
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design, preparation and characterization of recombinant Ag85B variants and their glycoconjugates with T-cell antigenic activity against Mycobacterium tuberculosis†‡
Francesca Rinaldi§
a,
Sara Tengattini§a,
Luciano Piubellibc,
Roberta Bernardinid,
Francesca Mangionee,
Teodora Bavaroa,
Gregorino Paonef,
Maurizio Matteid,
Loredano Pollegionibc,
Gaetano Filiceg,
Caterina Temporini *a and
Marco Terrenia
*a and
Marco Terrenia
aDepartment of Drug Sciences, University of Pavia, Viale Taramelli 12, 27100 Pavia, Italy. E-mail: caterina.temporini@unipv.it; Fax: +39-0382-422975; Tel: +39-0382-987788 ext. 7368
bDepartment of Biotechnology and Life Sciences, University of Insubria, Via Dunant 3, 21100 Varese, Italy
cThe Protein Factory Research Centre, Politecnico of Milan and University of Insubria, Via Mancinelli 7, 20131 Milan, Italy
dDepartment of Biology and Animal Technology Station, University of Rome “Tor Vergata”, Via Montpellier 1, 00133 Rome, Italy
eIRCCS San Matteo Hospital Foundation Microbiology and Virology Unit, Viale Camillo Golgi 19, 27100 Pavia, Italy
fDepartment of Cardiovascular, Respiratory, Nephrologic, Anesthesiologic and Geriatric Sciences, Sapienza University of Rome, Piazzale Aldo Moro 5, 00185 Rome, Italy
gDepartment of Internal Medicine and Therapeutics, University of Pavia and Unit of Infectious Diseases, IRCCS San Matteo Hospital Foundation, Viale Camillo Golgi 19, 27100 Pavia, Italy
First published on 26th June 2018
Abstract
Tuberculosis is the deadliest infectious disease in the world. The variable efficacy of the current treatments highlights the need for more effective agents against this disease. In the past few years, we focused on the investigation of antigenic glycoconjugates starting from recombinant Ag85B (rAg85B), a potent protein antigen from Mycobacterium tuberculosis. In this paper, structural modifications were rationally designed in order to obtain a rAg85B variant protein able to maintain its immunogenicity after glycosylation. Lysine residues involved in the main T-epitope sequences (namely, K30 and K282) have been substituted with arginine to prevent their glycosylation by a lysine-specific reactive linker. The effectiveness of the mutation strategy and the detailed structure of resulting neo-glycoconjugates have been studied by intact mass spectrometry, followed by peptide and glycopeptide mapping. The effect of K30R and K282R mutations on the T-cell activity of rAg85B has also been investigated with a preliminary immunological evaluation performed by enzyme-linked immunospotting on the different variant proteins and their glycosylation products. After glycosylation, the two variant proteins with an arginine in position 30 completely retain the original T-cell activity, thus representing adequate antigenic carriers for the development of efficient glycoconjugate vaccines against tuberculosis.
1. Introduction
The use of glycoconjugate vaccines is a promising strategy to fight various pathologies, among which different infectious diseases and forms of cancer.1 Several vaccines of this kind have been developed and those against Haemophilus influenzae type b, Streptococcus pneumoniae and Neisseria meningitidis are the most widespread.1–3In general, a glycoconjugate vaccine is composed of a carbohydrate moiety able to stimulate the B-cell immune response, coupled to an immunogenic carrier protein or peptide. The conjugation of glycans derived from bacterial surface polysaccharides to immunogenic proteins is today recognized as a valid approach to produce high-effective vaccines, suitable to induce long-term protection against encapsulated bacteria.1,4 Typically, saccharide–protein conjugates are recognized by antigen-presenting cells (APC) through the interaction with receptors specific for bacterial and viral polysaccharides. The antigen-receptor complex is then internalized into the cells, where various proteases digest the protein carrier. The ensuing fragment epitopes are exposed on the APC surface, and activate lymphocytes T and B. Thus, both protein/peptide and carbohydrate components contribute to vaccine immunogenicity.2 Accordingly, semi-synthetic glycoproteins (neo-glycoproteins) resulting from the chemical linkage between protein and saccharide, can provide carbohydrate antigens and immunogens from which therapeutic agents can be derived.5,6 In fact, neo-glycoproteins have been already developed7,8 to obtain vaccine products of clinical relevance for the prevention of infective diseases9 and have also been largely investigated as therapeutic anticancer vaccines.10 In addition, the conjugation of antigenic peptides/proteins with synthetic immunogenic carbohydrates might strongly increase their biological activity by improving the peptide/protein antigen uptake targeting receptors of APC.6,11 On the other hand, the glycosylation of antigenic peptides/proteins should exclude the amino acids involved in the epitopes to prevent a decrease or the loss of their biological activity.11
The chemical synthesis of neo-glycoproteins involves the linkage of glycans by a reactive linker, which in some cases targets specific amino acid residues.1,12–15 This process typically results in non-homogeneous products, namely glycoforms differing in glycan content and position, thus showing different features in terms of pharmacokinetics and immunogenicity. Therefore, the development of a neo-glycoprotein as therapeutic product requires well-characterized structure–activity relationships3,4 including a defined glycosylation profile (i.e. glycoform composition and glycosylation sites). This is usually achieved by a combination of analytical techniques such as liquid chromatography (LC) and mass spectrometry (MS) in different modalities and operating conditions.16–19
Tuberculosis (TB) is one of the main causes of death in the world20 and efficient vaccines suitable for replacing the old Mycobacterium bovis Bacillus Calmette-Guérin (BCG) are urgently needed21,22 due to its poor efficacy. Among the vaccines currently under investigation, most are based on Ag85B protein, one of the strongest Mycobacterium tuberculosis (MTB) antigens,23,24 that induces both antibody and T-cell mediated immune responses against MTB. The glycosylation of this protein has also been proposed25 based on the evidence that surface arabinomannan in MTB is able to stimulate the humoral immune response.26,27 However, in this case glycosylation of protein epitopes should be avoided in order to maintain the antigenic activity of Ag85B.
In previous studies, we investigated the glycosylation of a recombinant form of Ag85B (rAg85B), produced in E. coli with an additional pre-sequence of 7 amino acids.28 Thus, different saccharides activated with the 2-iminomethoxyethyl (IME) reactive linker were used for conjugation with this MTB protein, allowing the preparation of the corresponding glycoconjugates.29,30 By combining the analytical characterization of glycosylation sites with the biological evaluation of neo-glycoproteins, the effect of the glycosylation on rAg85B antigenic activity was investigated. In detail, the ex vivo immunological data revealed a reduction of the T-cell activity consequently to glycosylation, which was mainly ascribed to the glycan reaction with K30 and K282 residues (corresponding to K23 and K275 in the wild-type protein), both involved in T-cell epitope(s).30,31 The contribution of these two residues to the formation of Ag85 T-cell epitopes is supported by several experimental evidences including different immunized animal models in which epitope regions were defined by monitoring the responses to synthetic peptides covering the complete protein sequence.32–37 Interestingly, it was observed that the antigenic sequence 261-280 of Mycobacterium ulcerans, with an arginine residue in the position corresponding to K275 in MTB Ag85B, maintains the ability to stimulate the T-cell response, suggesting that K/R substitution might be introduced in the epitopic sequences without affecting the antigenic activity of the protein.37 Finally, the involvement of lysines 23 and 275 in T-cell epitopes of wild-type Ag85B has also been confirmed using human peripheral blood mononuclear cell (PBMC) cultures from BCG-vaccinated individuals and TB patients.38,39
In the present work, we describe the preparation of new antigenic proteins obtained by conservative substitutions of K30 and K282 with arginine residues in rAg85B, specifically designed to hinder the glycosylation at the main T-cell epitope sequences of this protein. A capillary reverse phase (RP)LC-UV-MS/MS system was used to confirm protein sequences after proteolytic digestion by chymotrypsin, while the T-cell antigenic activity of rAg85B and its variants was evaluated by ex vivo tests (enzyme-linked immunospot, ELISPOT).
