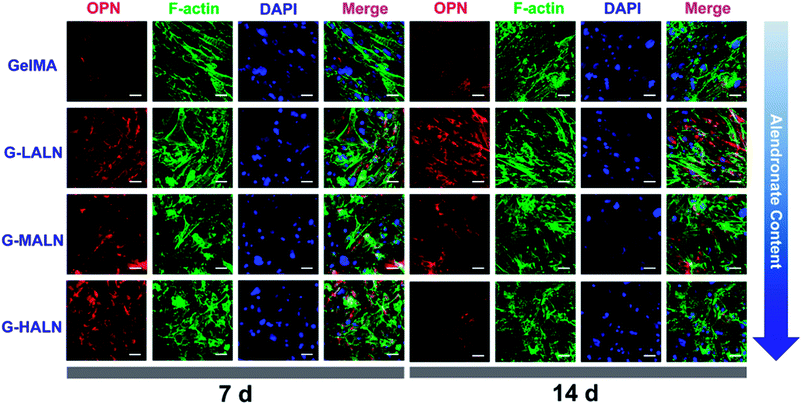 |
| | Fig. 10 Immunofluorescence staining of hFOB cells cultured on the GelMA, G-LALN, G-MALN and G-HALN hydrogels. OPN expression of hFOB cells cultured for 7 and 14 days. The scale bar is 50 μm. | |
3.8 Cells encapsulated in the 3D hydrogel
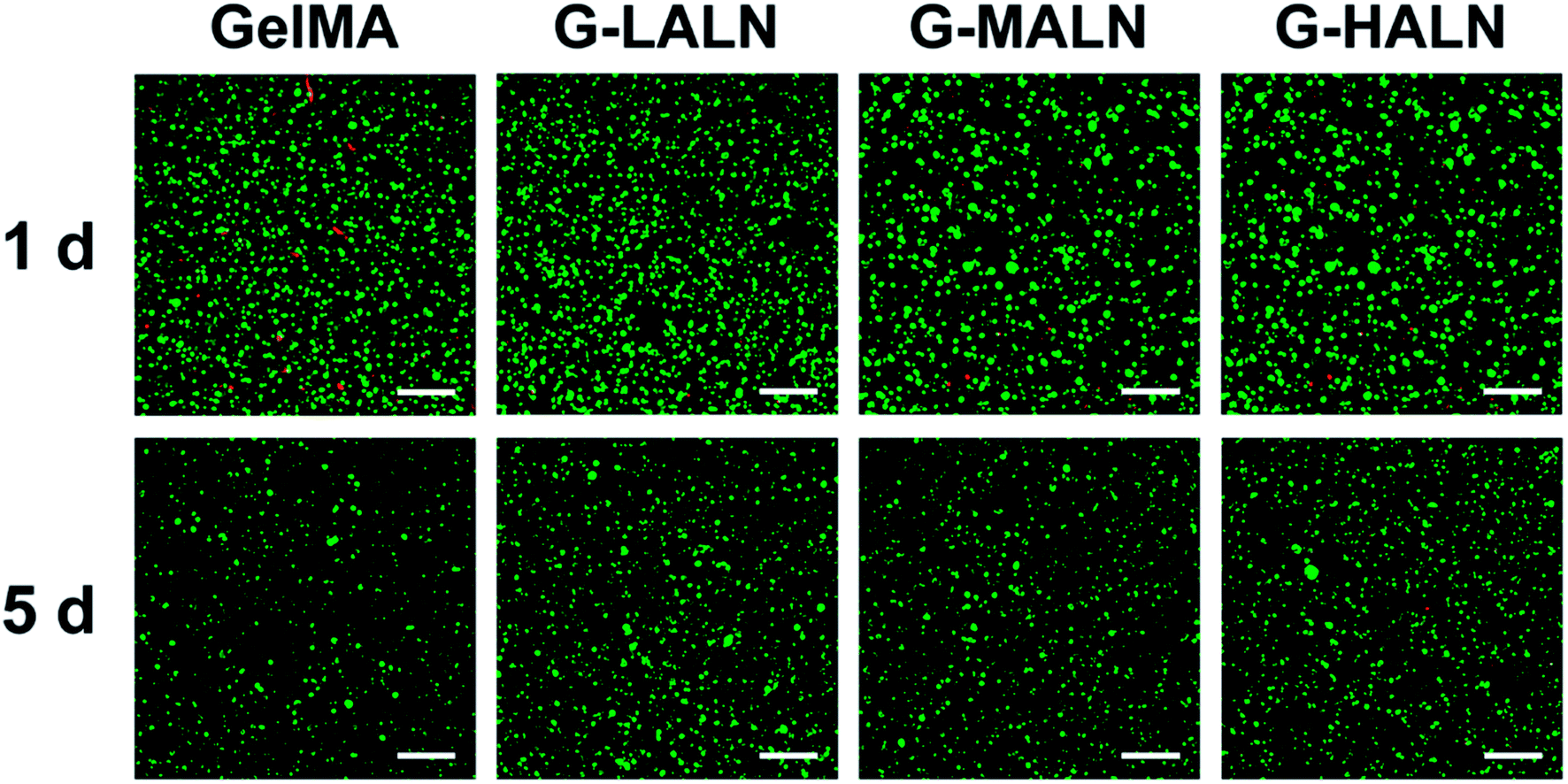
3.8.1 Cell activity. Hydrogels are promising tissue engineering scaffolds because they closely mimic the extracellular microenvironment. Maintaining normal cellular behaviour in a 3D microenvironment is an important criterion for the fabrication of tissue engineering scaffolds. When hFOB cells were encapsulated in the hydrogels, they maintained good activity for 5 days (Fig. 11). Together with the results for the cells cultured on the surfaces of the hydrogels, these results demonstrated that the alendronate-modified GelMA hydrogels exhibited no cytotoxicity compared to the GelMA group.
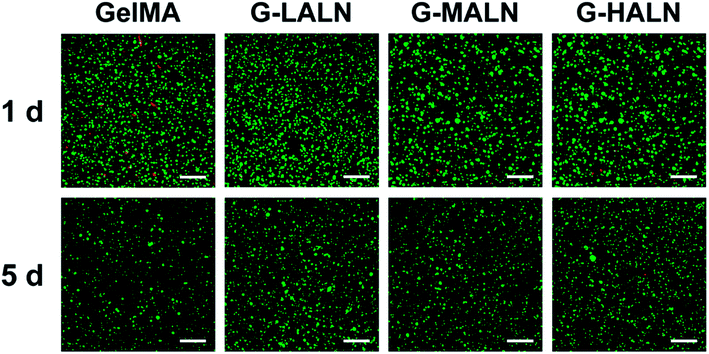 |
| | Fig. 11 Laser confocal images of hFOB cells encapsulated in the hydrogels for 1 and 5 days. The live and dead cells were stained green and red, respectively. The scale bar is 200 μm. | |
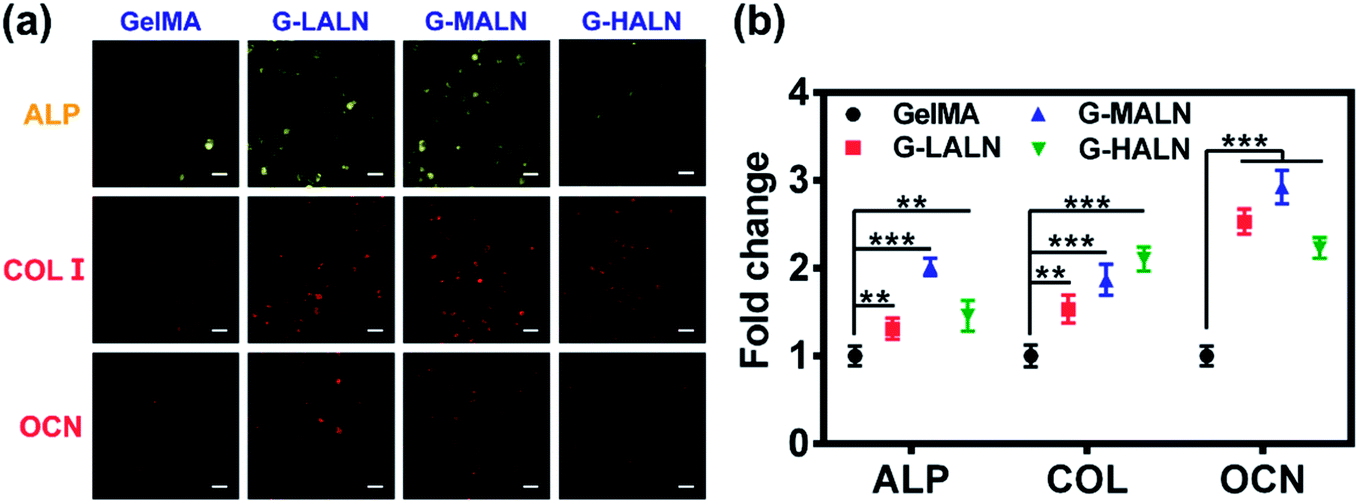
3.8.2 Immunofluorescence staining. Bone tissue engineering scaffolds must be able to mimic extracellular matrix characteristics, including the interconnected porous structures, 3D microenvironments and abundant cell adhesion sites, which have been shown to be essential factors.58 Similar to the results for the cells cultured on the surfaces of the hydrogels, the hFOB cells encapsulated in the GelMA-ALN hydrogels could be differentiated into osteoblasts. The cells secreted the osteogenesis proteins ALP, OCN and COL I in the 3D environment (Fig. 12a). A semi-quantitative analysis of the immunofluorescence staining showed that the fluorescence intensities of all the alendronate-modified GelMA hydrogels were significantly higher than that of the control group (Fig. 12b). This result indicated that the GelMA-ALN hydrogels exhibited a greater osteogenic differentiation effect than the control group in the 3D environment. The inner structures of the GelMA-ALN hydrogels appeared to be more conducive for osteogenesis. In summary, the GelMA-ALN hydrogels enhanced the expression of osteogenic marker proteins in hFOB cells in both 2D and 3D environments.
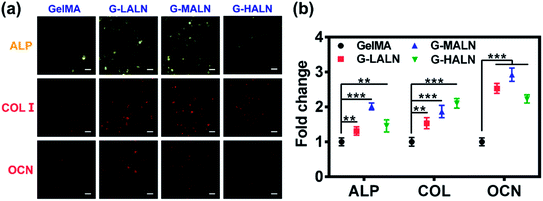 |
| | Fig. 12 Immunofluorescence staining images of hFOB cells encapsulated in the GelMA, G-LALN, G-MALN and G-HALN hydrogels for 7 days (ALP) and 14 days (COL I and OCN). The scale bar is 25 μm. | |
4 Conclusions
Methacryloyl gelatin (GelMA) was synthesized and then successfully grafted with alendronate without introducing any unrelated functional groups. The modification greatly improved the swelling ratio and equilibrium water absorption. The ALP activity and alizarin red S staining results revealed the up-regulation of alkaline phosphatase and mineralization, respectively, of hFOB cells cultured on the alendronate-modified GelMA hydrogels. The up-regulation of the osteogenesis-related genes Runx 2, ALP, OCN and OPN indicated that osteogenic differentiation occurred in the alendronate-modified GelMA hydrogels. Furthermore, the cells maintained their activity and differentiated into osteoblasts when they were encapsulated in the GelMA-ALN hydrogels. The alendronate-modified hydrogels are expected to be useful for the minimally invasive treatment of irregular bone defects.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge funding from the National Nature Science Foundation of China (51502095 and 81401191), Guangdong Natural Science Funds for Distinguished Young Scholar (2016A030306018), Guangdong Natural Science Funds (2017B090911008), Guangdong Young Pearl River Scholars and Science and Technology Research Foundation of Shenzhen Bureau of Science and Technology & Information (JCYJ20170306141716014).
References
- K. Yue, G. T. d. Santiago, M. M. Alvarez, A. Tamayol, N. Annabi and A. Khademhosseini, Biomaterials, 2015, 73, 254–271 CrossRef PubMed.
- A. I. V. D. Bulcke, B. Bogdanov, N. D. Rooze, E. H. Schacht, M. Cornelissen and H. Berghmans, Biomacromolecules, 2000, 1, 31–38 CrossRef.
- Y. X. Liu and M. B. Chan-Park, Biomaterials, 2010, 31, 1158–1170 CrossRef PubMed.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Biomaterials, 2010, 31, 5536–5544 CrossRef PubMed.
- E. Kaemmerer, F. P. W. Melchels, B. M. Holzapfel, T. Meckel, D. W. Hutmacher and D. Loessner, Acta Biomater., 2014, 10, 2551–2562 CrossRef PubMed.
- T. Billiet, E. Gevaert, T. D. Schryver, M. Cornelissen and P. Dubruel, Biomaterials, 2014, 35, 49–62 CrossRef PubMed.
- L. E. Bertassoni, J. C. Cardoso and V. Manoharan, Biofabrication, 2014, 6, 024105 CrossRef PubMed.
- A. Jain, A. Karadag, B. Fohr, L. W. Fisher and N. S. Fedarko, J. Biol. Chem., 2002, 277, 13700–13708 CrossRef PubMed.
- R. F. Loese, Exp. Cell Res., 1994, 211, 17–23 CrossRef PubMed.
- A. Veis, C. Sfeir and C. B. Wu, Crit. Rev. Oral Biol. Med., 1997, 8, 360–379 CrossRef.
- G. He, A. Ramachandran, T. Dahl, S. George, D. Schultz, D. Cookson, A. Veis and A. George, J. Biol. Chem., 2005, 280, 33109–33114 CrossRef PubMed.
- W. J. Landis, K. J. Hodgens, M. J. Song, J. Aren, S. Kiyonag, M. Marko, C. Owen and B. F. McEwen, J. Struct. Biol., 1996, 117, 24–35 CrossRef PubMed.
- A. L. Boskey, M. Maresca, S. Doty, B. Sabsay and A. Veis, Bone Miner., 1990, 11, 55–65 CrossRef.
- D. S. W. Benoit, M. P. Schwartz, A. R. Durney and K. S. Anseth, Nat. Mater., 2008, 7, 816–823 CrossRef PubMed.
- J. D. Kretlow, M. C. Hacker, L. Klouda, B. B. Ma and A. G. Mikos, Biomacromolecules, 2010, 11, 797–805 CrossRef PubMed.
- Z. F. Lin, S. Q. Cao, X. Y. Chen, W. Wu and J. S. Li, Biomacromolecules, 2013, 14, 2206–2214 CrossRef PubMed.
- X. K. Li and J. Chang, J. Biomed. Mater. Res., Part A, 2008, 85, 293–300 CrossRef PubMed.
- L. S. Gu, Y. K. Kim, Y. Liu, K. Takahashi, S. Arun, C. E. Wimmer, R. Osorio, J. Q. Ling, S. W. Looney, D. H. Pashley and F. R. Tay, Acta Biomater., 2011, 7, 268–277 CrossRef PubMed.
- C. R. Nuttelman, D. S. W. Benoit, M. C. Tripodi and K. S. Anseth, Biomaterials, 2006, 27, 1377–1386 CrossRef PubMed.
- P. M. Lopez-Perez, R. M. da Silva, R. A. Sousa, I. Pashkuleva and R. L. Reis, Acta Biomater., 2010, 6, 3704–3712 CrossRef PubMed.
- P. Datta, S. Dhara and J. Chatterjee, Carbohydr. Polym., 2012, 87, 1354–1362 CrossRef.
- J. Tan, R. A. Gemeinhart, M. Ma and W. M. Saltzman, Biomaterials, 2005, 26, 3663–3671 CrossRef PubMed.
- K. A. Conrads, M. Yi, K. A. Simpson, D. A. Lucas, C. E. Camalier, L. R. Yu, T. D. Veenstra, R. M. Stephens, T. P. Conrads and G. R. Beck Jr, Mol. Cell. Proteomics, 2005, 4, 1284–1296 CrossRef PubMed.
- G. R. Beck Jr, J. Cell. Biochem., 2003, 90, 234–243 CrossRef PubMed.
- G. R. Beck Jr and N. Knecht, J. Biol. Chem., 2003, 278, 41921–41929 CrossRef PubMed.
- M. Julien, S. Khoshniat, A. Lacreusette, M. Gatius, A. Bozec, E. F. Wagner, Y. Wittrant, M. Masson, P. Weiss, L. Beck, D. Magne and J. Guicheux, J. Bone Miner. Res., 2009, 24, 1856–1868 CrossRef PubMed.
- Y. J. Wang, G. L. Zhu, N. L. Li, J. Q. Song, L. Wang and X. T. Shi, Biotechnol. Adv., 2015, 33, 1626–1640 CrossRef PubMed.
- H. Fleisch, A. Reszka, G. Rodan and M. Rogers, Princ. Bone Biol., 2nd edn, 1998, vol. 19, pp. 80–100 Search PubMed.
- L. Y. Shi, H. Carstensen, K. Hölzl, M. Lunzer, H. Li, J. Hilborn, A. Ovsianikov and D. A. Ossipov, Chem. Mater., 2017, 29, 5816–5823 CrossRef.
- P. M. Lopez-Perez, R. M. P. da Silva, I. Strehin, P. H. J. Kouwer, S. C. G. Leeuwenburgh and P. B. Messersmith, Macromolecules, 2017, 50, 8698–8706 CrossRef PubMed.
- A. Polini, D. G. Petre, M. Iafisco, S. d. L. Schickert, A. Tampieri, J. v. d. Beucken and S. C. G. Leeuwenburgh, J. Biomed. Mater. Res., Part A, 2017, 105, 2335–2342 CrossRef PubMed.
- M. Diba, J. An, S. Schmidt, M. Hembury, D. Ossipov, A. R. Boccaccini and S. C. G. Leeuwenburgh, Macromol. Rapid Commun., 2016, 37, 1952–1959 CrossRef PubMed.
- M. T. Drake, B. L. Clarke and S. Khosla, Mayo Clin. Proc., 2008, 83, 1032–1045 CrossRef PubMed.
- H. J. Yoon, S. R. Shin, J. M. Ch, S.-H. Lee, J.-H. Kim, J. T. Do and H. B. Hyuk Song, PLoS One, 2016, 11, e0163902 CrossRef PubMed.
- S. T. Koshy, R. M. Desai, P. Joly, J. Li, R. K. Bagrodia, S. A. Lewin, N. S. Joshi and D. J. Mooney, Adv. Healthcare Mater., 2016, 5, 541–547 CrossRef PubMed.
- P. Jaipan, A. Nguyen and R. J. Narayan, MRS Commun., 2017, 7, 416–426 CrossRef.
- R. Gauvin, Y. C. Chen, J. W. Lee, P. Soman, P. Zorlutuna, J. W. Nichol, H. Bae, S. Chen and A. Khademhosseini, Biomaterials, 2012, 33, 3824–3834 CrossRef PubMed.
- M. A. Daniele, A. A. Adams, J. Naciri, S. H. North and F. S. Ligler, Biomaterials, 2014, 35, 1845–1856 CrossRef PubMed.
- G. Camci-Unal, D. Cuttica, N. Annabi, D. Demarchi and A. Khademhosseini, Biomacromolecules, 2013, 14, 1085–1092 CrossRef PubMed.
- A. C. Daly, G. M. Cunniffe, B. N. Sathy, O. Jeon, E. Alsberg and D. J. Kelly, Adv. Healthcare Mater., 2016, 5, 2353–2362 CrossRef PubMed.
- D. Loessner, C. Meinert, E. Kaemmerer, L. C. Martine, K. Yue, P. A. Levett, T. J. Klein, F. P. Melchels, A. Khademhosseini and D. W. Hutmacher, Nat. Protoc., 2016, 11, 727–746 CrossRef PubMed.
- M. Kazemzadeh-Narbat, J. Rouwkema, N. Annabi, H. Cheng, M. Ghaderi, B.-H. Cha, M. Aparnathi, A. Khalilpour, B. Byambaa, E. Jabbari, A. Tamayol and A. Khademhosseini, Adv. Healthcare Mater., 2017, 6, 1601122 CrossRef PubMed.
- B. Byambaa, N. Annabi, K. Yue, G. T.-d. Santiago, M. M. Alvarez, W. Jia, M. Kazemzadeh-Narbat, S. R. Shin, A. Tamayol and A. Khademhosseini, Adv. Healthcare Mater., 2017, 6, 1700015 CrossRef PubMed.
- Y. C. Zuo, X. L. Liu, D. Wei, J. Sun, W. Q. Xiao, H. Zhao, L. K. Guo, Q. R. Wei, H. S. Fan and X. D. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 10386–10394 CrossRef PubMed.
- N. Monteiro, G. Thrivikraman, A. Athirasala, A. Tahayeri, C. M. França, J. L. Ferracane and L. E. Bertassoni, Dent. Mater., 2018, 34, 389–399 CrossRef PubMed.
- X. T. Shi, S. Ostrovidov, Y. H. Zhao, X. B. Liang, M. Kasuya, K. Kurihara, K. Nakajima, H. Bae, H. K. Wu and A. Khademhosseini, Adv. Funct. Mater., 2015, 25, 2250–2259 CrossRef.
- N. R. Gandavarapu, P. D. Mariner, M. P. Schwartz and K. S. Anseth, Acta Biomater., 2013, 9, 4525–4534 CrossRef PubMed.
- Q. Fu, M. N. Rahaman, B. S. Bal and R. F. Brown, Mater. Sci. Eng., C, 2009, 29, 2147–2153 CrossRef.
- M. L. Zhu, S. Lin, Y. X. Sun, Q. Feng, G. Li and L. M. Bian, Biomaterials, 2016, 77, 44–52 CrossRef PubMed.
- P. F. Jiang, Z. W. Mao and C. Y. Gao, Acta Biomater., 2015, 19, 76–84 CrossRef PubMed.
- F. Meder, T. Daberkow, L. Treccani, M. Wilhelm, M. Schowalter, A. Rosenauer, L. Madler and K. Rezwan, Acta Biomater., 2012, 8, 1221–1229 CrossRef PubMed.
- S. Weiner and H. A. Lowenstam, Science, 1985, 227, 51–54 CrossRef PubMed.
- S. Pacelli, R. Maloney, A. R. Chakravarti, J. Whitlow, S. Basu, S. Modaresi, S. Gehrke and A. Paul, Sci. Rep., 2017, 7, 6577 CrossRef PubMed.
- F. von Knoch, C. Jaquiery, M. Kowalsky, S. Schaeren, C. Alabre, I. Martin, H. E. Rubash and A. S. Shanbhag, Biomaterials, 2005, 26, 6941–6949 CrossRef PubMed.
- Y. J. Wang, X. T. Shi, R. R. Varshney, L. Ren, F. Zhang and D.-A. Wang, Biomaterials, 2009, 30, 3996–4005 CrossRef PubMed.
- G. I. Im, S. A. Qureshi, J. Kenney, H. E. Rubash and A. S. Shanbhag, Biomaterials, 2004, 25, 4105–4115 CrossRef PubMed.
- C. Z. Wang, S. M. Chen, C. H. Chen, C. K. Wang, G. J. Wang, J. K. Chang and M. L. Ho, Biomaterials, 2010, 31, 8674–8683 CrossRef PubMed.
- I. G. Kim, M. P. Hwang, P. Du, J. Ko, C. W. Ha, S. H. Do and K. Park, Biomaterials, 2015, 50, 75–86 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03550d |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab and
Yingjun Wang
*ab and
Yingjun Wang