
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu(II) Schiff base complex intercalated into layered double hydroxide for selective oxidation of ethylbenzene under solvent-free conditions
Jagat Singh Kirar and
Savita Khare *
*
School of Chemical Sciences, Devi Ahilya University, Takshashila Campus, Khandwa Road, Indore, M. P. 452001, India. E-mail: kharesavita@rediffmail.com
First published on 23rd May 2018
Abstract
A new heterogeneous catalyst was prepared by intercalation of NNOO donor Cu(II) Schiff base complex derived from 2-hydroxy-1-naphthaldehyde and 4-amino benzoic acid into Zn–Al layered double hydroxide {LDH-[NAPABA–Cu(II)]}. Synthesized catalyst was characterized by Inductively coupled plasma atomic emission spectroscopy, energy dispersive X-ray analysis, scanning electron microscopy, X-ray diffraction, Transmission electron microscopy, BET surface area, Fourier transform infrared spectroscopy, thermo-gravimetric analysis, electron paramagnetic resonance spectroscopy and diffuse reflectance UV-visible spectroscopy. The catalytic performance of LDH-[NAPABA–Cu(II)] was studied for the liquid phase solvent-free oxidation of ethylbenzene at 393 K, using tert-butylhydroperoxide as an oxidant. In oxidation reaction ethylbenzene was oxidized to acetophenone, benzaldehyde and benzoic acid. The major product was acetophenone. A maximum, 80.54% conversion of ethylbenzene was observed after 7 hours. The catalyst was recycled seven times without significant loss of catalytic activity.
Introduction
Selective oxidation of ethylbenzene to acetophenone is an important industrial process. The oxidation product, acetophenone, is an important intermediate and has been widely used for the synthesis of many chemicals such as perfumes, pharmaceuticals, resins, alcohols and esters.1,2 The current industrial process involves oxidation of ethylbenzene to acetophenone in the presence of molecular oxygen using cobalt acetate as homogeneous catalyst in acetic acid.3 This method suffers from a corrosive and environmentally unfriendly nature.4 Furthermore, many homogeneous catalytic systems were developed for the oxidation of ethylbenzene which give high conversion and selectivity.5–8 Most of these catalysts are having certain drawbacks like difficulty in separation of catalyst at the end of reaction for reuse as well as decomposition during the catalytic reaction. Therefore, heterogenization of homogeneous metal complex on insoluble support to develop new heterogeneous catalyst using various methods has attracted a lot of attention. Considerable researches have devoted to find efficient heterogeneous catalysts for the selective oxidation of ethylbenzene using various supports such as silica, polymers, MCM-41, SBA-15.9–13A variety of copper(II) salts is suitable as catalyst or catalyst precursors14 because being a transition metal, Cu-based materials can promote and undergo a variety of reactions due to its variable oxidation states. In particular, Schiff base complexes of copper(II) have been used as catalysts in the oxidation of alkenes, alkanes, alcohols, alkyl aromatics and sulphides.15–20 Many Cu(II) based catalysts were used for the oxidation of ethylbenzene using various oxidants. T. H. Bennur et al.21 have reported copper tri- and tetraaza macrocyclic complexes encapsulated in zeolite-Y as heterogeneous catalysts for the oxidation of ethylbenzene to acetophenone using molecular oxygen. The system gave 13.4% conversion of ethylbenzene with 73.3% selectivity to acetophenone after 5 h. S. Gago et al.22 have reported catalytic activity of MCM-41 supported bis-chlorocopper(II) complex for oxidation of ethylbenzene with tert-butylhydroperoxide as an oxidant. The maximum 62% conversion of ethylbenzene was achieved after 24 h. M. Palanichamy et al.23 have prepared (Cu-BTC) metal organic framework (MOF) for oxidation of ethylbenzene with tert-butylhydroperoxide as an oxidant. The maximum 28.26% conversion of ethylbenzene with 95.7% selectivity of acetophenone was observed after 6 h. M. R. Maurya et al.24 have reported polymer anchored copper(II) complex of 2-(α-hydroxymethyl)benzimidazole, {PS-[Cu(hmbmz)2]} for oxidation of ethylbenzene. M. R. Maurya et al.25 have synthesized polymer supported oxovanadium(IV), dioxomolybdenum(VI) and copper(II) complexes for the oxidation of styrene, cyclohexene and ethylbenzene using 30% H2O2 as an oxidant.
The transition metal complex intercalated into layered compounds by intercalation method is better because it provides temperature and solvent stable entities. Layered double hydroxide (LDHs) is found to be interesting for us because it is inorganic material with a layered structure which acts as anionic clays or hydrotalcite-like compounds.26–28 The structures of LDH consist of brucite (MII(OH)2)-like layers with part of divalent cations (MII) substituted by the trivalent ions (MIII). Both divalent and trivalent ions are located at the center of octahedral (OH−) units of the brucite layers. The resulting layers are in an excess of positive charge acquired to balance the intercalated anions. The general formula of LDH is [MII1−xMIIIx(OH)2]x+(Ax/nn−)·mH2O, where An− is the interlayer anion.29,30 The coefficient x is equal to the molar ratio [MIII/(MII + MIII)], and m is the number of water molecules located in the interlayer region together with the anions. LDH an anion exchanger has been extensively studied for its intercalation chemistry,31–33 heterogeneous catalyst precursor34,35 and ion exchanger properties.36,37 LDH based catalysis is of high interest for green and sustainable chemistry38 since the LDHs can provide distinct nanometer-scaled layers for intercalation of transition metal complexes.39,40 In most of cases LDH has been prepared by co-precipitation methods.34,35 In previous studies we have reported layered material supported recyclable heterogeneous catalysts for the oxidation of cyclohexene, cyclohexane and styrene.41–46
In continuation of our interest, herein, we are reporting the synthesis of Cu(II) Schiff base complex derived from 2-hydroxy-1-naphthaldehyde and 4-amino benzoic acid NAPABA–Cu(II) intercalated into layered double hydroxide, abbreviated as LDH-[NAPABA–Cu(II)] and its catalytic behaviour was studied for oxidation of ethylbenzene using tert-butylhydroperoxide (TBHP) as an oxidant.
Experimental methods
General considerations
Zinc(II) nitrate hexahydrate (Zn(NO3)3·6H2O), aluminium(III) nitrate nonahydrate (Al(NO3)3·9H2O), copper acetate (Cu(OAc)2·H2O), 4-amino benzoic acid, 2-hydroxy-1-naphthaldehyde and sodium hydroxide were all of analytical grade and purchased from E. Merck. The 70% commercial aqueous solution of TBHP also purchased from E. Merck. HPLC grade ethylbenzene purchased from SDFCL and its purity was checked by gas chromatography (G. C.), to ensure that no oxidation products were present. A stock solution of TBHP in ethylbenzene (72% in ethylbenzene) was prepared by extraction of 55 ml of commercial TBHP (70% in water) into 15 ml of ethylbenzene. Phase separation was promoted by saturation of the aqueous layer with NaCl. The organic layer was dried over MgSO4, filtered, and stored at 5 °C. The molar ratio of ethylbenzene to TBHP in this solution was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.
:3.
Powder X-ray diffraction (XRD) patterns of the samples were recorded on a Rigaku diffractometer in the 2θ range of 2–70° using CuKα radiation (λ = 1.5418 Å) at scanning speed 2° per minute with step size 0.02°. Scanning electron microscopy (SEM) measurements were performed using a JEOL JSM 6100 electron microscope, operating at 20 kV. Transmission electron microscope (TEM) studies were performed on a Tecnai G2 20 microscope. The Fourier transform infrared (FTIR) spectra were recorded on Perkin Elmer model 1750 in KBr. Thermo-gravimetric analysis (TGA) were conducted on a Perkin Elmer USAA diamond system in the range 298–973 K, at heating rate of 10 °C min−1. Inductively coupled plasma atomic emission spectroscopy, Thermo Electron IRIS INTREPID II XSP DUO (ICP-AES) was used for estimation of copper. The electronic spectrum of solid was recorded on Varian, Cary 5000 Spectrophotometer. The electron paramagnetic resonance (EPR) spectrum was recorded on a JES-FA200 ESR, X band spectrometer operated at a microwave frequency of 9.65 GHz at room temperature. The g value is reported relative to a 2,2-diphenyl-1-picrylhydrazil (dpph) standard with g = 2.0036. The N2 adsorption data, measured at 77 K by volumetric adsorption set-up (Micromeritics ASAP-2010, USA), were used to determine BET surface area. Analytical gas chromatography was carried out on a Shimadzu Gas Chromatograph GC-14B with dual flame ionization detector (FID) and attached Shimadzu printer having XE-60 ss column. The products were identified by GC-MS (Perkin-Elmer Clasus 500 column; 30 m × 60 mm).
Preparation of catalyst
Preparation of ligand {[NAPABA]}
The ligand was prepared by mixing of the methanolic solution of 4-amino benzoic acid (10 mmol) and 2-hydroxy-1-naphthaldehyde (10 mmol) in 1:1 ratio; the solution became yellow due to imines formation. The resulting mixture was refluxed for 3 h with continuous stirring under nitrogen atmosphere. The yellow product was filtered off, washed with methanol followed by acetonitrile and then dried at 333 K overnight. The elemental percentage of CHN for C18H13NO3: anal. found: C, 74.17%; H, 4.43%; N, 4.75%. Calc. For C18H13NO3: C, 74.22%; H, 4.50%; N, 4.81%. FTIR (KBr pellet: cm−1): 3381 (br), 1721 (w), 1607 (m), 1547 (m), 1435 (w).
