
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled self-assembly into diverse stimuli-responsive microstructures: from microspheres to branched cylindrical micelles and vesicles†
Xiaoteng Zhou,
Lingxiao Li,
He Qin,
Bo Ning,
Junpei Li and
Chengyou Kan *
*
Department of Chemical Engineering, Key Laboratory of Advanced Materials of Ministry of Education, Tsinghua University, Beijing 100084, People’s Republic of China. E-mail: kancy@mail.tsinghua.edu.cn
First published on 13th June 2018
Abstract
A series of amphiphilic PDMAEMA–SS–PCL chains with variable ratios of hydrophilic poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) to hydrophobic poly(ε-caprolactone) (PCL) were prepared via ring-opening polymerization, in which the two different moieties were linked via a disulfide bond with reduction responsiveness. After cross-linking by the photodegradable o-nitrobenzyl linkage, the amphiphilic chains could self-assemble into microspheres, branched cylindrical micelles and vesicles, which were responsive to the reduction agent DL-dithiothreitol and UV light irradiation through different mechanisms.
Introduction
Recently, fabrication of microscale stimuli-responsive polymeric systems has increasingly become a hot research field in material, pharmaceutics, catalysis, etc.1 Various stimuli-responsive microstructures including microspheres, micro/nano-gels, micelles and especially vesicles, which are similar to organism structure, have shown a powerful methodology for applications in drug delivery, microreactors and biosensors.1–6 In the applications of these polymeric microstructures, stimuli-responsiveness is a necessary function which can make them recognize their microenvironment and react in a dynamic way, mimicking the responsiveness of living organisms.7 Besides traditional thermo- and pH-sensitive systems, reduction-sensitive architectures based on disulfide bonds, which are commonly found in organisms and can react with intra-cellular glutathione, have drawn more attention especially in drug delivery systems.8 Moreover, because light-responsive systems can be rapidly induced at a specific time and are affected less than other experimental variables, light-triggered polymeric systems are also attractive now to achieve diverse functions in response to illumination of a specific wavelength. Up to now, most investigations of light responsive systems have been focused on simple micelles and microstructures such as vesicles and branched micelles have not been reported widely.9Among the above applications, encapsulation of therapeutic agents into polymer microstructures has been successfully used in the development of new drug carriers. A number of design parameters which affect the functional behaviour of carriers, including the choice of polymer, particle size and surface chemistry, have been adjusted to optimize their performance in vivo.10 In recent years, some investigations have confirmed that the shape of the microstructures can significantly impact the performance of polymer drug carriers, and have indicated that different morphologies of micro/nano structures exhibit different solubilization capacities, blood circulation times, biodistribution, toxicity, cellular uptake and intracellular fate.11,12 Thus, more and more attention has been paid to control morphology in common drug delivery for anti-cancer applications.13–16 Most multiple stimuli-responsive drug delivery systems can only be self-assembled into a spherical shape,17–19 and how to make these systems into different controlled morphologies is indeed meaningful for real application. It has been demonstrated that amphiphilic block copolymers in solution could be self-assembled into highly organized microstructures of different morphologies such as spheres, rods, lamellas and vesicles because of their architecture with two disparate physical property blocks.20–22 Since the morphologies of these microstructures used to be accurately tuned by the design of relative hydrophobic/hydrophilic block lengths, block arrangement, solvent composition and so on,20 controlled self-assembly through these pathways can help to adjust an appropriate microstructure to meet our demands.
Herein, we demonstrate a novel amphiphilic polymeric system to obtain three microstructures of different morphologies including microspheres, branched cylindrical micelles and vesicles, which just originate from the similar chains with diverse hydrophobic/hydrophilic ratios. With the help of the disulfide bond between the disparate segments and the photodegradable cross-linker incorporated into the amphiphilic chain, the microstructures all have reduction- and light-responsive properties. In addition, it is well known that the hydrophilic PDMAEMA segment is responsive to pH and temperature,23,24 and the hydrophobic PCL segment is recognized by its enzymatic degradation property.25,26 Thus, these microstructures can be used in many applications with diverse requirements and the different structures may also provide various physical performances and loading capacities to disparate cargos.27
Experimental
Materials
5-Hydroxy-2-nitrobenzaldehyde (Adams, 98%), sodium tetrahydroborate (NaBH4, 98%, Tianjin Yongda Chemical Reagent Co. Ltd), bromoacetyl bromide (TCI, 98%), 4-dimethylaminopyridine (DMAP, Adams, 99%), ε-caprolactone (CL, Adams, 99%), and stannous octoate (Sn(Oct)2, Admas, 95%) were of analytical grade. PDMAEMA–SS–OH (Mw = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 708 g mol−1, PDI = 1.127) was synthesized through an ATRP method as described previously.28 DL-Dithiothreitol (DTT, Aladdin, 99%), tetrahydrofuran (THF), triethylamine (TEA), toluene, and dichloromethane were of analytical grade and purchased from Beijing Chemical Works (Beijing, China).
708 g mol−1, PDI = 1.127) was synthesized through an ATRP method as described previously.28 DL-Dithiothreitol (DTT, Aladdin, 99%), tetrahydrofuran (THF), triethylamine (TEA), toluene, and dichloromethane were of analytical grade and purchased from Beijing Chemical Works (Beijing, China).
Synthesis of the light-stimulation responsive cross-linker Br–ONB–Br
Firstly, 5-hydroxy-2-nitrobenzyl alcohol was synthesized by a reduction reaction as follows. To a 50 mL flask 5-hydroxy-2-nitrobenzaldehyde (0.5000 g, 3 mmol) and 20 mL of methanol were added and stirred. A methanol solution of NaBH4 (0.2300 g, 6 mmol in 20 mL methanol) was added dropwise into the flask at 0 °C in an ice bath. The reaction mixture was stirred for 4 h at 25 °C, and then poured into H2O (30 mL). After adjusting the pH to around 6 using 0.1 M HCl, the solution was extracted with diethyl ether three times, dried by MgSO4 overnight, filtered, and concentrated using a rotary evaporator to obtain the target compound (0.4346 g, 2.57 mmol, yield 85.7%). 1H NMR (DMSO-D6, δ, ppm, see Fig. S1†): 10.86 (s, 1H), 8.04, 7.21 and 6.76 (m, 3H), 5.5 (s, 1H), 4.77 (s, 2H).29Then, 5-hydroxy-2-nitrobenzyl alcohol (0.5070 g, 3 mmol), TEA (2.3 mL, 16 mmol) and DMAP (49.00 mg, 0.4 mmol) were dissolved in dry dichloromethane (30 mL) in a 50 mL flask under a N2 atmosphere, and the flask was immersed in an ice bath for 30 min. A solution of bromoacetyl bromide (1.05 mL, 12 mmol) in dichloromethane (10 mL) was then added dropwise into the flask over 15 min. After stirring at 25 °C for 22 h, the white solid was removed by filtration, and the reaction mixture was transferred to a separatory funnel and diluted with 10 mL of dichloromethane. After this, the mixture was washed sequentially with 1 M HCl (10 mL), a saturated solution of NaHCO3 (10 mL) and water (10 mL), and the resulting organic phase was then dried with anhydrous magnesium sulfate overnight. The mixture was concentrated under reduced pressure via a rotary evaporator, and the resulting crude product was purified by column chromatography with mixtures of 3:1 hexane/ethyl acetate (by volume). After being dried in a vacuum at 50 °C for 24 h, the pure photodegradable cross-linker Br–ONB–Br was obtained (0.8335 g, 2.03 mmol, yield 67.7%). 1H NMR (CDCl3, δ, ppm, see Fig. S2†): 8.25 (d, 1H), 7.46 (s, 1H), 7.32 (d, 1H), 5.61 (s, 2H), 4.07 (s, 2H), 3.95 (s, 2H).28
Synthesis of linear PDMAEMA–SS–PCL amphiphilic copolymers with diverse hydrophobic ratios by ROP polymerization
PDMAEMA–SS–OH (0.5000 g, 0.028 mmol), CL (0.3420 g, 3 mmol for sample AC1, 0.500 g, 4.38 mmol for sample AC2, 1.069 g, 9.375 mmol for sample AC3), Sn(Oct)2 (25.02 mg, 6.13 × 10−2 mmol), and toluene (8 mL) were charged into a 25 mL flask equipped with a magnetic stirrer. The flask was placed into an oil bath and the reaction was carried out at 95 °C for 24 h. The reaction mixture was poured into cold diethyl ether, and the precipitate was collected and dried under vacuum at 50 °C for 48 h to obtain the product.Self-assembly of the linear amphiphilic polymer chain
PDMAEMA–SS–PCL block copolymer (10 mg) was first dissolved in 50 mL of THF, and 10 mL of trimethylamine was then added dropwise to the solution. Then, the solution was concentrated via evaporation at room temperature to about 5 mL. After dialysis (molecule weight cutoff 3500 Da) of the solution with water, an aqueous dispersion (0.33 mg mL−1) of the self-assembled microstructures was obtained.Self-assembly of the cross-linked amphiphilic polymer chain with Br–ONB–Br
A typical procedure for the self-assembly of the cross-linked amphiphilic polymer chain with Br–ONB–Br is described as follows. PDMAEMA–SS–PCL block copolymer (10 mg) was first dissolved in 30 mL of THF, and a solution of Br–ONB–Br (10 μL) dissolved in 20 mL of THF was added dropwise into the polymer solution. After one-day reaction at 70 °C, 10 mL of TEA was added to the reaction system and the solution was concentrated via evaporation at room temperature to about 5 mL. After dialysis (molecular weight cutoff, 3500 Da) of the solution with water, the aqueous dispersion (0.33 mg mL−1) of the self-assembly cross-linked microstructures was obtained.Reduction stimuli-responsiveness of the microstructures self-assembled from the cross-linked PDMAEMA–SS–PCL
The DTT solution (20 mM) was first prepared by dissolving 31 mg of DTT in 10 mL of PBS buffer solution (pH = 7.4), and the dispersion of the self-assembled PDMAEMA–SS–PCL microstructures (0.33 mg mL−1) was then added into the solutions. After being stirred at 25 °C for different periods of time, the resulting microstructures were characterized by TEM and DLS analysis.Light stimuli-responsiveness of the microstructures self-assembled from the cross-linked PDMAEMA–SS–PCL
3 mL of the solution (0.33 mg mL−1) of the self-assembled PDMAEMA–SS–PCL microstructures was exposed to a UV-led point light source (IWATA UV-101D, 365 nm, 36 mW cm−2) for 0.5 h, and the resulting microstructures were characterized by TEM.Characterization
1H NMR spectra of the samples were recorded using a JEOL ECS-400 spectrometer with CDCl3 as the solvent, especially DMSO-D6 for the 5-hydroxy-2-nitrobenzyl alcohol. FTIR spectra were recorded on a Nicolet 560 fourier transform infrared analyzer in the range from 4000 to 400 cm−1 with a resolution of 4 cm−1. The average molecular weight was measured by gel permeation chromatography (GPC) (SHIMADZU LC-20A) using THF as eluent. The morphologies of the different structures were characterized on a HITACHI H-7650 transmission electron microscope (TEM), and the samples for TEM observation were prepared by spraying the dispersion (5 μL, 0.33 mg mL−1) onto 300-mesh copper grids and drying at room temperature. The hydrodynamic diameter, size distribution and zeta potential were determined on a Malvern Zetasizer Nano ZS90.Results and discussion
Design and characteristics of the amphiphilic PDMAEMA–SS–PCL chains with different hydrophobic/hydrophilic ratios
It is well known that poly(ε-caprolactone) (PCL) has great biodegradability and biocompatibility in the pharmaceutical field.30–33 Because the PCL segments can encapsulate hydrophobic anti-cancer drugs by hydrophobic interactions and its chain length can affect the morphology and stability of the copolymers which can be self-aggregated into particles or fibres used as a drug carrier, PCL has shown great potential in drug delivery in vivo when it makes up the hydrophobic part of the polymer chain.30,31 On the other hand, because of its pH- and thermo-sensitivity, hydrophilicity and ease of quaternisation, poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) has been utilized widely in drug delivery systems.23,24,34 For example, using the electrostatic interactions between its cationic groups and DNA or siRNA, PDMAEMA segments can stably package the gene, preventing that encapsulated from extracellular digestion and extending the opportunity for efficient delivery into target cells by endocytosis.11,34,35 The co-polymer PHA–PDMAEMA formed from the combination of PDMAEMA with polyhydroxyalkanoates (PHAs), which are a class of diverse biodegradable polyesters, has been synthesized.36 Researchers have found that this copolymer could co-deliver hydrophobic anti-cancer drugs by hydrophobic interactions and genes by electrostatic attraction and had a great result when entering liver cancer cells.37,38 Thus, in our work, amphiphilic PDMAEMA–SS–PCL block copolymers, in which the hydrophilic DMAEMA segment and hydrophobic CL segment were linked by –S–S–, were designed to synthesize stimuli-responsive microstructures with different morphologies. The copolymers were then cross-linked by Br–ONB–Br to obtain a light-stimuli-responsive property. The synthetic routes of the linear and cross-linked PDMAEMA–SS–PCL block copolymers as well as the photodegradable cross-linker Br–ONB–Br are shown in Scheme 1. | ||
| Scheme 1 The synthetic routes of (A) photodegradable cross-linker Br–ONB–Br, (B) amphiphilic linear PDMAEMA–SS–PCL, and (C) cross-linked PDMAEMA–SS–PCL with Br–ONB–Br. | ||
Designing the relative hydrophobic/hydrophilic block lengths to obtain diverse morphologies of the block copolymer has been used to obtain “Crew-Cut” aggregates with multiple morphologies via tuning of the hydrophilic part of polystyrene-block-polyacrylic acid (PSt-b-PAA),39 and this method shows potential for forming diverse architectures of copolymer by means of hydrophobic interactions. In addition, this type of self-assembly into micelles, vesicles and so on has an apparent resemblance to cells, because hydrophilic regions can form in a hydrophobic matrix which is surrounded by a hydrophilic surface. So according to Scheme 1(B), three types of the linear block copolymers PDMAEMAn–SS–PCLm with diverse hydrophobic/hydrophilic ratios were prepared through ring-opening polymerization (ROP) between PDMAEMA–SS–OH and ε-caprolactone, and their chemical structure and composition were confirmed by 1H NMR (see Fig. 1). Characteristic peaks of the PCL segments in the block copolymers were easily recognized, which presented at g, h, f and i. Moreover, on the basis of the integration of the (c + i) peak and (f + a) peak, the specific value of (3m + n):(m + n) was first obtained for each sample, and on the basis of the Mw of the initial PDMAEMA–SS–OH, then the relative value m:n of the hydrophobic/hydrophilic block lengths in the amphiphilic molecules was estimated, which agreed with the value of Mw measured by GPC (see Table 1).
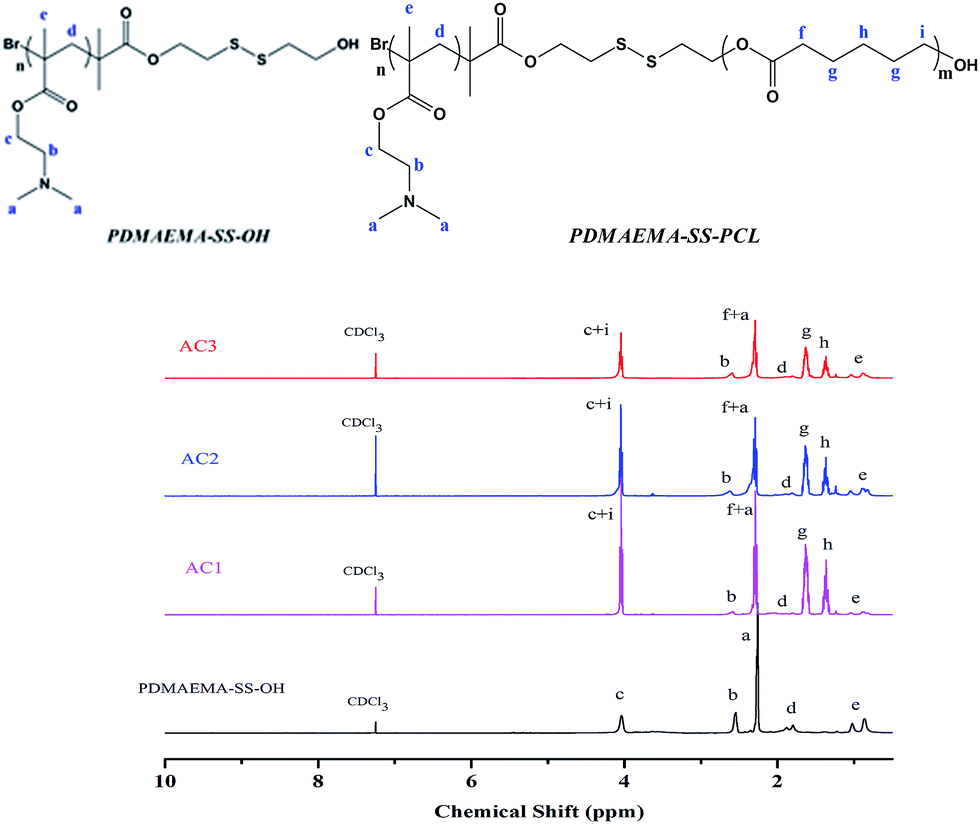 | ||
| Fig. 1 1H NMR spectra of PDMAEMAn–SS–PCLm with diverse hydrophobic ratios for AC1, AC2 and AC3 compared with that of the initial PDMAEMA–SS–OH. | ||
| Sample | m:na |
Br–ONB–Brb | Mwc | Dhd (nm) | DTe (μm) | Zeta (mV) | CAC (mg mL−1) | Morphology |
|---|---|---|---|---|---|---|---|---|
| a m:n is the hydrophobic/hydrophilic segments ratio calculated based on 1H NMR and GPC.b Photodegradable cross-linker.c Measured by GPC.d Measured by the DLS method as shown in Fig. S3.e Obtained from TEM images shown in Fig. S5. |
||||||||
| AC1 | 555:132 |
None | 88123 |
403.7 | 0.336 | 53.9 | 0.182 | Micelle |
| AC2 | 264:132 |
None | 51994 |
389.3 | 0.386 | 25.5 | 0.191 | Micelle |
| AC3 | 158:132 |
None | 36637 |
387.2 | 0.395 | 55.3 | 0.209 | Hollow micelle |
| x-AC1 | 555:132 |
Yes | — | — | 1.50 | 17.5 | 0.093 | Microsphere |
| x-AC2 | 264:132 |
Yes | — | — | 3.04–4.08 | 9.3 | 0.091 | Branched micelle |
| x-AC3 | 158:132 |
Yes | — | — | 3.22–7.26 | 16.0 | 0.079 | Vesicle |
Because its ester bond can be easily cleaved by UV light irradiation, o-nitrobenzyl ester has been used as a photosensitive protective group for carboxylic acids in organic synthesis40 and used to prepare photoresist materials.41 Recently, more attention has been paid to these kinds of photosensitive compounds in the fields of photoresists for nanolithography,42 photo-stimuli-responsive materials for controlled release43,44 and stimuli-regulated “traceless” crosslinking.45 In our previous work, a novel photodegradable cross-linker Br–ONB–Br based on o-nitrobenzyl ester was synthesized, and we found that it had an excellent stimuli-responsiveness to UV light irradiation in the releasing experiment.25 In this work, the cross-linker Br–ONB–Br was synthesized with a higher yield using an improved method as shown in Scheme 1(A), and the 1H NMR spectra of the precursor 5-hydroxy-2-nitrobenzyl alcohol and Br–ONB–Br are given in Fig. S1 and S2.† The linear PDMAEMA–SS–PCL was cross-linked by Br–ONB–Br in THF via a quaternisation reaction between the bromine and tertiary amine groups of the DMAEMA unit. After 24 h of reaction, the solution was placed in a dialysis bag to perform the self-assembly through changing the medium from THF to H2O and purifying by removing unreacted Br–ONB–Br simultaneously.27,42 As shown in Fig. 2, the peaks belonging to Br–ONB–Br appeared on the FTIR spectra of the cross-linked polymers, indicating that the photodegradable cross-linker actually makes up a new segment between a couple of PDMAEMA–SS–PCL chains.
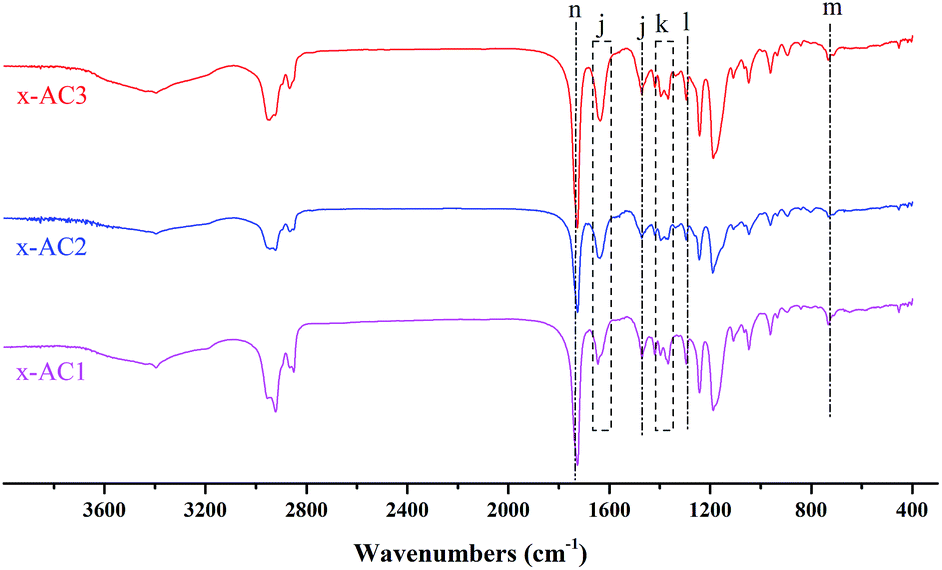 | ||
Fig. 2 FTIR spectra of the cross-linked PDMAEMAn–SS–PCLm. (j) 1645 cm−1, 1471 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of benzene), (k) 1367 cm−1 (NO stretching of –NO2), (l) 1296 cm−1 (C–N stretching) (m) 733 cm−1 (C–H bending of ortho-substituted benzene) and (n) 1725 cm−1 (CO stretching). C stretching of benzene), (k) 1367 cm−1 (NO stretching of –NO2), (l) 1296 cm−1 (C–N stretching) (m) 733 cm−1 (C–H bending of ortho-substituted benzene) and (n) 1725 cm−1 (CO stretching). | ||
Comparison of the microstructures self-assembled from linear and cross-linked PDMAEMA–SS–PCL chains
In order to confirm the influence of the cross-linking, the self-assemblies of the linear PDMAEMA–SS–PCL (sample AC1, AC2 and AC3) were carried out under the same conditions as the cross-linked ones, and their colloidal property and morphology were then characterized. DLS analysis (see Table 1 and Fig. S3†) indicated that the diameter of the micelles self-assembled from the linear PDMAEMA–SS–PCL was about 400 nm, which agreed with the TEM images (see Scheme 2: AC1, AC2, AC3).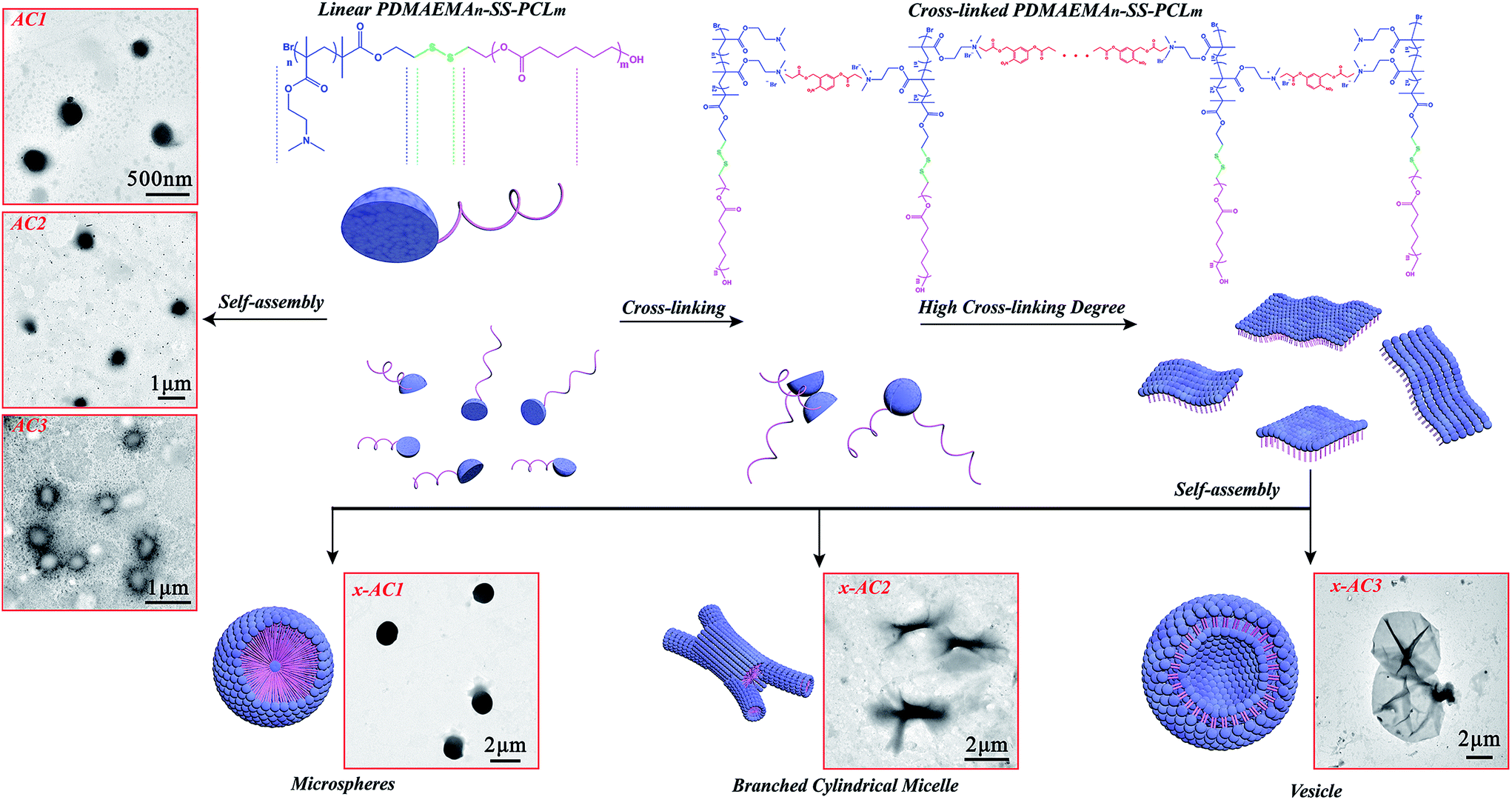 | ||
| Scheme 2 Self-assembly process of the cross-linked PDMAEMA–SS–PCL chains with different hydrophobic/hydrophilic ratios. | ||
The amphiphilic chains of AC3 might be self-assembled on the surface of THF droplets like in the process of styrene emulsification as reported before,46 in which a similar copolymer PCL–SS–PDMAEMA with a ratio of hydrophobic/hydrophilic lengths similar to the value of AC3 was synthesized and used as surfactant to prepare PS nanoparticles, so when the THF core was removed by the dialysis in water, the hollow micelles appeared (Scheme 2: AC3). As Table 1 and Fig. S4† indicate, the linear sample shows a higher zeta potential than the corresponding cross-linked one, and it may have been caused by the quarternisation reaction in the cross-linking step, in which the number of positive charges on the surface of the self-assembled microstructures decreased.
The self-assemblies of the cross-linked PDMAEMA–SS–PCL copolymers were very different from those of the linear ones, and specific microstructures (see Scheme 2: x-AC1, x-AC2, x-AC3) with morphologies such as microspheres, branched cylindrical micelles and vesicles were generated. The cross-linked PDMAEMA–SS–PCL chains could be restricted in one dimension because of the covalent interaction between the PDMAEMA–SS–PCL chain and Br–ONB–Br, so their aggregations depended not only on the hydrophobic interaction, but also on the covalent bond among every amphiphilic chain. It is clear that the diameters of the self-assembled microstructures from the cross-linked copolymers were greater than those from the linear ones. As indicated in Fig. S5,† the microspheres were about 1.5 μm in diameter, and the branched cylindrical micelles were 3–4 μm in one dimension but only 250–400 nm in the middle areas. The vesicles were not very homogeneous, and the diameters ranged from about 3.5 μm to 7.2 μm, which might result from the different cross-linking levels. In order to understand the formation mechanism of different microstructures, the critical aggregate concentration (CAC) was measured by the conductivity titration method47 (see Table 1 and Fig. S6†). Because of the cross-linking, the molecular weight and the original particle volume of PDMAEMA–SS–PCL increased, which certainly enhanced the probability of aggregation.48,49 The CAC values of the cross-linked PDMAEMA–SS–PCL solutions were obviously lower than that of the corresponding linear ones, and since the concentrations of the PDMAEMA–SS–PCL solutions were all beyond their CAC values, there is no doubt that the self-assembled copolymer chains existed in the system.
Based on the above results, the formation mechanisms of the different self-assembed microstructures are proposed. As shown in Scheme 2, the PDMAEMA segments of the PDMAEMA–SS–PCL chains could be covalently linked by Br–ONB–Br in THF which is a good solvent for both blocks, and ultimately formed into single layers like two-dimensional materials when the cross-linking reached a certain degree. The change of dissolving medium might drive them into three-dimensional shapes,50 like the solvent-responsive bilayer microstructures, the self-folding of which could be triggered by solvent after certain cross-linking as reported before.51,52 So, the sample x-AC1 with the longest hydrophobic chain could be self-assembled from single layers into a microsphere with one or numerous hydrophilic cores, like the reported large compound micelles in which the hydrophilic segments formed the corona of micelles and the core consisted of numerous reverse micelles with hydrophilic islands in continuous hydrophobic phase.53–55 In this study, the existence of hydrophilic cores was proved by encapsulating hydrophilic Rhodamine B (RhB) into the microspheres (see Fig. S7†). As for the branched cylindrical micelle originating from x-AC2, its formation process is similar to that of a reported poly(ferrocenyldimethylsilane)-b-poly(2-vinylpyridine) system.56 After the cross-linking, these single layers were not homogeneous, and the layers with a high degree of cross-linking formed a thicker core in the middle. Subsequent growth of multiple thinner cores by the other layers occurred to form a branched cylindrical architecture. Like the system of the similar amphiphilic polylactide-block-poly(N-isopropyl acrylamide) (PLA-b-PNIPAAm) shell-cross-linked by hexanediol diacrylate,57 the sample x-AC3 in which the hydrophilic shell was cross-linked by Br–ONB–Br could also aggregate into vesicles by hydrophobic interactions. Since different cross-linking levels led to different swelling behaviour in aqueous media,57 x-AC3 showed disparate morphology compared with uncross-linked AC3. Because of the cross-linking process, the self-assembled units of x-AC3 were much bigger than those of the uncross-linked system (AC3), resulting in the formation of larger vesicles than common ones.58 Moreover, by comparing x-AC3 with x-AC1, the sample x-AC3 was shown to have more tertiary amine groups for cross-linking, which made the initial amphiphilic layer much larger before the self-assembly process, and as a result, the vesicles were much larger than the microspheres self-assembled from x-AC1.
This phenomenon of the changes between the diverse morphologies is consistent with the packing parameter, P = v/(al), which was introduced by Israelachvili59 and utilized to predict the resulting morphologies, where (for an amphiphilic AB diblock co-polymer) v is the volume of the hydrophobic block, l is the chain length normal to the interface, and a (a = 3v/R, R is the packing radius) is the interfacial area between two blocks.59,60 It is generally believed that for the self-assembly of amphiphilic diblock copolymer, the copolymers with P < 1/3 will form spherical micelles, and those with 1/3 < P < 1/2 will favor the formation of cylindrical micelles, whereas when 1/2 < P < 1, enclosed membrane structures (vesicles, also known as polymersomes) will be preferred.59,61–63 In this study, as the length of PCL segment decreased, the values of l and v both decreased, but according to a = 3v/R, with an increase in R caused by the cross-linking process, a is reduced more drastically than v. Thus, the value of P increased with the length of the PCL segment decreasing and as results, the sample x-AC1 with the longest PCL segments self-assembled into a spherical shape, the sample x-AC2 with PCL segments of medium length preferred to form a branched architecture, and the sample x-AC3 with the shortest PCL segments self-aggregated into a vesicle structure.51,64
Reduction- and light-responsive properties of the microstructures self-assembled from cross-linked PDMAEMA–SS–PCL chains
As mentioned above, the disulfide bond and the photodegradable linkage in the amphiphilic PDMAEMA–SS–PCL copolymers could endow the self-assembled architectures with reduction- and light-responsive properties. To verify the reduction-responsive property, DL-dithiothreitol (DTT), a disulfide bond reducing agent, was used to treat the three microstructures at the same concentration for different lengths of time.As Fig. 3(a1–a3) indicates, the microspheres seemed to be destroyed in comparison with those of the untreated sample (see Scheme 2: x-AC1). Area I and II in Fig. 3(a1) were in an incomplete reduction process because of a limited treatment time of 2 days. Area I shows the initial stage that DTT molecules started to penetrate into the microstructure, and area II with the central core was closer to the final state in which the central core still existed but the edge was destroyed into small irregular fragments like area III (magnified in Fig. 3(a2)). When the treatment time was prolonged to 5 days, all of the microspheres were completely broken into small pieces (see Fig. 3(a3)) as in area III. For the branched cylindrical microstructure, the destruction first occurred in the two terminal parts of the branched micelle (Fig. 3(b1)), and the amplification of area IV is given in Fig. 3(b2). After 5 days treatment, most of the architectures were broken into pieces (Fig. 3(b3)), and only a few architectures were incompletely destroyed, leaving a small central part of the structures (see Fig. S8†). Furthermore, from the TEM images and DLS analysis (see Fig. S9†), we can see that the destruction occurred more violently for the branched micelles. After 5 days reduction, the size of the branched micelles decreased sharply to 200 nm, while the size of the microspheres only decreased from about 1.5 μm to 488 nm. This significant difference mainly came from the distinction of the intensity of the hydrophobic interactions based on the length of the hydrophobic chain, and another incentive was that the scale of the branched micelle microstructures was only about 250–400 nm in one dimension (see Fig. S5†), which was significantly less than the diameter of microspheres and made the branched micelles break down more thoroughly. The destruction of the vesicles was similar to that of the branched micelles. During the reduction process, small pieces were gradually separated from the microstructures (see Fig. 3(c1)) and the enlarged image of area V is shown in Fig. 3(c2), but the framework still existed in this process. When the treatment time increased to 5 days, most of the pieces became dissociative fragments away from the framework (see Fig. 3(c3)), and the size of the degradative vesicles was around 600–1000 nm with a wide size distribution (see Fig. S9†), which is much smaller than the original.
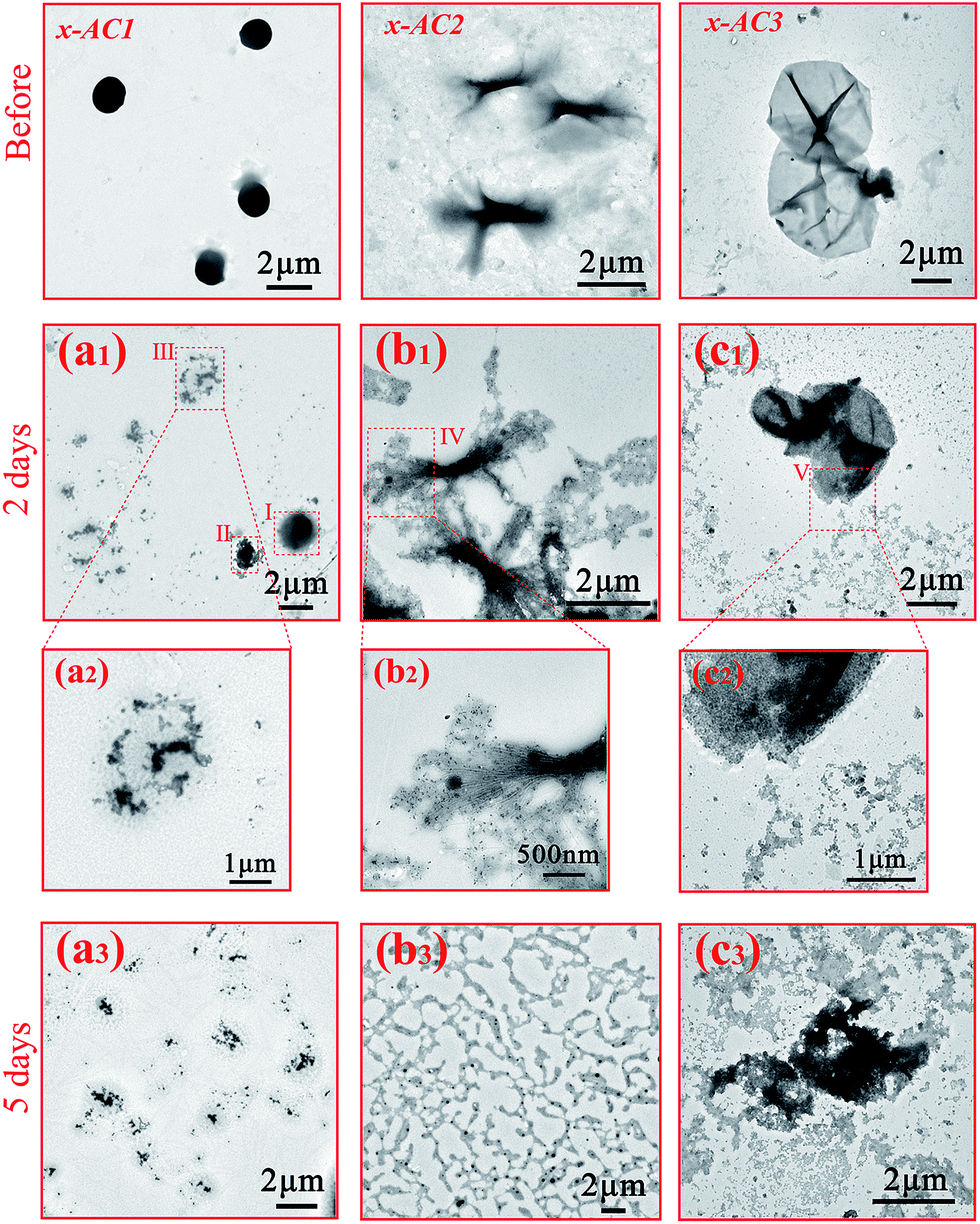 | ||
| Fig. 3 TEM images of the cross-linked PDMAEMA–SS–PCL treated with DTT for different lengths of time: (a1) and (a2) x-AC1, 2d, (a3) x-AC1, 5d; (b1) and (b2) x-AC2, 2d, (b3) x-AC2, 5d; (c1) and (c2) x-AC3, 2d, (c3) x-AC3, 5d. | ||
The light responsive property of the cross-linked architectures was evaluated by UV light irradiation for 0.5 h, and the results are given in Fig. 4(a)–(c). It is obvious that the UV stimulation didn’t cause dramatic destruction like the reduction stimulation, and some fragments appeared on the edges of the microspheres and on the tail of the branched micelles in one dimension after the irradiation (see Fig. 4(a), (b) and S10(a), (b)†). It was noted that the vesicles were broken into a number of lamellas (see Fig. 4(c) and S10(c)†), indicating that they were much easier to be destroyed by the UV light than other two architectures because of the different hydrophilic block lengths.
 | ||
| Fig. 4 TEM images of the cross-linked PDMAEMA–SS–PCL before and after UV light irradiation: (a) x-AC1 (b) x-AC2 (c) x-AC3. | ||
So as shown in Scheme 3, the reduction stimulation might destroy the –S–S– linkage between the hydrophobic and hydrophilic segments, and as a result, the hydrophilic chain is broken from the microstructures and dispersed or even dissolved in the aqueous phase.65 However, the UV light irradiation just cleaves an ester bond of the cross-linking structures, like scissors “cut” out some parts of the linking “wires” on the surface.66 Based on the different results between the two responsivenesses, this might provide a new system to release drugs in diverse intensities or a selective releasing system for disparate drugs.
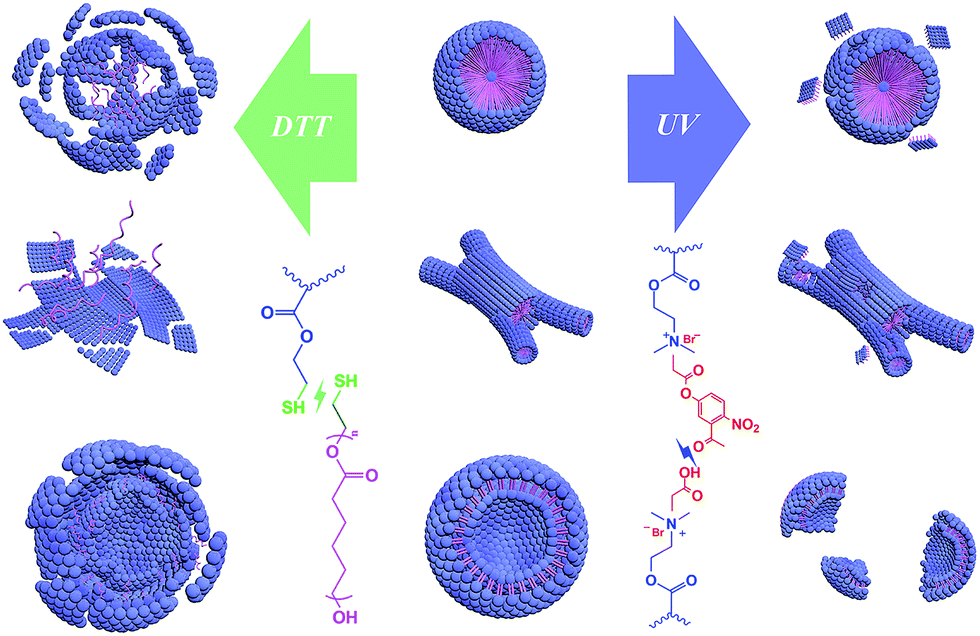 | ||
| Scheme 3 Schematic diagram for the destruction of three microstructures by reduction- and light-stimulations. | ||
Conclusions
In summary, we have designed and demonstrated a novel simple amphiphilic cross-linked PDMAEMA–SS–PCL system to controllably obtain three different microstructures including microspheres, branched cylindrical micelles and vesicles. The three types of microstructures were obtained just by varying the hydrophobic/hydrophilic ratios and cross-linking, and the results show that they were all responsive to reductant and UV light stimulation via two different mechanisms. This may provide a fresh idea for preparing smart systems which will be responsive to different stimulations and the three different morphologies may help us to pick out the best performance shape in specific applied scenarios. Combining the pH and temperature responsiveness of the PDMAEMA moiety and the enzymatic degradation property of the PCL moiety, this smart polymeric system can even have quintuple stimuli-responsive properties which we will continue to use for drug delivery and release in our follow-up research and we believe this system will meet a lot of requirements for its various morphologies and stimuli-responsiveness.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Key Research and Development Program of China (2017YFD0200704) and the Young Elite Scientists Sponsorship Program by CAST (2017QNRC001).Notes and references
- M. A. Stuart, T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. T. Tsukruk, M. Urban, F. Winnik, S. Zauscher and I. Luzinov, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef PubMed.
- Y. Zhu, B. Yang, S. Chen and J. Du, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef.
- N. Rapoport, Prog. Polym. Sci., 2007, 32, 962–990 CrossRef.
- S. Ahn, R. M. Kasi, S. C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 2012, 64, 72–82 CrossRef.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef PubMed.
- B. Deng, P. Ma and Y. Xie, Nanoscale, 2015, 7, 12773–12795 RSC.
- M. H. Li and P. Keller, Soft Matter, 2009, 5, 927–937 RSC.
- J. A. Champion, Y. K. Katare and S. Mitragotri, J. Controlled Release, 2007, 121, 3–9 CrossRef PubMed.
- N. P. Truong, W. Gu, I. Prasadam, Z. Jia, R. Crawford, Y. Xiao and M. J. Monteiro, Nat. Commun., 2013, 4, 1902 CrossRef PubMed.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- A. Kakkar, G. Traverso, O. C. Farokhzad, R. Weissleder and R. Langer, Nat. Rev. Chem., 2017, 1, 0063 CrossRef.
- Y. Li, J. Lin, Y. Huang, Y. Li, X. Yang, H. Wu, S. Wu, L. Xie, L. Dai and Z. Hou, ACS Appl. Mater. Interfaces, 2015, 7, 25553–25559 Search PubMed.
- S. Qin, A. Zhang, S. Cheng, L. Rong and X. Zhang, Biomaterials, 2017, 112, 234–247 CrossRef PubMed.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- T. Ramasamy, H. B. Ruttalaa, B. Guptaa, B. K. Poudela, H. Choic, C. S. Yonga and J. O. Kima, J. Controlled Release, 2017, 258, 226–253 CrossRef PubMed.
- X. Li, C. Liu, S. Wang, J. Jiao, D. Di, T. Jiang, Q. Zhao and S. Wang, Mater. Sci. Eng., C, 2017, 71, 594–603 CrossRef PubMed.
- X. Wu, Y. J. Tan, H. T. Toh, L. H. Nguyen, S. H. i. Kho, S. Y. Chew, H. S. Yoon and X. Liu, Chem. Sci., 2017, 8, 3980–3988 RSC.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- K. Kita-Tokarczyk, J. Grumelard, T. Haefele and W. Meier, Polymer, 2005, 46, 3540–3563 CrossRef.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef PubMed.
- Y. Xie, F. Yu, W. Tang, B. O. Alade, Z. Peng, Y. Wang, J. Li and D. Oupický, Macromol. Biosci., 2017, 1700194 Search PubMed.
- A. J. Convertine, C. Diab, M. Prieve, A. Paschal, A. S. Hoffman, P. H. Johnson and P. S. Stayton, Biomacromolecules, 2010, 11, 2904–2911 CrossRef PubMed.
- E. Murray, B. C. Thompson, S. Sayyar and G. Wallace, Polym. Degrad. Stab., 2015, 111, 71–77 CrossRef.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef.
- S. Venkataraman, J. L. Hedrick, Z. Y. Ong, C. Yanga, P. L. Rachel, P. T. Hammond and Y. Yang, Adv. Drug Delivery Rev., 2011, 63, 1228–1246 CrossRef PubMed.
- Z. Q. Cao, X. T. Zhou and G. J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 28888–28896 Search PubMed.
- J. Yin, H. Hu, Y. Wu and S. Liu, Polym. Chem., 2011, 2, 363–371 RSC.
- C. Zheng, H. Gao, D. Yang, M. Liu, H. Cheng, Y. Wu and X. J. Loh, Mater. Sci. Eng., C, 2017, 74, 110–116 CrossRef PubMed.
- P. Charoongchit, J. Suksiriworapong, K. Sripha, S. Mao, A. Sapin-Minet, P. Maincent and V. B. Junyaprasert, Mater. Sci. Eng., C, 2017, 72, 444–455 CrossRef PubMed.
- A. O. Basar, S. Castro, S. Torres-Giner, J. M. Lagaron and H. T. Sasmazel, Mater. Sci. Eng., C, 2017, 81, 459–468 CrossRef PubMed.
- M. Basko, M. Bednarek, L. Vlaminck, P. Kubisaa and F. E. Du Prez, Eur. Polym. J., 2017, 89, 230–240 CrossRef.
- X. Fan, S. Jiang, Z. Li and X. J. Loh, Mater. Sci. Eng., C, 2017, 73, 275–284 CrossRef PubMed.
- Y. Xiang, N. N. Linn Oo, J. P. Lee, Z. Li and X. J. Loh, Drug Discovery Today, 2017, 22, 1318–1335 CrossRef PubMed.
- Z. Li and X. J. Loh, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 9, e1429 Search PubMed.
- X. Wang, S. S. Liow, Q. Wu, C. Li, C. Owh, Z. Li, X. J. Loh and Y. Wu, Macromol. Biosci., 2017, 17, 1700186 CrossRef PubMed.
- H. Cheng, Z. Wu, C. Wu, X. Wang, S. S. Liow, Z. Li and Y. Wu, Mater. Sci. Eng., C, 2018, 83, 210–217 CrossRef PubMed.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 Search PubMed.
- B. Amit, U. Zehavi and A. Patchornik, Isr. J. Chem., 1974, 12, 103–113 CrossRef.
- H. Barzynski and D. Sänger, Macromol. Mater. Eng., 1981, 93, 131–141 Search PubMed.
- L. Chen, Y. K. Goh, H. Cheng, B. W. Smith, P. Xie, W. Montgomery, A. K. Whittaker and I. Blakey, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4255–4265 CrossRef.
- J. Jiang, X. Tong, D. Morris and Y. Zhao, Macromolecules, 2006, 39, 4633–4640 CrossRef.
- Z. Cao, H. Wu, J. Dong and G. Wang, Macromolecules, 2014, 47, 8777–8783 CrossRef.
- X. Wang, G. Liu, J. Hu, G. Zhang and S. Liu, Angew. Chem., Int. Ed., 2014, 126, 3202–3206 CrossRef.
- Y. Zhang and H. Zhao, Polymer, 2015, 64, 277–284 CrossRef.
- C. Wang, S. Yin, S. Chen, H. Xu, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2008, 120, 9189–9192 CrossRef.
- M. J. Tizzotti, M. C. Sweedman, C. Schäfer and R. G. Gilbert, Food Hydrocolloids, 2013, 31, 365–374 CrossRef.
- N. Jain, S. Trabelsi, S. Guillot, D. McLoughlin, D. Langevin, P. Letellier and M. Turmine, Langmuir, 2004, 20, 8496–8503 CrossRef PubMed.
- C. Ye and V. V. Tsukruk, Science, 2015, 347, 130–131 CrossRef PubMed.
- M. H. Stenzel, X. Fan, J. Y. Chung, Y. X. Lim, Z. Li and X. J. Loh, ACS Appl. Mater. Interfaces, 2016, 8, 33351–33370 Search PubMed.
- J. Guan, H. He, D. J. Hansford and L. J. Lee, J. Phys. Chem. B, 2005, 109, 23134–23137 CrossRef PubMed.
- C. Feng, G. Lu, Y. Li and X. Huang, Langmuir, 2013, 29, 10922–10931 CrossRef PubMed.
- B. Xu, G. Gu, C. Feng, X. Jiang, J. Hu, G. Lu, S. Zhang and X. Huang, Polym. Chem., 2016, 7, 613–624 RSC.
- W. Jiang, Y. Zhou and D. Yan, Chem. Soc. Rev., 2015, 44, 3874–3889 RSC.
- H. Qiu, V. A. Du, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2013, 135, 17739–17742 CrossRef PubMed.
- M. Hales, C. B. Kowollik and T. P. Davis, Langmuir, 2004, 20, 10809–10817 CrossRef PubMed.
- J. Du and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 12800–12801 CrossRef PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef PubMed.
- D. S. Choi, M. S. Jhon and H. Eyring, J. Chem. Phys., 1970, 53, 2608–2614 CrossRef.
- C. Lu and M. W. Urban, Prog. Polym. Sci., 2017, 78, 24–46 CrossRef.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef PubMed.
- M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935–2946 CrossRef PubMed.
- X. Hua, F. Chen, N. Li and W. Bai, Polym. Degrad. Stab., 2017, 142, 51–66 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03374a |
| This journal is © The Royal Society of Chemistry 2018 |