Subsequently, the reactivity of the proteins was assessed by evaluating the glycosylation sites after conjugation of mono-, di- and tri-saccharides of mannose activated with IME reactive linker. The preservation of substituted sites from glycan attachment was confirmed by on-line solid phase extraction–hydrophilic interaction liquid chromatography (SPE-HILIC)-MS analyses. Finally, the effect of the site-directed glycosylation on the T-cell antigenic activity of the neo-glycoconjugates was evaluated by ELISPOT assays performed on BCG vaccinated volunteers.
2. Materials and methods
2.1. Materials, reagents and chemicals
Acetic acid, acetonitrile (ACN), ammonium bicarbonate, benzamidine hydrochloride, α-chymotrypsin from bovine pancreas, dithiothreitol (DTT), methanol, NaCl, 3-(N-morpholino) propanesulfonic acid (MOPS), sodium tetraborate and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All the employed reagents were of analytical grade.Recombinant bovine enterokinase (rEK, purity > 99%) and the Cleavage Capture Kit for rEK removal were purchased from Merck KGaA (Darmstadt, Germany). One rEK unit (U) is defined as the amount of enzyme that cleaves > 95% of 50 μg of a control protein (supplied by the producer) in 16 hours at room temperature and in 20 mM Tris HCl, 50 mM NaCl, 2 mM CaCl2, pH 7.4.
Deionized water was obtained from a Milli-Q® Integral purification system (Merck KGaA, Darmstadt, Germany). Sample desalting required the use of Amicon® Ultra-0.5 mL centrifugal filters (Merck KGaA, Darmstadt, Germany).
2.2. Production of recombinant Ag85B protein and variants
The wild-type Ag85B antigen from MTB was produced in a recombinant form in Escherichia coli BL21(DE3) as a chimeric protein bearing thioredoxin (Trx), a His6-tag and a sequence recognized by rEK at its N-terminus. The mature protein was obtained through the cleavage of the fusion protein by rEK, which was subsequently removed from the solution by affinity-based capture on EKapture™ Agarose (Merck KGaA, Darmstadt, Germany). Ag85B and Trx generated by the enzymatic cleavage were separated by metal-chelating chromatography (HiTrap Chelating, GE Healthcare, Piscataway, NJ, USA). The complete production and purification process has already been described.28Single (K30R and K282R) and double (K30R/K282R) point variants of rAg85B were produced using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). Site-directed mutagenesis was carried out via a PCR reaction on rAg85B (for K30R and K282R) or rAg85B–K30R (for K30R/K282R) cDNA cloned into pET32b expression plasmids. For each reaction, two complementary oligonucleotide primers containing the desired mutation, flanked by unmodified nucleotide sequence, were used (mutated bases are underlined):
(1.1) rAg85B–K30R: 5′ GGGTCGTGATATTA![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) AGTTCAGTTTCAGAGCGG 3′;
AGTTCAGTTTCAGAGCGG 3′;
(1.2) rAg85B–K30R: 3′ CCCAGCACTATAAT![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) TCAAGTCAAAGTCTCGCC 5′;
TCAAGTCAAAGTCTCGCC 5′;
(2.1) rAg85B–K282R: 5′ GCTGAATGCAATGAAGGTGATCTGCAGAGC 3′;
(2.2) rAg85B–K282R: 3′ CGACTTACGTTACTTCCACTAGACGTCTCG 5′.
For the K30R/K282R double variant, the oligonucleotides for the production of rAg85B–K282R (2.1 and 2.2) were employed.
The presence of the desired mutations was confirmed by automated DNA sequencing.
The Trx-Ag85B variant proteins were produced as reported for the unmodified counterpart.28 Briefly, E. coli BL21(DE3) cells transformed with the pET32b–Ag85B plasmids encoding for the different protein variants were grown in SB medium at 37 °C, added of 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at an OD600 nm = 2 and grown at 18 °C for additional 16 hours (overnight). Using these conditions, ∼20% of the single point chimeric proteins were soluble vs. ∼10% for the double variant. Trx-Ag85B variants were isolated by a single chromatographic step on a nickel-affinity column (HiTrap Chelating, GE Healthcare, Piscataway, NJ, USA). Maturation of the fusion proteins was obtained incubating 3 U of rEK per mg of Trx-Ag85B, at 20 °C for 16 hours, followed by a further HiTrap Chelating chromatography. The overall yields were: rAg85B–K30R, 1.5 mg pure protein per L fermentation broth; rAg85B–K282R, 1.25 mg pure protein per L fermentation broth; rAg85B–K30R/K282R, 0.9 mg pure protein per L fermentation broth. The proteins were finally collected in 20 mM MOPS, 0.4 M NaCl, pH 7.0. Purity and conformation of the recombinant proteins used in this study were assessed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), MS and circular dicroism (CD), as previously described.28,40
2.3. Synthesis of mannose-based saccharides
Synthesis of 2-iminomethoxyethyl activated α-D-mannopyranoside (Man-IME), α-D-mannopyranosyl-(1→6)-α-D-mannopyranoside (Man2-IME) and α-D-mannopyranosyl-(1→6)-(α-D-mannopyranosyl)-(1→6)-α-D-mannopyranoside (Man3-IME) were performed according to the procedure previously reported.29,41 Briefly, the peracetylated mannose and acetylated mannose bearing a thiocyanomethyl group at the C-1 position were submitted to regioselective hydrolysis catalyzed by Candida rugosa lipase. The obtained mono-deprotected compounds in C-6 position have been considered as intermediates for the synthesis of cyanomethyl 2′,3′,4′,6′-tetra-O-acetyl-α-D-mannopyranosyl-(1→6)-2,3,4-tri-O-acetyl-α-D-thiomannopyranoside and cyanomethyl 2′′,3′′,4′′,6′′-tetra-O-acetyl-α-D-mannopyranosyl-(1→6)-α-D-2′,3′,4′-tri-O-acetyl-mannopyranosyl-(1→6)-2,3,4-tri-O-acetyl-α-D-thiomannopyranoside. The compounds were characterized by nuclear magnetic resonance (NMR) spectroscopy and MS. Then, the obtained products and cyanomethyl 2,3,4,6-tetra-O-acetyl-α-D-thiomannopyranoside were treated with sodium methoxide in methanol to remove the acetyl esters protecting groups and to functionalize the C-1 position as IME group. After 48 h, the reaction mixture was concentrated in vacuum and the solid formed was analyzed by direct infusion in electrospray ionization (ESI)-MS to evaluate the degree of conversion into IME thioglycosides. The yields of IME products were calculated as the ratio between the relative abundance of the activated form and the total ion intensities.2.4. Production of neo-glycoconjugates
Glycosylation of rAg85B and its variants was performed as previously described,16,42 with minimal adjustments. Proteins were dissolved in 100 mM sodium tetraborate buffer, pH 9.5 to reach a final concentration of 5.5 mg mL−1 and the solution was mixed with IME-glycosides to a glycoside/protein molar ratio of 200/1. Benzamidine hydrochloride was added to a final concentration of 1 mM to prevent the digestion by residual rEK derived from the protein production process. Reaction mixtures were vortexed for 1 min and incubated for 24 h at 25 °C under continuous stirring.2.5. Intact MS measurements
Proteins, activated saccharides and glycoconjugates were analyzed in a linear trap quadrupole (LTQ)-MS with an ESI source controlled by X-calibur software 2.0.7 (Thermo Fisher Scientific, Waltham, MA, USA).Before direct infusion in ESI-MS, protein and glycoprotein solutions were desalted by ultrafiltration using Amicon® Ultra centrifugal filters with a nominal molecular weight (MW) cut-off of 10 kDa and a 500 μL volume. Seven washing steps were performed at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g and 4 °C for 10 minutes to transfer the (glyco)proteins in deionized water.
