
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lindera cyclopentenedione intermediates from the roots of Lindera aggregata†
Lin Chena,
Bo Liub,
Jun-Jie Denga,
Jun-Sheng Zhanga,
Wei Lia,
Abrar Ahmeda,
Sheng Yina and
Gui-Hua Tang *a
*a
aSchool of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, Guangdong 510006, China. E-mail: tanggh5@mail.sysu.edu.cn; Fax: +86-20-39943043; Tel: +86-20-39943043
bThe Second Clinical Medical College, Guangzhou University of Chinese Medicine, Guangzhou 510006, China
First published on 16th May 2018
Abstract
Chromatographic fractionation of the roots of Lindera aggregata has led to the isolation of three new monomers of Lindera cyclopentenedione derivatives (1–3), a pair of new enantiomers of bi-linderone derivatives (4a/4b), and six known Lindera cyclopentenediones (5–8 and 9a/9b). Their structures were determined by NMR and MS data. The absolute configurations of the new bi-linderone derivative enantiomers (4a/4b) were determined by ECD calculation. (±)-Lindepentone A (1) presents the novel skeleton of 3,5-dioxocyclopent-1-enecarboxylate. Lindoxepines A (2) and B (3) present an unprecedented oxepine-2,5-dione derivative skeleton, which may be enlightening for the in vivo biosynthesis of the monomers of Lindera cyclopentenediones. A possible biosynthetic pathway for 1 and a plausible biosynthetic pathway from stilbenes to Lindera cyclopentenediones via the key intermediates 2 and 3 were postulated. The inhibitory activity of these compounds against three microorganisms was also evaluated.
Introduction
The dry roots of Lindera aggregata (Sims) Kosterm. (L. strychnifolia) (Lauraceae), Radix Linderae (Chinese name “Wuyao” and Japanese name “Tendai-Uyaku”) have been used for many years as a well-known traditional medicine for the treatment of several diseases such as rheumatism, chest and abdomen pain, regurgitation, and frequent urination.1,2 In China, these evergreen shrubs or small trees of L. aggregata are mainly distributed in Anhui, Fujian, Guangdong, Zhejiang, and other southern provinces.3 So far, various types of natural products such as sesquiterpenoids, alkaloids, flavonoids, and lucidones have been isolated from the plant of L. aggregata, some of which exhibit various types of bioactivity such as antioxidation, cytotoxicity, protection against postischemic myocardial dysfunction, and the improvement of insulin sensitivity.4,5 Lucidones, a rare group of cyclopentenediones currently reported only in five species of the genus Lindera (L. aggregata, L. lucida, L. erythrocarpa, L. oxyphylla, and L. pipericarpa), mainly consist of several monomers (e.g. linderone, methyllinderone, lucidone, and methyllucidone) and dimers (e.g. linderaspirone A and bi-linderone) with various biological activities such as antimicrobial activity.5–7 Herein, we use the term “Lindera cyclopentenediones” to represent this group of structures. The intriguing structures and diverse activity of Lindera cyclopentenedione dimers have attracted broad interest from synthetic chemists. To date, several research groups have totally synthesized linderaspirone A and bi-linderone through different synthetic methods, in which the preparation of the key precursor methyllinderone is a necessary route.8–10 However, the biosynthesis of the monomers of Lindera cyclopentenediones in vivo has not been reported. The novel structures of lindoxepines A and B (2 and 3) discovered from L. aggregata may shed some light on this issue (Fig. 1). In the present study, besides the two novel oxepine-2,5-dione derivatives (2 and 3), a novel 3,5-dioxocyclopent-1-enecarboxylate derivative (1) and a pair of enantiomers of bi-linderone derivatives (4a/4b), six known Lindera cyclopentenediones (5–8 and 9a/9b) were obtained from this plant during our ongoing search for structurally diverse and biologically significant compounds from medical plants.11–17 A possible biosynthetic pathway for 1 and a plausible biosynthetic pathway from stilbenes to Lindera cyclopentenediones via the key intermediates 2 and 3 were postulated. The inhibitory effects of these compounds on three microorganisms were also evaluated using the microplate reader method.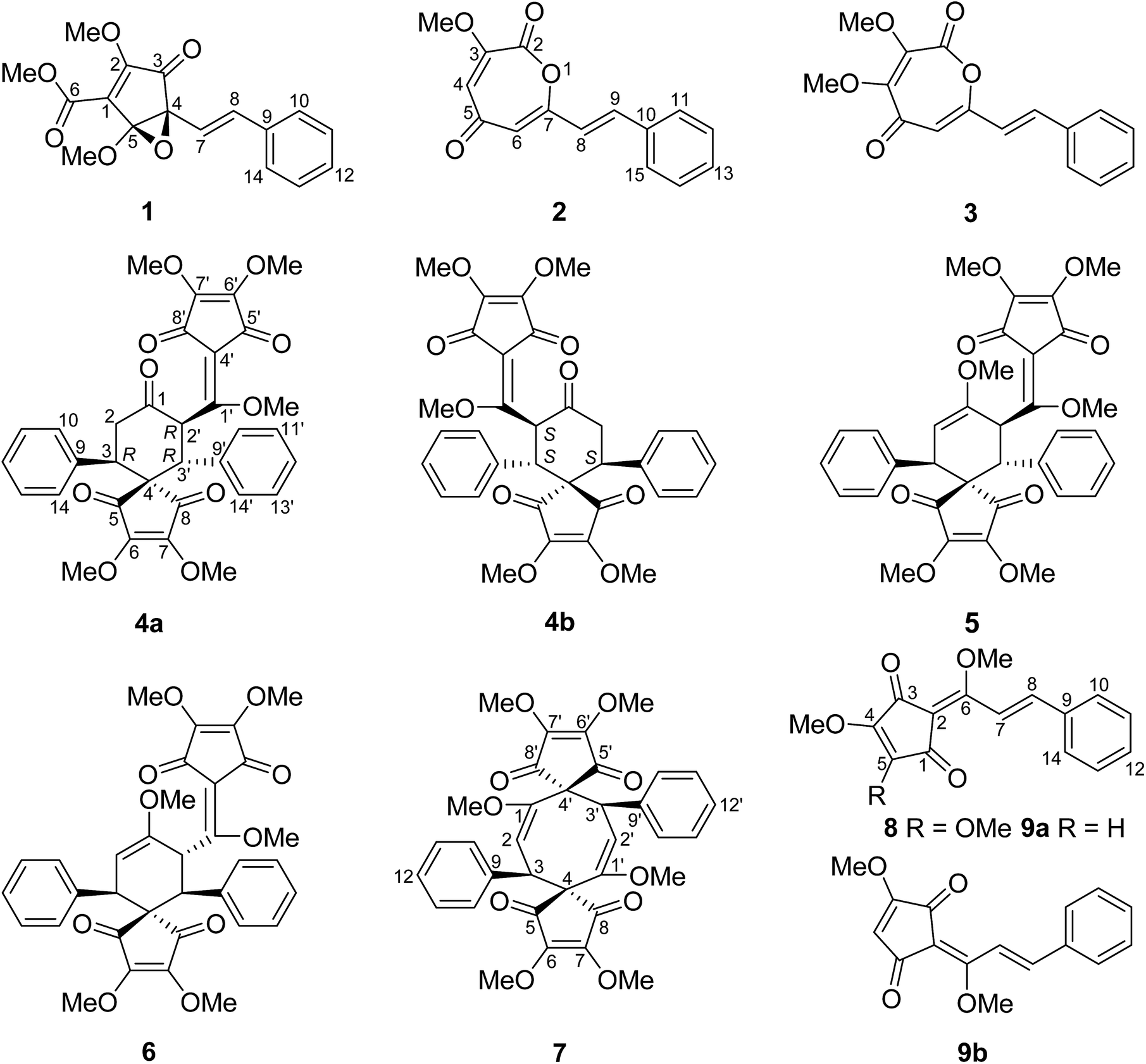 | ||
| Fig. 1 Structures of the compounds isolated from Lindera aggregata. | ||
Results and discussion
HRESIMS of 1 displayed a pseudomolecular ion peak at m/z 317.1011 [M + H]+ (calcd 317.1020), allowing the molecular formula C17H16O6 with ten degrees of unsaturation to be assigned. The IR spectrum exhibited absorptions for unsaturated ketone (1737 cm−1) and phenyl (1636, 1497, and 1449 cm−1) moieties. The 1H and 13C NMR data (Table 1) displayed signals for three methoxy groups [δH 4.26, 3.80, and 3.59 (each 3H, s); δC 60.2, 56.1, and 53.1 (each CH3)], a mono-substituted phenyl [δH 7.44 (2H, d, J = 7.5 Hz), 7.35 (2H, t, J = 7.5 Hz), and 7.27 (1H, t, J = 7.5 Hz); δC 138.0 (C), 129.7 (CH × 2), 128.9 (CH), and 127.6 (CH × 2)], a trans-double bond [δH 6.87 (1H, d, J = 16.1 Hz) and 6.33 (1H, d, J = 16.1 Hz); δC 133.0 (CH) and 127.9 (CH)], a tetrasubstituted double bond [δC 158.7 (C) and 136.6 (C)], a conjugated ketone [δC 196.0 (C)], one ester carbonyl group [δC 170.8 (C)], and two oxygenated quaternary carbons [δC 89.7 (C) and 83.3 (C)]. This collective information suggests that 1 is analogous to the cyclopentenedione derivatives, methyllinderone (8)18 and methyllucidone (9),19 co-isolated in the current study. The significant difference was that the formation of cyclopentenone had changed due to the presence of the ester carbonyl group and the two oxygenated quaternary carbons. The HMBC correlations of H-7 to C-9, H-8 to C-10 (14), and H-10 (14) to C-8 confirmed that the trans-double bond was linked to the mono-substituted phenyl to form an (E)-styryl moiety, which was located at the oxygenated quaternary carbon (C-4) by the HMBC correlations of H-8 to C-4 and H-7 to C-3, C-4, and C-5 (Fig. 2). The determination of the cyclopentenone characteristic of a conjugated system (ketone–tetrasubstituted double bond–ester carboxyl group, C-3–Δ1–C-6) with a methoxy group at C-2 and a methoxy group at C-6 was confirmed by the comparison of these carbon chemical shifts with those of the substructure of the synthesis, (4R,5R)-ethyl-4-(allyloxy)-2-methoxy-4-(4-methoxyphenyl)-3-oxo-5-phenylcyclopent-1-enecarboxylate,20 and the HMBC correlations of the methoxy protons (δH 4.26) to C-2 and the methoxy protons (δH 3.80) to C-6. As nine of the ten degrees of unsaturation were accounted for by a phenyl group, two double bonds, a ketone group, one ester carbonyl group, and a pentyl ring, the remaining one degree of unsaturation suggested that an epoxy ring was present between the two oxygenated quaternary carbons (C-4 and C-5). The HMBC correlation of the methoxy protons (δH 3.59) to C-5 determined that this methoxy group was located at the ketal carbon (C-5). Thus, the gross structure of 1 was deduced as shown, and represented a novel 3,5-dioxocyclopent-1-enecarboxylate derivative.| Position | 1a | Position | 2b | 3b | |||
|---|---|---|---|---|---|---|---|
| δH, multi (J in Hz) | δC, type | δH, multi (J in Hz) | δC, type | δH, multi (J in Hz) | δC, type | ||
| a In CD3OD.b In CDCl3. | |||||||
| 1 | 136.6, C | 2 | 151.9, C | 144.6, C | |||
| 2 | 158.7, C | 3 | 160.7, C | 160.8, C | |||
| 3 | 196.0, C | 4 | 7.05, d (2.3) | 119.3, CH | 149.9, C | ||
| 4 | 83.3, C | 5 | 179.7, C | 176.9, C | |||
| 5 | 89.7, C | 6 | 6.37, d (2.3) | 115.2, CH | 6.43, s | 114.7, CH | |
| 6 | 170.8, C | 7 | 162.5, C | 161.3, C | |||
| 7 | 6.33, d (16.1) | 127.9, CH | 8 | 6.72, d (16.0) | 118.9, CH | 6.69, d (16.1) | 118.7, CH |
| 8 | 6.87, d (16.1) | 133.0, CH | 9 | 7.561, d (16.0) | 138.0, CH | 7.50, d (16.1) | 138.0, CH |
| 9 | 138.0, C | 10 | 134.8, C | 134.8, C | |||
| 10/14 | 7.44, d (7.5) | 127.6, CH | 11/15 | 7.562, dd (7.6, 1.8) | 127.9, CH | 7.55, dd (8.0, 1.8) | 127.9, CH |
| 11/13 | 7.35, t (7.5) | 129.7, CH | 12/14 | 7.38–7.43, m | 129.2, CH | 7.38–7.43, m | 129.2, CH |
| 12 | 7.27, t (7.5) | 128.9, CH | 13 | 7.38–7.43, m | 130.2, CH | 7.38–7.43, m | 130.2, CH |
| 2-OMe | 4.26, s | 60.2, CH3 | 4-OMe | 4.03, s | 61.3, CH3 | ||
| 5-OMe | 3.59, s | 56.1, CH3 | 3-OMe | 4.00, s | 53.6, CH3 | 4.01, s | 53.3, CH3 |
| 6-OMe | 3.80, s | 53.1, CH3 | |||||
 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of compounds 1–4. | ||
The relative configuration of 1 was determined by NOESY experiment. The observed cross-peaks of H-7 and H-8 with the protons of 5-OCH3 suggested that the styryl moiety and 5-OCH3 were cofacial and were arbitrarily assigned as the α-orientation. This compound might be a racemic mixture because its specific rotation was zero. Therefore, compound 1 was named as (±)-lindepentone A.
Structurally, for the biosynthesis of the novel skeleton of 3,5-dioxocyclopent-1-enecarboxylate, a putative biogenetic pathway to 1 was proposed in Scheme 1 from a stilbene precursor, 2,5,6-trihydroxy-3-methoxystilbene, which could be modified through oxidation followed by hydroperoxy-mediated ring contraction21 to form the intermediate i. Enol-isomerization could convert i to ii which can be further oxidized to iii, which could be intraketally ketalized to form the epoxy ring, and finally methylated to give 1.
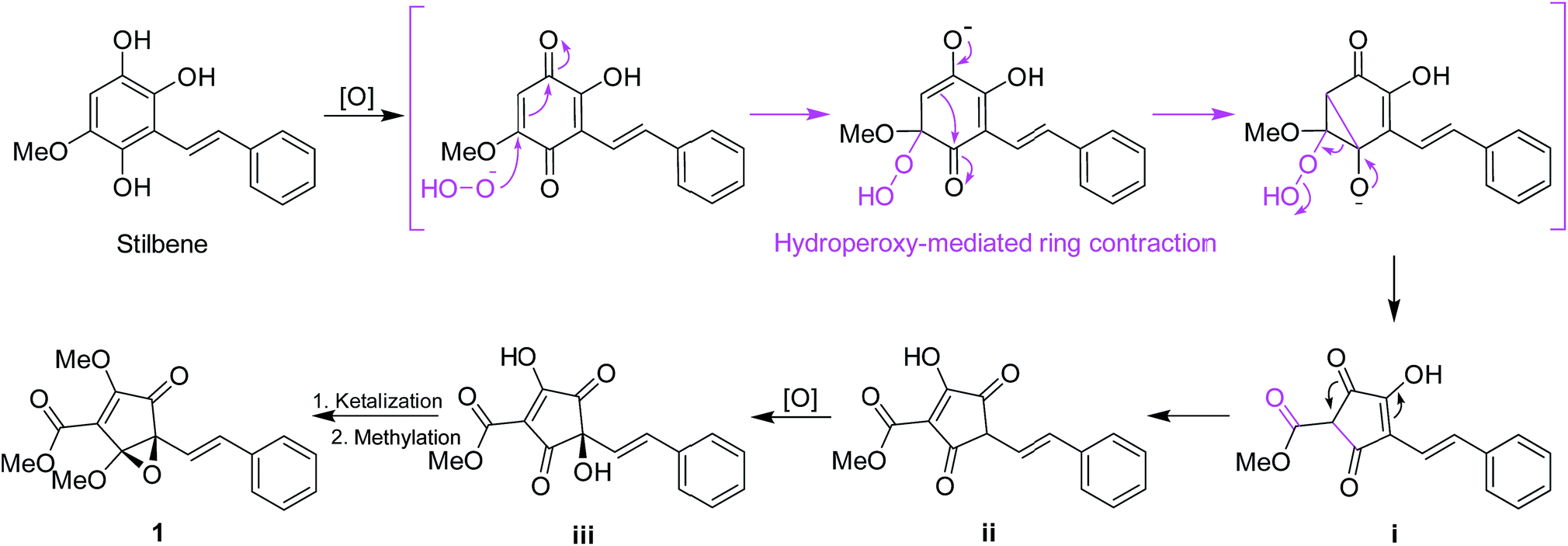 | ||
| Scheme 1 Plausible biosynthetic pathway for 1. | ||
Lindoxepine A (2) has the molecular formula C15H12O4 with ten degrees of unsaturation as established by the HRESIMS (m/z 257.0800 [M + H]+, calcd 257.0808) and 13C NMR data. The 1H and 13C NMR data of 2 (Table 1) exhibited signals for a methoxy group [δH 4.00 (3H, s); δC 53.6 (CH3)] and one (E)-styryl moiety [δH 7.561 and 6.72 (each 1H, d, J = 16.0 Hz, E-double bond protons), 7.562 (2H, dd, J = 7.6, 1.8 Hz, Ar–H), and 7.38–7.43 (3H, m, Ar–H); δC 138.0 and 118.9 (each CH, double bond carbons), 134.8 and 130.2 (each C, Ar–C), and 129.2 and 127.9 (each CH × 2, Ar–C)] as in 1, which was further supported by the HSQC and HMBC correlations (Fig. 2). The remaining resonances in the 1H and 13C NMR data of 2 (Table 1) were very similar to oxepine-2,5-dione derivatives.22 Analysis of its 2D NMR correlations (Fig. 2) and degrees of unsaturation further confirmed the presence of an oxepine-2,5-dione moiety with two substituents. The HMBC correlations of H-8 to C-6, H-9 to C-7, and H-6 to C-8 determined that the (E)-styryl group was located at C-7, and the methoxy group was placed at C-3 by the HMBC correlation of the protons to C-3. Thus, compound 2 was elucidated as shown, and represented a novel oxepine-2,5-dione derivative firstly isolated as a natural product.
The molecular formula of compound 3 was determined to be C16H14O5 by the HRESIMS and 13C NMR data. Comparison of the 1D NMR data (Table 1) and MS information of 3 with those of 2 suggested that 3 had an additional methoxy group at the down-shifted carbon C-4 [δC 149.9 (C) in 3; δC 119.3 (CH) in 2], which was further supported by the HMBC correlations of the protons (δH 4.03) of OCH3 to C-4 (Fig. 2). Thus, compound 3 was elucidated as lindoxepine B.
Lindoxepines A (2) and B (3) present an unprecedented oxepine-2,5-dione derivative skeleton, which may be enlightening for the in vivo biosynthesis of the monomers of Lindera cyclopentenediones. As shown in Scheme 2, the stilbene natural products may be oxidized to form p-benzoquinone derivatives (iv), followed by Baeyer–Villiger oxidation to obtain the key intermediates 2 and 3. Alcoholysis of 2 and 3 yielded v, which was then converted into the known Lindera cyclopentenediones, lucidone and linderone,5 by Dieckmann condensation. Moreover, the co-isolated methyllinderone (8)18 and methyllucidone (9)19 should be the methylated products of linderone and lucidone, respectively. Thus, lindoxepines A (2) and B (3) may be the key intermediates for the synthesis of Lindera cyclopentenediones in vivo.
 | ||
| Scheme 2 The plausible biosynthetic pathway from stilbenes to Lindera cyclopentenediones via the key intermediates (2 and 3) isolated from Lindera aggregata. | ||
Compound 4 was isolated as a white amorphous powder, and its molecular formula was established as C33H30O10 by the HRESIMS and 13C NMR data. The 1H and 13C NMR signals (Table 1) of 4 bore a high resemblance to those of epi-bi-linderone (5),8 a synthesized compound firstly isolated as a natural product in the current study, except that the characteristic signals for the enol ether moiety [δH 4.83 (1H, d, J = 5.1 Hz, H-2) and 3.58 (3H, s, 1-OMe); δC 155.2 (C, C-1), 95.6 (CH, C-2), and 55.0 (CH3, 1-OMe)] in 5 were not observed in 4 but signals were shown for an isolated ketone carbonyl (δC 206.4) and a methylene [δH 3.54 (1H, dd, J = 17.0 and 14.0 Hz) and 2.76 (1H, dd, J = 17.0 and 2.9 Hz); δC 41.5 (CH2)] in its 1D NMR spectrum. This information suggested that the enol ether at Δ1 was hydrolyzed to form a ketone at C-1, which was supported by the 1H–1H COSY correlations of H2-2 with H-3 and the HMBC correlations of H-2 and H-2′ to the ketone (C-1) (Fig. 2). Thus, the gross structure of 4 was elucidated as shown in Fig. 2.
The relative configuration of 4 was determined by the analysis of its coupling constant and NOE correlations. The big coupling constant (J2′/3′ = 13.3 Hz) of H-2′ and H-3′ in the 1H NMR spectrum suggested that H-2′ and H-3′ had a trans-configuration, and H-2′ and H-3′ were arbitrarily assigned as the α- and β-orientation, respectively. The NOE correlation of H-3/H-2′ in the NOESY spectrum indicated that H-3 was in the α-orientation (Fig. 3).
 | ||
| Fig. 3 Key NOE correlations of compound 4. | ||
Compound 4 was a racemic mixture because its specific rotation approached zero and it had no cotton effect in its ECD spectrum. Subsequently, 4 was subjected to HPLC with a chiral column to obtain the enantiomers 4a and 4b, which had opposite specific rotations ([α]25D = +274.4 for 4a and [α]25D = −286.8 for 4b) and mirror image-like ECD curves (Fig. 4). The experimental ECD spectra of 4a and 4b were compared with the calculated ECD spectra of (RRR)-4 or (SSS)-4 by the TDDFT method to determine the absolute configurations of the enantiomers. In Fig. 4, the experimental ECD spectrum of 4a matched the calculated ECD curve for (RRR)-4, indicating that 4a possessed the same absolute configuration as (RRR)-4. Meanwhile, the absolute configuration of 4b was deduced to be the same as that of (SSS)-4 on the basis of their matched ECD spectra. Thus, the enantiomers 4a and 4b were determined as shown and named (+)-demethoxy-epi-bi-linderone (4a) and (−)-demethoxy-epi-bi-linderone (4b), respectively.
 | ||
| Fig. 4 Experimental ECD spectra of 4a and 4b and TDDFT calculated ECD spectra for RRR-4 and SSS-4. | ||
The known compounds were identified as epi-bi-linderone (5),8 bi-linderone (6),6 linderaspirone A (7),7 methyllinderone (8),18 and methyllucidone (9) (a pair of cis–trans isomers, 9a and 9b)19 by comparing their NMR data with those reported in the literature.
Compounds 1–9 from Lindera aggregata were tested for their antimicrobial activity against the bacteria Bacillus subtilis and the fungi Candida albicans and Aspergillus niger by the microplate reader method. It is shown in Table 3 that compounds 1–4a/4b and 6–9 exhibited weak inhibitory activity against C. albicans (MIC values: 25 μg mL−1 for 6 and 7 and 50 μg mL−1 for the others) and compound 7 had a weak effect on A. niger with a MIC value of 50 μg mL−1. All of the compounds were inactive towards the bacteria B. subtilis (MIC values: >50 μg mL−1) and compound 5 and compounds 1–6, 8, and 9 were inactive towards the fungi C. albicans and A. niger, respectively.
| Position | δH, multi (J in Hz) | δC, type | Position | δH, multi (J in Hz) | δC, type |
|---|---|---|---|---|---|
| 1 | 206.4, C | 1′ | 169.2, C | ||
| 2 | β 3.54, dd (17.0, 14.0); α 2.76, dd (17.0, 2.9) | 41.5, CH2 | 2′ | 6.39, d (13.3) | 52.8, CH |
| 3 | 3.97, dd (14.0, 2.9) | 41.2, CH | 3′ | 4.23, d (13.3) | 45.0, CH |
| 4 | 58.7, C | 4′ | 110.7, C | ||
| 5, 8 | 195.9 and 195.7, each C | 5′, 8′ | 187.5 and 183.3, each C | ||
| 6, 7 | 153.2 and 153.1, each C | 6′, 7′ | 148.7 and 146.9, each C | ||
| 9 | 137.1, C | 9′ | 135.7, C | ||
| 10/14, 11/13, 12 | 7.02–7.24, m, overlapped | 128.8 × 2, 128.6 × 2, and 127.9, each CH | 10′/14′, 11′/13′, 12′ | 7.02–7.24, m, overlapped | 128.8 × 2, 128.4 × 2, and 127.8, each CH |
| 6-, 7-OMe | 3.74 and 3.66, each s | 59.8 and 59.6, each CH3 | 6′-, 7′-OMe | 4.20 and 4.12, each s | 60.2 and 59.7, each CH3 |
| 1′-OMe | 3.65, s | 65.8, CH3 |
Experimental section
General experimental procedures
Optical rotations were measured on a Rudolph Autopol I automatic polarimeter, UV spectra on a Shimadzu UV-2450 spectrophotometer, and IR spectra on Bruker Tensor 37 infrared spectrophotometers. NMR spectra were measured on Bruker AM-400 spectrometers at 25 °C. ESIMS was measured on a Finnigan LCQ Deca instrument, and HRESIMS was performed on a Waters Micromass Q-TOF spectrometer. Silica gel (300–400 mesh, Qingdao Haiyang Chemical Co., Ltd.), reversed-phase C18 (RP-C18) (12 nm, S-50 μm, YMC Co., Ltd.), and Sephadex LH-20 gel (Amersham Biosciences) were used for column chromatography (CC). A Shimadzu LC-20 AT equipped with a SPD-M20A PDA detector was used for HPLC. A YMC-pack ODS-A column (250 × 10 mm, S-5 μm, 12 nm) and a Phenomenex Lux cellulose-2 chiral column (10 × 250 mm, 5 μm) were used for semi-preparative HPLC separation. All of the solvents used were of analytical grade (Guangzhou Chemical Reagents Company, Ltd.).Plant material
The roots of L. aggregata were collected in the Kunming, Yunnan Province, P. R. China, in November 2016, and were authenticated by one of the authors (G. H. Tang). A voucher specimen (accession number: YLAC-201611) has been deposited at the School of Pharmaceutical Sciences, Sun Yat-sen University.Extraction and isolation
The air-dried powder of the roots of L. aggregata (2.5 kg) was extracted with 95% EtOH (3 × 10 L) at rt to give 362 g of crude extract. The extract was suspended in H2O (2 L) and successively partitioned with petroleum ether (PE, 3 × 2 L), EtOAc (3 × 2 L), and n-BuOH (3 × 2 L) to yield three corresponding portions. The EtOAc extract (120 g) was subjected to silica gel CC (PE/EtOAc, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 0:1) to afford Fr. I–Fr. IV. Fr. I (4.5 g) was subjected to silica gel CC (PE/CH2Cl2, 5:1 → 1:1) to get 8 (870 mg). Fr. III (3.2 g) was subjected to silica gel CC (PE/CH2Cl2, 5:1 → 0:1) to afford Fr. IIIa–Fr. IIIc. Fr. IIIb was loaded onto a Sephadex LH-20 column and eluted with MeOH to give Fr. IIIb1–Fr. IIIb3. Fr. IIIb2 was purified on a silica gel CC using PE/CH2Cl2, 10:1 → 0:1 to give 7 (6 mg). Fr. IIIb3 was subjected to silica gel CC (PE/acetone, 5:1 → 2:1) to give 1 (5.3 mg). Fr. IIIa was loaded onto a Sephadex LH-20 column and eluted with MeOH to give Fr. IIIa1–Fr. IIIa2, Fr. IIIa1 was subjected to silica gel CC (PE/EtOAc, 10:1 → 5:1) to afford Fr. IIIa1a–Fr. IIIa1b. Fr. IIIa1a was purified on a semi-preparative HPLC system equipped with a YMC column (MeOH/H2O, 7.5:2.5, 3 mL min−1), to give 5 (15 mg, tR 7 min). Fr. IIIa1b was subjected to silica gel CC (PE/EtOAc, 10:1 → 1:1) to give 6 (4 mg). Compound 4 (12 mg) was purified by using HPLC (MeOH/H2O, 8:2, 3 mL min−1) from Fr. IIIa1b, and this was followed by HPLC with a Phenomenex Lux chiral column (MeOH/H2O, 100:0, 3 mL min−1) to obtain 4a (3.3 mg, tR 15 min) and 4b (3.1 mg, tR 19 min), respectively. Fr. IIIa2 was chromatographed over a RP-C18 column eluted with MeOH/H2O (7:3 → 10:0) to afford Fr. IIIa2a–Fr. IIIa2c. Fr. IIIa2a was loaded onto a Sephadex LH-20 column and eluted with MeOH to give Fr. IIIa2a1–Fr. IIIa2a2. and Fr. IIIa2a1 was purified using HPLC (MeOH/H2O, 8:2, 3 mL min−1), to give 2 (10 mg, tR 10 min) and 3 (4 mg, tR 12 min). Fr. IIIa2a2 was subjected to silica gel CC (PE/CH2Cl2, 3:1 → 1:1) to give 9 (13 mg).
ε) 261 (4.20), 194 (4.31) nm; IR (KBr) νmax 1737, 1636, 1497, 1449, 1377, 1263, 1123 cm−1; for the 1H and 13C NMR data, see Table 1; ESIMS m/z 317.1 [M + H]+; HRESIMS m/z 317.1011 [M + H]+ (calcd for C17H17O6+, 317.1020).ε) 322 (4.28), 264 (4.12), 235 (4.07), 228 (4.06) nm; IR (KBr) νmax 1747, 1654, 1600, 1582, 1449, 1407, 1371, 1258, 1181, 1110 cm−1; for the 1H and 13C NMR data, see Table 1; ESIMS m/z 257.1 [M + H]+; HRESIMS m/z 257.0800 [M + H]+ (calcd for C15H13O4+, 257.0808).ε) 320 (4.30), 257 (4.06), 235 (4.10), 228 (4.12) nm; IR (KBr) νmax 1735, 1633, 1580, 1450, 1435, 1409, 1363, 1229, 1198, 1175, 1149 cm−1; for the 1H and 13C NMR data, see Table 1; ESIMS m/z 287.1 [M + H]+; HRESIMS m/z 287.0911 [M + H]+ (calcd for C16H15O5+, 287.0914).ε) 296 (4.14), 248 (4.14) nm; ECD (c 0.87 × 10−4 M, MeCN) λmax (Δε) 360 (+3.06), 290 (+12.38), 235 (+4.19) nm; IR (KBr) νmax 2955, 2923, 2854, 1721, 1670, 1639, 1460, 1322, 1279, 1208, 1117, 1019 cm−1; for the 1H and 13C NMR data, see Table 2; ESIMS m/z 587.1 [M + H]+; HRESIMS m/z 587.1912 [M + H]+ (calcd for C33H31O10+, 587.1912).
:1 → 0:1) to afford Fr. I–Fr. IV. Fr. I (4.5 g) was subjected to silica gel CC (PE/CH2Cl2, 5:1 → 1:1) to get 8 (870 mg). Fr. III (3.2 g) was subjected to silica gel CC (PE/CH2Cl2, 5:1 → 0:1) to afford Fr. IIIa–Fr. IIIc. Fr. IIIb was loaded onto a Sephadex LH-20 column and eluted with MeOH to give Fr. IIIb1–Fr. IIIb3. Fr. IIIb2 was purified on a silica gel CC using PE/CH2Cl2, 10:1 → 0:1 to give 7 (6 mg). Fr. IIIb3 was subjected to silica gel CC (PE/acetone, 5:1 → 2:1) to give 1 (5.3 mg). Fr. IIIa was loaded onto a Sephadex LH-20 column and eluted with MeOH to give Fr. IIIa1–Fr. IIIa2, Fr. IIIa1 was subjected to silica gel CC (PE/EtOAc, 10:1 → 5:1) to afford Fr. IIIa1a–Fr. IIIa1b. Fr. IIIa1a was purified on a semi-preparative HPLC system equipped with a YMC column (MeOH/H2O, 7.5:2.5, 3 mL min−1), to give 5 (15 mg, tR 7 min). Fr. IIIa1b was subjected to silica gel CC (PE/EtOAc, 10:1 → 1:1) to give 6 (4 mg). Compound 4 (12 mg) was purified by using HPLC (MeOH/H2O, 8:2, 3 mL min−1) from Fr. IIIa1b, and this was followed by HPLC with a Phenomenex Lux chiral column (MeOH/H2O, 100:0, 3 mL min−1) to obtain 4a (3.3 mg, tR 15 min) and 4b (3.1 mg, tR 19 min), respectively. Fr. IIIa2 was chromatographed over a RP-C18 column eluted with MeOH/H2O (7:3 → 10:0) to afford Fr. IIIa2a–Fr. IIIa2c. Fr. IIIa2a was loaded onto a Sephadex LH-20 column and eluted with MeOH to give Fr. IIIa2a1–Fr. IIIa2a2. and Fr. IIIa2a1 was purified using HPLC (MeOH/H2O, 8:2, 3 mL min−1), to give 2 (10 mg, tR 10 min) and 3 (4 mg, tR 12 min). Fr. IIIa2a2 was subjected to silica gel CC (PE/CH2Cl2, 3:1 → 1:1) to give 9 (13 mg).
ε) 261 (4.20), 194 (4.31) nm; IR (KBr) νmax 1737, 1636, 1497, 1449, 1377, 1263, 1123 cm−1; for the 1H and 13C NMR data, see Table 1; ESIMS m/z 317.1 [M + H]+; HRESIMS m/z 317.1011 [M + H]+ (calcd for C17H17O6+, 317.1020).ε) 322 (4.28), 264 (4.12), 235 (4.07), 228 (4.06) nm; IR (KBr) νmax 1747, 1654, 1600, 1582, 1449, 1407, 1371, 1258, 1181, 1110 cm−1; for the 1H and 13C NMR data, see Table 1; ESIMS m/z 257.1 [M + H]+; HRESIMS m/z 257.0800 [M + H]+ (calcd for C15H13O4+, 257.0808).ε) 320 (4.30), 257 (4.06), 235 (4.10), 228 (4.12) nm; IR (KBr) νmax 1735, 1633, 1580, 1450, 1435, 1409, 1363, 1229, 1198, 1175, 1149 cm−1; for the 1H and 13C NMR data, see Table 1; ESIMS m/z 287.1 [M + H]+; HRESIMS m/z 287.0911 [M + H]+ (calcd for C16H15O5+, 287.0914).ε) 296 (4.14), 248 (4.14) nm; ECD (c 0.87 × 10−4 M, MeCN) λmax (Δε) 360 (+3.06), 290 (+12.38), 235 (+4.19) nm; IR (KBr) νmax 2955, 2923, 2854, 1721, 1670, 1639, 1460, 1322, 1279, 1208, 1117, 1019 cm−1; for the 1H and 13C NMR data, see Table 2; ESIMS m/z 587.1 [M + H]+; HRESIMS m/z 587.1912 [M + H]+ (calcd for C33H31O10+, 587.1912).Computational section
The details of the ECD calculations for compounds 4a and 4b are presented in the ESI.†Antimicrobial assays
The test microorganisms obtained from the ATCC (American Type Culture Collection) in this bioassay were the bacteria Bacillus subtilis (ATCC 21332) (grows in beef-protein medium) and the fungi Candida albicans (ATCC 60193) (grows in YPD medium) and Aspergillus niger (GIM3.412) (grows in PDA medium). The minimum inhibitory concentrations (MICs) were determined using the microplate reader method according to the Clinical and Laboratory Standards Institute (CLSI) guidelines. For the antimicrobial assays, all of the compounds were dissolved in DMSO at a concentration of 4 mg mL−1, and were diluted with the corresponding medium to 100 μg mL−1 working solutions. The cultures were respectively inoculated with 1 × 106 CFU (colony-forming units) mL−1 for B. subtilis, 1 × 103 CFU mL−1 for C. albicans, and 0.4–5 × 104 CFU mL−1 for A. niger in a final volume of 200 μL in 96-well tissue culture-treated microtiter plates (Greiner Bio-One, CELLSTAR) and incubated at 37 °C. The microbial growth was determined turbidimetrically (OD595) using a BIO-RAD iMark microplate reader. The MIC values were obtained after 16 h for the bacteria or 24 h for the fungi. The resulting values were compared with the values for a positive control under the same conditions.Conclusions
Eleven Lindera cyclopentenedione derivatives including a novel 3,5-dioxocyclopent-1-enecarboxylate derivative (1), two novel oxepine-2,5-dione derivatives (2 and 3), a pair of new enantiomers of bi-linderone derivatives (4a/4b) as well as six known ones (5–8 and 9a/9b) were obtained from the dry roots of L. aggregata. Their structures, including the absolute configurations, were determined by comprehensive spectroscopic analysis and ECD calculations. Interestingly, lindoxepines A (2) and B (3) present an unprecedented oxepine-2,5-dione derivative skeleton, and may be the key intermediates for the synthesis of Lindera cyclopentenediones in vivo from the proposed precursor stilbenes. A putative pathway to the biosynthesis of a novel skeleton of 3,5-dioxocyclopent-1-enecarboxylate, (±)-lindepentone A (1), was also proposed from a stilbene precursor. To the best of our knowledge, the novel structures of 2 and 3 discovered from L. aggregata may be enlightening for the in vivo biosynthesis of the monomers of Lindera cyclopentenediones. In addition, the synthesized epi-bi-linderone (5) was also isolated and identified as a natural product, which provides evidence for the existence of the high biomimetic biosynthetic pathways for Lindera cyclopentenedione dimers proposed by Wang et al.8 Antimicrobial assays showed that compounds 6 and 7 exhibited weak inhibitory activity against Candida albicans with a MIC value of 25 μg mL−1, while other compounds were inactive (MIC values: >50 μg mL−1) against the tested microorganisms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 81722042 and 81573302), the Science and Technology Planning Project of Guangdong Province, China (No. 2015A020211007), the Fundamental Research Funds for the Central Universities (No. 17ykzd12) and the Research Project for Practice Development of National TCM Clinical Research Bases (No. JDZX2015207) for providing financial support for this work.Notes and references
- Editorial Committee of the Administration Bureau of Traditional Chinese Medicine, Chinese Materia Medica (Zhonghua BenCao), Shanghai Science and Technology Press, Shanghai, 1999, vol. 3, pp. 56–59 Search PubMed.
- N. Wang, S. Minatoguchi, M. Arai, Y. Uno, K. Hashimoto, X.-H. Chen, K. Fukuda, S. Akao, G. Takemura and H. Fujiwara, Am. J. Chin. Med., 2004, 32, 587–598 CrossRef PubMed.
- H. P. Tsui and H. van der Werff, Flora of China, 2008, vol. 7, pp. 142–159 Search PubMed.
- D. L. Custodio and V. Florencio da Veiga Jr, RSC Adv., 2014, 4, 21864–21890 RSC.
- Y. Cao, B. Xuan, B. Peng, C. Li, X. Chai and P. Tu, Phytochem. Rev., 2016, 15, 869–906 CrossRef.
- F. Wang, Y. Gao, L. Zhang and J. K. Liu, Org. Lett., 2010, 12, 2354–2357 CrossRef PubMed.
- F. Wang, Y. A. Gao, L. Zhang, B. Bai, Y. N. Hu, Z. J. Dong, Q. W. Zhai, H. J. Zhu and J. K. Liu, Org. Lett., 2010, 12, 3196–3199 CrossRef PubMed.
- H. B. Tan, C. Zheng, Z. Liu and D. Z. Wang, Org. Lett., 2011, 13, 2192–2195 CrossRef PubMed.
- G. Q. Wang, K. Wei, L. Zhang, T. Feng, F. Wang, Q. A. Wang and J. K. Liu, Tetrahedron Lett., 2011, 52, 2719–2721 CrossRef.
- F. F. Xiao, W. Liu, Y. X. Wang, Q. Zhang, X. Li and X. D. Hu, Asian J. Org. Chem., 2013, 2, 216–219 CrossRef.
- Z. Dong, Q. Gu, B. Cheng, Z. B. Cheng, G. H. Tang, Z. H. Sun, J. S. Zhang, J. M. Bao and S. Yin, RSC Adv., 2014, 4, 55036–55043 RSC.
- J. M. Bao, Z. Y. Su, L. L. Lou, J. Y. Zhu, G. H. Tang, L. S. Gan, X. Z. Bu and S. Yin, RSC Adv., 2015, 5, 62921–62925 RSC.
- Y. Q. Guo, G. H. Tang, Z. Z. Li, S. L. Lin and S. Yin, RSC Adv., 2015, 5, 103047–103063 RSC.
- J. Y. Zhu, B. Cheng, Y. J. Zheng, Z. Dong, S. L. Lin, G. H. Tang, Q. Gu and S. Yin, RSC Adv., 2015, 5, 12202–12208 RSC.
- J. Y. Zhu, L. L. Lou, Y. Q. Guo, W. Li, Y. H. Guo, J. M. Bao, G. H. Tang, X. Z. Bu and S. Yin, RSC Adv., 2015, 5, 47235–47243 RSC.
- J. S. Zhang, Y. H. Zou, Y. Q. Guo, Z. Z. Li, G. H. Tang and S. Yin, RSC Adv., 2016, 6, 53469–53476 RSC.
- W. Li, J. S. Zhang, J. L. Huang, M. H. Jiang, Y. K. Xu, A. Ahmed, S. Yin and G. H. Tang, RSC Adv., 2017, 7, 31061–31068 RSC.
- Y. W. Leong, L. J. Harrison, G. J. Bennett, A. A. Kadir and J. D. Connolly, Phytochemistry, 1998, 47, 891–894 CrossRef.
- S. Ng, H. H. Lee and G. J. Bennett, Magn. Reson. Chem., 1990, 28, 337–342 CrossRef.
- Z. Zhou and M. A. Tius, Angew. Chem., Int. Ed., 2015, 54, 6037–6040 CrossRef PubMed.
- E. M. K. Wijeratne, T. J. Turbyville, Z. G. Zhang, D. Bigelow, L. S. Pierson, H. D. VanEtten, L. Whitesell, L. M. Canfield and A. A. L. Gunatilaka, J. Nat. Prod., 2003, 66, 1567–1573 CrossRef PubMed.
- A. Fujimoto, Y. Shingai, M. Nakamura, T. Maekawa, Y. Sone and T. Masuda, Bioorg. Med. Chem. Lett., 2010, 20, 7393–7396 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of the 1D NMR data assignments of known compounds (5–9), 1D and 2D NMR spectra of new compounds, 1D NMR spectra of known ones, MS, HRMS, and IR spectra of new compounds and ECD computational details. See DOI: 10.1039/c8ra03094d |
| This journal is © The Royal Society of Chemistry 2018 |