
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReduced graphene oxide as a water, carbon dioxide and oxygen barrier in plasticized poly(vinyl chloride) films†
Ngoc Minh Nguyen Huynha,
Zhanna A. Boeva*a,
Jan-Henrik Småttb,
Markus Pesonenc and
Tom Lindfors *a
*a
aÅbo Akademi University, Faculty of Science and Engineering, Laboratory of Analytical Chemistry, (Johan Gadolin Process Chemistry Centre, PCC), 20500 Åbo, Finland. E-mail: tom.lindfors@abo.fi; jboyeva@gmail.com; Tel: +358 2 2154419
bÅbo Akademi University, Faculty of Science and Engineering, Laboratory of Physical Chemistry (Center for Functional Materials, FUNMAT), 20500 Åbo, Finland
cÅbo Akademi University, Faculty of Science and Engineering, Physics (Center for Functional Materials, FUNMAT), 20500 Åbo, Finland
First published on 15th May 2018
Abstract
Herein, we report the incorporation of a 10 μm thick reduced graphene oxide (RGO) barrier layer in a plasticized poly(vinyl chloride) (PVC) film as the main constituent in ion-selective membranes used in potentiometric solid-contact ion-selective electrodes (SCISE). Fourier transform infrared attenuated total reflection (FTIR-ATR) and oxygen transmission rate (OTR) measurements showed that the embedded RGO barrier efficiently impedes the diffusion of liquid water, carbon dioxide and oxygen (O2) through the 400 μm thick PVC film, which causes potential instability and irreproducibility of the SCISEs. The measurements revealed that the RGO layer completely blocks the carbon dioxide diffusion, while it fully blocks the water diffusion for 16 h and reduced the OTR by 85% on average. The μm-thick RGO films used in this study were easier to handle and incorporate into host polymers, and form more efficient and robust barriers compared to the mono-, few- and multilayer graphene commonly applied as barrier layers for liquids and gases. We also demonstrated that the FTIR-ATR technique employed in the permeability measurements is a versatile and very sensitive technique for studying the diffusion of small amounts of water and carbon dioxide through graphene-based thin films.
1. Introduction
Graphene has been studied extensively due to its exceptional mechanical properties,1 high thermal2 and electrical conductivity (high electron mobility),3 flexibility and transparency. These properties make graphene and reduced graphene oxide (RGO) useful in a wide range of applications such as gas sensors,4 transparent conductive films,5 corrosion protection6 and electronic devices.7 Although electrons can easily pass a defect-free monolayer of graphene,8 the penetration of ions, gases (including helium)9 and water10 through graphene is efficiently prevented by its dense electron cloud.8 This makes graphene one of the most promising materials for preparing thin, impermeable membranes and coatings, which was demonstrated by the excellent corrosion inhibition of copper and nickel substrates.6 It was shown that the metal substrates corroded only at cracks in the monolayer graphene film. Despite its superior barrier properties, it was also recently reported that monolayer graphene is highly permeable to thermal protons under ambient conditions, in contrast to bilayer graphene that is impermeable because of the AB stacking of the graphene sheets (i.e. the hexagonal rings of the second monolayer are centered on the carbon atoms of the first monolayer).8The synthesis of monolayer graphene is challenging and therefore it usually contains defects. First-principle density functional theory calculations have shown that small point defects conserving the sp2 hybridization of graphene (e.g. Stone–Wales defect, 555-777 and 585 divacancy, tetravacancy and hexavacancy) do not destroy the impermeability of the graphene towards He.11,12 Hence, only relatively large defects will make the graphene sheet permeable to small atoms and molecules. The graphene monolayer also forms a very efficient water barrier by trapping traces of moisture left on a single-crystal diamond surface encapsulated by graphene10 or liquid placed between two graphene layers in a graphene liquid cell used for studying colloidal platinum nanocrystal growth.13 The fabrication of monolayer graphene is generally a rather complicated bottom–up process, often including chemical vapor deposition (CVD) of graphene on nickel or copper14 substrates followed by PMMA wet transfer of the formed film to another metal or polymer substrate.15 The wet transfer often leaves PMMA impurities on the graphene surface, in addition to defects (holes) in the graphene film, resulting in a graphene surface that does not fully cover the substrate material, which decreases its barrier properties. Moreover, only rather small area graphene monolayers can be prepared with the relatively expensive CVD technique, limiting the practical applicability.
This limitation can be overcome with top–down approaches that enable the fabrication of large area graphene-based films and barrier layers. Most use either the hydrophilic graphene oxide (GO) or the more hydrophobic RGO,16 which is obtained by chemical reduction (e.g. with hydrazine,17–19 sodium borohydride,20 sodium hydroxide,21,22 ascorbic acid23) or electrochemical reduction of GO.24–26 GO films can be easily prepared by spraying,27 drop-casting,28 spin-4 and dip-coating29 on various substrates or prepared as freestanding films.30 However, the GO films usually contain defects seen as a D band in the Raman spectrum31,32 due to the graphite oxide exfoliation destroying the sp2 hybridization and electrical conductivity of graphene leaving defects and oxygen-containing groups on the surface of the graphene sheets (hydroxyl, epoxy, carbonyl and carboxyl groups).33 The reduction of GO removes most of these surface groups and partially restores the electrical conductivity, but cannot restore the hexagonal lattice of graphene.33,34 If the defects are large enough, they weaken the barrier properties of GO and RGO. In addition, the diffusion of ions, gases and water can also occur through sheet edges, inter-edge and interlayer spacing formed between the GO/RGO sheets.35–42 In the dry state, the GO films are vacuum-tight but act as a molecular sieve blocking all solutes larger than 4.5 Å when immersed in water.35,36 The partially lost barrier property of GO is caused by the formed nanocapillary network, which in the hydrated state increases the interlayer distance between the GO sheets allowing diffusion of, for example, Mg2+ and Cl− through the GO film but blocking larger ions and organic molecules.36 Nair et al. speculated that a layered GO film consists of separated non-oxidized pristine graphene regions and oxygen-functionalized regions acting as spacers keeping the sheets apart with an estimated distance of ca. 5 Å that is large enough for hosting a monolayer of water.36 Molecular dynamics simulations (MDS) showed that two monolayers of water are required between the GO sheets to facilitate the diffusion of larger ions. According to Nair et al., the pristine graphene regions form a network of channels that allow the low-friction flow of water while the oxidized regions retard the water diffusion. On the other hand, results obtained by atomistic simulations reveal a strong interaction of water with the oxidized regions reducing considerably the expected flow enhancement (boundary slip) in ultrathin GO films.42,43 This is in good accordance with MDS performed by Devanathan et al., showing that water diffuses slowly between the GO sheets due to strong interactions with hydroxy groups and only 21% of the water molecules are in the free form or as bulk-like water.44 Wei et al. attributed the reported fast water flow across GO membranes to its porous microstructure, i.e. the expanded interlayer spacing, wide channels formed at wrinkles, inter-edge spaces and holes (defects).42
The gas diffusion through graphene and GO follows the same principles as the water diffusion. The diffusion can occur at structural defects45 but selective gas diffusion can also be induced by varying the pore size and interlayer sheet distance with different GO stacking methods.46 Structural defects of GO give high separation selectivity of 3400 and 900 for hydrogen (H2) over carbon dioxide and nitrogen (N2), respectively, because only hydrogen gas molecules are small enough to penetrate the defects.45 In addition to increasing the GO thickness, humidity also decreases the gas permeance, indicating that condensed water accumulated between the GO sheets and in the pores of GO slows down the gas diffusion.46 Su et al. reported that the humidity-dependent interlayer distance of adjacent GO sheets decreases from ca. 7–13 Å to 4 Å upon reduction to RGO, which in combination with the increased hydrophobicity decreases the permeability of water, ions and gases.40 RGO was utilized as an oxygen ultra barrier for organic electronics cast on top of poly(3-hexylthiophene) (P3HT),41 which increased the lifetime of P3HT to 1300 h, demonstrating the good barrier properties of RGO. One of the most promising routes for the chemical reduction of GO is the use of hydroiodic acid as a reducing agent, which is an efficient and facile method to produce freestanding RGO films of high quality.40 Recently, it was suggested that iodine catalyzes the ring-opening reaction of the epoxy groups of GO and converts them into hydroxyl groups.47 It was shown that a 30 nm thick freestanding RGO film prepared by hydroiodic acid reduction was practically impermeable to water vapor.40 The hydroiodic acid reduction contributes to the excellent barrier properties by forming water as the reaction product, in comparison to the commonly used reducing agents that release carbon dioxide and cause wrinkles on the RGO surface and thus increase its permeability.
In this paper, we report how we first formed ca. 10 μm thick freestanding GO films at the liquid/air interface by heating an aqueous GO suspension at 80 °C for 1 h.30 The GO films were then reduced to RGO in hydroiodic acid and incorporated as barrier layers in 400 μm thick plasticized poly(vinyl chloride) (PVC) films, which are commonly used as the ion-selective membrane (ISM) matrix in potentiometric solid-contact ion-selective electrodes (SCISE). The role of the RGO barrier is to prevent the detrimental water layer formation at the inner interfaces of the SCISE and also the diffusion of oxygen and carbon dioxide to these interfaces, which have all been shown to induce the potential instability and irreproducibility of the SCISEs; this hampers their commercialization and the calibration-free applications of SCISEs. Here, we have purposely used thicker and more robust RGO films that are easier to handle in practical applications compared to mono- and few-layer graphene barrier layers. Our results obtained with Fourier transform infrared attenuated total reflection (FTIR-ATR) spectroscopy and oxygen transmission rate (OTR) measurements reveal that the buried RGO layer functions as a highly efficient barrier in plasticized PVC films by impeding the diffusion of liquid water, carbon dioxide and oxygen through the PVC film. In addition, we show that the FTIR-ATR spectroscopy is a very simple and sensitive technique for monitoring low levels of water and carbon dioxide diffusing through the RGO and RGO–PVC films compared to most other methods that are based on gravimetric principles.48
2. Experimental
2.1 Materials
The GO solution (5 mg mL−1; SKU-HCGO-W-175) was purchased from the Graphene Supermarket with the individual flake size varying between 0.5–5 μm. High molecular weight PVC (HMW PVC), bis(2-ethylhexyl) sebacate (DOS), tetrahydrofuran (THF) and concentrated hydroiodic acid (57 wt%) were purchased from Sigma-Aldrich.2.2 Preparation and reduction of GO films
Freestanding GO films were formed via the assembly process occurring in an aqueous GO solution at the liquid/air interface.30 We placed the GO solution (2 mg mL−1) in a glass beaker and after heating in a thermostated water bath at 80 °C for 1 h, a smooth and condensed film was formed at the liquid/air interface. The less concentrated suspension formed under the film was decanted to another beaker leaving the GO film at the bottom of the heated beaker. We then dried the membranes at 80 °C for 8 h before peeling them off from the bottom of the beaker. The reduction of the freestanding GO films was carried out by immersing them for 100 min in concentrated hydroiodic acid solution (57 wt%) kept at 95 °C. We did the reduction in a sealed Teflon container placed in a water bath to avoid the release of hydroiodic acid vapor to the ambient air. Finally, we washed the RGO films extensively with ethanol to remove excessive hydroiodic acid from the films. We determined all film thicknesses in this work including RGO, plasticized PVC and RGO–PVC with a micrometer with the accuracy of 1 μm (Digimatic Micrometer Series 293 MDC-MX Lite, Mitutoyo).2.3 FTIR spectroscopy
We used the Bruker IFS 66/S instrument to study the water diffusion through the RGO and the PVC films with the FTIR-ATR setup shown schematically in Fig. 1.49 The freestanding RGO films with a thickness of ca. 10 μm were pressed against a germanium reflection element (10 mm × 10 mm) by applying a force of 15 cNm to the Teflon grid and the cell above the film. The grid had four symmetrically placed holes to allow the deionized water in the Teflon cell to be in contact with the RGO film. We used zinc selenide reflection elements (10 mm × 10 mm) for studying the water diffusion through the plasticized PVC films with and without the built-in RGO barrier. The plasticized PVC films consisting of HMW PVC (33.3 wt%) and DOS (66.7 wt%) as a plasticizer were first dissolved in THF (20 wt%) with a vortex mixer and were left on an orbital shaker overnight. The pure plasticized PVC films (without the RGO barrier) were then prepared by drop casting the membrane solution (2 × 75 μL) on zinc selenide, resulting in a film thickness of ca. 300 μm. We recommend leaving the vials with the membrane solutions unstirred in an upright position for ca. 5–6 h before the drop casting to allow microscopic air bubbles to escape from the solution, in order to avoid the formation of small air pockets/bubbles in the PVC membranes during their overnight drying. | ||
| Fig. 1 Schematic view of the experimental setup used in the water uptake and carbon dioxide measurements of the freestanding RGO films and the plasticized PVC membranes with and without a RGO barrier layer. The Teflon grid was only used for the freestanding RGO films. | ||
We used a two-step drop casting protocol for the PVC films with the built-in RGO barrier. First, the membrane solution (2 × 50 μL) was drop cast on zinc selenide. It was allowed to dry for 2–3 min before the freestanding RGO barrier was applied on top of the still not completely dried first layer of plasticized PVC. After that, the second layer of plasticized PVC was prepared by applying the membrane solution (50 μL) on top of the RGO film. Drying overnight resulted in a thickness of ca. 400 μm for the RGO–PVC films. The upper and lower PVC films had the thickness of ca. 130 and 260 μm, respectively and the RGO layer of ca. 10 μm. We did not use the Teflon grid in the FTIR-ATR measurements with the PVC and RGO–PVC films because of their good adhesion to the underlying zinc selenide substrate. The sample compartment of the FTIR instrument was purged with dry air for at least 20 min before starting the measurement to remove moisture that could interfere with the water uptake studies. The background spectrum and the first spectrum were always measured identically with an empty cell, resulting in a straight horizontal line with zero absorbance as the first spectrum. After recording the first spectrum, we quickly filled the cell with water and during the next two hours the FTIR spectra were recorded at 60 s intervals, and at 15 min intervals for the rest of the total measurement time of 24 h. The spectral resolution was 4 cm−1 and each spectrum consisted of 16 interferograms measured with a signal gain of 16. The incident angle of the IR beam was 45° for both the germanium and the zinc selenide crystal. We estimated the penetration depth of the evanescent standing wave at the germanium/RGO and the zinc selenide/plasticized PVC interface with the Harrick equation50 in the wavenumber region of the OH stretching vibrations (3000–3700 cm−1), which was used to study the diffusion of water through the RGO and the PVC films. We obtain penetration depths of ca. 0.24–0.30 μm and 0.51–0.63 μm for germanium/RGO and zinc selenide/plasticized PVC, respectively, by using the refractive index of 4.05, 2.24, 2.43 and 1.5 for germanium, RGO (GO), zinc selenide and plasticized PVC, respectively. Hence, the evanescent standing wave senses in practice the presence of water only at the germanium/RGO, zinc selenide/PVC and the zinc selenide/RGO–PVC interfaces, and not in the bulk of the RGO and PVC films, making FTIR-ATR spectroscopy suitable for studying the barrier properties of RGO.
We measured the transmission mode FTIR spectra of the GO and RGO films with the Bruker IFS 66/S instrument connected to the Harrick's Video-MVPTM single reflection diamond ATR accessory (incidence angle: 45°) with a horizontal sampling area (d = 500 μm) and a built-in manual pressure regulator. The measurements were done with a resolution of 4 cm−1 in the wavenumber range of 300–4000 cm−1 by accumulating 16 interferograms with a signal gain of 16. We prepared the GO films by first drop casting the GO solution (140 μL) onto a polyethylene terephthalate (PET) substrate and then allowing it to dry in contact with the ambient atmosphere. After that, we peeled off the films from the substrate and measured the FTIR-ATR spectra.
2.4 Raman spectroscopy
The Raman spectra of the GO and RGO films were measured with the Renishaw Raman imaging microscope (using the WireTM v1.3 Raman software) in the back-scattering mode on the Leica DMLM microscope stage with 50× magnification. We used the 514 nm laser excitation wavelength to record the spectra in the wavenumber region of 300–4000 cm−1. The spectrometer was always calibrated against a silicon standard (520.0 cm−1) before recording the Raman spectra.2.5 Carbon dioxide and oxygen permeability
We detected the diffusion of carbon dioxide through the RGO and RGO–PVC films with FTIR-ATR spectroscopy by purging carbon dioxide gas through the Teflon cell shown in Fig. 1 and monitoring the intensity changes in the asymmetric stretching mode of carbon dioxide at 2335 and 2366 cm−1, respectively.51 We used the Clark electrode in a two-compartment Teflon cell to monitor the OTR through the plasticized PVC and the RGO–PVC films by placing the films between the two compartments. First, we filled both compartments of the Teflon cell with deionized water and the plasticized PVC films were then allowed to equilibrate overnight before purging the aqueous solutions with nitrogen gas to remove oxygen. The thickness of the films was ca. 300 μm and 0.17 cm2 of the films were in contact with the electrolyte solution. We placed the Clark electrode in the receiving solution that was protected with nitrogen gas and sealed with Parafilm® and purged the source solution continuously with oxygen (O2). The Clark electrode monitored the change in the oxygen concentration (in ppm) in the receiving solution resulting from the diffusion of oxygen through the PVC films with and without the RGO barrier.2.6 XRD, XPS, SEM and EDXA
We measured the X-ray diffraction (XRD) patterns with a Bruker AXS D8 Discover instrument (Karlsruhe, Germany) equipped with a Cu Kα X-ray source (λ = 0.154 nm) and a dynamic scintillation detector. The samples were measured with a grazing incident angle of 0.5° in the 2θ range of 2° to 35° and with the step size of 0.04°. The X-ray photoelectron spectra (XPS) were measured with the Phi Quantum 2000 instrument with the monochromatized A1 Kα irradiation line as the radiation source. We did the data fittings with the PHI Multipak Version 9.0.0 software by calibrating the spectra against 284.8 eV. The LEO 1530 Gemini FEG-SEM instrument (Oberkochen, Germany) equipped with a Thermo Scientific UltraDry Silicon Drift Detector (SDD, Thermo Scientific USA) was used for the scanning electron microscopic (SEM) measurements and energy dispersive X-ray analysis (EDXA) of the GO and RGO films.2.7 Electrical conductivity
We used the 4-point probe technique in a linear configuration with a tip spacing of 1.79 mm to measure the electrical conductivity of the GO and RGO films. The gold tips were spring-loaded to ensure good contact with the sample and a bias current between 6 × 10−7 and 1 × 10−3 A (depending on the film resistance) was applied over the GO and RGO films with a Keithley 2400 SourceMeter® until a stable and reproducible voltage was obtained. All the measurements were carried out in ambient conditions (relative humidity of 28.5% and at T = 24.6 °C) and the film conductivities were calculated using finite-sized corrections.522.8 Water contact angles
The apparent WCAs (θ) of the GO and RGO films were measured with the CAM200 contact angle goniometer (KSV Instruments, Ltd.) by applying a droplet of deionized water (2 μL) on the film surface and measuring its profile for 5 s with the image capture rate of 4 frames per s followed by 20 s with 1 frame per s. We always did three or four separate measurements (depending on the sample area) at different spots on the same sample surface. The average contact angle for the left and the right side of the droplets was calculated with the Young–Laplace fitting method.3. Results and discussions
3.1 Reduction of GO
Freestanding GO films were first synthesized from aqueous GO solution at the liquid/air interface by heating the solution at 80 °C for 1 h as described in the Experimental section.30 The chemical reduction of the GO films to RGO was then performed by immersing the films in concentrated hydroiodic acid solution (57 wt%) for 100 min at 95 °C. We used concentrated hydroiodic acid solution because the reduction with hydroiodic acid vapor proposed by Su et al.40 reduced the mechanical strength of the RGO films. Fig. S1† shows an image of the RGO film. The efficiency of the reduction was evaluated with XPS, which gives quantitative information about the chemical state of the GO/RGO surfaces and functional groups covalently attached to them.53 In Fig. 2a and c, the XPS spectra of the full binding energy range show that GO contains more oxygen (29.5 atomic%) than RGO (10.5%), indicating that the reduction removed a substantial amount of the oxygen-containing functional groups from the surface of the GO sheets. This was accompanied by the increase in the carbon content from 70.4% (GO) to 84.6% (RGO) giving C/O ratios of 2.4 (GO) and 8.0 (RGO). These values are in good accordance with the C/O ratios of 1.3 (GO) and 6.4 (RGO) determined with EDXA (Table S1†). The XPS spectrum in Fig. 2c also shows that ca. 1.7 atomic% iodine was bound to the RGO surface under the hydroiodic acid reduction (I 3d5/2 spectra not shown here). This is much lower than that obtained with EDXA (8.2%), which may depend on local differences in the iodine concentration or insufficient washing of the films with ethanol after the hydroiodic acid reduction. Moreover, the penetration depth of X-rays in XPS analysis is much smaller (only a few nm measuring the very surface of the RGO film) compared with EDXA. Therefore, the EDXA analysis gives a more representative elemental composition of the entire RGO film. In addition to iodine, the EDXA measurements showed that GO and RGO films contained minor silicon impurities (0.2–0.3%), probably originating from the glass beaker used in the GO synthesis (Table S1†).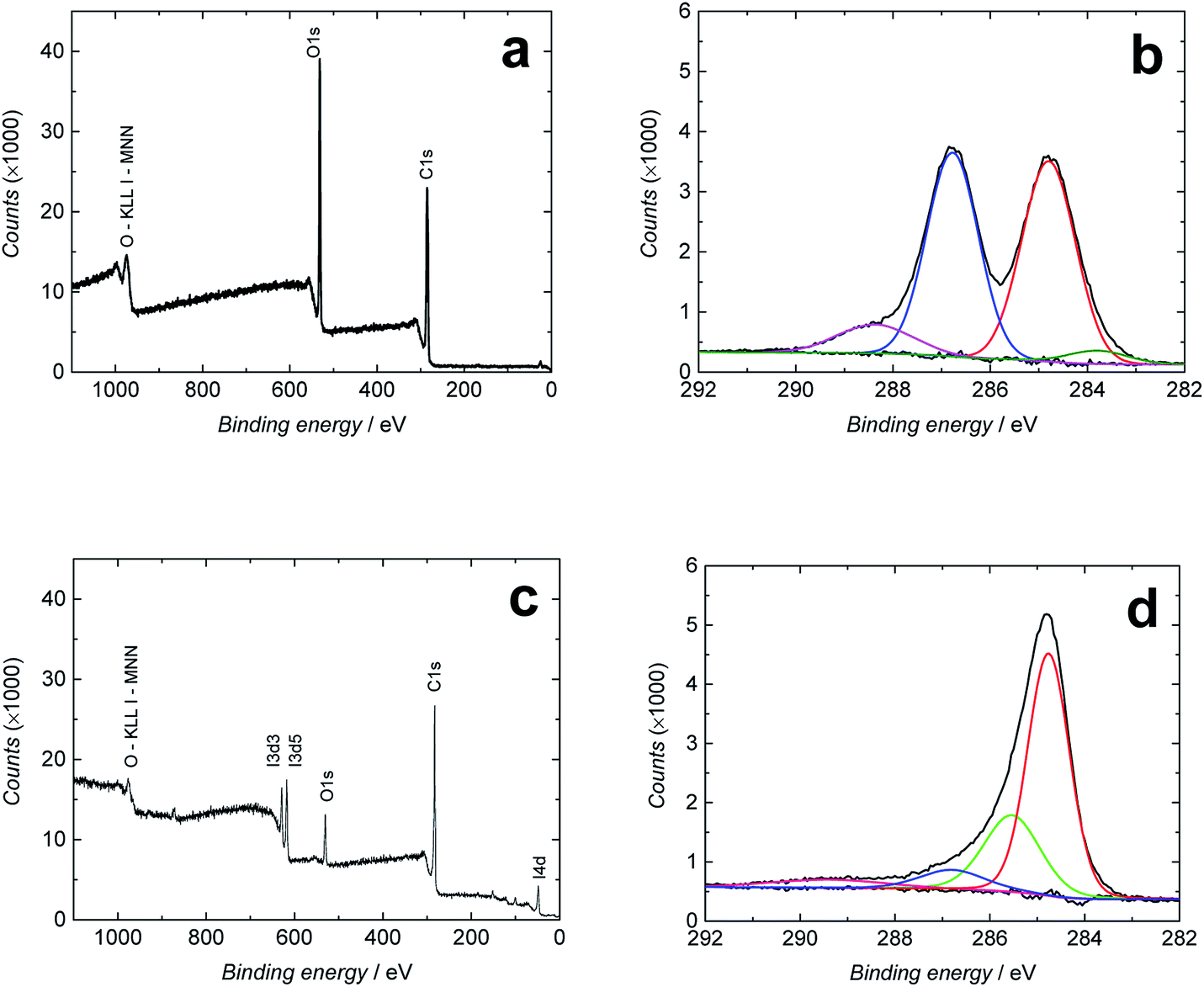 | ||
| Fig. 2 Full binding energy range (a and c) and C 1s XPS spectra (b and d) of GO (a and b) and RGO (c and d). | ||
Fig. 2b and d show the high-resolution C 1s XPS spectra of GO and RGO, respectively, fitted to four different bands. Both spectra contain a strong peak at 284.6 (GO)/284.8 eV (RGO) assigned to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in the carbon lattice (sp3/sp2 hybridization).23,54 The spectra of the GO film in Fig. 2b have additional bands at 286.7 eV (strong) characteristic of the C–O/C–O–C bonds (epoxy and hydroxyl),23,54,55 at 288.5 eV (carbonyl CO bonds),23,54,55 and a very weak band at 283.4 eV that we assign to C–Si bonds,56 although we are not fully sure of its origin due to its absence in the full energy range spectrum of GO in Fig. 2a. After the hydroiodic acid reduction, the XPS spectrum of RGO in Fig. 2d drastically changes its shape, indicating that the hydroxyl and epoxy groups (286.8 eV), which are the major oxygen-containing functional groups in GO, have been almost entirely removed. Also, the CO and C–Si bands cannot be distinguished in the spectrum, showing the efficiency of the hydroiodic acid reduction. On the other hand, two new bands appear in the XPS spectrum of RGO at 285.6 and 289.5 eV, which are assigned to C–I bonds56 and O–CO bonds (carboxyl),23,55,57 respectively. The presence of covalent C–I bonds explains why all iodine cannot be removed from the RGO film with the ethanol washing and why it is observed in the EDXA spectrum. We conclude that the C–C/CC band with a peak at 284.8 eV dominates the XPS spectra after the hydroiodic acid reduction, showing that the hydroiodic acid reduction efficiently removes most of the oxygen-containing functional groups from the RGO surface.
C bonds in the carbon lattice (sp3/sp2 hybridization).23,54 The spectra of the GO film in Fig. 2b have additional bands at 286.7 eV (strong) characteristic of the C–O/C–O–C bonds (epoxy and hydroxyl),23,54,55 at 288.5 eV (carbonyl CO bonds),23,54,55 and a very weak band at 283.4 eV that we assign to C–Si bonds,56 although we are not fully sure of its origin due to its absence in the full energy range spectrum of GO in Fig. 2a. After the hydroiodic acid reduction, the XPS spectrum of RGO in Fig. 2d drastically changes its shape, indicating that the hydroxyl and epoxy groups (286.8 eV), which are the major oxygen-containing functional groups in GO, have been almost entirely removed. Also, the CO and C–Si bands cannot be distinguished in the spectrum, showing the efficiency of the hydroiodic acid reduction. On the other hand, two new bands appear in the XPS spectrum of RGO at 285.6 and 289.5 eV, which are assigned to C–I bonds56 and O–CO bonds (carboxyl),23,55,57 respectively. The presence of covalent C–I bonds explains why all iodine cannot be removed from the RGO film with the ethanol washing and why it is observed in the EDXA spectrum. We conclude that the C–C/CC band with a peak at 284.8 eV dominates the XPS spectra after the hydroiodic acid reduction, showing that the hydroiodic acid reduction efficiently removes most of the oxygen-containing functional groups from the RGO surface.
The XPS results are supported by the FTIR, Raman and XRD measurements. The FTIR spectrum of RGO in Fig. S2† is almost featureless compared to GO, showing that the reduction removed almost all oxygen-containing surface groups from the GO film. We note that the bands at ca. 1900–2300 cm−1 are due to interferences caused by the Video-MVPTM single reflection diamond ATR setup that we used for recording the FTIR-ATR spectra of GO and RGO. In the otherwise featureless RGO spectrum, the distinct doublet bands of water vapor are distinguished at ca. 1300–1900 cm−1 (partially overlapping with the interferences from the Video-MVPTM setup) and ca. 3500–3900 cm−1.51 In addition, the Raman spectrum of RGO in Fig. 3 reveals that GO was reduced to RGO. The Raman spectrum of GO has the characteristic D and G bands, and the very weak 2D (G′), D + G and 2D′ (G′) bands at 1352 cm−1, 1591 cm−1, ∼2700 cm−1 (≅2 × 1352), ∼2930–2940 cm−1 (≅1352 + 1591) and ∼3180 cm−1, respectively.32 Upon the hydroiodic acid reduction of GO, the peak positions of the D and G bands shift to 1345 and 1582 cm−1, respectively, followed by the decrease in the full width at half maximum (FWHM) of both peaks, which is typical for GO reduction.32 The FWHM of the D band, which is easier to determine compared to the G band, decreased from ca. 140 cm−1 to 63 cm−1. We note that the ID/IG ratio (peak height ratio of the D and G band) increased during the reduction (100 min in hydroiodic acid) from ca. 0.94 to 1.46. A similar increase in the ID/IG ratio has been reported to occur during reduction with hydrazine,16,32 but this ratio decreased considerably at reduction times longer than 2 h and the G band became dominant as the hexagonal graphene lattice was better restored at longer reduction times.32 The Raman spectrum of RGO in Fig. 3 also reveals that the 2D band at ∼2692 cm−1, characteristic of graphene and graphite, grows in intensity upon the reduction of GO, which further indicates the restoration of the graphene lattice.58 The shape of this band is dependent on the number of graphene layers and can be used to distinguish between 1–5 layers of graphene. For more than five layers, like the RGO in this work, the Raman spectrum is in practice similar to graphite.58 Graphene has a single sharp 2D band that has approximately three times higher intensity than the G band.58 However, our RGO films are relatively thick and the intensity of the 2D band is therefore much lower than for the G band.58 The D + G band at ∼2930 cm−1 becomes more peak shaped upon reduction and the D′ band at ∼1605 cm−1 becomes visible due to the decreased peak width of the G band and its slight shift to lower wavenumbers (9 cm−1). The D, D′, D + G and 2D′ (≅2 × 1605 cm−1 ≅ 3210 cm−1) bands are all associated with the disorder in the graphene lattice59 and show that the hydroiodic acid reduction still leaves the RGO sheets with some imperfections.
 | ||
| Fig. 3 Raman spectra of (a) GO and (b) RGO. | ||
The XRD data in Fig. S3† show that the 002 diffraction peak of GO shifts from 12.2° (2θ) to 24.4° for RGO due to the hydroiodic acid reduction. We note here that the peak intensity of RGO is typically lower and the peak broader compared to GO,60 which Dresselhaus showed was typical for randomly ordered turbostratic graphitic platelets.61 The interlayer spacing (d) of the GO and RGO sheets was calculated using Bragg's law, nλ = 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ (where n = 1, λ = 0.154 nm (CuKα) and θ is the diffraction angle) and was found to be 0.724 nm and 0.364 nm for GO and RGO, respectively.23 This shows that the interlayer spacing of RGO is much smaller than for GO and very close to single crystal graphite (0.335 nm),62 thus impeding the interlayer diffusion of water, gaseous and ionic species, which is advantageous for the barrier applications of RGO. The hydroiodic acid reduction also increased the water contact angle (WCA) of the GO film from 58 ± 3° to 95 ± 1° (RGO), thus further increasing its barrier properties (Fig. S4†). In addition, the hydroiodic acid reduction increased the electrical conductivity by more than eight orders of magnitude from 8 × 10−6 S cm−1 (GO) to 140–220 S cm−1 (RGO), which is similar to the conductivity of 298 S cm−1 for RGO reduced in hydroiodic acid reported by Pei et al.34 This proves that the applied reduction method efficiently restores the electrical conductivity of the graphene lattice, despite its defects.
sinθ (where n = 1, λ = 0.154 nm (CuKα) and θ is the diffraction angle) and was found to be 0.724 nm and 0.364 nm for GO and RGO, respectively.23 This shows that the interlayer spacing of RGO is much smaller than for GO and very close to single crystal graphite (0.335 nm),62 thus impeding the interlayer diffusion of water, gaseous and ionic species, which is advantageous for the barrier applications of RGO. The hydroiodic acid reduction also increased the water contact angle (WCA) of the GO film from 58 ± 3° to 95 ± 1° (RGO), thus further increasing its barrier properties (Fig. S4†). In addition, the hydroiodic acid reduction increased the electrical conductivity by more than eight orders of magnitude from 8 × 10−6 S cm−1 (GO) to 140–220 S cm−1 (RGO), which is similar to the conductivity of 298 S cm−1 for RGO reduced in hydroiodic acid reported by Pei et al.34 This proves that the applied reduction method efficiently restores the electrical conductivity of the graphene lattice, despite its defects.
3.2 Morphology of the GO and RGO films
First, we measured the thickness of the GO and RGO films with a digital micrometer to ca. 13 and 10 μm, respectively, indicating that the GO film slightly shrank during the hydroiodic acid reduction due to the removal of oxygen-containing functional groups. This is in good accordance with the decreased interlayer spacing of RGO observed with XRD, but also with the film thickness of ca. 10 μm measured with SEM. Cross-sectional SEM images of the RGO film reveal that it consists of ca. 30 nm thick RGO bundles made of approximately 30–85 individual RGO sheets by assuming a sheet thickness of 0.35–1 μm. Fig. 4 shows that the RGO bundles have a compact layered structure with the basal planes of the individual sheets stacked on each other with some wrinkles (surface corrugation) visible on the uppermost RGO sheets, which is typical for GO and RGO.55,63 The interlayer distance between the bundles is relatively large, which may weaken the barrier properties of the RGO film, although the bundles are connected together by some shorter RGO bundles functioning as “pillars” between the horizontally oriented bundles, thus forming an electrically conducting RGO network. Despite the larger interlayer spacing between the RGO bundles, we expect that the individual bundles would function as multiple barriers for liquid water, gases, molecules and ionic substances, which strongly retards the diffusion through the RGO film by considerably increasing their diffusion path length compared to single- or few-layer RGO/graphene barriers. | ||
| Fig. 4 Cross-sectional SEM image of the RGO film (magnification: 50000×). The thickness of the RGO bundles is ca. 30 nm, indicating that they consist of ca. 30–85 individual RGO sheets. | ||
3.3 Water uptake of freestanding RGO films
We studied the barrier properties of the freestanding RGO film (10 mm × 10 mm) to liquid water with the experimental setup shown in Fig. 1. We observed the typical OH stretching bands at ca. 3000–3700 cm−1 with the absorbance maximum at ca. 0.13 A.U. for liquid water placed in contact with the bare germanium reflection element (spectrum (a) in Fig. 5). In this wavenumber region, the penetration depth of the evanescent standing wave formed at the germanium/RGO interface is ca. 240–300 nm, calculated with the Harrick equation.50 Therefore, the RGO film must be at least two times thicker than 300 nm to ensure that the evanescent standing wave senses only water in the RGO film and that it is not exposed to deionized water in the Teflon cell. The spectra (b) in Fig. 5 measured for 24 h during the water uptake reveal that the 10 μm thick RGO film is impermeable to water due to the absence of OH stretching bands in the FTIR spectra. We distinguished only two very weak water vapor bands at ca. 3500–3950 cm−1, originating from traces of water vapor in the sample compartment of the FTIR instrument.51 It was concluded that the hydroiodic acid reduction effectively converts the hydrophilic GO into hydrophobic RGO, decreasing the interlayer spacing between the RGO sheets, which blocks the water diffusion through the ca. 10 μm thick RGO film for at least 24 hours. This also indicates that the film does not have any interconnected holes, voids or inter-edge spaces that could facilitate the diffusion of water through the film. The relatively large interlayer spacing between the RGO bundles (Fig. 4) does not seem to be a critical issue for water diffusion through the RGO films. | ||
| Fig. 5 (a) FTIR-ATR spectrum of the bare germanium reflection element in contact with liquid water and (b) the FTIR spectra recorded during the water uptake of the ca. 10 μm thick RGO film in deionized water after a contact time of 1 min, 30 min, 1, 2, 4, 6, 9, 12, 18 and 24 h. | ||
3.4 RGO as a water barrier in plasticized PVC membranes
We expect that due to its water impermeability, the RGO film will also function as an efficient barrier embedded in thin polymer films. We recently used FTIR-ATR spectroscopy to show that a 120 nm thick amorphous hydrogenated carbon (a:C–H) layer deposited with radio-frequency plasma-enhanced CVD on 50 μm thick poly(lactic acid) films (PLA) decreased the water uptake of PLA by 55% after a contact time of 24 h, and even more at shorter times.64 The FTIR-ATR technique was previously used for studying the water uptake of plasticized PVC,49,65,66 poly(acrylate)67,68 and silicone rubber based ion-selective membranes66,68,69 to prevent the detrimental water layer or pool formation at the interfaces of potentiometric SCISEs.69–71 Although plasticized PVC is rather hydrophobic, we have shown with FTIR-ATR measurements that it cannot prevent the water layer formation49,66 that results in the potential instability and irreproducibility of these devices.65 The embedded RGO layer is therefore essential for improving the SCISE performance by preventing the water layer formation, which is an important step towards maintenance-free and calibration-free SCISEs. In addition, the FTIR-ATR technique is especially powerful for studying the water diffusion in polymers since it distinguishes between different types of water ranging from isolated (monomeric), small clusters (weakly hydrogen bonded) to larger clusters, and to bulk-like water (more strongly hydrogen bonded) forming a continuous extended water network,72–74 which indicates the formation of water pools at the electrode interfaces.Fig. 6a shows the FTIR-ATR spectra measured during the water uptake of 24 h for a ca. 300 μm thick plasticized PVC film (without the RGO barrier) prepared on a zinc selenide reflection element (10 mm × 10 mm). The intensity of the H–O–H stretching band at 3000–3700 cm−1 (ref. 75) increased quickly during the first 15 min to ca. 0.025 and then gradually to ca. 0.14 during the rest of the measurement time of 24 h with the water uptake slowing down during the last 6 hours. Characteristic bands for small (dimeric) and larger water clusters, and bulk-like water are distinguished in the spectra at ∼3600, 3365 and ∼3250 cm−1, respectively. In addition, the band at 1643 cm−1 assigned to H–O–H deformation vibrations grew in intensity during the water uptake.75 The water uptake of plasticized PVC is in good accordance with the water uptake of similar membranes,49 showing that water diffuses rather easily through plasticized PVC, which makes it suitable for studying the water barrier properties of the embedded RGO film. We have previously shown with FTIR-ATR spectroscopy that the water diffusion coefficients in plasticized PVC are (9.4 ± 0.2) × 10−8 cm s−1 (smaller water clusters) and (9.2 ± 0.1) × 10−9 cm s−1 (larger water clusters and aggregates).65 The water diffusion coefficients were therefore not mathematically modelled in this work. We also note that the downward pointing bands with negative intensity at 1149, 1172, ∼1240, 1733, 2857, 2929 and 2958 cm−1 are all related to vibrational modes of the plasticizer DOS and PVC.49,76 Their intensities decrease due to the minor swelling of the plasticized PVC film during its water uptake, which decreases the amount of both plasticizer and PVC at the zinc selenide/plasticized PVC interface.49
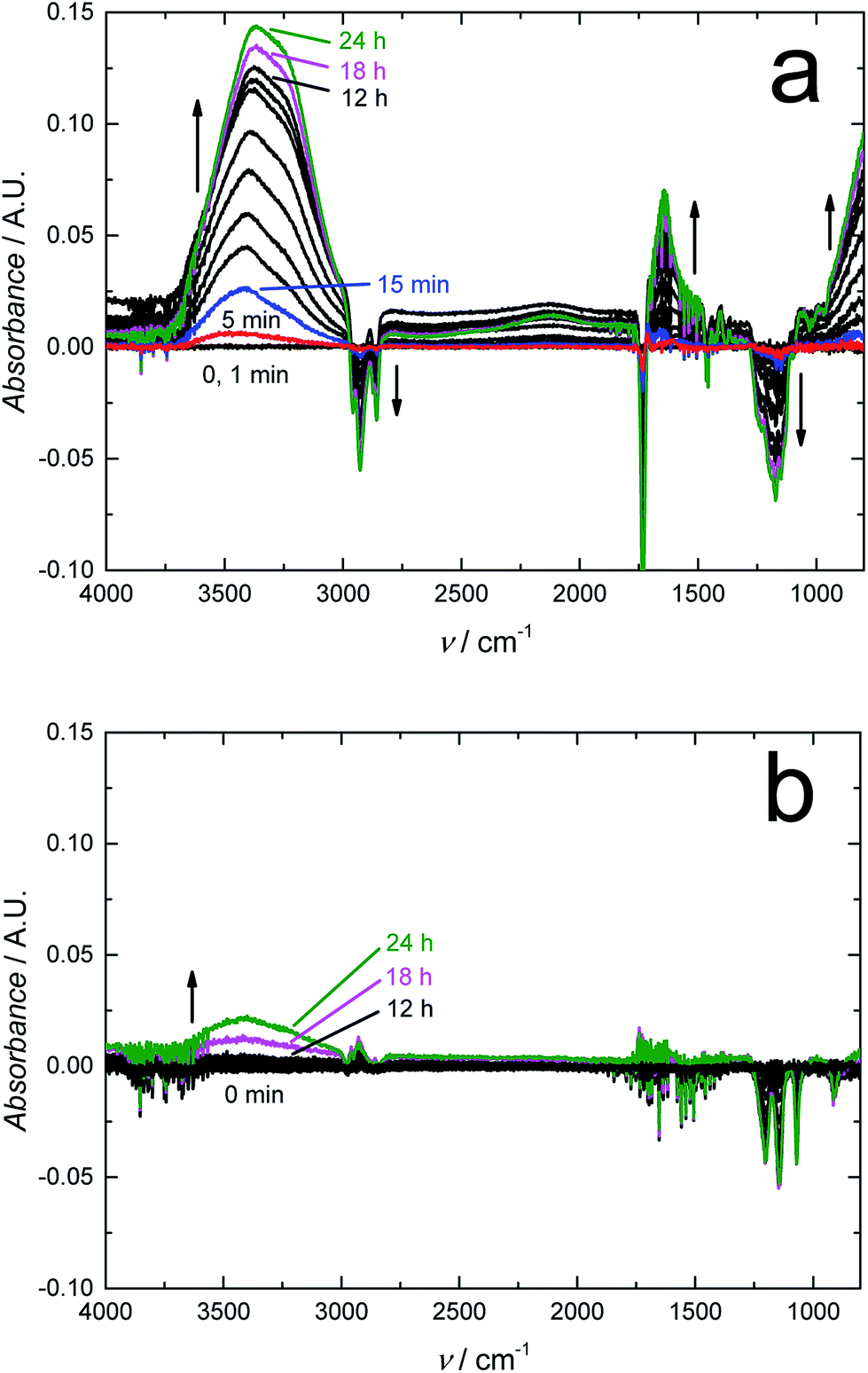 | ||
| Fig. 6 FTIR spectra measured during the water uptake of the plasticized PVC membranes (a) without and (b) with the RGO barrier layer. We recorded the FTIR spectra in deionized water after a contact time of 1, 5, 15 and 30 min, and 1, 2, 4, 6, 9, 12, 18 and 24 h. The plasticized PVC membranes had a thickness of ca. 300 μm (without RGO) and 400 μm (with RGO). | ||
Fig. 6b shows that the RGO barrier blocks the water diffusion through the ca. 400 μm thick plasticized PVC films for at least 16 h (only 0–12, 18 and 24 h shown in the figure). Although RGO–PVC is ca. 100 μm thicker than the PVC film without the RGO barrier (Fig. 6a), it cannot explain the complete absence of water at the underlying zinc selenide interface.49 The only reasonable explanation is that the RGO film functions as an effective water barrier in the plasticized PVC film. After 16 h, the intensity of the OH stretching bands (3000–3700 cm−1) increased indicating that the RGO barrier properties became weaker. We do not know the exact reason for this, but we speculate that the slight swelling of the PVC film during the water uptake causes cracks in the RGO film facilitating the diffusion of water through the barrier. When inspecting the RGO–PVC film after the water uptake measurements, we visually observed a few small cracks at the edges of the RGO layer where the circular inner part of the water uptake cell had been pressed against the RGO–PVC film. In addition, the absorbance maximum of larger water clusters after 24 h of water uptake (ca. 0.025 A.U. at 3400 cm−1) appeared at a wavenumber that is 35 cm−1 higher compared to the PVC film without the barrier. This reveals that water forms a network to a lesser extent in the RGO–PVC film (consisting of smaller clusters) compared to the plasticized PVC film lacking the barrier layer. The absence of a distinguishable bulk water maximum at ca. 3250 cm−1 supports this assumption. We conclude that the FTIR-ATR measurements confirm that the 10 μm RGO film functions as an efficient water barrier in plasticized PVC.
3.5 Carbon dioxide and oxygen barrier properties of RGO
We used also FTIR-ATR spectroscopy to determine the carbon dioxide barrier properties of the RGO–PVC films by purging the FTIR cell in Fig. 1 with carbon dioxide gas and measuring the FTIR spectra after 1, 2 and 24 h. In Fig. 7, the intensity increase in the carbon dioxide bands at 2335 and 2366 cm−1 assigned to asymmetric stretching modes of carbon dioxide51 shows that it takes more than 1 h for carbon dioxide to diffuse through the plasticized PVC film while the RGO–PVC film is impermeable to carbon dioxide for at least 24 h. We assume that the very weak downward pointing bands of DOS and PVC observed for both PVC films are related to drying and subsequent shrinking of the PVC films, although the exact reason is unclear. We speculate also that the slight swelling of the upper PVC layer during the water uptake may be the reason for the negative bands at 1070, 1146 and 1203 cm−1 observed in Fig. 6b. | ||
| Fig. 7 FTIR spectra showing the carbon dioxide diffusion through the plasticized PVC membrane with (1) and without (2) the RGO barrier layer. We measured the spectra after exposing the membranes to carbon dioxide for (a) 1, (b) 2 and (c) 24 h. | ||
We also utilized RGO to explore its barrier properties toward oxygen permeation. Fig. S5,† including the membrane-free control measurement, shows that the RGO–PVC has much better oxygen barrier properties compared to the plasticized PVC lacking the RGO barrier. The oxygen concentration in the receiving solution (given in ppm) is much lower for RGO–PVC than for the PVC film without the RGO barrier, indicating that less oxygen diffuses through the RGO–PVC film. As shown in Table 1, the OTR after 15, 30, 45 and 60 min of purging the source solution with oxygen was 1220 ± 90, 1190 ± 50, 1280 ± 70 and 1250 ± 20 cm3 m−2 per day for RGO–PVC (mean value: 1240 cm3 m−2 per day) and 13500 ± 100, 7900 ± 300, 6600 ± 100 and 5700 ± 200 cm3 m−2 per day (mean value: 8400 cm3 cm−2 per day) for the PVC film without the barrier. On average, the RGO barrier reduced the OTR by 85.2%, although the OTR for the barrierless PVC film decreased continuously during the 1 h measurement, which is currently not well understood. The OTR is usually measured at 0% relative humidity (RH) and 23 °C; however, we did the OTR measurements in (deionized) liquid water, which was placed in direct contact with the plasticized PVC films because of their intended use as ISM matrices in SCISEs designed for the determination of inorganic analytes in aqueous solutions. It is therefore not straightforward to compare our OTR values with literature data. The OTR for three different 13–15 μm thick PVC films used as the overwrap for fresh produce varied between 628 ± 19 (5 °C) and 7010 ± 106 cm3 m−2 per day (40 °C) in the temperature range of 5–40 °C, whereas the OTR was not influenced by the RH (measured at 0% or > 90–95%).77 At 20 °C, the OTR was from 1609 ± 43 to 2779 ± 68 cm3 m−2 per day, which is slightly lower but still in good accordance with our results obtained at 21–23 °C (Table 1), especially when considering the differences in the experimental setup (water vapor vs. liquid water) and the high amount of plasticizer (66 wt%) present in our PVC films.
| t/min | OTR (cm3 m−2 per day) | ||
|---|---|---|---|
| Control | PVC | RGO–PVC | |
| 15 | 21000 ± 1000 |
13500 ± 100 |
1220 ± 90 |
| 30 | 16000 ± 4000 |
7900 ± 300 | 1190 ± 50 |
| 45 | 18000 ± 3000 |
6600 ± 100 | 1280 ± 70 |
| 60 | 20000 ± 2000 |
5700 ± 200 | 1250 ± 20 |
In a recent study, a 70 nm thick electrochemically exfoliated GO barrier layer was fabricated by mechanically pressing it against a 49 μm thick PVC film.78 It was shown that the thin GO barrier reduced the OTR by ca. 40% from 668 ± 1 to 266 ± 6 mol m−2 s−1 Pa−1 at 0% RH and 35 °C, and even up to ca. 93% when the GO barrier was deposited on polypropylene. The decrease in the latter case is comparable to the OTR reduction of 85.2% to 1240 cm3 m−2 per day in liquid water induced by the RGO film embedded in the plasticized PVC film in this work. Despite the significant OTR decrease, the OTR value indicates that RGO–PVC is still only a moderate oxygen transmitter (∼1000 cm3 m−2 per day). In contrast, a high oxygen barrier layer with an OTR of only 0.12 cm3 m−2 per day was fabricated from a mixture of 0.1 wt% polyethyleneimine and 0.2 wt% GO deposited on PET.79 However, the OTR measurement was conducted at 23 °C and 0% RH, and it has been shown for ethylene vinyl alcohol, which is a high barrier material for oxygen, that increasing the RH markedly reduced its barrier properties.80 In summary, we speculate that the smaller oxygen molecules (d = 0.12 nm) can more easily diffuse via the defects, voids, inter-edge spaces and interlayer spacings of RGO compared to the two times larger carbon dioxide molecules (d = 0.23 nm), which could explain the differences in their diffusion through the RGO–PVC films. In addition, the plasticized PVC films are in direct contact with liquid water in the OTR measurements, in contrast to the carbon dioxide diffusion measurements, thus complicating the comparison of the carbon dioxide and oxygen diffusion through the PVC films.
4. Conclusion
We show with FTIR-ATR spectroscopy and OTR measurements that a 10 μm thick RGO film embedded in plasticized PVC commonly used as the ISM matrix in SCISEs functions as an efficient barrier for liquid water, carbon dioxide and oxygen, which causes potential instability and irreproducibility of the SCISEs. The barrier layer impedes carbon dioxide diffusion, blocks the water diffusion for 16 h and decreases the OTR by 85%. The RGO barrier prepared by the reduction of GO with hydroiodic acid is robust and easy to handle and therefore has greater practical relevance compared to the mono- and few-layer RGO or graphene barriers. Cross-sectional SEM images show that the barrier films are composed of ca. 30 nm thick RGO bundles consisting of approximately 30–85 individual RGO sheets. The good barrier properties of RGO are due to its multi-layer structure and the shorter interlayer distance (0.364 nm) of the RGO sheets induced by the hydroiodic acid reduction. The multi-layer structure increases the diffusion path length of water, carbon dioxide and oxygen, and efficiently reduces the negative effect of holes, voids and inter-edge spacing on the permeability of RGO. In addition, the relatively high hydrophobicity (WCA: 95 ± 1°) of RGO counteracts the water diffusion. It is shown here that FTIR-ATR spectroscopy is a very sensitive, simple and suitable technique for studying low levels of water and carbon dioxide diffusing through RGO and RGO–PVC films, compared to the commonly used gravimetric techniques. We aim to report the application of RGO as a barrier layer in SCISEs in a follow-up paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Zhanna A. Boeva gratefully acknowledges the Jane and Aatos Erkko Foundation (Finland) for financial support.References
- C. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef PubMed.
- J. T. Robinson, F. K. Perkins, E. S. Snow, Z. Q. Wei and P. E. Sheehan, Nano Lett., 2008, 8, 3137–3140 CrossRef PubMed.
- W. W. Cai, Y. W. Zhu, X. S. Li, R. D. Piner and R. S. Ruoff, Appl. Phys. Lett., 2009, 95, 123115 CrossRef.
- D. Prasai, J. C. Tuberquia, R. R. Harl, G. K. Jennings and K. I. Bolotin, ACS Nano, 2012, 6, 1102–1108 CrossRef PubMed.
- J. Park, W. H. Lee, S. Huh, S. H. Sim, S. B. Kim, K. Cho, B. H. Hong and K. S. Kim, J. Phys. Chem. Lett., 2011, 2, 841–845 CrossRef PubMed.
- S. Hu, M. Lozada-Hidalgo, F. C. Wang, A. Mishchenko, F. Schedin, R. R. Nair, E. W. Hill, D. W. Boukhvalov, M. I. Katsnelson, R. A. W. Dryfe, I. V. Grigorieva, H. A. Wu and A. K. Geim, Nature, 2014, 516, 227–230 CrossRef PubMed.
- J. S. Bunch, S. S. Verbridge, J. S. Alden, A. M. van der Zande, J. M. Parpia, H. G. Craighead and P. L. McEuen, Nano Lett., 2008, 8, 2458–2462 CrossRef PubMed.
- C. Lim, A. Sorkin, Q. L. Bao, A. Li, K. Zhang, M. Nesladek and K. P. Loh, Nat. Commun., 2013, 4, 1556 CrossRef PubMed.
- O. Leenaerts, B. Partoens and F. M. Peeters, Appl. Phys. Lett., 2008, 93, 193107 CrossRef.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef PubMed.
- J. M. Yuk, J. Park, P. Ercius, K. Kim, D. J. Hellebusch, M. F. Crommie, J. Y. Lee, A. Zettl and A. P. Alivisatos, Science, 2012, 336, 61–64 CrossRef PubMed.
- X. S. Li, W. W. Cai, J. H. An, S. Kim, J. Nah, D. X. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef PubMed.
- A. Reina, X. T. Jia, J. Ho, D. Nezich, H. B. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2009, 9, 30–35 CrossRef PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef PubMed.
- S. Stankovich, R. D. Piner, X. Q. Chen, N. Q. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. S. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef.
- H. L. Wang, Q. L. Hao, X. J. Yang, L. D. Lu and X. Wang, Nanoscale, 2010, 2, 2164–2170 RSC.
- X. B. Fan, W. C. Peng, Y. Li, X. Y. Li, S. L. Wang, G. L. Zhang and F. B. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef.
- J. L. Zhang, H. J. Yang, G. X. Shen, P. Cheng, J. Y. Zhang and S. W. Guo, Chem. Commun., 2010, 46, 1112–1114 RSC.
- G. K. Ramesha and S. Sampath, J. Phys. Chem. C, 2009, 113, 7985–7989 Search PubMed.
- A. Österholm, T. Lindfors, J. Kauppila, P. Damlin and C. Kvarnström, Electrochim. Acta, 2012, 83, 463–470 CrossRef.
- Y. Y. Shao, J. Wang, M. Engelhard, C. M. Wang and Y. H. Lin, J. Mater. Chem., 2010, 20, 743–748 RSC.
- S. Gilje, S. Han, M. Wang, K. L. Wang and R. B. Kaner, Nano Lett., 2007, 7, 3394–3398 CrossRef PubMed.
- M. Krueger, S. Berg, D. Stone, E. Strelcov, D. A. Dikin, J. Kim, L. J. Cote, J. X. Huang and A. Kolmakov, ACS Nano, 2011, 5, 10047–10054 CrossRef PubMed.
- X. Wang, L. J. Zhi and K. Mullen, Nano Lett., 2008, 8, 323–327 CrossRef PubMed.
- C. M. Chen, Q. H. Yang, Y. G. Yang, W. Lv, Y. F. Wen, P. X. Hou, M. Z. Wang and H. M. Cheng, Adv. Mater., 2009, 21, 3007–3011 CrossRef.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef.
- X. Diez-Betriu, S. Alvarez-Garcia, C. Botas, P. Alvarez, J. Sanchez-Marcos, C. Prieto, R. Menendez and A. de Andres, J. Mater. Chem. C, 2013, 1, 6905–6912 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- S. F. Pei and H. M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef PubMed.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef PubMed.
- F. Guo, G. Silverberg, S. Bowers, S. P. Kim, D. Datta, V. Shenoy and R. H. Hurt, Environ. Sci. Technol., 2012, 46, 7717–7724 CrossRef PubMed.
- D. Kang, J. Y. Kwon, H. Cho, J. H. Sim, H. S. Hwang, C. S. Kim, Y. J. Kim, R. S. Ruoff and H. S. Shin, ACS Nano, 2012, 6, 7763–7769 CrossRef PubMed.
- L. Huang, M. Zhang, C. Li and G. Q. Shi, J. Phys. Chem. Lett., 2015, 6, 2806–2815 CrossRef PubMed.
- Y. Su, V. G. Kravets, S. L. Wong, J. Waters, A. K. Geim and R. R. Nair, Nat. Commun., 2014, 5, 4843 CrossRef PubMed.
- H. Yamaguchi, J. Granstrom, W. Y. Nie, H. Sojoudi, T. Fujita, D. Voiry, M. W. Chen, G. Gupta, A. D. Mohite, S. Graham and M. Chhowalla, Adv. Energy Mater., 2014, 4, 1300986 CrossRef.
- N. Wei, X. S. Peng and Z. P. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 5877–5883 Search PubMed.
- N. Wei, X. S. Peng and Z. P. Xu, Phys. Rev. E, 2014, 89, 012113 CrossRef PubMed.
- R. Devanathan, D. Chase-Woods, Y. Shin and D. W. Gotthold, Sci. Rep., 2016, 6, 29484 CrossRef PubMed.
- H. Li, Z. N. Song, X. J. Zhang, Y. Huang, S. G. Li, Y. T. Mao, H. J. Ploehn, Y. Bao and M. Yu, Science, 2013, 342, 95–98 CrossRef PubMed.
- H. W. Kim, H. W. Yoon, S. M. Yoon, B. M. Yoo, B. K. Ahn, Y. H. Cho, H. J. Shin, H. Yang, U. Paik, S. Kwon, J. Y. Choi and H. B. Park, Science, 2013, 342, 91–95 CrossRef PubMed.
- J. M. Coxon and M. A. E. Townsend, Tetrahedron, 2007, 63, 5665–5668 CrossRef.
- R. Dorati, I. Genta, C. Colonna, T. Modena, F. Pavanetto, P. Perugini and B. Conti, Polym. Degrad. Stab., 2007, 92, 1660–1668 CrossRef.
- T. Lindfors, F. Sundfors, L. Höfler and R. E. Gyurcsányi, Electroanalysis, 2009, 21, 1914–1922 CrossRef.
- N. J. Harrick, Internal reflection spectroscopy, Interscience Publishers, New York, 1967 Search PubMed.
- http://webbook.nist.gov/chemistry/.
- F. A. Smits, Bell Syst. Tech. J., 1958, 37, 711–718 CrossRef.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef.
- S. Yumitori, J. Mater. Sci., 2000, 35, 139–146 CrossRef.
- T. Szabó, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- http://srdata.nist.gov/xps/.
- C. Kozlowski and P. M. A. Sherwood, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2099–2107 RSC.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef PubMed.
- R. J. Nemanich and S. A. Solin, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 20, 392–401 CrossRef.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabo, A. Szeri and I. Dekany, Langmuir, 2003, 19, 6050–6055 CrossRef.
- M. S. Dresselhaus, Supercarbon: Synthesis, Properties and Applications, ed. S. Yoshimura and R. P. H. Chang, Springer, New York, 1998, vol. 33, p. 9 Search PubMed.
- Y. Baskin and L. Meyer, Phys. Rev. B: Condens. Matter Mater. Phys., 1955, 100, 544 Search PubMed.
- S. K. Deng and V. Berry, Mater. Today, 2016, 19, 197–212 CrossRef.
- Z. A. Boeva, A. Catena, L. Hofler, S. Wehner, C. B. Fischer and T. Lindfors, Carbon, 2017, 120, 157–164 CrossRef.
- T. Lindfors, F. Sundfors, L. Höfler and R. E. Gyurcsanyi, Electroanalysis, 2011, 23, 2156–2163 CrossRef.
- N. He and T. Lindfors, Anal. Chem., 2013, 85, 1006–1012 CrossRef PubMed.
- F. Sundfors, L. Höfler, R. Gyurcsányi and T. Lindfors, Electroanalysis, 2011, 28, 1769–1772 CrossRef.
- F. Sundfors, T. Lindfors, L. Höfler and R. E. Gyurcsányi, Anal. Chem., 2009, 81, 5925–5934 CrossRef PubMed.
- T. Lindfors, L. Höfler, G. Jágerszki and R. E. Gyurcsányi, Anal. Chem., 2011, 83, 4902–4908 CrossRef PubMed.
- N. He, S. Papp, T. Lindfors, L. Hofler, R. M. Latonen and R. E. Gyurcsanyi, Anal. Chem., 2017, 89, 2598–2605 CrossRef PubMed.
- M. Fibbioli, W. E. Morf, M. Badertscher, N. F. de Rooij and E. Pretsch, Electroanalysis, 2000, 12, 1286–1292 CrossRef.
- G. T. Fieldson and T. A. Barbari, Polymer, 1993, 34, 1146–1153 CrossRef.
- P. Sutandar, D. J. Ahn and E. I. Franses, Macromolecules, 1994, 27, 7316–7328 CrossRef.
- C. Sammon, C. Mura, J. Yarwood, N. Everall, R. Swart and D. Hodge, J. Phys. Chem. B, 1998, 102, 3402–3411 CrossRef.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, Tables and Charts, Wiley, Chichester, 3rd edn, 2001, p. 301 Search PubMed.
- R. Kellner, G. Fischbock, G. Gotzinger, E. Pungor, K. Toth, L. Polos and E. Lindner, Fresenius' Z. Anal. Chem., 1985, 322, 151–156 CrossRef.
- G. Doyon, J. Gagnon, C. Toupin and F. Castaigne, Packag. Technol. Sci., 1991, 4, 157–165 CrossRef.
- D. Pierleoni, Z. Y. Xia, M. Christian, S. Ligi, M. Minelli, V. Morandi, F. Doghieri and V. Palermo, Carbon, 2016, 96, 503–512 CrossRef.
- Y. H. Yang, L. Bolling, M. A. Priolo and J. C. Grunlan, Adv. Mater., 2013, 25, 503–508 CrossRef PubMed.
- Z. B. Zhang, I. J. Britt and M. A. Tung, J. Appl. Polym. Sci., 2001, 82, 1866–1872 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03080d |
| This journal is © The Royal Society of Chemistry 2018 |