Preparation of neat NAPABA–Cu(II) complex
The Cu(II) Schiff complex was synthesized by dissolving 4-amino benzoic acid (10 mmol) into a 50 ml the methanolic solution of NaOH (20 mmol) followed by addition of the methanolic solution of 2-hydroxy-1-naphthaldehyde (10 mmol). The mixture becomes yellow, immediately Cu(OAc)2·H2O (5 mmol) was added to this mixture and mixture is kept under continuous stirring for 4 h at room temperature. After 4 h the greenish brown precipitate of NAPABA–Cu(II) complex was formed which was filtered, washed with petroleum ether and dried in air. The obtained complex was characterized by elemental analysis and FTIR spectroscopy. The elemental percentage of CHN for C36H22N2O6Cu: anal. found: C, 67.2%; H, 3.2%; N, 4.3%. Calc. For C36H22N2O6Cu: C, 67.34%; H, 3.2%; N, 4.4%. FTIR (KBr pellet: cm−1): 3385 (br), 1612 (m), 1541 (m), 1437 (w), 1387 (m), 751 (w), 519 (w).Preparation of support {LDH-[NH2–C6H4COO]}
The LDH-[NH2–C6H4COO] was prepared by co-precipitation method according to previously reported procedure.47 A solution of zinc(II) nitrate hexahydrate (14.8 g) and aluminium(III) nitrate nonahydrate (6.25 g) was prepared in decarbonised water having Zn–Al molar ratio 3. To this mixture, a solution of 4-amino benzoic acid (10.86 g) and NaOH (7.8 g) in decarbonised water was added with continuous stirring. Immediately a gel like mixture was obtained, which was digested at 348 K for 48 h. Upon cooling, the product was isolated by filtration, washed with water followed by methanol and the solid was dried at 333 K overnight.Preparation of ligand {LDH-[NAPABA]}
The ligand was prepared by mixing LDH-[NH2–C6H4COO] (1.0 g) with the methanolic solution of 2-hydroxy-1-naphthaldehyde (0.48 g, 2.8 mmol); the solution became yellow due to imines formation. The resulting mixture was refluxed for 3 h with continuous stirring under nitrogen atmosphere. The yellow product was filtered off, washed with methanol followed by acetonitrile and then dried at 333 K overnight.Preparation of catalyst {LDH-[NAPABA–Cu(II)]}
The catalyst LDH-[NAPABA–Cu(II)] was prepared by incorporation of Cu(II) into the ligand {LDH-[NAPABA]}. Firstly ligand, LDH-[NAPABA] (1 g) was suspended in 50 ml methanol followed by addition methanolic solution of Cu(OAc)2·H2O (0.25 g) under continuous stirring. The mixture was refluxed for 4 h under nitrogen atmosphere. After cooling, the solid was filtered and washed with methanol. The resulting solid was soxhlet extracted using methanol followed by acetone and acetonitrile to remove excessive ligand and metal salt remained uncomplexed in the host layer as well as on the surface and dried at 333 K overnight.Oxidation of ethylbenzene
Oxidation of ethylbenzene over LDH-[NAPABA–Cu(II)] catalyst using tert-butylhydroperoxide (TBHP) as an oxidant was carried out under solvent-free condition in a three-necked round-bottom flask. In a typical run, a three-necked round bottom flask was loaded with ethylbenzene, TBHP (72% TBHP in ethylbenzene) and catalyst at required temperature in an oil bath for 7 h with continuous stirring using Teflon needle on magnetic hot plate. After completion of the reaction, the catalyst was separated by filtration and washed several time with methanol followed by acetone. Then the catalyst was dried at 333 K for further use. The oxidation products were qualitatively as well as quantitative analysed by GC-MS spectrometer and GC spectrometer using XE-60 ss column. The GC-MS analysis revealed that the main products were acetophenone and benzaldehyde. Selectivity of products was calculated with respect to the converted ethylbenzene using internal standard method. The internal standard was dodecane. The percentage conversion of the substrate, the percentage selectivity and turn over number of the products in the oxidation reaction are calculated as.45,48| Substrate conversion (%) = substrate converted (moles)/substrate used (moles) × 100 |
| Product selectivity (%) = product formed (moles)/substrate used (moles) × 100 |
| Turn over number = mmol of products/mmol of catalyst |
Separation of acetophenone
The reaction mixture of ethylbenzene oxidation was collected and concentrated under reduced pressure. The concentrated mixture was dissolved in 15 ml of ethanol. Then 1 mmol Zn granules were added, and the mixture was stirred for 30 min at room temperature and filtered. After filtration, the solvent was removed under reduced pressure followed by extraction of concentrated mixture with saturated NaHCO3 solution to remove benzoic acid, if any. Then mixture was washed with water, followed by extraction with ether. After removing the aqueous layer, ethereal layer was dried over anhydrous MgSO4. Yellowish oil was obtained which was 80.2% acetophenone with 92% purity; it was characterized by FTIR and LC-MS analysis m/z = 21, 57, 77, 105, 121. FTIR (KBr pellet: cm−1): 2978 (w), 1690 (m), 1263 (w). Separation of acetophenone has been done on the basis of previously reported method.49Results and discussion
Characterization of catalyst
Cu(II) Schiff base intercalated layered double hydroxide, LDH-[NAPABA–Cu(II)] was synthesized by intercalation of NNOO donor Cu(II) complex derived from 2-hydroxy-1-naphthaldehyde and 4-amino benzoic acid into Zn–Al layered double hydroxide. A schematic representation of the preparation procedure of LDH ligand and LDH-[NAPABA–Cu(II)] catalyst is shown in Scheme 1.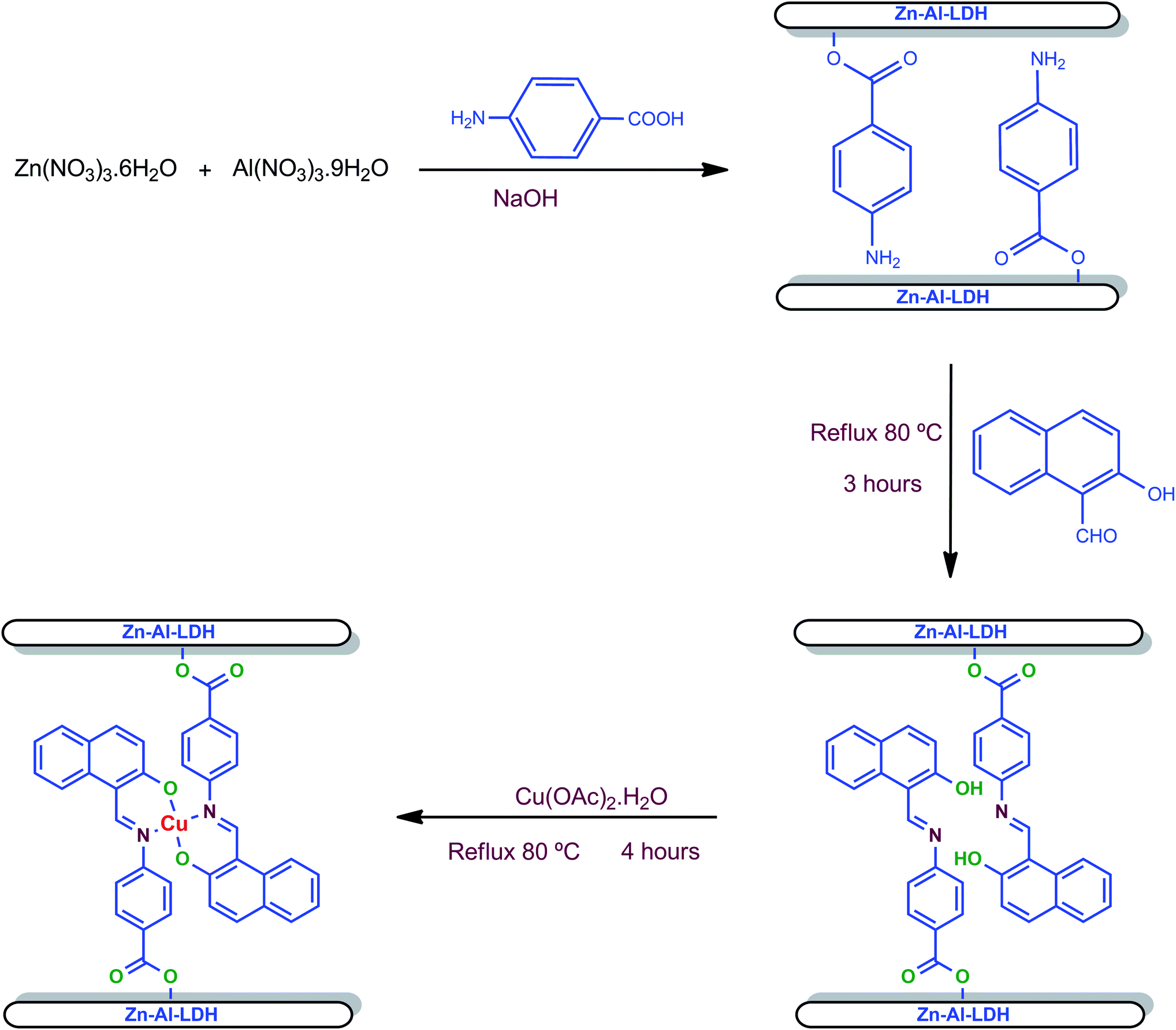 | ||
| Scheme 1 Synthesis of heterogeneous catalyst, LDH-[NAPABA–Cu(II)]. | ||
The physical and analytical data of compounds including chemical composition at different stages of synthesis of catalyst, viz. LDH-[NH2–C6H4COO], LDH-[NAPABA–Cu(II)]b and LDH-[NAPABA–Cu(II)]a (b = before and a = after catalytic reaction) are given in Table 1. The fresh catalyst LDH-[NAPABA–Cu(II)]b contains 8.6% copper contents before catalytic reaction, whereas LDH-[NAPABA–Cu(II)]a contain 7.4% copper contents after seven catalytic cycles, which was determined by ICP-AES. The reduction in the copper contents may be due to several washings of the catalyst after each cycle.
| Catalyst* | Metal contents* (%) | EDX data (wt%) | BET surface area (m2 g−1) | d-spacing (Å) | Particle size (nm) | C/Cu | N/Cu | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zn | Al | C | O | N | Cu | |||||||
| a a: After catalysis, b: before catalysis; *ICP-AES analysis, c: the theoretical value. | ||||||||||||
| LDH-[NH2–C6H4COO] | — | 26.39 | 7.73 | 32.49 | 29.12 | 4.27 | - | 6.92 | 15.71 | 2.4 | — | — |
| LDH-[NAPABA–Cu(II)]b | 8.6 | 20.51 | 6.24 | 47.62 | 15.43 | 3.98 | 6.22 | 19.84 | 22.17 | 2.6 | 29.30 | 2.10 |
| (35.99)c | (2.80)c | |||||||||||
| LDH-[NAPABA–Cu(II)]a | 7.4 | 20.49 | 6.17 | 47.53 | 15.37 | 3.86 | 5.19 | 19.77 | 22.17 | 2.6 | 33.98 | 2.80 |
The elemental composition of the catalyst has determined by EDX analysis. The EDX measurements results of LDH-[NH2–C6H4COO], LDH-[NAPABA–Cu(II)]b and LDH-[NAPABA–Cu(II)]a and C/Cu and N/Cu ratio are listed in Table 1. The presence of carbon, nitrogen, oxygen, zinc, aluminium along with copper metal in LDH-[NAPABA–Cu(II)] confirms the formation of heterogeneous catalyst. The calculation of C/Cu and N/Cu ratio is performed on the basis of elemental analysis. The experimental data of C/Cu and N/Cu ratio in LDH-[NAPABA–Cu(II)] is lesser than the theoretical value which indicates that some metal ions were not coordinated with the ligand ions in LDH-[NAPABA–Cu(II)]. The ideal formula for LDH-[NAPABA–Cu(II)], based on the elemental content and Zn–Al ratio is [Zn0.73Al0.27(OH)2][NAPABA–Cu(II)]0.13[NH2–C6H4COO]0.08·1.46H2O.
The SEM image shown in Fig. 1 of LDH-[NH2–C6H4COO] and LDH-[NAPABA–Cu(II)] clearly shows that both have similar structure, having flake-like aggregates,50 but with different sizes of the flakes. It is clearly indicating that the morphology of the host LDH-[NH2–C6H4COO] is not significantly influenced by the intercalation of the copper complex into the LDH host.
 | ||
| Fig. 1 SEM image of (A) LDH-[NH2–C6H4COO] and (B) LDH-[NAPABA–Cu(II)]. | ||
Fig. 2 shows the XRD patterns of LDH-[NH2–C6H4COO], LDH-[NAPABA] ligand, LDH-[NAPABA–Cu(II)]b and LDH-[NAPABA–Cu(II)]a the d-spacing corresponding to the plane (003). The XRD pattern of LDH-[NH2–C6H4COO] shows the most intense basal reflection at 15.71 Å (003). The characteristic reflections corresponded to (110) plane shows atomic distribution density depending on molar ratio of Zn–Al.51 The results have incorporated in Table 1. The basal spacing of (003) plane increases from 15.71 to 22.17 Å in LDH-[NAPABA–Cu(II)]b, while during the intercalation no change is observed at (110) plane. The gallery height of the catalyst is 17.47 Å when the thickness of the brucite layers (4.7 Å) was subtracted. The increase in gallery height clearly indicates successful intercalation of [NAPABA–Cu(II)] Schiff base complex in the LDH layers. Furthermore, the size of NAPABA–Cu(II) Schiff base complex in the LDH layers is 13.67 Å which was determined by subtracting the bond length of linkage bond between LDH and Cu from gallery height (17.47 Å). The particle size of support, LDH-[NH2–C6H4COO] and heterogeneous catalyst, LDH-[NAPABA–Cu(II)] was calculated using Debye–Scherrer equation.
| D = 0.9λ/βcosθ |
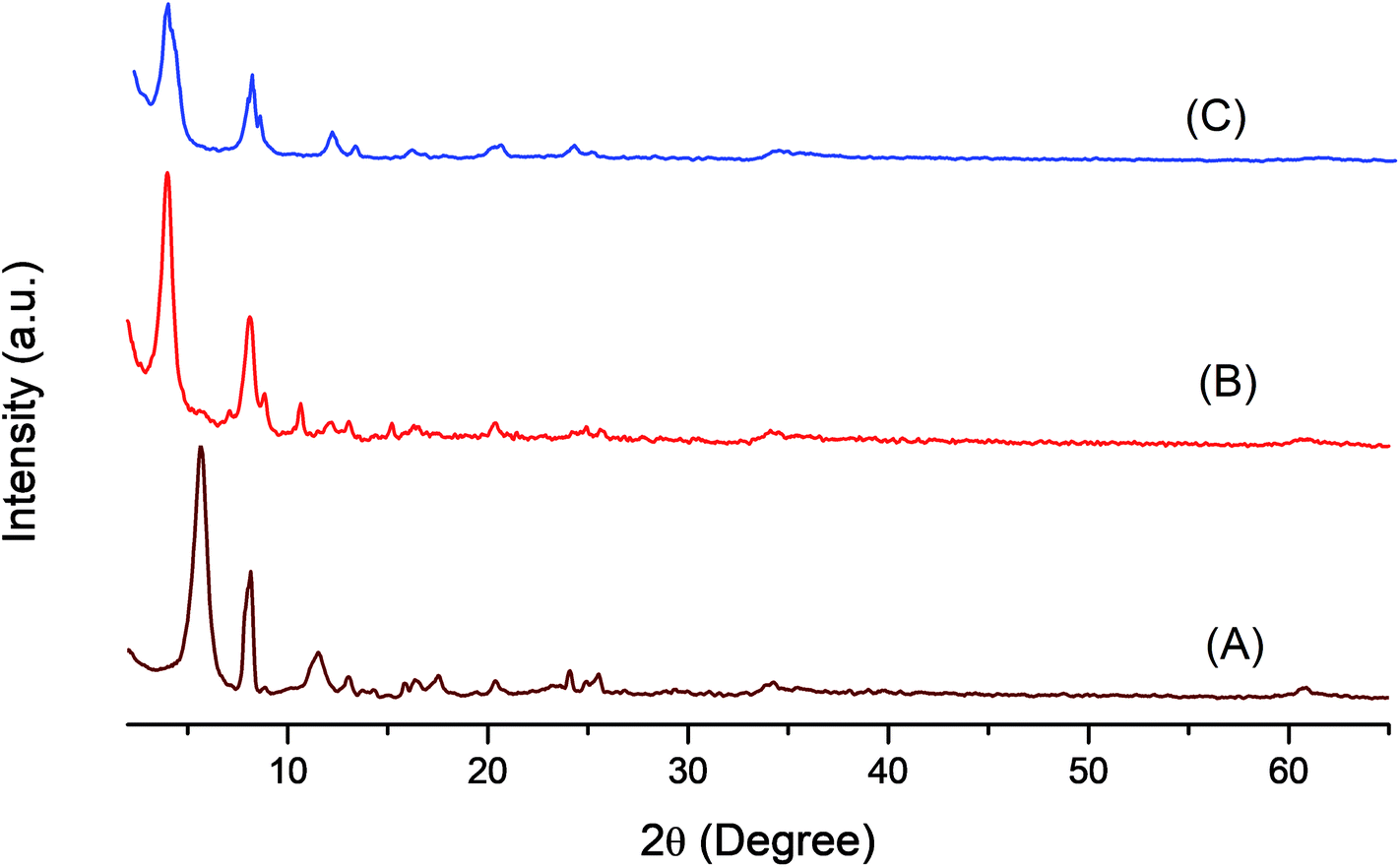 | ||
| Fig. 2 XRD pattern of (A) LDH-[NH2–C6H4COO], (B) LDH-[NAPABA–Cu(II)b and (C) LDH-[NAPABA–Cu(II)]a {a = after and b = before catalytic reactions}. | ||
The average particle size was calculated for most intense peak corresponding to the (003) plane of LDH-[NH2–C6H4COO] and LDH-[NAPABA–Cu(II)] and it was found to be 2.4 and 3.1 nm respectively. The results are incorporated in Table 1. The increase in gallery height and average particle size of LDH clearly indicates successful intercalation of NAPABA–Cu(II) Schiff base complex into the LDH layers.
The TEM images of LDH-[NH2–C6H4COO] and LDH-[NAPABA–Cu(II)] are shown in Fig. 3 having hexagonal flake like structure with aggregates. The particle size varied from 2 to 10 nm, which was consistent with the results of XRD. This average particle size is similar to the range calculated using the Scherrer equation.
 | ||
| Fig. 3 TEM image of (A) LDH-[NH2–C6H4COO] and (B) LDH-[NAPABA–Cu(II)]. | ||
The surface area of support LDH-[NH2–C6H4COO] is 6.915 m2 g−1 whereas surface is of heterogeneous catalyst LDH-[NAPABA–Cu(II)] is 19.84 m2 g−1 (Table 2). The increase in surface area indicates successful intercalation of Cu(II) Schiff base complex into the LDH layers.51 After seven cycles, there is no notable change in BET surface area (19.77 m2 g−1) of the catalyst, which justifies the recyclability of the catalyst.
| Catalysts | Oxidant/solvent | Ethylbenzene conversion (%) | Products selectivity (%) | TON | ||
|---|---|---|---|---|---|---|
| BZ | AP | BA | ||||
| a AP: acetophenone, Bz: benzaldehyde, BA: benzoic acid; *72% TBHP in ethylbenzene; reaction conditions: ethylbenzene (13 mmol); TBHP (39 mmol), temperature (393 K), time 7 h. | ||||||
| No catalyst | *TBHP/solvent-free | — | — | — | — | — |
| LDH-[NH2–C6H4COO] | *TBHP/solvent-free | — | — | — | — | — |
| NAPABA–Cu(II) | *TBHP/solvent-free | 58.4 | 0.44 | 99.56 | — | 531 |
| LDH-[NAPABA–Cu(II)] | H2O2/solvent-free | — | — | — | — | — |
| LDH-[NAPABA–Cu(II)] | O2/solvent-free | 25.1 | 0.63 | 95.60 | 3.77 | 227 |
| LDH-[NAPABA–Cu(II)] | 70% TBHP/solvent-free | 28.5 | 0.44 | 98.41 | 1.15 | 259 |
| LDH-[NAPABA–Cu(II)] | *TBHP/solvent-free | 80.5 | 0.40 | 99.60 | — | 732 |
| LDH-[NAPABA–Cu(II)] | *TBHP/dichloromethane | 55.0 | 48.31 | 36.31 | 15.38 | 499 |
| LDH-[NAPABA–Cu(II)] | *TBHP/methanol | 5.8 | 55.95 | 26.52 | 17.53 | 53 |
| LDH-[NAPABA–Cu(II)] | *TBHP/acetonitrile | 53.8 | 2.74 | 60.16 | 37.10 | 488 |
| LDH-[NAPABA–Cu(II)] | *TBHP/acetone | 23.3 | 0.26 | 57.90 | 41.84 | 211 |
The FTIR spectrum of LDH-[NH2–C6H4COO], NAPABA–Cu(II) and LDH-[NAPABA–Cu(II)] are shown in Fig. 4. In LDH-[NH2–C6H4COO] spectrum, the broad absorption bands at 3390 and 3255 cm−1 are attributed to stretching modes of OH and NH2 groups respectively. The strong band at 1549 cm−1 is attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration. A band at 1445 cm−1 is due to CC stretching of aromatic ring while absorption band at 1399 cm−1 is attributed to the NO3− stretching vibration.52 The bands in the range of 428–784 cm−1 are due to stretching vibration of M–O and O–M–O (M = Zn, Al)53 which includes translation vibrations of Zn–OH at 637 cm−1 and Al–OH at around 517 cm−1 and deformation vibration of HO–Zn–Al–OH at 428 cm−1 which are typical of this class of materials.54,55 In the spectrum of homogeneous catalyst, NAPABA–Cu(II), the broad absorption bands in the range of 3390–3255 cm−1 is attributed to stretching modes of OH and NH2 groups. The band at 1612 cm−1 is due to CN stretching of imines while bands 751 cm−1 and 519 cm−1 arises due to the Cu–N and Cu–O frequency respectively. The spectrum of heterogeneous catalyst, LDH-[NAPABA–Cu(II)] shows all the absorption bands corresponding to LDH-[NH2–C6H4COO] and NAPABA–Cu(II). The band due to CN stretching of imines was shifted to lower frequencies at 1609 cm−1 while the bands due to the Cu–N and Cu–O stretching shifted to higher frequencies i.e. at 759 and 522 cm−1 in the LDH-[NAPABA–Cu(II)].56 The presence of these bands clearly indicates the intercalation of NAPABA–Cu(II) into LDH.
O stretching vibration. A band at 1445 cm−1 is due to CC stretching of aromatic ring while absorption band at 1399 cm−1 is attributed to the NO3− stretching vibration.52 The bands in the range of 428–784 cm−1 are due to stretching vibration of M–O and O–M–O (M = Zn, Al)53 which includes translation vibrations of Zn–OH at 637 cm−1 and Al–OH at around 517 cm−1 and deformation vibration of HO–Zn–Al–OH at 428 cm−1 which are typical of this class of materials.54,55 In the spectrum of homogeneous catalyst, NAPABA–Cu(II), the broad absorption bands in the range of 3390–3255 cm−1 is attributed to stretching modes of OH and NH2 groups. The band at 1612 cm−1 is due to CN stretching of imines while bands 751 cm−1 and 519 cm−1 arises due to the Cu–N and Cu–O frequency respectively. The spectrum of heterogeneous catalyst, LDH-[NAPABA–Cu(II)] shows all the absorption bands corresponding to LDH-[NH2–C6H4COO] and NAPABA–Cu(II). The band due to CN stretching of imines was shifted to lower frequencies at 1609 cm−1 while the bands due to the Cu–N and Cu–O stretching shifted to higher frequencies i.e. at 759 and 522 cm−1 in the LDH-[NAPABA–Cu(II)].56 The presence of these bands clearly indicates the intercalation of NAPABA–Cu(II) into LDH.
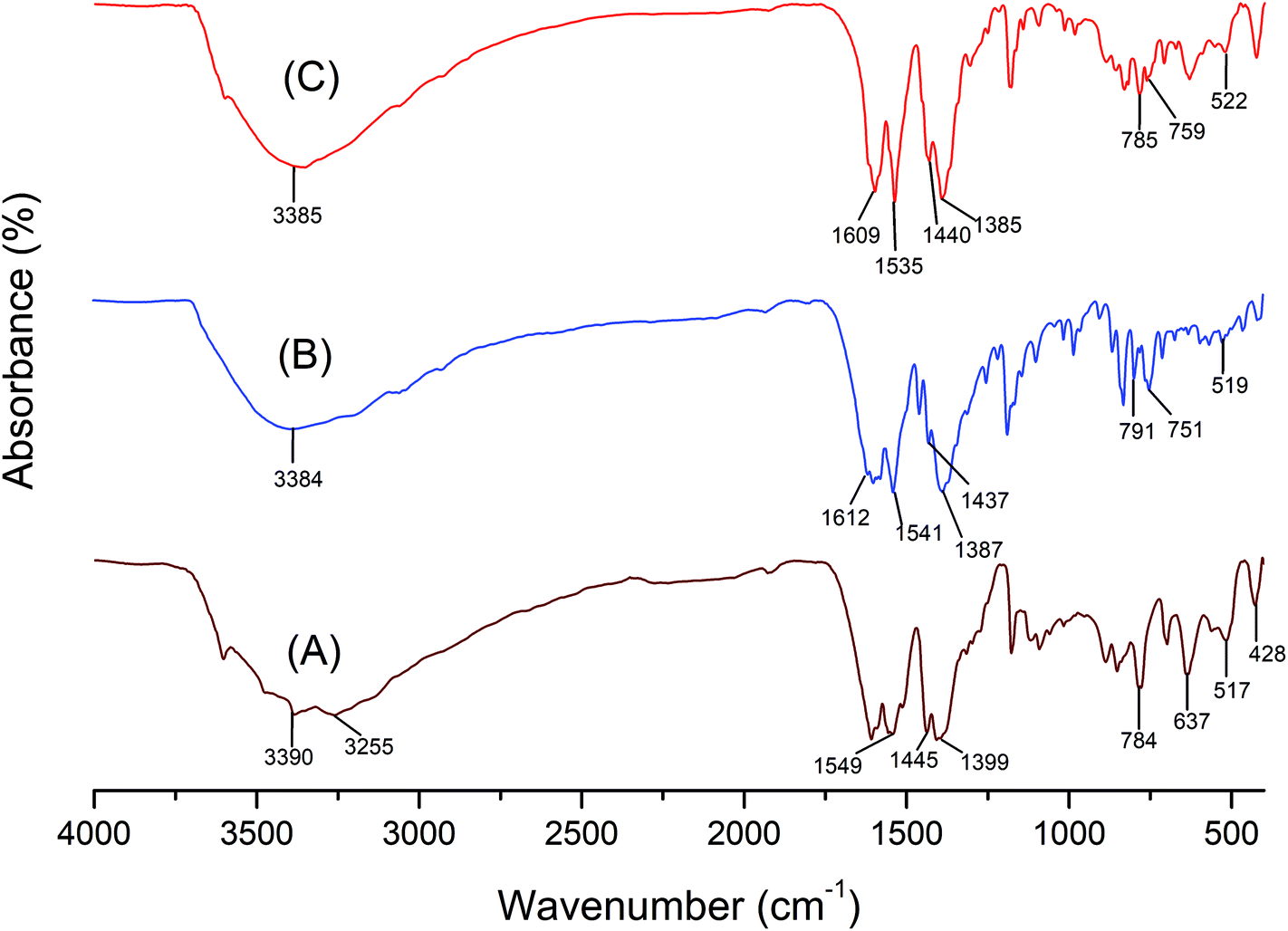 | ||
| Fig. 4 FTIR spectra of (A) LDH-[NH2–C6H4COO], (B) NAPABA–Cu(II) and (C) LDH-[NAPABA–Cu(II)]. | ||
The thermal decomposition behavior of LDH-[NH2–C6H4COO] and LDH-[NAPABA–Cu(II)] was evaluated by TGA analysis and are shown in Fig. 5. The TGA curve of LDH-[NH2–C6H4COO] shows first weight loss from 50 to 170 °C due to the removal of adsorbed water. The second stage corresponds to the degradation of the brucite like layer and removal of inter layer benzoate anions in temperature range of 200–500 °C. In the TGA curve of LDH-[NAPABA–Cu(II)], shows first weight loss from room 50 to 175 °C. A second step in the range 213 to 330 °C is assigned to partial dehydroxylation of the double hydroxide layers. The third weight loss in the temperature range 359 to 550 °C was due to complete decomposition of the organic part. On the basis of decomposition temperatures, it can be inferred that LDH-[NH2–C6H4COO] and LDH-[NAPABA–Cu(II)] both are thermal stable up to 550 °C.
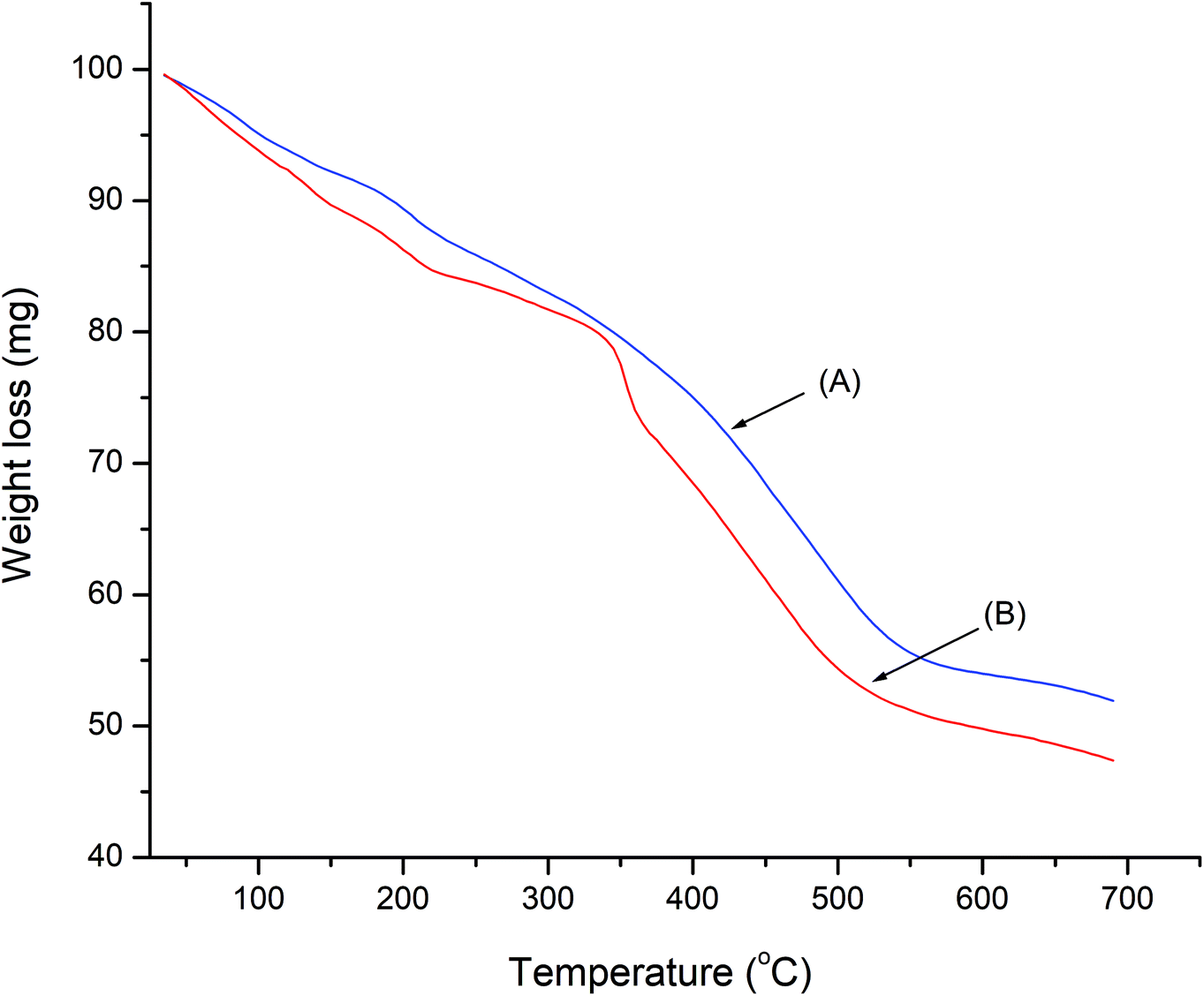 | ||
| Fig. 5 TGA curve of (A) LDH-[NH2–C6H4COO] and (B) LDH-[NAPABA–Cu(II)]. | ||
The EPR spectra of heterogeneous catalyst, LDH-[NAPABA–Cu(II)] recorded at room temperature shown in Fig. 6. The spectra exhibit anisotropic signal, without any hyperfine splitting, with g∥ = 2.13, g⊥ = 2.03 and gav = 2.08. The trend g∥ > g⊥ > 2.00 indicates the covalent environment of metal ion with the Schiff base in which the unpaired electron is localized in dx2−y2 orbital of the Cu(II) ion and copper is present in +2 oxidation state.57 Furthermore, the Cu(II) complex exhibits a G value [G = (g∥ − 2.0023)/(g⊥ − 2.0023)] of 4.33, which is slightly higher than 4, indicating no exchange interaction in the copper(II) complex.58,59 Thus, a square planar environment was proposed for the complexes.57,60
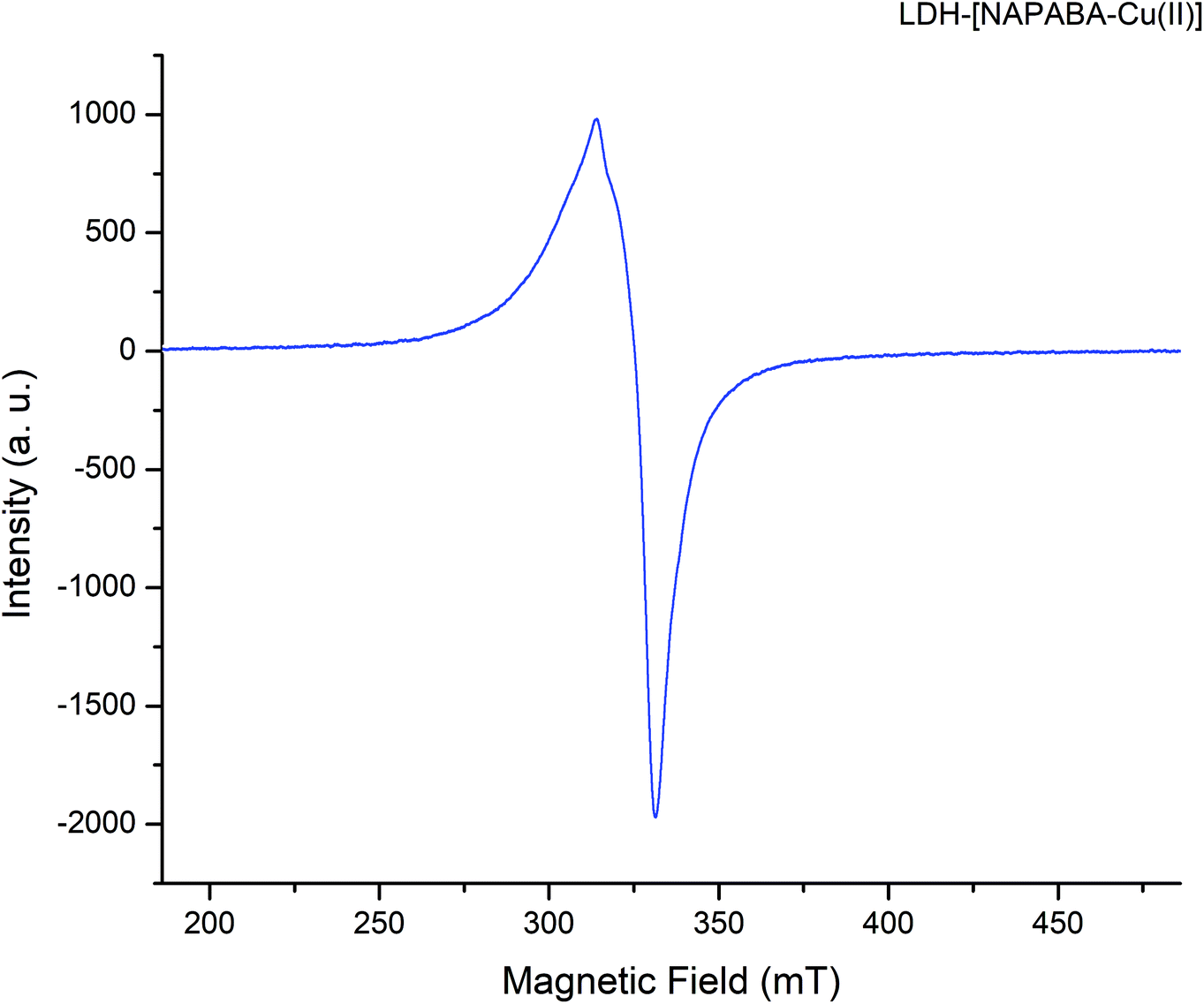 | ||
| Fig. 6 EPR spectra of LDH-[NAPABA–Cu(II)]. | ||
The DRUV-Vis spectra of LDH-[NH2–C6H4COO], NAPABA–Cu(II) and LDH-[NAPABA–Cu(II)] are shown in Fig. 7. The electronic spectrum of homogeneous complex, NAPABA–Cu(II) shows three bands at 262, 318 and 435 nm. The first band at 262 nm may be assigned to π → π* of aromatic rings. The second band at 318 nm is due to n → π* transitions of imines group (CN). The third band in the range of 362–490 nm is assigned due to ligand to metal charge transfer (LMCT) transition in the metal complex. An additional weak and broad band in the range of 550–710 nm in metal complexes is due to the d–d transition. In heterogeneous catalyst, LDH-[NAPABA–Cu(II)], after heterogenization of homogeneous catalyst the intensity of all bands get reduced and all absorption bands appear at almost similar position. The band due to the d–d transition also appears with very low intensity.
 | ||
| Fig. 7 DRUV-visible spectra of (A) LDH-[NH2–C6H4COO], (B) NAPABA–Cu(II) and (C) LDH-[NAPABA–Cu(II)]. | ||
Catalytic activity studies
Oxidation of ethylbenzene
The oxidation of ethylbenzene was investigated using LDH-[NAPABA–Cu(II)] under solvent-free conditions with TBHP as an oxidant. We have considered various aspects of catalytic oxidation of ethylbenzene for 7 h of reaction. When reaction carried out under following conditions, reaction did not take place; (i) in the presence of support, LDH-[NH2–C6H4COO] and TBHP, (ii) in the presence of heterogeneous catalyst LDH-[NAPABA–Cu(II)] and without TBHP and (iii) in the presence of only TBHP (without catalyst). Hence, both the copper catalyst and TBHP are required for the catalytic oxidation of ethylbenzene and (iii) also ruled out auto-oxidation. The reaction of ethylbenzene with TBHP in presence of LDH-[NAPABA–Cu(II)] and NAPABA–Cu(II), under similar reaction conditions, gave acetophenone, benzaldehyde and benzoic acid. It is notable, no oxidation products were observed in the aromatic ring of the ethylbenzene. The catalytic performance of catalysts was assessed based on conversion of substrate and selectivity of the products.It was observed that LDH-[NAPABA–Cu(II)] gives more conversion of ethylbenzene than that of NAPABA–Cu(II) (Table 2). The inferior activity shown by homogeneous catalyst may be attributed to its partial solubility in reaction mixture. The LDH shows high adsorption capacity which make them effective supports for the immobilization of catalytically active species, NAPABA–Cu(II) in to the layers.61 Furthermore uniform dispersion of M(II) and M(III) cations in the LDH layers, and preferred orientation of anions in the interlayer, is useful properties to use LDH as precursors for the preparation of stable supported catalysts.62
The catalytic oxidation of ethylbenzene, catalysed by LDH-[NAPABA–Cu(II)]/TBHP system is explained in Scheme 2.
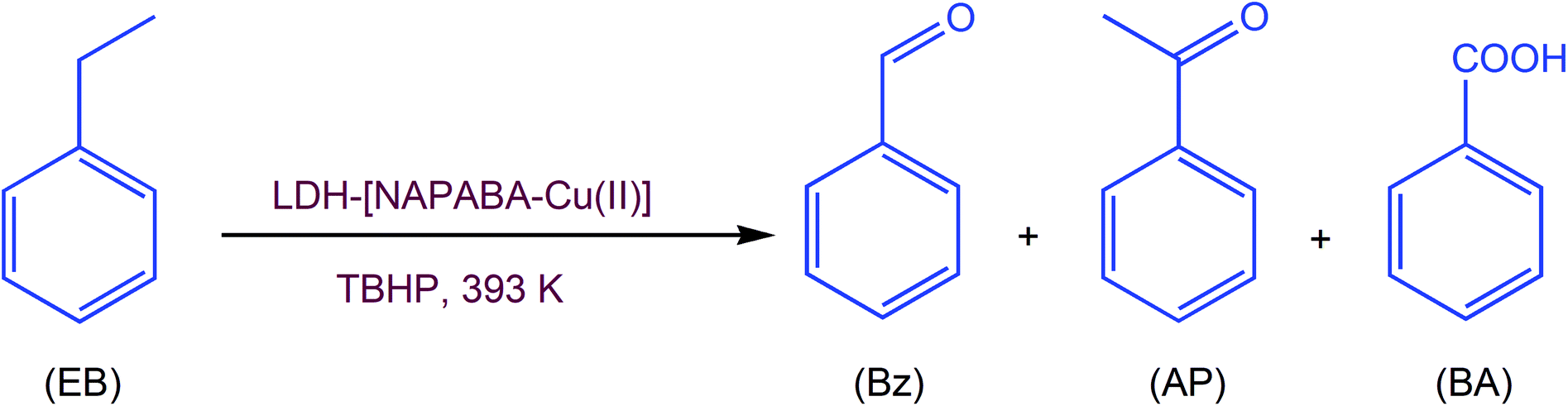 | ||
| Scheme 2 Oxidation of ethylbenzene. | ||
To obtain maximum conversion of ethylbenzene, optimization of various parameters viz. effects of oxidants, solvents, concentration of TBHP, catalyst concentration and reaction temperature have been studied in detail.
In order to optimize ethylbenzene oxidation various parameters viz. effects of oxidants, solvents, concentration of TBHP, catalyst concentration and reaction temperature have been studied in detail.
The effect of various oxidants viz, H2O2, O2, 70% TBHP and 72% TBHP in ethylbenzene was investigated for the oxidation of ethylbenzene. The results are listed in Table 2. When molecular oxygen (O2) was used as the oxidant, the conversion of ethylbenzene (25.1%) was less due to easy expulsion of O2 from the reaction mixture. When H2O2 was used as the oxidant, no conversion was observed due to the high exothermic nature it was decomposed easily and evolved oxygen rapidly. In case of 70% TBHP the conversion of ethylbenzene (28.5%) was less due to interference of water in the reaction whereas 72% TBHP in ethylbenzene gave maximum conversion of ethylbenzene (80.5%). Hence, it was considered as best oxidant for our catalytic system.
The effect of various solvents (acetonitrile, acetone, methanol and dichloromethane) on the oxidation of ethylbenzene catalyzed by LDH-[NAPABA–Cu(II)] was studied to develop an efficient solvent system. It was observed that the conversion of ethylbenzene decreased in the following order with different solvents; 55.0% dichloromethane > 53.8% acetonitrile > 23.3% acetone > 5.8% methanol (Table 2). The result indicates that the aprotic and chlorinated solvents favoured the conversion of ethylbenzene. The major product in the oxidation of ethylbenzene in all solvents except dichloromethane and methanol was acetophenone. The acetophenone selectivity on different solvents decreased in the following order; acetonitrile (60.16%) > acetone (57.90%) > dichloromethane (36.31%) >methanol (26.52%) (Table 2). The selectivity of benzaldehyde and benzoic acid were favoured in protic and chlorinated solvent while the selectivity of acetophenone was favoured in aprotic solvents.63 These results indicate that dipole moments of the solvent may play an essential role in the oxidation of ethylbenzene. It was observed that in presence of solvents conversion of ethylbenzene was decreased due to the blocking of active sites by solvent molecules or may be diffusion competition between solvent and substrate molecules for the active sites. However, the reaction under solvent-free conditions gave higher selectivity and more catalytic activity than in the presence of a solvent.
The effect of concentration of TBHP was studied by considering three different molar ratios (1:1, 1:2 and 1:3) of ethylbenzene to TBHP on the oxidation of ethylbenzene under solvent-free condition at 393 K for 7 hours. The results are given in Table 3 and in Fig. 8A. It was observed that the conversion of ethylbenzene gradually increases with increasing ethylbenzene to TBHP molar ratio from 1:1 to 1:2, the conversion increases from 45.9 to 67.5%. Further increasing molar ratio to 1:3 conversions also increases to 80.5%. At all molar ratios acetophenone (99.60%) was the major product. The conversion of ethylbenzene was increases by 13.1% when ethylbenzene to TBHP molar ratio from 1:2 to 1:3. Therefore, we have considered 1:3 molar ratio of ethylbenzene to TBHP as optimum.
| Entry | Substrate:oxidant ratio |
Catalyst amount (mg) | Temperature (K) | Conversion (%) | Selectivity of products | TON | ||
|---|---|---|---|---|---|---|---|---|
| Bz | AP | BA | ||||||
| a AP: acetophenone, Bz: benzaldehyde, BA: benzoic acid; *72% TBHP in ethylbenzene; reaction conditions: ethylbenzene (13 mmol); TBHP (39 mmol), temperature (393 K), time 7 h. | ||||||||
| 1 | 1:1 |
100 | 393 | 45.9 | 0.37 | 94.57 | 7.06 | 417 |
| 2 | 1:2 |
100 | 393 | 67.5 | 0.24 | 99.49 | 0.27 | 613 |
| 3 | 1:3 |
100 | 393 | 80.5 | 0.16 | 99.60 | 0.24 | 732 |
| 4 | 1:3 |
75 | 393 | 73.0 | 0.15 | 99.28 | 0.57 | 879 |
| 5 | 1:3 |
50 | 393 | 65.7 | 0.23 | 99.24 | 0.53 | 1195 |
| 6 | 1:3 |
100 | 373 | 77.5 | 0.35 | 99.41 | 0.24 | 703 |
| 7 | 1:3 |
100 | 353 | 69.6 | 0.51 | 99.22 | 0.27 | 632 |
 | ||
| Fig. 8 Illustration of various parameters on the oxidation of cyclohexane: (A) effect of ethylbenzene: TBHP molar ratio, (B) effect of catalyst concentration, (C) effect of temperature and (D) recycling of catalyst. | ||
The effect of catalyst concentration on the oxidation of ethylbenzene under solvent-free condition was studied by considering four different concentration of catalyst (50, 75 and 100 mg) at 393 K for 7 hours. The results are incorporated in Table 3 and in Fig. 8B. It was observed that when the concentration of catalyst increases for 50 to 100 mg, the conversion of ethylbenzene was increases from 65.7 to 80.5%. The conversion and selectivity was low at lower catalyst concentration due to the insufficient active sites of catalyst, while the conversion and selectivity increases when the catalyst concentration reaches to 100 mg. It was probably due to the active sites released great amount of nascent oxygen, which ultimately proceeded to vigorous oxidation reaction. Acetophenone was the major product at all catalyst concentrations. Thus, 100 mg catalyst concentration considered as optimum.
The temperature plays an important role in catalytic reactions hence we have studied the oxidation of ethylbenzene at three different temperatures (353, 373 and 393 K). The results are incorporated in Table 3 and in Fig. 8C. It was observed that initially when temperature increases from 353 to 373 K conversion of ethylbenzene increases from 69.6 to 77.5%. Further increase in temperature to 393 K conversions also increases to 80.54%. It may be due to the increase of effective collision of substrate at higher temperature. Thus, 393 K is considered best temperature for reaction. At all reaction temperatures acetophenone was the major product. Thus, overall optimum conditions to obtain maximum conversion of ethylbenzene are 1:3 molar ratio of ethylbenzene to TBHP, 100 mg catalyst and 393 K temperature.
We have also compared our present catalytic system with the literature catalysts to find out the novelty of our catalyst. In Table 4, we have compared present work with the literature catalysts.21–25 The catalyst and applied method in this paper has the advantages in terms of heterogeneous nature, better conversions, selectivity and reusability of the catalyst.
| Catalytic system | Temperature | Oxidant/solvent | Time (h) | Conversion (%) | Product | Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a AP: acetophenone. | |||||||
| Cu(cyclam)-Y | 60 °C | TBHP | 10 | 44.4 | AP | 98 | 21 |
| MCM-41-L/CuCl2 | 30 °C | TBHP | 48 | 47.0 | AP | 97 | 22 |
| Cu-BTC | 150 °C | O2 | 6 | 28.26 | AP | 95.8 | 23 |
| PS-[Cu(hmbmz)2] | 60 °C | TBHP | 6 | 43.5 | AP | 96.6 | 24 |
| PS-[Cu(ligand)n] | 80 °C | TBHP | 8 | 15.0 | AP | 4.5 | 25 |
| Present work | 120 °C | TBHP | 7 | 80.54 | AP | 99.6 | |
Mechanism for ethylbenzene oxidation
Mechanism for the ethylbenzene oxidation was investigated by quenching experiment using radical scavenger 2,6-di-tert-butyl-4-methylphenol (BHT), as a quenching reagent. The reaction was carried out under optimized reaction conditions after completion of 1 h of reaction, BHT was added and the reaction was continued for next 7 h. The gas chromatographic analysis showed no further increase in conversion of ethylbenzene after the addition of BHT. This clearly indicates that oxidation of ethylbenzene by tert-butylhydroperoxide appears to be a radical process because addition of BHT, a radical scavenger, inhibited the formation of any product in the oxidation reaction. Oxidation of ethylbenzene with TBHP catalyzed by LDH-[NAPABA–Cu(II)] probably proceeds via free radical mechanism (Scheme 3). In first step metal centre of catalyst LDH-[NAPABA–Cu(II)] acts as an initiator in the homolytic cleavage of t-BuOOH into free alkoxy (t-BuO˙) and alkylperoxy radicals (t-BuOO˙). In second step, alkylperoxy radicals attack on ethylbenzene to convert it into 1-tert-butylperoxyethylbenzene. In the next step, 1-tert-butylperoxy ethylbenzene leads to the formation of acetophenone, benzaldehyde and benzoic acid. Acetophenone is formed by dehydration while benzaldehyde by losing methanol from 1-tert-butylperoxyethylbenzene. Benzaldehyde on further oxidation gives benzoic acid. Dehydration reaction involves lower energy pathway than the elimination reaction therefore acetophenone was major product (Scheme 3).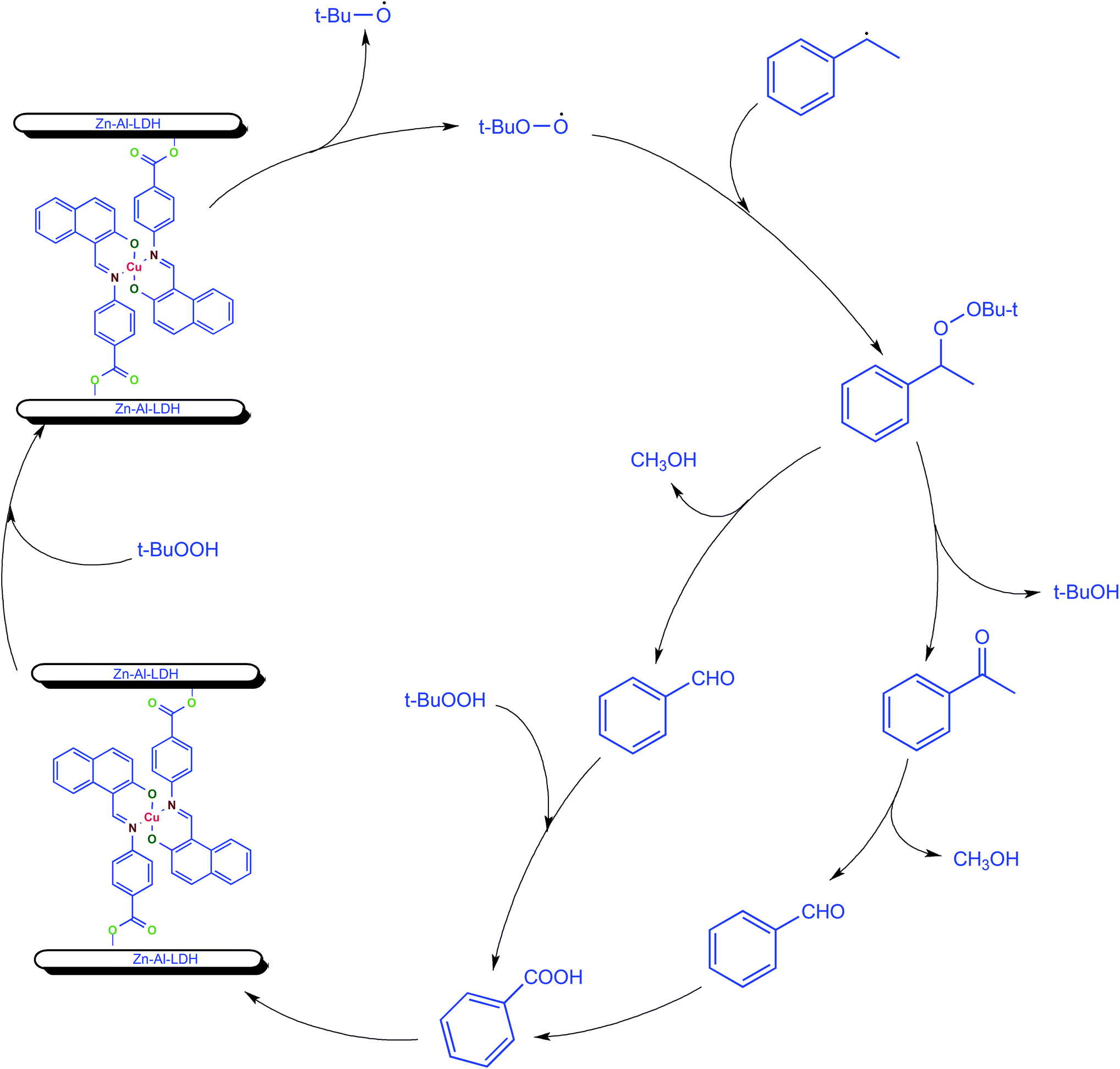 | ||
| Scheme 3 The probable free radical mechanism for oxidation of ethylbenzene with TBHP catalyzed by the LDH-[NAPABA–Cu(II)] catalyst. | ||
Hot filter experiment
The heterogeneity of the catalyst has tested for the oxidation of ethylbenzene by hot filter experiment. The catalytic reaction was carried out under optimized reaction condition during reaction the catalyst was filtered out after 1 h in first cycle at 393 K to avoid re-adsorption of leached copper onto the catalyst surface. The filtrate collected after 1 hour of first cycle was placed again into the reaction flask and the reaction was continued for next 7 h. The gas chromatographic analysis showed no further increase in conversion of ethylbenzene. The reaction mixture did not exhibit any colour indicating the absence of copper, which was estimated using ICP-AES. It suggested that no copper leaching occurred during the catalytic reaction. This observation indicates that the catalyst is heterogeneous in nature.Recycling of catalyst
The reusability of the heterogeneous catalyst is a key factor from commercial and economic points of view. In order to check the reusability of the catalyst recycling experiment was performed under optimized reaction conditions. After the completion of reaction, the catalyst was separated from the reaction mixture by filtration, washed several times with methanol, acetonitrile followed by acetone. The washed catalyst was dried at 333 K overnight to remove the adsorbed solvent molecules from the catalyst surface and then it is further subjected for consecutive catalytic cycles. The catalyst, LDH-[NAPABA–Cu(II)] was recycled for seven cycles for the oxidation of ethylbenzene. The conversion of ethylbenzene was dropped from 80.5 to 77.4% after seven cycles (Fig. 8D). The conversion of ethylbenzene was reduced by 3.1% from first to seven cycles. The recycling results indicate that the catalyst was stable during catalytic reaction and suitable for recycling. The recycled catalyst was characterized by BET surface area, ICP-AES, XRD and EDX.Conclusions
Cu(II) Schiff base complex intercalated into LDH, LDH-[NAPABA–Cu(II)] is a novel heterogeneous catalyst for the liquid phase selective oxidation of ethylbenzene with TBHP under solvent-free condition. The heterogeneous catalyst was found to be highly active as compared to its homogeneous counter parts in our experimental conditions. In the oxidation of ethylbenzene, acetophenone was obtained with 99.60% of selectivity and 80.5% conversion of ethylbenzene under optimized reaction conditions, representing a very promising result. Acetophenone was the major product and can be isolated from reaction mixture with 92% purity. Essentially pure acetophenone was isolated in 80.21% yield. The catalyst was heterogeneous, stable and can be recycled up to seven cycles without significant loss of catalytic activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to University Grant Commission New Delhi, India, for financial support {(UGC-SAP: No. F.540/13/DRS-I/2016 (SAP-I)) and (UGC-RGNF: F1-17.1/2016-17/RGNF-2015-17-SC-MAD-19994/SAIII/Website)}, Head, School of Chemical Sciences, UGC-DAE Consortium of Scientific Research, Devi Ahilya University Indore for providing SEM, XRD, and EDX facilities and Central Salt and Marines Chemical Research Institute (CSMCRI), Bhavnagar, Gujarat for providing BET surface area, GC-MS facilities and STIC Cochin for providing ICP-AES, DRUV-Vis facility.References
- R. A. Sheldon and J. K. Kochi, Metal-catalyzed oxidation of organic compounds, Academic, New York, 1981, p. 34 Search PubMed.
- W. Partenheimer, Catal. Today, 1995, 23, 69–158 CrossRef.
- T. Maeda, A. K. Pee, D. Haa, JP 7.196573, 1995.
- R. H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2437–2450 RSC.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef PubMed.
- P. Karthikeyan, P. R. Bhagat and S. S. Kumar, Chin. Chem. Lett., 2012, 23, 681–684 CrossRef.
- S. M. Islama, A. S. Roya, P. Mondala, M. Mubaraka, S. Mondala, D. Hossaina, S. Banerjeeb and S. C. Santra, J. Mol. Catal. A: Chem., 2011, 336, 106–114 CrossRef.
- S. Velusamy and T. Punniyamurthy, Tetrahedron Lett., 2003, 44, 8955–8957 CrossRef.
- Z. G. Liu, L. T. Ji, J. Liu, L. L. Fu and S. F. Zhao, J. Mol. Catal. A: Chem., 2014, 395, 315–321 CrossRef.
- S. T. Man, C. Ramshaw, K. Scott, J. Clark, D. J. Macquarrie and R. Jachuck, Org. Process Res. Dev., 2001, 5, 204–210 CrossRef.
- B. Gao, Y. Li and N. Shi, React. Funct. Polym., 2013, 73, 1573–1579 CrossRef.
- S. Vetrivel and A. Pandurangan, J. Mol. Catal. A: Chem., 2004, 217, 165–174 CrossRef.
- R. L. Brutchey, I. J. Drake, A. T. Bell and T. D. Tilley, Chem. Commun., 2005, 3736–3738 RSC.
- M. B. Gawande, A. Goswami, F. X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef PubMed.
- C. Adhikary, R. Bera, B. Dutta, S. Jana, G. Bocelli, A. Cantoni, S. Chaudhuri and S. Koner, Polyhedron, 2008, 27, 1556–1562 CrossRef.
- L. H. Abdel-Rahman, A. M. Abu-Dief, M. S. S. Adam and S. K. Hamdan, Catal. Lett., 2016, 146, 1373–1396 CrossRef.
- P. Roy and M. Manassero, Dalton Trans., 2010, 39, 1539–1545 RSC.
- P. Sarmah, R. K. Barman, P. Purkayastha, S. J. Bora, P. Phukan and B. K. Das, Indian J. Chem., Sect. A: Inorg., Phys., Theor. Anal., 2009, 48, 637–644 Search PubMed.
- A. Ghorbani-Choghamarani, Z. Darvishnejad and M. Norouzi, Appl. Organomet. Chem., 2015, 29, 170–175 CrossRef.
- B. Li, X. Luo, Y. Zhu and X. Wang, Appl. Surf. Sci., 2015, 359, 609–620 CrossRef.
- T. H. Bennur, D. Srinivas and S. Sivasanker, J. Mol. Catal. A: Chem., 2004, 207, 163–171 CrossRef.
- S. Gago, S. M. Bruno, D. C. Queiros, A. A. Valente, I. S. Goncalves and M. Pillinger, Catal. Lett., 2011, 141, 1009–1017 CrossRef.
- M. M. Peng, H. T. Jang and M. Palanichamy, Int. J. Control Autom., 2013, 6, 1–12 CrossRef.
- M. R. Maurya, S. Sikarwar, T. Joseph, P. Manikandan and S. B. Halligudi, React. Funct. Polym., 2005, 63, 71–83 CrossRef.
- M. R. Maurya, A. Arya, P. Adao and J. C. Pessoa, Appl. Catal., A, 2008, 351, 239–252 CrossRef.
- D. Carriazo, M. del Arco, E. García-López, G. Marcì, C. Martín, L. Palmisano and V. Rives, J. Mol. Catal. A: Chem., 2011, 342–343, 83–90 CrossRef.
- W. T. Reichle, Solid State Ionics, 1986, 22, 135–141 CrossRef.
- D. Carriazo, M. Del Arco, C. Martín and V. Rives, Appl. Clay Sci., 2007, 37, 231–239 CrossRef.
- A. Seron and F. Delorme, J. Phys. Chem. Solids, 2008, 69, 1088–1090 CrossRef.
- D. Y. Wang, F. R. Costa, A. Vyalikh, A. Leuteritz, U. Scheler, D. Jehnichen, U. Wagenknecht, L. Haussler and G. Heinrich, Chem. Mater., 2009, 21, 4490–4497 CrossRef.
- A. Aguzzi, V. Ambrogi, U. Costantino and F. Marmottini, J. Phys. Chem. Solids, 2007, 68, 808–812 CrossRef.
- R. Marangoni, L. P. Ramos and F. Wypych, J. Colloid Interface Sci., 2009, 330, 303–309 CrossRef PubMed.
- N. Wang, J. Sun, H. Fan and S. Ai, Talanta, 2016, 148, 301–307 CrossRef PubMed.
- X. Wang, G. Wu, X. Liu, C. Zhang and Q. Lin, Catal. Lett., 2016, 146, 620–628 CrossRef.
- K. M. Parida, M. Sahoo and S. Singha, J. Mol. Catal. A: Chem., 2010, 329, 7–12 CrossRef.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef.
- Z. Meng, X. Li, F. Lv, Q. Zhang, P. K. Chu and Y. Zhang, Colloids Surf., B, 2015, 135, 339–345 CrossRef PubMed.
- C. H. Zhou, Appl. Clay Sci., 2011, 53, 87–96 CrossRef.
- Z. P. Xu, J. Zhang, M. O. Adebajo, H. Zhang and C. Zhou, Appl. Clay Sci., 2011, 53, 139–150 CrossRef.
- K. M. Parida, M. Sahoo and S. Singha, J. Catal., 2010, 276, 161–169 CrossRef.
- S. Khare, R. Chokhare, P. Shrivastava, J. S. Kirar and S. Parashar, J. Porous Mater., 2017, 24, 855–866 CrossRef.
- S. Khare, R. Chokhare, P. Shrivastava, J. S. Kirar and S. Parashar, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2016, 55, 1449–1457 Search PubMed.
- S. Khare, P. Shrivastava, J. S. Kirar and S. Parashar, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2016, 54, 403–412 Search PubMed.
- S. Khare, R. Chokhare, P. Shrivastava and J. S. Kirar, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2015, 54, 1032–1038 Search PubMed.
- S. Khare and P. Shrivastava, J. Mol. Catal. A: Chem., 2016, 411, 279–289 CrossRef.
- S. Khare and P. Shrivastava, Catal. Lett., 2016, 146, 319–332 CrossRef.
- S. Bhattcharjee and J. A. Anderson, Catal. Lett., 2004, 95, 119–125 CrossRef.
- S. Khare and R. Chokhare, J. Mol. Catal. A: Chem., 2012, 353–354, 138–147 CrossRef.
- B. Gutmann, P. Elsner, D. Roberge and C. O. Kappe, ACS Catal., 2013, 3, 2669–2676 CrossRef.
- S. Bhattacharjee, K. E. Jeong, S. Y. Jeong and W. S. Ahn, New J. Chem., 2010, 34, 156–162 RSC.
- G. Wu, X. Wang, J. Li, N. Zhao, W. Wei and Y. Sun, Catal. Today, 2008, 131, 402–407 CrossRef.
- M. Mamat, E. Kusrini, A. H. Yahaya, M. Z. Hussein and Z. Zainal, Int. J. Technol., 2013, 1, 73–80 Search PubMed.
- P. Ding and B. Qu, J. Colloid Interface Sci., 2005, 291, 13–18 CrossRef PubMed.
- F. Li, L. Zhang, D. G. Evans, C. Forano and X. Duan, Thermochim. Acta, 2004, 424, 15–23 CrossRef.
- Y. Feng, D. Li, Y. Wang, D. G. Evans and X. Duan, Polym. Degrad. Stab., 2006, 91, 789–794 CrossRef.
- B. G. Jeong, C. P. Rim, H. N. Chae, K. H. Chjo, K. C. Nam and Y. K. Choi, Bull. Korean Chem. Soc., 1996, 17, 688–693 Search PubMed.
- A. Sreekanth and M. R. P. Kurup, Polyhedron, 2003, 22, 3321–3332 CrossRef.
- G. R. Reddy, S. Balasubramaniana and K. Chennakesavulu, J. Mater. Chem. A, 2014, 2, 15598–15610 Search PubMed.
- T. Rosu, E. Pahontu, C. Maxim, R. Georgescu, N. Stanica and A. Gulea, Polyhedron, 2011, 30, 154–162 CrossRef.
- N. Raman, Y. P. Raja and A. Kulandaisamy, Proc. – Indian Acad. Sci., Chem. Sci., 2001, 113, 183–189 CrossRef.
- S. Nishimura, A. Takagakib and K. Ebitani, Green Chem., 2013, 15, 2026–2042 RSC.
- J. T. Feng, Y. J. Lin, D. G. Evans, X. Duan and D. Q. Li, J. Catal., 2009, 266, 351–358 CrossRef.
- D. Habibi and A. R. Faraji, C. R. Chim., 2013, 16, 888–896 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |