
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChitosan-based self-assembled nanocarriers coordinated to cisplatin for cancer treatment
Ronny Trummera,
Worranan Rangsimawongb,
Warayuth Sajomsangc,
Mont Kumpugdee-Vollratha,
Praneet Opanasopitb and
Prasopchai Tonglairoum *b
*b
aFaculty of Pharmaceutical and Chemical Engineering, Beuth Hochschule für Technik Berlin, University of Applied Sciences, 13353 Berlin, Germany
bPharmaceutical Development of Green Innovations Group (PDGIG), Faculty of Pharmacy, Silpakorn University, Nakhon Pathom 73000, Thailand. E-mail: tonglairoum_p@su.ac.th
cNational Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), Thailand Science Park, Pathum Thani 12120, Thailand
First published on 22nd June 2018
Abstract
Polymeric nanocarriers were prepared via a dialysis method using three chitosan derivatives, N-benzyl-N,O-succinyl chitosan (BSCT), N-naphthyl-N,O-succinyl chitosan (NSCT), and N-octyl-N-O-succinyl chitosan (OSCT) and were coordinated to cisplatin. The nanocarrier properties and cytotoxicity on the human carcinoma cells, HN22 (head and neck), were investigated. In addition, intracellular cisplatin accumulation, apoptosis induction and toxicity on renal cells were also evaluated. The findings revealed that the succinyl groups of the polymers were perfectly deprotonated and bound with cisplatin by co-ordinate bonds at pH 8.5. Among the derivatives, BSCT exhibited the highest cisplatin loading and release in simulated physiological medium. The cytotoxicities on HN22 cells of cisplatin-loaded BSCT nanocarriers were lower than that of free cisplatin, however, they presented a greater percentage of early apoptosis in HN22 cells and could decrease cisplatin induced renal cell death. In conclusion, the BSCT self-assembly nanocarrier might be a cisplatin carrier for sustained release, which provides prolonged antitumour treatment and reduced nephrotoxicity.
Introduction
Cisplatin or cis-dichlorodiammineplatinum(II) (CDDP) has been reported as an effective anti-cancer drug for many solid tumours (head and neck, lung, testicular, ovarian, and breast cancer). However, its side effects on normal tissue, especially nephrotoxicity, limit the use of cisplatin.1 Because it is excreted by the kidneys, high amounts of the drug may accumulate in renal cells, leading to renal injury.2 In aqueous media, cisplatin undergoes step-wise aquation reactions. The replacement of chloride ions with water molecules leads to formation of a cationic aquated structure. Cisplatin experiences a high chloride concentration (100 mM) that prevents its hydrolysis to the aquated form. In contrast, the low content of chloride in cells (4–12 mM) provides the cationic aquated form. Furthermore, the exchange rate between chloride ligands and water molecules relates to the efficacy and cytotoxicity of cisplatin.3Carrier-based delivery systems for anti-cancer treatment have been widely developed over several years, and possess many advantages such as enhancing drug effectiveness, prevention of side effects, and circumvention of cellular accumulation mediated drug resistance. Platinum complex drug-loaded polymeric carriers have been formulated for passive targeting (major) and active targeting (minor).4 Preparation methods of cisplatin-loaded polymeric carriers have been reported, such as encapsulation, covalent attachment (conjugation), and complexation (co-ordination bound). Recently, drug delivery systems for cisplatin have been developed that utilize polymer–platinum conjugates or prodrugs,5,6 complexes in nanotubes7 and micro/nanoparticulates such as liposomes,8–10 nanocomplexes,3 gold nanoparticles,11 and polymeric micelles.12–15 The complex of cisplatin and poly(ethylene glycol)–poly(aspartic acid) block copolymer (PEG–P(Asp)) in an aqueous medium has been reported as a successful system for drug delivery,12,13 which also reduced nephrotoxicity.14 This system was developed with the name of NC-6004 (Nanoplatin; NanoCarrier Co., Ltd.; Japan) and has been studied at the final clinical stage in Asia.
Chitosan is an economic, biocompatible and biodegradable polymer for formulating drug or gene carriers. Because its applications are restricted by its solubility in biological fluids, soluble chitosan derivatives were developed.16 The fabrication of nanocarriers from chitosan derivatives offers an effective drug delivery system. Modifications of the chitosan structure to produce derivatives such as N-lauryl-carboxymethylchitosan, N-phthaloylchitosan-g-mPEG, and N-octyl-O-sulfate chitosan were successfully used to prepare nanocarrier systems.17 Our group has previously synthesized N-naphthyl-N,O-succinyl chitosan (NSCT) to incorporate meloxicam via physical entrapment methods such as dialysis, o/w emulsion, dropping, and evaporation. The particle properties depend on entrapment method and initial concentrations of drugs.18 In addition, N-octyl-N,O-succinyl chitosan (OSCT) that contains octyl groups in the chitosan backbone, as a hydrophobic part, can form polymeric self-assembled structures.
Herein, we report the utilization of BSCT, NSCT and OSCT polymers to form self-assembled nanocarriers. The BSCT, NSCT and OSCT can generate self-assembled nanocarriers due to their modified structures. The carboxylic moieties available on the polymer backbones were employed to form coordinate bonds with cisplatin. The physicochemical properties and drug loading efficiencies of the nanocarriers were characterized. Moreover, the cytotoxicity, apoptosis induction of cancer cells, intracellular cisplatin accumulation and toxicity on renal cells were evaluated.
Experimental
Materials
Chitosan was obtained from OliZac Technologies Co., Ltd. (Bangkok, Thailand). Cisplatin, benzaldehyde, 2-naphthaldehyde, and octaldehyde were obtained from Sigma Aldrich Chemical Company (St. Louis, MO, USA). Nitric acid was purchased from Merck & Co. (Darmstadt, Germany). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS) L-glutamine, non-essential amino acids, penicillin–streptomycin, and trypsin–EDTA were acquired from Gibco BRL (Rockville, MD, USA). The reagents used in the assay were of analytical reagent grade.Cell culture
HN22 and HT29 cells are a human oral squamous cancer cell line and a human colon adenocarcinoma cell line, respectively. HN22 cells were provided by the Faculty of Dentistry, Naresuan University (Phitsanulok, Thailand), and HT29 cells were obtained from the American Type Culture Collection (Rockville, USA).Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 and stirred in the dark for at least 12 h to allow the formation of coordinate bonds. Dialysis against distilled water was performed for 3 h (water exchanged every 30 min) to remove the free cisplatin.
:5 and stirred in the dark for at least 12 h to allow the formation of coordinate bonds. Dialysis against distilled water was performed for 3 h (water exchanged every 30 min) to remove the free cisplatin.Loading efficiency and loading capacity. Cisplatin-loaded polymeric nanocarriers were frozen and lyophilized. Then, the dry powder was mixed with 2 mL of nitric acid at 90 °C for 60 min. Ultrapure water was used for adjusting the mixture volume up to 10 mL. The concentration of cisplatin on the polymeric nanocarriers was determined by Inductively Coupled Plasma-Mass Spectrometry (ICP-MS, Agilent Technologies, 1100 series, Santa Clara, CA, USA).15 A platinum (Pt(II)) atomic absorption standard was used in the concentration ranges of 10 to 100 ng mL−1 for generating the standard curve. The percent loading efficiency (% LE) and loading capacity (LC) were calculated according to eqn (1) and (2), respectively:
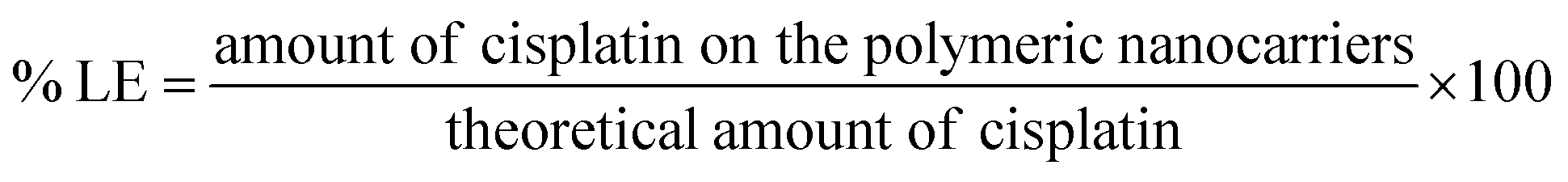 | (1) |
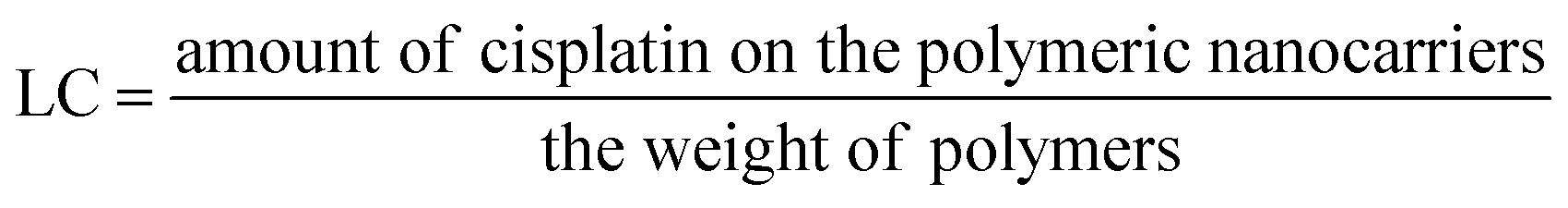 | (2) |
pH measurement. The pH values were measured at room temperature using a pH meter (Metler Toledo, Seven compact S220). All measurements were performed in triplicate.
Particle size and zeta potential. The sizes and zeta potentials of the polymeric nanocarriers were measured using a DLS particle size analyser (Zetasizer Nano ZS, Malvern Instruments, Malvern, UK) at 25 °C. The samples were diluted with water (1
:9) and then placed in a quartz cuvette. The laser was operated at the wavelength of 633 nm and was a 4 mW He–Ne laser. The scattering angle was set at 173°. Each sample was measured in triplicate.
000 cells per well in 96-well plates. The cells were kept in a humidified atmosphere of 5% CO2 and 95% air and at 37 °C for 24 h. The cells were incubated with cisplatin in a serum-free medium for 24 h. Afterwards, the treated cells were separated from the medium and incubated with MTT-containing medium (1 mg mL−1) for 4 h. After that, the cells were washed with phosphate buffer saline pH 7.4 to remove the residual medium. The formed formazan crystals in the living cells were solubilized in DMSO (100 μL per well). The absorbance at 550 nm was determined using a microplate reader (Universal Microplate Analyser, Model AI53601, Packard BioScience, CT, USA). The relative cell viability (%) was calculated by comparing with non-treated control cells.Flow cytometry was performed to investigate the killing effect of the cisplatin-loaded nanocarriers compared with free cisplatin. An apoptosis assay was carried out using a flow cytometer (BD, FACSC Canto, Becton Dickinson, CA, USA) with FITC-conjugated Annexin V/propidium iodide (PI) (BD Bioscience) co-staining. Briefly, the HN22 cells were seeded in 6-well plates and incubated in a humidified 5% CO2 incubator at 37 °C until 60–70% confluence. The cells were treated with the nanocarriers or free cisplatin at 37 °C for 24 h. After treatment, the cells were collected by trypsinization and subsequently washed with medium and gently pipetted to detach the cells. The treated cells were stained with Annexin V-FITC and PI in the dark for 15 min at ambient temperature, and immediately subjected to flow cytometry analysis. The data were evaluated using FACS diva software (BD Bioscience, USA). Both early apoptotic (Annexin V+/PI−) and late apoptotic (Annexin V+/PI+) cells were included in the cell apoptosis evaluation.
To determine whether the killing activity of the cisplatin-loaded nanocarriers was time dependent, the cytotoxicity of the cisplatin-loaded nanocarriers on the HN22 cells was investigated using the MTT assay, as previously described, at a cisplatin concentration ranging from 1–80 μg mL−1 (equivalent concentration). The cytotoxicity was assessed at 24 h and 48 h after the treatment.
 | (3) |
Results and discussion
Characterization of cisplatin-loaded nanocarriers
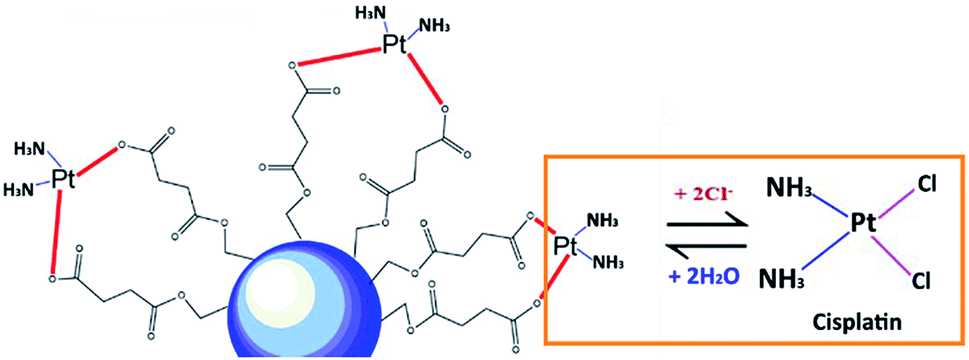
In this study, the pH values of the nanocarrier formulations were fixed at 8.5. This is because huge improvements in cisplatin loading onto these micellar systems were observed at pH 8 to 8.5. As shown in Fig. 1, acidic sites (R–COOH) of the polymer chain at the pH value up to 8.5 were perfectly deprotonated (R–COO–) and bound with Pt(II) of the cisplatin structure. By formation of the aqueous complex, cisplatin, therefore, was able to form two bonds with the polymer by cleavage of the water ligands. However, the binding ability between cisplatin and the polymer decreased at pH > 9, and the co-ordinate bonds of the cisplatin–nanocarrier structure were unable to form. Moreover, these cisplatin-loaded nanocarriers were stable under physiological conditions and broken at high chloride (Cl−) conditions.20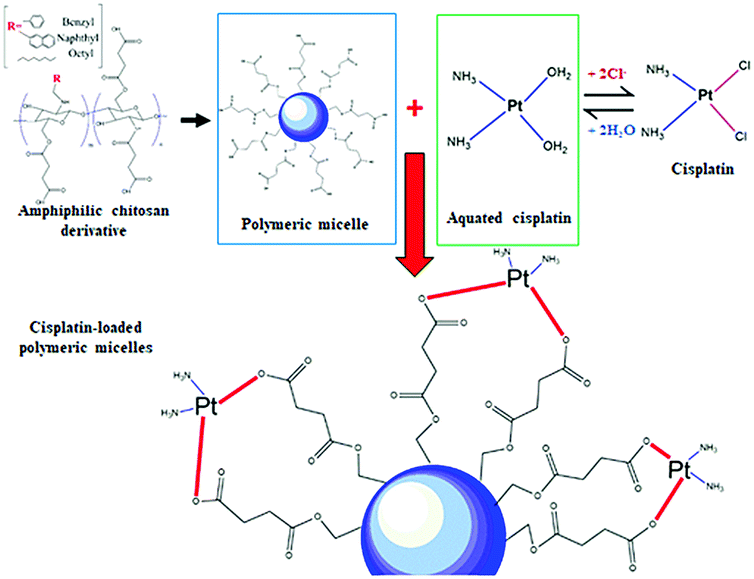 | ||
| Fig. 1 Schematic representation of the polymeric nanocarriers coordinated to cisplatin. | ||
The BSCT, NSCT, and OSCT nanocarriers were successfully coordinated with cisplatin, of which the BSCT nanocarriers exhibited the highest % EE and LC compared with the NSCT and OSCT nanocarriers (Fig. 2). In fact, the % EE of BSCT, NSCT and OSCT nanocarriers were 48.69%, 29.76% and 31.8%, respectively. On the other hand, the LC (mg mg−1) of BSCT, NSCT and OSCT nanocarriers were found to be 0.74, 0.45 and 0.48, respectively. The higher ability of the BSCT nanocarriers to load cisplatin may be due to the higher degree of substitution of succinic groups on the BSCT polymer chain (1.12%) when compared to the NSCT (0.52%) and OSCT (1.07%) polymer chains. The succinic moieties perfectly deprotonate at pH 8.5 and are able to bind with cisplatin via coordinate bonds. The higher amount of succinic moieties on the BSCT may result in the higher drug loading ability. The zeta potential values of the nanocarriers were in the range of −38.5 to −41 mV, while cisplatin-loaded nanocarriers exhibited negative charges between −16.7 and −19.7 mV. After the loading of cisplatin, the zeta potentials of the nanocarriers were increased, suggesting that the deprotonated carboxylic groups of the succinic acid moieties bound to cisplatin by co-ordinate bonds resulted in the reduction of the negative surface charge.20 Similarly, in the case of a PEG linker that contained carboxylic groups, significant quenching between the negatively charged carboxylic groups and cisplatin was reported.21
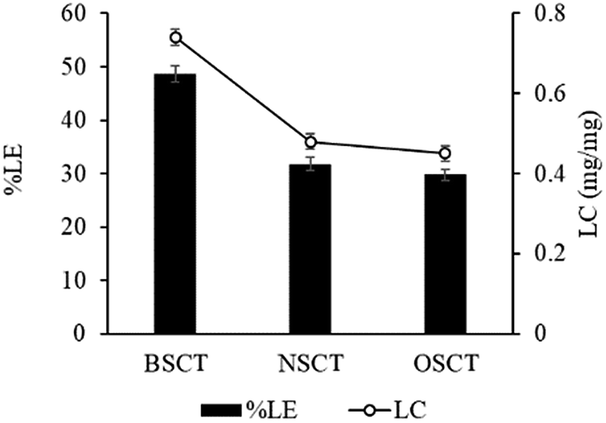 | ||
| Fig. 2 The percent loading efficacy (% LE) and loading capacity (LC) of cisplatin-loaded BSCS, NSCS, and OSCS nanocarriers. Each value represents the mean ± SD (N = 3). | ||
The particle sizes and surface charges of the nanocarriers are presented in Table 1. The increase in size correlated with the loading of cisplatin on the nanocarriers and the types of polymer used. The particle sizes of all the empty polymeric nanocarriers were between 294.2 and 325.2 nm. The size of the nanocarriers significantly increased (295–591 nm) after cisplatin was loaded onto the nanocarriers. To examine the relation between the effect of drug loading on the size of the particles, the particle sizes were measured again after the release study. Interestingly, it was found that after the cisplatin was completely released from the polymeric nanocarriers, the particle size was decreased to the initial size of the blank nanocarriers. This may be because the cisplatin loaded on the surface of the nanocarriers via coordinate bonds led to bigger size particles. Moreover, agglomeration, by coordinate bonds forming between one cisplatin molecule and two neighbouring particles, might increase the measured particle sizes.22
| Polymer | Particle size (nm) | Zeta potential (mV) | ||
|---|---|---|---|---|
| Before drug loading | After drug loading | Before drug loading | After drug loading | |
| NSCT | 325.2 ± 51.1 | 590.6 ± 65.8 | −38.5 ± 5.8 | −19.7 ± 2.4 |
| OSCT | 294.2 ± 22.4 | 295.4 ± 32.9 | −40.9 ± 4.0 | −19.5 ± 1.9 |
| BSCT | 302.7 ± 47.5 | 356.6 ± 23.0 | −30.3 ± 4.3 | −16.7 ± 0.2 |
Drug release characteristic
As shown in Fig. 3, the cumulative release of cisplatin from all the polymeric nanocarriers exhibited the same release behaviour for 7 days. The release of cisplatin tended to reach the highest value on day 4. Then, the release rate was steady on days 5 to 7. However, the BSCT nanocarriers showed significantly higher cisplatin release than that of the NSCT and OSCT nanocarriers, and the mass of cisplatin release on day 7 was in the order: BSCT nanocarriers (0.228 mg) > NSCT nanocarriers (0.187 mg) > OSCT nanocarriers (0.175 mg). This indicated that the highest amount of cisplatin loaded onto the BSCT nanocarriers represented the highest amount of cisplatin released into the release medium (pH 7.4). However, the highest percent of cumulative release of cisplatin from BSCT nanocarriers was 43% on day 7, indicating incomplete release and a slow release rate. Although these amphiphilic chitosan derivatives are pH-sensitive polymers with succinyl moieties in the polymer chain that can be ionized, swollen, and dissociated in media with a pH higher than the pKa,19 these succinyl moieties formed co-ordinate bonds with cisplatin. Therefore, the strong co-ordinate bonds provide highly stable links between the carriers and the drug. A biphasic release profile of cisplatin was found for the chitosan polymeric nanocarriers, for which a rapid release was observed within 4 days, followed by a sustained release profile for 1 week. Moreover, the release of cisplatin from the cisplatin-loaded polymeric nanocarriers was dependent on the relative chloride ion concentration, and thus followed the substitution equilibrium as shown in Fig. 4.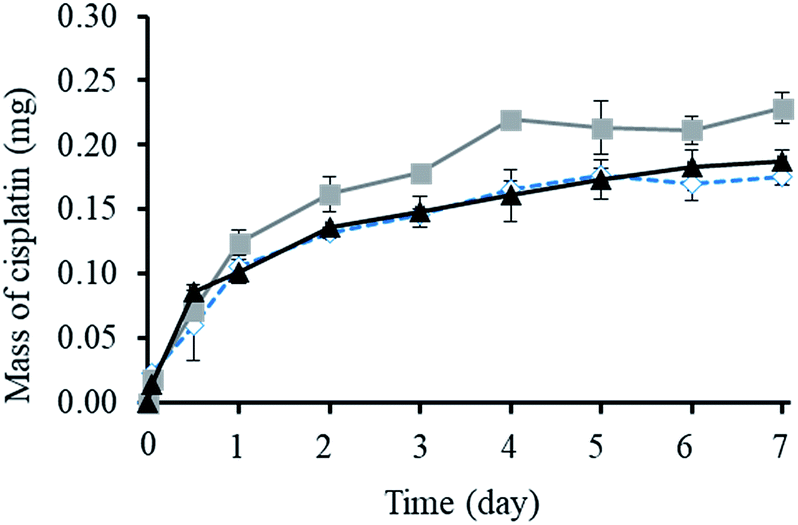 | ||
| Fig. 3 The mass of cisplatin released from the (■) BSCT, (▲) NSCT, and (◊) OSCT nanocarriers in release medium at pH 7.4 containing 10 mM phosphate buffer and 150 mM NaCl. Each value represents the mean ± SD (N = 3). | ||
 | ||
| Fig. 4 Schematic illustrating the release of cisplatin from the cisplatin-loaded polymeric nanocarriers in the presence of Cl− ions. | ||
Furthermore, the release of chemotherapy drugs by the loading of drugs in polymers has been widely developed. Subsequently, the release of the drug is required close to the tumour tissue. The co-ordination bonds between the carboxylic groups of the polymer and the Pt(II) of cisplatin were broken at the low pH value of tumour tissue (pH 5).20 Therefore, these systems might be targeted drug delivery systems, that lack properties of systemic toxicity.
FT-IR
The ATR-FTIR spectra of the blank BSCT nanocarriers, the cisplatin-loaded BSCT nanocarriers, the blank BSCT nanocarriers–cisplatin mixture and the cisplatin powder are presented in Fig. 5. The spectrum of empty BSCT nanocarriers exhibited absorption peaks at 3388 cm−1 (O–H stretching), 2924 cm−1 (C–H stretching vibration), 1637 cm−1 (primary amide), and 1553 cm−1 (N–H bending vibration). The peaks at 1159 cm−1, 1050 cm−1, and 869 cm−1 correspond to C–O–C stretching, C–O stretching, and pyranoid ring stretching of the saccharide backbone. The medium intensity peaks at around 1690 cm−1 correspond to the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bending of the benzyl groups. The peak at 1397 cm−1 indicated asymmetric stretching of the –COO− ions.
C bending of the benzyl groups. The peak at 1397 cm−1 indicated asymmetric stretching of the –COO− ions.
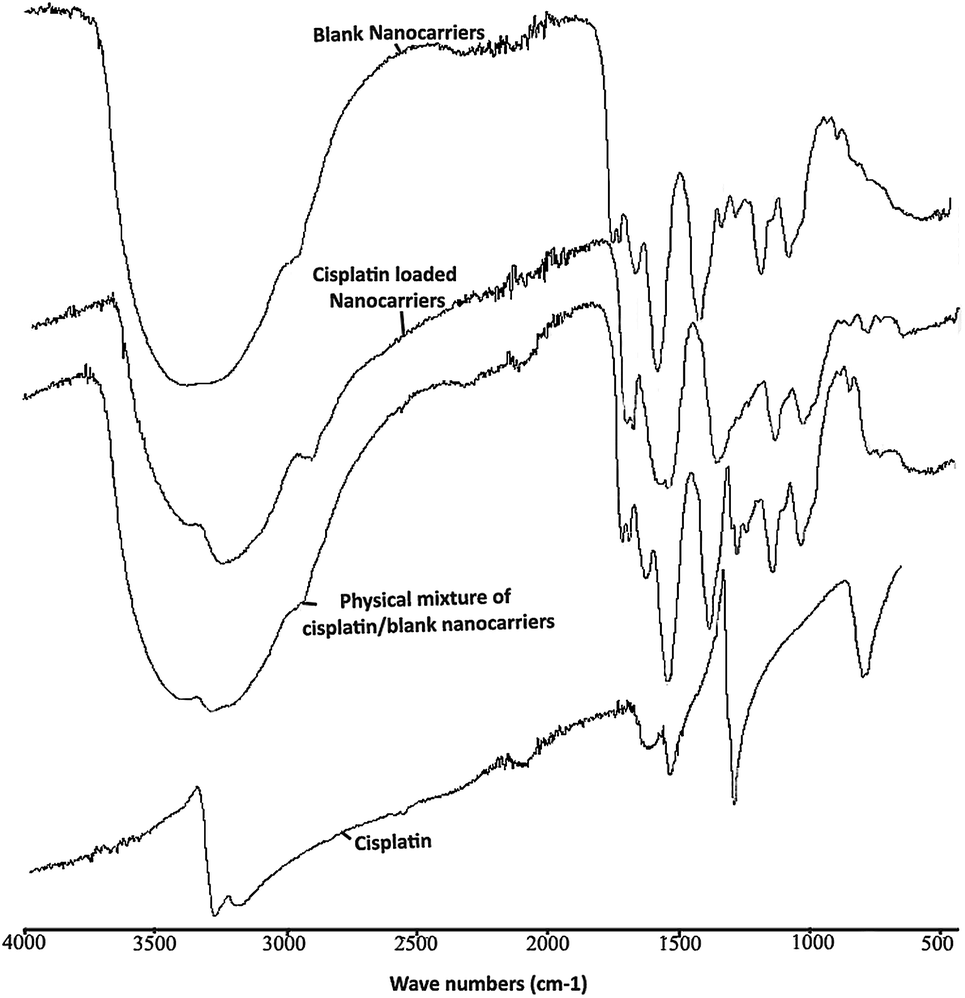 | ||
| Fig. 5 ATR-FTIR spectra of empty BSCT nanocarriers, cisplatin-loaded BSCT nanocarriers, blank BSCT nanocarriers–cisplatin physical mixture, and cisplatin powder. | ||
After loading cisplatin onto the nanocarriers, there was a decrease in the intensity of the peak at 3388 cm−1 which was due to the reduction in –OH functionalities after the COOH groups of the succinic acid moieties formed coordinate bonds with cisplatin. However, the peak did not disappear because of the OH groups of chitosan backbone which were still present (3408 cm−1). In addition, the decrease in the intensity of the –OH peak at 3388 cm−1 allowed the primary amine peak of cisplatin to present at 3265 cm−1. Moreover, the CO stretching of the COOH groups of the succinic moieties at 1699 cm−1 was shifted toward 1598 cm−1 after drug loading, representing the generation of coordinate bonds between the carboxylic groups of the BSCT nanocarriers and cisplatin. However, no change was found in the ATR-FTIR spectrum of the blank nanocarriers–cisplatin mixture. Therefore, these results suggested that the co-ordinate bonds play an important role in cisplatin loading onto BSCT nanocarriers.
In vitro cytotoxicity
The effects of cisplatin on the cell lines HT29 and HN22 were investigated. The IC50 values of free cisplatin on HT29 and HN22 were 47.74 μg mL−1 and 9.12 μg mL−1, respectively, representing high cytotoxicity for these cells. The greater effect of cisplatin on HN22 cells than HT29 cells indicated a strong killing effect on head and neck cancer cells. According to Wilmes et al. cisplatin is commonly used as an anticancer drug to treat head and neck, lung, testicular, ovarian, and breast cancer.1 Therefore, HN22 cells were employed for the evaluation of the cancer cell-killing properties of cisplatin-loaded nanocarriers compared with free cisplatin using flow cytometry. The flow cytometry results of HN22 cells treated with free cisplatin and cisplatin-loaded BSCT nanocarriers are displayed in Fig. 6a.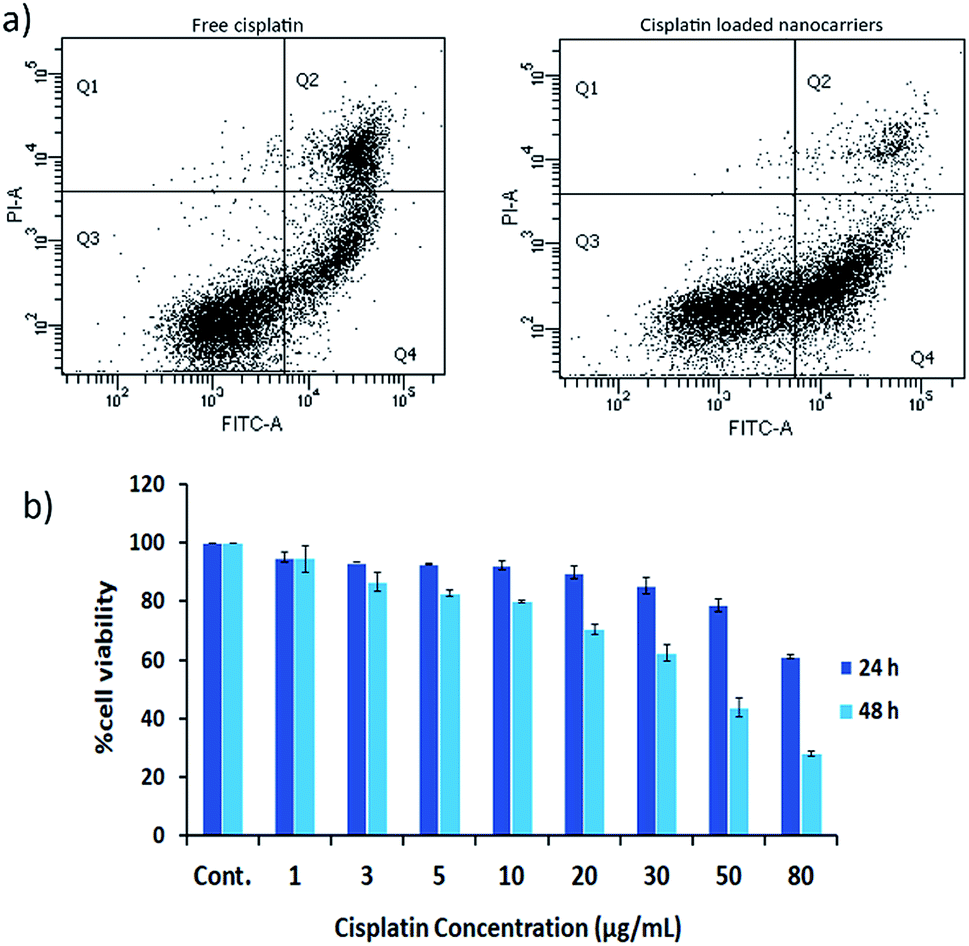 | ||
| Fig. 6 (a) Flow cytometry of HN22 cells treated with free cisplatin and cisplatin-loaded BSCT nanocarriers. (b) Cell viability of HN22 cells treated with the cisplatin-loaded nanocarriers (equivalent to various concentration of cisplatin) for 24 and 48 h. | ||
As shown in Fig. 6a, the HN22 cells treated with the cisplatin-loaded nanocarriers presented a greater percentage of early apoptosis (40.60%) compared with late apoptosis (4.6%) and necrosis (0.2%), in comparison to those of the cells treated with the cisplatin-loaded nanocarriers. The cells treated with free cisplatin demonstrated less of a percentage of early apoptosis (23.50%) and a higher percentage of late apoptosis (17.60%) and necrosis (0.5%). Apoptosis is a dynamic energy-dependent process that mostly takes place without inflammation or injury to neighbouring tissues.23 The inflammation process is inhibited by rapid and efficient clearance of apoptotic cells when their cell membranes are still intact (early apoptosis). On the other hand, the late apoptotic cells (apoptotic cells that have lost membrane integrity), and necrotic cells release their inflammatory intracellular contents and can generate inflammatory responses.24 Therefore, treating the cancer cells with the cisplatin-loaded nanocarriers could lead to less of an inflammatory response after the treatment compared to treatment with free cisplatin.
Due to the sustained release of cisplatin from the nanocarriers, the killing activity may be increased when the treatment time is increased. Therefore, the cell viability of the NH22 cells after treatment with the cisplatin-loaded nanocarriers was evaluated at 24 and 48 h. The results revealed that the killing effect of the cisplatin-loaded nanocarriers was found to be time and concentration dependent as shown in Fig. 6b.
Intracellular Pt(II) accumulation
The incorporation of cisplatin into living cells was investigated by intracellular Pt(II) accumulation (ng) as displayed in Fig. 7. Free cisplatin exhibited a very low amount of Pt(II) uptake in living cells (HN22 cells) compared to the uptake caused by cisplatin-loaded BSCT nanocarriers. Because cisplatin is widely used to induce tumour cell death,25 the inclusion of free cisplatin in the living cells provided a strong cytotoxic effect on these cancer cells. Cell death or poor penetration of the aqueous complex of cisplatin into cells (poor membrane permeability) might decrease the value of Pt(II) accumulated in the living cells.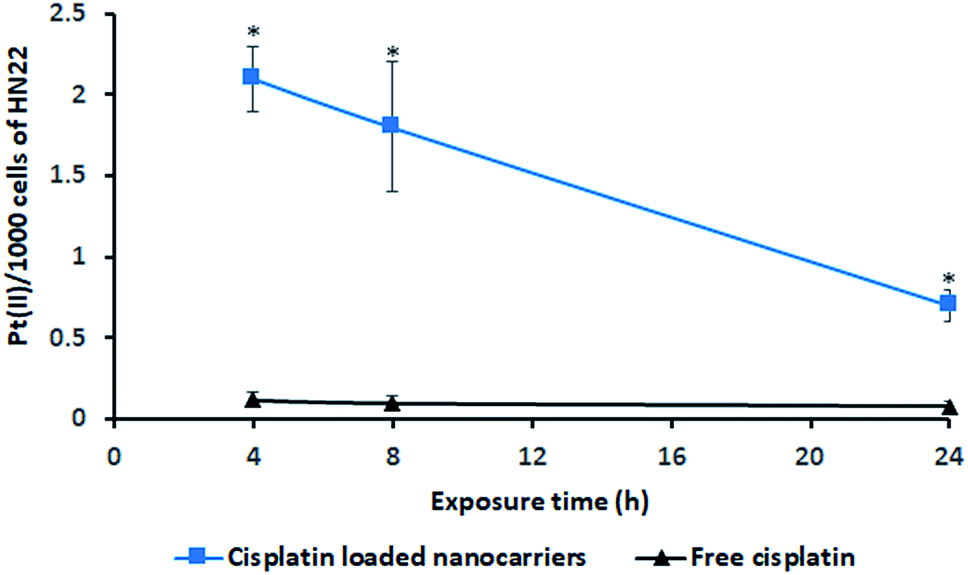 | ||
| Fig. 7 Intracellular Pt(II) accumulation (ng) in the cell line (HN22) after treatment with cisplatin-loaded BSCT nanocarriers (■) and free cisplatin (▲). *Statistically significant difference from free cisplatin. | ||
In the case of the cisplatin-loaded BSCT nanocarriers, a slower killing rate and greater permeability of cisplatin-loaded polymeric nanocarriers might result in an increased amount of entrapped cisplatin in the living cells. The incorporation of Pt(II) in the living cells was observed to be a higher value at the times of 4 to 8 h and decreased at 24 h, suggesting that the release of cisplatin from the BSCT nanocarriers at the time between 8 h and 24 h led to the killing of cancer cells. Therefore, the cisplatin-loaded BSCT nanocarriers were accumulated in the cancer cells and subsequently sustained the release of the drug, leading to a longer period in which to kill cancer cells. However, the prolonged treatment by the sustained release rate might also decrease the inflammatory response and reduce the systemic toxicity of cisplatin.
Apoptosis and necrosis detection in renal cells
The rapid drug distribution throughout the whole body via intravenous administration of cisplatin provided severe side effects such as acute nephrotoxicity and chronic nephrotoxicity.26 The sustained release of the drug from the nanocarriers has a tendency to reduce the adverse side effects of cisplatin in the whole body. Flow cytometry analysis was performed to investigate the effect of cisplatin-loaded nanocarriers on the normal kidney cells, RPTEC/TERT1, compared with free cisplatin. The amount of cell death was then determined, and the results are displayed in Fig. 8. The results revealed that the cells treated with cisplatin showed a higher percentage of necrotic or late apoptotic cells (32.23%) compared with the cisplatin-loaded nanocarriers (15.57%). Therefore, delivering cisplatin via the nanocarriers could reduce the renal toxicity of cisplatin. This may be due to the gradual release of cisplatin from the nanocarriers which could reduce the toxicity of cisplatin on the cells. On the other hand, for the cells treated with free cisplatin, a high concentration of cisplatin was exposed to the renal cells straight away, and this led to greater cell death. Therefore, using BSCT nanocarriers might be an appropriate carrier of cisplatin for decreasing toxicity and prolonging antitumour treatment. | ||
| Fig. 8 Flow cytometry of RPTEC/TERT1 cells exposed to different treatments (a) control, (b) cisplatin, and (c) cisplatin-loaded BSCT. | ||
Conclusions
In this study, chitosan-based nanocarriers were successfully fabricated and bound with cisplatin by co-ordinate bonds between the carboxylic group of the polymer and the Pt centre of cisplatin. The BSCT nanocarriers exhibited the highest cisplatin loading efficiency. Cisplatin was gradually released from the cisplatin-loaded nanocarriers by slow exchange with chloride ions. The sustained release of cisplatin from the nanocarriers reduced the toxicity toward kidney cells and decreased the inflammatory response without losing the anticancer activity. Therefore, BSCT nanocarriers might be promising cisplatin carriers for drug delivery to solid tumours which provide desirable antitumour efficacy and reduced toxicity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Thailand Research Funds through the Research Team Promotion Grant (RTA6180003), the Silpakorn University Research and Development Institute (SURDI610110), the Faculty of Pharmacy, Silpakorn University and the Beuth Hochschule für Technik Berlin, University of Applied Sciences for financial support. We also give special thanks to the staff in the laboratory for their assistance on this research.References
- A. Wilmes, C. Bielow, C. Ranninger, P. Bellwon, L. Aschauer, A. Limonciel, H. Chassaigne, T. Kristl, S. Aiche, C. G. Huber, C. Guillou, P. Hewitt, M. O. Leonard, W. Dekant, F. Bois and P. Jennings, Toxicol. In Vitro, 2015, 30, 117–127 CrossRef PubMed
.
- R. P. Miller, R. K. Tadagavadi, G. Ramesh and W. B. Reeves, Toxins, 2010, 2, 2490–2518 CrossRef PubMed
- J. Song, T. Xu, Y. Zhang, H. Guo, S. Zhu and L. Yang, Drug Delivery, 2016, 23, 3285–3293 CrossRef PubMed
- H. S. Oberoi, N. V. Nukolova, A. V. Kabanov and T. K. Bronich, Adv. Drug Delivery Rev., 2013, 65, 1667–1685 CrossRef PubMed
- E. Gianasi, R. G. Buckley, J. Latigo, M. Wasil and R. Duncan, J. Drug Targeting, 2002, 10, 549–556 CrossRef PubMed
- Z. Xu, Z. Wang, S. M. Yiu and G. Zhu, Dalton Trans., 2015, 44, 19918–19926 RSC
- K. Ajima, T. Murakami, Y. Mizoguchi, K. Tsuchida, T. Ichihashi, S. Iijima and M. Yudasaka, ACS Nano, 2008, 2, 2057–2064 CrossRef PubMed
- A. D. Carvalho Junior, F. P. Vieira, V. J. Melo, M. T. Lopes, J. N. Silveira, G. A. Ramaldes, A. Garnier-Suillerot, E. C. Pereira-Maia and M. C. Oliveira, Braz. J. Med. Biol. Res., 2007, 40, 1149–1157 CrossRef PubMed
- E. A. Leite, C. M. Souza, A. D. Carvalho-Junior, L. G. Coelho, A. M. Lana, G. D. Cassali and M. C. Oliveira, Int. J. Nanomed., 2012, 7, 5259–5269 Search PubMed
- J. Song, W. Ren, T. Xu, Y. Zhang, H. Guo, S. Zhu and L. Yang, Drug Des., Dev. Ther., 2017, 11, 441–449 CrossRef PubMed
- C. G. England, M. C. Miller, A. Kuttan, J. O. Trent and H. B. Frieboes, Eur. J. Pharm. Biopharm., 2015, 92, 120–129 CrossRef PubMed
- M. Yokoyama, T. Okano, Y. Sakurai, S. Suwa and K. Kataoka, J. Controlled Release, 1996, 39, 351–356 CrossRef
- N. Nishiyama and K. Kataoka, J. Controlled Release, 2001, 74, 83–94 CrossRef PubMed
- H. Uchino, Y. Matsumura, T. Negishi, F. Koizumi, T. Hayashi, T. Honda, N. Nishiyama, K. Kataoka, S. Naito and T. Kakizoe, Br. J. Cancer, 2005, 93, 678–687 CrossRef PubMed
- H. S. Oberoi, F. C. Laquer, L. A. Marky, A. V. Kabanov and T. K. Bronich, J. Controlled Release, 2011, 153, 64–72 CrossRef PubMed
- R. A. A. Muzzarelli, J. Boudrant, D. Meyer, N. Manno, M. DeMarchis and M. G. Paoletti, Carbohydr. Polym., 2012, 87, 995–1012 CrossRef
- C. Zhang, P. Qineng and H. Zhang, Colloids Surf., B, 2004, 39, 69–75 CrossRef PubMed
- T. Woraphatphadung, W. Sajomsang, P. Gonil, S. Saesoo and P. Opanasopit, Carbohydr. Polym., 2015, 121, 99–106 CrossRef PubMed
- W. Sajomsang, P. Gonil, S. Saesoo, U. R. Ruktanonchai, W. Srinuanchai and S. Puttipipatkhachorn, Int. J. Pharm., 2014, 477, 261–272 CrossRef PubMed
- J. Comenge, C. Sotelo, F. Romero, O. Gallego, A. Barnadas, T. G. Parada, F. Dominguez and V. F. Puntes, PLoS One, 2012, 7, e47562 CrossRef PubMed
- G. E. Craig, S. D. Brown, D. A. Lamprou, D. Graham and N. J. Wheate, Inorg. Chem., 2012, 51, 3490–3497 CrossRef PubMed
- T. Ngawhirunpat, N. Wonglertnirant, P. Opanasopit, U. Ruktanonchai, R. Yoksan, K. Wasanasuk and S. Chirachanchai, Colloids Surf., B, 2009, 74, 253–259 CrossRef PubMed
- G. Majno and I. Joris, Am. J. Pathol., 1995, 14, 3–15 Search PubMed
- V. A. Patel, A. Longacre, K. Hsiao, H. Fan, F. Meng, J. E. Mitchell, J. Rauch, D. S. Ucker and J. S. Levine, J. Biol. Chem., 2006, 281, 4663–4670 CrossRef PubMed
- A.-M. Florea and D. Büsselberg, Cancers, 2011, 3, 1351–1371 CrossRef PubMed
- J. H. Kim, Y. S. Kim, K. Park, S. Lee, H. Y. Nam, K. H. Min, H. G. Jo, J. H. Park, K. Choi, S. Y. Jeong, R. W. Park, I. S. Kim, K. Kim and I. C. Kwon, J. Controlled Release, 2008, 127, 41–49 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2018 |