
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new and informative [a,b,c,d] nomenclature for one-pot multistep transformations: a simple tool to measure synthetic efficiency
Satrajit Indu
and
Krishna P. Kaliappan
 *
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai-400076, India. E-mail: kpk@chem.iitb.ac.in
First published on 11th June 2018
Abstract
Domino, cascade and tandem reactions constitute the most efficient and creative chemical transformations with a huge domain of synthetic utility and applications. A number of reactions may be achieved in a single pot, accompanied by the formation of new rings and new bonds, leading towards higher molecular complexity. A lack of one unified, yet informative descriptor often understates the synthetic ingenuity of certain highly creative transformations. In this review, we propose a new tetra-coordinated [a,b,c,d] nomenclature which takes into account and displays the basic parameters which generally indicate the level of efficiency of a chemical transformation. An almost exhaustive set of one-pot multistep reactions may be described by this system and this review is an attempt to display the one-pot multistep transformations reported from our group and to classify them based on our proposed descriptor.
 Satrajit Indu | Satrajit Indu received his BSc degree from the erstwhile Presidency College in Kolkata in 2010 and then pursued his MSc in Chemistry at the Indian Institute of Technology Kanpur, from where he graduated in 2012. He then joined Indian Institute of Technology Bombay, Mumbai for his PhD under the supervision of Prof. Krishna P. Kaliappan where he has been working on the total synthesis of biologically active natural products and development of new synthetic methods. |
 Krishna P. Kaliappan | Krishna P. Kaliappan received his PhD (Prof. G. S. R. Subba Rao) in 1997 at Indian Institute of Science, Bangalore. Subsequently, he spent two years (97–98) each at University of Geneva (Prof. E. Peter Kündig) and at Duke University (99-00 with Prof. Michael C. Pirrung) for his post-doctoral studies before joining IIT Bombay as an Assistant Professor in 2001, where he is a full Professor since 2009. He served as Associate Dean (R & D) at IIT Bombay (2012–2015) & Head of Chemistry Department (2016–2018). He is currently Dean (Faculty Affairs) and Institute Chair Professor at IIT Bombay. He is on the Editorial Advisory Boards of Organic & Biomolecular Chemistry and Chemistry An Asian Journal since 2014. He is a Fellow of the Royal Society of Chemistry (FRSC) and Fellow of Indian Academy of Sciences, Bangalore (FASc). He has received Prof. C. N. R. Rao National Prize in Chemical Sciences, Swarnajayanti Fellowship and B. M. Birla prize in recognition of his research contributions and also received Excellence in Teaching award from IIT Bombay. His current research interests include total synthesis of natural products, domino strategy to synthesis of complex natural products and natural product like molecules. |
1 Introduction
Over the last few decades, one-pot multistep reactions have evolved into one of the major tools for synthetic organic chemists to execute the construction of highly complex molecular architectures.1 With the isolation of more and more new natural products of significant complexity every year, the need for assembling a complicated molecular framework using less resources and in a short time period has gained utmost importance. An ideal synthetic route demands: (i) quantitative yields, (ii) fewest possible steps, (iii) shortest possible reaction times, (iv) zero by-product formation and (v) easy purification. Of course, an ideal synthesis is far from realistic, but difficult as the planning and execution may be, many groups have succeeded in exhibiting reactions where a number of processes are wonderfully orchestrated into a single well-controlled sequence.With gradual increase in the number of one-pot, multistep and multicomponent reactions, there arises a definite need to define and classify them according to some general representative system. The terms “domino”, “cascade” and “tandem” have all been interspersed in their use to describe one-pot multistep reactions. Tietze defined “domino” reactions as “a process involving two or more consecutive reactions in which subsequent reactions result as a consequence of the functionality formed by bond formation or fragmentation in the previous step”.1a,1b In other words, any multistep process where the reaction conditions were altered after formation of a primary product would be counted as “consecutive reactions”.1b Denmark defined “tandem reactions” as an all-encompassing term to refer to reactions that occurred “one after the other” to classify cycloaddition reactions. He used the terms “cascade” (or domino), “consecutive” and “sequential” to determine how the two (or more) reactions follow.1d Nicolaou also described all the three terms as broadly interchangeable and used “cascade reactions” as the common descriptor.1e
In more recent times, the efficiency of a synthetic route is measured in terms of “pot economy”, “atom economy”, “step economy” and “redox economy” as summed up very precisely by Hayashi.2 “Atom economy”, a term first coined by Trost ensures that most of the material used in a process ends up getting utilized as part of the final product(s), which minimizes by-product formation.3 “Step economy” was introduced by Wender, which focuses on the elegance and creativity of a designed synthetic route.4 Lesser number of steps leads to lesser amount of materials used and less time and energy spent in the quest towards completing an “ideal synthesis”.4c “Redox economy” proposed first by Baran and Hoffmann reduces the unnecessary changes in oxidations states of isolable intermediates, which in turn reduces the number of steps.5 Finally “pot economy” reduces the number of intermittent work-up and purification processes as multiple reactions may be carried out in the same vessel.6
But even after all these terms have been defined and applied to reactions, there would always be some reactions which get excluded from a certain set or subset of such domains of scientific descriptors. Most of these terms would not encompass an exhaustive (or comparable to) set of one-pot, multistep reactions and would also fail to provide many of the significant details about the efficiency of the same. A very pertinent case in point would be the tandem inter- and intramolecular Diels–Alder reactions in the key step of Roush's enantioselective total synthesis of (−)-chlorothricolide (Scheme 1). Starting from an acyclic substrate 1, the formation of two different bicyclic units (a fused system and a spiro system) was accomplished in the same pot and under the same reaction conditions, in the presence of a chiral dienophile 2.7 Tietze in his book titled “Domino Reactions in Organic Synthesis” has classified the above transformation as a “tandem process”, rather than a “domino process” with a lot of clarity.1a
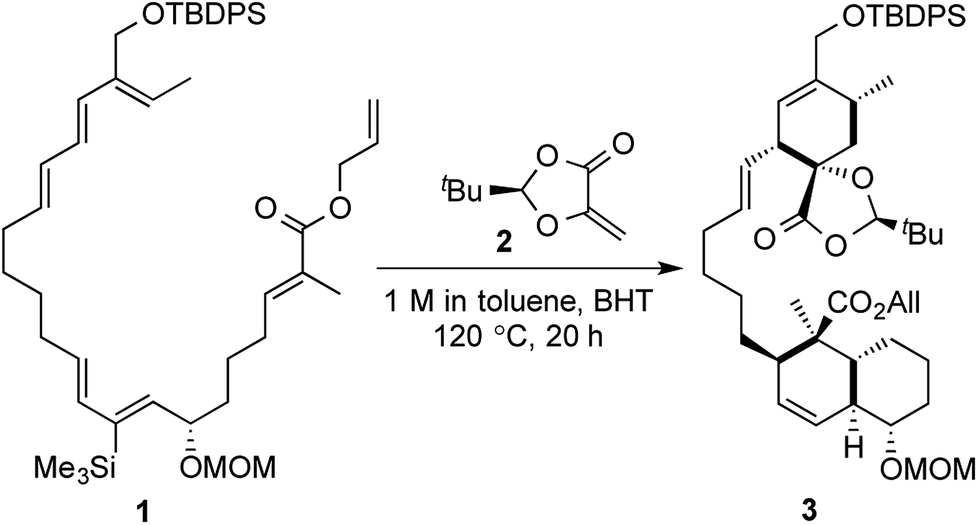 | ||
| Scheme 1 Roush's tandem inter- and Intramolecular Diels–Alder strategy towards (−)-chlorothricolide. | ||
Heathcock and co-workers devised another equally brilliant chemical transformation during their synthesis of daphnilactone A, where they assembled the hexacyclic core structure of the molecule in one-pot starting from a much simpler bicyclic intermediate.8a,8b
The diol 4 underwent oxidation to generate the corresponding dialdehyde, followed by condensation with ammonia to give the 2-azabutadiene intermediate 5. This was followed by a [4 + 2] cycloaddition and an aza ene-type cyclization to obtain the highly complex hexacyclic intermediate 7 (Scheme 2).8 While both of these ingenious transformations are “atom economic”, “pot economic” and “step economic”, unfortunately there is a lack of a unified technical descriptor, which can serve justice to the beauty and ingenuity of the overall transformations mentioned above.
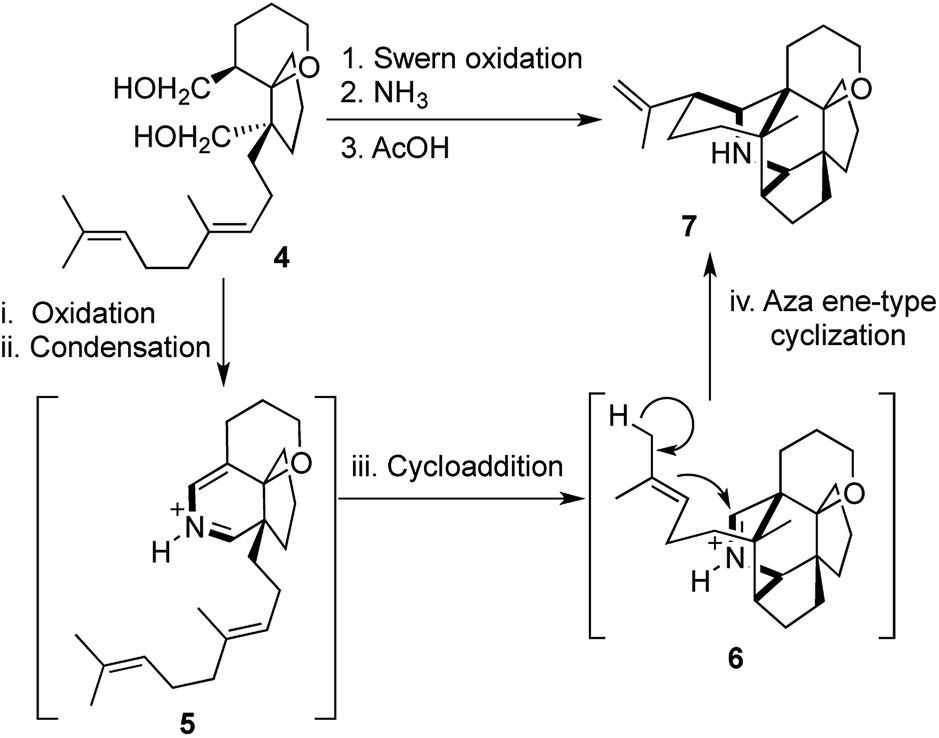 | ||
| Scheme 2 Heathcock's aza diene hetero Diels–Alder strategy towards daphnilactone A. | ||
Hence a general and more informative nomenclature is required to classify the plethora of multistep as well as multi-component reactions. This review would be directed towards the proposal of such a system where most one-pot reactions may be included, irrespective of the working field in which they are applied. Once introduced with clarity, we would classify a few examples of one-pot multistep reactions reported from our group according to this new nomenclature.
2 The [a,b,c,d] nomenclature for one-pot multistep reactions
We herein propose the use of a tetra-coordinated [a,b,c,d] nomenclature to describe multistep or even multi-component one-pot reactions, where the four alphabets denote the following:(a) Number of pots.
(b) Number of reactions taking place in one-pot.
(c) Number of rings formed in one-pot.
(d) Number of bonds formed in the same one-pot sequence.
The value of ‘(a)’ would generally be 1, as we are broadly interested in one-pot sequences, although for describing a multistep synthetic route, the same model might be extrapolated, but that would be outside the scope of this review. The number of distinct reactions ‘(b)’ taking place in the same pot would exclude the steps where a reactive intermediate might be formed as one of the “steps”. So, the formation of a carbocation or a reactive radical or the formation of an enolate would not count as a “reaction”, although it might involve bond breaking and/or bond making. A higher value of ‘(b)’ would automatically make a synthesis “step economic”. The value of ‘(c)’ gives a very good indication about the ingenuity of the reaction, as the formation of two or more rings in a single step would generally be a very commendable achievement. Lastly, the number of new bonds formed ‘(d)’ would also provide valuable information about the amount of complexity, which is generated in a particular step. Radical or cation mediated domino polyene cyclizations, domino metathesis reactions, cycloadditions, transition metal catalyzed multi-fold coupling, C–H activation or even reactions involving a combination of two or more of the above may be described by the [a,b,c,d] system of nomenclature. Even multi-component reactions, which are generally not considered under the domain of domino/cascade/tandem reactions, may be described efficiently through this nomenclature. In light of this new system, Roush's tandem inter- and intramolecular Diels–Alder transformation mentioned earlier may be highlighted as a [1,2,3,4] process (Scheme 1). Likewise for Heathcock's transformation from a bicyclic intermediate to the core structure of daphnilactone A, 4 new rings and 5 new bonds are formed, through a domino sequence of 4 reactions and this highly efficient one-pot transformation may be termed as a [1,4,4,5] process, thereby throwing light on the synthetic value of the transformation (Scheme 2). The high values of ‘(c)’ and ‘(d)’ underline the efficiency of these processes.
Nicolaou's one-pot total syntheses of the endiandric acids family also provide a monumental achievement in the field of cascade transformations to arrive at natural product scaffolds.9 The one-pot total syntheses involved thermally allowed pericyclic reactions using the mildest of reagents (Lindlar catalyst, H2 gas and heat), and the strategy was based on a hypothesis for their biosynthesis proposed by Black and co-workers.10 The polyunsaturated methyl ester containing a conjugated diyne with cis-oriented double bonds 8 underwent a thermal 8π electrocyclization followed by another thermal 6π electrocyclization to obtain a mixture of the interconvertible bicyclic compounds endiandric acid D and E methyl esters ([1,2,2,2] transformations). Endiandric acid E methyl ester 10 underwent another facile intramolecular Diels–Alder reaction with the pendant diene to form endiandric acid A methyl ester 12. The overall synthesis of endiandric acid A methyl ester involved the formation of 3 independent pericyclic reactions with the subsequent formation of 4 rings and 4 bonds, thereby classifying it as a [1,3,4,4] transformation (Scheme 3).
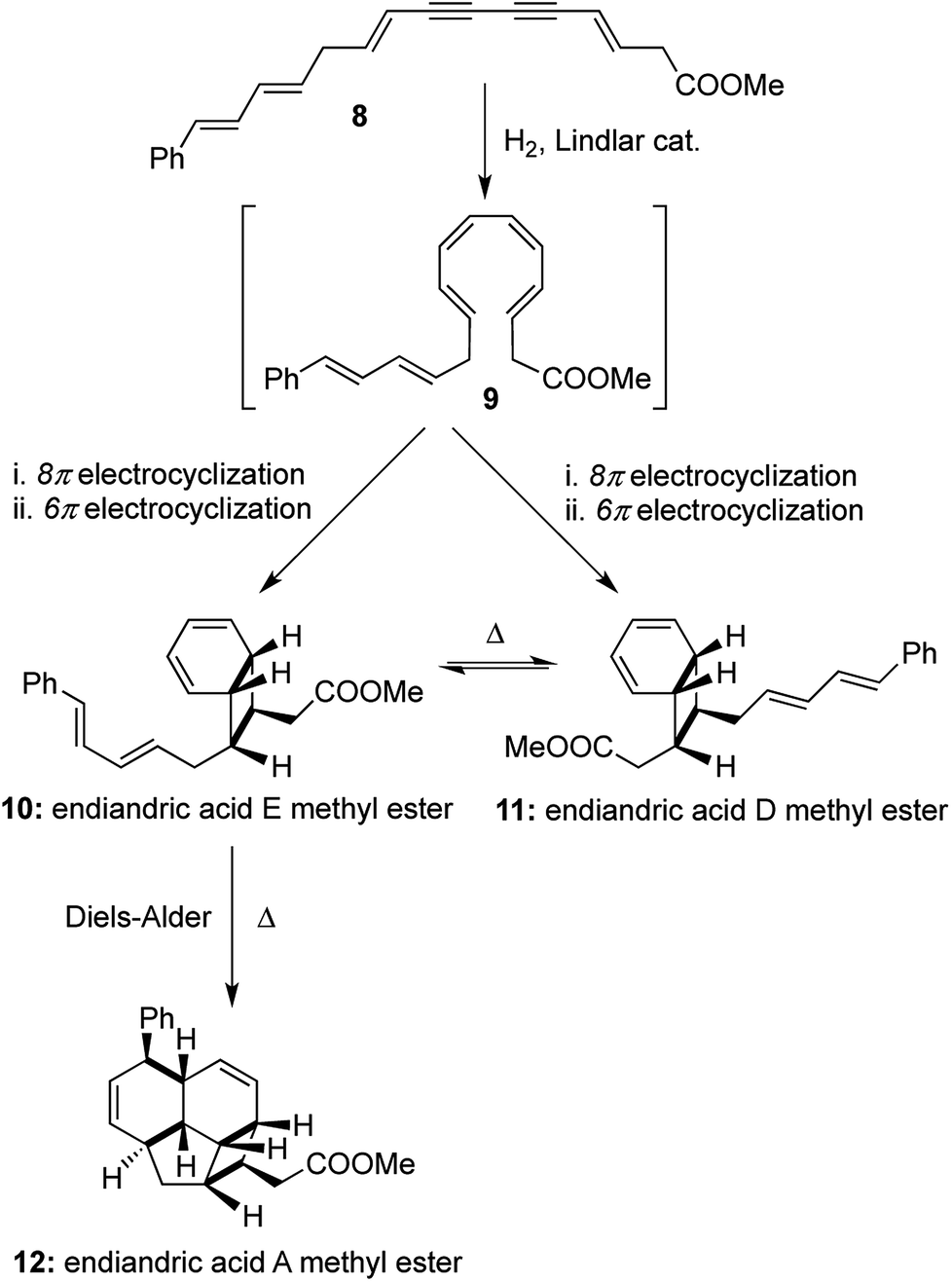 | ||
| Scheme 3 Nicolaou's total synthesis of endiandric acid A, D and E methyl esters. | ||
A similar strategy was followed with a higher vinylogous homologue of the conjugated diyne 13 to obtain an interconvertible mixture of endiandric acid F and G methyl esters ([1,2,2,2] transformations). Each of them underwent thermally allowed intramolecular Diels–Alder reactions to afford endiandric acid B methyl ester 16 and endiandric acid C methyl ester 17 respectively ([1,3,4,4] transformations). The excellent degree of stereocontrol in setting up of 8 new stereocenters in the one-pot transformations is indeed remarkable in its synthetic efficiency and operational simplicity (Scheme 4). Nicolaou's syntheses of endiandric acids along with the two earlier one-pot transformations cited here serve as very instructive examples for the application of our proposed [a,b,c,d] nomenclature, where high values of ‘(b)’, ‘(c)’ and ‘(d)’ equates to higher efficiency of a synthetic process. Also terming the one-pot syntheses of endiandric acid A, B and C methyl esters as [1,3,4,4] transformations adds more value to the ingenuity of the reactions than merely terming it as a “cascade” process.
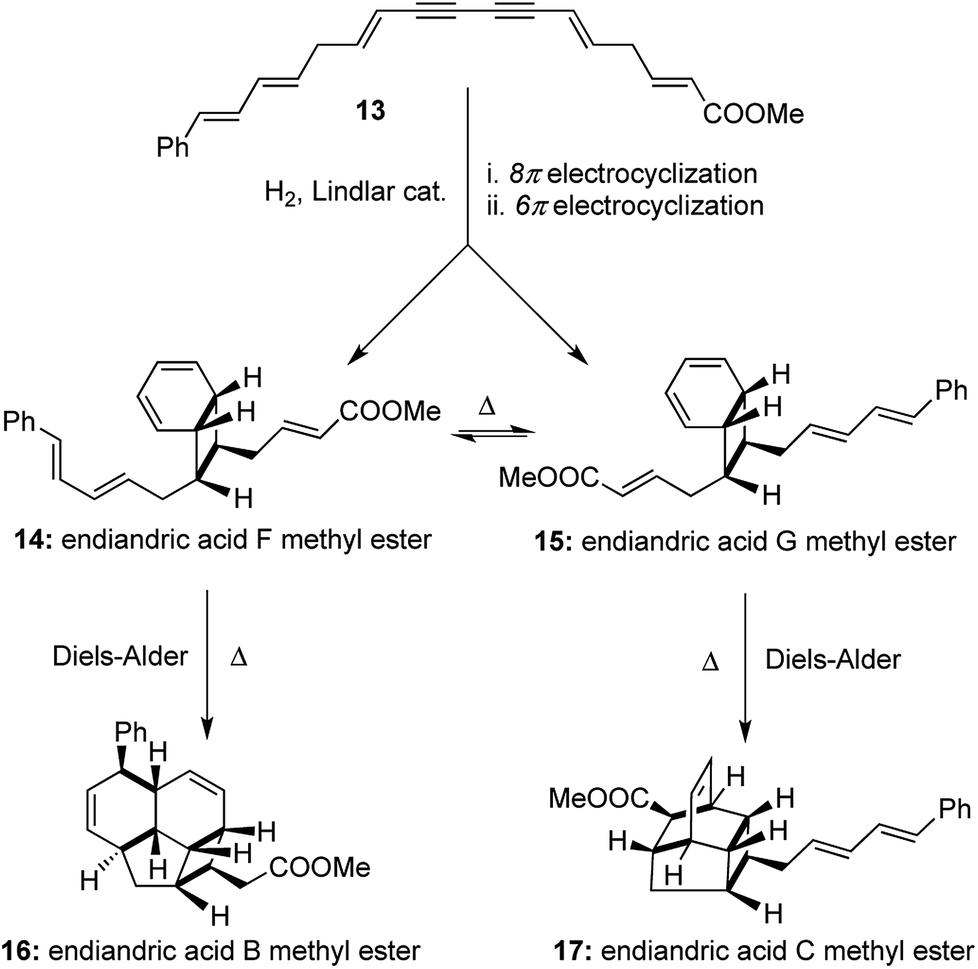 | ||
| Scheme 4 Nicolaou's total syntheses of endiandric acid B, C, F and G methyl esters. | ||
In general, synthetic efficiency is broadly measured by the number of steps involved in achieving a particular transformation and the respective yields of each of the steps involved therein. Subjective terms like “step economy” and “pot economy” sometimes tend to overlook the complexity that is achieved during a particular synthetic transformation. Although our proposed system does not take into account the yield of a particular transformation, but the tetra-coordinated nomenclature would provide a very good idea about the amount of complexity achieved through a particular chemical reaction. A low value of ‘(a)’ (preferably 1) suggests a “pot economic” process, while high values of ‘(b)’, ‘(c)’ and ‘(d)’ in most cases, would indicate a high degree of “step economy”. Most importantly, this provides a quantitative estimation about the synthetic process, which provides a better tool, in comparison to any existing parameter for describing synthetic efficiency.
Having defined this new system of nomenclature and cited a few classical examples from literature, we would like to classify a more or less exhaustive list of one-pot reactions reported from our group according to the [a,b,c,d] system. We would be focusing entirely on one-pot reactions and hence the values of ‘(a)’ would be 1 for all our examples cited in this review. While it is easy to count the number of reactions taking place in succession and identify the number of rings formed in the process, it might sometimes be a little confusing to identify all the new bonds formed during the one-pot transformation. In order to make things easier for the reader, we would highlight the new bonds formed during any process with bold bonds.
The key domains of organic synthesis where we have explored one-pot domino reactions may be listed as follows: (i) enyne metathesis (EM)/ring closing metatheses (RCM) cascades, (ii) tandem Enyne metathesis/Diels–Alder reactions, (iii) multifold transition metal catalyzed coupling (iv) transition metal catalyzed coupling followed by annulation. In addition to these, we would also sum up some other reaction combinations, which have been utilized in one-pot, domino fashion to facilitate complicated transformations.
3 Enyne Metathesis/RCM cascades
3.1. [1,2,2,2] transformations
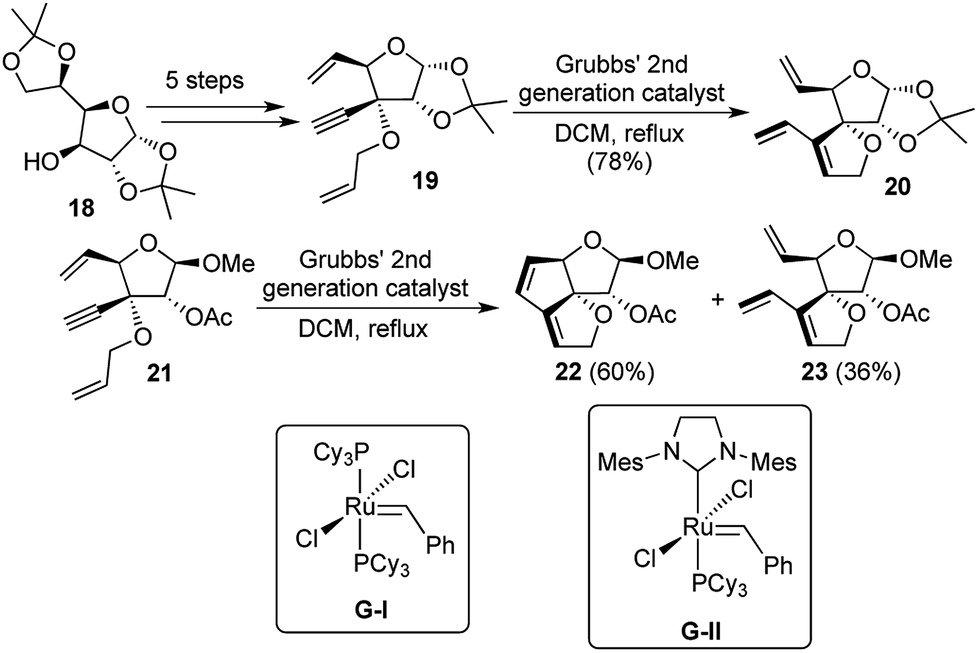
Polyquinanes, both angular as well as linear have always generated a lot of synthetic interest from natural product chemists, because of their aesthetically pleasing structures, besides exhibiting important biological activities. While the synthesis of carbocyclic triquinanes had a lot of literature precedence, there was a dearth of reports regarding the formation of their related oxa-analogues. In 2004, our group reported the use of sugar backbones to accomplish the synthesis of angularly fused dioxatriquinanes through a novel domino metathesis strategy.11Glucose diacetonide 18 was converted to the potential domino metathesis precursor 19 over 5 steps. But unfortunately all attempts to carry out the sequential EM/RCM transformation failed as the enyne metathesis product 20 was obtained as the sole product. At this point, it was envisaged that removal of the acetonide group might reduce the strain in the tetrahydrofuran sugar unit, which would bring the reacting double bonds closer to each other after enyne metathesis had been accomplished. To this end, the acetonide was solvolyzed under methanolic conditions, to generate a diastereomeric mixture of the methylated lactols. The major product of the two was separated and the 2-hydroxyl group was acylated to generate compound 21 as the precursor for the domino reaction.
Treatment of 21 with Grubbs' 2nd generation catalyst G-II (Grubbs' 1st generation catalyst G-I had provided only traces of the enyne metathesis product) under ethylene atmosphere could provide only the enyne metathesis product 23, while the domino reaction product 22 could be generated under argon atmosphere with 60% yield (Scheme 5). Results did not vary much upon changing the solvent from DCM to toluene. A closer look at the tricyclic product revealed that in the one-pot transformations, 2 new bonds were formed in each of the products. Hence the classifications may be described as [1,2,2,2] for 22 and [1,1,1,2] for 23. From the above studies, it was evident that solvent does not play a major role in the overall distribution of the two possible products, but the catalyst used and the reaction atmosphere made a huge difference in product selectivity.
 | ||
| Scheme 5 Domino enyne metathesis/RCM strategy towards angular dioxatriquinanes. | ||
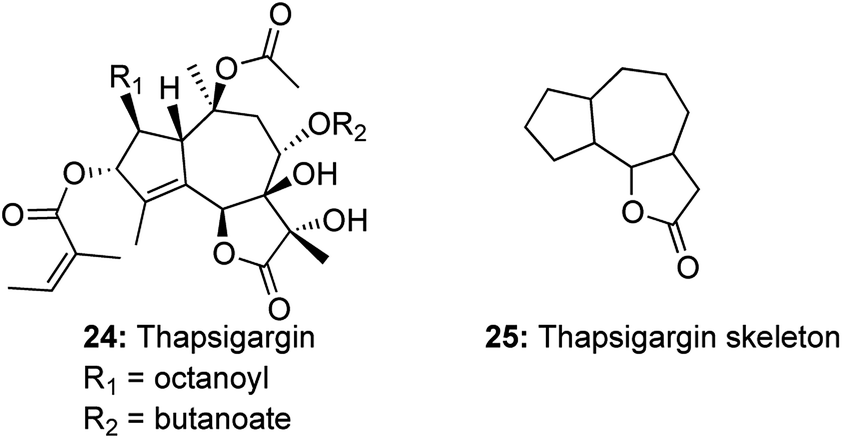
A similar domino metathetic strategy was utilized to prepare the thapsigargin skeleton starting from D-glucose diacetonide.12 Thapsigargins, a family of closely related guaianolides, isolated from the Mediterranean species Thapsia, exhibit a densely oxygenated tricyclic core, with more than 7 stereogenic centers and are functionalized with an array of acyl groups (Fig. 1). Thapsigargins were known to be potent histamine liberators and selective inhibitors of sarcoendoplasmic reticulum Ca2+ ATP dependent pumps (SERCAs).13 Thapsigargins also show remarkable specificity for the SERCA isozymes, and so became powerful tools to manipulate and study intracellular Ca2+-dependent signaling pathways.14 Thapsigargin 24 and its family of guaianolide natural products have aroused a lot of synthetic interest due to their remarkable biological activity and limited availability from natural resources.15
 | ||
| Fig. 1 Thapsigargin and its core structure. | ||
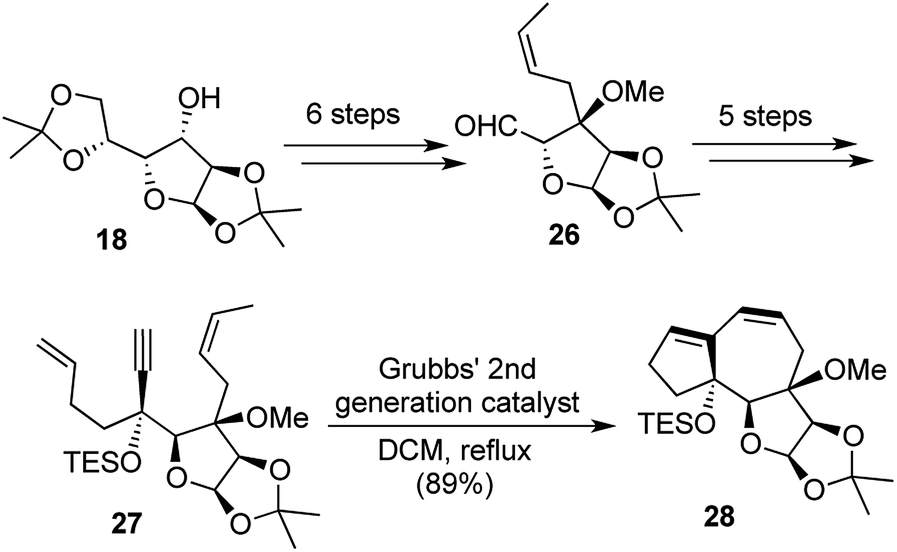
Our synthetic strategy commenced with glucose diacetonide and the domino EM/RCM precursor 27 was reached through a sequence of 11 steps. The precursor 27 when treated with G-I catalyst yielded 79% of the 5,7,5-fused tricyclic framework 28 over 10 h, whereas the use of G-II facilitated the transformation further with an excellent yield of 89% in just 5 h (Scheme 6).
 | ||
| Scheme 6 Domino enyne metathesis/RCM strategy towards the thapsigargin skeleton. | ||
Much like the one-pot synthesis of dioxatriquinanes earlier, this was a two-reaction sequence, accompanied by the formation of 2 rings and 2 new bonds formed during the transformation and hence this could be termed as a [1,2,2,2] strategy.
4 Enyne metathesis/Diels–Alder cascades
4.1. [1,2,2,4] transformations
Construction of motifs containing the bicyclo[5.3.1]undecene system 29 is a very useful and attractive prospect as it forms a part of many complex natural products including taxol 30 (Fig. 2).16 As a part of our research on the development of domino reactions in conjunction with metatheses, a very elegant enyne cross metathesis/intramolecular Diels–Alder (IMDA) strategy for the synthesis of this privileged[5.3.1]undecene scaffold was reported.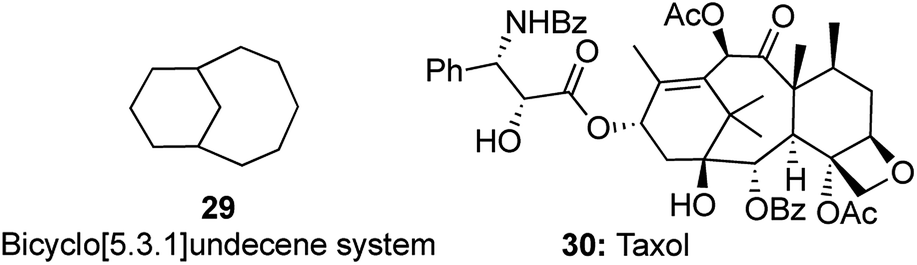 | ||
| Fig. 2 Taxol and its core structure. | ||
The key transformation was carried out on the enyne 31 using 10 mol% of G-II in toluene at 80 °C to obtain the desired bicyclic scaffold 33. The primary cross enyne metathesis formed two new bonds, while the sequential intramolecular Diels–Alder reaction formed two more new bonds in the process of constructing two new rings (Scheme 7).
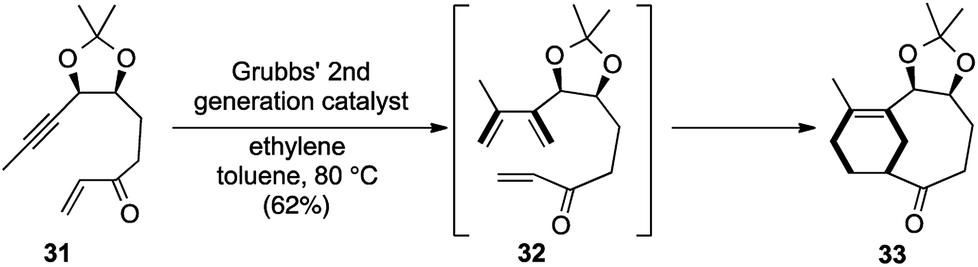 | ||
| Scheme 7 Domino cross enyne metathesis/Diels–Alder route to bicyclo[5.3.1]undecene framework. | ||
So, according to our proposed template of nomenclature, this could be classified as a [1,2,2,4] strategy. This was the first report of an intermolecular enyne metathesis in sequence with an IMDA reaction for the preparation of the AB ring system of taxol.17
In relation to our interest in the synthesis of taxol and its related structures,17,18 we also developed the design and synthesis of hybrid natural products related to taxol. Hybrid molecules are functional adducts generated from two or more natural or unnatural molecules, each retaining their own structural features.19 The various components of each entity may be modified or tuned to enhance their characteristic biological activities and hence the construction of such hybrid natural products opens up a lot of scope for structure activity relationship studies. We envisaged the construction of a taxa-oxa sugar hybrid core structure utilizing a domino enyne cross metathesis/Diels–Alder sequence.20
Starting from D-glucose diacetonide 18, the free hydroxyl was propargylated and the terminal alkyne was methylated in good yields to obtain the intermediate 34. A further 4-step sequence yielded the precursor 35 for the domino enyne metathesis/Diels–Alder transformation. Treatment of the methylated alkyne 35 with Grubbs' 2nd generation catalyst G-II under ethylene atmosphere triggered off the enyne cross metathesis and the intermediate diene reacted spontaneously with the pendant enone to form the Diels–Alder adduct 36 in 57% yield (Scheme 8).
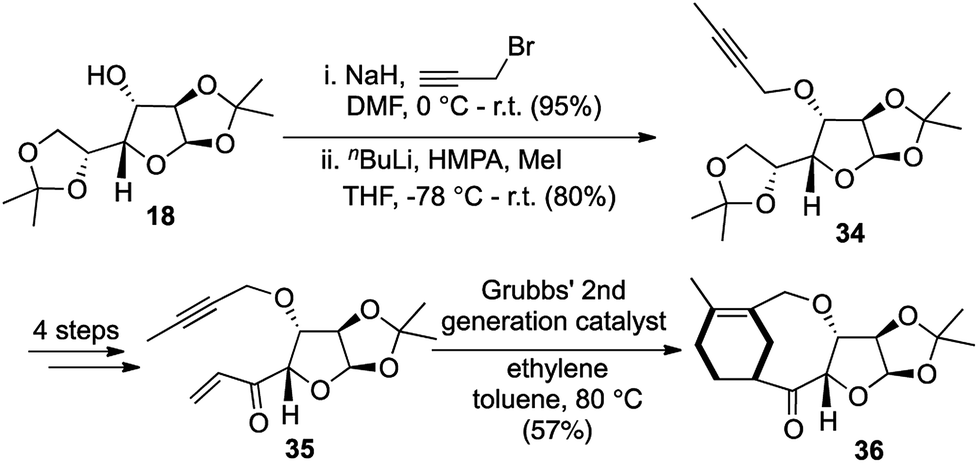 | ||
| Scheme 8 Cross enyne metathesis/Diels–Alder cascade for the construction of taxa-oxa sugar hybrids. | ||
This proved to be a highly general and efficient protocol for constructing taxa-oxa sugar hybrids, where the hybrids could be varied by the use of different sugar moieties. The key one-pot domino process involved two reactions resulting in the formation of two new rings and four new bonds, thereby being established as a [1,2,2,4] process.
4.2. [1,3,1,4] transformations
The C-aryl glycosides are an important variety of carbohydrates, where the sugar unit is attached directly to aromatic rings, which makes them much less susceptible to acid and enzymatic hydrolysis (Fig. 3).21 Much attention is generally given towards the construction of the key C–C bond to forge the sugar unit and the aromatic group, but the direct use of quinones via Diels–Alder/aromatization routes to set up the aromatic moiety was much less explored.22 So in 2007, our group reported a general strategy for the preparation of C-aryl glycosides utilizing an enyne cross metathesis of sugar derived alkynes 41 with ethylene followed by a Diels–Alder/aromatization sequence.23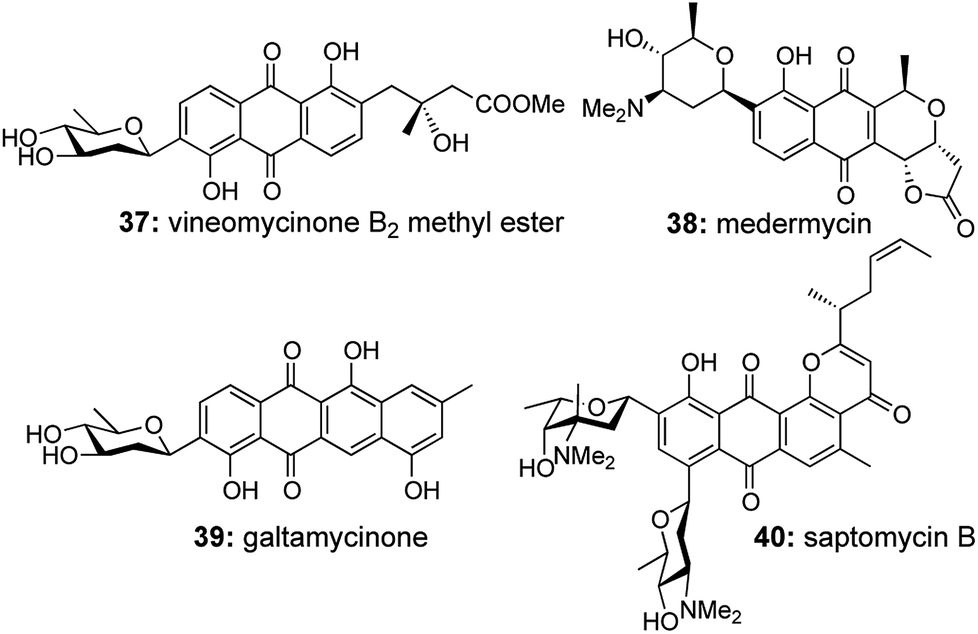 | ||
| Fig. 3 Naturally occurring C-aryl glycosides. | ||
While the standard conditions for carrying out the above mentioned transformation consisted of a 2-step sequence, a single attempt at the one-pot metathesis/DA/aromatization transformation using Grubbs' 2nd generation catalyst G-II under ethylene atmosphere also yielded the required C-aryl glycosides, albeit with lower yields (30–35%), in comparison to the 2-step sequence (more than 50% combined yields over 2 steps) (Scheme 9).
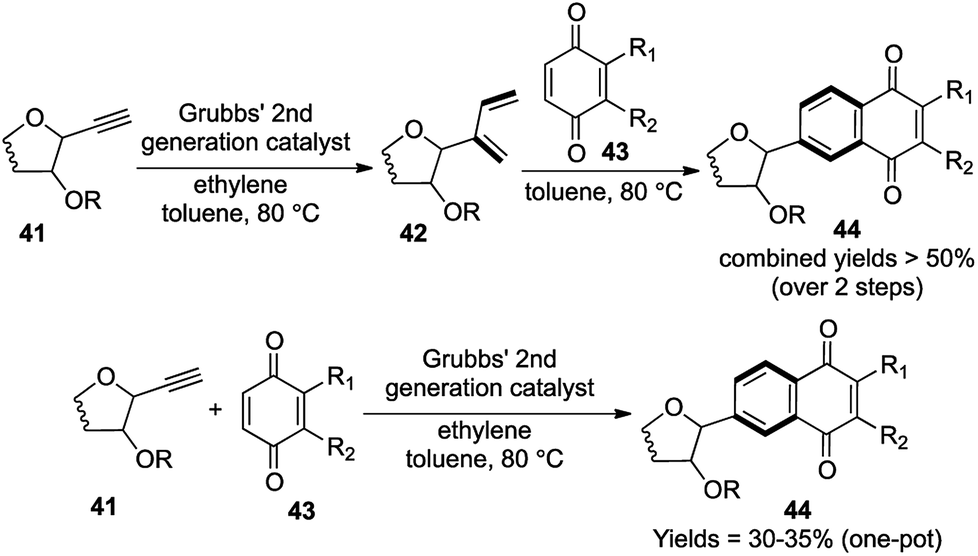 | ||
| Scheme 9 Domino cross enyne metathesis/Diels–Alder/aromatization route to C-aryl glycosides. | ||
A detailed analysis of the structural transformation demonstrates the formation of only 1 ring over the 3 successive reactions assisted by the formation of 4 new bonds (Scheme 9). So this versatile synthetic strategy towards the formation of C-aryl glycosides offering a lot of scope for diversification should be termed as a [1,3,1,4] strategy with respect to our proposed nomenclature.
4.3. [1,2,1,2] transformations
The growing utilities of enyne cross metathesis/Diels–Alder cascades led to a similar strategy being adopted for an efficient construction of 4-methylene-2-cyclohexanone moieties, which form an integral part of natural products like otteliones and loloanolides.24Otteliones 45 were isolated from the fresh-water plant Ottelia alismoides collected in the Nile Delta and they show impressive antitubercular activity and cytotoxicity at nM to pM levels against a panel of 60 human cancer cell lines.25 Loloanolides 46, 47 exhibit cytotoxicity against the HepG2 cell line, with GI50 values of nanomolar level.26 It is believed that these biological activities arise from the presence of the unique 4-methylene-2-cyclohexanone motifs in these natural products (Fig. 4).
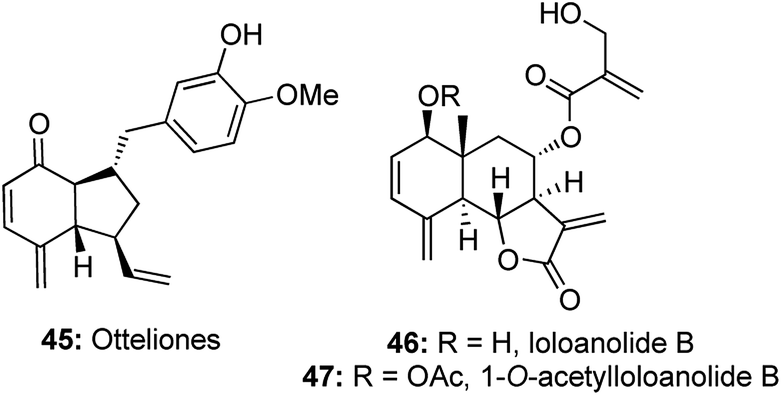 | ||
| Fig. 4 Otteliones and loloanolides. | ||
The synthetic journey started with the introduction of the triple bond through an addition of Li-acetylide on glucose diacetonide derived ketone. A sequence of 4 further steps provided the enyne precursor 48. Treatment with 8 mol% of G-II under ethylene atmosphere yielded the compound 40 with good yields for each of the differently functionalized tertiary alcohols. 2 subsequent reactions forming 1 ring and 2 new bonds, classifies this transformation as a [1,2,1,2] strategy (Scheme 10).
 | ||
| Scheme 10 Construction of 4-methylene-2-cyclohexenone scaffold. | ||
Having completed the construction of cis-fused 4-methylene-2-cyclohexanone scaffolds 50, a similar precursor was set up for the trans-fused scaffold in 9 steps from the glucose diacetonide derived ketone. Upon treatment with the same conditions as before, the trans-fused scaffold was obtained in 81% yield through another [1,2,1,2] strategy. The main significance of this strategy lies in the scope for diversification by employing different sugar units.
4.4. [1,2,3,4] transformations
The efficiency of utilizing a one-pot strategy reached its pinnacle when an intramolecular enyne metathesis followed by an intramolecular Diels–Alder (IMDA) reaction was developed to construct the tricyclic core structure of vinigrol in one step, starting from a completely acyclic molecule (Fig. 5).27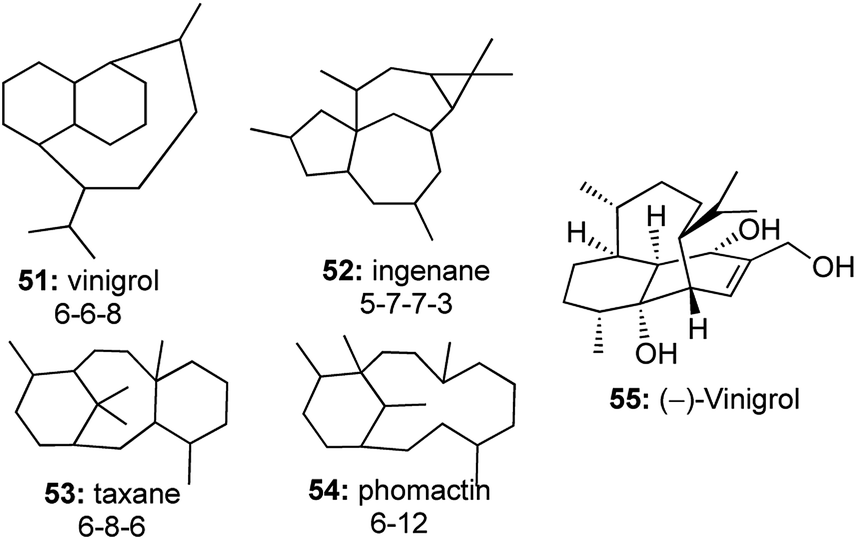 | ||
| Fig. 5 Vinigrol in comparison with other complex core structures. | ||
Vinigrol 55, a unique diterpene having a decahydro-1,5-butanonaphthalene skeleton forms a part of an elite class of diterpenoid molecules including taxanes, ingenanes and phomactins, which have posed the biggest challenge to synthetic chemists.28 Only Baran29 and Njardarson30 had completed the total synthesis of this daunting molecule, but there existed no enantioselective approaches to construct the tricyclic core structure of vinigrol.
Our synthesis started with the diastereoselective 1,4-addition of the Grignard reagent 57 on the known Michael acceptor 56.31 This was followed by the diastereoselective allylation of 58 followed by removal of the chiral auxiliary and introduction of the TMS-protected alkyne to generate the terminal olefin 60. Oxidative cleavage of the terminal olefin followed by Brown's allylation provided the chiral secondary alcohol 62. The primary alcohol protecting group was swapped from MOM to TBS and the secondary alcohol was pivaloylated to obtain the intermediate 64. A further sequence of 4 steps furnished the enone 65, which would be the precursor for the key step (Scheme 11).
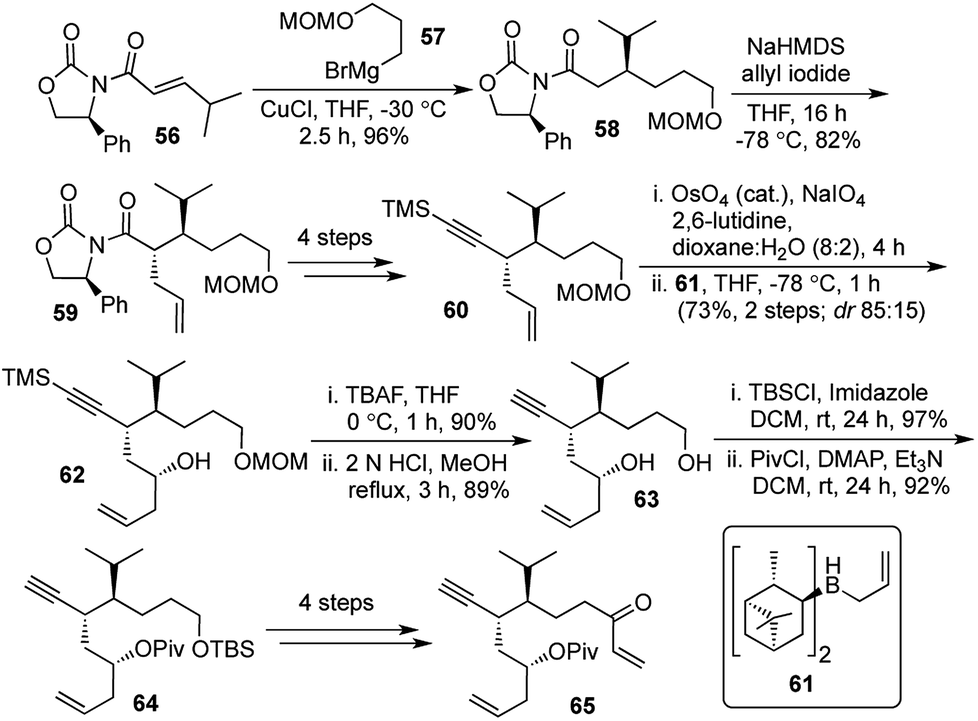 | ||
| Scheme 11 Synthesis of domino enyne metathesis/Diels–Alder precursor. | ||
Our key intermediate 65 was structurally designed so as to have an olefin and a triple bond positioned suitably for an intramolecular enyne metathesis and another enone moiety, which would be at a perfect orientation for the subsequent IMDA reaction with the resulting diene. From our earlier experience during the synthesis of angularly fused dioxatriquinanes, we had observed that the use of an ethylene atmosphere halted the reaction after the initial enyne metathesis and did not facilitate a further RCM in sequence.9 Indeed, treatment of the acyclic key intermediate 65 with G-II (5 mol%) under ethylene atmosphere provided the required tricyclic intermediate as the major product 67, although an enyne metathesis/RCM cascade produced the bicyclic compound 68 as a substantial minor product (1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). However, replacement of the ethylene atmosphere with nitrogen after complete conversion of 65, followed by reduction of the temperature to −78 °C and addition of SnCl4 as a Lewis acid facilitated the IMDA reaction and afforded our desired product in 4:1 proportion with the unwanted bicyclic compound 58 (Scheme 12). The tricyclic structure was then homologated and reduced stereoselectively to obtain the core structure of vinigrol, which was identical to Barriault's key intermediate 69 in their formal synthesis of vinigrol.32
:1). However, replacement of the ethylene atmosphere with nitrogen after complete conversion of 65, followed by reduction of the temperature to −78 °C and addition of SnCl4 as a Lewis acid facilitated the IMDA reaction and afforded our desired product in 4:1 proportion with the unwanted bicyclic compound 58 (Scheme 12). The tricyclic structure was then homologated and reduced stereoselectively to obtain the core structure of vinigrol, which was identical to Barriault's key intermediate 69 in their formal synthesis of vinigrol.32
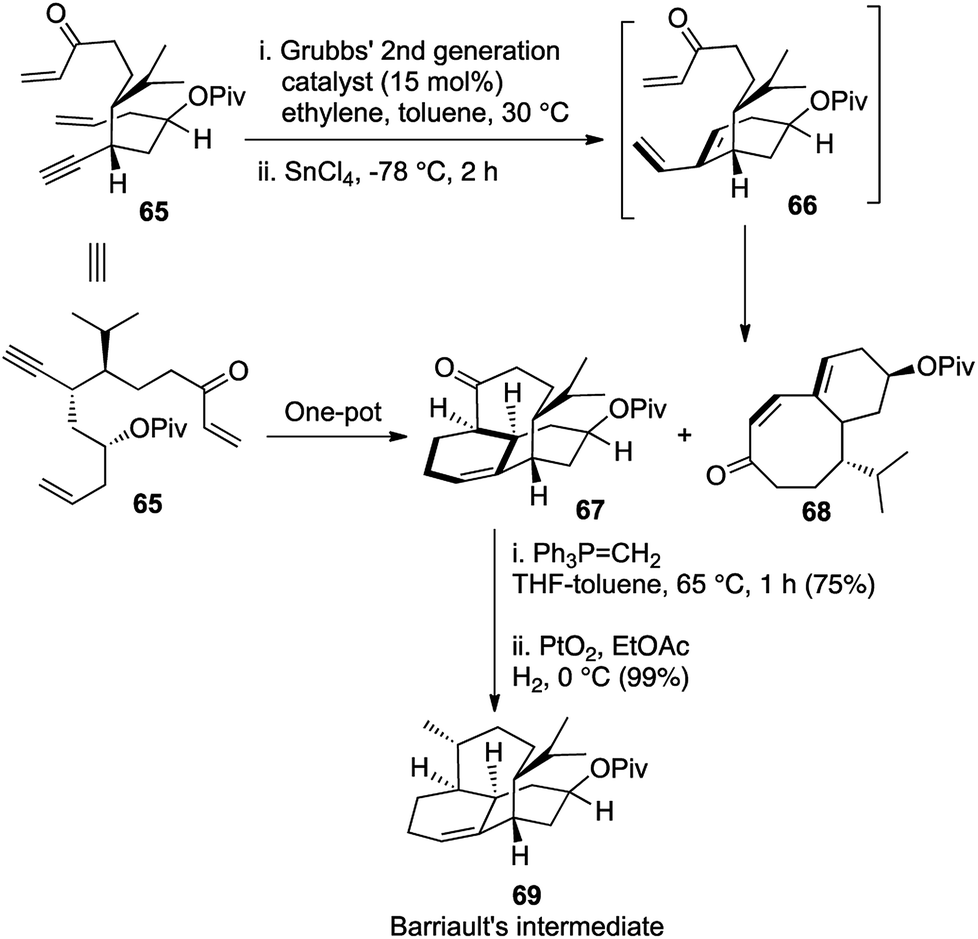 | ||
| Scheme 12 First enantioselective formal synthesis of (−)-vinigrol. | ||
This powerful [1,2,3,4] strategy holds huge importance in displaying the strength of one-pot domino reactions if tuned properly to one's desire. The high values of ‘(c)’ and ‘(d)’ in the nomenclature relates to the tandem formation of 3 rings and 4 new bonds in the same pot and this first ever enantioselective approach towards the core structure heralded a whole new domain in the synthetic history of (−)-vinigrol.
5 Multifold transition metal catalyzed coupling
5.1. [1,2,1,2] transformations
The scope and applications of one-pot domino transformations is not only limited to the construction of complex natural products, but they have also been widely used in heterocyclic chemistry as well. A large number of important pharmaceutical compounds,33 functional materials34 and agrochemical products35 contain a variety of nitrogen containing heterocycles. In 2014, we reported a one-pot Cu-catalyzed route to N-aryl imidazobenzimidazoles via a two-fold cascade C–N bond formation involving an sp2 C–H activation (Scheme 13).36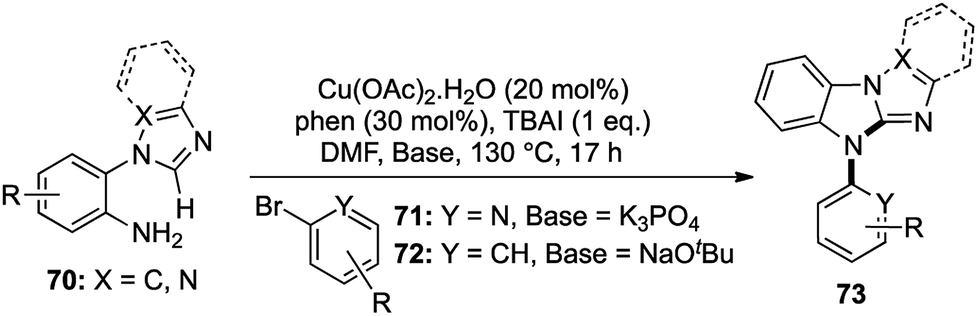 | ||
| Scheme 13 2-fold cascade C–N bond formation leading to N-aryl imidazobenzimidazoles. | ||
A range of N-(o-aminoaryl)azoles 70 were treated with 2-bromopyridines 71 in the presence of Cu(OAc)2·H2O, 1,10-phenanthroline as a ligand and potassium phosphate as a base in DMF at 130 °C. A library of N-pyridyl imidazobenzimidazoles was synthesized with very good yields, exhibiting a wide functional group tolerance.
For the synthesis of N-phenyl imidazobenzimidazoles, the same conditions were used by only altering the base to sodium tert-butoxide to generate another group of these interesting tetracyclic compounds with good to excellent yields (Scheme 13). This short and crisp access to complicated heterocyclic skeletons in a single pot represented an efficient [1,2,1,2] transformation, which was not just straightforward, but also highly scalable. A number of these imidazobenzimidazoles could potentially act as useful ligands in various catalytic reactions, which added to the scope of the methodology.
A related class of nitrogen containing fused heterocycles is represented by benzimidazoquinazolinones 75, known to exhibit anti-cancer37 and anti-tumor activities by truncating the proliferation of human cell lines.38
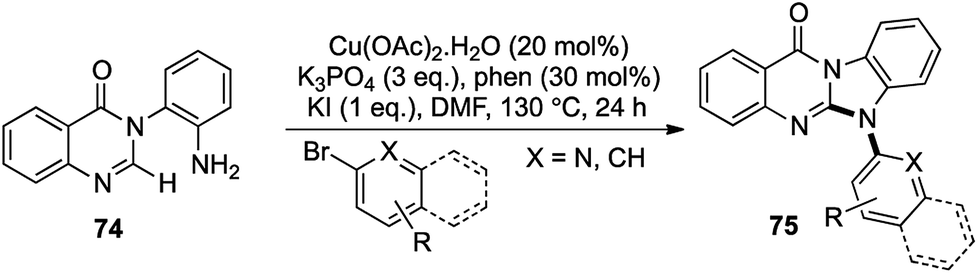
Using very similar conditions as before, N-(o-aminoaryl)quinazolinones 74 were treated with aryl or heteroaryl bromide derivatives to afford N-aryl benzimidazoquinazolinones 75 in good to excellent yields (Scheme 14).39 Special emphasis was laid on the fluorine containing analogues as they are known to be biologically much more potent than their hydrogen analogues40 and under the standard Cu-catalyzed conditions, a variety of fluorine substituted N-aryl benzimidazoquinazolinones were prepared.
 | ||
| Scheme 14 2-fold C–N bond formation leading to N-aryl benzimidazoquinazolinones. | ||
This direct [1,2,1,2] amination route towards the formation of such complex heterocyclic scaffolds with no side product formation and high yield was the first report of such a 2-fold C–N bond formation route towards the N-aryl benzimidazoquinazolinone scaffolds. This was another example of an azole ring formation with the construction of 2 new bonds via a domino C–N bond coupling followed by intramolecular C–H activation.
6. Transition metal catalyzed coupling followed by cyclization
6.1. [1,2,1,2] transformations
Selective catalysis leading to different products from a common intermediate under the influence of different catalytic conditions is a boon in the field of divergent synthesis.41 2-substituted benzofurans and indoles form versatile building blocks for the synthesis of various biologically active compounds and natural products. In 2014, a selective cyclization strategy using 2-alkynyl anilines and derivatives of 2-iodophenol was established to undergo N-cyclization to afford 2-substituted indoles and O-cyclization under different conditions to form 2-substituted benzofurans (Scheme 15).42 Sonogashira coupling of the terminal alkyne with the aromatic iodide was followed in situ by selective cyclization from the common alkynic intermediate 78.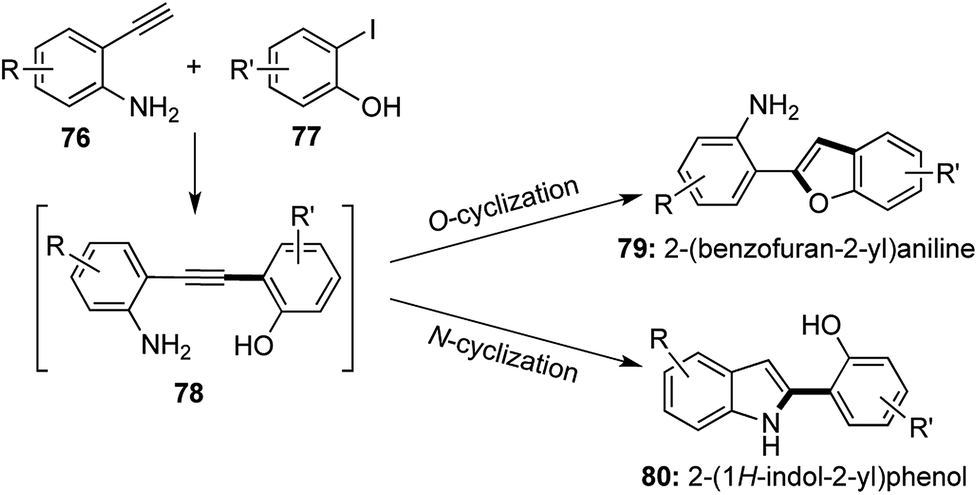 | ||
| Scheme 15 Selective O- and N-cyclization strategy towards benzofurans and indoles. | ||
The benzofuran derivatives 79 were synthesized using the same Cu(I) catalyst devised by Venkataraman43 and the use of a hard inorganic base like Cs2CO3 at 110 °C was required to achieve O-cyclization. Benzofuran derivatives were prepared via a [1,2,1,2] transformation, where 2 new bonds were formed in the process of constructing the furan ring.
To achieve N-cyclization, softer conditions were required and the aniline–NH2 group was functionalized with electron withdrawing groups (Ts, Piv) in order to attribute a softer nature to the nitrogen center. Pd(PPh3)2Cl2 was used along with a soft organic base Et3N at room temperature to obtain a further range of 2-substituted indole derivatives 80 via yet another [1,2,1,2] transformation (Scheme 16).
 | ||
| Scheme 16 Sonogashira coupling followed by cyclization to form 2-substituted benzofurans and indoles. | ||
In continuation with our earlier report on the formation of 2-indolyl-C-glycosides, this was another important addition in the development of one-pot transformations towards the construction of substituted bicyclic aromatics.
6.2. [1,3,1,2] transformations
2-Indolyl-C-glycosides,44 a subset of C-aryl glycosides form an important class of biologically active compounds having a heteroaryl aglycon (Fig. 6).45 | ||
| Fig. 6 Some naturally occurring indolyl-C-glycosides. | ||
Most methods for the preparation of indolyl-C-glycosides were known through addition of lithio-indoles onto sugar lactones or lactols.46 Other multi-step routes had also been achieved, but there was a lack of direct one-pot approaches towards the synthesis of the same.47,48 In order to develop an efficient one-pot route towards the formation of this special class of C-aryl glycosides, we developed a domino Sonogashira coupling/cyclization strategy to arrive at 2-indolyl-C-glycosides.49
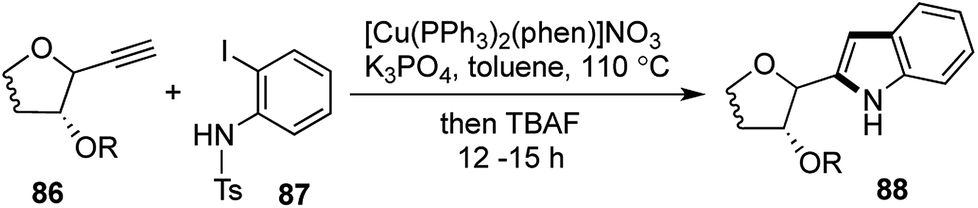
In all the attempts to achieve cyclization with a free –NH2 in the aniline partner, all that was obtained was the Sonogashira coupling product, having an internal triple bond (analogous to 68). It was thus envisaged that attachment of an electron withdrawing group on the aniline nitrogen would increase the acidity of the remaining N–H bond, which was imperative to facilitate the cyclization. The cyclization was ultimately achieved by treating a range of sugar derived alkynes 86 and N-tosyl-2-iodoaniline 87 with Venkataraman's Cu(I) catalyst43 and K3PO4 in toluene at elevated temperatures. The remaining tosyl group in the resulting N-tosyl indoles were removed using TBAF in the same pot and a library of indolyl-C-glycosides were prepared using this protocol in good to excellent yields (Scheme 17). This was a process exhibiting 3 reactions in succession, namely Sonogashira coupling, subsequent deprotection and finally tosyl deprotection upon addition of TBAF. For all the substrates barring the biologically important β-thymidine derived indolyl-glycoside (restricted to a [1,2,1,2] transformation), the tosyl group could be deprotected successfully in one-pot fashion. The general [1,3,1,2] transformation was a direct and quite efficient route towards the formation of 2-indolyl-C-glycosides 88 and related natural products.
 | ||
| Scheme 17 Domino Sonogashira coupling followed by cyclization towards indolyl-C-glycosides. | ||
7. Other categories of one-pot multistep reactions
7.1. [1,2,1,2] transformations
Cope–House cyclization is the reverse of Cope elimination and offers a direct route to the formation of 5 membered heterocycles with a hydroxylamine unit,50,51 but had seldom been utilized in the synthesis of natural products or their analogues (Scheme 18).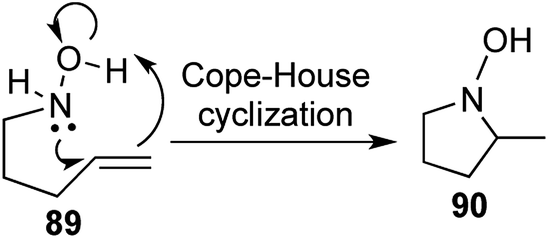 | ||
| Scheme 18 Cope–House elimination. | ||
Taking interest in studying the synthetic utility of sugar derived cyclic nitrones, we designed a one-pot protocol to construct 5-epi-hycynthacine A3 and 5-epi-hyacinthacine A5 along with a few closely related analogous alkaloids (Fig. 7).52
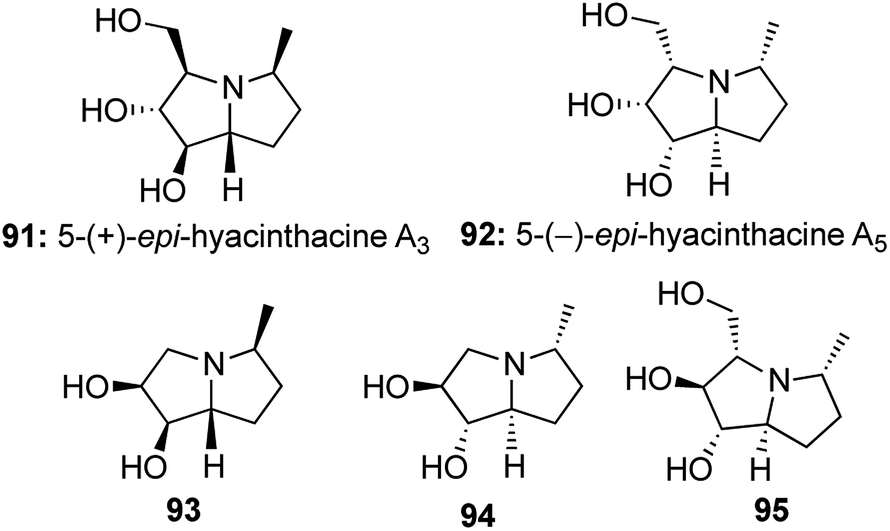 | ||
| Fig. 7 Synthesized family of polyhydroxy pyrrolizidine alkaloids. | ||
Various sugar derived cyclic nitrones 96 were utilized and treatment of the same with homoallyl Grignard and exposure to chloroform produced the Cope–House cyclization products 98 in excellent yields.
Cleavage of the resulting N–O bonds and global benzyl deprotection was effected under hydrogenation conditions to furnish a total of 5 pyrrolizidine alkaloids (Scheme 19 and Fig. 7). The concerted nature of the cyclization ensures complete stereoselectivity in the 5,5-fused bicyclic products, which provided the key feature of this 2-step [1,2,1,2] transformation.
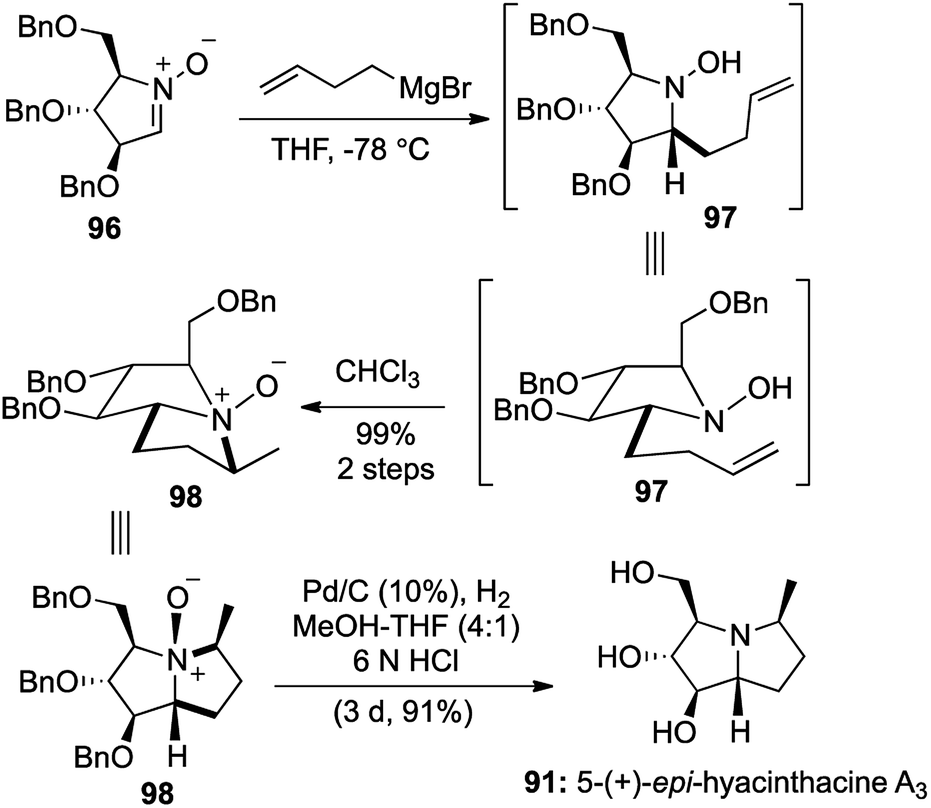 | ||
| Scheme 19 Stereocontrolled synthesis of 5-(+)-epi-hyacinthacine A3. | ||
7.2. [1,2,2,4] transformations
Triazole based molecules are known to have very interesting biological profiles such as anti-HIV53 and anti-microbial activities.54 The biocompatible nature and inertness of the triazole ring towards metabolic transformations55 increases its potential as a covalent bioconjugate linker. As a continuation of our interest in the synthesis of C-aryl glycosides, we sought to devise a method for the preparation of one novel class of this unique family of compounds.Owing to the high selectivity and functional group tolerance of “click” reactions, these transformations have found enormous applications in the field of drug discovery and materials science.56 Click reactions involving azides and sugar derived alkynes have been widely explored, including tandem “click–click approaches” leading to the formation of more than one triazole at a time, by inter and intramolecular click reactions on azido alkynes.56e,57 However, back in 2009, there had been no reports on the formation of more than one triazole ring in tandem fashion using intermolecular click reaction between an azido alkyne, an alkyne and an azide. This prompted us to develop a tandem “click–click approach” using the sequential addition of an external alkyne 99, a sugar derived azido alkyne 100 and an external azide to form a group of bis-triazoles 101 in the presence of CuI and DIPEA with moderate yields (Scheme 20).58
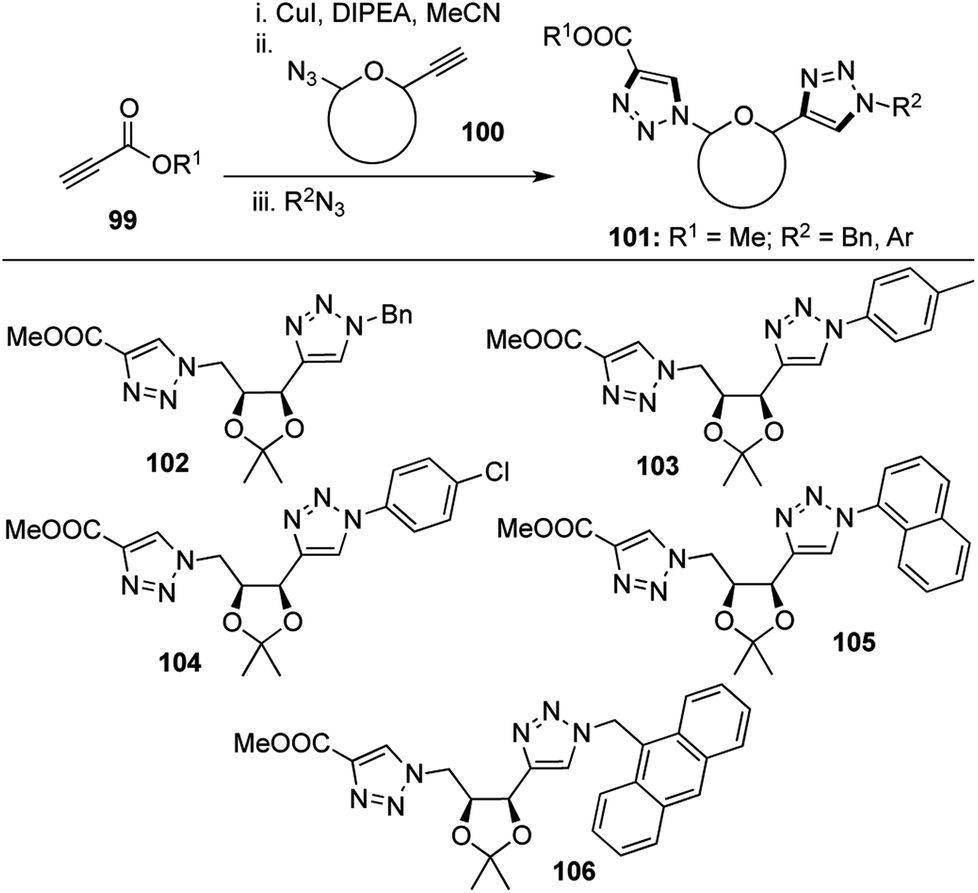 | ||
| Scheme 20 Formation of bis-triazoles using tandem “click–click” strategy. | ||
4 new bonds and 2 new rings were formed during this [1,2,2,4] transformation which has great merit in the fact that two triazole rings were obtained in the same pot and the external alkyne could easily be tuned in order to link two carbohydrate units together.
7.3. [1,4,1,3] transformations
Another subset of C-aryl glycosides is the family of coumarin glycosides where the anomeric carbon of the sugar unit is linked with a coumarin pharmacophore (Fig. 8).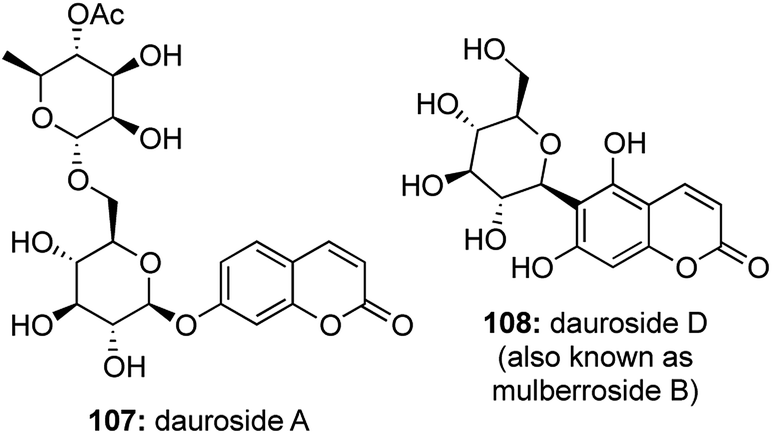 | ||
| Fig. 8 Naturally occurring coumarin glycosides. | ||
A large number of coumarin O-glycosides are known, while the only naturally occurring coumarin C-glycoside is reported to be Dauroside D59 (also known as mulberroside B60). Such compounds are known to possess anti-inflammatory,61 anticoagulant,62 anticancer,63 anti bacterial activities64 and they also act as useful fluorescent probes for studies on ultrafast DNA dynamics.65
Iminocoumarins have been shown to be inhibitors of protein tyrosine kinase in cancer research,66 but there existed little knowhow about the potential biological activities of glycosyl iminocoumarins. Inspired by Wang's synthesis of iminocoumarins through a copper catalysed multicomponent reaction of alkynes, sulfonyl azides and 2-hydroxybenzaldehydes or 2-hydroxyacetophenones,67 we devised a multicomponent route to synthesize 3 C-linked glycosyl iminocourains using a similar strategy.68
Sugar derived alkynes 109 in the presence of CuI and Et3N react with tosyl azide to afford the triazolyl copper species 112, which undergoes facile elimination of nitrogen to form the highly reactive ketenimine 113. The salicaldehyde derived phenolate 114 adds to the ketenimine followed by subsequent cyclization to the intermediate 115, which finally undergoes dehydration to provide the target glycosyl iminocoumarin 111 via a [1,4,1,3] strategy (Scheme 21).67,68
 | ||
| Scheme 21 Multicomponent synthesis and postulated mechanism for the formation of glycosyl iminocoumarins. | ||
6 Conclusion
State-of-the-art research in organic synthesis demands the assembly of complex structures in short time, involving lesser number of steps and fewer intermediate purifications. With the advent of new reactions and one-pot strategies with every passing year, there lies a requirement to classify this plethora of reactions in a systematic manner, which would be concise, yet informative. Through this review, we have intended to bring forward a simple method for the much-required classification of one-pot multistep reactions. We have also underlined all such one-pot reactions reported from our group, whose applications lie towards the synthesis of various natural products and their analogues, a variety of hybrid natural products and a number of interesting heterocyclic scaffolds.Our tetra-coordinated nomenclature system lays emphasis on some of the most important parameters required for appreciating the synthetic efficiency of a multi-step one-pot reaction. Higher number of reactions in sequence increases the probability of achieving more complex transformations, while higher number of rings and new bonds formed indicates the formation of a more advanced structure. We strongly believe that our [a,b,c,d] descriptor would at a glance, shed more light on the important aspects of some of the more ingeniously designed chemical transformations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. P. K thanks SERB, New Delhi India for funding (EMR/2017/000578) and S. I thanks CSIR, New Delhi for research fellowship.Notes and references
- (a) L. F. Tietze, G. Brasche, and K. M. Gericke, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed; (b) L. F. Tietze and U. Beifuss, Angew. Chem., Int. Ed., 1993, 32, 131–163 CrossRef; (c) L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef PubMed; (d) S. E. Denmark and A. Thorarensen, Chem. Rev., 1996, 96, 137–165 CrossRef PubMed; (e) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef PubMed; (f) H. Pellissier, Chem. Rev., 2013, 113, 442–524 CrossRef PubMed.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 Search PubMed; (b) B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259–281 CrossRef.
- (a) P. A. Wender, M. P. Croatt and B. Witulski, Tetrahedron, 2006, 62, 7505–7511 CrossRef; (b) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef PubMed; (c) P. A. Wender, Nat. Prod. Rep., 2014, 31, 433–440 RSC.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 RSC.
- (a) P. A. Clarke, S. Santos and W. H. C. Martin, Green Chem., 2007, 9, 438–440 RSC; (b) C. Vaxelaire, P. Winter and M. Christmann, Angew. Chem., Int. Ed., 2011, 50, 3605–3607 CrossRef PubMed.
- (a) W. R. Roush and R. J. Sciotti, J. Am. Chem. Soc., 1994, 116, 6457–6458 CrossRef; (b) W. R. Roush and R. J. Sciotti, J. Am. Chem. Soc., 1998, 120, 7411–7419 CrossRef.
- (a) R. B. Ruggeri, K. F. McClure and C. H. Heathcock, J. Am. Chem. Soc., 1989, 111, 1530–1531 CrossRef; (b) C. H. Heathcock, R. B. Ruggeri and K. F. McClure, J. Org. Chem., 1992, 57, 2585–2594 CrossRef.
- (a) K. C. Nicolaou, N. A. Petasis, R. E. Zipkin and J. Uenishi, J. Am. Chem. Soc., 1982, 104, 5555–5557 CrossRef; (b) K. C. Nicolaou, N. A. Petasis, J. Uenishi and R. E. Zipkin, J. Am. Chem. Soc., 1982, 104, 5557–5558 CrossRef; (c) K. C. Nicolaou, R. E. Zipkin and N. A. Petasis, J. Am. Chem. Soc., 1982, 104, 5558–5560 CrossRef; (d) K. C. Nicolaou, N. A. Petasis and R. E. Zipkin, J. Am. Chem. Soc., 1982, 104, 5560–5562 CrossRef; (e) K. C. Nicolaou and J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993–3009 RSC.
- W. M. Bandaranayake, J. E. Banfield and D. S. C. Black, J. Chem. Soc., Chem. Commun., 1980, 902–903 RSC.
- (a) K. P. Kaliappan and R. S. Nandurdikar, Chem. Commun., 2004, 2506–2507 RSC; (b) K. P. Kaliappan, R. S. Nandurdikar and M. M. Shaikh, Tetrahedron, 2006, 62, 5064–5073 CrossRef.
- K. P. Kaliappan and R. S. Nandurdikar, Org. Biomol. Chem., 2005, 3613–3614 Search PubMed.
- (a) S. A. Patkar, U. Rasmussen and B. Diamant, Agents Actions, 1979, 9, 53–57 CrossRef PubMed; (b) J. Lytton, M. Westlin and M. Hanley, J. Biol. Chem., 1991, 266, 17067–17071 Search PubMed; (c) C. Caspersen and M. Treiman, FEBS Lett., 1995, 377, 31–36 CrossRef PubMed.
- M. Treiman, C. Caspersen and S. B. Christensen, Trends Pharmacol. Sci., 1998, 19, 131–135 CrossRef PubMed.
- (a) S. F. Oliver, K. Högenauer, O. Simic, A. Antonello, M. D. Smith and S. V. Ley, Angew. Chem., Int. Ed., 2003, 42, 5996–6000 CrossRef PubMed; (b) M. Ball, S. P. Andrews, F. Wierschem, E. Cleator, M. D. Smith and S. V. Ley, Org. Lett., 2007, 9, 663–666 CrossRef PubMed; (c) S. P. Andrews, M. Ball, F. Wierschem, E. Cleator, S. Oliver, K. Högenauer, O. Simic, A. Antonello, A. U. Hünger, M. D. Smith and S. V. Ley, Chem.–Eur. J., 2007, 13, 5688–5712 CrossRef PubMed; (d) H. Chu, J. M. Smith, J. Felding and P. S. Baran, ACS Cent. Sci., 2017, 3, 47–51 CrossRef PubMed; (e) D. Chen and P. A. Evans, J. Am. Chem. Soc., 2017, 139, 6046–6049 CrossRef PubMed; (f) S. Borman, Chem. Eng. News, 2017, 95, 7 Search PubMed.
- (a) G. Mehta and V. Singh, Chem. Rev., 1999, 99, 881–930 CrossRef PubMed; (b) J. Oh, J.-R. Choi and J. K. Cha, J. Org. Chem., 1992, 57, 6664–6667 CrossRef; (c) G. A. Kraus and D. Zheng, Synlett, 1993, 71–72 CrossRef; (d) T. Mukaiyama, I. Shiina, K. Kimura, Y. Akiyama and H. Iwadare, Chem. Lett., 1995, 229–230 CrossRef; (e) I. Shiina, H. Iwadare, M. Saitoh, N. Ohkawa, T. Nishimura and T. Mukaiyama, Chem. Lett., 1995, 781–782 CrossRef; (f) D. Crich and S. Natarajan, J. Chem. Soc., Chem. Commun., 1995, 85–86 RSC; (g) M. G. Banwell, P. Darmos, M. D. McLeod and D. C. R. Hockless, Synlett, 1998, 897–899 CrossRef; (h) M. Wenz, D. Grobach, M. Beitzel and S. Blechert, Synthesis, 1999, 607–614 CrossRef; (i) Y. Shimada, M. Nakamura, T. Suzuka, J. Matsui, T. Tatsumi, K. Tsutsumi, T. Morimoto, H. Kurosawa and K. Kaiuch, Tetrahedron Lett., 2003, 44, 1401–1403 CrossRef.
- K. P. Kaliappan, V. Ravikumar and S. A. Pujari, Tetrahedron Lett., 2006, 47, 981–984 CrossRef.
- K. P. Kaliappan, V. Ravikumar and S. A. Pujari, J. Chem. Sci., 2008, 120, 205–216 CrossRef.
- (a) G. Mehta and V. Singh, Chem. Soc. Rev., 2002, 31, 324–334 RSC; (b) L. F. Tietze, H. P. Bell and S. Chandrasekhar, Angew. Chem., Int. Ed., 2003, 42, 3996–4028 CrossRef PubMed; (c) K. Palanichamy and K. P. Kaliappan, Pure Appl. Chem., 2013, 85, 1185–1202 CrossRef.
- R. S. Nandurdikar, A. V. Subrhamanyam and K. P. Kaliappan, Eur. J. Org. Chem., 2010, 2788–2799 CrossRef.
- For some general reviews on C-aryl glycosides, see: (a) K. Kitamura, Y. Ando, T. Matsumoto and K. Suzuki, Chem. Rev., 2018, 118, 1495–1598 CrossRef PubMed; (b) Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281–12356 CrossRef PubMed; (c) K. W. Wellington and S. A. Benner, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 1309–1333 CrossRef PubMed; (d) Y. Du and R. J. Linhardt, Tetrahedron, 1998, 54, 9913–9959 CrossRef; (e) K. Krohn and J. Rohr, Top. Curr. Chem., 1997, 188, 127–195 CrossRef.
- For some of the approaches, see: (a) T. Hosoya, E. Takashiro, T. Matsumoto and K. Suzuki, J. Am. Chem. Soc., 1994, 116, 1004–1015 CrossRef; (b) K. A. Parker and Y. H. Koh, J. Am. Chem. Soc., 1994, 116, 11149–11150 CrossRef; (c) E. R. Palmacci and P. H. Seeberger, Org. Lett., 2001, 3, 1547–1550 CrossRef PubMed; (d) F. M. Hauser and X. Hu, Org. Lett., 2002, 4, 977–978 CrossRef PubMed; (e) S. Vijayasaradhi and I. S. Aidhen, Org. Lett., 2002, 4, 1739–1742 CrossRef PubMed.
- K. P. Kaliappan and A. V. Subrahmanyam, Org. Lett., 2007, 9, 1121–1124 CrossRef PubMed.
- V. V. Betkekar, S. Panda and K. P. Kaliappan, Org. Lett., 2012, 14, 198–201 CrossRef PubMed.
- S. E. N. Ayyad, A. S. Judd, W. T. Shier and T. R. Hoye, J. Org. Chem., 1998, 63, 8102–8106 CrossRef.
- C. S. Li, H. W. Yu, X. Z. Chen, X. Q. Wu, G. Y. Li and G. L. Zhang, Helv. Chim. Acta, 2011, 94, 105–110 CrossRef.
- V. V. Betkekar, A. A. Sayyad and K. P. Kaliappan, Org. Lett., 2014, 16, 5540–5543 CrossRef PubMed.
- (a) T. J. Maimone and P. S. Baran, Nat. Chem. Biol., 2007, 3, 396–407 CrossRef PubMed; (b) T. J. Maimone, A.-F. Voica and P. S. Baran, Angew. Chem., Int. Ed., 2008, 47, 3054–3056 CrossRef PubMed.
- T. J. Maimone, J. Shi, S. Ashida and P. S. Baran, J. Am. Chem. Soc., 2009, 131, 17066–17067 CrossRef PubMed.
- (a) Q. Yang, J. T. Njaradarson, C. Draghici and F. Li, Angew. Chem., Int. Ed., 2013, 52, 8648–8651 CrossRef PubMed; (b) Q. Yang, C. Draghici, J. T. Njardarson, F. Li, B. R. Smith and P. Das, Org. Biomol. Chem., 2014, 12, 330–344 RSC.
- W. Hu, F. Zhang, Z. Xu, Q. Liu, Y. Cui and Y. Jia, Org. Lett., 2010, 12, 956–959 CrossRef PubMed.
- J. Poulin, C. M. Grise-Bard and L. Barriault, Angew. Chem., Int. Ed., 2012, 51, 2111–2114 CrossRef PubMed.
- (a) M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685–714 CrossRef PubMed; (b) J. Kim and M. Movassaghi, Chem. Soc. Rev., 2009, 38, 3035–3050 RSC.
- (a) E. Hao, B. Fabre, F. R. Fronczek and M. G. H. Vicente, Chem. Mater., 2007, 19, 6195–6205 CrossRef; (b) Y. G. Lee, Y. Koyama, M. Yonekawa and T. Takata, Macromolecules, 2009, 42, 7709–7717 CrossRef.
- (a) Q. Du, W. Zhu, Z. Zhao, X. Qian and Y. Xu, J. Agric. Food Chem., 2012, 60, 346–353 CrossRef PubMed; (b) N. Jacobsen and L.-E. K. Pedersen, Pestic. Sci., 1983, 14, 90–97 CrossRef.
- P. Subramanian and K. P. Kaliappan, Eur. J. Org. Chem., 2014, 5986–5997 CrossRef.
- (a) A. Cagir, S. H. Jones, R. Gao, B. M. Eisenhauer and S. M. Hecht, J. Am. Chem. Soc., 2003, 125, 13628–13629 CrossRef PubMed; (b) M. Viji and R. Nagarajan, J. Chem. Sci., 2014, 126, 1075–1080 CrossRef.
- (a) W. H. W. Lunn, R. W. Harper and R. L. Stone, J. Med. Chem., 1971, 14, 1069–1071 CrossRef PubMed; (b) L. Dalla Via, O. Gia, S. Marciano Magno, A. Da Settimo, A. M. Marini, G. Primofiore, F. Da Settimo and S. Salerno, Farmaco, 2001, 56, 159–167 CrossRef PubMed.
- A. Banerjee, P. Subramanian and K. P. Kaliappan, J. Org. Chem., 2016, 81, 10424–10432 CrossRef PubMed.
- (a) K. R. Gore, S. Harikrishna and P. I. Pradeepkumar, J. Org. Chem., 2013, 78, 9956–9962 CrossRef PubMed; (b) R. K. Khangarot and K. P. Kaliappan, Eur. J. Org. Chem., 2013, 2692–2698 CrossRef; (c) D. M. Lemal, J. Org. Chem., 2004, 69, 1–11 CrossRef PubMed; (d) P. Liu, A. Sharon and C. K. Chu, J. Fluorine Chem., 2008, 129, 743–766 CrossRef PubMed.
- (a) J. Mahatthananchai, A. M. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954–10990 CrossRef PubMed; (b) J. N. Johnston, H. Muchalski and T. L. Troyer, Angew. Chem., Int. Ed., 2010, 49, 2290–2298 CrossRef PubMed; (c) S. Uesugi, Z. Li, R. Yazaki and T. Ohshima, Angew. Chem., Int. Ed., 2014, 53, 1611–1615 CrossRef PubMed; (d) P.-H. Chen, T. Xu and G. Dong, Angew. Chem., Int. Ed., 2014, 53, 1674–1678 CrossRef PubMed.
- S. Indu, P. Subramanian and K. P. Kaliappan, Eur. J. Org. Chem., 2014, 7193–7202 CrossRef.
- C. G. Bates, P. Saijueng, J. M. Murphy and D. Venkataraman, Org. Lett., 2002, 4, 4727–4729 CrossRef PubMed.
- (a) D. E. Levy, The Chemistry of C-glycosides, 1st edn, Elsevier, Oxford, 1995 Search PubMed; (b) F. Nicotra, Top. Curr. Chem., 1997, 187, 55–83 CrossRef; (c) L. Liu, M. McKee and M. H. D. Postema, Curr. Org. Chem., 2001, 5, 1133–1167 CrossRef; (d) S. Serra, C. Fuganti and E. Brenna, Chem.–Eur. J., 2007, 13, 6782–6791 CrossRef PubMed.
- (a) E. C. Cadman, D. E. Dix and R. E. Handschumacher, Cancer Res., 1978, 38, 682–688 Search PubMed; (b) Y. Yamashita, N. Fujii, C. Murakata, T. Ashizawa, M. Okabe and H. Nakano, Biochemistry, 1992, 31, 12069–12075 CrossRef PubMed; (c) Y. Wu, Z.-X. Zhang, H. Hu, D. Li, G. Qiu, X. Hu and X. He, Fitoterapia, 2011, 82, 288–292 CrossRef PubMed; (d) A. Yepremyan and T. G. Minehan, Org. Biomol. Chem., 2012, 10, 5194–5196 RSC.
- (a) M. Cornia, G. Casiraghi and L. Zetta, J. Org. Chem., 1991, 56, 5466–5468 CrossRef; (b) W. He, H. Togo and M. Yokoyama, Tetrahedron Lett., 1997, 38, 5541–5544 CrossRef; (c) W. He, H. Togo, Y. Waki and M. Yokoyama, J. Chem. Soc., Perkin Trans. 1, 1998, 2425–2434 RSC; (d) D. P. Steinhuebel, J. J. Flemming and J. Du Bois, Org. Lett., 2001, 3, 293–295 Search PubMed; (e) D. Guianvarc'h, J.-L. Fourrey, M.-E. Tran Huu Dau, V. Guerineau and R. Benhida, J. Org. Chem., 2002, 67, 3724–3732 CrossRef; (f) J. Barbaric, C. Wanninger-Weiβ and H.-A. Wagenknecht, Eur. J. Org. Chem., 2009, 364–370 CrossRef.
- (a) T. Nishikawa, T. Nishikawa, M. Ishikawa and M. Isobe, Synlett, 1999, 123–125 CrossRef; (b) T. Nishikawa, Y. Koide, S. Kajii, K. Wada, M. Ishikawa and M. Isobe, Org. Biomol. Chem., 2005, 3, 687–700 RSC.
- D. Necas, D. Hidasova, M. Hocek and M. Kotora, Org. Biomol. Chem., 2011, 9, 5934–5937 Search PubMed.
- P. Subramanian and K. P. Kaliappan, Eur. J. Org. Chem., 2013, 595–604 CrossRef.
- N. J. Cooper and D. W. Knight, Tetrahedron, 2004, 60, 243–269 CrossRef; N. P. Brainbridge, A. C. Currie, N. J. Cooper, J. C. Muir, D. W. Knight and J. M. Walton, Tetrahedron Lett., 2007, 48, 7782–7787 CrossRef.
- (a) H. O. House, D. T. Manning, D. G. Melillo, L. F. Lee, O. R. Haynes and B. E. Wilkes, J. Org. Chem., 1976, 41, 855–863 CrossRef; (b) H. O. House and L. F. Lee, J. Org. Chem., 1976, 41, 863–869 CrossRef.
- K. P. Kaliappan and P. Das, Synlett, 2008, 841–844 CrossRef.
- R. Alvarez, S. Velazquez, A. San-Felix, S. Aquaro, E. De Clercq, C.-F. Perno, A. Karlsson, J. Balzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4185–4194 CrossRef PubMed.
- M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A. Garmon, D. R. Graber, K. C. Grega, J. B. Hester, D. K. Hutchinson, J. Morris, R. J. Reischer, C. W. Ford, G. E. Zurenko, J. C. Hamel, R. D. Schaadt, D. Stapert and B. H. Yagi, J. Med. Chem., 2000, 43, 953–970 CrossRef PubMed.
- D. K. Dalvie, A. S. Kalgutkar, S. C. Khojasteh-Bakht, R. S. Obach and J. P. O'Donnell, Chem. Res. Toxicol., 2002, 15, 269–299 CrossRef PubMed.
- (a) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef PubMed; (b) J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef PubMed; (c) V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef PubMed; (d) M. S. Singh, S. Chowdhury and S. Koley, Tetrahedron, 2016, 72, 5257–5283 CrossRef; (e) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- (a) A. Marra, L. Moni, D. Pazzi, A. Corallini, D. Bridi and A. Dondoni, Org. Biomol. Chem., 2008, 6, 1396–1409 RSC; (b) C. Ornelas, J. R. Aranzaes, L. Salmon and D. Astruc, Chem.–Eur. J., 2008, 14, 50–64 CrossRef PubMed ; (c) A. Dondoni and A. Marra, J. Org. Chem., 2006, 71, 7546–7557 CrossRef PubMed; (d) E. Fernandez-Megia, J. Correa, I. Rodriguez-Meizoso and R. Riguera, Macromolecules, 2006, 39, 2113–2120 CrossRef; (e) S. Chandrasekhar, C. L. Rao, C. Nagesh, C. R. Reddy and B. Sridhar, Tetrahedron Lett., 2007, 48, 5869–5872 CrossRef; (f) S. Hotha, R. I. Anegundi and A. A. Natu, Tetrahedron Lett., 2005, 46, 4585–4588 CrossRef.
- K. P. Kaliappan, P. Kalanidhi and S. Mahapatra, Synlett, 2009, 13, 2162–2166 CrossRef.
- (a) D. Batsuren, E. K. Batirov and V. M. Malikov, Chem. Nat. Compd., 1982, 18, 616–617 CrossRef; (b) A. D. Vdovin, D. Batsuren, E. K. Batirov, M. R. Yagudaev and V. M. Malikov, Chem. Nat. Compd., 1983, 19, 413–416 CrossRef.
- K. Hirakura, I. Saida, T. Fukai and T. Nomura, Heterocycles, 1985, 23, 2239–2242 CrossRef.
- M.-A. Dubois, M. Wierer and H. Wagner, Phytochemistry, 1990, 29, 3369–3371 CrossRef.
- Y. L. Garazd, E. M. Kornienko, L. N. Maloshtan, M. M. Garazd and V. P. Khilya, Chem. Nat. Compd., 2005, 41, 508–512 CrossRef.
- J. A. Burlison, C. Avila, G. Vielhauer, D. J. Lubbers, J. Holzbeierlein and B. S. J. Blagg, J. Org. Chem., 2008, 73, 2130–2137 CrossRef PubMed.
- (a) B. Musicki, A.-M. Periers, L. Piombo, P. Laurin, M. Klich, C. Dupuis-Hamelin, P. Lassaigne and A. Bonnefoy, Tetrahedron Lett., 2003, 44, 9259–9262 CrossRef; (b) A. C. Scatigno, S. S. Garrido and R. Marchetto, J. Pept. Sci., 2004, 10, 566–577 CrossRef PubMed.
- (a) R. S. Coleman and M. L. Madaras, J. Org. Chem., 1998, 63, 5700–5703 CrossRef; (b) R. S. Coleman, M. A. Berg and C. J. Murphy, Tetrahedron, 2007, 63, 3450–3456 CrossRef.
- (a) C. N. O'Callaghan and M. L. Conalty, Proc. R. Ir. Acad., Sect. B, 1979, 6, 87–98 Search PubMed; (b) T. R. Jr Burke, B. Lim, V. E. Marquez, Z.-H. Li, J. B. Bolen, I. Stefanova and I. D. Horak, J. Med. Chem., 1993, 36, 425–432 CrossRef.
- S.-L. Cui, X.-F. Lin and Y.-G. Wang, Org. Lett., 2006, 8, 4517–4520 CrossRef PubMed.
- K. Palanichamy, S. R. Suravarapu and K. P. Kaliappan, Synthesis, 2012, 44, 1841–1848 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |