DOI:
10.1039/C8RA03545H
(Paper)
RSC Adv., 2018,
8, 27546-27555
Reduction-responsive diblock copolymer-modified gold nanorods for enhanced cellular uptake†
Received
25th April 2018
, Accepted 14th July 2018
First published on 2nd August 2018
Abstract
Reduction-responsive polymer micelles are highly promising drug carriers with better tumor therapeutic effect, which can be achieved by controlled drug release under stimulation. Gold nanorods (AuNRs) have attracted considerable attention due to their unique optical and electronic properties when used for biomedical applications. Herein, the lipoic-acid-functionalized reduction-responsive amphiphilic copolymer poly(ε-caprolactone)-b-poly[(oligoethylene glycol) acrylate] (LA–PCL–SS–POEGA) with a disulfide group between the two blocks was prepared to modify AuNRs via Au–S bonds. The size and morphology of AuNRs@LA–PCL–SS–POEGA were measured by dynamic laser light scattering (DLS) and transmission electron microscopy (TEM) methods. The stabilities of AuNRs@LA–PCL–SS–POEGA in different types of media were studied by UV/vis spectroscopy and DLS techniques. The results show that AuNRs@LA–PCL–SS–POEGA gradually aggregate in a concentrated salt solution containing 150 mM dithiothreitol (DTT), but exhibit high stability in a non-reducing environment. Near infrared (NIR)-induced heating of AuNRs@LA–PCL–SS–POEGA was investigated in an aqueous solution under NIR laser irradiation (808 nm), revealing that AuNRs@LA–PCL–R–POEGA maintain excellent photothermal conversion efficiency after modification. When compared with non-reduction responsive AuNRs@LA–PCL–CC–POEGA, the in vitro internalization of AuNRs@LA–PCL–SS–POEGA demonstrates that the reduction-responsive polymer could enhance the cellular uptake of nanoparticles measured by inductively coupled plasma mass spectrometry (ICP-MS) and TEM.
Introduction
Over the past few decades, micelles formed by amphiphilic block copolymers have received considerable attention for drug delivery applications, as they could reduce the side effects of drugs, improve drug tolerance, prolong circulation time, and enhance accumulation in tumor sites via the enhanced permeability and retention (EPR) effect.1–4 In particular, stimuli-responsive micelles have emerged as promising nanosystems for drug delivery due to their distinct advantages such as controlled drug delivery, improved drug release, and superior anti-cancer activity as compared to traditional micelles.5,6 Until now, various stimuli-responsive micelles have been designed to achieve a better therapeutic effect in response to specific stimuli, such as pH, temperature, and thiols.7–12 Due to the higher concentration of the reducing glutathione (GSH) in the cell interior and tumor tissues than extracellular and normal tissues, a number of works about reduction-sensitive polymer nanomaterials containing disulfide groups have recently been developed to accelerate drug release at tumor sites.13–18 For example, Zhong et al. have conducted a series of studies on the use of reduction-responsive polymers as anti-cancer drug carriers including disulfide-linked dextran-b-poly(ε-caprolactone) (Dex–SS–PCL), poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG–SS–PCL), and poly(ethyleneglycol)-b-poly(2,4,6-trimethoxybenzylidene-pentaerythritol carbonate) (PEG–SS–PTMBPEC). They stated that the reduction-responsive micelles efficiently delivered and rapidly released doxorubicin (DOX) into cancer cells, resulting in better anti-tumor activity when compared with conventional micelles.19–21
Moreover, reduction-responsive micelles can be used in combination with other materials to obtain more therapeutic possibilities.22,23 Recently, some works about integrating photothermal materials such as gold nanorods (AuNRs) into nanomedicine carriers have attracted widespread attention because of the tunable surface plasmon and photothermal effects of AuNRs. For example, Chen and coworkers have successfully prepared a single nanocomposite by sheltering AuNRs@SiO2 with a thermo-responsive polymer shell poly(N-isopropylacrylamide-co-acrylic acid) (PNIPAM-co-PAA), which could increase the blood circulation time and inhibit tumor growth by water bath heating and NIR laser irradiation.24
In addition, they studied the influence of aspect ratio and surface chemistry of AuNRs on internalization and cytotoxicity. They stated that both aspect ratio and surface chemistry affected the cellular uptake, while the cytotoxicity was highly dependent on the surface chemistry.25 Besides, some other works about the toxicology of Au nanoparticles, including AuNRs, have been reported.26–35 Wang and coworkers systematically investigated the cellular uptake behavior and cytotoxicity of AuNRs with various surface coatings, including organic and inorganic materials. Their results indicated that AuNRs with organic polymer surface coatings showed lower cellular uptake and photothermal effects in U-87 MG cells.26 Additionally, Zhong et al. prepared AuNR-cored biodegradable poly(ethylene glycol)-b-poly(ε-caprolactone) block copolymer micelles for drug delivery. Their results have shown that the micelles efficiently delivered and released DOX into the nuclei of multidrug-resistant (MDR) cancer cells, resulting in high efficiency reversal of drug resistance.36 However, Duguet et al. have mentioned that AuNRs coated with poly(ε-caprolactone)-b-poly(ethylene glycol) had a relatively low cellular uptake, with an averaged uptake of 1250 ± (110) particles per melanoma MEL-5 cells after incubation at 50 μg mL−1 for 12 h.37 Thus, it would be interesting to investigate if the cellular uptake of AuNRs can be improved by modification with reduction-sensitive amphiphilic polymers.
In this study, reduction-responsive amphiphilic copolymer poly(ε-caprolactone)-b-poly[(oligoethylene glycol) acrylate] (LA–PCL–SS–POEGA) with a disulfide group and a control copolymer without a disulfide group were first prepared, as shown in Scheme 1. Due to the approved use of PCL in drug delivery by US Food and Drug Administration (FDA) and good biocompatibility of POEGA, we chose PCL as the hydrophobic block and POEGA as the hydrophilic segment.38,39 CTAB on the surface of AuNRs was replaced with reduction-sensitive amphiphilic copolymer or reduction-nonsensitive copolymer to reduce cytotoxicity. The stabilities of AuNRs coated with an amphiphilic polymer in different environments, including the reducing environment, were investigated by UV/vis spectrophotometer and DLS. Furthermore, the effect of disulfide-linked copolymer on the thermal property, cytotoxicity, and cellular uptake of AuNRs@LA–PCL–SS–POEGA was systematically investigated.
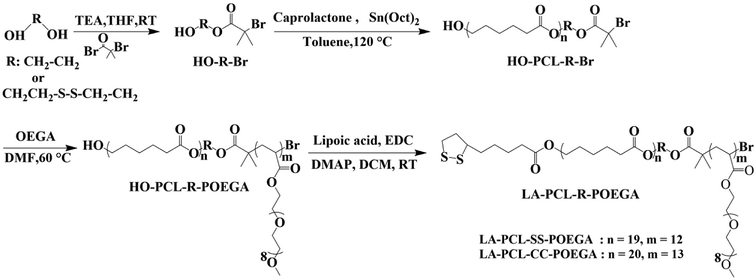 |
| | Scheme 1 Synthetic routes for LA–PCL–SS–POEGA and LA–PCL–CC–POEGA. | |
Experimental section
Materials and methods
Oligo(ethylene glycol) methyl ether acrylate (OEGA, Mn = 480 g mol−1, Sigma-Aldrich) was passed through a short column of alkaline alumina to remove the inhibitor. ε-Caprolactone (CL, Aladdin, 99%) was distilled under reduced pressure after drying over calcium hydride (CaH2). 2-Hydroxyethyl disulfide (Aldrich, 90%), 2-bromo-2-methyl-propionyl bromide (Energy Chemical, 98%), N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDETA, Sigma-Aldrich, 99%), stannous octoate (Sn(EH)2, Sigma-Aldrich, 95%), lipoic acid (LA, Damas-Beta, 99%), 4-dimethyl aminopyridine (DMAP, Aladdin, 99%), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC, Aladdin, 98%), cetyltrimethylammonium bromide (CTAB, Sinopharm, 99%), hydrochloric acid (HCl, Sinopharm), concentrated nitric acid (HNO3, Sinopharm), sodium borohydride (NaBH4, Sinopharm, 98%), silver nitrate (AgNO3, Sinopharm, 99%), L-ascorbic acid (AA, Sinopharm, 99%), hydrogen tetrachloroauric acid (HAuCl4·3H2O, Energy Chemical, 99.99%), and dithiothreitol (DTT, Aladdin, 99%) were used without any treatment. Dimethyl sulfoxide (DMSO, Sinopharm, 97%) and dimethylformamide (DMF, Sinopharm, 97%) were dried over anhydrous magnesium sulfate and then purified by distillation under reduced pressure. Dichloromethane (DCM, Sinopharm, 97%) and triethylamine (TEA, Sinopharm, 99%) were distilled over CaH2. Tetrahydrofuran (THF, Sinopharm, 97%) and toluene (Sinopharm, 97%) were distilled after refluxing over metal sodium for 24 h.
Preparation of the initiator HO–SS–Br
The small molecule initiator was synthesized according to previous works with certain modifications as follows.40,41 2-Hydroxyethyl disulfide (5.1 g, 33.1 mmol) and TEA (4.63 mL, 33.3 mmol) were dissolved in 80 mL purified THF and DCM (30/50, v/v) at 25 °C. Under a nitrogen flow, α-bromoisobutyryl bromide (4.12 mL, 33.3 mmol) was dissolved in purified DCM (10 mL) and was added dropwise into the reaction mixture at 0 °C within 30 min. Then, the mixture was stirred for 10 h at room temperature. After that, the solvent was removed by rotary evaporation under reduced pressure, and the mixture was redissolved in THF and filtered to remove the salts. The crude product was purified by column chromatography after removing THF (eluent: n-hexane/ethyl acetate ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give a pale-yellow clear oil (HO–SS–Br, 4.9 g) in a 49% yield.
:1) to give a pale-yellow clear oil (HO–SS–Br, 4.9 g) in a 49% yield.
Preparation of HO–SS–PCL–Br
HO–PCL–SS–Br was prepared by ring-opening polymerization (ROP) using HO–SS–Br as the initiator and Sn(EH)2 as the catalyst.42,43 HO–SS–Br (0.44 g, 1.45 mmol), Sn(EH)2 (0.118 g, 0.29 mmol), CL (3.00 g, 26.30 mmol), and anhydrous toluene (30 mL) were added into a three-necked, round-bottomed flask. The reaction mixture was refluxed for 6 h at 120 °C under a nitrogen atmosphere. Then, the mixture was redissolved in THF after toluene was removed by distilling under reduced pressure. The polymers dissolved in THF were precipitated into an excess of the mixture of cold methanol/water (2/1, v/v). The polymer HO–PCL–SS–Br (Mn = 2.2 × 103 g mol−1, 2.8 g, yield: 93%) was collected by filtration and dried under a vacuum for 24 h.
Preparation of HO–PCL–SS–POEGA
The amphiphilic polymers were synthesized by atom transfer radical polymerization (ATRP) as follows. Macroinitiator HO–SS–PCL–Br (0.5 g, 0.2 mmol), monomer OEGA (2.4 g, 5 mmol), ligand PMDETA (34.66 mg, 0.2 mmol), and 3 mL of anhydrous DMF were added into a 10 mL glass tube with a magnetic stirrer. Catalyst CuBr (28.8 mg, 0.2 mmol) was added into the tube with a hot funnel after degassing three times by freeze–pump–thaw cycles; subsequently, the tube was sealed under a vacuum. After that, the tube was placed in a 60 °C oil bath and stirred for 3.5 h. Then, the reaction was rapidly terminated by freezing with liquid nitrogen. To remove the copper salts, the mixture was diluted with THF and passed through a neutral alumina column using THF as the eluent. The mixture was concentrated by rotary evaporation and then precipitated into an excess amount of cold diethyl ether/n-hexane (1/2, v/v). A sticky solid of HO–PCL–SS–POEGA was collected by filtration and dried under a vacuum for 24 h. HO–PCL–SS–POEGA was obtained as a sticky white solid (1.2 g, yield: 80%) after the reaction of ATRP.
Preparation of LA–PCL–SS–POEGA
The hydroxyl groups at the end of the PCL chains were modified with LA by esterification modification. HO–PCL–SS–POEGA (0.8 g, 0.1 mmol) was dissolved in 20 mL of purified DCM and then LA (0.113 g, 0.5 mmol), DMAP (5.7 mg, 0.044 mmol), and TEA (15 μL, 0.1 mmol) were added step by step. EDC (0.1 g, 0.5 mmol) was added into the reaction mixture under a nitrogen atmosphere at 0 °C. The reaction was stopped after stirring for 24 h at room temperature. Thereafter, DCM was removed by a rotary evaporator, and the residues were dissolved in THF; the salt precipitate was then removed by centrifugation. Finally, the supernatant was precipitated into an excess amount of cold diethyl ether/n-hexane (1/2, v/v). LA–PCL–SS–POEGA (0.6 g, yield: 79%) was obtained as a pale yellow sticky solid by modifying the hydroxyl groups with LA and drying under a vacuum at room temperature for 24 h. The reduction-nonresponsive copolymer LA–PCL–CC–POEGA was prepared by a similar procedure, as shown in Scheme 1.
Preparation of AuNRs by seed-growth methods
AuNRs were synthesized based on a seed-growth procedure with some modification.44 Au seeds were prepared by adding HAuCl4·3H2O (25 mM, 0.1 mL) into CTAB (0.1 M, 10 mL) solution, followed by thorough mixing. Then, freshly prepared ice-cold NaBH4 (0.01 M, 600 μL) was rapidly added under vigorous stirring, resulting in the formation of a brownish-yellow solution. This seed solution was kept at 28 °C in a water bath for 2 h. After that, the growth solution was prepared in a 100 mL conical flask as follows: 100 mL of CTAB (3.6445 g, 0.1 M) solution, 1.2 mL of AgNO3 (16.987 mg, 10 mM) solution, 2 mL of aqueous HAuCl4·3H2O (19.69 mg, 25 mM), and a certain amount of HCl (1 M) solution were thoroughly mixed in a conical flask. Then, 0.7 mL solution of AA (9.72 mg, 78.8 mM) was slowly added into the solution mentioned above under gentle stirring. Finally, the solution was left undisturbed at 28 °C overnight after 120 μL seed solution was added. The AuNRs@CTAB were obtained after centrifuging at 8000 rpm for 15 min and redispersed in deionized water.
Preparation of AuNRs@LA–PCL–R–POEGA nanoparticles
Typically, AuNRs@CTAB (0.25 mg) were diluted with water to 10 mL and centrifuged at 8000 rpm for 15 min, the supernatant was discarded, and the precipitate was redispersed in 2.5 mL of ultrapure water. Further, 5 mg LA–PCL–SS–POEGA diblock copolymer was dissolved in 1 mL purified DMSO, and added dropwise into AuNRs (2.5 mL, 0.1 mg mL−1) solution using an injection pump at a speed of 1 mL h−1 under stirring. The mixture was kept at room temperature for 24 h and then centrifuged at 8000 rpm for 15 min and washed with DMSO (2.5 mL) and water (2.5 mL) to remove the excess polymers. In the end, the precipitate was redispersed in 2.5 mL of deionized water and dialyzed against 1 L deionized water in a dialysis bag (Green Bird; molecular weight cut-off (MWCO) = 3500 Da) for 24 h. The water was changed every 6 h to remove the organic solvent.
Characterization
1H NMR
The 1H NMR spectra were conducted on a Bruker AV400 spectrometer using tetramethylsilane (TMS) as the internal reference. Each sample was dissolved in deuterated chloroform (CDCl3, 0.6 mL) at a concentration of ∼20 mg mL−1.
Gel permeation chromatography (GPC)
The macromolecular weight information including number-average molar mass (Mn), weight-average molar mass (Mw), and dispersity (Mw/Mn) of the polymers were measured at 35 °C by a Waters 1515 GPC instrument equipped with three Waters Styragel columns (HR2, HR4, and HR6) and a refractive index detector (RI, Wyatt WREX-02). THF was used as the eluent at a flow rate of 1.0 mL min−1, and a series of linearly narrow distribution of polystyrene with different molecular weights were used as the standard samples.
Characterization of AuNRs@CTAB and AuNRs@LA–PCL–R–POEGA nanoparticles
The absorption spectra of AuNRs@CTAB and the modified AuNRs@LA–PCL–R–POGEA nanoparticles were measured in the visible to near-infrared light region using a UV/vis spectrophotometer (UNICO2802PCS). The morphologies of AuNRs@CTAB and AuNRs@LA–PCL–R–POEGA nanoparticles were observed by a transmission electron microscope (TEM, Hitachi H-7650, 100 kV). The average hydrodynamic diameters and zeta potentials of micelles were measured at 25 °C on a Zetasizer Nano-ZS (Malvern Instruments) with a 633 nm He-Ne laser as the light source.
Stability of AuNRs@LA–PCL–R–POEGA nanoparticles
The stabilities of AuNRs@LA–PCL–R–POGEA were probed by a UV/vis spectrophotometer in different types of dispersion media, including PBS, 2 M NaCl, and high concentration of salt solution containing DTT. The DTT competition test was also used to assess the stability of AuNRs@LA–PCL–R–POGEA, according to the ref. 45 and 46. Firstly, a certain amount of DTT (23.13 mg) was added into a degassed solution of NaCl (100 μL, 4 M) in a quartz cuvette with a thread capsule and thoroughly mixed. After that, the whole solution was degassed with high purity of nitrogen for 1 min after a certain amount of AuNRs@LA–PCL–R–POEGA and water were added to make the longitudinal absorption band around 800 nm of ∼0.3–0.4 under the concentration of DTT ∼ 150 mM and NaCl ∼ 400 mM. Then, a series of absorption spectra were measured at various time intervals to monitor the changes in the absorption peaks. In addition, we determined the average hydrodynamic diameters of the AuNRs@LA–PCL–R–POEGA at 37 °C over time by DLS, which were dispersed in the cell culture medium containing DMEM, 10% FBS, 1% sodium pyruvate, and 1% penicillin–streptomycin.
Photothermal heating experiments
The aqueous solutions of AuNRs@CTAB and AuNRs@LA–PCL–R–POEGA were diluted to 100 μg mL−1 (200 μL) and irradiated with an 808 nm NIR laser (New Industries Optoelectronics, Changchun, China) for 10 min at a power density of 200 mW cm−2. The change in temperature was monitored by an IR camera (ICI7320, Infrared Camera Inc., Texas, USA) every 10 s and analyzed using IR Flash thermal imaging analysis software. Similarly, various concentrations (100, 50, and 20 μg mL−1) of AuNRs@LA–PCL–SS–POEGA in water were also irradiated with an 808 nm NIR laser at the same power density and detected under the same conditions. Deionized water was used as the control.
Cytotoxicity tests
The standard MTT assay was used to assess the cell viability under different conditions. Briefly, the Hela cells were seeded in a 96-well plate with a density of 5000 cells per well and cultivated in a 5% CO2 atmosphere at 37 °C for 24 h. Then, the medium was replaced with different concentrations of AuNRs@LA–PCL–R–POEGA dispersed in the medium containing DMEM, 10% FBS, 1% sodium pyruvate, and 1% penicillin–streptomycin, and the cells were incubated for another 24 h. Thereafter, the MTT solution (120 μL, 0.5 mg mL−1) was added to each well after the medium containing nanoparticles was removed. After incubation for an additional 4 h, the MTT solution was removed and 150 μL of DMSO was added. The absorbance of solubilized formazan was measured after thoroughly mixing at 595 nm by a spectrophotometric plate reader (Bio-Rad iMark). The measurement for each treatment was repeated six times.
Cell internalization of AuNRs@LA–PCL–R–POEGA
A total of 1 × 105 Hela cells per well were plated in 24-well plates. After cultivating for 24 h, the medium was replaced by AuNRs@LA–PCL–R–POEGA (400 μL, 75 μg mL−1) dispersed in a culture medium and then placed in an incubator for 24 h or 36 h. Thereafter, the cells were washed three times with PBS to remove the nanoparticles adsorbed on the cells surface and then detached with trypsin. After 5 min, a DMEM medium (1 mL) was added to homogeneously disperse the cells; the cells were then collected and the number of cells in the solution per well were counted. After that, the cells were collated by centrifugation at a speed of 1000 rpm for 5 min, and HNO3 (2 mL) was added into the tube for pre-digestion overnight. After 12 h, HCl (6 mL) was mixed with HNO3, and the mixture was boiled at 140 °C until the solution became colorless and clear; then, the residual solution was diluted with deionized water to 10 mL. The concentration of Au ions of liquid (10 mL) was determined using an inductively coupled plasma mass spectrometer (ICP-MS; Plasma Quad 3). The number of AuNRs per cell was calculated based on the mass of the individual AuNR and the number of cells. The measurement for each treatment was repeated in triplicate.
TEM analysis for cellular uptake
The morphology and location of AuNRs in fixed cells was obtained using a TEM (JEM-2100). Hela cells were incubated in 2 mL of medium (DMEM) at a density of 1.0 × 106 cells. After 24 h, the medium was discarded and replaced with 2 mL of 75 μg mL−1 AuNRs@LA–PCL–R–POEGA dispersed in a DMEM medium and then placed in an incubator for 36 h. After removing the medium containing AuNRs@LA–PCL–R–POEGA, the cells were washed with PBS for three times and detached with trypsin; they were then collected by centrifugation at 1500 rpm for 8 min. After discarding the supernatant, the cells were fixed by 3% glutaraldehyde solution overnight; they were then embedded in resin, cut into ultrathin sections, and stained by osmic acids. Finally, the location and morphology of AuNRs in the cells were detected by TEM.
Results and discussion
Synthesis of LA–PCL–R–POEGA copolymers
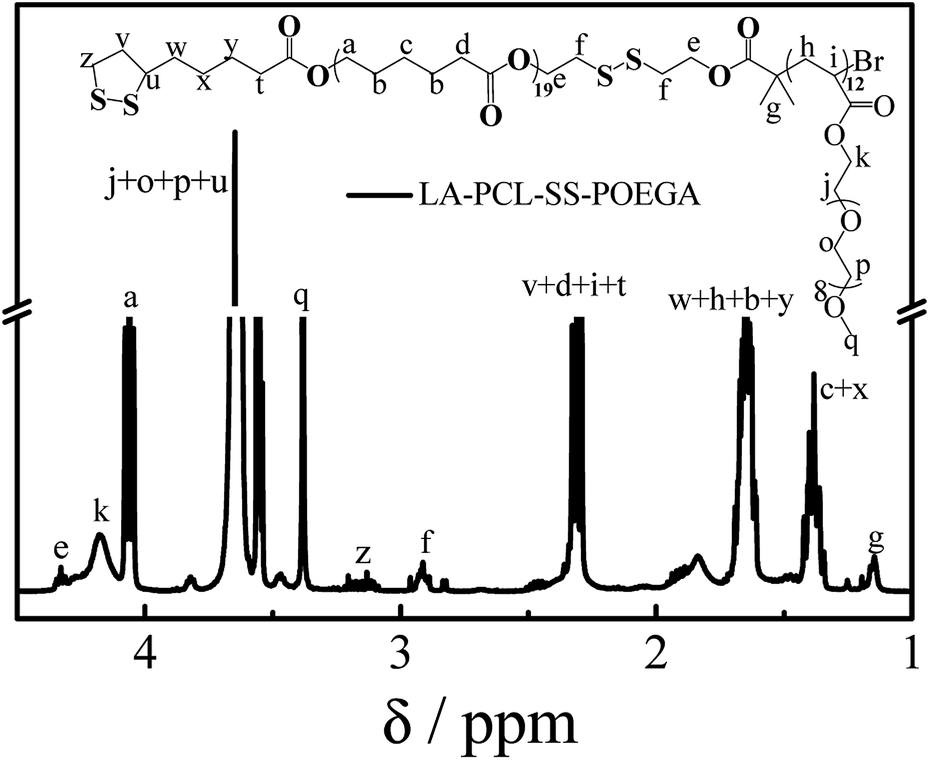
As shown in Scheme 1, LA–PCL–R–POEGA was prepared by ROP and ATRP. First of all, the initiator HO–SS–Br was synthesized through an esterification reaction. The purity and structure of the initiator were confirmed by 1H NMR (Fig. S1†). Secondly, we prepared HO–PCL–R–Br by ROP of ε-caprolactone using HO–R–Br as the initiator and Sn(EH)2 as a catalyst in toluene. The degree of polymerization (DP) was measured by 1H NMR (Fig. S2†). Fig. S2† shows that the protons at –COO–CH2–CH2–SS–CH2–CH2–OOC, –OOC–CH2–CH2–CH2–CH2–CH2O–, –OOC–CH2–CH2–CH2–CH2–CH2O–, and –OOC–CH2–CH2–CH2–CH2–CH2O– on the carbon chain backbone of the PCL block are located at 2.87–2.98 ppm (f), 2.18–2.40 ppm (d), 4.01–4.10 ppm (a), and 1.50–1.72 ppm (b), respectively. According to the integral area ratio of the peak (Ad/Af, Aa/Af, or Ab/Af), the DP of PCL was calculated as 19. Meanwhile, the end retention of the terminal bromine and hydroxyl in the polymer HO–PCL–R–Br are ∼99% according to the integral area ratio of the peak area (Ag/Af) and (Aa′/Af).
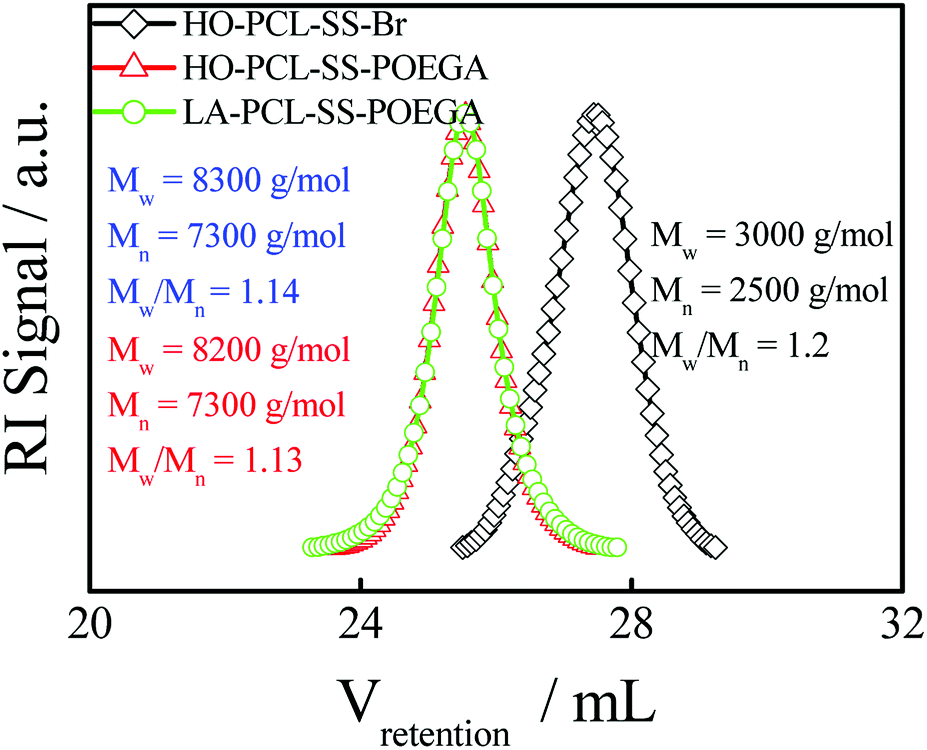
In addition, we used HO–PCL–R–Br as the macroinitiator, CuBr as the catalyst, and PMEDTA as the ligand in DMF to obtain the copolymer HO–PCL–R–POEGA. Similarly, the DP of the hydrophilic segments was calculated as 12 according to the area ratio of the characteristic peaks in the 1H NMR spectrum (Fig. S3†). According to the integral area ratio of the characteristic peaks of the protons at –COO–CH2–CH2–SS–CH2–CH2–OOC, OOC–CH2–CH2–CH2–CH2–CH2O–, –COO–CH2–CH2–O–CH2–, and –O–CH3 are located at 2.87–2.98 ppm (f), 1.29–1.48 ppm (c), 4.10–4.28 ppm (k), and 3.32–3.42 ppm (q), respectively; the DP of the hydrophilic segments was obtained as 12, as shown in Fig. S3†. It is difficult to calculate the degree of functionality of the hydroxyl group after ATRP due to the overlapping of the peaks. At the end, the final product was obtained by modifying the hydroxyl groups at the end of the PCL chains with LA. After precipitation for five times, we concluded that there was no change in the integral area of the characteristic peaks of LA before and after precipitation, indicating the complete removal of the free LA molecules. Moreover, the characteristic protons peaks of LA can be observed from Fig. 1, such as the peaks (z, v, w, y). The degree of functionality of LA is 99% according to the integral area ratio of the peak area (Az/Af). Fig. 1 shows the 1H NMR spectrum of LA–PCL–SS–POEGA, and the integral area ratio of peak (z), peak (f), peak (c + x), and peak (q) is almost 1.00/2.05/20.0/20, which is consistent with the theoretical ratio. Therefore, Mn calculated from the 1H NMR spectrum for the copolymer LA–PCL–SS–POEGA is approximately 8000 g mol−1. The GPC curves of the polymers shown in Fig. 2 reveal that the retention time of HO–PCL–SS–POEGA is obviously shorter than that of HO–PCL–SS–Br, which confirms the success of the ATRP process. The results involving the reduction-nonsensitive initiator and copolymers HO–CC–Br, HO–PCL–CC–Br, HO–PCL–CC–POEGA, and LA–PCL–CC–POEGA are shown from Fig. S4 to S8, respectively, in the ESI.† Moreover, Mn calculated from the 1H NMR spectrum for the copolymer LA–PCL–CC–POEGA is approximately 8500 g mol−1.
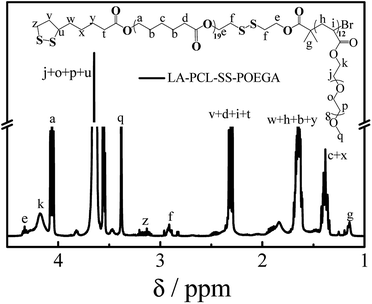 |
| | Fig. 1 1H NMR spectrum of disulfide-linked polymer LA–PCL–SS–POEGA measured in CDCl3 with TMS as an internal reference. | |
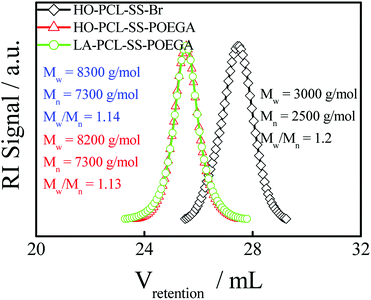 |
| | Fig. 2 GPC chromatograms (in THF) of HO–PCL–SS–Br, HO–PCL–SS–POEGA, and LA–PCL–SS–POEGA. The concentration of these polymers was ∼5 mg mL−1. | |
Preparation and characterization of AuNRs@CTAB and AuNRs@LA–PCL–R–POEGA nanoparticles
A number of methods for synthesizing AuNRs have been reported, such as the template method, electrochemical method, and seed-growth method.44,47,48 We synthesized AuNRs using the seed-growth method reported by El-Sayed et al. with some modifications.44,49 The TEM image illustrates that AuNRs are monodisperse in water with an average length of 60 ± 8 nm and a width of 16 ± 3 nm. As shown in Fig. 3(c), the distribution of the relaxation time for AuNRs@CTAB exhibit two peaks, as measured by DLS: the first peak at 7–40 μs is attributed to rotational diffusion and the other one with a longer relaxation time is attributed to translational diffusion. The average hydrodynamic diameter of AuNRs@CTAB is 67 nm obtained from the translational diffusion coefficient.50,51 The diameter of AuNRs@CTAB measured by DLS is a little longer than that detected by TEM due to the CTAB bilayer absorbed on the surface of AuNRs.51 As shown in the normalized UV/vis absorption spectra [Fig. 3(b)], AuNRs have a strong absorption peak at approximately 802 nm, which is located in the therapeutic window, since biological tissue and water have a lower absorption at 650–900 nm.52 According to the absorbance of the longitudinal absorption at 802 nm, the concentration of AuNRs can be determined with a molar extinction coefficient of 4.4 ± 0.5 × 109 L mol−1 cm−1.53,54 Meanwhile, the absorption shape of AuNRs@LA–PCL–R–POEGA is consistent with AuNRs@CTAB, suggesting the monodispersity of nanoparticles after modification.
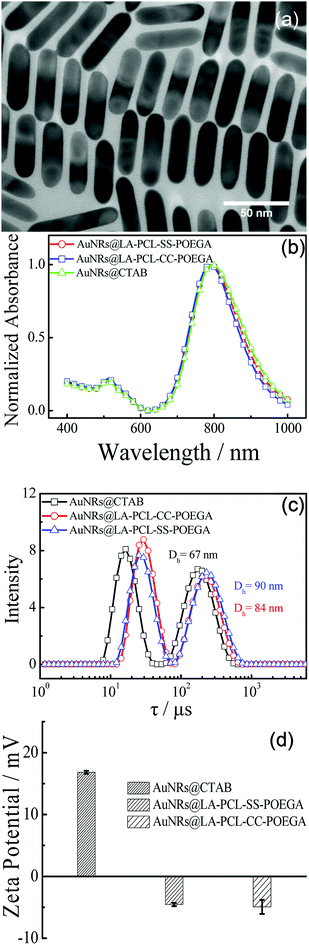 |
| | Fig. 3 (a) TEM image of AuNRs@CTAB in an aqueous solution. (b) UV/vis absorption spectra of AuNRs@CTAB and AuNRs@LA–PCL–R–POEGA dispersed in water. (c) Distribution of relaxation time obtained from DLS measurement for AuNRs@CTAB and AuNRs@LA–PCL–R–POEGA in water. (d) Zeta potentials of AuNRs@CTAB and AuNRs@LA–PCL–R–POEGA dispersed in 10 mM phosphate buffer (pH = 7.4) solution. | |
AuNRs@ LA–PCL–R–POEGA were prepared by forming Au–S bonds between LA and AuNRs according to the method reported by Zhong and coworkers with some modifications.36 In the current method, the nanoparticles were washed with DMSO and centrifuged to remove excessive polymers that have not combined with AuNRs. As shown in Fig. 3(c), the relaxation time of translational diffusion for AuNRs@LA–PCL–R–POEGA is longer than that for AuNRs@CTAB. The average hydrodynamic diameters of AuNRs@LA–PCL–R–POEGA calculated from the relaxation time of translational diffusion [Fig. 3(c)] are larger than those of AuNRs@CTAB, with an average diameter of about 87 ± 3 nm. Since the amphiphilic polymer chains are much longer than the bilayer of CTAB absorbed on AuNRs, the average diameter becomes larger after the copolymer replaced CTAB on the AuNRs. However, a change in the particle size of AuNRs@LA–PCL–R–POEGA cannot be observed from the TEM images due to the low electron density and collapse of the polymer chain after drying. Moreover, the zeta potentials of AuNRs@LA–PCL–R–POEGA in 10 mM PB solution at pH = 7.4 are −4.5 ± 1 mV [Fig. 3(d)], but the zeta potentials of AuNRs@CTAB in the same medium are +16.8 ± 1 mV due to the presence of CTAB molecules with positive charge. In addition, Fig. S9† shows that AuNRs@LA–PCL–R–POEGA exhibit good stability and can be well dispersed in organic solvents such as THF and DMSO, while AuNRs@CTAB aggregate or even precipitate in organic solvents. All these results suggest that our copolymers have been successfully grafted onto AuNRs.
Stability of AuNRs@LA–PCL–R–POEGA nanoparticles
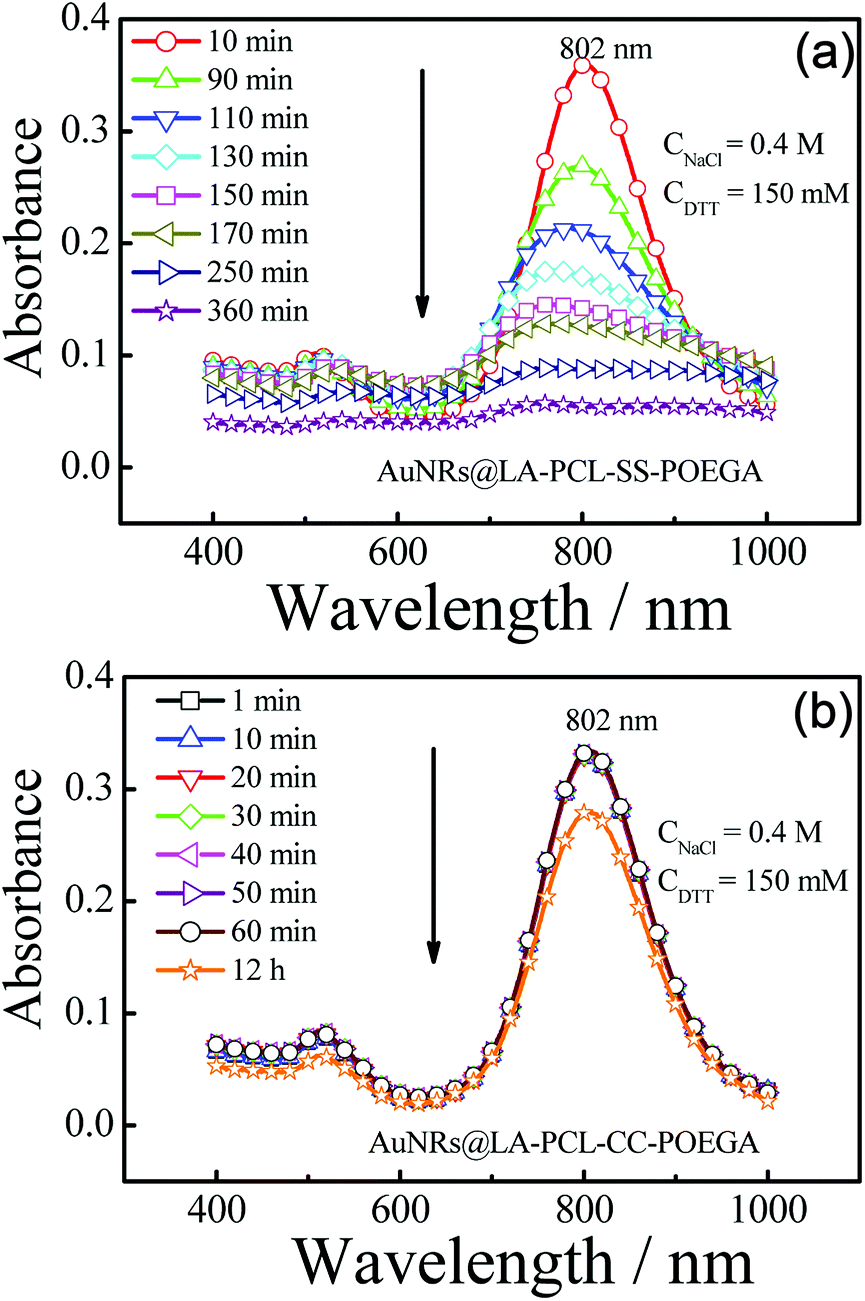
We probed the stabilities of AuNRs@LA–PCL–R–POEGA under different conditions, including PBS, cell culture media, high concentration of sodium chloride solution, and reducing environments. Fig. S10† shows that the UV/vis absorption peak positions and shapes of AuNRs@LA–PCL–R–POEGA remain constant under all non-reducing environments (a). Meanwhile, the average hydrodynamic diameter of AuNRs@LA–PCL–R–POEGA shown in Fig. S11† is essentially constant with low dispersity at 37 °C in DMEM, containing 10% FBS, 1% sodium pyruvate, and 1% penicillin–streptomycin, which indicates that AuNRs@LA–PCL–R–POEGA could be stable for biological applications. Furthermore, the DTT competition test was used to explore the difference between the stability of AuNRs@LA–PCL–SS–POEGA and AuNRs@LA–PCL–CC–POEGA under the reduction condition.45 The UV/vis spectra of AuNRs capped with LA–PCL–R–POEGA in an aqueous solution containing 150 mM DTT and 400 mM NaCl were collected. As shown in Fig. 4(b), AuNRs@LA–PCL–CC–POEGA exhibit fantastic colloidal stability without aggregation after 12 h. The steric hindrance of the amphiphilic polymer grafted on AuNRs prevent the aggregation of AuNRs. In contrast, obvious aggregation was observed for AuNRs@LA–PCL–SS–POEGA at 90 min after mixing with the DTT solution, which is attributed to the disulfide bond cleavage, as shown in Fig. 4(a). The hydrophilic POEGA blocks depart from the AuNRs surface after this cleavage in the backbone; then, the nanoparticles become more hydrophobic and the steric hindrance of the copolymer decreases. The main difference between AuNRs@LA–PCL–SS–POEGA and the control group is the disulfide bond among the copolymer chain, resulting in the aggregation of AuNRs@LA–PCL–SS–POEGA under the reduction condition.
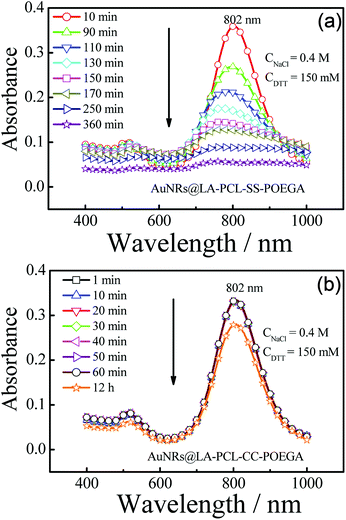 |
| | Fig. 4 Time-dependent absorption spectra of (a) AuNRs@LA–PCL–SS–POEGA and (b) AuNRs@LA–PCL–CC–POEGA aqueous solutions containing 150 mM DTT and 400 mM NaCl with an initial absorbance ≈ 0.3–0.4. | |
Photothermal effect of AuNRs@LA–PCL–R–POEGA under NIR irradiation
AuNRs have shown to be a promising photothermal material owing to their high photothermal conversion efficiency.55,56 In order to evaluate the photothermal efficiency of AuNRs, we tested the temperature changes of AuNRs@CTAB and AuNRs@LA–PCL–R–POEGA in aqueous solutions under 808 nm laser irradiation with different concentrations. Fig. 5(a) shows the rapid temperature changes (∼10 °C) within 5 min for AuNRs@LA–PCL–R–POEGA, which is similar for AuNRs@CTAB at the same concentration of 0.1 mg mL−1; as a control, water exhibits almost no change. Furthermore, the dependence of photothermal conversion efficiency on the concentration of AuNRs@LA–PCL–SS–POEGA is also shown in Fig. 5(b). These results reveal that AuNRs modified with LA–PCL–R–POEGA exhibit good photothermal conversion efficiency.
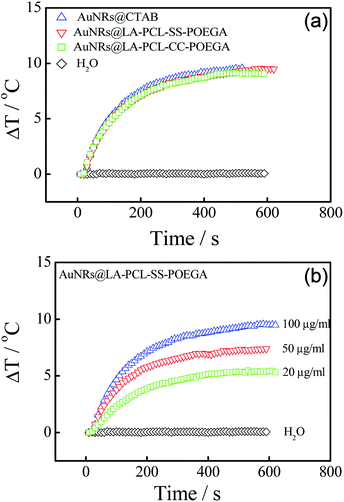 |
| | Fig. 5 (a) Photothermal heating curves of water, AuNRs@CTAB, and AuNRs@LA–PCL–R–POEGA irradiated by an 808 nm laser at a power density of 200 mW cm−2 for 10 min. The concentration of AuNRs was 100 μg mL−1. (b) Concentration-dependent heating profiles of an aqueous suspension of AuNRs@LA–PCL–SS–POEGA after exposure to 808 nm NIR laser irradiation (200 mW cm−2) for 10 min. | |
Cell viability and cellular uptake of AuNRs@LA–PCL–R–POEGA
Fig. 6 shows that AuNRs@CTAB have obvious cytotoxicity on Hela cells before modification with LA–PCL–R–POEGA owing to the presence of CTAB, which is consistent with the results reported in previous studies.25,45 Therefore, it is necessary to modify the AuNRs' surfaces with biocompatible materials to improve the consequent biocompatibility. In our work, we capped AuNRs with the biocompatible amphiphilic polymer LA–PCL–R–POEGA, which can simultaneously work as a drug carrier. The cell viability of AuNRs@LA–PCL–R–POEGA remained greater than 85% even at high concentrations of 100 μg mL−1 for Hela cells. The results confirm that the modification with LA–PCL–R–POEGA successfully reduces the cytotoxicity of AuNRs.
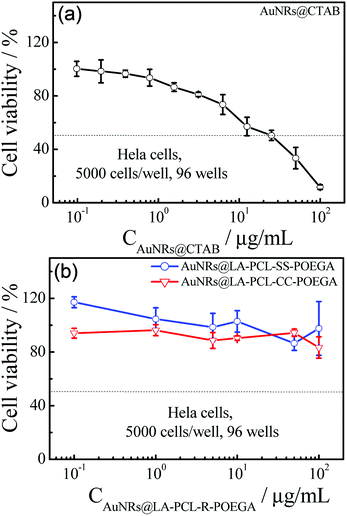 |
| | Fig. 6 Cell viability of Hela cells after incubation with (a) AuNRs@CTAB and (b) AuNRs@LA–PCL–R–POEGA at different concentrations for 24 h was assessed by the standard MTT assay. Error bars represent the mean ± SD (n = 6). | |
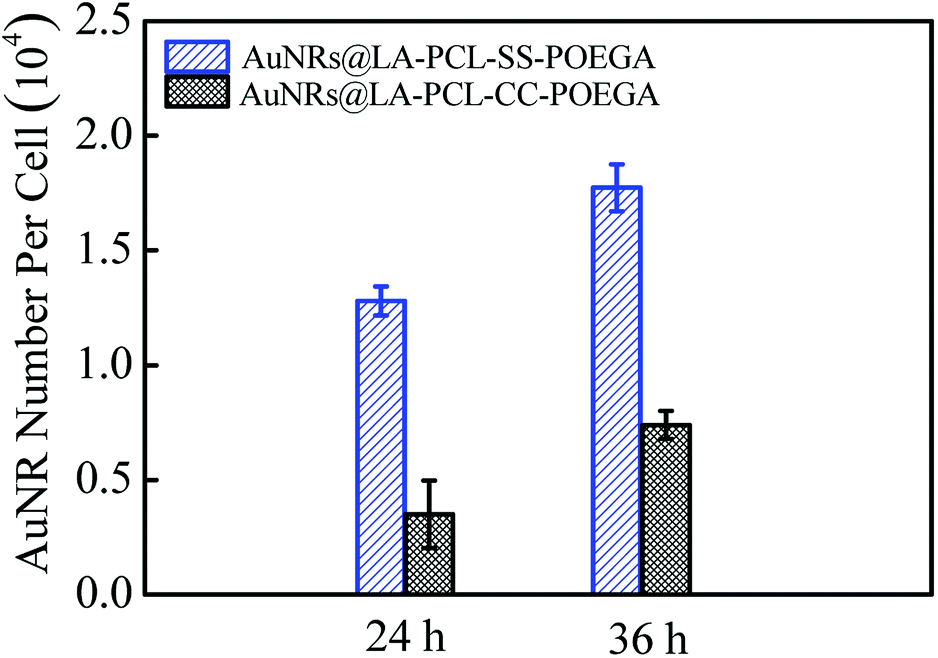
The amount of AuNRs internalized within Hela cells was detected by ICP-MS measurements. The concentration of Au ions was measured by ICP-MS after digesting in aqua regia. The number of AuNRs per cell (Fig. 7) was calculated according to the quality of a single AuNR (the volume was obtained from TEM images) and the number of cells in each well. The result shows that the cellular uptake of AuNRs is greatly improved by introducing a disulfide linkage between the PCL and POEGA blocks. The amount of cellular uptake for AuNRs@LA–PCL–SS–POEGA coated with polymers containing disulfide linkage was 2–3 times higher than that of AuNRs@LA–PCL–CC–POEGA after being incubated for 24 h or 36 h. Accurately, the internalized number per cell of AuNRs@LA–PCL–SS–POEGA is 12800 ± 640 and 17700 ± 1030 after being incubated for 24 h and 36 h, respectively. However, the cellular uptake number per cell of AuNRs@LA–PCL–CC–POEGA is 3500 ± 1500 and 7400 ± 610 after being incubated for 24 h and 36 h, respectively. It might be related to the poor stability of AuNRs@LA–PCL–SS–POEGA under the reducing condition in tumor cells. AuNRs@LA–PCL–SS–POEGA aggregate obviously due to the cleavage of disulfide bonds under the high concentration of DTT, as shown in Fig. 4. We speculated that AuNRs@LA–PCL–SS–POEGA might aggregate as the disulfide bond can be cleaved by the high concentration of GSH in tumor cells, leading to the aggregation of AuNRs. The aggregates formed in tumor cells are not easily excluded from cells, so the uptake of AuNRs@LA–PCL–SS–POEGA nanoparticles was enhanced.
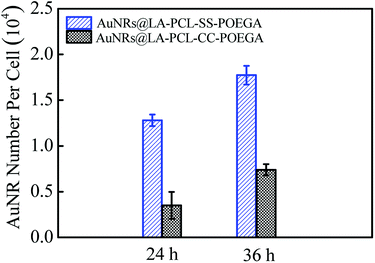 |
| | Fig. 7 Internalized number of AuNRs per cell after incubation with AuNRs@LA–PCL–SS–POEGA or AuNRs@LA–PCL–CC–POEGA at 75 μg mL−1 for 24 h or 36 h. Error bars represent the mean ± SD (n = 3). | |
To further identify the AuNRs@LA–PCL–R–POEGA internalized by the cells, we observed the morphology and location of the intracellular AuNRs with TEM, as shown in Fig. 8. According to the TEM images of the cells treated with AuNRs@LA–PCL–SS–POEGA, we found that certain AuNRs formed aggregates in cells and were located in the cytoplasm. However, fewer AuNRs and aggregates were observed in cells from the TEM images after cultivating with AuNRs@LA–PCL–CC–POEGA. Therefore, AuNRs@LA–PCL–SS–POEGA nanoparticles might tend to aggregate after entering into the Hela cells as compared with AuNRs@LA–PCL–CC–POEGA. Thus, these results also indicate the reduction-responsive polymers modified with AuNRs might enhance the cellular uptake for tumor cells.
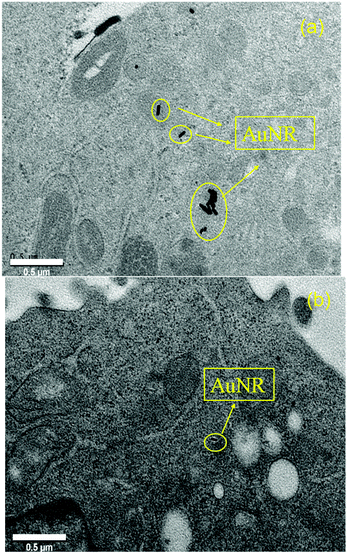 |
| | Fig. 8 Location and morphology of AuNRs accumulated in the cells after incubation with (a) AuNRs@LA–PCL–SS–POEGA at 75 μg mL−1 and (b) AuNRs@LA–PCL–CC–POEGA at the same concentration for 36 h. Scale bar is 0.5 μm. | |
Conclusions
In this study, we have successfully synthesized reduction-responsive and terminal functionalized amphiphilic polymer LA–PCL–SS–POEGA with good biocompatibility through ROP and ATRP. The copolymer could be used to replace CTAB and conjugate on AuNRs via the formation of Au–S bonds to improve the stability and biocompatibility of AuNRs. The stabilities of AuNRs@LA–PCL–R–POEGA were investigated under different conditions. The results reveal that both AuNRs@LA–PCL–SS–POEGA and AuNRs@LA–PCL–CC–POEGA could be stable under non-reducing conditions. However, under a reducing environment containing 150 mM DTT, AuNRs@LA–PCL–SS–POEGA aggregated obviously according to the UV/vis spectrum changes of AuNRs, while the control group remains stable. In addition, the cell viability obtained from the MTT assay shows that the biocompatibility of AuNRs@LA–PCL–R–POEGA is much better than AuNRs@CTAB. Furthermore, the internalization of AuNRs@LA–PCL–R–POEGA in vitro measured by ICP-MS measurement and TEM demonstrate that the reduction-responsive copolymer might enhance the cellular uptake, presumably due to the aggregation of AuNRs@LA–PCL–SS–POEGA under reduction conditions. We believe that these findings will have a significant impact on the application of AuNRs modified with stimuli-responsive polymers.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Professor Yucai Wang for generously providing Near-Infrared laser. The financial support of the National Natural Scientific Foundation of China (NNSFC) Project (21674107) and the Fundamental Research Funds for the Central Universities (WK2340000066) is gratefully acknowledged.
Notes and references
- A. Mandal, R. Bisht, I. D. Rupenthal and A. K. Mitra, J. Controlled Release, 2017, 248, 96–116 CrossRef PubMed.
- S. Biswas, P. Kumari, P. M. Lakhani and B. Ghosh, Eur. J. Pharm. Sci., 2016, 83, 184–202 CrossRef PubMed.
- A. Gothwal, I. Khan and U. Gupta, Pharm. Res., 2016, 33, 18–39 CrossRef PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef PubMed.
- Y. P. Li, K. Xiao, W. Zhu, W. B. Deng and K. S. Lam, Adv. Drug Delivery Rev., 2014, 66, 58–73 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef PubMed.
- K. Engin, D. B. Leeper, J. R. Cater, A. J. Thistlethwaite, L. Tupchong and J. D. McFarlane, Int. J. Hyperthermia, 1995, 11, 211–216 CrossRef PubMed.
- E. S. Lee, Z. G. Gao and Y. H. Bae, J. Controlled Release, 2008, 132, 164–170 CrossRef PubMed.
- J. Liu, Y. R. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X. J. Liang, Biotechnol. Adv., 2014, 32, 693–710 CrossRef PubMed.
- M. Stubbs, P. M. J. McSheehy, J. R. Griffiths and C. L. Bashford, Mol. Med. Today, 2000, 6, 15–19 CrossRef PubMed.
- Y. Li, J. J. Li, B. Chen, Q. X. Chen, G. Y. Zhang, S. Y. Liu and Z. S. Ge, Biomacromolecules, 2014, 15, 2914–2923 CrossRef PubMed.
- S. Panja, G. Dey, R. Bharti, K. Kumari, T. K. Maiti, M. Mandal and S. Chattopadhyay, ACS Appl. Mater. Interfaces, 2016, 8, 12063–12074 CrossRef PubMed.
- P. Kuppusamy, H. Q. Li, G. Ilangovan, A. J. Cardounel, J. L. Zweier, K. Yamada, M. C. Krishna and J. B. Mitchell, Cancer Res., 2002, 62, 307–312 Search PubMed.
- F. H. Meng, W. E. Hennink and Z. Y. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef PubMed.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef PubMed.
- Y. F. Wen and J. K. Oh, RSC Adv., 2014, 4, 229–237 RSC.
- K. Rahimian, Y. F. Wen and J. K. Oh, Polymer, 2015, 72, 387–394 CrossRef.
- A. Cunningham, N. R. Ko and J. K. Oh, Colloids Surf., B, 2014, 122, 693–700 CrossRef PubMed.
- W. Chen, P. Zhong, F. H. Meng, R. Cheng, C. Deng, J. Feijen and Z. Y. Zhong, J. Controlled Release, 2013, 169, 171–179 CrossRef PubMed.
- H. L. Sun, B. N. Guo, R. Cheng, F. H. Meng, H. Y. Liu and Z. Y. Zhong, Biomaterials, 2009, 30, 6358–6366 CrossRef PubMed.
- H. L. Sun, B. N. Guo, X. Q. Li, R. Cheng, F. H. Meng, H. Y. Liu and Z. Y. Zhong, Biomacromolecules, 2010, 11, 848–854 CrossRef PubMed.
- J. J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef PubMed.
- S. Parida, C. Maiti, Y. Rajesh, K. K. Dey, I. Pal, A. Parekh, R. Patra, D. Dhara, P. K. Dutta and M. Mandal, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 3039–3052 CrossRef PubMed.
- Z. J. Zhang, J. Wang, X. Nie, T. Wen, Y. L. Ji, X. C. Wu, Y. L. Zhao and C. Y. Chen, J. Am. Chem. Soc., 2014, 136, 7317–7326 CrossRef PubMed.
- Y. Qiu, Y. Liu, L. M. Wang, L. G. Xu, R. Bai, Y. L. Ji, X. C. Wu, Y. L. Zhao, Y. F. Li and C. Y. Chen, Biomaterials, 2010, 31, 7606–7619 CrossRef PubMed.
- X.-M. Zhu, C. H. Fang, H. L. Jia, Y. Huang, C. H. K. Cheng, C.-H. Ko, Z. Y. Chen, J. F. Wang and Y.-X. J. Wang, Nanoscale, 2014, 6, 11462–11472 RSC.
- L. Vigderman, P. Manna and E. R. Zubarev, Angew. Chem., Int. Ed., 2012, 51, 636–641 CrossRef PubMed.
- Z. B. Li, S. Y. Tang, B. K. Wang, Y. Li, H. Huang, H. Y. Wang, P. H. Li, C. Z. Li, P. K. Chu and X.-F. Yu, ACS Biomater. Sci. Eng., 2016, 2, 789–797 CrossRef.
- Y. Jiang, S. D. Huo, T. Mizuhara, R. Das, Y. W. Lee, S. Hou, D. F. Moyano, B. Duncan, X. J. Liang and V. M. Rotello, ACS Nano, 2015, 9, 9986–9993 CrossRef PubMed.
- C. Grabinski, N. Schaeublin, A. Wijaya, H. D'Couto, S. H. Baxamusa, K. Hamad-Schifferli and S. M. Hussain, ACS Nano, 2011, 5, 2870–2879 CrossRef PubMed.
- T. B. Huff, M. N. Hansen, Y. Zhao, J. X. Cheng and A. Wei, Langmuir, 2007, 23, 1596–1599 CrossRef PubMed.
- T. Mizuhara, K. Saha, D. F. Moyano, C. S. Kim, B. Yan, Y.-K. Kim and V. M. Rotello, Angew. Chem., Int. Ed., 2015, 54, 6567–6570 CrossRef PubMed.
- A. M. Alkilany, P. K. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701–708 CrossRef PubMed.
- E. Oh, J. B. Delehanty, K. E. Sapsford, K. Susumu, R. Goswami, J. B. Blanco-Canosa, P. E. Dawson, J. Granek, M. Shoff, Q. Zhang, P. L. Goering, A. Huston and I. L. Medintz, ACS Nano, 2011, 5, 6434–6448 CrossRef PubMed.
- T. S. Hauck, A. A. Ghazani and W. C. W. Chan, Small, 2008, 4, 153–159 CrossRef PubMed.
- Y. N. Zhong, C. Wang, L. Cheng, F. H. Meng, Z. Y. Zhong and Z. Liu, Biomacromolecules, 2013, 14, 2411–2419 CrossRef PubMed.
- J. Liu, C. Detrembleur, B. Grignard, M.-C. De Pauw-Gillet, S. Mornet, M. Treguer-Delapierre, Y. Petit, C. Jérôme and E. Duguet, Chem.–Asian J., 2014, 9, 275–288 CrossRef PubMed.
- Z. G. Tang, J. T. Callaghan and J. A. Hunt, Biomaterials, 2005, 26, 6618–6624 CrossRef PubMed.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413–3431 CrossRef PubMed.
- L. W. Li, X. Wang, J. X. Yang, X. D. Ye and C. Wu, Macromolecules, 2014, 47, 650–658 CrossRef.
- Z. Q. Cao, H. Wu, J. Dong and G. J. Wang, Macromolecules, 2014, 47, 8777–8783 CrossRef.
- J. X. Yang, L. W. Li, Z. Y. Jing, X. D. Ye and C. Wu, Macromolecules, 2014, 47, 8437–8445 CrossRef.
- H. Y. Fan, Y. X. Li, J. X. Yang and X. D. Ye, J. Phys. Chem. B, 2017, 121, 9708–9717 CrossRef PubMed.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef.
- X. S. Liu, N. Huang, H. Li, H. B. Wang, Q. Jin and J. Ji, ACS Appl. Mater. Interfaces, 2014, 6, 5657–5668 CrossRef PubMed.
- M. H. Stewart, K. Susumu, B. C. Mei, I. L. Medintz, J. B. Delehanty, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, J. Am. Chem. Soc., 2010, 132, 9804–9813 CrossRef PubMed.
- C. R. Martin, Science, 1994, 266, 1961–1966 CrossRef PubMed.
- Y. Y. Yu, S. S. Chang, C. L. Lee and C. R. C. Wang, J. Phys. Chem. B, 1997, 101, 6661–6664 CrossRef.
- Q. S. Wei, J. Ji and J. C. Shen, J. Nanosci. Nanotechnol., 2008, 8, 5708–5714 CrossRef PubMed.
- M. Glidden and M. Muschol, J. Phys. Chem. C, 2012, 116, 8128–8137 CrossRef.
- J. Rodríguez-Fernández, J. Pérez-Juste, L. M. Liz-Marzán and P. R. Lang, J. Phys. Chem. C, 2007, 111, 5020–5025 CrossRef.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef PubMed.
- H. W. Liao and J. H. Hafner, Chem. Mater., 2005, 17, 4636–4641 CrossRef.
- C. J. Orendorff and C. J. Murphy, J. Phys. Chem. B, 2006, 110, 3990–3994 CrossRef PubMed.
- S. Shen, H. Y. Tang, X. T. Zhang, J. F. Ren, Z. Q. Pang, D. G. Wang, H. L. Gao, Y. Qian, X. G. Jiang and W. L. Yang, Biomaterials, 2013, 34, 3150–3158 CrossRef PubMed.
- R. F. Zhao, X. X. Han, Y. Y. Li, H. Wang, T. J. Ji, Y. L. Zhao and G. J. Nie, ACS Nano, 2017, 11, 8103–8113 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03545h |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab