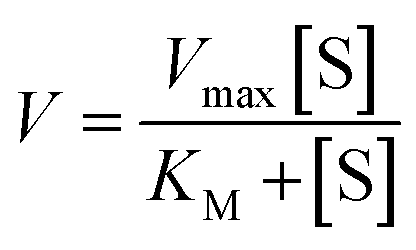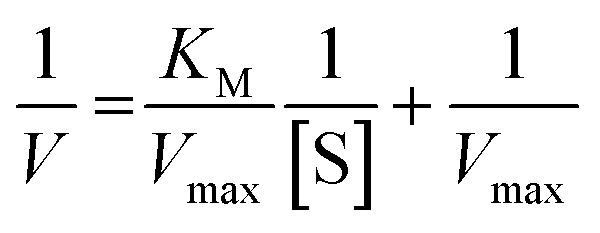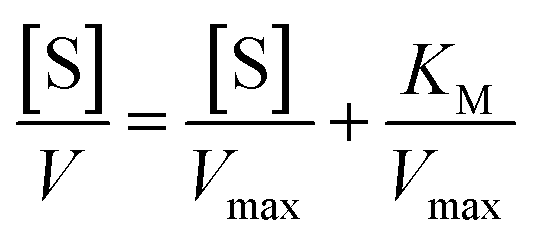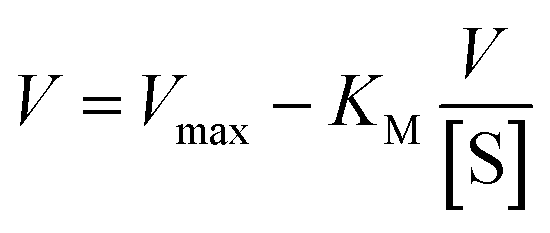DOI:
10.1039/C8RA03035A
(Paper)
RSC Adv., 2018,
8, 28216-28237
One pot synthesis of two cobalt(III) Schiff base complexes with chelating pyridyltetrazolate and exploration of their bio-relevant catalytic activities†
Received
9th April 2018
, Accepted 16th July 2018
First published on 7th August 2018
Introduction
Transition metals are known to play various significant roles in living systems.1 They are present in many metalloenzymes and metalloproteins and the roles of these enzymes and proteins are primarily dependant on these metal ions.2 If the metal ions are removed from these enzymes, the corresponding apo-enzymes are found to lose their catalytic activity, either partially or completely.3 The group of enzymes that is capable of processing molecular oxygen under ambient conditions has attracted the attention of synthetic inorganic chemists and they designed many catalysts acting as mimics of various oxidase enzymes.4 Mimics of these oxidase metalloenzymes are of particular interest for the development of bio-inspired catalysts for oxidation reactions.5 For example, atmospheric dioxygen is used by catechol oxidase, a type-3 di-copper enzyme, for the oxidation of catechols to ortho-quinones.6 A variety of di-copper(II), di-nickel(II) and manganese(II/III) containing functional models of these metalloenzymes have been shown to mediate such catechol oxidation.7 Phenoxazinone synthase, another multicopper oxidase enzyme, is known to catalyze the oxidative condensation of two molecules of a substituted o-aminophenol to the phenoxazinone chromophore.8 Many transition metal complexes of Schiff bases have been showing to have the ability to mimic these two enzyme.9 Focusing to cobalt(III) Schiff base complexes, extensive attention into their metallo-organic and coordination chemistry arises from their synthetic accessibility, fascinating structural features and diverse range of applications in photochemistry,10 electrochromism,11 and catalysis.12 They also have significant biological applications.13 Cobalt(III) Schiff base complexes have been used as precursor for the synthesis of Co3O4 nanoparticles via thermal decomposition method.14
The chemistry of tetrazoles and their complexes, on the other hand, has received great attention now-a-days15 for their wide range of applications in coordination chemistry,16 medicinal chemistry17 and material science.18 Although several synthetic methods have been reported so far, synthesis by 1,3-dipolar cycloaddition of cyanopyridine/cyanopyrazines and metal coordinated azide is probably the best method.19 However, although few metal tetrazolate complexes containing Schiff bases as blocking ligands exist in literature,20 best to our knowledge there is no report for the synthesis of any cobalt(III) tetrazolate complexes with Schiff base coligands. Keeping this in mind, we plan to synthesize and characterize few cobalt(III) tetrazolato complexes with Schiff base co-ligands. Supramolecular extended networks generated through weak non-covalent forces (hydrogen bonding and C–H⋯π interaction) have also been examined in detail using several theoretical models at the DFT-D3 level, estimating the contribution of each interaction to the formation of the assembly. Finally, catechol oxidase and phenoxazinone synthase like activities of both cobalt(III) tetrazolato complexes have been explored.
Experimental section
Materials
All starting materials were commercially available, reagent grade, and used as purchased from Sigma-Aldrich without further purification. The reactions and all manipulations of the samples were carried out under aerobic conditions.
Caution!!! Metal complexes containing azides and organic ligands are potentially explosive. Only a small amount of the material should be prepared and they should be handled with care. The experimental work is safe with the proper precautions.
Preparations
Preparation of [Co(L1)(PTZ)(N3)] (1). A methanol solution of (20 mL) N-methyl-1,3-diaminopropane (0.11 mL, 1 mmol) and 3-methoxysalicylaldehyde (152 mg, 1 mmol) was refluxed for ca. 1 h to form a Schiff base, HL1. A methanol solution (5 mL) of 2-cyanopyridine (1 mmol, 105 mg) was then added to it, followed by the addition of a methanol solution (5 mL) of cobalt(II) nitrate hexahydrate (1 mmol, 291 mg) with constant stirring. A solution of sodium azide (1 mmol, 65 mg) in methanol-water was added to it and the stirring was continued for additional 2 h. A dark brown crystalline material was started to separate out on standing the resulting solution at room temperature. It was collected by filtration and dissolved in acetonitrile. Dark brown single crystals, suitable for X-ray diffraction started to separate after ca. 7 days.Yield: 347 mg (∼74%); based on cobalt(III). Anal. calc. for C18H21CoN10O2 (FW = 468.38): C, 46.12; H, 4.48; N, 29.89%. Found: C, 46.2; H, 4.4; N, 29.9%. FT-IR (KBr, cm−1): 3182 (υN–H), 2990–2890 (υC–H), 2022 (υN3), 1632 (υC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1472 (υtetrazolate), 1446 (υtetrazolate). UV-Vis, λmax (nm), [εmax (dm3 mol−1 cm−1)] (CH3CN), 670 (1.12 × 102), 345 (6.16 × 103), 240 (3.95 × 104). Magnetic moment: 0.095 BM (diamagnetic).
N), 1472 (υtetrazolate), 1446 (υtetrazolate). UV-Vis, λmax (nm), [εmax (dm3 mol−1 cm−1)] (CH3CN), 670 (1.12 × 102), 345 (6.16 × 103), 240 (3.95 × 104). Magnetic moment: 0.095 BM (diamagnetic).
Preparation of [Co(L2)(PTZ)(N3)] (2). Complex 2 was prepared using a similar method as that used for complex 1 except that HL2 was used instead of HL1. HL2 was synthesized by refluxing a methanol solution of N,N-dimethyl-1,3-diaminopropane (0.125 mL, 1 mmol) and 3-ethoxysalicylaldehyde (166 mg, 1 mmol) for ca. 1 h. X-ray quality dark brown single crystals started to grow from acetonitrile solution after ca. 7 days.Yield: 360 mg (∼72%); based on cobalt(III). Anal. calc. for C20H25CoN10O2 (FW = 496.43): C, 48.35; H, 5.04; N, 28.20%. Found: C, 48.4; H, 5.0; N, 28.3%. FT-IR (KBr, cm−1): 3000–2928 (υC–H), 2015 (υN3), 1616 (υCN), 1468 (υtetrazolate), 1442 (υtetrazolate). UV-Vis, λmax (nm), [εmax (dm3 mol−1 cm−1)] (CH3CN), 710 (3.02 × 102), 352 (9.85 × 103), 240 (5.20 × 104). Magnetic moment: 0.098 BM (diamagnetic).
Crystallographic data collection and refinement
‘Bruker SMART APEX II’ diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) was used for data collection from suitable single crystals of both complexes. Direct methods were used to solve the molecular structures and full-matrix least squares on F2 were used to refine the structures using SHELX-2014 package.21 Non-hydrogen atoms were refined with anisotropic thermal parameters. The hydrogen atoms were placed in their geometrically idealized positions and constrained to ride on their parent atoms. Multi-scan empirical absorption corrections were applied to the data using the program SADABS.22 Details of the crystallographic data and refinement details of both complexes are given in Table 1.
Table 1 Crystal data and refinement details of complexes 1 and 2
| |
1 |
2 |
| Formula |
C18H21CoN10O2 |
C20H25CoN10O2 |
| Formula weight |
468.38 |
496.43 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
| a (Å) |
7.9648 (3) |
16.1282 (4) |
| b (Å) |
7.9932 (3) |
7.7871 (2) |
| c (Å) |
31.0201 (11) |
18.6891 (5) |
| β (°) |
96.409 (2) |
104.133 (1) |
| d(calc.) [g cm−3] |
1.585 |
1.449 |
| μ [mm−1] |
0.915 |
0.794 |
| F(000) |
968 |
1032 |
| Total reflections |
25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 838 838 |
31148 |
| Unique reflections |
3491 |
3995 |
| Observed data [I > 2σ(I)] |
3158 |
3493 |
| No. of parameters |
280 |
298 |
| R (int) |
0.04 |
0.031 |
| R1, wR2 (all data) |
0.0607, 0.1219 |
0.0325, 0.0745 |
| R1, wR2 [I > 2 σ(I)] |
0.0547, 0.1194 |
0.0271, 0.0714 |
| Residual electron density (e Å−3) |
0.683, −0.919 |
0.199, −0.227 |
Details of instrumentation
Elemental analyses (carbon, hydrogen and nitrogen) were performed using a Perkin Elmer 240C elemental analyzer. IR spectra in KBr (4500–500 cm−1) were recorded with a Perkin Elmer Spectrum Two spectrophotometer. Electronic spectra in acetonitrile–methanol (2:1) mixture were recorded on a SHIMADZU UV-1700(E) Pharma Spec UV-Vis spectrophotometer. Steady-state photoluminescence spectra in acetonitrile–methanol (2:1) mixture were obtained with a Shimadzu RF-5301PC spectrofluorometer at room temperature. Time-dependent photoluminescence spectra were recorded using a Hamamatsu MCP photomultiplier (R3809) and were analyzed using IBHDAS6 software. The cyclic voltammetry experiments were performed in DMF solution containing tetrabutylammonium perchlorate (TBAP) as supporting electrolyte using glassy carbon, Ag/Ag+ (nonaqueous) and platinum wire as working, reference and auxiliary electrodes respectively at ambient temperature (300 K) with no trace of decomposition as reflected in the smooth curve, in the potential range from −2 to +2 V and were uncorrected for junction contribution. The magnetic susceptibility measurements were done with an EG & PAR vibrating sample magnetometer, model 155 at room temperature and diamagnetic corrections were made using Pascal's constants. X-ray diffraction of the powder samples were performed on a Bruker D8 instrument with Cu Kα radiation (λ = 1.54184 Å). Electrospray ionization mass spectra were recorded using Waters QTOF Micro YA263 mass analyzer. Electron paramagnetic resonance (EPR) spectra were recorded in standard quartz EPR tubes using JEOL JES-FA200 X-band spectrometer.
Hirshfeld Surface analysis
Hirshfeld surfaces23 and the fingerprint24 (2D) plots were calculated using Crystal Explorer.25 The details could be found in many previous papers.26,27
Theoretical methods
The calculations of the noncovalent interactions were carried out using the TURBOMOLE version 7.0 (ref. 28) using the BP86-D3/def2-TZVP level of theory. To evaluate the interactions in the solid state, the crystallographic coordinates were used. This procedure and level of theory was successfully used to evaluate similar interactions.29 The interaction energies were computed by calculating the difference between the energies of isolated monomers and their assembly. The interaction energies were corrected for the Basis Set Superposition Error (BSSE) using the counterpoise method.30 The molecular electrostatic potential surfaces were computed using the SPARTAN software.31 The Bader's “atoms-in-molecules” analysis was performed at the same level of theory, using the AIMAll program.32
Kinetics of the catalytic oxidations of 3,5-DTBC and o-aminophenol
With the aim of study the catechol oxidase and phenoxazinone synthase mimicking activity of cobalt(III) tetrazolato complexes, 10−4 M solution of each complex in acetonitrile–methanol (2:1) mixture was treated with at least ten equivalents of substrate solution separately (3,5-DTBC for catechol oxidase mimicking activity and o-aminophenol for phenoxazinone synthase mimicking activity) under aerobic conditions at room temperature. All catalytic reactions were monitored spectrophotometrically for ∼2 h in the wavelength range 300–500 nm. In the catalytic oxidations of 3,5-DTBC and o-aminophenol the increase in the absorbance as a function of time around 401 and 433 nm respectively, confirms the formation of corresponding chromophores. In addition, to check the rate dependency on catalyst concentration, similar sets of experiments were performed at a fixed concentration of substrate with variable amounts of catalyst concentrations. Rate of the reactions was derived from the initial rate method, and the average initial rate over three independent measurements was recorded.
Results and discussion
Synthesis
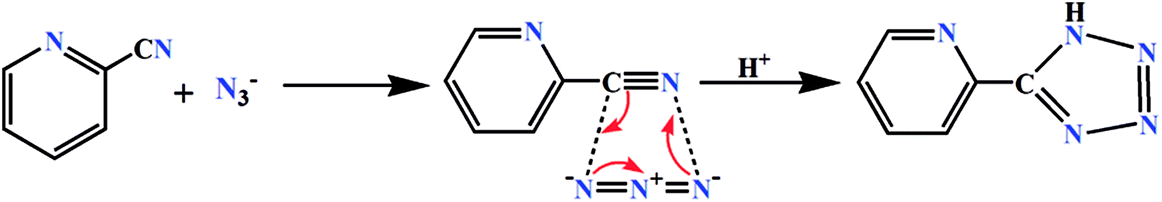
In current investigation, two Schiff base ligands HL1 and HL2 were used as blocking ligands to prepare two mononuclear cobalt(III) tetrazolato complexes. HL1 was synthesized by the 1:1 condensation of N-methyl-1,3-diaminopropane with the 3-methoxysalicylaldehyde whereas, HL2 was synthesized by similar 1:1 condensation of N,N-dimethyl-1,3-propanediamine with 3-ethoxysalicylaldehyde in methanol following the literature method.33 The ligands were not isolated and the yellow coloured methanol solutions were used directly for the synthesis of cobalt(III) complexes. On the other hand, the tetrazolate was prepared in situ by 1,3-dipolar cycloaddition of 2-cyanopyridine and azide (Scheme 1).34
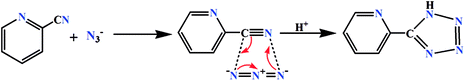 |
| | Scheme 1 In situ formation of tetrazolate via 1,3-dipolar cycloaddition reaction. | |
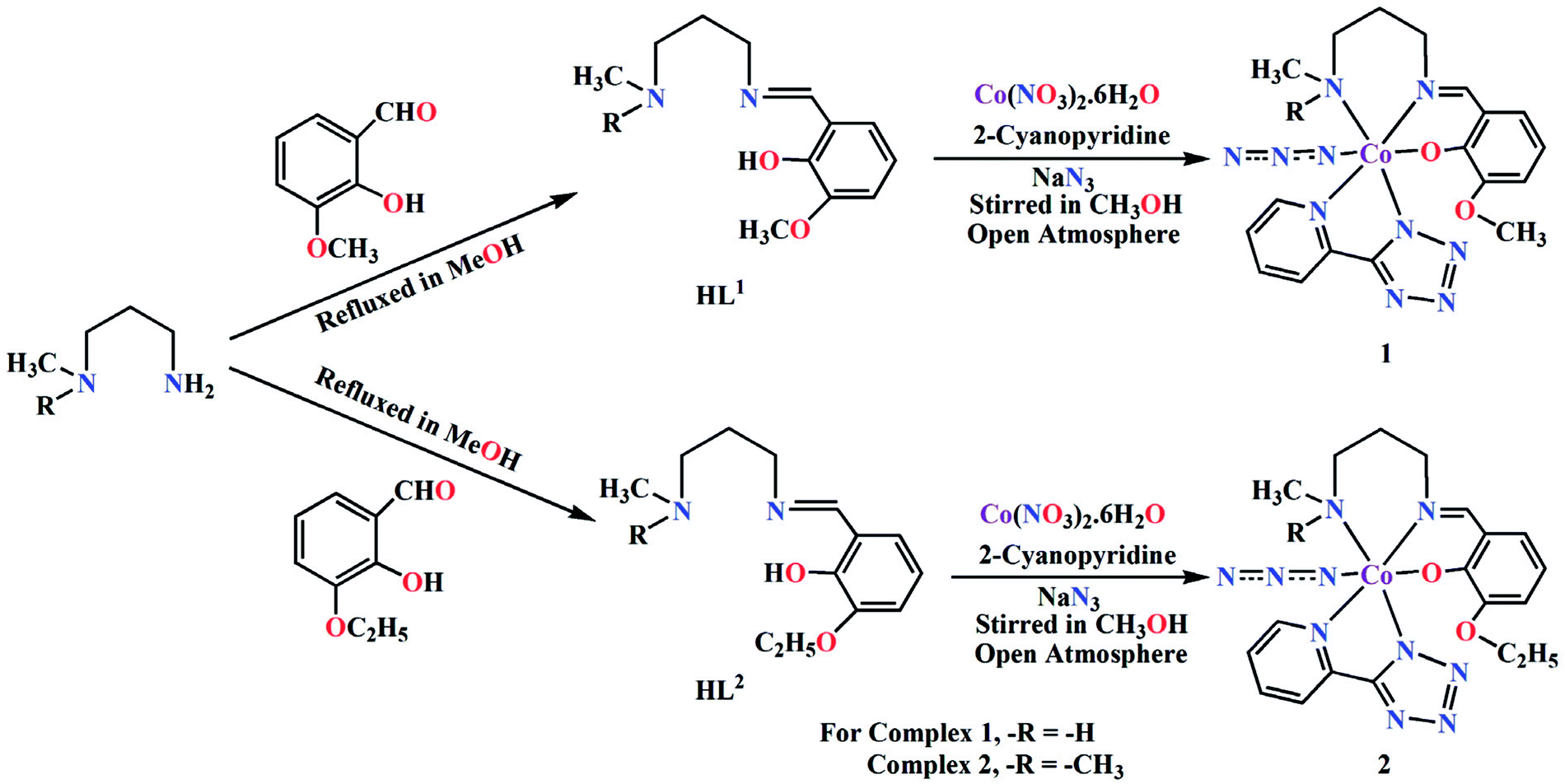
The complexes were prepared in situ by the reaction of cobalt(II) nitrate hexahydrate, 2-cyanopyridine and sodium azide with the corresponding Schiff base ligands (HL1 for 1 and HL2 for 2) under stirring condition using methanol as solvent. Cobalt(II) is readily oxidised to cobalt(III) by aerial oxygen in presence of strong field Schiff base ligands, as was also observed in similar complexes.35 Cobalt(II) is quite stable in aqueous (or methanol) medium and remains as high spin Co(L)62+ (L = H2O, CH3OH), which assumes a pale pink colour. The electronic configuration of this species is t52ge2g. On adding a strong field ligand, the spin state changes to LS with concomitant change in the electronic configuration to t62ge1g. If the strong field ligand produces sufficient strong field, 10 Dq becomes very high. This leads to severe destabilization of the eg electron, which, in turn, is responsible for the easy removal of this electron, leading to the oxidation of cobalt(II) to cobalt(III).
We have repeated the reaction under N2 atmosphere, but no complex could be isolated. The formation of both complexes is shown in Scheme 2.
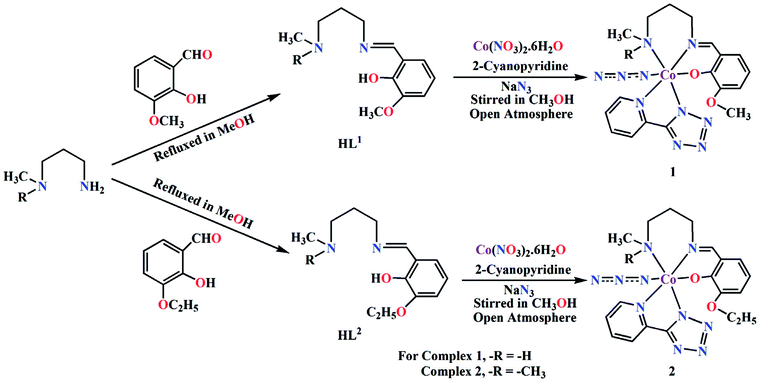 |
| | Scheme 2 Synthetic route to complexes 1 and 2. | |
Description of crystal structures
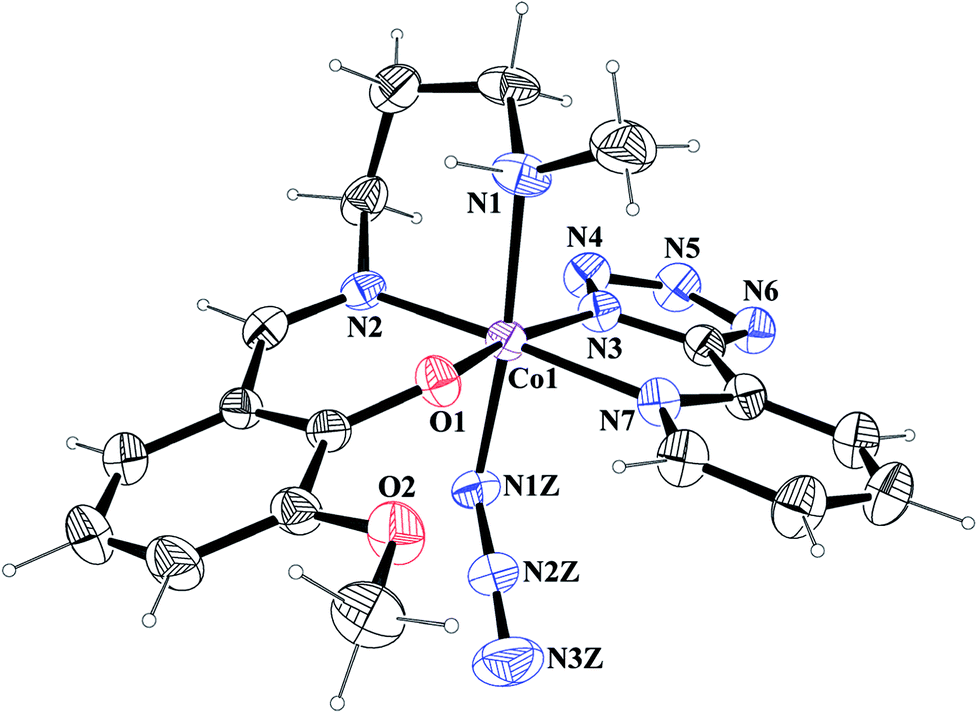
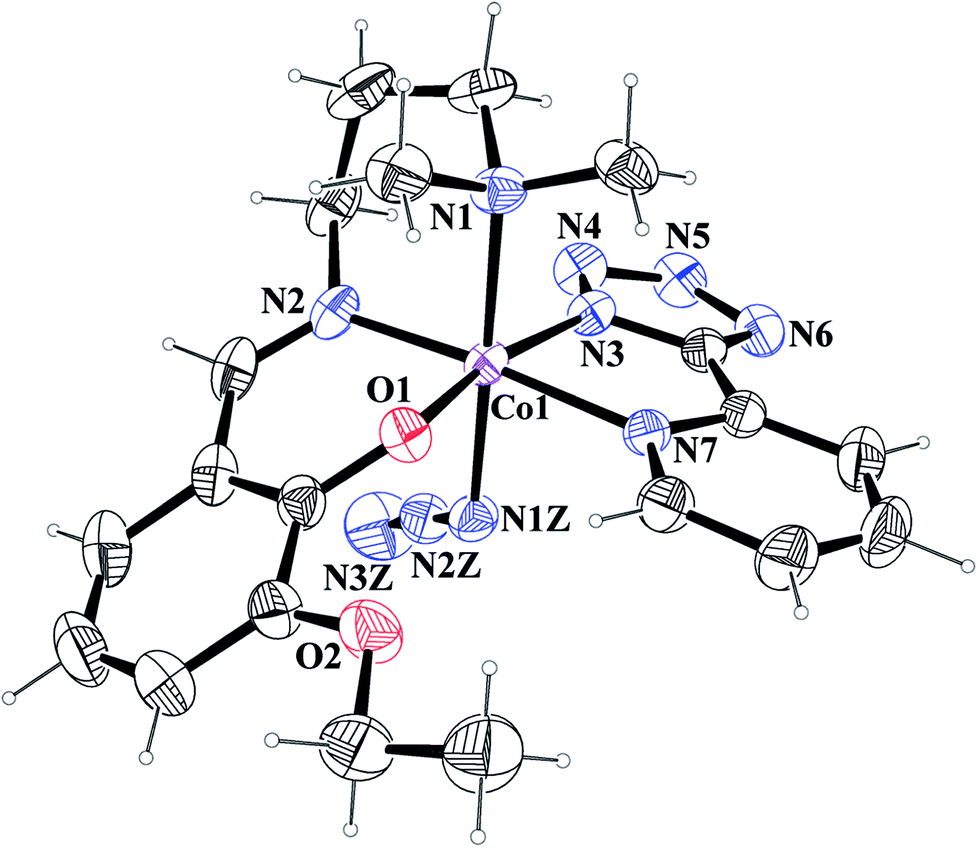
[Co(L1)(PTZ)(N3)] (1) and [Co(L2)(PTZ)(N3)] (2). Both complexes crystallize in the monoclinic space group P21/c. The structures consist of discrete mononuclear units in each of which cobalt(III) exhibits similar coordination environment. The perspective views of complexes 1 and 2 together with selected atom-numbering schemes are shown in Fig. 1 and 2 respectively. In each complex, cobalt(III) is in a distorted octahedral environment being coordinated by the phenoxo oxygen atom, O(1), the amine nitrogen atom, N(1), and the imine nitrogen atom, N(2), from the deprotonated Schiff base ligand {(L1)− for complex 1 and (L2)− for complex 2} and two additional nitrogen atoms, N(3) and N(7), from 2-pyridyltetrazolate, (PTZ)− in the equatorial plane and by the amine nitrogen atom, N(1) of the tridentate Schiff base and another nitrogen atom, N(1Z) of a terminal azide in the axial positions. It is to be noticed here that in each case, the Schiff base is occupying one facial position. Thus the important point may be the bending of the Schiff base. In the equatorial plane, pyridyltetrazolate and Schiff base moieties are not co-planar, but they form dihedral angles of 32.650 (in 1) and 43.190 (in 2). Azide moieties are differently oriented in the coordination sphere of the two complexes that lead to different packing patterns in their solid state structures. The bond lengths and bond angles of the complexes (given in Tables 2 and 3, respectively) are comparable with previously reported octahedral cobalt(III) Schiff base complexes.20c,34 The distortion of the octahedral geometry can be easily observed from the bond angles, which deviate from the ideal values of 90° (for the cis angles) or 180° (for the trans angles). The Co(III)–Nimine distances are shorter than the Co(III)–Namine ones, due to the different hybridization of nitrogen atoms. This is a common phenomenon observed in many other cobalt(III) Schiff base complexes.36 The saturated six member chelate rings, Co(1)–N(1)–C(2)–C(3)–C(4)–N(2) (for complex 1) and Co(1)–N(1)–C(3)–C(4)–C(5)–N(2) (for complex 2) are present in chair conformations with puckering parameters37a q = 0.674(3) Å, θ = 13.3(3), ϕ = 352.2(15)° (for complex 1) and q = 0.6724(18) Å, θ = 12.55(17), ϕ = 4.8(8)° (for complex 2).
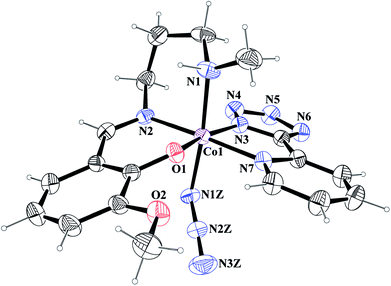 |
| | Fig. 1 ORTEP presentation of complex 1 (ellipsoids are drawn at the 40% probability level) with selected atom-numbering scheme. | |
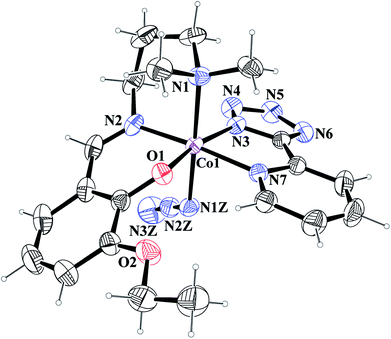 |
| | Fig. 2 ORTEP presentation of complex 2 (ellipsoids are drawn at the 40% probability level) with selected atom-numbering scheme. | |
Table 2 Selected bond lengths (Å) of complexes 1 and 2
| |
1 |
2 |
| Co(1)–O(1) |
1.871(2) |
1.8651(14) |
| Co(1)–N(1) |
2.002(3) |
2.0720(15) |
| Co(1)–N(1Z) |
1.952(3) |
1.9521(16) |
| Co(1)–N(2) |
1.911(3) |
1.9076(17) |
| Co(1)–N(3) |
1.955(3) |
1.9419(15) |
| Co(1)–N(7) |
1.981(3) |
1.9911(15) |
Table 3 Selected bond angles (°) of complexes 1 and 2
| |
1 |
2 |
| O(1)–Co(1)–N(1) |
86.56(12) |
90.60(6) |
| O(1)–Co(1)–N(1Z) |
93.19(12) |
89.33(6) |
| O(1)–Co(1)–N(2) |
92.00(12) |
91.15(6) |
| O(1)–Co(1)–N(3) |
168.01(12) |
168.59(6) |
| O(1)–Co(1)–N(7) |
87.57(12) |
87.67(6) |
| N(1)–Co(1)–N(1Z) |
173.85(14) |
179.27(7) |
| N(1)–Co(1)–N(2) |
85.18(14) |
88.19(7) |
| N(1)–Co(1)–N(3) |
93.94(13) |
92.72(6) |
| N(1)–Co(1)–N(7) |
97.44(13) |
96.19(6) |
| N(1Z)–Co(1)–N(2) |
88.69(13) |
91.08(7) |
| N(1Z)–Co(1)–N(3) |
87.58(13) |
87.48(6) |
| N(1Z)–Co(1)–N(7) |
88.69(13) |
84.53(6) |
| N(2)–Co(1)–N(3) |
99.98(13) |
99.85(7) |
| N(2)–Co(1)–N(7) |
177.31(14) |
175.47(7) |
| N(3)–Co(1)–N(7) |
80.48(13) |
81.12(6) |
Theoretical study of noncovalent interactions
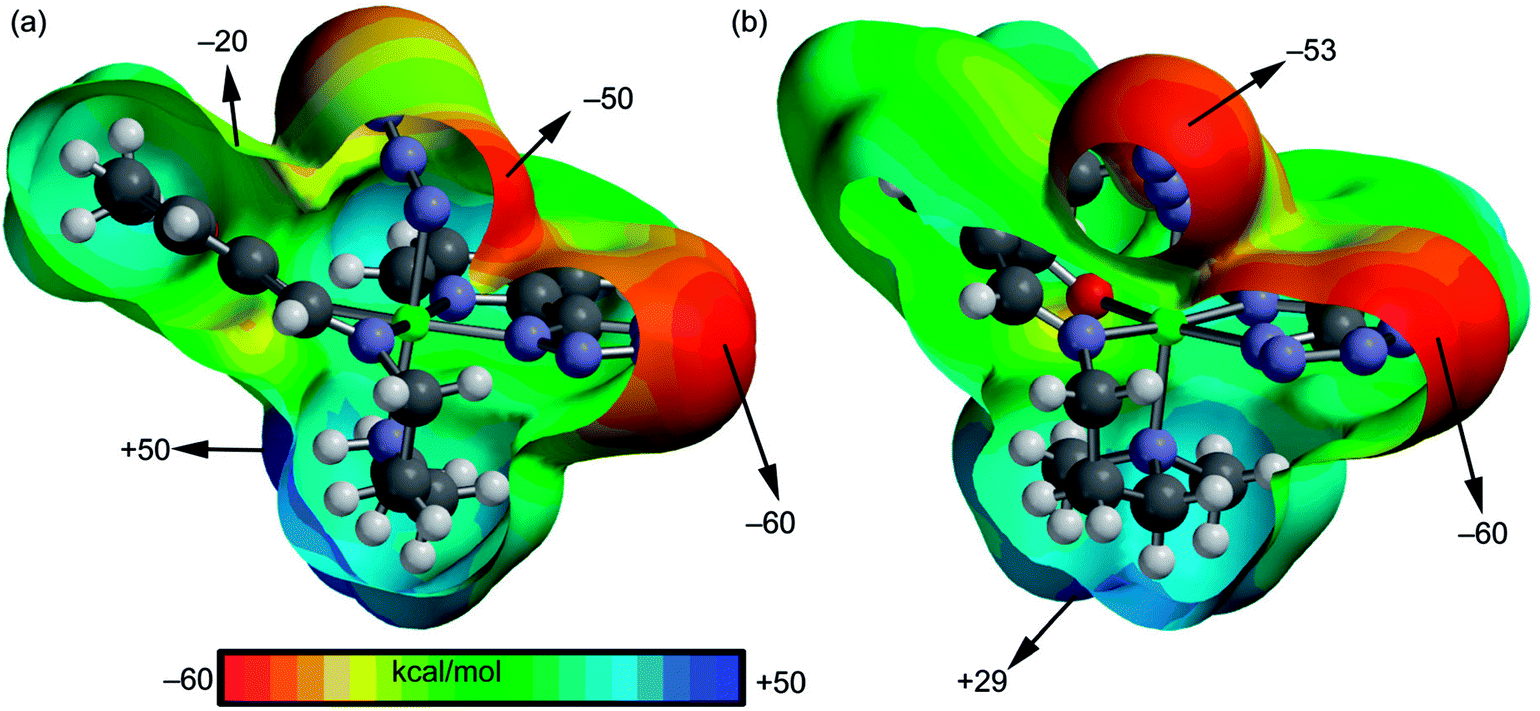
We have focused our attention to the study of relevant hydrogen bonding and C–H⋯π interactions observed in the solid state architecture of both complexes. At first, the MEP surfaces of both complexes were computed to investigate the most positive and negative regions in order to rationalize the interactions observed in their crystal packing. The MEP surfaces are given in Fig. 3 and it can be observed that the most negative region is located at the nitrogen atoms of the tetrazole ring in both complexes (−60 kcal mol−1) and the most positive one at the NH group in complex 1 (+50 kcal mol−1) and at the hydrogen atoms of the coordinated N(CH3)2 group in 2 (+29 kcal mol−1). The nitrogen atoms of the azide are also present in strongly negative MEP values. Therefore interactions involving the NH/N(CH3)2 groups as hydrogen bond donors and tetrazole/azide as acceptors are electrostatically favored.
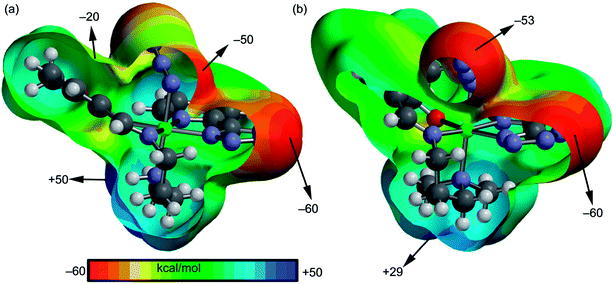 |
| | Fig. 3 MEP surfaces of complexes 1 (a) and 2 (b) with indication of the energetic values in selected point of the surface (isosurface at 0.002 a.u.). | |
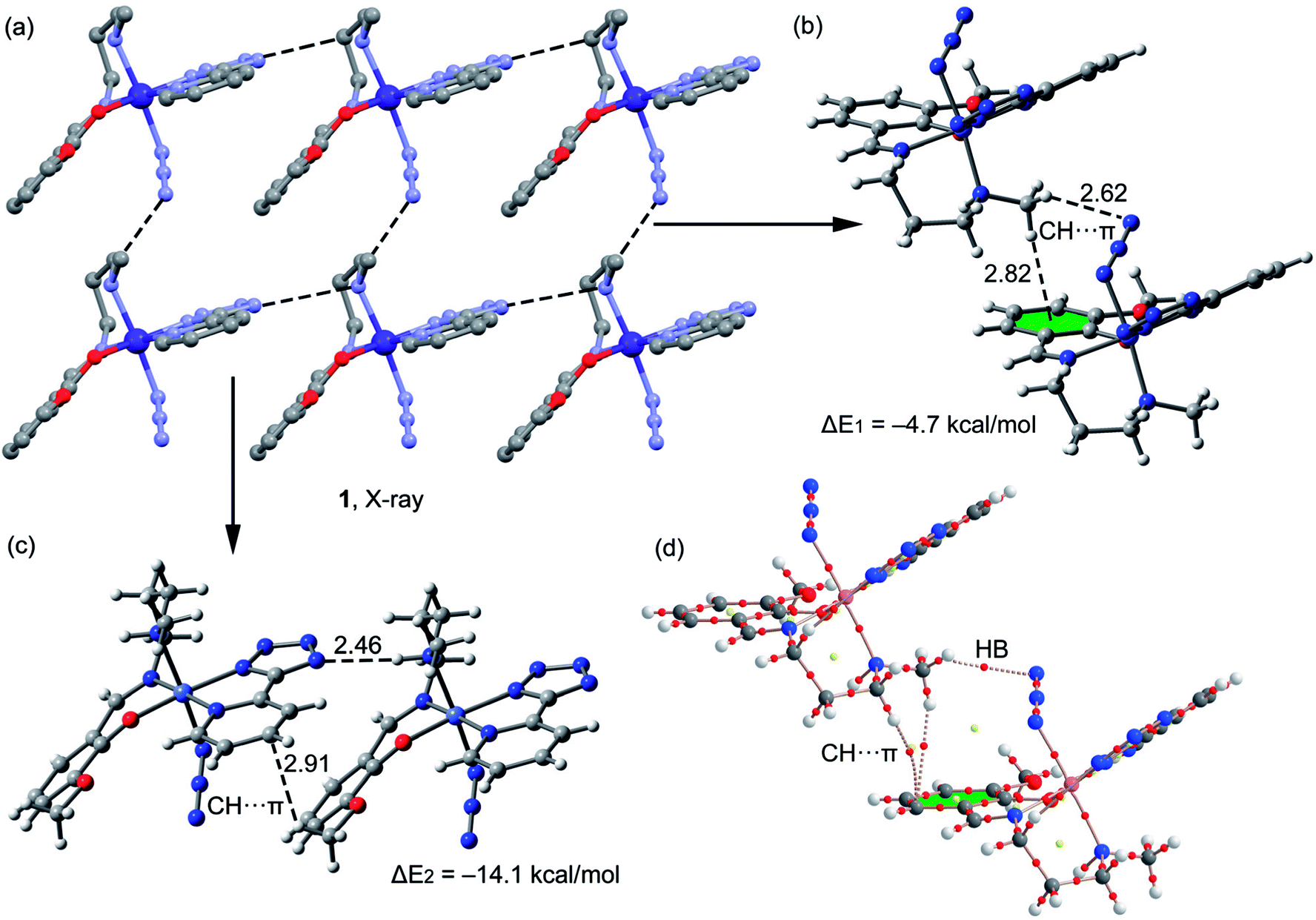
A fragment of the crystal packing of complex 1 is shown in Fig. 4a, where the formation of 2D layer is highlighted. In one direction the chain is governed by hydrogen bonding interactions (Fig. 4c) and in the perpendicular direction the molecules interact by means of C–H⋯π interactions (Fig. 4b). The interaction energies of both assemblies were evaluated using the models shown in Fig. 4c. It can be easily observed that the hydrogen bonding dimer is energetically more favorable (ΔE2 = −14.1 kcal mol−1) than the dimer (ΔE1 = −4.7 kcal mol−1) governed by CH⋯π interactions. In the latter, two different π-systems are involved, the conventional aromatic ring (at 2.82 Å to the ring centroid) and the π-system of the azide, since the directionality of the interaction (hydrogen bond) indicates that the hydrogen atom is pointing to the π instead of the σ-electrons of the pseudohalide. The hydrogen bonded dimer presents stronger interaction energy in agreement to the MEP surface, since the interaction of N–H and tetrazole is very favored electrostatically. The C–H⋯π interaction described in Fig. 4c (2.91 Å to one C atom of the ring) is similar to that previously highlighted by N. Nishio in the crystal structure of levopimaric acid (2.94 Å to one carbon atom)37b and also in phenyl-substituted tetrahydroisoquinoline derivatives (2.86 Å).37c Similar C–H⋯π interactions have also been commonly described in supramolecular assemblies of several complexes, reported in previous journals.37d,e The Bader's theory of atoms-in-molecules was used to further confirm the existence of the CH⋯π interactions. The existence of a bond critical point (CP) and bond path connecting two atoms is an unambiguous evidence of interaction. The distributions of critical points and bond paths for the dimer of complex 1 are shown in Fig. 4d. It can be observed that the conventional C–H⋯π(phe) interaction is characterized by the presence of two bond CPs (red spheres) and bond paths connecting two hydrogen atoms to the phenyl ring. Moreover, the unconventional C–H⋯π(azide) interaction is characterized by the presence of a bond CP and bond path connecting the hydrogen atom to a nitrogen atom of the ligand, thus confirming the existence of the interaction.
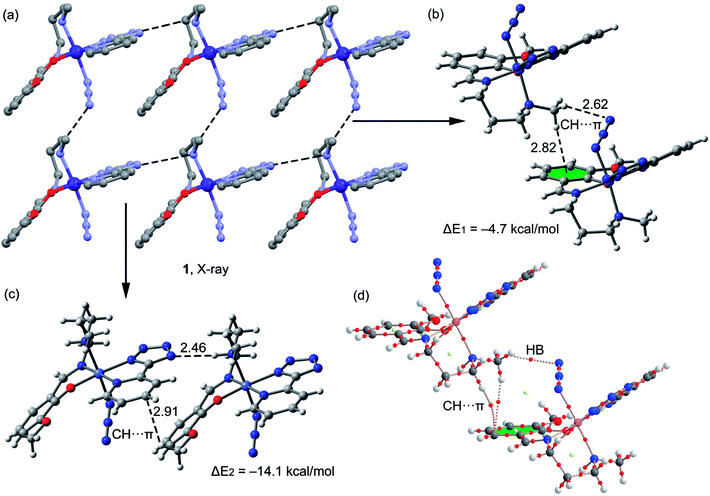 |
| | Fig. 4 (a) Crystal packing of 1 (hydrogen atoms omitted); (b and c) theoretical models used to evaluate the noncovalent interactions (distances in Å); (d) distribution of bond and ring (red and yellow spheres) critical points in one dimer of complex 1. | |
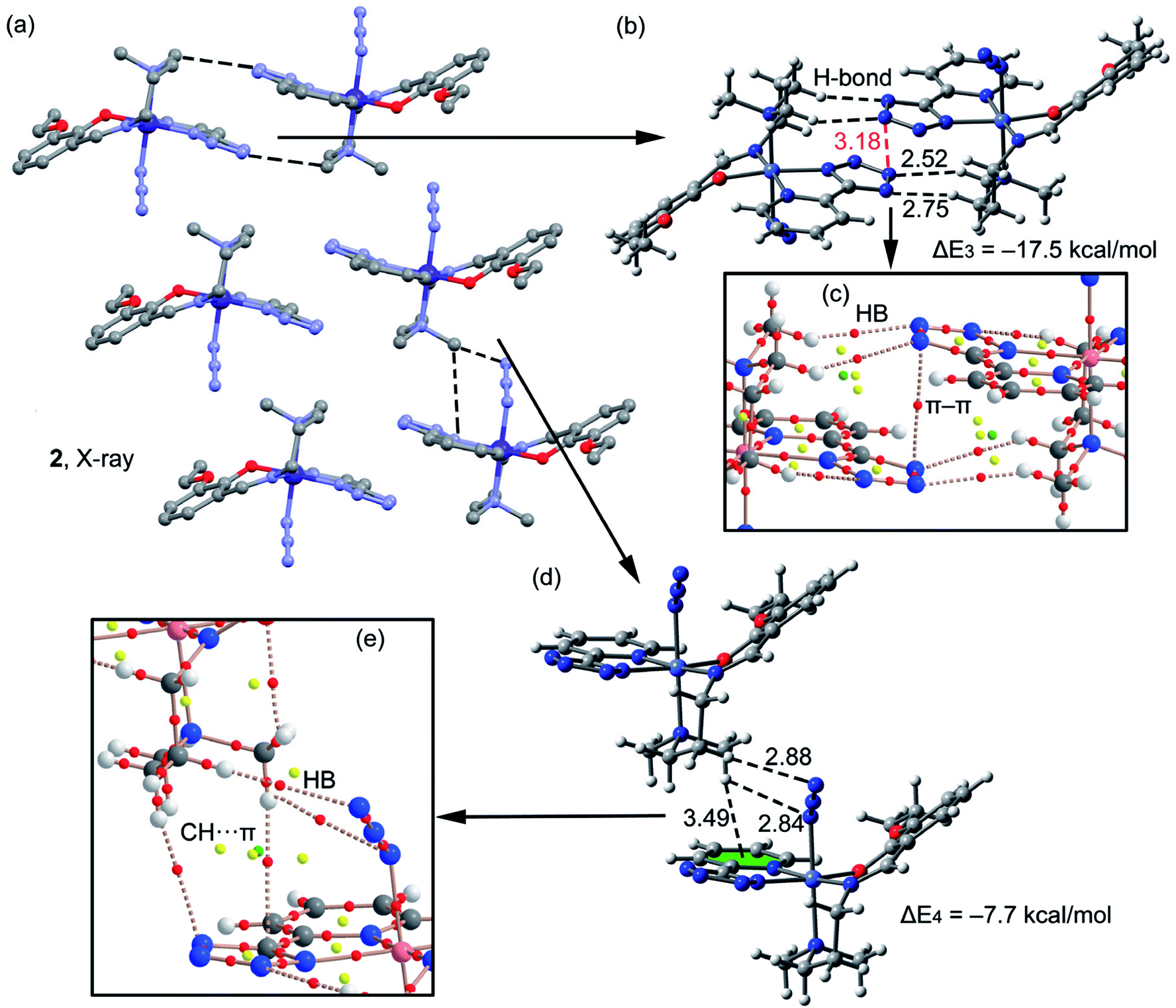
A fragment of the crystal packing of complex 2 is shown in Fig. 5a, where the formation of 2D layers is highlighted. Complex 2 forms self-assembled dimers governed by hydrogen bonding and π–π stacking interactions (see black and red dashed lines, respectively, Fig. 5b). This self-assembled dimer interacts with the adjacent one by means of C–H⋯π interactions (see Fig. 5d) involving the π-systems of phenyl and azide moieties, as previously observed in complex 1. The interaction energies of both assemblies were evaluated using the models shown in Fig. 5b and d. It can be observed that the self-assembled hydrogen bonding dimer is energetically very favorable (ΔE3 = −17.5 kcal mol−1) due to the formation of several hydrogen bonds. The interaction energy of the other dimer governed by C–H⋯π interactions is more modest (ΔE4 = −7.7 kcal mol−1). In complex 2 the conventional aromatic ring involved in the C–H⋯π(phe) interaction belongs to the PTZ ligand instead to the Schiff-base ligand. In addition, two H-atoms of the N(CH3)2 group interact with π-system of the azide (2.8 Å). The distributions of critical points and bond paths for the dimers of 2 are shown in Fig. 5c and e. In the self-assembled dimer, it can be observed that each hydrogen bonding interaction is characterized by the presence of one bond CPs (red spheres) and bond path that connects the hydrogen atom to the nitrogen atom of the tetrazole ring (Fig. 5c). The interaction is further characterized by the presence of a ring CP (yellow sphere) due to the formation of a supramolecular ring. Moreover, the π⋯π interaction is characterized by the presence of a bond CP and bond path interconnecting two nitrogen atoms of the tetrazole rings, thus confirming the existence of the interaction. Finally, in Fig. 5e it is shown the distribution of CPs for the C–H⋯π assembly. It can be observed that the conventional C–H⋯π(phe) interaction is characterized by the presence of two bond CPs (red spheres) and bond paths connecting two hydrogen atoms of the methyl groups to the phenyl ring and tetrazole rings. Moreover, the unconventional C–H⋯π(azide) interaction is characterized by the presence of two bond CPs and bond paths connecting two hydrogen atoms of the N(CH3)2 group to two N-atoms of the pseudohalide.
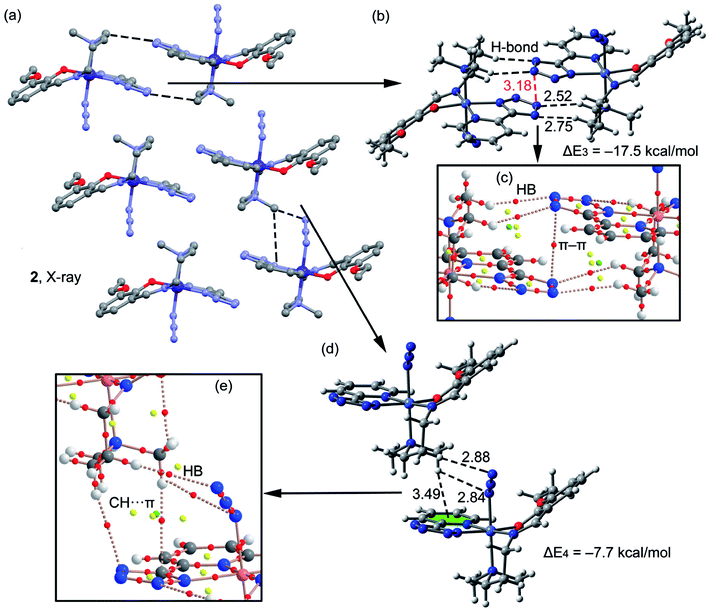 |
| | Fig. 5 (a) Crystal packing of 2 (hydrogen atoms omitted); (b and c) theoretical models used to evaluate the noncovalent interactions (distances in Å); (d and e) distribution (partial views) of bond, ring and cage (red, yellow and green spheres) critical points in two dimers of complex 2. | |
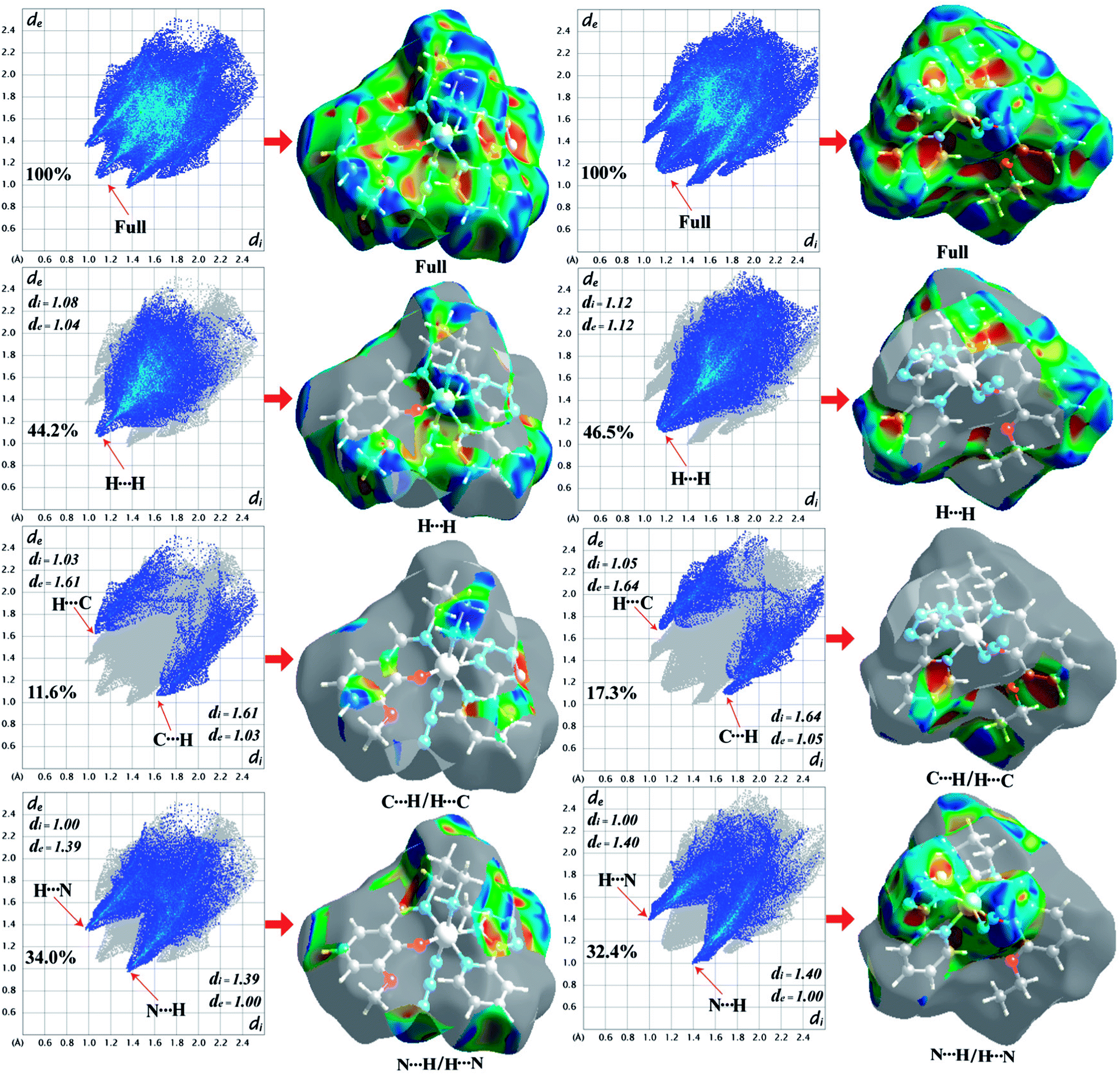
Hirshfeld surface analysis
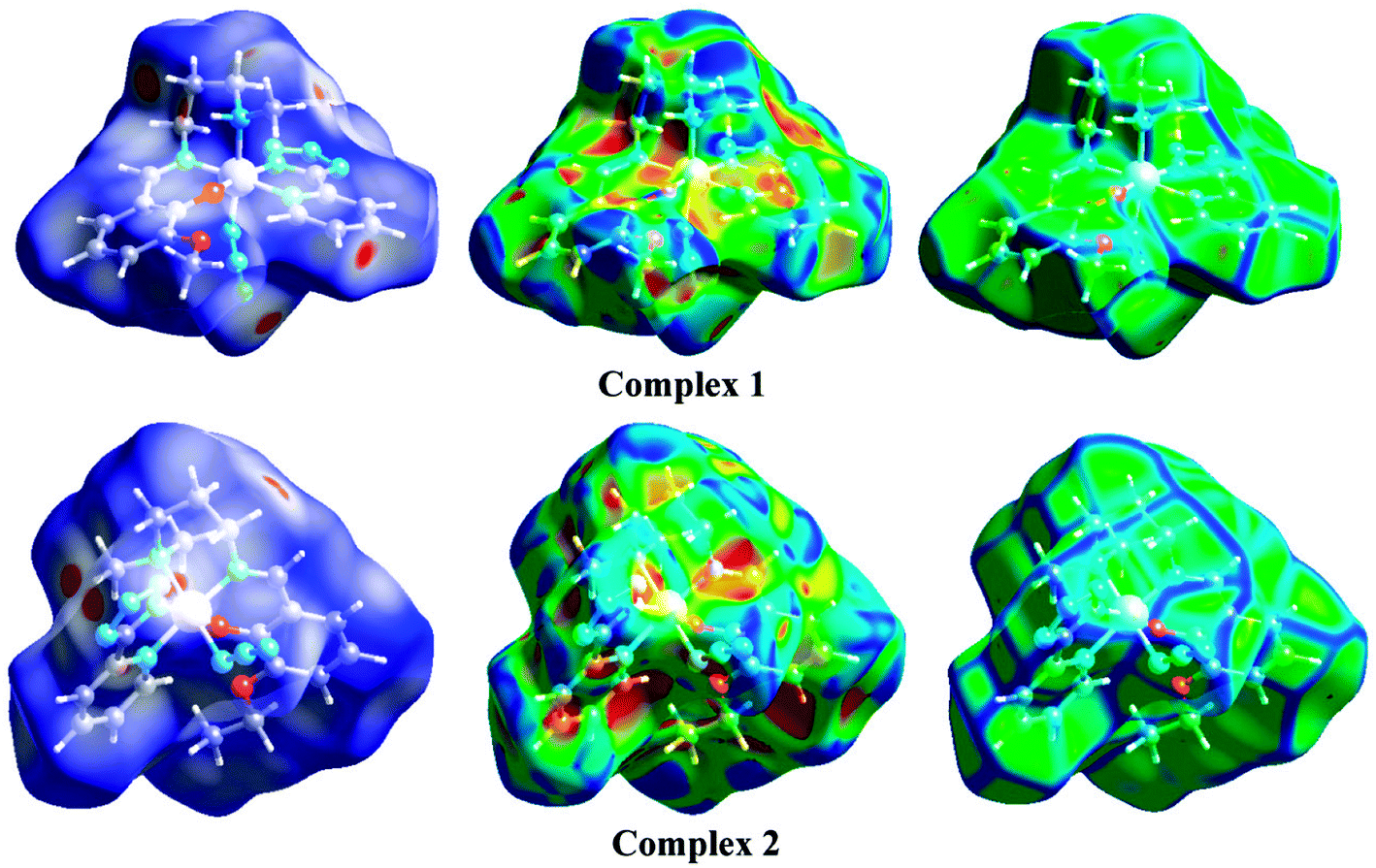
The Hirshfeld surfaces of the two complexes, mapped over dnorm (range of −0.1 to 1.5 Å), shape index and curvedness, are illustrated in Fig. 6. The dominant interaction between N⋯H/H⋯N atoms can be seen in the Hirshfeld surfaces as red spots on the dnorm surface in Fig. 6. Other visible spots in the Hirshfeld surfaces correspond to mainly C⋯H/H⋯C and H⋯H contacts. Additionally, the 2D fingerprint plots (Fig. 7) illustrate the difference between the intermolecular interaction patterns and the relative contributions (in percentage) for the major intermolecular interactions associated with the complex. In the 2D fingerprint plot (Fig. 7) intermolecular interactions appear as distinct spikes. The fingerprint plots can also be decomposed to emphasize selected atoms pair close contacts. This decomposition enables separation of contributions from different interaction types, which overlap in the full fingerprint. Relative contributions to the Hirshfeld surface area for the various intermolecular contacts of both complexes are shown in Fig. S1 (ESI†).
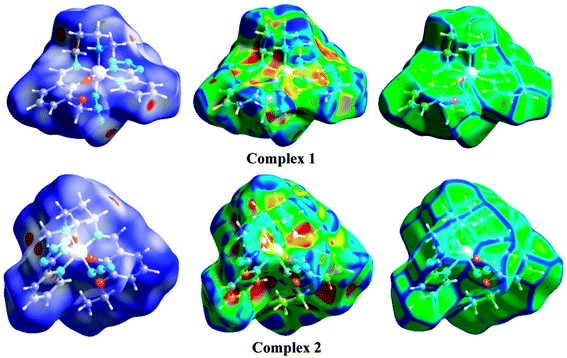 |
| | Fig. 6 Hirshfeld surfaces mapped with dnorm (left-side), shape index (middle) and curvedness (right-side). | |
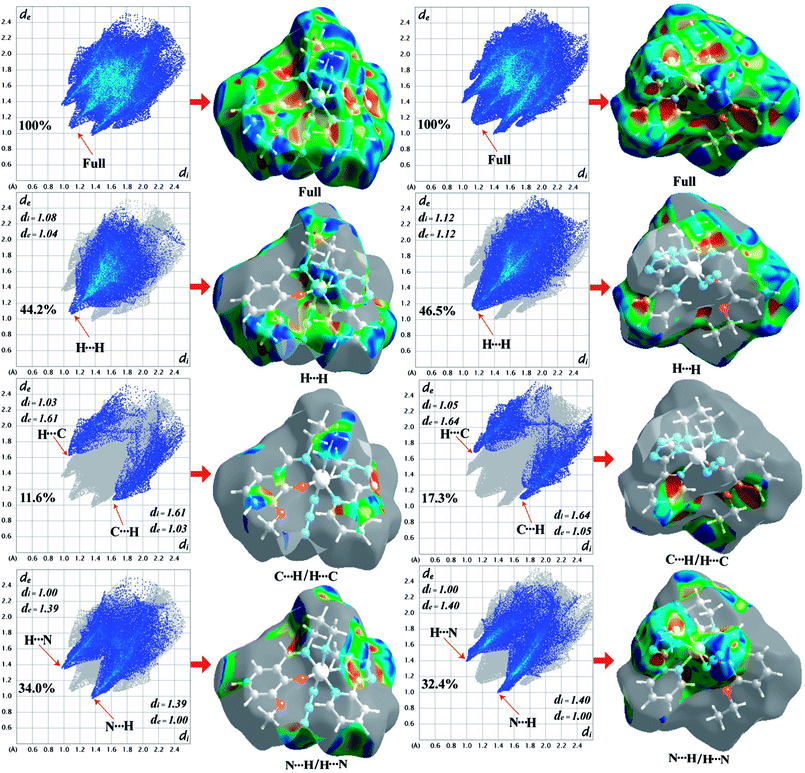 |
| | Fig. 7 Fingerprint plots of 1 (left-side) and 2 (right-side): full and resolved into H⋯H, C⋯H/H⋯C, N⋯H/H⋯N contacts showing the percentages of contacts contributed to the total Hirshfeld surface area of both complexes. Surfaces in the right hand columns highlight the relevant surface patches associated with the specific contacts in the total Hirshfeld surface area of complexes 1 and 2. | |
X-ray diffraction of powdered sample
The X-ray diffraction patterns of the powdered samples are in excellent agreement with those simulated from single crystal X-ray diffraction, confirming the purity of the bulk samples. The simulated patterns of the complexes were calculated from the single crystal structural data using the CCDC Mercury software. Experimental and simulated powder XRD patterns of complexes 1 and 2 are given in Fig. S2 and S3 respectively (ESI†).
Spectral and magnetic properties
The IR spectrum of complex 1 exhibits a moderately strong band at 3182 cm−1, which may be ascribed to the N–H stretching vibration. No such band is observed in case of complex 2, because of the absence of N–H bond in it. The bands in the range of 3000–2900 cm−1 due to alkyl C–H stretching vibrations are routinely noticed in IR spectra of both complexes.38 Sharp bands at 2021 and 2015 cm−1 indicate the presence of monodentate azide coligand in the IR spectra of complexes 1 and 2, respectively.39 The distinct bands at ∼1625 cm−1 due to the azomethine (CN) groups are noticed in the IR spectra of both complexes.40 The bands at 1446, 1472 cm−1 (for 1) and 1444, 1469 cm−1 (for 2) are due to the tetrazolate moiety and also observed in other tetrazolate-based metal complexes in previous reports.41 IR spectra of complexes 1 and 2 are given in Fig. S4 and S5 respectively (ESI†).
The electronic spectra (200–900 nm) of the complexes are similar to each other in acetonitrile–methanol (2:1) mixture at room temperature. Usually low-spin octahedral cobalt(III) complexes show two broad absorption bands at low energy region due to spin allowed 1A1g → 1T1g and 1A1g → 1T2g transitions.42 In the present case, complexes 1 and 2 show broad absorption bands at 670 nm and 710 nm, respectively, attributable to one of the two expected transitions for any low-spin cobalt(III) in octahedral geometry, as mentioned above.43 The intense absorption bands of the complexes, at around 350 nm may be assigned as charge transfer transitions from the coordinated ligands to the cobalt(III) centers (LMCT).44a High energy absorption bands around 240 nm in both complexes may be assigned to intra ligand π–π* transition.44b Electronic spectra of 1 and 2 are given in Fig. S6 and S7 respectively (ESI†).
Both complexes show strong photoluminescence upon the irradiation of ultraviolet light. Upon excitation at around 240 nm, complex 1 emits at 350 nm whereas complex 2 emits at 380 nm. The excited state mean lifetimes of complexes 1 and 2 are 7.24 ns and 7.91 ns, respectively (Table 4) Photoluminescence spectra and time dependent photoluminescence decay profile of both complexes is shown in Fig. S8–S10 (ESI†).
Table 4 The details data of the photoluminescence and time-resolved photoluminescence decays of complexes 1 and 2
| Complexes |
Absorption (nm) |
Emission (nm) |
Time (ns) |
χ2 |
| 1 |
240 |
350 |
7.24 |
1.042081 |
| 2 |
240 |
380 |
7.91 |
1.067222 |
Room temperature magnetic susceptibility measurements show that both complexes are diamagnetic as expected for low-spin cobalt(III) complexes.45a
Electrochemical studies
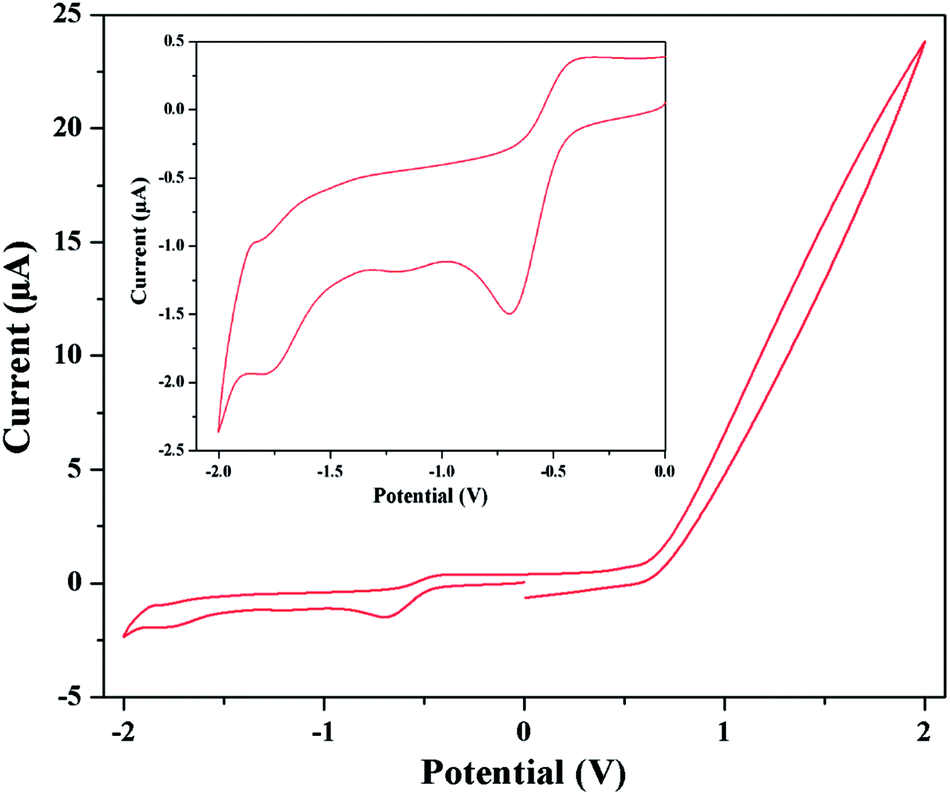
By scanning in the negative direction initiated at 0.00 V, two reduction peaks are observed. The starting reduction peaks corresponding to Co(III) → Co(II) reduction processes are observed at about potential −0.70 and −0.62 V for complexes 1 and 2 respectively. Similarly the subsequent reduction peaks of complexes 1 and 2 are observed at −1.80 and −1.81 V respectively, which indicate Co(II) → Co(I) transformation. The corresponding oxidation peaks are observed during the reverse scan process. The oxidation of Co(I) → Co(II) is suggested by the presence of the peaks at −1.84 and −1.44 V for complexes 1 and 2 respectively. Co(II) → Co(III) oxidation processes are observed at −0.38 and −0.30 V for complexes 1 and 2 respectively. The cyclic voltammogram of complex 1 is shown in Fig. 8. The quasi-reversible redox reaction may be rationalized by the expected structural changes by elongation of axial bond lengths or losing of axial ligands because of the addition of one electron to dz2 orbital and vice versa.45b Repeated scans lead to nearly superposable cyclic voltammograms, which suggest all three oxidation states of cobalt are markedly stable. The reduction peak potentials are strongly depend on donor strength of the coordinate ligands. Presence good electron withdrawing groups such as 5-(2-pyridyl)tetrazole and azide causes more electron affinity on cobalt center and so a more negative cathodic peak than cobalt Schiff base complexes.45c The results of cyclic voltammetry also closely resemble that of the similar reported complexes, which serve as further evidences for similar structural and electronic properties.45b–d The detailed cyclic voltammetry data (V) for both complexes are given in Table 5.
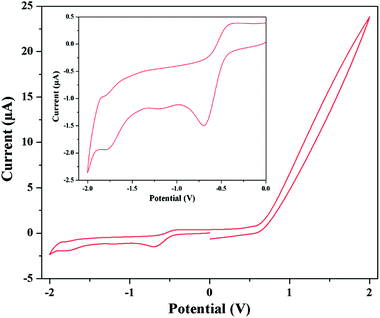 |
| | Fig. 8 Cyclic voltammogram of complex 1 in DMF medium. Inset shows the voltammogram in between −2 to 0 V. | |
Table 5 Cyclic voltammetry data (V) for both complexes in DMF medium (scan rate 25 mV s−1)
| Complexes |
EIII→II |
EII→III |
ΔEIII↔II |
E1/2 III↔II |
EII→I |
EI→II |
ΔEII↔I |
E1/2 II↔I |
| 1 |
−0.70 |
−0.38 |
0.32 |
−0.54 |
−1.80 |
−1.84 |
0.04 |
−1.82 |
| 2 |
−0.62 |
−0.30 |
0.32 |
−0.46 |
−1.81 |
−1.44 |
0.37 |
−1.63 |
Catalytic activity and kinetic study
The catalytic experiments were conducted in aerobic condition at room temperature with model substrates 3,5-di-tert-butylcatechol (3,5-DTBC) for catechol oxidase like activity and OAPH (o-aminophenol) for phenoxazinone synthase like activity. Whole catalytic activity was performed in acetonitrile–methanol (2:1) mixture because of very good solubility of the complexes, substrates and their products. No additional base was added in the reaction medium.
Catechol oxidase mimicking activity
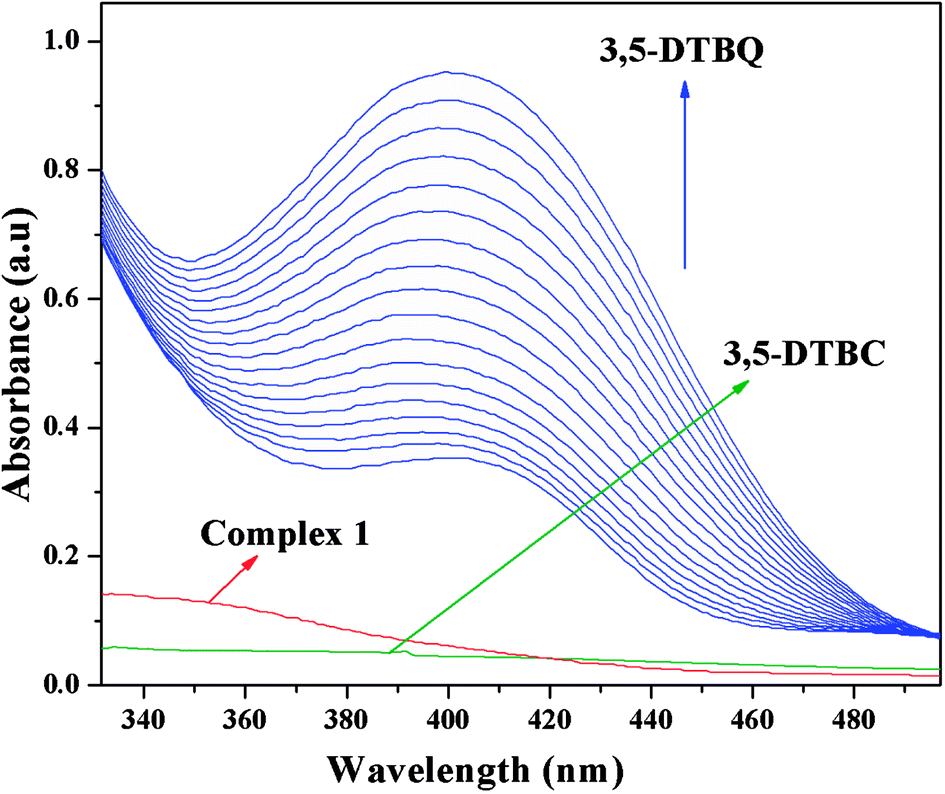
The catechol oxidase mimicking activity of both complexes was investigated with model substrate 3,5-DTBC. Among the different catechols used in catechol oxidase model studies, 3,5-DTBC is the most widely used substrate because of its low redox potential for the quinone–catechol couple, which makes it easy to be oxidized to the corresponding quinone (3,5-DTBQ), and its bulky substituents help to stop further oxidation reactions, such as ring opening etc. The product (3,5-DTBQ) is significantly stable and it exhibits a strong absorption around 400 nm. So, catechol oxidase mimicking activity and the respective reaction kinetics can be easily followed by UV-visible spectroscopy by following the appearance of the characteristic absorption of the 3,5-DTBQ. The band intensity around 400 nm is gradually increased as a function of time, suggesting cobalt(III) tetrazolato complexes are capable of exhibiting prominent bio-mimetic catalytic activity related to catechol oxidase. Time resolved UV-Vis spectral profiles indicating the increment of 3,5-DTBQ characteristic peak of complex 1 is shown in Fig. 9. It was found that in the absence of catalyst, no catalytic oxidation of the substrate occurs.
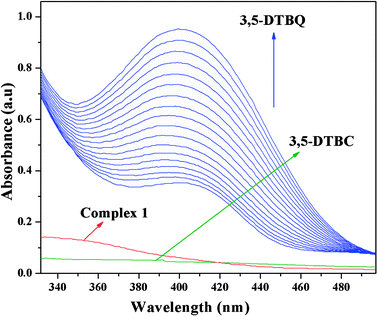 |
| | Fig. 9 Time resolved UV-Vis spectral profiles indicating the increment of 3,5-DTBQ peak at ∼400 nm upon addition of 10−2 M 3,5-DTBC to the 10−4 M of complex 1 in acetonitrile–methanol (2:1) mixture at room temperature. | |
Phenoxazinone synthase mimicking activity
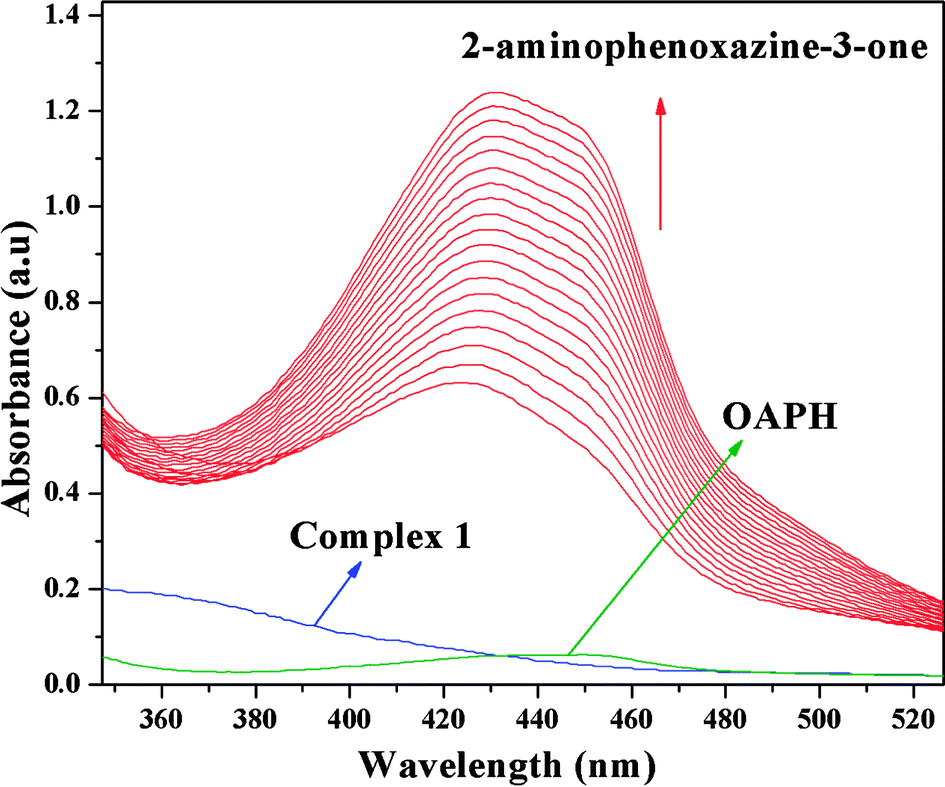
It is generally agreed phenoxazinone synthase catalyzes the penultimate step in the biosynthesis of the antibiotic actinomycin D by Streptomyces antibioticus, where oxidative condensation of o-aminophenol substituents occurs. Therefore, o-aminophenol is used as model substrate for investigating phenoxazinone synthase mimicking activity of both complexes. The oxidative condensation of o-aminophenol catalyzed by the complexes was investigated by monitoring the increase of the absorption peak around 433 nm, characteristic of the phenoxazinone chromophore. The spectral scan for both complexes reveal the progressive increase of peak intensity at ca. ∼433 nm suggesting catalytic oxidation of o-aminophenol in aerobic condition. The time dependent spectral change for the period of 2 h after the addition of o-aminophenol to the acetonitrile–methanol (2:1) mixture of complex 1 is depicted in Fig. 10. It should be noted at this point that a blank experiment without catalyst (complex) does not show significant growth of the band at ∼433 nm, characteristic of phenoxazinone chromophore, in an identical reaction condition.
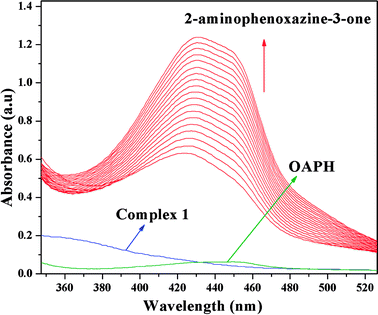 |
| | Fig. 10 Time resolved UV-Vis spectral profiles indicating the increment of o-aminophenoxazine-3-one peak at ∼433 nm upon addition of 10−2 M o-aminophenol to the 10−4 M of complex 1 in acetonitrile–methanol (2:1) mixture at room temperature. | |
Kinetic parameters and explanation of the turnover numbers
Michaelis–Menten model is an important tool to treat this type of saturation rate dependency on the concentration of the substrate. The Michaelis–Menten equation:| |
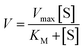 | (1) |
where, V = initial rate; [S] = concentration of the substrates; KM = Michaelis–Menten constant for the metal complex and Vmax = maximum initial rate attained for a particular concentration of the metal complex in the presence of a large excess of the substrate.
On the other hand, rearrangement of Michaelis–Menten equation produces Lineweaver–Burk equation (eqn (2)), Hanes equation (eqn (3)) and Eadie–Hofstee equation (eqn (4)).
| |
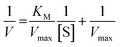 | (2) |
| |
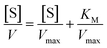 | (3) |
| |
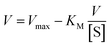 | (4) |
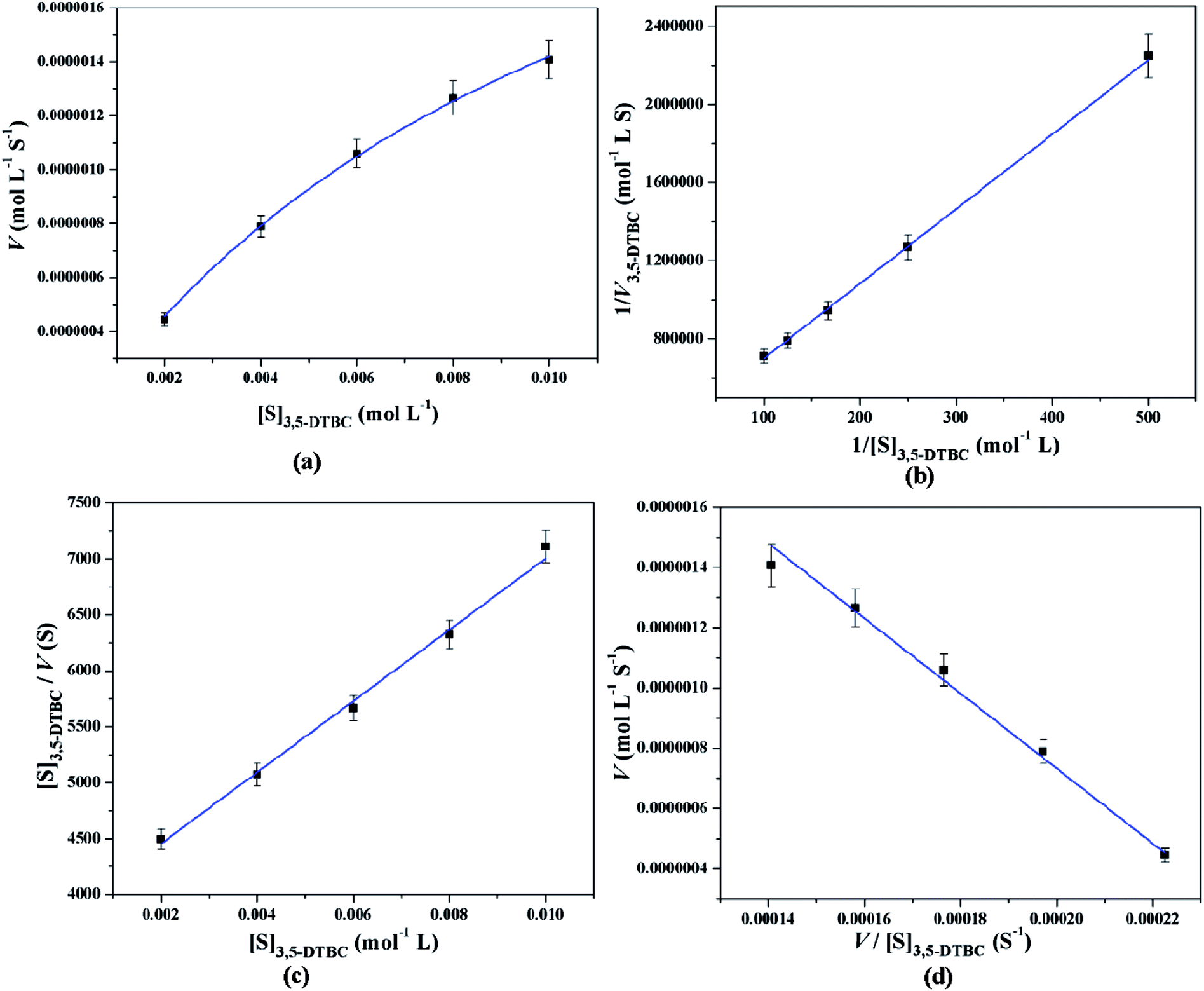
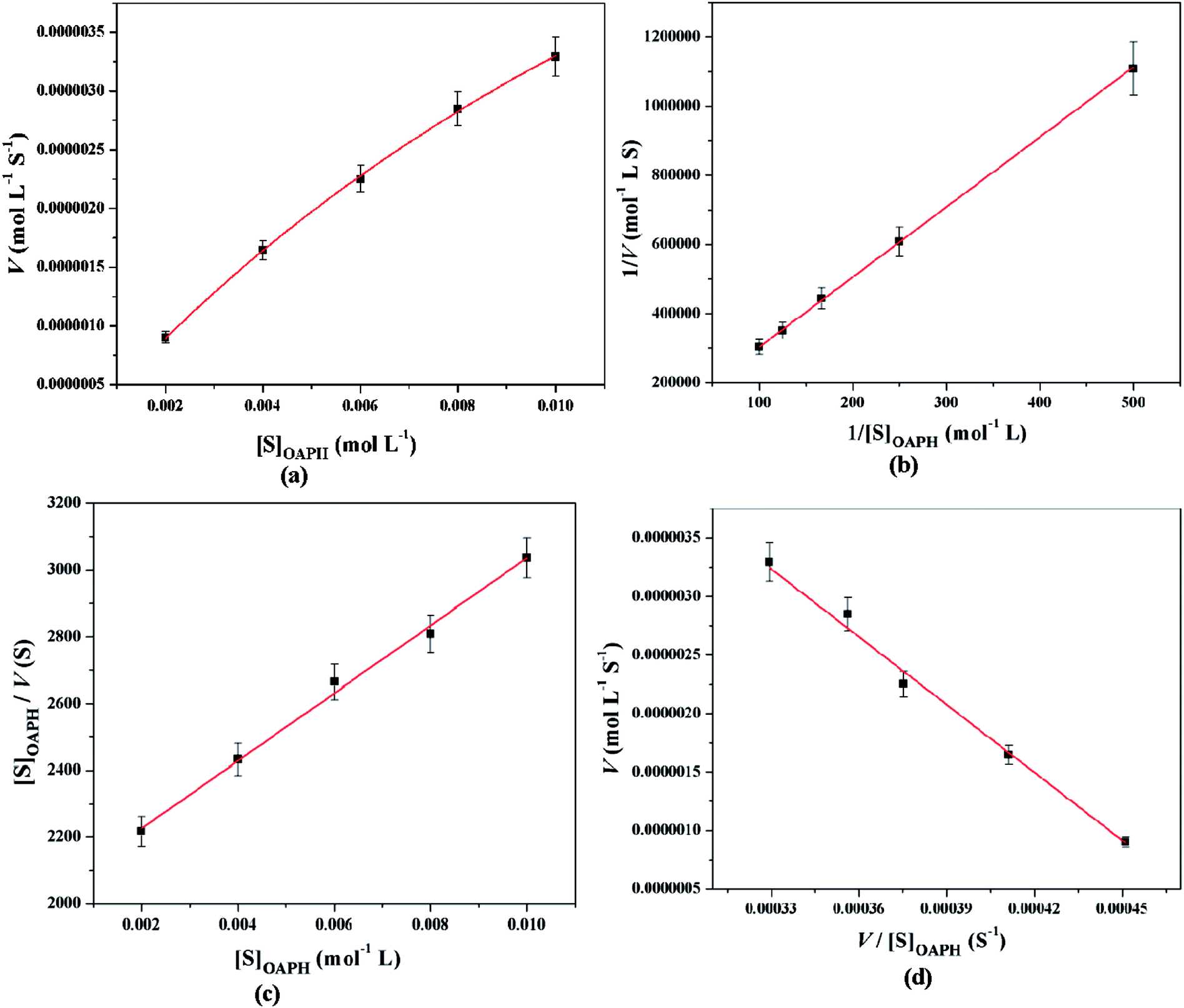
All these equations are used to analyze a variety of kinetic parameters, including turnover number (kcat) and specificity constant (kcat/KM) for catechol oxidase and phenoxazinone synthase mimicking activity of synthesized complexes. Kinetic parameters like Vmax and KM were calculated by the analysis of entire experimental data. The turnover number value (kcat) is obtained by dividing the Vmax by the concentration of the catalyst (complex) used. Fig. 11 and 12 represent the Michaelis–Menten plot, Lineweaver–Burk plot, Hanes plot and Eadie–Hofstee plot of complex 1 for catalytic oxidation of 3,5-DTBC and OAPH, respectively. Similarly, the Michaelis–Menten plot, Lineweaver–Burk plot, Hanes plot and Eadie–Hofstee plot of complex 2 for catalytic oxidation of 3,5-DTBC and OAPH are given in Fig. S11 and S12 (ESI†), respectively. Tables 6 and 7 contain all kinetic parameters of the complexes for catechol oxidase and phenoxazinone synthase like activity, respectively. In order to calculate kinetic parameters more accurately, several enzyme kinetic plots have been used. More or less similar kinetic parameters have been obtained in all cases and this indicates the accuracy of the experimental results. The kcat value suggest that both complexes are very effective catalyst for the conversation of 3,5-DTBC to 3,5-DTBQ and o-aminophenol to 2-aminophenoxazine-3-one.
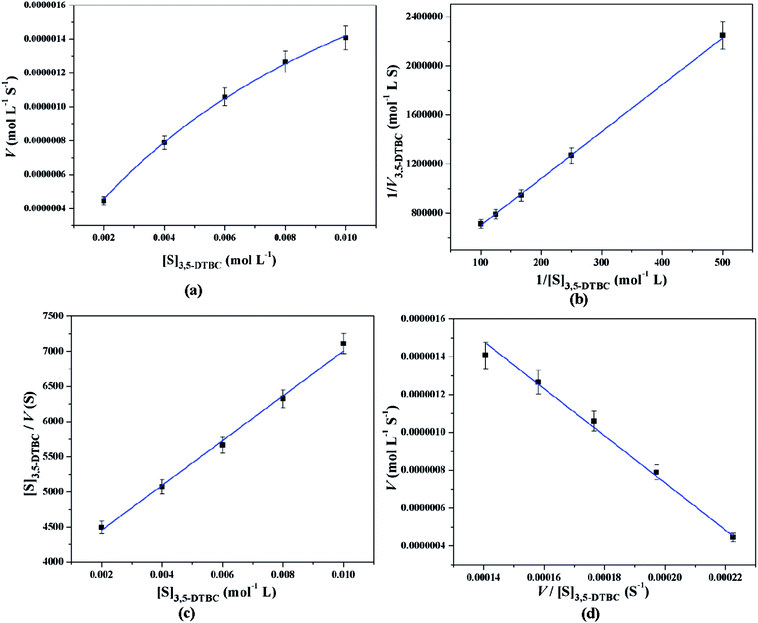 |
| | Fig. 11 Michaelis–Menten plot (a), Lineweaver–Burk plot (b), Hanes plot (c) and Eadie–Hofstee plot (d) of complex 1 for catalytic oxidation of 3,5-DTBC in acetonitrile–methanol (2:1) mixture at room temperature. | |
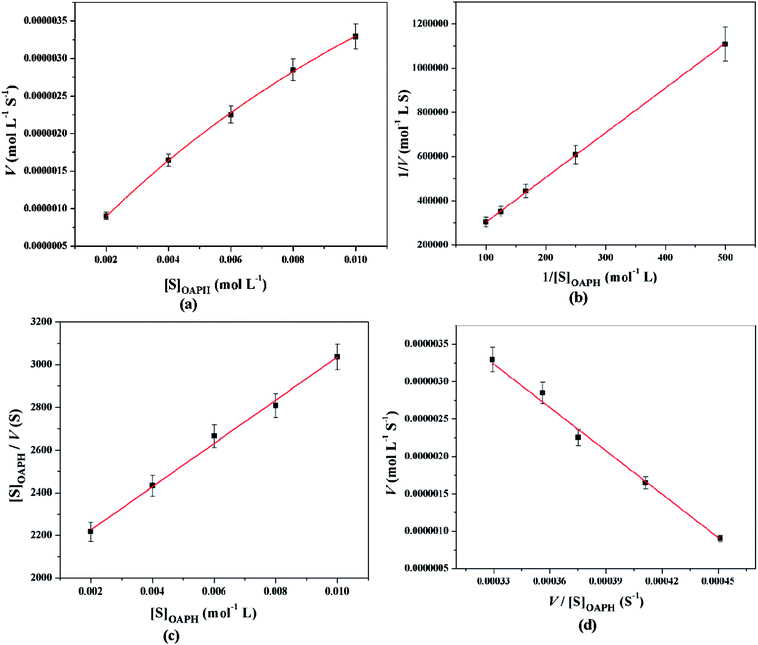 |
| | Fig. 12 Michaelis–Menten plot (a), Lineweaver–Burk plot (b), Hanes plot (c) and Eadie–Hofstee plot (d) of complex 1 for catalytic oxidation of OAPH in acetonitrile–methanol (2:1) mixture at room temperature. | |
Table 6 Kinetic parameters of complexes 1 and 2 for catechol oxidase mimicking activity using various enzyme kinetic plots at 25 °C in acetonitrile–methanol (2:1) mixture
| Enzyme kinetic plots |
Vmax ± SE (M S−1) |
KM ± SE (M) |
kcat ± SE (S−1) |
kcat/KM ± SE (S−1 M−1) |
| 1 |
2 |
1 |
2 |
1 |
2 |
1 |
2 |
| Michaelis–Menten plot |
(3.009 ± 0.123) × 10−6 |
(3.065 ± 0.115) × 10−6 |
(112.300 ± 7.500) × 10−4 |
(102.300 ± 5.400) × 10−4 |
(3.009 ± 0.123) × 10−2 |
(3.065 ± 0.115) × 10−2 |
(0.027 ± 0.001) × 102 |
(0.030 ± 0.001) × 102 |
| Lineweaver–Burk plot |
(3.139 ± 0.453) × 10−6 |
(3.162 ± 0.415) × 10−6 |
(119.843 ± 8.922) × 10−4 |
(104.669 ± 6.226) × 10−4 |
(3.139 ± 0.453) × 10−2 |
(3.162 ± 0.415) × 10−2 |
(0.026 ± 0.002) × 102 |
(0.030 ± 0.002) × 102 |
| Hanes–Woolf plot |
(3.139 ± 0.356) × 10−6 |
(3.163 ± 0.163) × 10−6 |
(119.843 ± 3.506) × 10−4 |
(104.670 ± 2.456) × 10−4 |
(3.139 ± 0.356) × 10−2 |
(3.163 ± 0.163) × 10−2 |
(0.026 ± 0.002) × 102 |
(0.030 ± 0.001) × 102 |
| Eadie–Hofstee plot |
(3.225 ± 0.135) × 10−6 |
(3.227 ± 0.135) × 10−6 |
(124.600 ± 6.558) × 10−4 |
(107.900 ± 5.864) × 10−4 |
(3.225 ± 0.135) × 10−2 |
(3.227 ± 0.135) × 10−2 |
(0.026 ± 0.001) × 102 |
(0.030 ± 0.001) × 102 |
Table 7 Kinetic parameters of complexes 1 and 2 for phenoxazinone synthase mimicking activity using various enzyme kinetic plots at 25 °C in acetonitrile–methanol (2:1) mixture
| Enzyme kinetic plots |
Vmax ± SE (M S−1) |
KM ± SE (M) |
kcat ± SE (S−1) |
kcat/KM ± SE (S−1 M−1) |
| 1 |
2 |
1 |
2 |
1 |
2 |
1 |
2 |
| Michaelis–Menten plot |
(9.724 ± 0.314) × 10−6 |
(9.716 ± 0.132) × 10−6 |
(202.200 ± 9.482) × 10−4 |
(100.784 ± 7.500) × 10−4 |
(9.724 ± 0.314) × 10−2 |
(9.716 ± 0.132) × 10−2 |
(0.048 ± 0.004) × 102 |
(0.096 ± 0.001) × 102 |
| Lineweaver–Burk plot |
(9.869 ± 0.906) × 10−6 |
(9.769 ± 1.248) × 10−6 |
(199.682 ± 18.486) × 10−4 |
(101.684 ± 5.708) × 10−4 |
(9.869 ± 0.306) × 10−2 |
(9.769 ± 1.248) × 10−2 |
(0.049 ± 0.003) × 102 |
(0.096 ± 0.005) × 102 |
| Hanes–Woolf plot |
(9.869 ± 0.808) × 10−6 |
(9.769 ± 0.493) × 10−6 |
(199.683 ± 11.384) × 10−4 |
(101.681 ± 2.253) × 10−4 |
(9.869 ± 0.808) × 10−2 |
(9.769 ± 0.493) × 10−6 |
(0.049 ± 0.001) × 102 |
(0.096 ± 0.003) × 102 |
| Eadie–Hofstee plot |
(9.605 ± 0.401) × 10−6 |
(9.800 ± 0.408) × 10−6 |
(193.100 ± 9.403) × 10−4 |
(102.200 ± 5.616) × 10−4 |
(9.605 ± 0.401) × 10−2 |
(9.800 ± 0.408) × 10−2 |
(0.050 ± 0.001) × 102 |
(0.096 ± 0.001) × 102 |
Origin and mechanistic pathway of catalytic activity
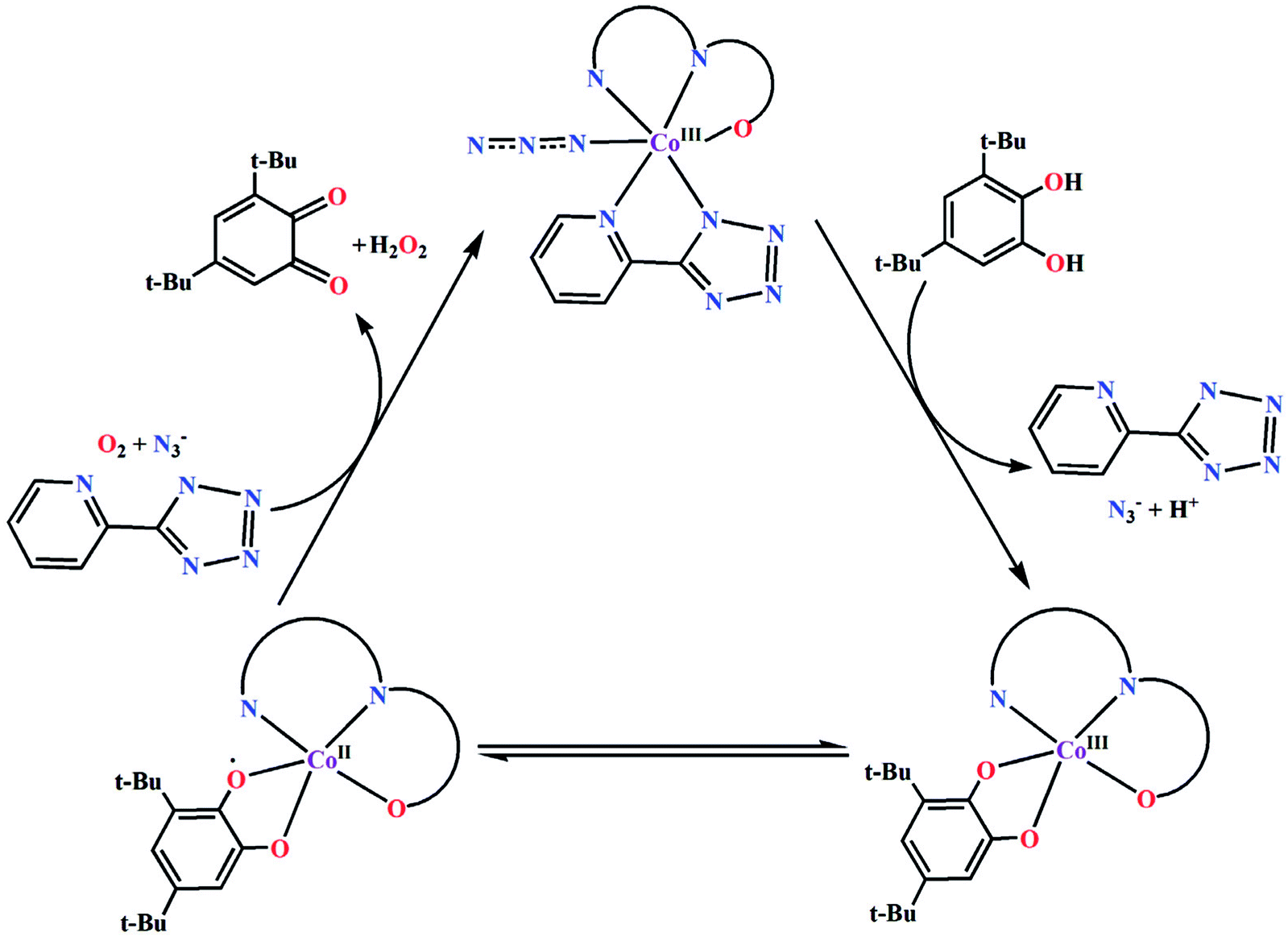
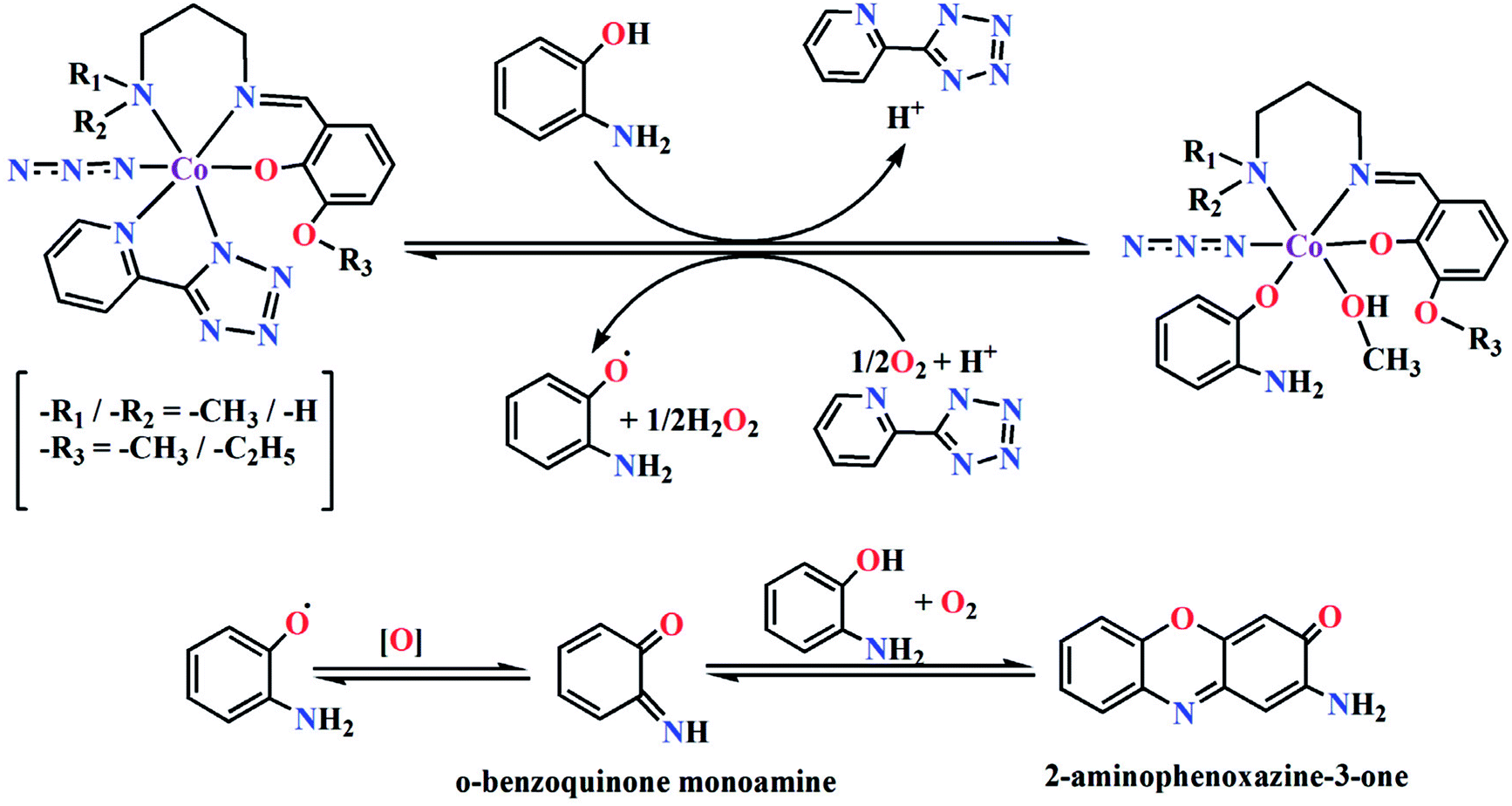
Based on the above experimental results, it is clear that both complexes are very reactive towards the catechol oxidase and phenoxazinone synthase mimicking activity. Schemes 3 and 4 represent possible mechanism for catalytic conversation of 3,5-DTBC to 3,5-DTBQ and o-aminophenol to 2-aminophenoxazine-3-one, respectively. It is easily understandable that at initial step of the catalytic cycles, the tetrazole moiety and/or monodentate azide co-ligand were replaced by the substrate, thereby forming a substrate–catalyst adduct. Hence it may be stated that the replacement of tetrazole moiety and/or monodentate azide co-ligand initiated the catalytic cycles. This substrate–catalyst adduct yields a substrate radical in the rate determining step. Finally, quinone chromophore was formed from this substrate radical via a number of oxidative dehydrogenation processes.
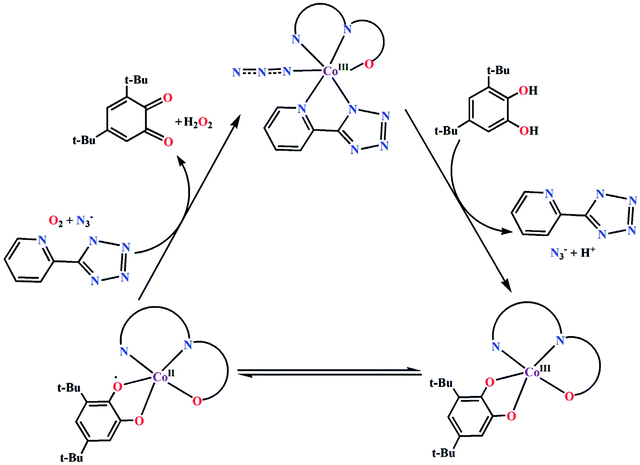 |
| | Scheme 3 Proposed mechanistic pathway for the aerial oxidation of 3,5-DTBC catalyzed by complexes 1 and 2. | |
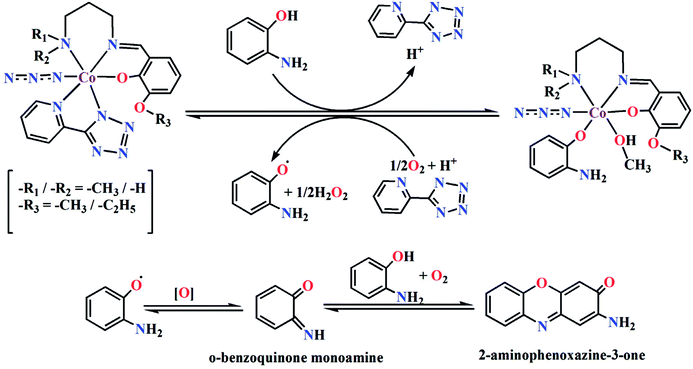 |
| | Scheme 4 Proposed mechanistic pathway for the aerial oxidation of o-aminophenol catalyzed by complexes 1 or 2. | |
The catalytic activity depends on the interaction between substrate and catalyst. Higher the interaction between substrate and catalyst higher the kcat values (Tables 6 and 7). Both complexes have comparable turnover numbers. Similar types of tentative catalytic cycles have also been proposed by several other groups.46
A detailed literature survey with cobalt based model complexes for catechol oxidase and phenoxazinone synthase mimicking activities has shown that several factors may affect their catalytic activity, such as the oxidation state of the metal, coordination geometry around the metal center, metal–metal distance, ligand flexibility, exogenous bridging ligand etc.47,48 All these cobalt based model complexes showing catechol oxidase and phenoxazinone synthase mimicking activity are gathered in Tables 8 and 9 respectively along with several kinetic parameters such as KM, Vmax and kcat. As the turnover numbers (kcat values) of both complexes fall in between the range, they may be considered as moderate catalysts.
Table 8 A comparison of catechol oxidase mimicking activity of previously reported cobalt based complexesa
| Complexes |
Solvent |
Vmax (M s−1) |
KM (M) |
kcat (h−1) |
Ref. |
| Where, H2L3 = N-(2-hydroxyethyl)-3-methoxysalicylaldimine; H4L4 = condensation product of 2,3-dihydroxynaphthalene-1,4-dicarbaldehyde and 2-[O-(1-ethyloxyamide)]oxime-4,6-dibromophenol; HL5 = 2-{(3-ethoxypropylimino)methyl}-6-methoxyphenol; H2L6 = 1:2 condensation product of 1,2,4,5-tetra-amino benzene and 2-hydroxy benzaldehyde; H2L7 = 1:2 condensation product of 1,2,4,5-tetra-amino benzene and 2,4-dihydroxy benzaldehyde; H2L8 = 1:2 condensation product of 1,2,4,5-tetra-amino benzene and 2-hydroxy naphthaldehyde; H4L9 = 3,5-di-tert-butyl-2-hydroxybenzylideneamino)-2-(hydroxymethyl)propane-1,3-diol; H4L10 = 2-(3,5-di-tert-butyl-2-hydroxybenzylideneamino)-2-(hydroxymethyl)propane-1,3-diol; L11 = N,N,N′,N′-tetrakis(2′-benzimidazolylmethyl)-1,4-diethylene amino glycol ether; phen = 1,10-phenanthroline; H2L12 = [2 + 2] condensation product of 2,6-diformyl-4-methylphenol and 2,2-dimethyl-1,3-diaminopropane; H3L13 = 3-[(2-hydroxy-benzylidene)-amino]-propane-1,2-diol; H3L14 = 3-[(2-hydroxy-3-methoxy-benzylidene)-amino]-propane-1,2-diol; H2L15 = [2 + 2] condensation of 4-ethyl-2,6-diformylphenol and 2,2′-dimethyl-1,3-diaminopropane; HL16 = 2,6-bis(N-ethylpiperazine-iminomethyl)-4-methyl-phenolato; HL17 = 2,6-bis(2-ethylpyridine-iminomethyl)-4-methyl-phenolato; HL18 = 2,6-bis(N-ethylpiperidine-iminomethyl)-4-methyl-phenolato; H2L22 = N,N′-bis(salicylidene)-1,3-propanediamine; H2L23 = N,N′-bis(2-hydroxybenzyl)-1,3-propanediamine; H2L24 = N,N′-ethylenebis(3-ethoxysalicylaldiimine); L25 = condensation product of 1,3-propanediamine and 2-benzoylpyridine; HL26 = 2((2(piperidin-1-yl)ethylimino)methyl)-6-ethoxyphenol; HL27 = 1-acetyl-2-naphthol; HL28 = 2-((3(dimethylamino)propylimino)methyl)-6-methoxyphenol; HL29 = 2,4-pentanedione. |
| [CoIII(HL3)2](OAc)·H2O |
CH3OH |
(3.36 ± 0.339) × 10−5 |
(7.38 ± 0.597) × 10−4 |
1210 |
47a |
| CH3CN |
(5.99 ± 0.198) × 10−5 |
(4.90 ± 0.597) × 10−4 |
2160 |
| DCM |
(4.06 ± 0.571) × 10−5 |
(1.25 ± 0.229) × 10−3 |
1460 |
| [CoII3(L4)(OAc)2(CH3OH)2]·2CHCl3 |
CH3CN |
1.02 × 10−7 |
5.20 × 10−3 |
14.7 |
47b |
| [CoII3(L4)(OAc)2(H2O)2] |
CH3CN |
1.83 × 10−7 |
7.09 × 10−3 |
26.4 |
| [CoII3L52(μ-L5)2(μ-OH2)2(CF3CO2)2] |
CH3CN |
(10.54 ± 0.742) × 10−7 |
(24.55 ± 0.072) × 10−5 |
379 |
47c |
| [CoII7(μ-L5)6(μ-OMe)6]Cl2 |
CH3CN |
(20.59 ± 0.988) × 10−7 |
(43.23 ± 0.098) × 10−5 |
741 |
| [CoII2L6]·3H2O |
CH3OH |
4.50 × 104 |
9.76 × 102 |
270 |
47d |
| [CoII2L7]·4H2O |
CH3OH |
3.70 × 104 |
8.57 × 102 |
222 |
| [CoII2L8]·5H2O |
CH3OH |
2.50 × 104 |
5.47 × 102 |
150 |
| [(CoIII2(H2L9)2(OAc)2)]·CH3OH |
CH3CN:DMF |
(1.33 ± 0.136) × 10−7 |
(8.70 ± 1.470) × 10−3 |
79.75 |
47e |
| [CoII3CoIII3(HL10)2(H2L10)2(OAc)2]·9CH3OH·H2O |
CH3CN:CH3OH |
1.20 × 10−5 |
5.60 × 10−3 |
72.2 |
47f |
| CoII(OAc)2·4H2O |
CH3CN:CH3OH |
9.30 × 10−8 |
1.00 × 10−2 |
32 |
47g |
| [CoII2(L11)Cl2](ClO4)2·5H2O {at pH 7.3} |
CH3OH Tris–HCL buffer |
2.50 × 10−8 |
2.47 × 10−3 |
0.9 |
47h |
| [CoII2(L11)Cl2](ClO4)2·5H2O {at pH 7.6} |
| 3.16 × 10−8 |
2.51 × 10−3 |
1.14 |
| [CoII2(L11)Cl2](ClO4)2·5H2O {at pH 8.0} |
| 1.95 × 10−7 |
2.77 × 10−3 |
7.02 |
| [CoIII(phen)2(Cl)2]+ |
CH3OH |
2.68 × 10−5 |
1.77 × 10−3 |
965 |
47i |
| [CoIIICoIIL12(N3)3]·0.5CH3CN·0.27H2O |
CH3CN |
(7.93 ± 0.631) × 10−7 |
(1.18 ± 0.214) × 10−3 |
114.24 |
47j |
| [CoIIICoII(HL13)2(H2O)(HOCH2CH3)]Cl·2H2O |
CH3OH |
5.946 × 10−4 |
8.815 × 10−3 |
21.408 |
47k |
| DMF |
8.25 × 10−4 |
2.014 × 10−4 |
29.700 |
| [CoIIICoII2(H2L14)2(L2)Cl2]·3H2O |
CH3OH |
6.764 × 10−4 |
8.972 × 10−3 |
24.353 |
| DMF |
5.57 × 10−5 |
12.00 × 10−4 |
20.054 |
| [CoIIICoIIL15(N3)3]·CH3CN |
CH3OH |
(3.152 ± 0.695) × 10−7 |
(1.576 ± 0.702) × 10−3 |
45.38 |
47l |
| CH3CN |
(3.348 ± 0.858) × 10−6 |
(3.011 ± 1.227) × 10−3 |
482.16 |
| [CoII2(L16H)(H2O)2(OAc)2](OAc)2 |
CH3OH |
1.24 × 10−4 |
2.45 × 10−3 |
447 |
47m |
| [CoII2(L17)(H2O)2(OAc)2](OAc) |
CH3OH |
1.28 × 10−5 |
1.78 × 10−3 |
45.9 |
| [CoII2(L18)(H2O)2(OAc)2](OAc) |
CH3OH |
1.19 × 10−5 |
2.39 × 10−3 |
42.9 |
| [CoIIICoIIIL19(N3)4]·6H2O |
CH3CN |
(1.477 ± 0.259) × 10−6 |
(1.678 ± 0.578) × 10−3 |
212.6 |
47n |
| [CoIIICoIIIL20(N3)4]·3.5H2O |
CH3CN |
(1.306 ± 0.268) × 10−6 |
(3.300 ± 1.045) × 10−3 |
188.0 |
| [CoIIICoIIL21(N3)3]·H2O |
CH3CN |
(1.328 ± 0.329) × 10−6 |
(4.523 ± 1.578) × 10−3 |
191.3 |
| cis-[Co3L222(MeOH)2(N3)2(μ1,1-N3)2] |
CH3CN |
(1.967 ± 0.113) × 10−6 |
(1.89 ± 0.059) × 10−3 |
142 |
47o |
| trans-[Co3L222(H2O)2(N3)2(μ1,1-N3)2]·(H2O)2 |
CH3CN |
(1.195 ± 0.010) × 10−6 |
(1.44 ± 0.008) × 10−3 |
85 |
| [Co3L232(N3)3(μ1,3-N3)] |
CH3CN |
(1.382 ± 0.065) × 10−6 |
(1.65 ± 0.048) × 10−3 |
99 |
| [CoIIIL24(N3)2(H3O+)]·2MeOH |
CH3CN |
(6.967 ± 0.858) × 10−8 |
(1.26 ± 0.328) × 10−3 |
10.0 |
47p |
| [CoIIIL24(NCS)(H2O)]·DMF·H2O |
DMF |
(7.80 ± 0.592) × 10−8 |
(1.24 ± 0.208) × 10−3 |
11.2 |
| [CoIII(L25)2](ClO4)3 |
CH3OH |
(1.45 ± 1.24) × 10−4 |
(1.90 ± 0.880) × 10−3 |
5020 |
47q |
| [CoIII(L26)(L27)(N3)] |
CH3CN |
3.412 × 10−6 |
2.553 × 10−4 |
122.83 |
47r |
| [CoIII(L28)(L29)(NCS)] |
CH3CN |
12.965 × 10−6 |
6.834 × 10−4 |
466.74 |
| [CoIII(L1)(PTZ)(N3)] |
CH3CN:CH3OH |
(3.139 ± 0.453) × 10−6 |
(1.198 ± 0.089) × 10−6 |
113.00 |
This work |
| [CoIII(L2)(PTZ)(N3)] |
CH3CN:CH3OH |
(3.162 ± 0.415) × 10−6 |
(1.047 ± 0.623) × 10−6 |
113.83 |
Table 9 A comparison of phenoxazinone synthase mimicking activity of previously reported cobalt based complexesa
| Complexes |
Solvent |
Vmax (M s−1) |
KM (M) |
kcat (h−1) |
Ref. |
| Where, HL30 = condensation product of N,N-dimethyldipropylenetriamine and o-vanillin; H2L31 = 3,5,6-tribromo-4-pyridiniumcatechol; L32 = pyridine; H2L33 = N-(2-hydroxyethyl)-3-methoxysalicylaldimine; H2L34 = N,N′-bis(3-methoxysalicylidehydene)cyclohexane-1,2-diamine); L35 = 4-aminopyridine; L36 = 1-methylimidazole; L37 = 2-aminomethylpyridine; L38 = 2-iminomethylpyridine anion; L39 = bis(2-pyridylmethyl)amine; L40 = (2-pyridylmethyl)(2-pyridylethyl)amine; L41 = 1:2 condensation product of 3,3′-bisaminopropylamine and 2-pyridinecarboxaldehyde; L42 = 1:2 condensation product of 3,3′-bisaminopropylamine and 2-acetylpyridine; L43 = 1:2 condensation product of diethylenetriamine and 2-pyridinecarboxaldehyde; L44 = 1:2 condensation product of 3,3′-bisaminopropylamine and 6-methyl-2-pyridinecarboxaldehyde; L45 = 1:2 condensation product of 3,3′-diamino-N-methyldipropylamine and 6-methyl-2-pyridinecarboxaldehyde; L46 = N,N′-bis(pyridin-2-ylmethylene)-2,2-dimethylpropane-1,3-diamine; L47 = N,N′-bis(6-methylpyridin-2-ylmethylene)-2,2-dimethylpropane-1,3-diamine; HL48 = 2-(3-(dimethylamino)propyliminomethyl)-6-ethoxyphenol; HL49 = 1-benzoylacetone; HL50 = 2((2(2-hydroxyethylamino)ethylimino)methyl)-6-ethoxyphenol, HL51 = 2-acetyl-1-naphthol; HL52 = 1((2(diethylamino)ethylimino)methyl)naphthalen-2-ol. |
| [CoIII(L25)2](ClO4)3 |
CH3OH |
(1.27 ± 0.915) × 10−4 |
(3.84 ± 0.847) × 10−3 |
4590 |
47q |
| CH3CN |
(1.42 ± 0.985) × 10−4 |
(6.04 ± 1.04) × 10−4 |
5120 |
| [CoIII2CoIIL232(μ2-C6H5CO2−)2 (C6H5CO2−)2]·(CH3CN)2 |
CH3CN |
(2.13 ± 0.412) × 10−6 |
(9.66 ± 0.198) × 10−3 |
153.60 |
48a |
| [CoIII(L30)(N3)2]·0.5CH3CN |
CH3OH |
3.00 × 10−7 |
1.35 × 10−2 |
54.0 |
48b |
| [CoIII(L30)(NCS)2]·0.5H2O |
CH3OH |
2.70 × 10−7 |
1.24 × 10−2 |
48.6 |
| [Co(HL30)2][Co(NCS)4]·NCS |
CH3OH |
4.65 × 10−8 |
8.87 × 10−3 |
26.1 |
| [Co(L30)2]2[Co(NCO)4] |
CH3OH |
1.45 × 10−7 |
1.02 × 10−2 |
8.37 |
| [CoIII(L31)2(L32)2]-Cl·8H2O |
CH3OH |
1.70 × 10−7 |
6.08 × 10−3 |
30.6 |
48c |
| [CoIII(HL33)2](OAc)·H2O |
CH3OH |
(7.87 ± 0.541) × 10−4 |
(4.31 ± 0.775) × 10−4 |
28300 |
48d |
| [CoIIIL34(L35)2]ClO4·MeOH·H2O |
CH3OH |
3.19 × 10−8 |
6.785 × 10−4 |
11.48 |
48e |
| [CoIIIL34(L36)2Na(ClO4)2]·0.5H2O |
CH3OH |
7.75 × 10−8 |
2.356 × 10−3 |
27.90 |
| [CoIII2(L37)2(μ-L38)2Cl2]Cl2·2H2O |
CH3OH |
1.91 × 10−7 |
1.57 × 10−3 |
13.752 |
9f |
| [CoIII(L39)(N3)3] |
CH3OH |
(5.66 ± 0.32) × 10−8 |
(7.12 ± 0.72) × 10−3 |
20.37 |
46b |
| [CoIII(L40)(N3)3] |
CH3OH |
(9.24 ± 0.52) × 10−8 |
(8.58 ± 0.96) × 10−3 |
33.26 |
| [CoIII2(L41)2(μ-O2)](ClO4)4·2CH3CN |
CH3OH |
(8.60 ± 0.09) × 10−8 |
(1.01 ± 0.15) × 10−2 |
30.09 |
48f |
| [CoIII2(L42)2(μ-O2)](ClO4)4 |
CH3OH |
(6.40 ± 0.08) × 10−8 |
(1.47 ± 0.16) × 10−2 |
23.04 |
| [CoII(L43)(CH3CN)](ClO4)2 |
CH3OH |
(1.87 ± 0.06) × 10−8 |
(1.01 ± 0.14) × 10−2 |
3.36 |
| [CoII(L44)(H2O)](ClO4)2 |
CH3OH |
(3.54 ± 0.06) × 10−8 |
(1.28 ± 0.15) × 10−2 |
6.37 |
| [CoII(L45)(H2O)](ClO4)2 |
CH3OH |
(4.60 ± 0.08) × 10−8 |
(1.32 ± 0.17) × 10−2 |
8.28 |
48f |
| [CoII(L46)Cl(H2O)]Cl·H2O |
CH3OH |
(3.80 ± 0.31) × 10−7 |
(2.0 ± 0.30) × 10−2 |
13.68 |
48g |
| [CoII(L46)(NCS)2] |
CH3OH |
(2.05 ± 0.22) × 10−7 |
(1.9 ± 0.30) × 10−2 |
7.38 |
| [CoII(L47)Cl2] |
CH3OH |
(1.14 ± 0.05) × 10−7 |
(1.6 ± 0.90) × 10−2 |
4.10 |
| [CoIII(L48)(L49)(N3)] |
CH3CN |
6.73 × 10−6 |
5.08 × 10−4 |
247.2 |
48h |
| [CoIII(L48)(L49)(NCS)] |
CH3CN |
7.15 × 10−6 |
5.75 × 10−4 |
257.4 |
| [CoIII(L50)(L51)(N3)] |
CH3CN |
2.153 × 10−6 |
1.613 × 10−5 |
77.52 |
45 |
| [Co(L52)(L49)(N3)] |
CH3CN |
2.003 × 10−6 |
1.565 × 10−5 |
72.12 |
| [CoIII(L26)(L27)(N3)] |
CH3CN |
4.629 × 10−6 |
0.676 × 10−4 |
166.64 |
47r |
| [CoIII(L28)(L29)(NCS)] |
CH3CN |
17.400 × 10−6 |
3.238 × 10−4 |
626.40 |
| [CoIII(L1)(PTZ)(N3)] |
CH3CN:CH3OH |
(9.869 ± 0.906) × 10−6 |
(199.682 ± 18.486) × 10−4 |
355.28 |
This work |
| [CoIII(L2)(PTZ)(N3)] |
CH3CN:CH3OH |
(9.769 ± 1.248) × 10−6 |
(101.684 ± 5.708) × 10−4 |
351.68 |
Experimental and theoretical proof behind proposed mechanistic pathways
Experimental proof
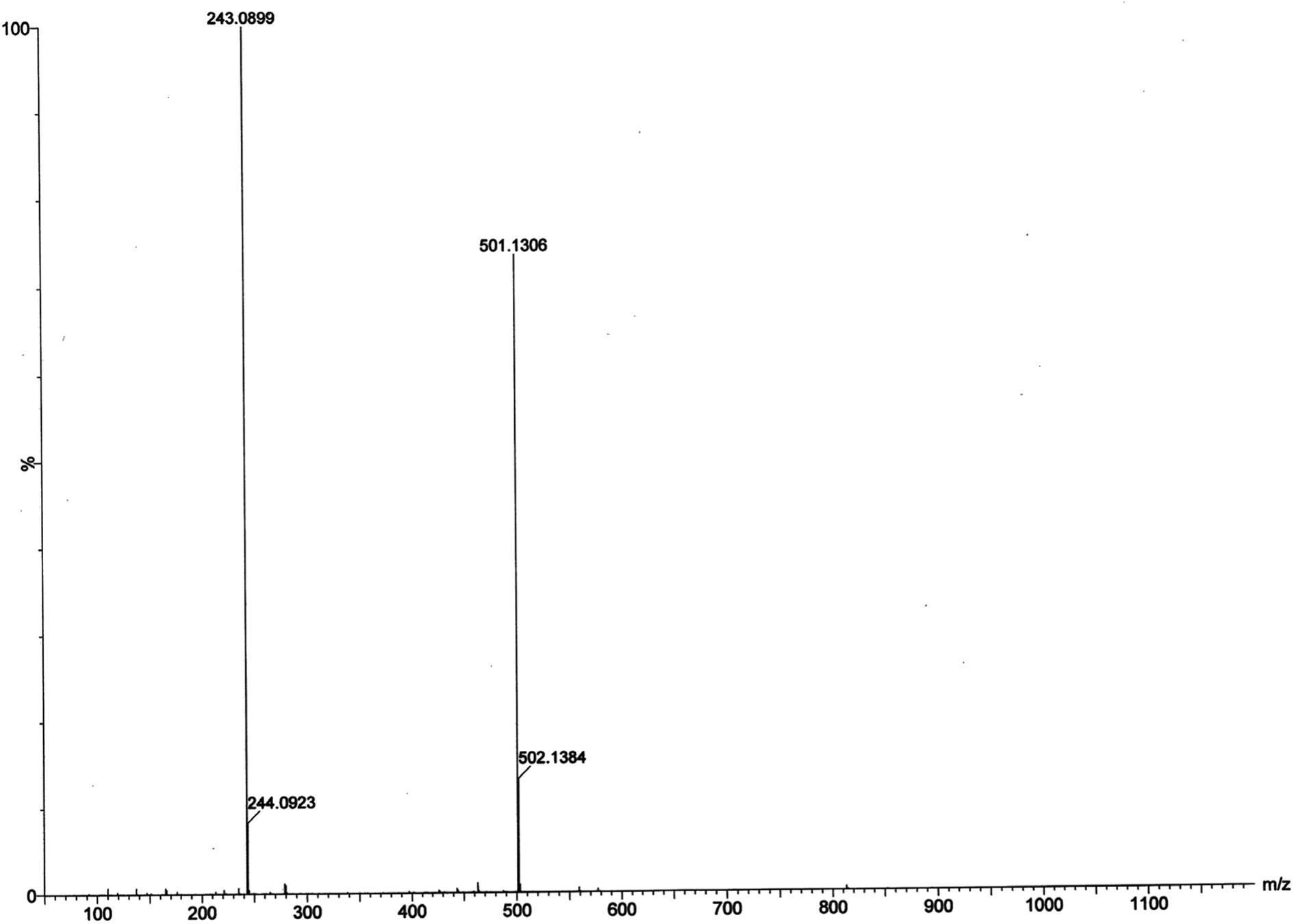
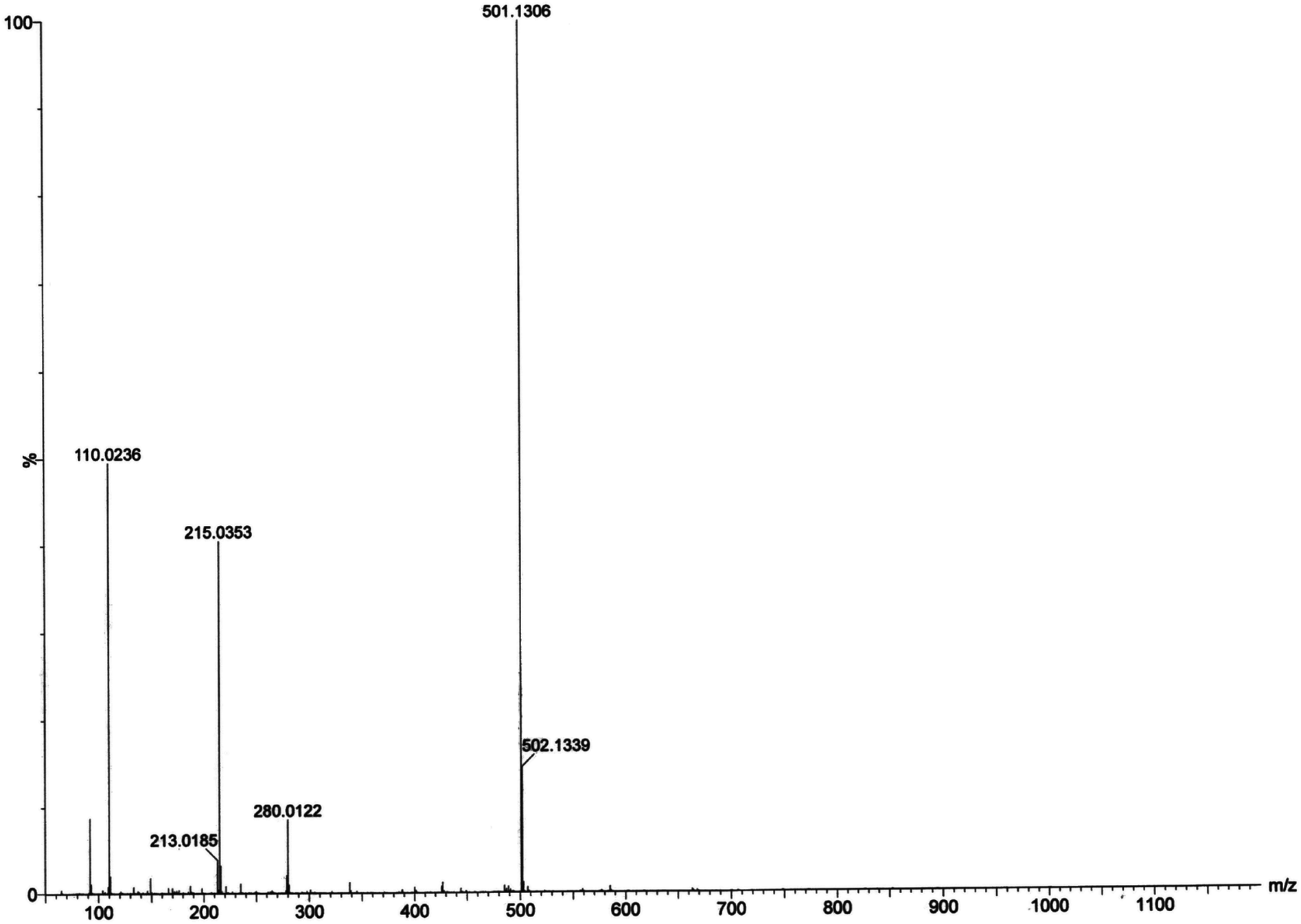
Electrospray ionization mass spectral analysis. The electrospray ionization mass spectral analysis (ESI-MS positive) of complex 1 in presence of substrate (3,5-DTBC for catechol oxidase mimicking activity and o-aminophenol for phenoxazinone synthase mimicking activity) in acetonitrile–methanol (2:1) mixture were recorded to get inside the catalytic cycles. Complex 1 was taken as a model complex. ESI-MS positive spectra of a 1:50 mixture of the complex 1 with 3,5-DTBC and o-aminophenol were recorded separately after 5 min of mixing in acetonitrile–methanol (2:1) mixture and the results are depicted in Fig. 13 and 14, respectively. The most attention-grabbing observation from the mass spectral study is the appearance of peaks related to substrate–catalyst intermediate species. For catechol oxidase and phenoxazinone synthase mimicking activity of the complex 1, mass peaks around m/z ∼ 501 indicate the substrate–catalyst intermediate species, [Co-L1-(3,5-DTBC)-H]+ and [Co-L1-N3-(OAPH)-CH3OH-K]+, respectively. These peaks verify that the catalytic cycles proceed through a stable substrate–catalyst intermediate formation.
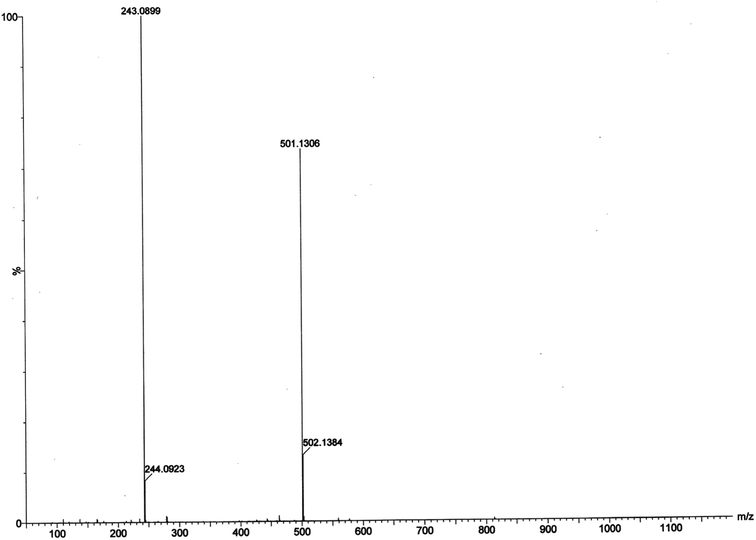 |
| | Fig. 13 ESI-MS positive spectrum of 1:50 mixture of complex 1 and 3,5-DTBC in acetonitrile–methanol (2:1) mixture at room temperature. | |
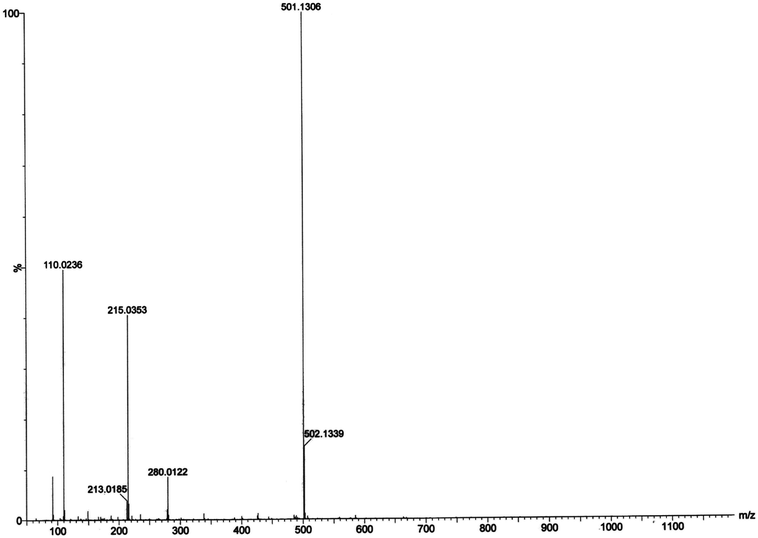 |
| | Fig. 14 ESI-MS positive spectrum of 1:50 mixture of complex 1 and o-aminophenol in acetonitrile–methanol (2:1) mixture at room temperature. | |
In presence of 3,5-DTBC, the mass spectrum exhibits a peak at m/z = 243.0899 corresponding to quinone sodium aggregate [3,5-DTBQ-Na]+, confirming the catechol oxidase mimicking activity of complex 1. On the other hand, in presence of o-aminophenol a peak appears at m/z = 215.0353 which corresponds the formation of [phenoxazinone-H]+. This confirms phenoxazinone synthase mimicking activity of the complex. Electrospray ionization mass spectral analysis (ESI-MS positive) of complex 1 in acetonitrile–methanol (2:1) mixture is given in S13 (ESI†). Peak around m/z ∼ 477 indicates the formation of [Co-L1-N3-OCH3-2(CH3CN)-K]+ (loss of tetrazole moiety) which is the initial step of catalyses. The proposed mechanistic pathways (Schemes 3 and 4) are therefore justified by the mass spectral analysis.
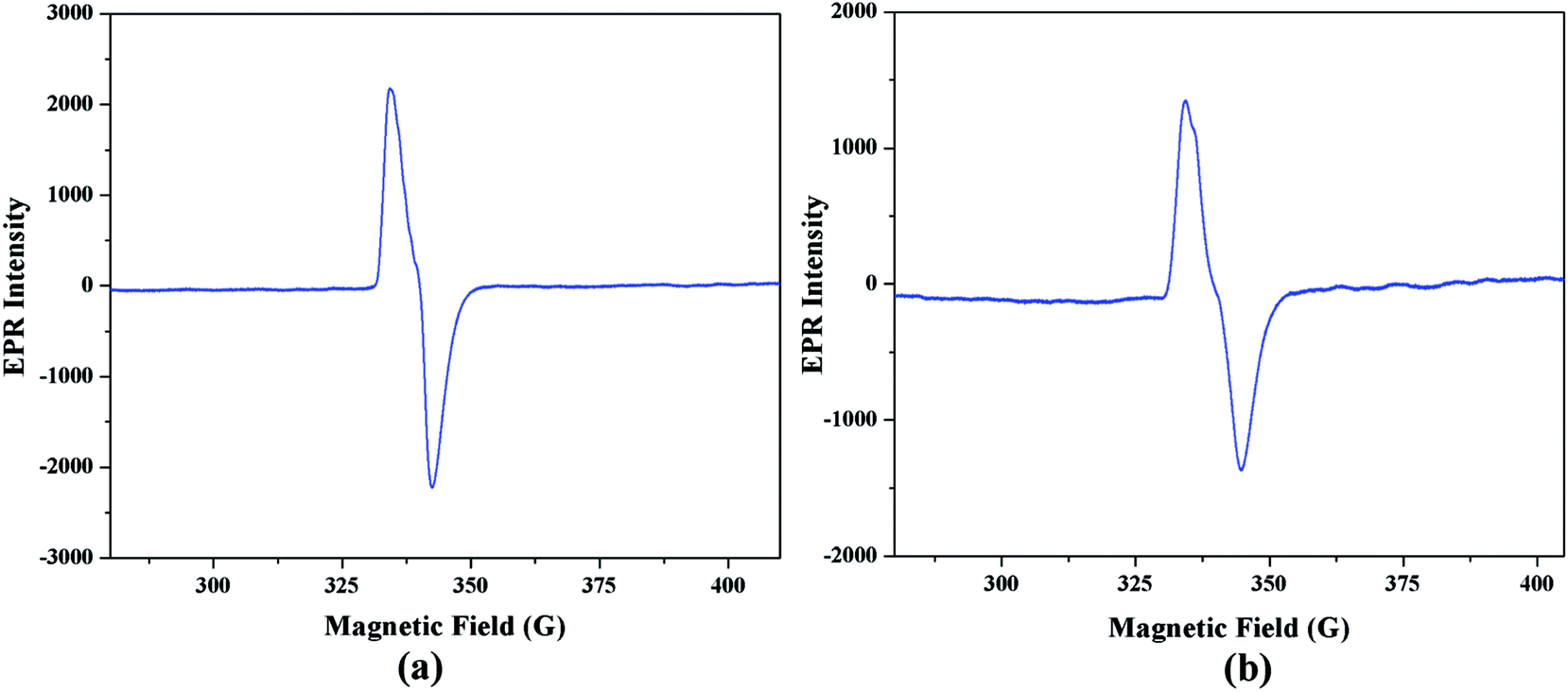
Electron paramagnetic resonance spectral analysis. Electron paramagnetic resonance spectroscopy is a very helpful tool to monitor the generation of radical intermediate during catalytic cycles. As radical is formed as an intermediate species in our proposed mechanistic pathways, EPR measurements at room temperature have been performed on the mixture of the complex 1 with 3,5-DTBC and o-aminophenol separately. Fig. 15(a) and (b) illustrate the EPR spectra of the mixture containing complex 1 and 3,5-DTBC/o-aminophenol, respectively in acetonitrile medium. Both the EPR spectra is dominated by an isotropic signal around giso ∼ 2.00 (for instance, giso = 1.974 in case of 3,5-DTBC and giso = 1.973 in case of o-aminophenol), with a peak-to-peak line width of ca. 20 G. The g value of the signal is very much closed to 2.0023 (value for a free electron49), a value that is characteristic for organic radicals, such as a phenoxyl-type radical.50 Hence it can be concluded that cobalt(III) tetrazolato complexes are capable of oxidizing 3,5-DTBC and o-aminophenol through a radical pathway, as shown in Schemes 3 and 4, respectively.
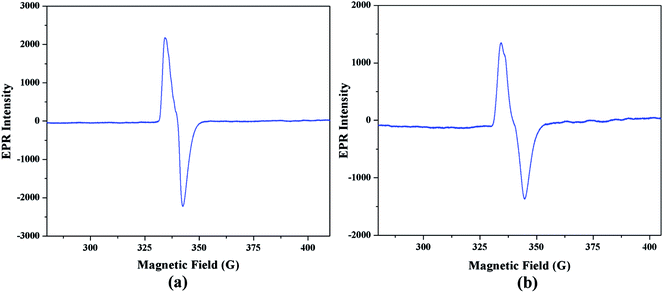 |
| | Fig. 15 EPR spectra of acetonitrile solution of complex 1 (10−4 M) at 77 K immediately after addition of (a) 3,5-DTBC or (b) o-aminophenol. | |
Theoretical proof
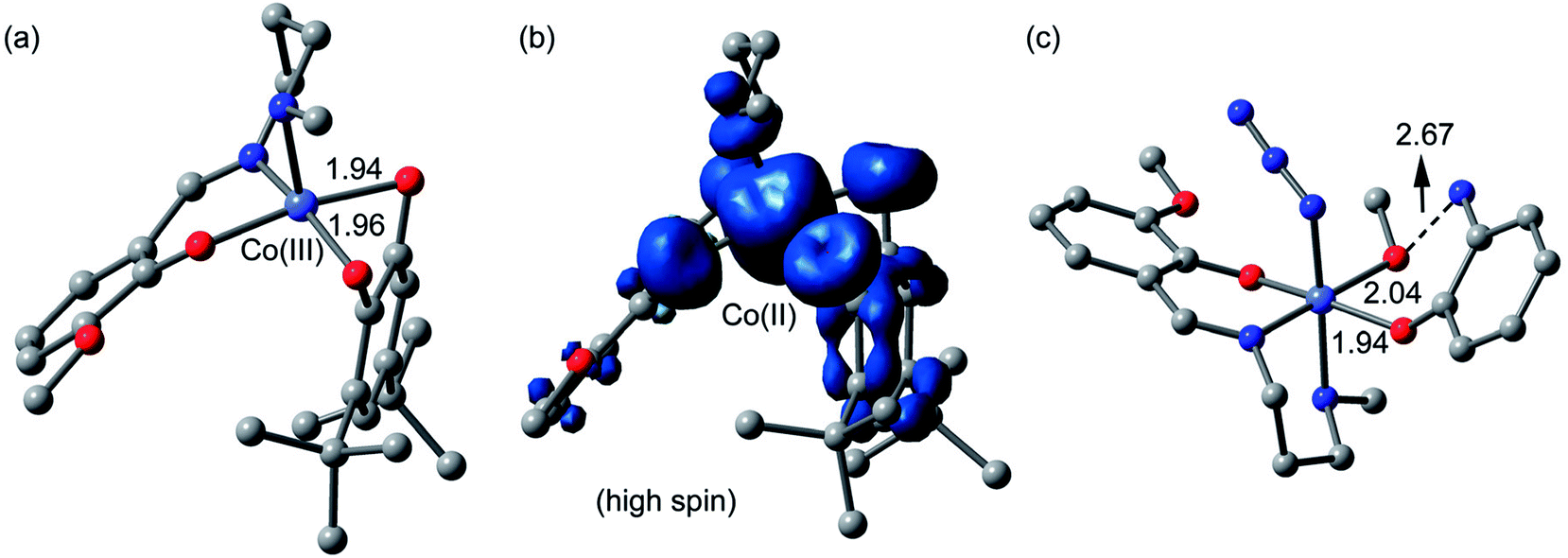
Catechol oxidase and phenoxazinone synthase mimicking activities have been extensively analyzed previously by theoretical studies.9g,46c,47o,51 Therefore, we have mainly focused in the theoretical study to analyze the energetic features of the supramolecular interactions and how they influence the crystal packing. However, in an effort to give some theoretical support to the mechanism and species proposed in the ESI-MS experiments, we have computed both cobalt(III) and cobalt(II) intermediates proposed in Scheme 3 using DFT calculations (BP86-D3/def2-TZVP). The fully optimized geometries and spin density analysis of the cobalt(II) complex have been given in Fig. 16. The catechol is doubly coordinated to the cobalt(III) with similar Co⋯O distances. Upon the single electron transfer, the spin density plot reveals that the unpaired electron is equally distributed in both O-atoms and also the aromatic ring. The spin density at the cobalt(II) atom is 2.7 e, thus confirming the cobalt(II) oxidation state, with some spin density delocalized into the ligand (to the atoms directly bonded to cobalt). Finally, we have also optimized using DFT calculations the proposed complex [Co-L1-N3-(OAPH)-CH3OH] as the first mechanistic step shown in Scheme 4 (see Fig. 16c). The substrate is mono-coordinated to cobalt, and the NH2 group is H-bonded to the MeOH group (O⋯N distance 2.67 Å).
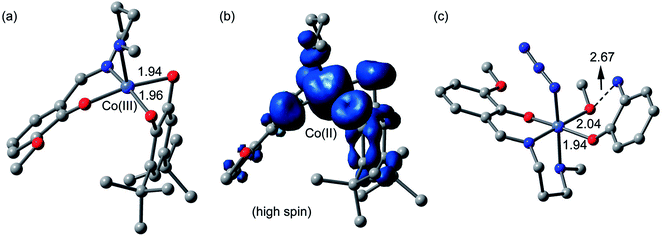 |
| | Fig. 16 (a) DFT optimized geometry of the complex between the 3,5-DTBC substrate and cobalt(III) metal center. (b) Spin density plot of the cobalt(II) complex (upon SET). (c) DFT optimized geometry of the complex between the OAPH substrate and cobalt(III) metal center. H-atoms omitted for clarity. Distances in Å. | |
Concluding remarks
Two new cobalt(III) tetrazolato complexes with Schiff bases and azide co-ligands have been synthesized under non-hydrothermal and non-microwave reaction conditions using a simple in situ 1,3-dipolar cycloaddition reaction. The simple domino synthesis of these complexes therefore opens a new convenient synthetic route for such types of cobalt(III) tetrazolato complexes with Schiff base and azide as co-ligands. The structures of the complexes were confirmed by single crystal X-ray diffraction technique. The formation of hydrogen bonding and C–H⋯π interactions have been studied using DFT calculations. Catechol oxidase and phenoxazinone synthase mimicking activity of the complexes have been studied spectrophotometrically. Due to the similarity of molecular structures, the complexes show comparable results as far as catechol oxidase and phenoxazinone synthase activity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
K. G. expresses his gratitude to UGC, India, for awarding a Senior Research Fellowship (Sr. No. 2121410149; Roll No.: 132586; Ref. No.: 21/12/2014(ii)EU-V; Dated: 25.08.2015). Crystallographic data were collected at the DST-FIST, India funded Single Crystal Diffractometer Facility at the Department of Chemistry, Jadavpur University. A. F. and A. B. gratefully acknowledge the financial support of this work by the MINECO of Spain (project CTQ2014-57393-C2-1-P, FEDER funds). We thank the CTI (UIB) for free allocation of computer time.
Notes and references
-
(a) Y. Fu, F.-M. J. Chang and D. P. Giedroc, Acc. Chem. Res., 2014, 47, 3605–3613 CrossRef PubMed;
(b) H. Zhu, J. Fan, B. Wang and X. Peng, Chem. Soc. Rev., 2015, 44, 4337–4366 RSC;
(c) T. McKee and J. R. McKee, Biochemistry: An Introduction, McGraw-Hill Companies, Inc., China Science Press, New York, 2nd edn, 1999 Search PubMed;
(d) C. S. Bonnet and É. Tóth, Future Med. Chem., 2010, 2, 367–384 CrossRef PubMed;
(e) C. K. Mathews and K. E. Van Holde, Biochemistry, The Benjamin/Cummings Publishing Company, Inc., Menlo Park, 2nd edn, 1996 Search PubMed;
(f) J. J. Braymer and D. P. Giedroc, Curr. Opin. Chem. Biol., 2014, 19, 59–66 CrossRef PubMed;
(g) C. Blériot, M. Gault, E. Gueguen, P. Arnoux, D. Pignol, M. A. M. Berthelota and A. Rodrigue, Metallomics, 2014, 6, 1400–1409 RSC.
-
(a) D. A. Quist, D. E. Diaz, J. J. Liu and K. D. Karlin, J. Biol. Inorg Chem., 2017, 22, 253–188 CrossRef PubMed;
(b) M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef PubMed;
(c) H.-J. Thierse, S. Helm, M. Pink and H. U. Weltzien, J. Immunol. Methods, 2007, 328, 14–20 CrossRef PubMed;
(d) M. M. Bernardo, S. Brown, Z. H. Li, R. Fridman and S. Mobashery, J. Biol. Chem., 2002, 277, 11201–11207 CrossRef PubMed;
(e) P. A. Lindahl and M. J. Moore, Biochemistry, 2016, 55, 4140–4153 CrossRef PubMed.
-
(a) M. Hoaraua, C. Hureaua, E. Gras and P. Faller, Chem. Soc. Rev., 2016, 308, 445–459 Search PubMed;
(b) T. Ye, X. Bai, X. Jiang, Q. Wu, S. Chen, A. Qu, J. Huang, J. Shenc and W. Wu, Polym. Chem., 2016, 7, 2847–2857 RSC;
(c) R. L. D'Ordine, R. S. Linger, C. J. Thai and V. J. Davisson, Biochemistry, 2012, 51, 5791–5803 CrossRef PubMed;
(d) E. L. Kovrigin and J. P. Loria, Biochemistry, 2006, 45, 2636–2647 CrossRef PubMed.
-
(a) J. Fritsch, O. Lenz and B. Friedrich, J. Bacteriol., 2011, 193, 2487–2497 CrossRef PubMed;
(b) K. S. Hewitson, L. A. McNeill, J. M. Elkins and C. J. Schofield, Biochem. Soc. Trans., 2003, 31, 510–515 CrossRef PubMed;
(c) P. A. Cobine, F. Pierrel and D. R. Winge, Biochim. Biophys. Acta, 2006, 1763, 759–772 CrossRef PubMed.
-
(a) K. Ladomenou, G. Charalambidis and A. G. Coutsolelos, Inorg. Chim. Acta, 2010, 363, 2201–2208 CrossRef;
(b) S. K. Dey and A. Mukherjee, Coord. Chem. Rev., 2016, 310, 80–115 CrossRef;
(c) A. A. Vernekar, T. Das, S. Ghosh and G. Mugesh, Chem.–Asian J., 2016, 11, 72–76 CrossRef PubMed;
(d) T. Ueno, Angew. Chem., Int. Ed., 2010, 49, 3868–3869 CrossRef PubMed;
(e) P. Chaudhuri, K. Wieghardt, T. Weyhermüller, T. K. Paine, S. Mukherjee and C. Mukherjee, Biol. Chem., 2005, 386, 1023–1033 Search PubMed.
-
(a) F. T. Ferre, J. A. L. C. Resende, J. Schultz, A. S. Mangrich, R. B. Faria, A. B. Rocha and M. Scarpellini, Polyhedron, 2017, 123, 293–304 CrossRef;
(b) A. Panja, N. C. Jana, S. Adak and K. Pramanik, Inorg. Chim. Acta, 2017, 459, 113–123 CrossRef;
(c) A. T. Fiedler and A. A. Fischer, J. Biol. Inorg Chem., 2017, 22, 407–424 CrossRef PubMed;
(d) H. Yang, J. Zha, P. Zhang, Y. Qin, T. Chen and F. Ye, Sens. Actuators, B, 2017, B247, 469–478 CrossRef;
(e) N. Sarkar, P. K. Bhaumik and S. Chattopadhyay, Polyhedron, 2016, 115, 37–46 CrossRef.
-
(a) F. Zippel, F. Ahlers, R. Werner, W. Haase, H. F. Nolting and B. Krebs, Inorg. Chem., 1996, 35, 3409–3419 CrossRef PubMed;
(b) T. Kurahashi and H. Fujii, J. Am. Chem. Soc., 2011, 133, 8307–8316 CrossRef PubMed;
(c) P. Setha, M. G. B. Drew and A. Ghosh, J. Mol. Catal. A: Chem., 2012, 365, 154–161 CrossRef;
(d) M. Mondal, P. M. Guhaa, S. Giri and A. Ghosh, J. Mol. Catal. A: Chem., 2016, 424, 54–64 CrossRef;
(e) C. Fernandes, A. Neves, A. J. Bortoluzzi, A. S. Mangrich, E. Rentschler, B. Szpoganicz and E. Schwingel, Inorg. Chim. Acta, 2001, 320, 12–21 CrossRef.
-
(a) M. L. Roes-Hill, C. Goodwin and S. Burton, Trends Biotechnol., 2009, 27, 248–268 CrossRef PubMed;
(b) C. E. Barry, P. G. Nayar and T. P. Begley, Biochemistry, 1989, 28, 6323–6333 CrossRef PubMed;
(c) A. W. Smith, A. Camara-Artigas, M. Wang, J. P. Allen and W. A. Francisco, Biochemistry, 2006, 45, 4378–4387 CrossRef PubMed.
-
(a) I. A. Koval, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814–840 RSC;
(b) R. Than, A. A. Feldmann and B. Krebs, Coord. Chem. Rev., 1999, 182, 211–241 CrossRef;
(c) C. Gerdemann, C. Eicken and B. Krebs, Acc. Chem. Res., 2002, 35, 183–191 CrossRef PubMed;
(d) K. Selmeczi, M. Reglier, M. Giorgi and G. Speier, Coord. Chem. Rev., 2003, 245, 191–201 CrossRef;
(e) N. Sarkar, M. Das and S. Chattopadhyay, Inorg. Chim. Acta, 2017, 457, 19–28 CrossRef;
(f) A. Panja and P. Guionneau, Dalton Trans., 2013, 42, 5068–5075 RSC;
(g) N. Sarkar, K. Harms, A. Frontera and S. Chattopadhyay, New J. Chem., 2017, 41, 8053–8065 RSC.
-
(a) S. Kumar, D. N. Dhar and P. N. Saxena, J. Sci. Ind. Res., 2009, 68, 181–187 Search PubMed;
(b) T. Suzuki, T. Moriya, R. Endo and N. Iwasaki, Polym. Chem., 2017, 8, 761–768 RSC;
(c) U. Schatzschneider, Eur. J. Inorg. Chem., 2010, 1451–1467 CrossRef.
-
(a) R. Bikas, H. H. Monfared, T. Lis and M. Siczek, Inorg. Chem. Commun., 2012, 15, 151–155 CrossRef;
(b) A. H. Kianfar, M. Sedighipoor, G. Mohammadnezhad, H. Görls, W. Plass and M. Roushani, J. Iran. Chem. Soc., 2017, 14, 313–322 CrossRef;
(c) S. K. Gupta, N. Sen, J. A. Ganaie, R. J. Butcher and J. P. Jasinski, J. Coord. Chem., 2017, 70, 3147–3170 CrossRef;
(d) H. Iranmanesh, M. Behzad, G. Bruno, H. A. Rudbari, H. Nazari, A. Mohammadi and O. Taheri, Inorg. Chim. Acta, 2013, 395, 81–88 CrossRef.
-
(a) S. Shit, D. Saha, D. Saha, T. N. G. Row and C. Rizzoli, Inorg. Chim. Acta, 2014, 415, 103–110 CrossRef;
(b) A. Nishinaga, T. Yamada, H. Fujisawa, K. Ishizaki, H. Ihara and T. Matsuura, J. Mol. Catal., 1988, 48, 249–264 CrossRef;
(c) K. Maruyama, Y. Murakami, K. Yoda, T. Mashino and A. Nishinaga, J. Chem. Soc., Chem. Commun., 1992, 1617–1618 RSC.
-
(a) A. Biswas, L. K. Das, M. G. B. Drew, G. Aromí, P. Gamez and A. Ghosh, Inorg. Chem., 2012, 51, 7993–8001 CrossRef PubMed;
(b) P. Kavitha, M. R. Chary, B. V. V. A. Singavarapu and K. L. Reddy, J. Saudi Chem. Soc., 2016, 20, 69–80 CrossRef;
(c) N. Shahabadi, S. Kashanian and F. Darabi, Eur. J. Med. Chem., 2010, 45, 4239–4245 CrossRef PubMed.
-
(a) A. D. Khalaji, M. Nikookar, K. Fejfarova and M. Dusek, J. Mol. Struct., 2014, 1071, 6–10 CrossRef;
(b) A. D. Khalajia, R. Rahdaria, F. Ghariba, J. S. Matalobos and D. Das, J. Ceram. Process. Res., 2015, 16, 486–489 Search PubMed;
(c) L. Kafi-Ahmadi and L. Shirmohammadzadeh, J. Nanostruct. Chem., 2017, 7, 179–190 CrossRef.
-
(a) Q. Yang, X. Song, W. Zhang, L. hou, Q. Gong, G. Xie, Q. Wei, S. Chen and S. Gao, Dalton Trans., 2017, 46, 2626–2634 RSC;
(b) A. Mrowiec, A. Jurowska, M. Hodorowicz and J. Szklarzewicz, Dalton Trans., 2017, 46, 4030–4037 RSC;
(c) A. Gualandi, E. Matteucci, F. Monti, A. Baschieri, N. Armaroli, L. Sambri and P. G. Cozzi, Chem. Sci., 2017, 8, 1613–1620 RSC.
-
(a) C. C. Du, J. Z. Fan, X. F. Wang, S. B. Zhou and D. Z. Wang, J. Mol. Struct., 2017, 1133, 348–357 CrossRef;
(b) X.-L. Wang, T.-J. Li, A.-X. Tian and N. Xu, Inorg. Chim. Acta, 2016, 443, 78–85 CrossRef;
(c) J. Yang, J. Wang, B. Wei, L. Shen, Y.-T. Min, H.-J. Cheng, Y. Xu, J. Huang, C.-C. Wang, X. Zhang, M.-L. Wu, Q.-Y. Li and G.-W. Yang, Transition Met. Chem., 2016, 41, 35–43 CrossRef;
(d) M. Taheriha, M. Ghadermazi and V. Amani, J. Mol. Struct., 2016, 1107, 57–65 CrossRef;
(e) L. Shen1, M. J. Cao, F. F. Zhang, Q. Wu, L. Y. Zhao, Y. M. Lu, Q. Y. Li, G. W. Yang, B. Wei and J. H. Zou, Transition Met. Chem., 2016, 41, 125–131 CrossRef.
-
(a) R. Imai, S. Komeda, M. Shimura, S. Tamura, S. Matsuyama, K. Nishimura, R. Rogge, A. Matsunaga, I. Hiratani, H. Takata, M. Uemura, Y. Iida1, Y. Yoshikawa, J. C. Hansen, K. Yamauchi, M. T. Kanemaki and K. Maeshima, Sci. Rep., 2016, 6, 24712 CrossRef PubMed;
(b) S. Kuwata and T. Ikariya, Chem. Commun., 2014, 50, 14290–14300 RSC;
(c) G. E. Büchel, I. N. Stepanenko, M. Hejl, M. A. Jakupec, B. K. Keppler and V. B. Arion, Inorg. Chem., 2011, 50, 7690–7697 CrossRef PubMed;
(d) B. C. Losantos, A. A. Krokhin, I. N. Stepanenko, R. Eichinger, M. A. Jakupec, V. B. Arion and B. K. Keppler, Inorg. Chem., 2007, 46, 5023–5033 CrossRef PubMed.
-
(a) P. Neves, A. B. Lysenko, A. C. Gomes, M. Pillinger, I. S. Gonçalves and A. A. Valente, Catal. Lett., 2017, 147, 1133–1143 CrossRef;
(b) L. Peters, R. Fröhlich, A. S. F. Boyd and A. Kraft, J. Org. Chem., 2001, 66, 3291–3298 CrossRef PubMed;
(c) M. Das, S. Chatterjee, K. Harms, T. K. Mondal and S. Chattopadhyay, Dalton Trans., 2014, 43, 2936–2947 RSC.
-
(a) B. G. Mukhopadhyay, S. Mukhopadhyay, M. F. C. G. da Silva, M. A. J. Charmier and A. J. L. Pombeiro, Dalton Trans., 2009, 4778–4785 RSC;
(b) A. Hantzsch and A. Vagt, Justus Liebigs Ann. Chem., 1901, 314, 339–369 CrossRef;
(c) R. Nasani, M. Saha, S. M. Mobin and S. Mukhopadhyay, Polyhedron, 2013, 55, 24–36 CrossRef.
-
(a) M. Das, K. Harms and S. Chattopadhyay, Dalton Trans., 2014, 43, 5643–5647 RSC;
(b) R.-J. Xu, D.-W. Fu, J. Dai, Y. Zhang, J.-Z. Ge and H.-Y. Ye, Inorg. Chem. Commun., 2011, 14, 1093–1096 CrossRef;
(c) A. D. Bond, A. Fleming, J. Gaire, F. Kelleher, J. McGinley, V. McKee and U. Sheridan, Polyhedron, 2012, 33, 289–296 CrossRef;
(d) Z.-H. Sun, L.-B. Meng and H. Lin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, E65, m280 Search PubMed.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef PubMed;
(b) G. M. Sheldrick, SHELXS-97 and SHELXL-97: Program for Structure Solution, University of Göttingen, Institute fur Anorganische Chemieder Universitat, Gottingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SADABS: Software for Empirical Absorption Correction, University of Göttingen, Institute fur Anorganische Chemieder Universitat, Gottingen, Germany, 1999-2003 Search PubMed.
-
(a) M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC;
(b) F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef;
(c) H. F. Clausen, M. S. Chevallier, M. A. Spackman and B. B. Iversen, New J. Chem., 2010, 34, 193–199 RSC;
(d) M. A. Spackman and P. G. Byrom, Chem. Phys. Lett., 1997, 267, 215–220 CrossRef.
-
(a) A. L. Rohl, M. Moret, W. Kaminsky, K. Claborn, J. J. McKinnon and B. Kahr, Cryst. Growth Des., 2008, 8, 4517–4525 CrossRef;
(b) A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC;
(c) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D Jayatilaka, M. A. Spackman, Crystal Explorer 2.0; University of Western Australia: Perth, Australia, 2007. http://hirshfeldsurfacenet.blogspot.com/ Search PubMed.
-
(a) A. Bhattacharyya, S. Roy, J. Chakraborty and S. Chattopadhyay, Polyhedron, 2016, 112, 109–117 CrossRef;
(b) S. Roy, T. Basak, S. Khan, M. G. B. Drew, A. Bauzá, A. Frontera and S. Chattopadhyay, ChemistrySelect, 2017, 2, 9336–9343 CrossRef;
(c) T. Basak, K. Ghosh and S. Chattopadhyay, Polyhedron, 2018, 146, 81–92 CrossRef.
- A. Bhattacharyya, A. Bauzá, A. Frontera and S. Chattopadhyay, Polyhedron, 2016, 119, 451–459 CrossRef.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef.
-
(a) A. Bauzá, A. Terrón, M. Barceló-Oliver, A.García-Raso and A. Frontera, Inorg. Chim. Acta, 2016, 452, 244–250 CrossRef;
(b) D. Sadhukhan, M. Maiti, G. Pilet, A. Bauzá, A. Frontera and S. Mitra, Eur. J. Inorg. Chem., 2015, 11, 1958–1972 CrossRef;
(c) M. Mirzaei, H. Eshtiagh-Hosseini, Z. Bolouri, Z. Rahmati, A. Esmaeilzadeh, A. Hassanpoor, A. Bauza, P. Ballester, M. Barceló-Oliver, J. T. Mague, B. Notash and A. Frontera, Cryst. Growth Des., 2015, 15, 1351–1361 CrossRef;
(d) P. Chakraborty, S. Purkait, S. Mondal, A. Bauzá, A. Frontera, C. Massera and D. Das, CrystEngComm, 2015, 17, 4680–4690 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–555 CrossRef.
- Spartan ’10, v. 1.1.0, Wavefunction Inc, Irvine CA, www.wavefun.com Search PubMed.
- T. A. Keith, AIMAll (Version 17.01.25), Gristmill Software, Overland Park KS, USA, 2013 Search PubMed.
-
(a) S. Roy, K. Harms and S. Chattopadhyay, Polyhedron, 2015, 91, 10–17 CrossRef;
(b) A. Bhattacharyya, P. K. Bhaumik, P. P. Jana and S. Chattopadhyay, Polyhedron, 2014, 78, 40–45 CrossRef;
(c) W.-J. Lian, X.-T. Wang, C.-Z. Xie, H. Tian, X.-Q. Song, H.-T. Pan, X. Qiao and J.-Y. Xu, Dalton Trans., 2016, 45, 9073–9087 RSC.
- M. Das, K. Harms, B. N. Ghosh, K. Rissanen and S. Chattopadhyay, Polyhedron, 2015, 87, 286–292 CrossRef.
-
(a) S. Chattopadhyay, G. Bocelli, A. Musatti and A. Ghosh, Inorg. Chem. Commun., 2006, 9, 1053–1057 CrossRef;
(b) L. Pogány, J. Moncol, M. Gál, I. Šalitroš and R. Boča, Inorg. Chim. Acta, 2017, 462, 23–29 CrossRef;
(c) K. Ghosh, A. Banerjee, S. Roy, A. Bauzá, A. Frontera and S. Chattopadhyay, Polyhedron, 2016, 112, 86–95 CrossRef.
-
(a) J. Müller, C. Würtele, O. Walter and S. Schindler, Angew. Chem., Int. Ed., 2007, 46, 7775–7777 CrossRef PubMed;
(b) D. Bandyopadhyay, M. Layek, M. Fleck, R. Saha and C. Rizzoli, Inorg. Chim. Acta, 2017, 461, 174–182 CrossRef;
(c) H. A. Rudbari, M. R. Iravani, V. Moazam, B. Askari, M. Khorshidifard, N. Habibi and G. Bruno, J. Mol. Struct., 2016, 1125, 113–120 CrossRef.
-
(a) D. Cremern and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef;
(b) N. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC;
(c) H. Suezawa, T. Yuzuri, Y. Kohno, Y. Yamaguchi and M. Asami, Bull. Chem. Soc. Jpn., 2010, 83, 802–808 CrossRef;
(d) M. Nishio, Y. Umezawa, J. Fantini, M. S. Weiss and P. Chakrabarti, Phys. Chem. Chem. Phys., 2014, 16, 12648–12683 RSC;
(e) M. Nishio, J. Mol. Struct., 2012, 1018, 2–7 CrossRef.
-
(a) M. A. Spackman and P. G. Byrom, Chem. Phys. Lett., 1997, 267, 215–220 CrossRef;
(b) S. Roy and S. Chattopadhyay, Inorg. Chim. Acta, 2015, 433, 72–77 CrossRef;
(c) M. Das and S. Chattopadhyay, Polyhedron, 2013, 50, 443–451 CrossRef;
(d) T. Basak, K. Ghosh, C. J. Gómez-García and S. Chattopadhyay, Polyhedron, 2018, 146, 42–54 CrossRef.
-
(a) B. K. Shaw, M. Das, A. Bhattacharyya, B. N. Ghosh, S. Roy, P. Mandal, K. Rissanen, S. Chattopadhyay and S. K. Saha, RSC Adv., 2016, 6, 22980–22988 RSC;
(b) P. Schmid, M. Maier, H. Pfeiffer, A. Belz, L. Henry, A. Friedrich, F. Schönfeld, K. Edkins and U. Schatzschneider, Dalton Trans., 2017, 46, 13386–13396 RSC.
-
(a) M. Das, B. K. Shaw, B. N. Ghosh, K. Rissanen, S. K. Saha and S. Chattopadhyay, RSC Adv., 2015, 5, 46869–46872 RSC;
(b) M. Das, K. Harms, B. N. Ghosh, K. Rissanen and S. Chattopadhyay, Polyhedron, 2015, 87, 286–292 CrossRef;
(c) S. Roy, K. Harms, A. Bauzá, A. Frontera and S. Chattopadhyay, Polyhedron, 2017, 121, 199–205 CrossRef;
(d) K. Ghosh, K. Harms, A. Bauzá, A. Frontera and S. Chattopadhyay, Dalton Trans., 2018, 47, 331–347 RSC.
-
(a) P. K. Bhaumik, S. Roy, K. Harms and S. Chattopadhyay, Polyhedron, 2014, 81, 168–179 CrossRef;
(b) S. Bhattacharya, S. Roy and S. Chattopadhyay, J. Coord. Chem., 2016, 69, 915–925 CrossRef.
- S. Chattopadhyay, M. G. B. Drew and A. Ghosh, Eur. J. Inorg. Chem., 2008, 1693–1701 CrossRef.
- A. Kufelnicki, S. V. Tomyn, Y. S. Moroz, M. Haukka, J. J. Rosińska and I. O. Fritsky, Polyhedron, 2012, 33, 410–416 CrossRef.
-
(a) M. Galini, M. Salehi, M. Kubicki, A. Amiri and A. Khaleghian, Inorg. Chim. Acta, 2017, 461, 167–173 CrossRef;
(b) A. H. Kianfar, W. A. K. Mahmood, M. Dinari, M. H. Azarian and F. Z. Khafri, Spectrochim. Acta, Part A, 2014, 127, 422–428 CrossRef PubMed.
-
(a) K. Ghosh, K. Harms and S. Chattopadhyay, Polyhedron, 2017, 123, 162–175 CrossRef;
(b) A. Bottcher, T. Takeuchi, K. I. Hardcastle, T. J. Meade and H. B. Gray, Inorg. Chem., 1997, 36, 2498–2504 CrossRef;
(c) A. A. Khandar, B. Shaabani, F. Belaj and A. Bakhtiari, Polyhedron, 2006, 25, 1893–1900 CrossRef;
(d) M. Salehi and M. Hasanzadeh, Inorg. Chim. Acta, 2015, 426, 6–14 CrossRef.
-
(a) M. Das, B. N. Ghosh, A. Bauzá, K. Rissanen, A. Frontera and S. Chattopadhyay, RSC Adv., 2015, 5, 73028–73039 RSC;
(b) A. Panja, M. Shyamal, A. Saha and T. K. Mandal, Dalton Trans., 2014, 43, 5443–5452 RSC;
(c) N. Sarkar, K. Harms, A. Bauzá, A. Frontera and S. Chattopadhyay, ChemistrySelect, 2017, 2, 2975–2984 CrossRef;
(d) M. Mitra, T. Kundu, G. Kaur, G. Sharma, A. R. Choudhury, Y. Singh and R. Ghosh, RSC Adv., 2016, 6, 58831–58838 RSC.
-
(a) M. Mitra, P. Raghavaiah and R. Ghosh, New J. Chem., 2015, 39, 200–205 RSC;
(b) X.-Y. Li, L. Chen, L. Gao, Y. Zhang, S. F. Akogun and W.-K. Dong, RSC Adv., 2017, 7, 35905–35916 RSC;
(c) T. S. Mahapatra, D. Basak, S. Chand, J. Lengyel, M. Shatruk, V. Bertolasic and D. Ray, Dalton Trans., 2016, 45, 13576–13589 RSC;
(d) M. I. Ayad, Arabian J. Chem., 2016, 9, S1297–S1306 CrossRef;
(e) S. K. Dey and A. Mukherjee, New J. Chem., 2014, 38, 4985–4995 RSC;
(f) S. K. Dey, P. Mitra and A. Mukherjee, Cryst. Growth Des., 2015, 15, 706–717 CrossRef;
(g) S. K. Dey and A. Mukherjee, J. Mol. Catal. A: Chem., 2015, 407, 93–101 CrossRef;
(h) J.-H. Qiu, Z.-R. Liao, X.-G. Meng, L. Zhu, Z.-M. Wang and K.-B. Yu, Polyhedron, 2005, 24, 1617–1623 CrossRef;
(i) D. Dey, A. B. Roy, A. Ranjani, L. Gayathri, S. Chandraleka, D. Dhanasekaran, M. A. Akbarsha, C.-Y. Shen, H.-L. Tsai, M. Maji, N. Kole and B. Biswas, J. Chem. Sci., 2015, 127, 649–661 CrossRef;
(j) L. Mandal, S. Sasmal, H. A. Sparkes, J. A. K. Howard and S. Mohanta, Inorg. Chim. Acta, 2014, 412, 38–45 CrossRef;
(k) R. Modak, Y. Sikdar, S. Mandal and S. Goswami, Inorg. Chem. Commun., 2013, 37, 193–196 CrossRef;
(l) S. Majumder, S. Mondal, P. Lemoine and S. Mohanta, Dalton Trans., 2013, 42, 4561–4569 RSC;
(m) A. Banerjee, A. Guha, J. Adhikary, A. Khan, K. Manna, S. Dey, E. Zangrando and D. Das, Polyhedron, 2013, 60, 102–109 CrossRef;
(n) L. Mandal and S. Mohanta, Dalton Trans., 2014, 43, 15737–15751 RSC;
(o) A. Hazari, L. Kanta Das, R. M. Kadam, A. Bauza, A. Frontera and A. Ghosh, Dalton Trans., 2015, 44, 3862–3876 RSC;
(p) P. Chakraborty and S. Mohanta, Inorg. Chim. Acta, 2015, 435, 38–45 CrossRef;
(q) A. K. Maji, A. Chatterjee, S. Khan, B. K. Ghosh and R. Ghosh, J. Mol. Struct., 2017, 1146, 821–827 CrossRef;
(r) K. Ghosh, K. Harms and S. Chattopadhyay, ChemistrySelect, 2017, 2, 8207–8220 CrossRef.
-
(a) A. Hazari, A. Das, P. Mahapatra and A. Ghosh, Polyhedron, 2017, 134, 99–106 CrossRef;
(b) A. Panja, N. C. Janaa and P. Brandão, New J. Chem., 2017, 41, 9784–9795 RSC;
(c) A. Panja and A. Frontera, Eur. J. Inorg. Chem., 2018, 924–931 CrossRef;
(d) M. Mitra and R. Ghosh, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2016, 55A, 681–685 Search PubMed;
(e) M. Mahato, D. Mondal and H. P. Nayek, ChemistrySelect, 2016, 1, 6777–6782 CrossRef;
(f) A. Panja, Dalton Trans., 2014, 43, 7760–7770 RSC;
(g) A. Panja, Polyhedron, 2014, 80, 81–89 CrossRef;
(h) K. Ghosh, S. Roy, A. Ghosh, A. Banerjee, A. Bauzá, A. Frontera and S. Chattopadhyay, Polyhedron, 2016, 112, 6–17 CrossRef.
- P. H. Rieger, in Electron Spin Resonance: Analysis and Interpretation; Royal Society of Chemistry, Cambridge, U.K., 2007 Search PubMed.
-
(a) K. Ghosh, M. G. B. Drew and S. Chattopadhyay, Inorg. Chim. Acta, 2018, 482, 23–33 CrossRef;
(b) A. Biswas, L. K. Das, M. G. B. Drew, G. Aromí, P. Gamez and A. Ghosh, Inorg. Chem., 2012, 51, 7993–8001 CrossRef PubMed;
(c) R. Wanke, L. Benisvy, M. L. Kuznetsov, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Chem.–Eur. J., 2011, 17, 11882–11892 CrossRef PubMed.
-
(a) P. Chakraborty, J. Adhikary, B. Ghosh, R. Sanyal, S. K. Chattopadhyay, A. Bauzá, A. Frontera, E. Zangrando and D. Das, Inorg. Chem., 2014, 53, 8257–8269 CrossRef PubMed;
(b) P. Adak, B. Ghosh, A. Bauzá, A. Frontera, A. J. Blake, M. Corbella, C. D. Mukhopadhyay and S. K Chattopadhyay, RSC Adv., 2016, 6, 86851–86861 RSC;
(c) S. Dasgupta, I. Majumder, P. Chakraborty, E. Zangrando, A. Bauzá, A. Frontera and D. Das, Eur. J. Inorg. Chem., 2017, 133–145 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1548132 and 1548133 contain the supplementary crystallographic data for 1 and 2 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra03035a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Antonio Frontera
b,
Antonio Frontera