
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImido-substituted triazines as dehydrative condensing reagents for the chemoselective formation of amides in the presence of free hydroxy groups†
Masanori Kitamura ,
Suguru Sasaki,
Riho Nishikawa,
Kohei Yamada and
Munetaka Kunishima*
,
Suguru Sasaki,
Riho Nishikawa,
Kohei Yamada and
Munetaka Kunishima*
Faculty of Pharmaceutical Sciences, Institute of Medical, Pharmaceutical, and Health Sciences, Kanazawa University, Kakuma-machi, Kanazawa 920-1192, Japan. E-mail: kunisima@p.kanazawa-u.ac.jp
First published on 20th June 2018
Abstract
In this paper, we discuss the synthesis of imido-substituted chlorotriazines and demonstrate their use in dehydrative condensation reactions. Chemoselective amide-forming reactions of amino alcohols using succinimido-substituted chlorotriazine (2A) proceeded smoothly. Occasionally, nonselectivity was problematic during the synthesis of hydroxy-substituted amides. Moreover, it was noteworthy that this method was applicable to hydroxy-substituted carboxylic acids that could have formed a lactone or an ester during the carboxylic acid activation step. The imido-substituted chlorotriazine (2A) was superior to the amido-substituted chlorotriazine and 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) in terms of reaction rates and yields.
Introduction
Amides are important functional groups that can be found in many materials, pharmaceuticals, and natural products. Therefore, they are routinely synthesized from carboxylic acids and amines in both the laboratory and industry.1 Chemoselective amide-forming reactions of amino alcohols have long been investigated2–4 because many important compounds contain this combination of functional groups, including anti-inflammatory drugs such as acetaminophen,2 cosmetics,5 and surfactants.6 However, nonselective N- and O-acylation of amino alcohols is often problematic during amide synthesis.2,3,7 In addition, the activation of a hydroxy-substituted carboxylic acid is difficult using conventional methods because lactone or ester formation is possible during carboxylic acid activation.We have developed a class of trizaine-based dehydrative condensing reagents [(4,6-dimethoxy-1,3,5-triazin-2-yl)trialkylammonium salts, DMT-Ams], including 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM), which can be employed in aqueous or alcoholic solvents (Scheme 1. See the ESI† for the detailed reaction mechanism).8,9 These reagents were prepared from 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) and N-methylmorpholine (NMM) or tertiary amines (tert-amines).9 For the preparation of functional DMT-Ams that selectively activate carboxylic acids, various tert-amines were studied.10,11 However, in most cases, the substituents on the triazine ring were limited to dimethoxy groups. Considering that substituents on the triazine would be important for the development of a diverse family of dehydrative condensing reagents, we studied chlorotriazines and their ammonium salts. As part of this project, we have recently reported that electron-withdrawing amido-substituted triazines (1 and 3) showed a higher reactivity for dehydrative condensing reactions.12 According to the Hammett substituent constants,13 imido-substituents (σm = 0.34 for the succinimido group) are more electron-withdrawing than methoxy- (σm = 0.12) and amido-substituents (σm = 0.21 for the acetamido group); therefore, a higher reactivity for condensation was expected. In this study, triazines with even more electron-withdrawing imido-substituents (2 and 4) were investigated, including the chemoselective acylation of amino alcohols and activation of hydroxy-substituted carboxylic acids.
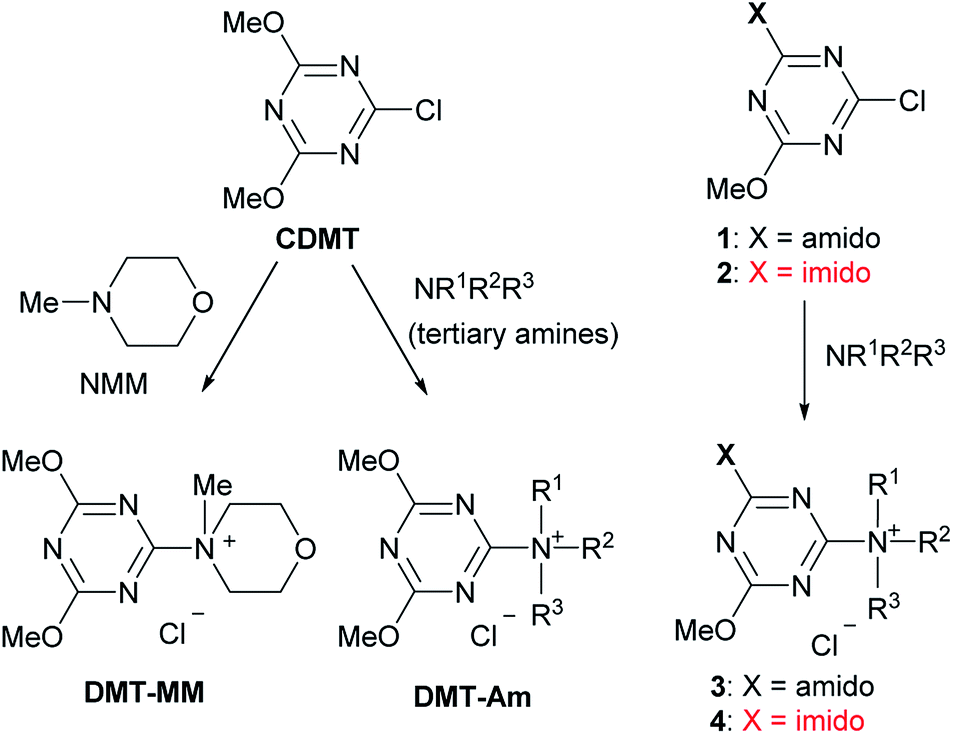 | ||
| Scheme 1 Triazine-based condensing reagents. | ||
Results and discussion
Rapidly available imides, such as succinimide, phthalimide, maleimide, and glutarimide, as substituents for triazine, were studied because condensing reagents should be inexpensive, especially if they are to be used in large-scale syntheses (2A–2D in Fig. 1). Moreover, instead of cyclic imides, linear diacetamide was also considered (2E in Fig. 1). For simplification, only one methoxy group on CDMT was replaced with an imido-substituent (Fig. 1).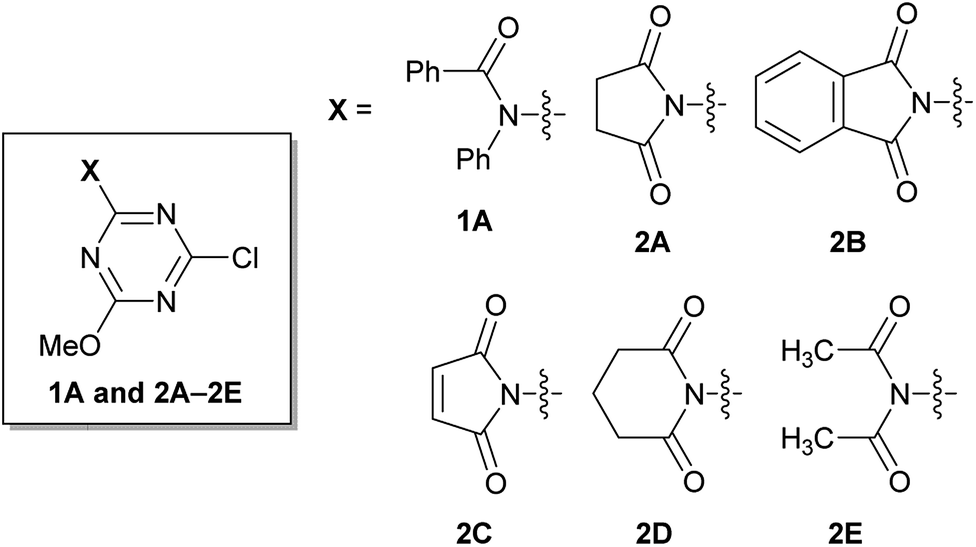 | ||
| Fig. 1 Structures of amido- or imido-substituted chlorotriazines. | ||
It was difficult to determine the general reaction conditions for the synthesis of the above-mentioned imido-substituted chlorotriazines because the different imides possessed unequal reactivities (Scheme 2). The succinimido-substituted compound 2A was obtained from the reaction of 2,4-dichloro-6-methoxy-1,3,5-triazine (DCMT) with the succinimide anion in tetrahydrofuran (THF) at 55 °C (55% isolated yield). 2B was prepared in a 58% isolated yield using the commercially available phthalimide potassium salt and DCMT in the presence of 18-crown-6 at ambient temperature. However, 2C could not be obtained under similar reaction conditions using DCMT in the presence of various bases. Compared to the five-membered ring succinimide, the nucleophilicity of the six-membered glutarimide anion toward DCMT was unexpectedly low. Despite the decreased delocalization of the negative charge for the glutarimide anion as expected from their pKa values (11.4 for glutarimide and 9.6 for succinimide), the yield of 2D was low.14 Shielding of the nitrogen lone pairs of the imide anion by the two adjacent carbonyl oxygen groups, as expected from X-ray crystal structure analysis of glutarimide15 and succinimide,16 may explain the reduced reactivity. Glutarimide had a smaller O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N bond angle (119.60° for glutarimide and 123.86° for succinimide) and larger C–N–C bond angle (126.90° for glutarimide and 112.62° for succinimide). 2D was successfully crystalized and the structure was confirmed by X-ray single crystal structure analysis (see the ESI†). In the case of the linear diacetamide, 2E was difficult to prepare. Despite the consumption of DCMT, only side products were detected.
C–N bond angle (119.60° for glutarimide and 123.86° for succinimide) and larger C–N–C bond angle (126.90° for glutarimide and 112.62° for succinimide). 2D was successfully crystalized and the structure was confirmed by X-ray single crystal structure analysis (see the ESI†). In the case of the linear diacetamide, 2E was difficult to prepare. Despite the consumption of DCMT, only side products were detected.
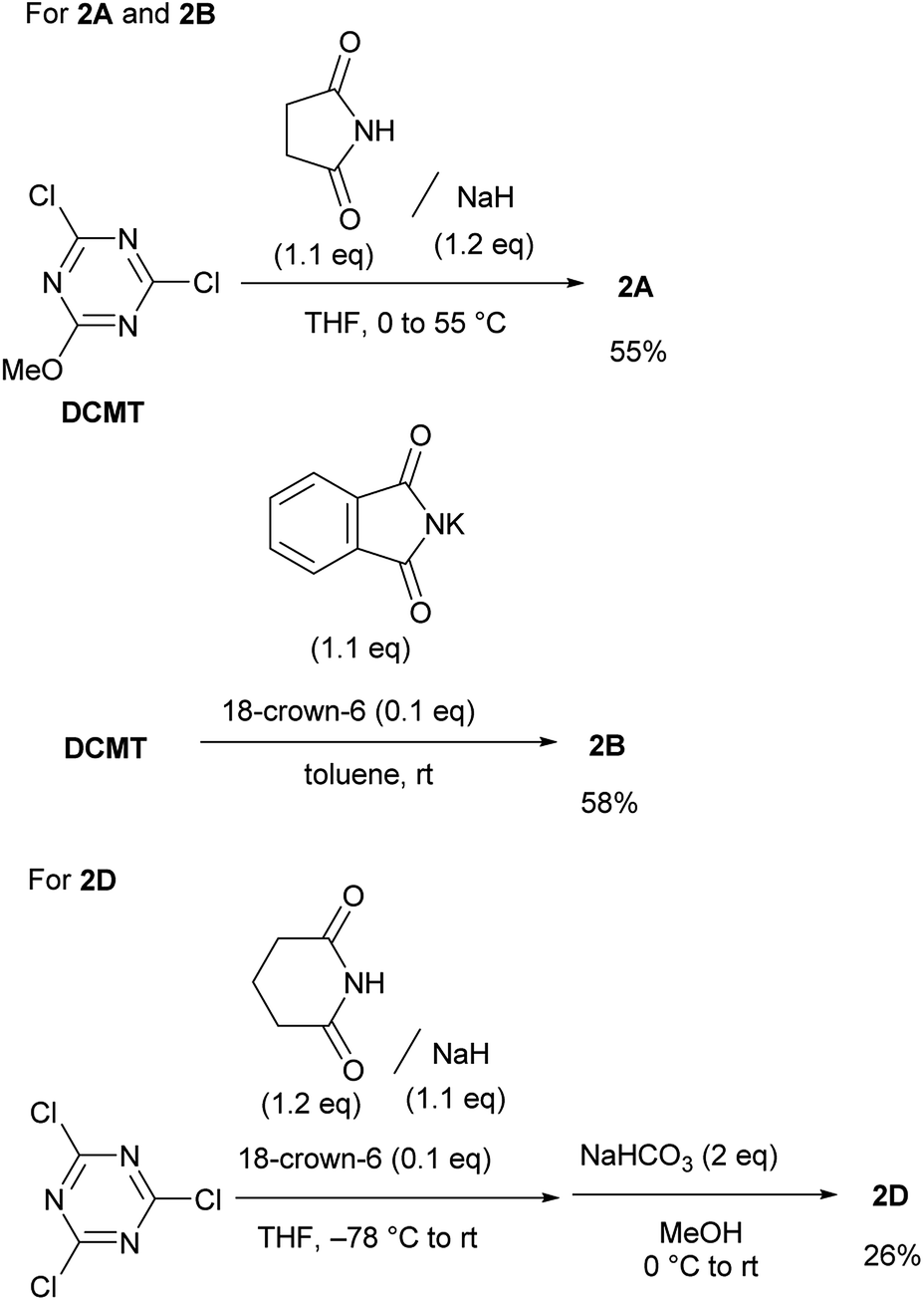 | ||
| Scheme 2 Synthesis of chlorotriazines (2A, 2B, and 2D). | ||
Next, we examined the preparation of the imido-substituted condensing reagents (4). Although the desired 4 would be formed using NMM and N-methylpiperidine (NMP) as the tert-amines, the purities of 4 were low because demethylation (by the chloride anion) and hydrolysis occurred during the isolation steps. To prevent demethylation, the synthesis of 4 was attempted in the presence of nonnucleophilic perchlorate anions; however, hydrolysis was still problematic. It was assumed that the strong electron-withdrawing effect of the imido group made isolation difficult.
For convenient evaluation of the reactivity of these prepared chlorotriazines (2A, 2B, and 2D), the corresponding DMT-Ams (10) were generated in situ and used for coupling of carboxylic acid 5a and amine 6a (Scheme 3 and Table 1).9 Chlorotriazine 2A was added to a mixture of 5a, 6a, and NMM at ambient temperature in MeOH (Table 1, entry 1).17 Amide 7a was only obtained in a 69% yield. The formation of side product 8A (3%) and methyl ester 9a (11%) was also observed. The yield of 9a using 2A was larger than that using CDMT (2% for CDMT), indicating that the electrophilicity of triazinyl ester 11 toward methanol was increased because of the succinimido group on the triazine ring. In THF, the increased yield of the side product 8A (35% for 2A) was observed, in contrast to CDMT (18% for CDMT), implying a similar electron-withdrawing effect of the succinimido group.18 To prevent the alcoholysis of 11, the reaction was conducted in iPrOH and the yield of 7a was improved to 84% (Table 1, entry 1). Due to the fact that similar results were obtained for 2B and 2D (Table 1, entries 2 and 3), the influence from the kind of imido-structures was thought to be small for the condensing reactions. Moreover, the phthalimido-substituted chlorotriazine (2B) was not very soluble in THF and alcohol; therefore, only succinimido-substituted chlorotriazine 2A and glutarimido-substituted chlorotriazine 2D were selected for further studies.
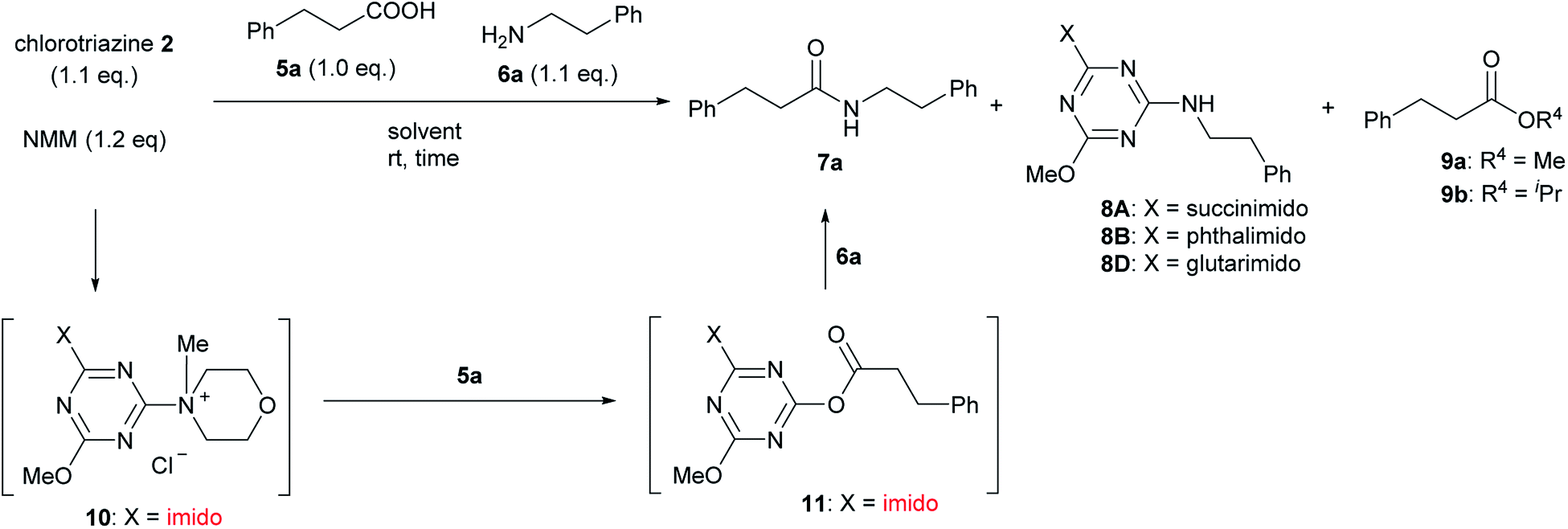 | ||
| Scheme 3 Amide-forming reactions of carboxylic acid 5a and amine 6a using imido-substituted chlorotriazines 2. | ||
The formation of the side product 8 was assumed to result from chlorotriazine 2 being more electrophilic than 10 or 11. Thus, the addition of amine 6 after the formation of 11 should prevent the formation of 8 (Table 2). Accordingly, amine 6 was added to the preformed mixture of chlorotriazine, NMM, and carboxylic acid 5 in THF. In this solvent, ester 9 would never be formed. In the amide-forming reactions of 2-phenylpropionic acid (5a) with 2-phenethylamine (6a), aniline (6b), or 4-(ethoxycarbonyl)aniline (6c) (the nucleophilicity of these amines decreases in this order), imido-substituted 2A and 2D afforded better yields than CDMT (Table 2, entries 1–9). From the viewpoints of atom economy, starting material costs, and synthetic yields, 2A was selected for further studies. In the case of sterically hindered isobutyric acid (5b, entries 10 and 11), 2A also afforded amide 7d in a better yield than that using CDMT. When the more sterically hindered pivalic acid (5c) was used (entries 12–17), different reactivities between 2A and CDMT were observed for aniline 6b (entry 16 versus 17). Although it was thought that the benzoic acid derivative (5d, with electron-withdrawing nitro groups) might form intermediate 11 slowly, 2A afforded product 7h in an excellent yield compared to CDMT (Table 2, entry 18 versus 19). This method is also effective for formation of a peptide bond, i.e., condensation of Boc-Leu-OH (5e) and H-Phe-OMe (6e) afforded the corresponding dipeptide (7i) in 94% NMR yield as a single diastereomer (Table 2, entry 20). As indicated in entry 18 of Table 2, purification by only extraction and recrystallization without column chromatography was also possible.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Chlorotriazine | Carboxylic acid 5 | Amine 6 | Amide 7 | Time | Yielda (%) |
| a NMR yields. Isolated yields are given in the parentheses.b Reaction time of the first step was 15 min.c The desired product was isolated in 81% yield only by extraction and recrystallization without column chromatography.d Boc-Leu-OH (1.0 eq.), 2A (1.05 eq.), NMM (1.2 eq.), H-Phe-OMe·HCl (1.2 eq.), and EtNiPr2 (1.2 eq.) were used. | ||||||
| 1 | 2A | 5a | 6a | 7a | 40 min | 91 (83) |
| 2 | CDMT | 40 min | 86 | |||
| 3 | 2A | 5a | H2N–Ph 6b | 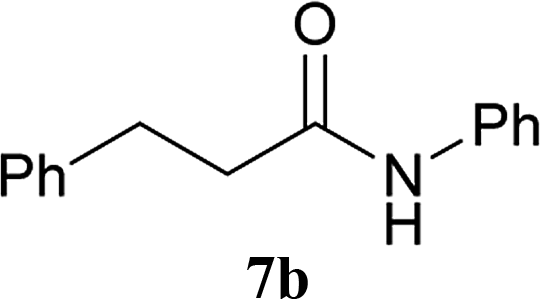 |
30 min | 91 (90) |
| 4 | 2D | 20 min | 91 | |||
| 5 | CDMT | 30 min | 65 | |||
| 6 | 2A | 5a | 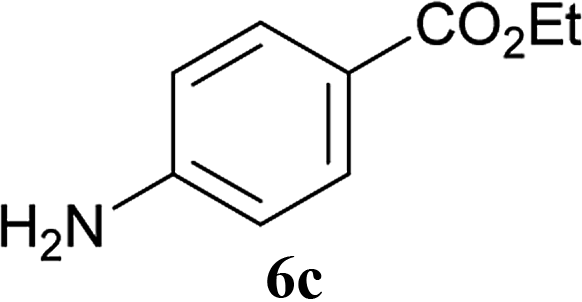 |
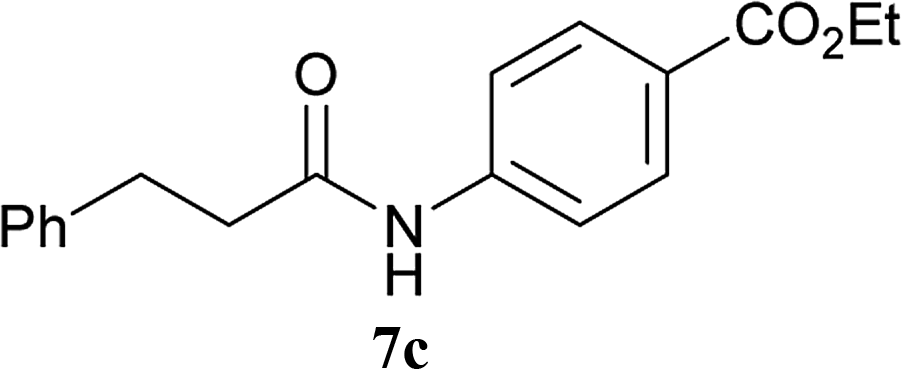 |
2 h | 92 (90) |
| 7 | 2D | 2 h | 71 | |||
| 8 | 2D | 7 h | 93 | |||
| 9 | CDMT | 2 h | 34 | |||
| 10 | 2A | iPr–CO2H 5b | H2N–iPr 6d | 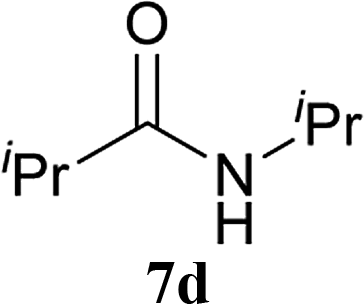 |
5 h | 91 (69) |
| 11 | CDMT | 5 h | 82 | |||
| 12 | 2A | tBu–CO2H 5c | 6a | 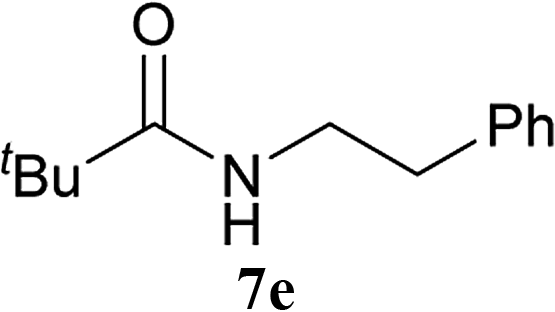 |
5 h | 78 (63) |
| 13 | CDMT | 5 h | 79 | |||
| 14b | 2A | 5c | 6d | 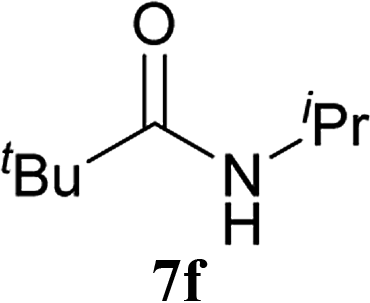 |
24 h | 77 (75) |
| 15b | CDMT | 24 h | 84 | |||
| 16b | 2A | 5c | 6b | 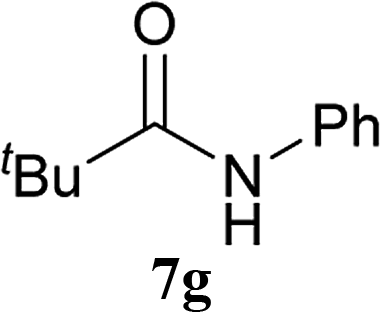 |
8 h | 69 (60) |
| 17b | CDMT | 8 h | 25 | |||
| 18 | 2A | 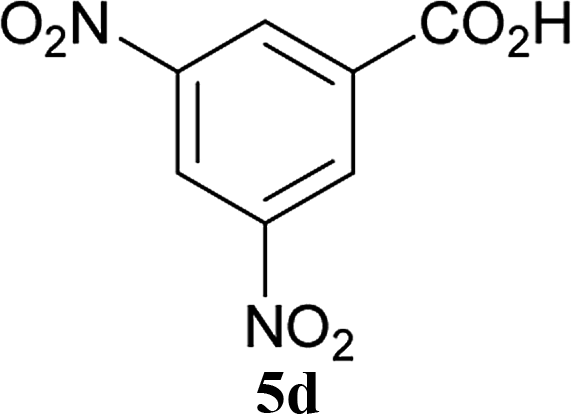 |
6a | 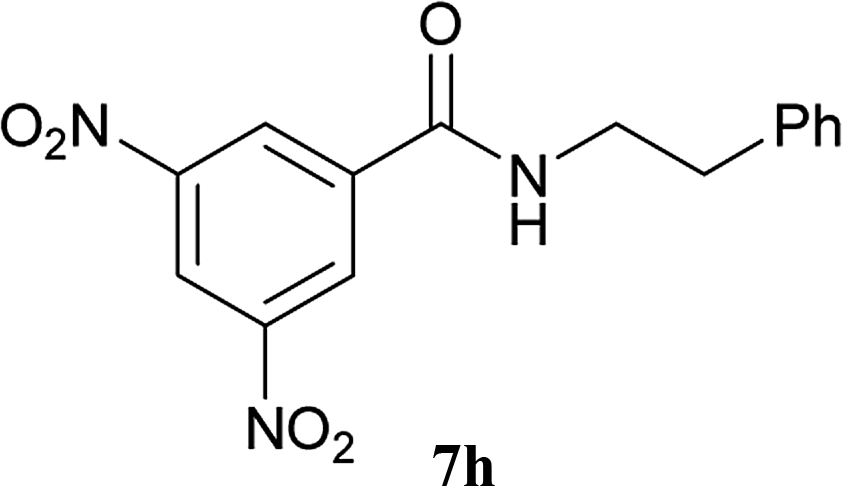 |
5 h | 96 (94c) |
| 19 | CDMT | 5 h | 86 | |||
| 20d | 2A | Boc-Leu-OH 5e | H-Phe-OMe 6e | Boc-Leu-Phe-OMe 7i | 1 h | 94 (90) |
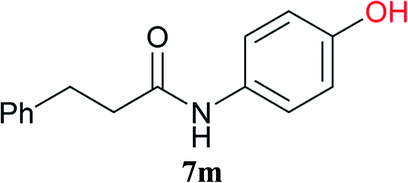
From these results, it is unambiguous that the succinimido-substituted chlorotriazine 2A was more reactive than CDMT, and less sterically hindered alcoholic solvents (especially methanol) are unsuitable for carrying out the condensing reactions because of alcoholysis of 11. However, we anticipated that the competing alcoholysis against aminolysis would be reduced when lower concentrations of alcohols were used. Namely, chemoselective amide forming reactions could be effective even using carboxylic acids 5 and amines 6 having free hydroxy groups. Thus, we tested 6-hydroxyhexanoic acid (5f) as it would be susceptible to an intramolecular lactone-formation or an intermolecular ester-formation during the carboxylic acid activation step (Table 3, entries 1 and 2). The desired amide 7j was obtained in a good yield compared to CDMT under the reaction conditions used in Table 2. When amino alcohols such as benzyl(2-hydroxyethyl)amine (6f, Table 3, entries 3 and 4) and N-(2-hydroxyethyl)aniline (6g, Table 3, entries 5 and 6) were employed, better yields of amide products (7k and 7l) were observed, even in the presence of the competing free hydroxy groups. Although the chemoselective acylation of 4-aminophenol (6h) using EDC and HOBt in the presence of a surfactant was insufficient,2 2A afforded the desired product 7m chemoselectively. Even though Boc-Tyr-OH having the phenolic hydroxy group was used as the starting material, the desired dipeptide (7n) was obtained in 80% NMR yield and 71% isolated yield (Table 3, entry 9).
| Entry | Chlorotriazine | Carboxylic acid 5 | Amine 6 | Amide 7 | Time | Yielda (%) |
|---|---|---|---|---|---|---|
| a NMR yields. Isolated yields are given in the parentheses.b 3-Phenylpropionic acid (1.1 eq.), 2A or CDMT (1.0 eq.), NMM (1.05 eq.), and 4-aminophenol (1.0 eq.) were used.c Boc-Tyr-OH (1.0 eq.), 2A (1.0 eq.), NMM (1.2 eq.), H-Gly-OtBu·HCl (2.0 eq.), and EtNiPr2 (2.0 eq.) were used. | ||||||
| 1 | 2A | 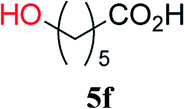 |
6c | 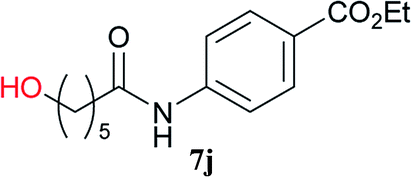 |
4 h | 81 (75) |
| 2 | CDMT | 4 h | 30 | |||
| 3 | 2A | 5a | 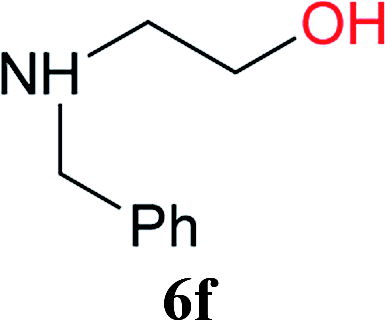 |
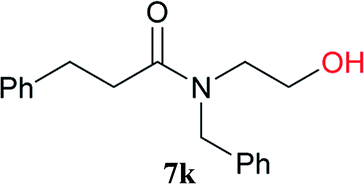 |
15 min | 88 (84) |
| 4 | CDMT | 15 min | 71 | |||
| 5 | 2A | 5a | 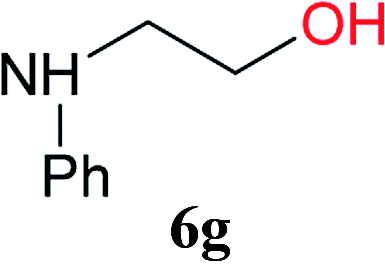 |
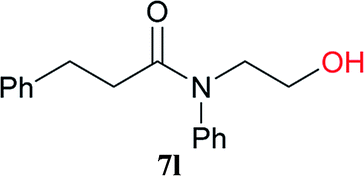 |
6 h | 97 (85) |
| 6 | CDMT | 6 h | 73 | |||
| 7b | 2A | 5a | 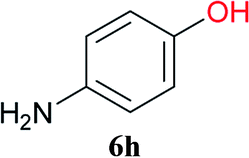 |
 |
15 min | 90 (73) |
| 8b | CDMT | 15 min | 54 | |||
| 9c | 2A | 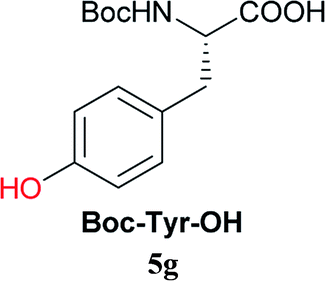 |
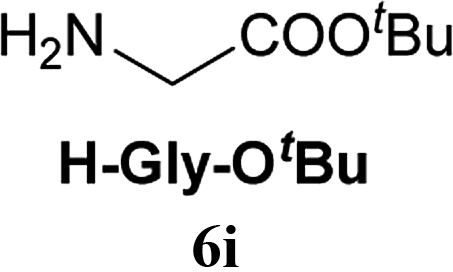 |
Boc-Tyr-Gly-OtBu 7n | 1 h | 80 (71) |
To clarify the reactivity of 2A compared with the amido-substituted chlorotriazine 1A and CDMT, the formation of 7l under similar conditions used in Tables 2 and 3 was investigated kinetically with 1H NMR spectroscopy (Fig. 2). The reaction rate with 2A was faster than that with 1A or CDMT.
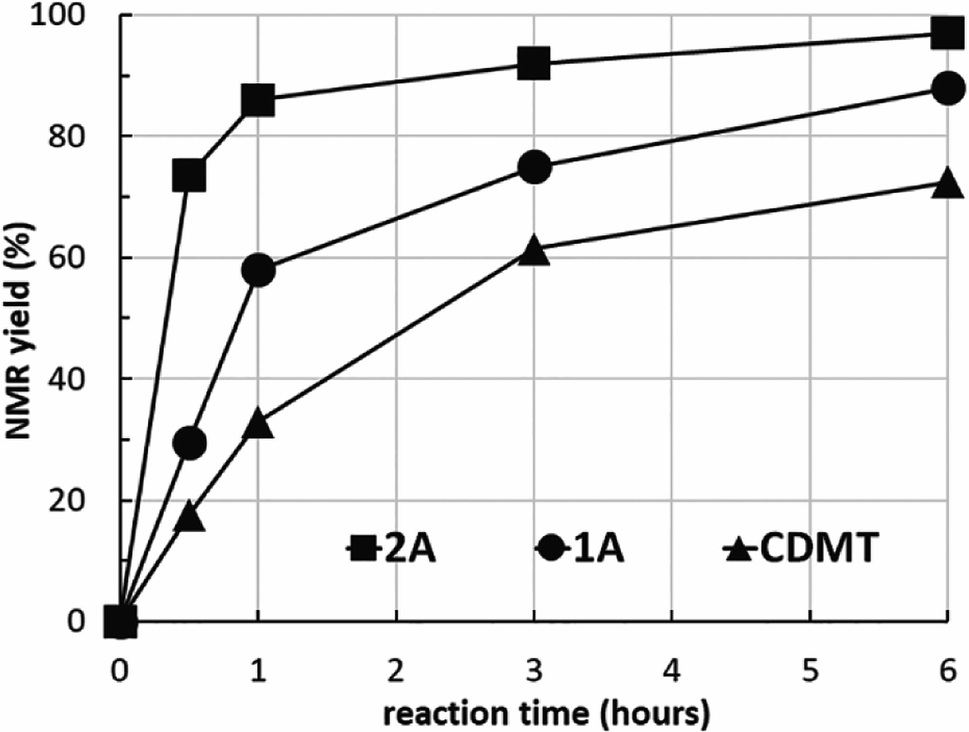 | ||
| Fig. 2 Kinetic study of amide 7l formation using 2A, 1A, and CDMT. | ||
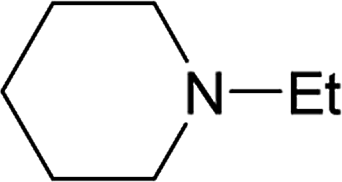
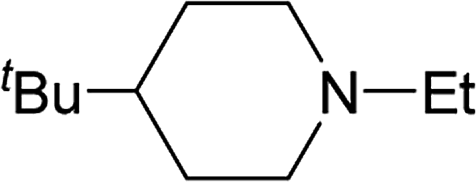
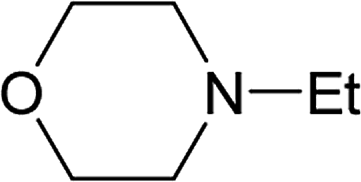
From the study of the DMT-Ams, we previously elucidated that the reactivity of tert-amines toward CDMT was strongly influenced by their steric hinderance;9 that is, a β-alkyl group toward the nitrogen lone pair in a gauche relationship remarkably hampers the nucleophilic attack on CDMT. Because of this effect, N-ethylmorpholine, N-ethylpiperidine, and triethylamine are unable to react with CDMT to afford DMT-Ams at ambient temperature. Contrastingly, the imido-substituted chlorotriazine 2A reacted with these tert-amines, resulting in the formation of 7a (Table 4). The strong electron-withdrawing ability of the succinimido group was thought to be responsible for overcoming this difficulty, thus enabling the use of various tert-amines for condensing reagents.10,11
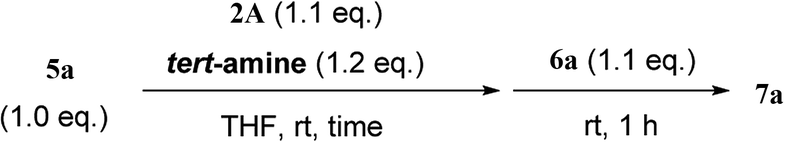
Conclusion
We have developed imido-substituted chlorotriazines (2) and demonstrated their use in efficient dehydrative condensing reactions between carboxylic acids and amines. Chemoselective amide-forming reactions using amino alcohols with succinimido-substituted 2A were successfully achieved. In addition, we demonstrated that 2A was an effective reagent for the chemoselective condensing reaction of hydroxy-substituted carboxylic acids that could form a lactone or an ester during the activation step. In terms of reaction rates and amide yields, 2A was superior to the amido-substituted chlorotriazine 1A and CDMT. Sterically hindered tert-amines that would never react with CDMT at ambient temperature, such as N-ethylmorpholine, N-ethylpiperidine, and triethylamine, were successfully employed in the formation of DMT-Ams with 2A. These results will contribute to the development of novel functionalized triazine-based condensing reagents and tert-amine-catalyzed amide-forming reactions.Experimental
General methods
Nuclear magnetic resonance [1H NMR (400 or 600 MHz) and 13C NMR (100 or 150 MHz)] spectroscopy was carried out on JEOL JNM-ECS400 and JEOL JNM-ECA600 spectrometers. Chemical shifts for 1H NMR were reported as δ values relative to tetramethylsilane (TMS). The coupling constants for 1H NMR were reported in hertz (Hz). The following abbreviations were used for reporting the spin multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, m = multiplet, and br = broad. Chemical shifts for 13C NMR were reported as δ values relative to the center line of the deuterochloroform triplet at 77.16 ppm. Mass spectra were measured on a JEOL JMS-T100TD spectrometer (DART- and ESI-MS). Analytical thin-layer chromatography (TLC) was performed on Merck precoated analytical plates, 0.25 mm thick, silica gel 60 F254. Flash chromatography separation was performed using Kanto Chemical silica gel 60 N (spherical, neutral, 40–100 mesh) unless otherwise noted. All Reagents were of commercial grade and were used without further purification unless otherwise noted. Anhydrous THF and toluene were purchased from commercial sources. All reactions that are sensitive to oxygen or moisture were conducted under a N2 atmosphere. The 1H NMR spectral data for 2-methoxy-4-phenethylamino-6-succinimido-1,3,5-triazine (8A) and 2-glutarimido-4-methoxy-6-phenethylamino-1,3,5-triazine (8D) were consistent with those of authentic samples prepared from 2A and 2D (see the ESI† for synthetic details). 1H NMR spectral data for methyl 3-phenylpropanoate (9a)19,20 and 2-propyl 3-phenylpropanoate (9b)20,21 were consistent with the referenced chemical shift values. 1H NMR spectral data of the isolated amide products 7a,22 7b,23,24 7d,25 7e,26 7f,27 7g,28 7i,29 7m,30 and 7n (ref. 31) were identical to known literature values (see the ESI†).Experimental procedure and characterization data
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford 2A (4.00 g, 55% yield) as a white solid. Mp 169–171 °C; 1H NMR (400 MHz, CDCl3): δ 4.14 (s, 3H), 2.94 (s, 4H); 13C NMR (100 MHz, CDCl3): δ 173.63, 173.27, 172.71, 163.20, 56.96, 28.92; IR (KBr): 2956, 1736, 1560, 1415, 1369, 1288, 1163, 991, 918, 812 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C8H8ClN4O3 243.0285; found: 243.0301; anal. calcd for C8H7ClN4O3: C, 39.60; H, 2.91; N, 23.09. Found: C, 39.43; H, 2.95; N, 22.89.:1) to afford 2B (1.68 g, 58% yield) as a white solid. Mp 163–165 °C; 1H NMR (400 MHz, CDCl3): δ 8.02 (dd, J = 5.5, 3.0 Hz, 2H), 7.87 (dd, J = 5.5, 3.0 Hz, 2H), 4.17 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 173.19, 172.40, 163.94, 162.76, 135.46, 131.29, 124.67, 56.74; IR (KBr): 3095, 3064, 2958, 1797, 1739, 1558, 1419, 1353, 1271, 1082, 935 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C12H8ClN4O3 291.0285; found: 291.0282; anal. calcd for C12H7ClN4O3: C, 49.59; H, 2.43; N, 19.28. Found: C, 49.41; H, 2.50; N, 19.23.:1) to afford 2D (135 mg, 26% yield) as a white solid. Mp 125–127 °C; 1H NMR (400 MHz, CDCl3): δ 4.14 (s, 3H), 2.79 (t, J = 6.4 Hz, 4H), 2.14 (quin, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 173.85, 173.13, 171.19, 167.12, 57.00, 32.35, 17.11; IR (KBr): 3016, 2970, 1743, 1697, 1560, 1539, 1498, 1408, 1358, 1282, 1248, 1144, 1016, 920, 760, 561, 442 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C9H10ClN4O3 257.0441; found: 257.0444; anal. calcd for C9H9ClN4O3: C, 42.12; H, 3.53; N, 21.83. Found: C, 42.05; H, 3.47; N, 21.83.:2) as a white solid. Mp 136–137 °C; 1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 8.8 Hz, 2H), 7.52 (d, J = 8.8 Hz, 2H), 7.52 (br, 1H), 7.30–7.20 (m, 5H), 4.34 (q, J = 7.2 Hz, 2H), 3.04 (t, J = 7.6 Hz, 2H), 2.69 (t, J = 7.6 Hz, 2H), 1.37 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 170.85, 166.32, 142.03, 140.49, 130.85, 128.81, 128.47, 126.60, 125.97, 118.93, 61.03, 39.60, 31.50, 14.45; IR (KBr): 3317, 3060, 3030, 2983, 2904, 1709, 1670, 1608, 1593, 1523, 1454, 1410, 1363, 1315, 1277 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C18H20NO3 298.1443; found: 298.1464; anal. calcd for C18H19NO3: C, 72.71; H, 6.44; N, 4.71. Found: C, 72.41; H, 6.34; N, 4.70.:1 to CHCl3, CHCl3/AcOEt = 7:3) as a white solid. Mp 155–156 °C; 1H NMR (400 MHz, CDCl3): δ 9.14 (t, J = 2.0 Hz, 1H), 8.86 (d, J = 2.0 Hz, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.29–7.24 (m, 3H), 6.43 (br, 1H), 3.80 (td, J = 6.9, 6.9 Hz, 2H), 3.00 (t, J = 6.9 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 162.91, 148.76, 138.24, 138.16, 129.09, 128.89, 127.22, 127.16, 121.19, 41.83, 35.50; IR (KBr): 3415, 3097, 2945, 1672, 1630, 1603, 1541, 1527, 1487, 1452, 1435, 1346, 1327, 1306, 1282 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C15H14N3O5 316.0934; found: 316.0936; anal. calcd for C15H13N3O5: C, 57.14; H, 4.16; N, 13.33. Found: C, 56.92; H, 4.15; N, 13.31.:1 to 95:5) as a white solid. 1H NMR (400 MHz, CDCl3/CD3OD = 1:1): δ 7.98 (d, J = 8.7 Hz, 2H), 7.68 (d, J = 8.7 Hz, 2H), 4.36 (q, J = 7.2 Hz, 2H), 3.59 (t, J = 6.6 Hz, 2H), 2.42 (t, J = 7.6 Hz, 2H), 1.75 (quin, J = 7.6 Hz, 2H), 1.60 (quin, J = 7.1 Hz, 2H), 1.45 (quin, J = 7.1 Hz, 2H), 1.40 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3/CD3OD = 1:1): δ 173.20, 166.53, 142.76, 130.16, 124.79, 118.62, 61.40, 60.63, 36.70, 31.70, 25.02, 24.99, 13.64; IR (KBr) 3315, 2935, 2868, 1712, 1657, 1610, 1535, 1462, 1439, 1408, 1367, 1282, 1259, 1176, 1103, 1074, 1024, 962, 858, 769, 725, 694, 501 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C15H21NNaO4 302.1368; found: 302.1350.:1) as a colorless liquid. 1H NMR (400 MHz, CDCl3): δ 7.33–7.05 (m, 10H, major and minor), 4.65 (s, 0.6H, minor), 4.49 (s, 1.4H, major), 3.71 (t, J = 5.0 Hz, 1.4H, major), 3.63 (t, J = 5.6 Hz, 0.6H, minor), 3.53 (t, J = 5.0 Hz, 1.4H, major), 3.32 (t, J = 5.6 Hz, 0.6H, minor), 3.01 (t, J = 7.8 Hz, 0.6H, minor), 2.98 (t, J = 7.8 Hz, 1.4H, major), 2.79 (t, J = 7.8 Hz, 0.6H, minor), 2.66 (t, J = 7.8 Hz, 1.4H, major); 13C NMR (100 MHz, CDCl3): δ 174.88, 173.29, 141.31, 140.95, 137.63, 136.29, 129.03, 128.65, 128.57, 128.51, 127.94, 127.77, 127.37, 126.31, 126.26, 126.17, 61.87, 59.93, 52.54, 49.95, 48.97, 48.70, 35.17, 35.06, 31.68, 31.56; IR (KBr) 3400, 3086, 3060, 3028, 2931, 2875, 1623, 1496, 1473, 1452, 1421, 1363, 1209, 1076, 1030, 733, 700 cm−1; HRMS (DART) m/z: [M + Na]+ calcd for C18H21NNaO2 306.1470; found: 306.1463.:1 to 2:3) as a colorless liquid. 1H NMR (400 MHz, CDCl3): δ 7.38–7.30 (m, 3H), 7.26–7.15 (m, 3H), 7.05 (d, J = 7.3 Hz, 2H), 7.02 (d, J = 7.3 Hz, 2H), 3.86 (t, J = 5.3 Hz, 2H), 3.74 (d, J = 4.6 Hz, 2H), 3.38 (br s, 1H), 2.90 (t, J = 7.8 Hz, 2H), 2.36 (t, J = 7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 174.37, 142.66, 140.99, 129.92, 128.52, 128.46, 128.26, 128.06, 126.21, 61.45, 52.92, 36.29, 31.78; IR (KBr) 3400, 3060, 3028, 2931, 2873, 1635, 1595, 1495, 1454, 1408, 1273, 1192, 1074, 1059, 1028, 858, 769, 752, 700, 552, 513 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C17H19NNaO2 292.1314; found: 292.1288.
:1) to afford 2A (4.00 g, 55% yield) as a white solid. Mp 169–171 °C; 1H NMR (400 MHz, CDCl3): δ 4.14 (s, 3H), 2.94 (s, 4H); 13C NMR (100 MHz, CDCl3): δ 173.63, 173.27, 172.71, 163.20, 56.96, 28.92; IR (KBr): 2956, 1736, 1560, 1415, 1369, 1288, 1163, 991, 918, 812 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C8H8ClN4O3 243.0285; found: 243.0301; anal. calcd for C8H7ClN4O3: C, 39.60; H, 2.91; N, 23.09. Found: C, 39.43; H, 2.95; N, 22.89.:1) to afford 2B (1.68 g, 58% yield) as a white solid. Mp 163–165 °C; 1H NMR (400 MHz, CDCl3): δ 8.02 (dd, J = 5.5, 3.0 Hz, 2H), 7.87 (dd, J = 5.5, 3.0 Hz, 2H), 4.17 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 173.19, 172.40, 163.94, 162.76, 135.46, 131.29, 124.67, 56.74; IR (KBr): 3095, 3064, 2958, 1797, 1739, 1558, 1419, 1353, 1271, 1082, 935 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C12H8ClN4O3 291.0285; found: 291.0282; anal. calcd for C12H7ClN4O3: C, 49.59; H, 2.43; N, 19.28. Found: C, 49.41; H, 2.50; N, 19.23.:1) to afford 2D (135 mg, 26% yield) as a white solid. Mp 125–127 °C; 1H NMR (400 MHz, CDCl3): δ 4.14 (s, 3H), 2.79 (t, J = 6.4 Hz, 4H), 2.14 (quin, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 173.85, 173.13, 171.19, 167.12, 57.00, 32.35, 17.11; IR (KBr): 3016, 2970, 1743, 1697, 1560, 1539, 1498, 1408, 1358, 1282, 1248, 1144, 1016, 920, 760, 561, 442 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C9H10ClN4O3 257.0441; found: 257.0444; anal. calcd for C9H9ClN4O3: C, 42.12; H, 3.53; N, 21.83. Found: C, 42.05; H, 3.47; N, 21.83.:2) as a white solid. Mp 136–137 °C; 1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 8.8 Hz, 2H), 7.52 (d, J = 8.8 Hz, 2H), 7.52 (br, 1H), 7.30–7.20 (m, 5H), 4.34 (q, J = 7.2 Hz, 2H), 3.04 (t, J = 7.6 Hz, 2H), 2.69 (t, J = 7.6 Hz, 2H), 1.37 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 170.85, 166.32, 142.03, 140.49, 130.85, 128.81, 128.47, 126.60, 125.97, 118.93, 61.03, 39.60, 31.50, 14.45; IR (KBr): 3317, 3060, 3030, 2983, 2904, 1709, 1670, 1608, 1593, 1523, 1454, 1410, 1363, 1315, 1277 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C18H20NO3 298.1443; found: 298.1464; anal. calcd for C18H19NO3: C, 72.71; H, 6.44; N, 4.71. Found: C, 72.41; H, 6.34; N, 4.70.:1 to CHCl3, CHCl3/AcOEt = 7:3) as a white solid. Mp 155–156 °C; 1H NMR (400 MHz, CDCl3): δ 9.14 (t, J = 2.0 Hz, 1H), 8.86 (d, J = 2.0 Hz, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.29–7.24 (m, 3H), 6.43 (br, 1H), 3.80 (td, J = 6.9, 6.9 Hz, 2H), 3.00 (t, J = 6.9 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 162.91, 148.76, 138.24, 138.16, 129.09, 128.89, 127.22, 127.16, 121.19, 41.83, 35.50; IR (KBr): 3415, 3097, 2945, 1672, 1630, 1603, 1541, 1527, 1487, 1452, 1435, 1346, 1327, 1306, 1282 cm−1; HRMS (DART) m/z: [M + H]+ calcd for C15H14N3O5 316.0934; found: 316.0936; anal. calcd for C15H13N3O5: C, 57.14; H, 4.16; N, 13.33. Found: C, 56.92; H, 4.15; N, 13.31.:1 to 95:5) as a white solid. 1H NMR (400 MHz, CDCl3/CD3OD = 1:1): δ 7.98 (d, J = 8.7 Hz, 2H), 7.68 (d, J = 8.7 Hz, 2H), 4.36 (q, J = 7.2 Hz, 2H), 3.59 (t, J = 6.6 Hz, 2H), 2.42 (t, J = 7.6 Hz, 2H), 1.75 (quin, J = 7.6 Hz, 2H), 1.60 (quin, J = 7.1 Hz, 2H), 1.45 (quin, J = 7.1 Hz, 2H), 1.40 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3/CD3OD = 1:1): δ 173.20, 166.53, 142.76, 130.16, 124.79, 118.62, 61.40, 60.63, 36.70, 31.70, 25.02, 24.99, 13.64; IR (KBr) 3315, 2935, 2868, 1712, 1657, 1610, 1535, 1462, 1439, 1408, 1367, 1282, 1259, 1176, 1103, 1074, 1024, 962, 858, 769, 725, 694, 501 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C15H21NNaO4 302.1368; found: 302.1350.:1) as a colorless liquid. 1H NMR (400 MHz, CDCl3): δ 7.33–7.05 (m, 10H, major and minor), 4.65 (s, 0.6H, minor), 4.49 (s, 1.4H, major), 3.71 (t, J = 5.0 Hz, 1.4H, major), 3.63 (t, J = 5.6 Hz, 0.6H, minor), 3.53 (t, J = 5.0 Hz, 1.4H, major), 3.32 (t, J = 5.6 Hz, 0.6H, minor), 3.01 (t, J = 7.8 Hz, 0.6H, minor), 2.98 (t, J = 7.8 Hz, 1.4H, major), 2.79 (t, J = 7.8 Hz, 0.6H, minor), 2.66 (t, J = 7.8 Hz, 1.4H, major); 13C NMR (100 MHz, CDCl3): δ 174.88, 173.29, 141.31, 140.95, 137.63, 136.29, 129.03, 128.65, 128.57, 128.51, 127.94, 127.77, 127.37, 126.31, 126.26, 126.17, 61.87, 59.93, 52.54, 49.95, 48.97, 48.70, 35.17, 35.06, 31.68, 31.56; IR (KBr) 3400, 3086, 3060, 3028, 2931, 2875, 1623, 1496, 1473, 1452, 1421, 1363, 1209, 1076, 1030, 733, 700 cm−1; HRMS (DART) m/z: [M + Na]+ calcd for C18H21NNaO2 306.1470; found: 306.1463.:1 to 2:3) as a colorless liquid. 1H NMR (400 MHz, CDCl3): δ 7.38–7.30 (m, 3H), 7.26–7.15 (m, 3H), 7.05 (d, J = 7.3 Hz, 2H), 7.02 (d, J = 7.3 Hz, 2H), 3.86 (t, J = 5.3 Hz, 2H), 3.74 (d, J = 4.6 Hz, 2H), 3.38 (br s, 1H), 2.90 (t, J = 7.8 Hz, 2H), 2.36 (t, J = 7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 174.37, 142.66, 140.99, 129.92, 128.52, 128.46, 128.26, 128.06, 126.21, 61.45, 52.92, 36.29, 31.78; IR (KBr) 3400, 3060, 3028, 2931, 2873, 1635, 1595, 1495, 1454, 1408, 1273, 1192, 1074, 1059, 1028, 858, 769, 752, 700, 552, 513 cm−1; HRMS (ESI) m/z: [M + Na]+ calcd for C17H19NNaO2 292.1314; found: 292.1288.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by JSPS KAKENHI Grant (No. 26293003, 17H03970, 16K08161).Notes and references
- P. D. Baily, I. D. Collier and K. M. Morgan, in Comprehensive Organic Functional Group Transformations, ed. A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Pergamon, Oxford, 1995, vol. 5, p. 264 Search PubMed.
- M. Parmentier, M. K. Wagner, K. Magra and F. Gallou, Org. Process Res. Dev., 2016, 20, 1104–1107 CrossRef.
- D.-H. Kim, H.-S. Rho, J. W. You and J. C. Lee, Tetrahedron Lett., 2002, 43, 277–279 CrossRef.
- A. R. Katritzky, S. K. Singh, C. Cai and S. Bobrov, J. Org. Chem., 2006, 71, 3364–3374 CrossRef PubMed.
- H. Maag, J. Am. Oil Chem. Soc., 1984, 61, 259–267 CrossRef.
- T. Maugard, M. Remaud-Simeon, D. Petre and P. Monsan, Tetrahedron, 1997, 53, 5185–5194 CrossRef.
- Y. Park, X. Fei, Y. Yuan, S. Lee, J. Hur, S. J. Park, J.-K. Jung and S.-Y. Seo, RSC Adv., 2017, 7, 41955–41961 RSC.
- M. Kunishima, C. Kawachi, K. Hioki, K. Terao and S. Tani, Tetrahedron, 2001, 57, 1551–1558 CrossRef.
- M. Kunishima, T. Ujigawa, Y. Nagaoka, C. Kawachi, K. Hioki and M. Shiro, Chem.–Eur. J., 2012, 18, 15856–15867 CrossRef PubMed.
- M. Kunishima, K. Yoshimura, H. Morigaki, R. Kawamata, K. Terao and S. Tani, J. Am. Chem. Soc., 2001, 123, 10760–10761 CrossRef PubMed.
- M. Kunishima, H. Imada, K. Kikuchi, K. Hioki, J. Nishida and S. Tani, Angew. Chem., Int. Ed., 2005, 44, 7254–7257 CrossRef PubMed.
- M. Kunishima, D. Kato, N. Kimura, M. Kitamura, K. Yamada and K. Hioki, Beilstein J. Org. Chem., 2016, 12, 1897–1903 CrossRef PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef.
- F. Milletti, L. Storchi, L. Goracci, S. Bendels, B. Wagner, M. Kansy and G. Cruciani, Eur. J. Med. Chem., 2010, 45, 4270–4279 CrossRef PubMed.
- C. S. Petersen, Acta Chem. Scand., 1971, 25, 379–389 CrossRef.
- R. Mason, Acta Crystallogr., 1961, 14, 720–724 CrossRef.
- Without NMM, the reaction in MeOH did not proceed smoothly..
- There is a possibility that 8 could be formed from 2, 10, or 11..
- P. J. Black, M. G. Edwards and J. M. J. Williams, Eur. J. Org. Chem., 2006, 2006, 4367–4378 CrossRef.
- C. Salomé and H. Kohn, Tetrahedron, 2009, 65, 456–460 CrossRef PubMed.
- T. Takuwa, T. Minowa, H. Fujisawa and T. Mukaiyama, Chem. Pharm. Bull., 2005, 53, 476–480 CrossRef PubMed.
- M. Kunishima, K. Yamamoto, Y. Watanabe, K. Hioki and S. Tani, Chem. Commun., 2005, 2698–2700 RSC.
- I. Shiina and Y. Kawakita, Tetrahedron, 2004, 60, 4729–4733 CrossRef.
- K. Hioki, S. Kameyama, S. Tani and M. Kunishima, Chem. Pharm. Bull., 2007, 55, 825–828 CrossRef PubMed.
- V. P. Srivastava, A. K. Yadav and L. D. S. Yadav, Synlett, 2014, 25, 665–670 CrossRef.
- C. M. Boehner, D. M. Marsden, H. F. Sore, D. S. Norton and D. R. Spring, Tetrahedron Lett., 2010, 51, 5930–5932 CrossRef.
- L. S. Baugh and J. A. Sissano, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1633–1651 CrossRef.
- K. Sasaki and D. Crich, Org. Lett., 2011, 13, 2256–2259 CrossRef PubMed.
- R. A. Rodriguez, P.-S. Pan, C.-M. Pan, S. Ravula, S. Lapera, E. K. Singh, T. J. Styers, J. D. Brown, J. Cajica, E. Parry, K. Otrubova and S. R. McAlpine, J. Org. Chem., 2007, 72, 1980–2002 CrossRef PubMed.
- T. Xu, F. Sha and H. Alper, J. Am. Chem. Soc., 2016, 138, 6629–6635 CrossRef PubMed.
- L. Wang, B. P. Lieberman, K. Ploessl and H. F. Kung, Nucl. Med. Biol., 2014, 41, 58–67 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1834905. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra03057j |
| This journal is © The Royal Society of Chemistry 2018 |