000 × g and 4 °C for 10 minutes to transfer the (glyco)proteins in deionized water.
(Glyco)protein samples were prepared in water/ACN/acetic acid (49/50/1 v/v/v) to a final concentration of 300 μg mL−1, while lyophilized saccharides were dissolved in methanol. Samples were introduced into the mass spectrometer with a syringe pump at a flow rate of 10 μL min−1 and full scan intact MS experiments were carried out under the following instrumental conditions: positive ion mode, mass range 700–2000 m/z or 150–1000 m/z, source voltage 4.5 or 4.6 kV, capillary voltage 35 or 36 V, sheath gas flow rate 15 or 6 (arbitrary units), auxiliary gas flow rate 2 or 1 (arbitrary units), capillary temperature 220 or 250 °C, tube lens voltage 140 or 80 V for (glyco)proteins and carbohydrates, respectively. Deconvolution of the (glyco)protein spectra was performed using Bioworks Browser (Thermo Fisher Scientific, revision 3.1) and the abundance of the different species was defined by the relative intensity of the corresponding peaks in the deconvoluted spectra. The accuracy of mass determination was calculated comparing the experimental value with the one calculated from the amino acid sequence by “Peptide Mass Calculator” on IonSource Mass Spectrometry Educational Resource (http://www.ionsource.com/). Identification of protein truncated forms was carried out by MS-NonSpecific database search program of ProteinProspector v 5.20.0 (http://prospector.ucsf.edu).
2.6. Chymotryptic digestion of (glyco)proteins
All the (glyco)proteins were enzymatically digested with chymotrypsin prior to LC-MS analyses. Briefly, 100 μL of 100 μM (glyco)protein solution in water were added with 90 μL of 100 mM ammonium bicarbonate, pH 8.5, and 10 μL of 100 mM DTT solution in 100 mM ammonium bicarbonate, pH 8.5. The solution was heated at 60 °C for 30 minutes for disulfide bridge reduction and then added of chymotrypsin to a final (glyco)protein/enzyme ratio of 50/1 (w/w). The reaction mixture was incubated for 3 h at 37 °C under continuous stirring and the reaction was stopped by adding 2.5% TFA (v/v).2.7. Capillary RPLC-UV-MS peptide mapping
Peptide solutions deriving from the chymotriptic digestion of non-glycosylated proteins were filtered and diluted in water/ACN (95/5 v/v) to a final concentration of 8 μM before LC-UV analyses.The separation of peptides was performed on a Dionex UltiMate 3000 HPLC system (Thermo Fisher Scientific, Waltham, MA, USA) equipped with an UltiMate 3000 Autosampler and an UltiMate 3000 Variable Wavelength Detector and controlled by Chromeleon software (version 6.8). The analytical column was a C18 Acclaim® PepMap RSLC (2 μm, 100 Å, 300 μm × 150 mm) from Dionex (Thermo Fisher Scientific, Waltham, MA, USA). The mobile phases consisted of water + 0.05% TFA (v/v) (solvent A) and water/ACN 20/80 + 0.04% TFA (all v/v) (solvent B). The gradient was from 4 to 55% B in 30 min with a flow rate of 4 μL min−1. UV detection was performed at a wavelength of 214 nm.
MS detection was carried out on a LTQ-MS with an ESI source controlled by X-calibur software 2.0.7 (Thermo Fisher Scientific, Waltham, MA, USA), under the following instrumental conditions: positive ion mode, source voltage 4.5 kV, capillary voltage 31 V, sheath gas flow rate 40 (arbitrary units), auxiliary gas flow rate 10 (arbitrary units), capillary temperature 250 °C, tube lens voltage 95 V. Full scan mass range was set up from 300 to 2000 Da. MS2 and MS3 spectra were obtained by collision induced dissociation (CID) with normalized collision energy of 35.0.
Data processing was performed using Bioworks Browser (Thermo Fisher Scientific, revision 3.1) by comparing experimental data with protein sequences in FASTA. Peptides selection was achieved using “Xcorr vs. Charge State filter” (+1: 1.50, +2: 2.00, +3: 2.50).
2.8. On-line SPE-HILIC-MS analysis of neo-glycopeptides
The neo-glycoprotein digestion mixtures were analyzed according to a method previously described.6 Briefly, glycopeptides were on-line extracted on a porous graphitized carbon (PGC) trap column Hypersil Hypercarb (4.6 × 50 mm) from Alltech Ass. (Deerfield, IL, USA) by applying the following conditions: 10 min of desorption from the trap column with 80% solvent A (ACN + 0.05% v/v TFA) and 20% solvent B (water + 0.05% v/v TFA) at a flow rate of 100 μL min−1. Glycopeptides separation was performed on a Dionex UltiMate 3000 HPLC system (Thermo Fisher Scientific, Waltham, MA, USA) controlled by Chromeleon software, using a TSK-gel Amide-80 column (3 μm, 80 Å, 2 × 150 mm) from Tosoh Bioscience LLC (Montgomeryville, PA, USA). The gradient for chromatographic analyses was from 30% to 57% B in 11 min with a flow rate of 200 μL min−1. The analytes were revealed by an ESI-LTQ-MS (Thermo Fisher Scientific, Waltham, MA, USA) controlled by X-calibur software. Mass spectra were generated in positive ion mode under the following instrumental conditions: source voltage 4.0 kV, capillary voltage 46 V, sheath gas flow rate 40 (arbitrary units), auxiliary gas flow rate 10 (arbitrary units), capillary temperature 250 °C and tube lens voltage 105 V. MS2 and MS3 spectra were obtained by CID with normalized collision energy of 35.0. Glycopeptides were identified on the basis of MS2 and MS3 spectra by Bioworks Browser (Thermo Fisher Scientific, revision 3.1). Only identifications with a Xcorr higher than 1 were considered and, to avoid false positives, MS2 and MS3 spectra of all the species recognized as glycopeptides were manually evaluated.2.9. Biological ex vivo evaluation of T-cell activity by ELISPOT
The study population included 23 subjects. 15 were healthy BCG-vaccinated individuals and 8 were healthy controls without any history of TB exposure. T-cell responses directed against the rAg85B proteins and their glycoconjugates in both the studied group were determined by ELISPOT experiments as previously described.28Data are expressed using mean and standard deviation of the mean or median and percentiles, as appropriate. Groups were compared using Mann–Whitney and χ2 tests. A p value below 0.05 was considered significant. All tests were performed using the GraphPad Prism 4.0 (Graphpad software, San Diego, CA, USA) software package.
3. Results and discussion
3.1. Rational design and analytical characterization of rAg85B variants
In previous studies, a reduction in the T-cell immunogenic activity was observed for mannosylated rAg85B compared to the non-glycosylated protein. This effect was ascribed to the glycosylaton of K30 and K282, which are involved in important epitopes and were also found to be the most reactive sites in the conjugation reaction with Man-IME.30 The substitution of lysine with arginine residues can be considered as chemically conservative (pKa of the ε amino-group of lysine = 10.54; pKa of the guanidine-side group of arginine = 12.48), and naturally occurring K/R variation in some regions of Ag85 protein was reported as not affecting its T-cell antigenic activity.37Taking into account these experimental evidences, in this work conservative K/R substitutions (K30R, K282R and K30R/K282R) in rAg85B protein have been planned in order to hinder the glycosylation at these positions. To verify also the preservation of the B-cell antigenicity, a preliminary in silico evaluation of the effect induced by the planned modifications was carried out. The study was performed by analyzing the three-dimensional structure of the query protein (PDB 1F0N for Ag85B, substitutions were manually inserted in the sequence) using the EPCES prediction system of antigenic Epitopes on Protein surfaces by ConsEnsus Scoring server43 (http://sysbio.unl.edu/EPCES/) and the SEPPA method44 for spatial epitope prediction (http://lifecenter.sgst.cn/seppa/). The EPCES system employs six different scoring functions (residue epitope propensity, conservation score, side-chain energy score, contact number, surface planarity score, and secondary structure composition), while SEPPA attributes to each residue a score according to its neighborhood residues; higher scores correspond to a higher probability for the residue to be part of an epitope (the default threshold value is set at 1.80). Outputs indicate that K30R and K282R substitutions do not alter the original antigenicity of rAg85B. K30 residue was identified as antigenic only by SEPPA method, giving an antigenic prediction score of 1.83 that was not altered introducing K30R mutation (1.83). Similarly, K282 and R282 were predicted as antigenic residues by EPCES and gave comparable scores using SEPPA method (1.59 for lysine and 1.63 for arginine).
Two single point variants (K30R and K282R) and a double variant (K30R/K282R) of rAg85B protein were thus produced by site-directed mutagenesis and overexpressed in E. coli under the same conditions employed for the unmodified counterpart.28 SDS-PAGE was used to preliminary assess purity of the products (data not shown), followed by intact MS measurement. Deconvoluted spectra are reported in Fig. S1 (ESI‡). For rAg85B and the two single point variants, the experimental mass corresponded to the expected MW of 31346.0 and 31374.0 Da, respectively (Fig. S1A–C‡ and Table 1). The mass spectrum of the double point variant rAg85B–K30R/K282R (Fig. S1D‡ and Table 1) showed a main peak at 31401.0 Da, which well agrees with the expected MW of 31402.0 Da. However, a second species was observed at 30480.0 Da with a relative abundance of around 20%. Based on its mass, this species was identified as the truncated by-product R10-G292, previously detected as impurity also for different batches of rAg85B.28
| Protein | Intact MW (relative abundance %) | Number of identified peptides | Protein sequence coverage (% aa) |
|---|---|---|---|
| rAg85B | 31345.0 (100) |
44 | 79.11 |
| rAg85B–K30R | 31375.0 (100) |
49 | 75.00 |
| rAg85B–K282R | 31376.0 (100) |
68 | 97.26 |
| rAg85B–K30R/K282R | 31401.0 (80) |
40 | 86.64 |
| 30480.0 (20) |
In order to confirm protein sequences, the presence and position of the desired mutations, rAg85B and its variants were digested with chymotrypsin and the resulting peptide mixtures were analyzed by capillary RPLC-UV-MS/MS. Results allowed to verify the identity of all the four recombinant proteins, being the sequence coverage (percentage of amino acids) over 75% in all cases (Table 1). Peptides containing each expected amino acid substitution were found in the corresponding variant sample, confirming the presence of the introduced changes at the desired positions (Table S1‡). The extracted ion chromatograms (EIC) of the m/z values corresponding to peptides comprising K30, K282, R30 and R282 further confirmed the presence/absence of the substitutions in the analyzed proteins (Fig. S2–5‡).
The T-cell activity of the three variant proteins was evaluated by ex vivo ELISPOT assays performed on samples obtained from two groups of selected volunteers,30 including a group of 15 people vaccinated with Mycobacterium bovis BCG (Fig. 1A) and healthy non-BCG vaccinated subjects without any history of TB exposure. The last group was used as a control (Fig. S6‡). Ag85B is one of the most important protein antigens in BCG vaccine. Vaccinated volunteers were used as they represent a homogeneous population of clinical cases. Indeed, contrary to patients with active TB, vaccinated individuals can be properly selected in order to avoid the occurrence of additional pathologies, which might interfere with the specific response to MTB antigens.
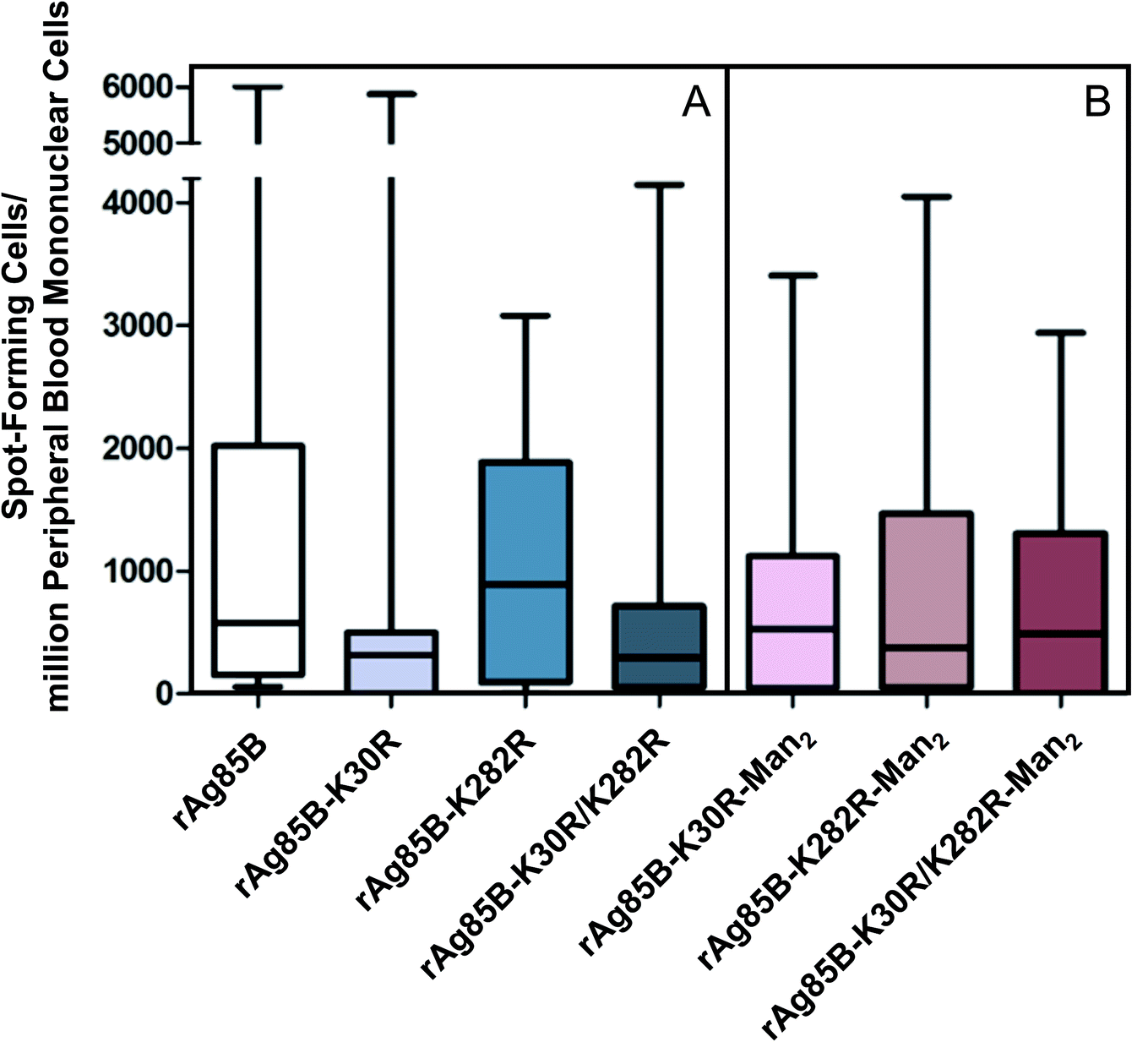 | ||
| Fig. 1 T-cell responses to rAg85B antigens (A) and their glycoconjugates (B). Data are presented as min to max value and boxplot of 25th–75th percentile of the spot-forming cells (SFC) per million PBMCs obtained by ELISPOT in BCG-vaccinated subjects. | ||
The analysis of the T-cell activity observed towards rAg85B protein and its variants shows that response was relevant for all proteins in BCG vaccinated subjects compared to healthy controls, showing a non-significant inter-individual variability (Fig. 1A and S6‡). These results demonstrate that the site-directed substitutions introduced in the putative epitope sequences do not statistically affect the antigenic activity of the protein. In fact, even if a reduction in the T-cell activity was observed for K30R variant, it was not statistically significant (p > 0.05 all comparisons).
3.2. Synthesis and characterization of glycoconjugates
The surface reactivity of rAg85B and its variants during synthetic conjugation reactions was carefully investigated. Mono-, di- and tri-saccharides of mannose (Fig. 2) were synthesized and activated with the IME functional group as already described.16,29,42 Direct infusions of the carbohydrates in ESI-MS allowed to define their activation degree before the conjugation reactions. The percentage of the activated products was found to decrease with the elongation of the chain, as previously reported in literature45 (Fig. S7A–C‡). | ||
| Fig. 2 IME-activated saccharides considered in the study. | ||
The characterization of glycoconjugates was carried out on rAg85B using the three IME-glycans, in order to assess the effect of the glycan length on the conjugation reaction outcomes (yield and position).
Glycosylation reactions were performed considering the purity of each activated saccharide, under previously optimized experimental conditions16 with minimal adjustments (see paragraph 2.4). Conjugation yield and glycan reactivity were derived by analysis in ESI-MS of intact neo-glycoconjugates. The relative abundance of each glycoform (%) was calculated as the ratio between the relative abundance indicated in the deconvoluted spectrum and the total ion intensities of the pattern, assuming the same ionization efficiency for all the detected proteoforms.46 Although complete glycosylation of rAg85B was achieved in every reaction, a change of the glycoform profile was observed depending on the size of the glycan, with a reduction of the overall glycan loading with the extension of the chain (Table 2).
| Glycoconjugate | Number of incorporated glycosides (%) | Y (%) | Glycoside/protein (mol mol−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| rAg85B-Man | — | — | — | 1.2 | 9.7 | 25.7 | 34.9 | 21.9 | 6.7 | 100.0 | 5.9 |
| rAg85B-Man2 | — | — | 11.7 | 25.1 | 31.4 | 21.9 | 9.8 | — | — | 100.0 | 3.9 |
| rAg85B-Man3 | — | 8.2 | 13.9 | 28.5 | 29.1 | 20.3 | — | — | — | 100.0 | 3.4 |
The results of the glycopeptide mapping by SPE-HILIC-MS/MS (Fig. 3) highlight that lysines 30, 103, 123 and 282 of rAg85B remain the most reactive with all the tested glycans, with the only exception of K123 in rAg85B-Man3 which results less glycosylated than K246. However, in general, the relative abundance order of glycosylated residues was not affected by the glycan type and the chain length. Therefore, in order to assess lysine reactivity and to characterize the obtained neo-glycoconjugates preliminarily to their ex vivo immunological evaluation, rAg85B single (K30R and K282R) and double (K30R/K282R) variants were glycosylated only with Man-IME moiety (selected as a reference).
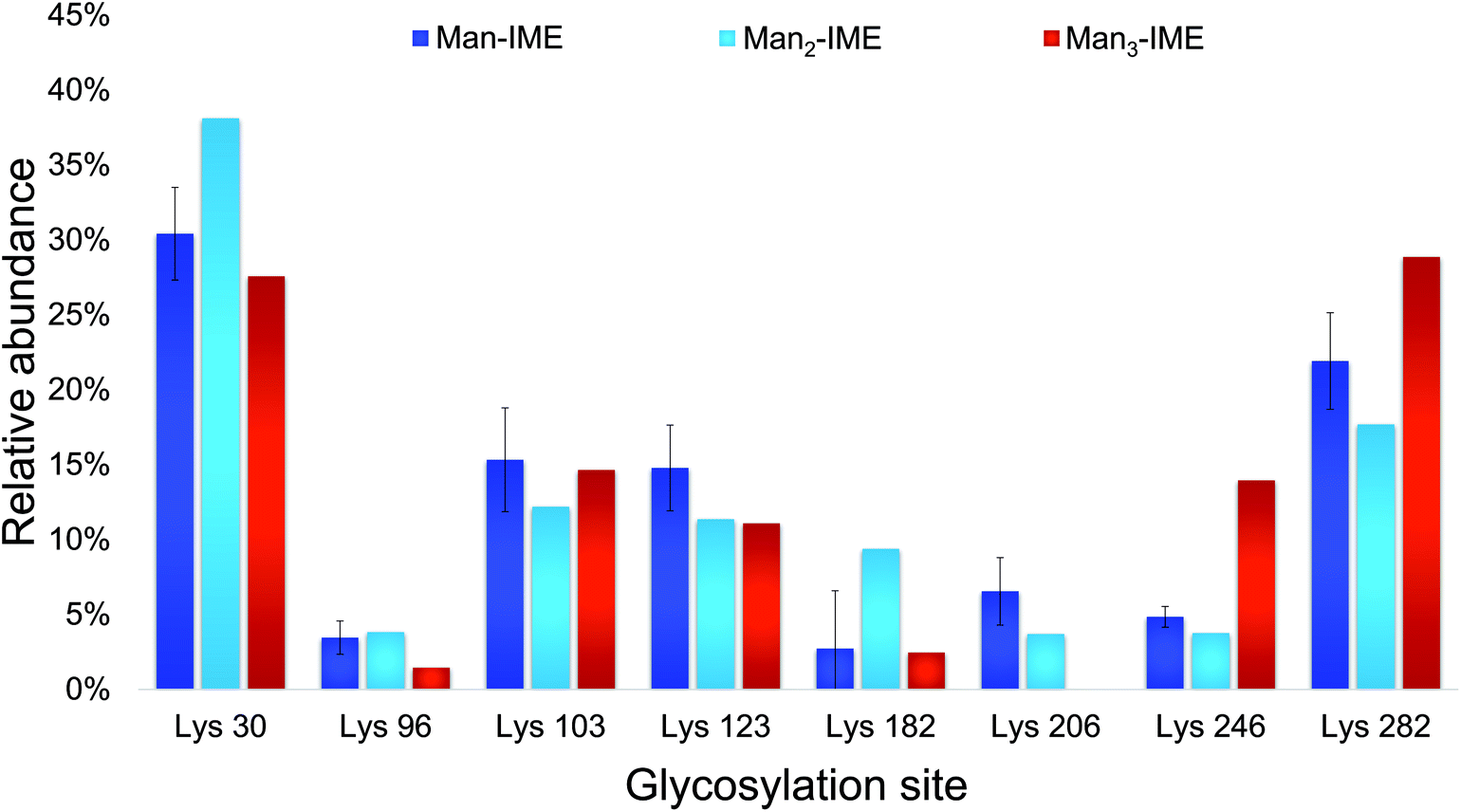 | ||
| Fig. 3 Lysine reactivity (relative abundance %) in rAg85B protein conjugated with Man-IME, Man2-IME and Man3-IME. | ||
Concerning the reactivity of the different protein variants, intact MS measurements of the purified reaction mixtures revealed reaction yields (Y) of 100% for all the coupling reactions, as unmodified proteins were not detected (Table 3). The maximum number of incorporated mannose units was equal to the total number of lysine residues in each protein sequence (8 for rAg85B, 7 for K30R and K282R single variants and 6 for K30R/K282R double variant), indicating that all lysine residues are involved in the glycosylation. Nevertheless, the Man/protein ratio was lower for the double variant, in agreement with the lack of the two most reactive lysines (Table 3).
| Glycoconjugate | Number of incorporated glycosides (%) | Y (%) | Glycoside/protein (mol mol−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| rAg85B–K30R-Man | — | — | — | 9.4 | 24.5 | 34.4 | 24.5 | 7.3 | — | 100.0 | 5.0 |
| rAg85B–K282R-Man | — | — | — | 5.6 | 20.3 | 35.4 | 29.1 | 9.7 | — | 100.0 | 5.2 |
| rAg85B–K30R/K282R-Man | — | — | 9.2 | 26.9 | 37.4 | 22.0 | 4.4 | — | — | 100.0 | 3.8 |
The glycosylation sites of rAg85B variants were then characterized in detail and compared to data obtained for the unmodified protein (Fig. 4). Analyses were performed by on-line SPE-HILIC-MS/MS following the chymotriptic digestion of the neo-glycoconjugates and the relative abundance of each glycosylation site was calculated as percentage of area of all the relative glycopeptides (Fig. 4A). No glycosylated peptides including R30 and R282 were detected in digestion mixtures obtained from the variant proteins, confirming that the substitutions of lysine residues with arginines effectively prevented their glycosylation. Experimental data suggested that amino acid reactivity reflects the absence of the substituted lysines, with a resulting predominant glycosylation of K282 in rAg85B–K30R and of K30 in rAg85B–K282R. In the rAg85B variant containing both amino acids substitutions, the major glycosylated site became K123, followed by K103, which were thus recognized as the most reactive lysines after K30 and K282. Moreover, the relative abundance order was generally respected in the four samples. Minimal variations in K206–K246 and K123–K103 relative orders were observed (Fig. 4). Even if the relative abundance of each occupied site significantly differs among the samples (Fig. 4A), the absolute abundance (calculated as the sum of mass peak areas of all the relative glycopeptides) is comparable between rAg85B and its variants (Fig. 4B). This finding suggested that the chemical reactivity of the individual lysines remains unaltered even in the absence of the most reactive groups and consequently the coupling reaction does not possess a competitive behaviour. Thus, the elimination of the most reactive lysines does not induce alterations in the level of glycosylation of less reactive ones, further supporting the success of the mutagenesis approach. These data are of utmost importance for other antigenic lysine residues such as K103 and K123 of rAg85B (corresponding to K96 and K116 of the wild-type protein), which have been reported to be included in potential T-epitopes of latent TB infected subjects.47–50
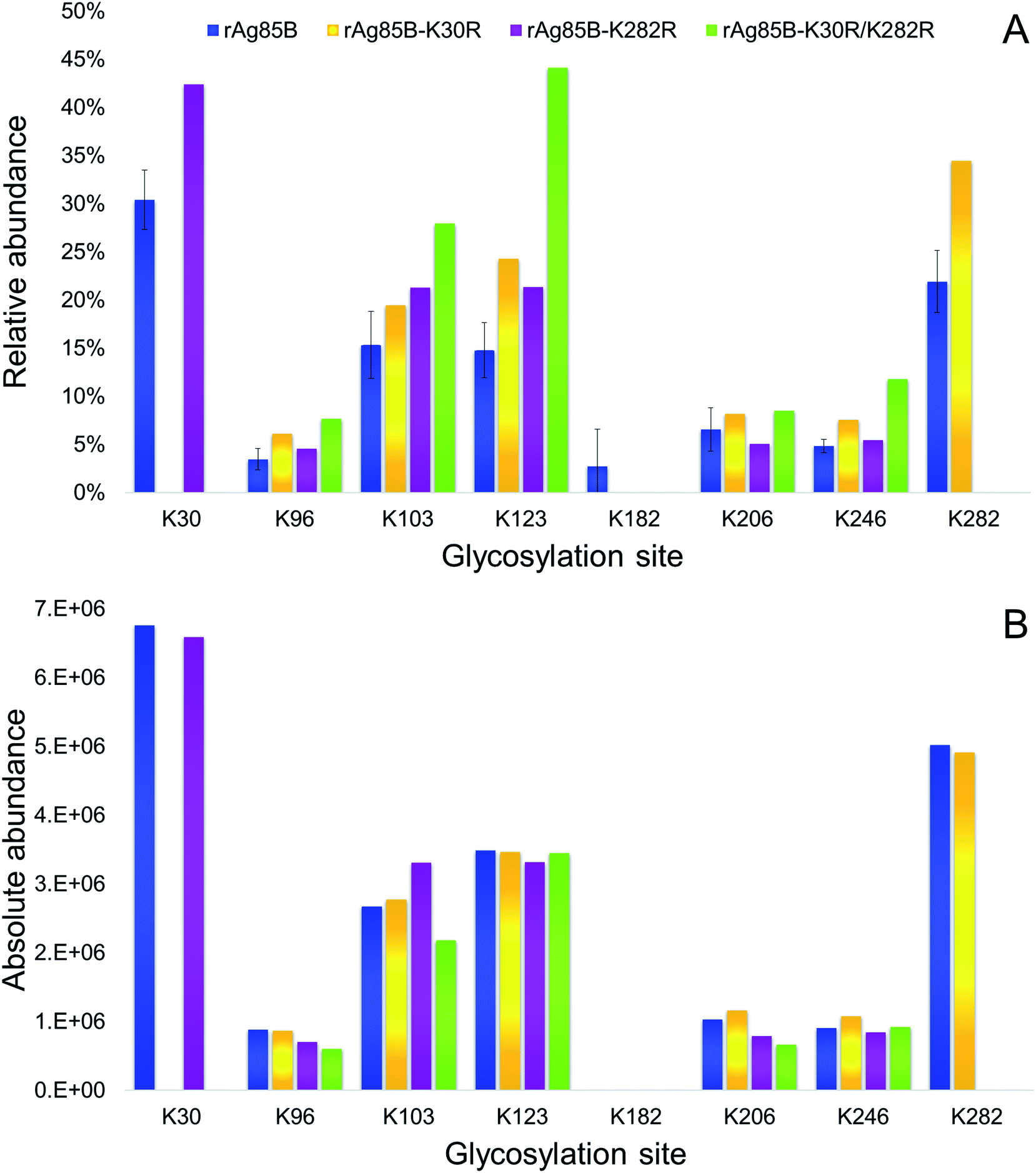 | ||
| Fig. 4 Relative (A) and absolute (B) abundances of glycosylated lysines in rAg85B and its variants conjugated with Man-IME. | ||
These evidences were reflected in the results obtained by the evaluation of T-cell activity (ELISPOT) performed for the glyco-derivative of the different protein species obtained by glycosylation with Man2-IME. All the tested glycoconjugates were characterized by direct infusion in ESI-MS, revealing that the conjugation reactions resulted in complete or almost complete protein glycosylation (Table S2‡). The data obtained from the ELISPOT assay showed that all glyco-derivatives maintained a good level of antigenic activity (Fig. 1B) as compared with the control group (Fig. S6‡). A reduction was observed only in the case of glycosylated rAg85B–K282R probably because of an abundant glycosylation of position 30. In fact, considering the median value observed in ELISPOT in the group of vaccinated volunteers tested, the response decreased from 800 SFC per million PBMCs as measured for the non-glycosylated rAg85B–K282R (Fig. 1A) to about 380 after glycosylation (Fig. 1B). These results agree with the essential role of the protein portion including K30 (K23 in wild-type protein) in the T-cell epitope of Ag85B in BCG.38
4. Conclusions
In this work, the substitution of amino acids involved in the main T-cell epitopes of Ag85B has been investigated in order to obtain MTB proteins suitable as antigenic carriers for the design of efficient glycoconjugate vaccines. Accordingly, substitution of lysines in position 30 and 282 of a recombinant Ag85B protein28 (corresponding to lysines 23 and 275 involved in important epitopes of the wild-type protein) has been performed by introducing arginine residues.Data obtained by analytical characterization allowed to verify the success of the designed site-directed mutagenesis, since arginine residues were identified in the predicted position. These substitutions were also confirmed to be conservative for protein conformation since all protein variants maintained the original T-cell immunogenic activity showed by rAg85B, as observed by preliminary ex vivo experiments performed by ELISPOT on biological samples obtained from a group of vaccinated volunteers.
The comparison of the results obtained from the glycosylation of the non-mutated rAg85B using mono-, di- and tri-saccharides of mannose activated with IME reactive linker demonstrated that lysines 30 and 282 remain the most reactive glycosylation sites regardless of the glycan used and suggested that protein surface reactivity is not influenced by the change in the sugar moiety.
Thus, glycosylation of the different variant proteins with Man-IME (selected as a reference) has been investigated. Notably, the substitutions introduced in rAg85B prevent the glycosylation of the main epitopes on this MTB protein without altering the glycosylation degree of the remaining lysine residues. As a consequence, the variant proteins carrying an arginine in position 30 (K30R and K30R/K282R variants), after glycosylation maintain the original antigenic activity of the rAg85B protein.
According to the obtained results, these variants of Ag85B may represent adequate antigenic carriers for the development of multivalent glycoconjugate vaccines against TB characterized by a high T-cell activity induced by the protein antigen.
Authors contribution
F. R. and S. T.: Glycoprotein synthesis and LC-MS proteins/glycoproteins structural characterization. Lu P.: rAg85B and its variants preparation. R. B. and F. M.: T-cell reactivity tests. T. B.: Synthesis of functionalized glycans. G. P. and M. M.: Immunological data elaboration. Lo P.: Head of Department of Biotechnology and Life Sciences, University of Insubria and The Protein Factory Research Centre. G. F.: Director of Unit of Infectious diseases, IRCCS San Matteo Hospital Foundation. M. T.: Project units coordinator. C. T.: Coordinator of the analytical characterization of proteins and glycoproteins; manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Fondazione Banca del Monte di Lombardia (FBML, Italy). Luciano Piubelli and Loredano Pollegioni were supported from Fondo di Ateneo per la Ricerca (University of Insubria). Professor Massimo Amicosante made a substantial contribution, sharing his knowledge and providing valuable suggestions. His busy schedule did not prevent him to assist us with great humanity and professionalism. This work is dedicated to his memory.References
- R. D. Astronomo and D. R. Burton, Nat. Rev. Drug Discovery, 2010, 9, 308–324 CrossRef PubMed.
- R. Adamo, A. Nilo, B. Castagner, O. Boutureira, F. Berti and G. J. Bernardes, Chem. Sci., 2013, 4, 2995–3008 RSC.
- M. Vella and D. Pace, Expert Opin. Biol. Ther., 2015, 15, 529–546 CrossRef PubMed.
- V. L. Campodónico, N. J. Llosa, L. V. Bentancor, T. Maira-Litran and G. B. Pier, Infect. Immun., 2011, 79, 3455–3464 CrossRef PubMed.
- T. Bavaro, L. Piubelli, M. Amicosante and M. Terreni, Curr. Org. Chem., 2016, 20, 1150–1168 CrossRef.
- E. W. Adams, D. M. Ratner, P. H. Seeberger and N. Hacohen, ChemBioChem, 2008, 9, 294–303 CrossRef PubMed.
- P. Stallforth, B. Lepenies, A. Adibekian and P. H. Seeberger, J. Med. Chem., 2009, 52, 5561–5577 CrossRef PubMed.
- O. Finco and R. Rappuoli, Front. Immunol., 2014, 5, 1–6 Search PubMed.
- V. Verez-Bencomo, V. Fernández-Santana, E. Hardy, M. E. Toledo, M. C. Rodríguez, L. Heynngnezz, A. Rodriguez, A. Baly, L. Herrera, M. Izquierdo, A. Villar, Y. Valdés, K. Cosme, M. L. Deler, M. Montane, E. Garcia, A. Ramos, A. Aguilar, E. Medina, G. Toraño, I. Sosa, I. Hernandez, R. Martínez, A. Muzachio, A. Carmenates, L. Costa, F. Cardoso, C. Campa, M. Diaz and R. Roy, Science, 2004, 305, 522–525 CrossRef PubMed.
- R. M. Wilson and S. J. Danishefsky, J. Am. Chem. Soc., 2013, 135, 14462–14472 CrossRef PubMed.
- J. D. McIntosh, M. A. Brimble, A. E. S. Brooks, P. Rod Dunbar, R. Kowalczyk, Y. Tomabechicd and A. J. Fairbanks, Chem. Sci., 2015, 6, 4636–4642 RSC.
- E. J. Grayson, G. J. L. Bernardes, J. M. Chalker, O. Boutureira, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2011, 50, 4127–4132 CrossRef PubMed.
- G. J. L. Bernardes, B. Castagner and P. H. Seeberger, ACS Chem. Biol., 2009, 4, 703–713 CrossRef PubMed.
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef PubMed.
- J. Zhu, Q. Wan, G. Ragupathi, C. M. George, P. O. Livingston and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 4151–4158 CrossRef PubMed.
- C. Temporini, T. Bavaro, S. Tengattini, I. Serra, G. Marrubini, E. Calleri, F. Fasanella, L. Piubelli, F. Marinelli, L. Pollegioni, G. Speranza, G. Massolini and M. Terreni, J. Chromatogr. A, 2014, 1367, 57–67 CrossRef PubMed.
- A. Staub, D. Guillarme, J. Schappler, J. L. Veuthey and S. Rudaz, J. Pharm. Biomed. Anal., 2011, 55, 810–822 CrossRef PubMed.
- K. Sandra, I. Vandenheede and P. Sandra, J. Chromatogr. A, 2014, 1335, 81–103 CrossRef PubMed.
- G. Brusotti, E. Calleri, R. Colombo, G. Massolini, F. Rinaldi and C. Temporini, Chromatographia, 2018, 81, 3–23 CrossRef.
- World Health Organization, Global tuberculosis report 2017, WHO Press, Geneva, Switzerland, 2017 Search PubMed.
- K. Bourzac, Nature, 2014, 507, S4–S7 CrossRef PubMed.
- P. Andersen and S. H. Kaufmann, Cold Spring Harbor Perspect. Med., 2014, 4, a018523 CrossRef PubMed.
- J. Dietrich, C. Aagaard, R. Leah, A. W. Olsen, A. Stryhn, T. M. Doherty and P. Andersen, J. Immunol., 2005, 174, 6332–6339 CrossRef.
- C. Aagaard, T. Hoang, J. Dietrich, P. J. Cardona, A. Izzo, G. Dolganov, G. K. Schoolnik, J. P. Cassidy, R. Billeskov and P. Andersen, Nat. Med., 2011, 17, 189–194 CrossRef PubMed.
- G. Källenius, A. Pawlowski, B. Hamasur and S. B. Svenson, Trends Microbiol., 2008, 16, 456–462 CrossRef PubMed.
- U. Demkow, B. Białas-Chromiec, M. Filewska, M. Sobiecka, J. Kuś, M. Szturmowicz, T. Zielonka, E. Augustynowicz-Kopeć, Z. Zwolska, M. Wasik and E. Rowińska-Zakrzewska, J. Physiol. Pharmacol., 2005, 56, 79–84 Search PubMed.
- A. K. Mishra, N. N. Driessen, B. J. Appelmelk and G. S. Besra, FEMS Microbiol. Rev., 2011, 35, 1126–1157 CrossRef PubMed.
- L. Piubelli, M. Campa, C. Temporini, E. Binda, F. Mangione, M. Amicosante, M. Terreni, F. Marinelli and L. Pollegioni, Microb. Cell Fact., 2013, 12, 115 CrossRef PubMed.
- T. Bavaro, M. Filice, C. Temporini, S. Tengattini, I. Serra, C. F. Morelli, G. Massolini and M. Terreni, RSC Adv., 2014, 4, 56455–56465 RSC.
- T. Bavaro, S. Tengattini, L. Piubelli, F. Mangione, R. Bernardini, V. Monzillo, S. Calarota, P. Marone, M. Amicosante, L. Pollegioni, C. Temporini and M. Terreni, Molecules, 2017, 22, 1081 CrossRef PubMed.
- K. Huygen, Front. Immunol., 2014, 5, 321 CrossRef PubMed.
- K. Huygen, E. Lozes, B. Gilles, A. Drowart, K. Palfliet, F. Jurion, I. Roland, M. Art, M. Dufaux, J. Nyabenda, J. De Bruyn, J. P. Van Vooren and R. Deleys, Infect. Immun., 1994, 62, 363–370 Search PubMed.
- A. Geluk, V. Taneja, K. E. van Meijgaarden, E. Zanelli, C. Abou-Zeid, J. E. Thole, R. R. de Vries, C. S. David and T. H. Ottenhoff, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10797–10802 CrossRef.
- B. Y. Lee and M. A. Horwitz, Infect. Immun., 1999, 67, 2665–2670 Search PubMed.
- S. D'Souza, V. Rosseels, M. Romano, A. Tanghe, O. Denis, F. Jurion, N. Castiglione, A. Vanonckelen, K. Palfliet and K. Huygen, Infect. Immun., 2003, 71, 483–493 CrossRef.
- V. Rosseels, S. Marché, V. Roupie, M. Govaerts, J. Godfroid, K. Walravens and K. Huygen, Infect. Immun., 2006, 74, 202–212 CrossRef PubMed.
- A. Tanghe, J. P. Dangy, G. Pluschke and K. Huygen, PLoS Neglected Trop. Dis., 2008, 2, e199 CrossRef PubMed.
- P. Launois, R. DeLeys, M. N. Niang, A. Drowart, M. Andrien, P. Dierckx, J. L. Cartel, J. L. Sarthou, J. P. Van Vooren and K. Huygen, Infect. Immun., 1994, 62, 3679–3687 Search PubMed.
- P. W. Roche, P. W. Peake, H. Billman-Jacobe, T. Doran and W. J. Britton, Infect. Immun., 1994, 62, 5319–5326 Search PubMed.
- L. Caldinelli, S. Iametti, A. Barbiroli, F. Bonomi, D. Fessas, G. Molla, M. S. Pilone and L. Pollegioni, J. Biol. Chem., 2005, 280, 22572–22581 CrossRef PubMed.
- S. Tengattini, E. Domínguez-Vega, C. Temporini, T. Bavaro, F. Rinaldi, L. Piubelli, L. Pollegioni, G. Massolini and G. W. Somsen, Anal. Chim. Acta, 2017, 981, 94–105 CrossRef PubMed.
- F. Rinaldi, S. Tengattini, E. Calleri, T. Bavaro, L. Piubelli, L. Pollegioni, G. Massolini and C. Temporini, J. Pharm. Biomed. Anal., 2017, 144, 252–262 CrossRef PubMed.
- S. Liang, D. Zheng, C. Zhang and M. Zacharias, BMC Bioinf., 2009, 10, 302 CrossRef PubMed.
- J. Sun, D. Wu, T. Xu, X. Wang, X. Xu, L. Tao, Y. X. Li and Z. W. Cao, Nucleic Acids Res., 2009, 37, W612–W616 CrossRef PubMed.
- B. G. Davis, S. I. Van Kasteren, D. Anthony and N. Sibson, Imaging agent, International Patent, WO 2007/020450 A2, 22 February 2007.
- A. Pedrali, S. Tengattini, G. Marrubini, T. Bavaro, P. Hemström, G. Massolini, M. Terreni and C. Temporini, Molecules, 2014, 19, 9070–9088 CrossRef PubMed.
- A. S. Mustafa, F. A. Shaban, A. T. Abal, R. Al-Attiyah, H. G. Wiker, K. Lundin, F. Oftung and K. Huygen, Infect. Immun., 2000, 68, 3933–3940 CrossRef PubMed.
- M. T. Valle, A. M. Megiovanni, A. Merlo, G. Li Pira, L. Bottone, G. Angelini, L. Bracci, L. Lozzi, K. Huygen and F. Manca, Clin. Exp. Immunol., 2001, 123, 226–232 CrossRef PubMed.
- R. F. Silver, R. S. Wallis and J. J. Ellner, J. Immunol., 1995, 154, 4665–4674 Search PubMed.
- C. S. Lindestam Arlehamn, A. Gerasimova, F. Mele, R. Henderson, J. Swann, J. A. Greenbaum, Y. Kim, J. Sidney, E. A. James, R. Taplitz, D. M. McKinney, W. W. Kwok, H. Grey, F. Sallusto, B. Peters and A. Sette, PLoS Pathog., 2013, 9, e1003130 CrossRef PubMed.
Footnotes |
| † This manuscript is dedicated to the memory of Massimo Amicosante. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03535k |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |