
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlack TiO2 nanotube arrays fabricated by electrochemical self-doping and their photoelectrochemical performance
Liyan Zhu,
Hongchao Ma†
 *,
Huibin Han,
Yinghuan Fu†*,
Chun Ma,
Zhihui Yu and
Xiaoli Dong
*,
Huibin Han,
Yinghuan Fu†*,
Chun Ma,
Zhihui Yu and
Xiaoli Dong
School of Light Industry & Chemical Engineering, Dalian Polytechnic University, No. 1 Qinggongyuan, Ganjingzi District, Dalian, P. R. China. E-mail: m-h-c@sohu.com; fuyinghuan@sina.com; Fax: (+86)-411-86323736; Tel: (+86)-411-86323508
First published on 23rd May 2018
Abstract
Herein, black TiO2 nanotube arrays (NTAs) were fabricated using electrochemical self-doping approaches, and characterized systemically by scanning electron microscopy (SEM), powder X-ray diffraction (XRD), UV-visible absorption spectroscopy and photoluminescence spectroscopy (PL). The as-obtained black TiO2 nanotube arrays (NTAs) exhibited stronger absorption in the visible-light region, a better separation rate of light-induced carriers, and higher electrical conductivity than TiO2 nanotube arrays (NTAs). These characteristics cause black TiO2 nanotube array (NTA) electrodes to have higher photoelectrocatalytic activity for degrading anthraquinone dye (reactive brilliant blue KN-R) than the TiO2 nanotube array (NTA) electrode. Furthermore, a synergetic action between photocatalysis and electrocatalysis was also observed. The black TiO2 nanotube array (NTA) electrode is considered to be a promising photoanode for the treatment of organic pollutants.
1. Introduction
Recently, TiO2-based active materials (especially, TiO2 nanotube arrays (NTAs)) have attracted a great deal of attention for the photocatalytic/photoelectrocatalytic degradation of organic pollutants, and energy storage, because their ordered nanotube architecture can provide a unidirectional electric channel for the photogenerated electrons, a large internal surface area, and high ion changeability.1–7 However, some problems associated with their wide band gap (3.0–3.2 eV) and the fast recombination of photogenerated carriers still needs to be solved to enable practical application of TiO2 NTAs.More effort has been made to enhance the light absorption, and to suppress the recombination of electron/hole pairs, such as, doping (non-metal and metal elements),8–12 noble metal particle decoration,13–18 and heterostructure construction.19–21 Other than the above strategies, photoelectrocatalysis (PEC) has received the most attention as a facile and efficient route to overcome the fast recombination between the photoinduced carriers in TiO2 NTAs.22,23 During the PEC process, a low positive bias was applied to the TiO2 NTAs, which significantly facilitated the transfer of photocarriers and improved the separation of the photoinduced carriers. However, it should be noted that the electrooxidation features of TiO2 NTAs for the degradation of organic pollutants have not attracted more attention, which may be attributed to their semiconducting nature and poor electronic conductivity. Thus, the development of methods to improve the electrocatalytic performance of TiO2 NTAs, to achieve synergy between photocatalysis and electrocatalysis, and to enhance the photoelectrocatalytic ability of TiO2 NTAs has become a significant challenge in the field of environmental remediation.
In recent years, reduced titanium oxide (or black TiO2) has attracted increasing attention for use in photovoltaics and photocatalysis, due to its narrower bandgap and higher electrical conductivity.24–26 Some reports indicated that reduced TiO2 NTAs not only exhibited excellent photocatalytic activity, but also demonstrated capacitive/oxidant generating properties due to the self-doping of TiO2 with Ti3+/oxygen vacancies.27–30 Therefore, reduced TiO2 NTAs are considered to be a promising photoanode material to achieve synergetic photoelectrochemical action in the PEC process. Nevertheless, thus far no information has been reported regarding their electrooxidation or PEC properties in the degradation of organic pollutants. Previous reports have mainly focused on electro-assisted photocatalytic processes of photocatalysts, in which electro-oxidation does not occur; only a few have focused on the electrooxidation processes during the PEC process. Thus, the aim of present work was to integrate the photooxidation and electrooxidation for black TiO2 NTAs as photoanode in the PEC degradation of organic pollutants. Our results indicated that the efficient removal of organic pollutants over the black TiO2 NTAs photoanode can be ascribed to the synergetic action between photocatalysis and electrocatalysis in the PEC process.
2. Experimental section
2.1 Materials
Titanium sheets (99.7% pure) were obtained from Yunjie Metal Company (China). The ethylene glycol, acetone, ethanol, sodium sulfate, NH4F, and reactive brilliant blue KN-R (from Tianjin Chemical Reagents Company) were of analytical grade. Deionized water was used as a solvent to prepare all solutions.2.2 Preparation of pristine TiO2 nanotube arrays
First, Ti sheets (10 mm × 10 mm × 1 mm in size) were cleaned in acetone and ethanol under ultrasonication, subsequently immersed in an aqueous oxalic acid solution (10%, 80 °C) for 2 h, and then washed using deionized water. A pretreated Ti sheet was used as the anode and an untreated Ti sheet was used as the cathode. The electrode sheets were placed vertically with a separation of 30 mm in an ethylene glycol electrolyte containing H2O (5 wt%) and NH4F (0.27 wt%) under a constant potential (40 V) for 2 h at room temperature (25 °C). Subsequently, the as-anodized TiO2 NTAs were washed in ethanol for 10 min and calcined at 400 °C for 2 h under ambient conditions.2.3 Preparation of black TiO2 nanotube arrays
Preparation of black TiO2 was performed using the electrochemical doping procedure suggested in ref. 31. In a typical procedure, the as-obtained pristine TiO2 NTAs were used as the cathode, and a Ti sheet with the same area was selected as the counter electrode. To obtain the black TiO2 NTAs, electrochemical reduction was performed at a set voltage (−30, −35, −40, −45 and −50 V) for a certain time (15, 20, 30, 60 and 120 min) in an ethylene glycol solution containing H2O (5 wt%) and NH4F (0.27 wt%).2.4 Activity test
The degradation of reactive brilliant blue KN-R in an aqueous solution with a concentration of 60 mg L−1 (200 mL of solution with 0.1 M Na2SO4 as a supporting electrolyte) in a quartz reactor was used to evaluate the photo-electrocatalytic activity of the electrodes. The black TiO2 NTAs anode and the titanium sheet cathode were immersed vertically in the solution, with a distance of 3 cm between the electrodes. Before the experiments, an adsorption–desorption equilibrium of the dye on the surface of the electrodes was established by magnetic stirring for 30 min under dark conditions. Electrochemical degradation was performed using a DC power supply, and a 175 W xenon lamp was selected as the light-source for the photocatalytic procedure. The xenon lamp was placed in a circulating water jacket to cool the reaction vessel. The reaction solution was sampled at given time intervals and centrifuged to remove the photocatalyst particles. A UV-visible spectrophotometer (UV-1800PC, MAPADA, China) was used for measuring the absorbance of solution and the degradation rate (R0) of KN-R was estimated by the following equation:| R0 = (A0 − At)/A0 × 100% |
2.5 Characterizations
Electrochemical tests were carried out using a CHI 660E potentiostat/galvanostat (ChenHua Instruments Co. Shanghai, China) with a standard three-electrode system. A platinum sheet and a saturated calomel electrode were selected as counter and reference electrodes. The morphologies of the samples were observed using a Hitachi-1510 scanning electron microscope (SEM) (Hitachi, Japan). X-Ray Diffraction (XRD) of the electrodes was performed using a SHIMADZU XRD-6100 X-ray diffractometer with Cu Kα radiation (λ = 0.15406 nm) at 40 kV and 30 mA. X-Ray photoelectron spectroscopy (XPS) experiments were carried out using a VG EscaLab 250 electron spectrometer (Thermo VG) with Al Kα X-ray radiation (1486.6 eV). The calibration of the binding energies was performed using the C 1s peak (284.6 eV). The UV-vis absorption spectra of the electrodes were obtained using a spectrophotometer (Varian Cary-100) with BaSO4 as a standard. In this work, the fluorescence spectrophotometry was used to measure the amount of hydroxyl radical (˙OH) originating from the photoelectrocatalytic process.32–34 The fluorescence experiments were carried out using a fluorescence spectrophotometer (Hitachi F-7000, Japan).3. Results and discussion
Fig. 1 shows the XRD patterns of the black TiO2-NTAs as a function of electrochemical reduction times and voltages. For reference, the XRD pattern of the TiO2-NTAs is also included. As shown in Fig. 1, all the diffraction peaks of the TiO2-NTAs matched well with the anatase phase of TiO2 (JCPDS file no 21-1272) and with metallic Ti (JCPDS file no. 44-1294). No obvious change to the diffraction peaks of the pristine TiO2-NTAs was observed after electrochemical reduction. The crystal structure of the pristine TiO2-NTAs (crystalline anatase TiO2) was also unchanged by the electrochemical reduction. Nevertheless, the XPS spectra of the pristine TiO2 NTAs and black TiO2 NTAs (see Fig. 1c) exhibited visible differences. The positions of the Ti 2p and O 1s peaks for the black TiO2 NTAs are shifted to lower binding energy than that of the pristine TiO2 NTAs. The negative shift in binding energy of the black TiO2 NTAs can be attributed to the increased presence of Ti3+ and oxygen vacancies compared with pristine TiO2 NTAs.29,35 Fig. 1d shows the UV-Vis absorption spectra of the TiO2 NTAs and black TiO2 NTAs samples. The inset of Fig. 1d showed that the pristine TiO2 NTAs is gray color, and its corresponding coloration is blue-black color after cathodic polarization. The pristine TiO2 NTAs showed intrinsic absorption of TiO2 at wavelengths below 400 nm. The visible-light absorption of the TiO2 NTAs may be ascribed to the light scattering caused by pores or cracks in the nanotube arrays.36–38 In contrast, the black TiO2 NTAs sample exhibited stronger light-absorption than the pristine TiO2 NTAs over the entire ultraviolet-visible region. Additionally, a notable red shift was observed for the black TiO2 NTAs. The absorption edge of the black TiO2 NTAs was shifted to 396 nm, whereas the TiO2 NTAs exhibited an absorption edge of 376 nm. The black TiO2 NTAs also showed an additional absorption peak from 400 nm to the near-infrared region. Obviously, the distinct differences between optical absorption behavior of the TiO2 NTAs and black TiO2 NTAs samples may be correlated with the presence of vacancies (Ti3+ sites formed by cathodic polarization39,40) as a result of a trap state.41,42 These results are also in agreement with those of previous studies.27,43–45 Nevertheless, the strong light-absorption of the black TiO2 NTAs is beneficial to improve their photocatalytic performance.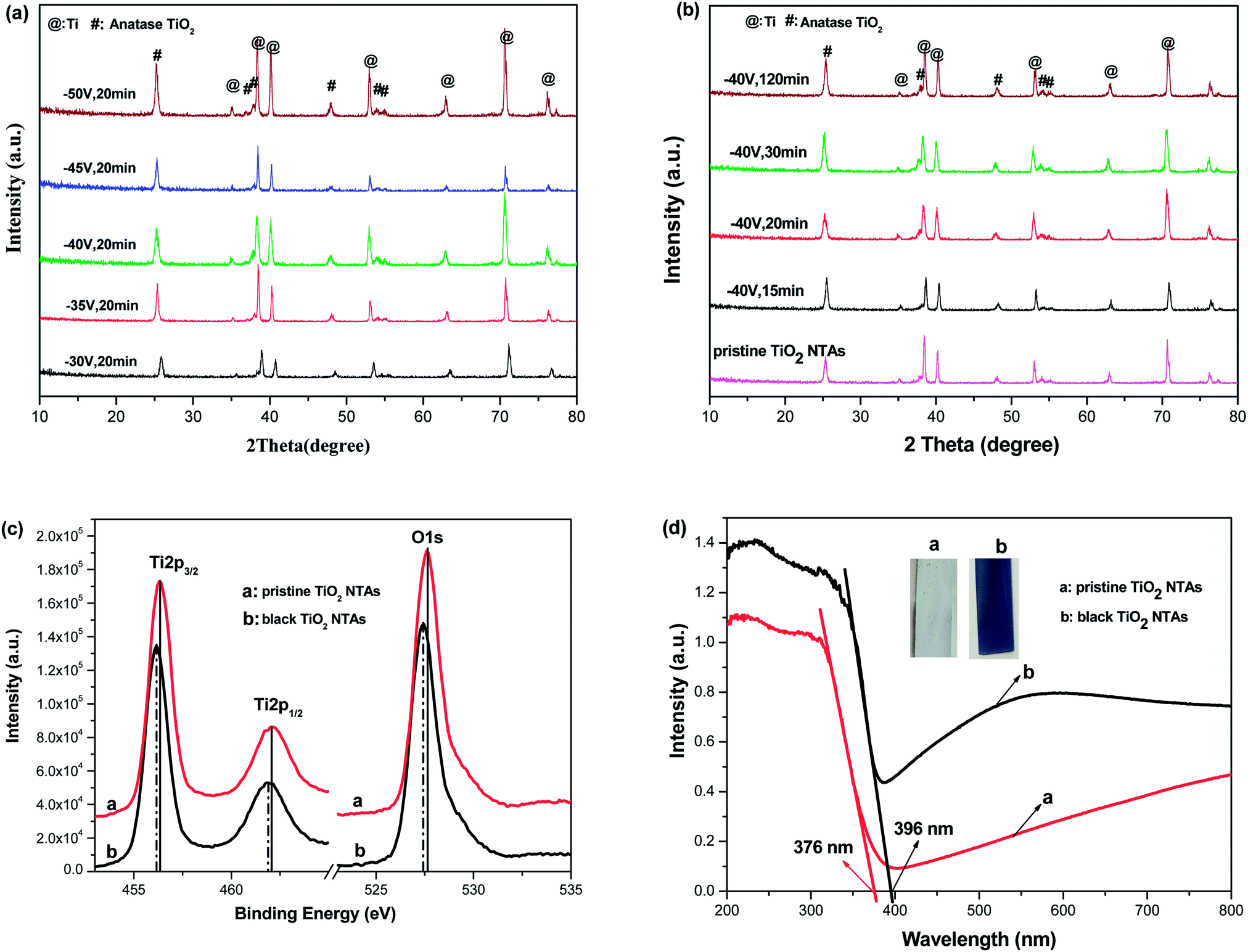 | ||
| Fig. 1 XRD patterns of black TiO2 NTAs prepared by electrochemical reduction under various reducing potential (a) and time (b); XPS (c) and UV-visible absorption spectra (d) of the pristine TiO2 NTAs and black TiO2 NTAs samples. | ||
In order to identify the effect of electrochemical reduction on the morphology and structure of the TiO2-NTAs, SEM measurement was performed (see Fig. 2). Fig. 2 shows that the TiO2 NTAs had a regular nanotube-structure with a pore diameter of ca. 100 nm, and a length and wall thickness of about 20 nm and 1.0 μm, respectively. The diameter and length of the TiO2 nanotubes did not change after electrochemical reduction. However, apparent damage to the pore structure of the TiO2 NTAs was observed when the reduction time and potential were increased to 20 min at −50 V and 60 min at −40 V. The damage to the pore structure should result in a decrease of electrocatalytic performance for the TiO2 NTAs due to their reduced electrochemically active areas. The composition of each TiO2 NTA was determined using energy-dispersive X-ray spectroscopy (EDX), and the results are also shown in Fig. 2. As can be seen the Ti/O ratio gradually increased as the time and voltage of the electrochemical reduction were increased. This result implied that electrochemical reduction should change the conductive properties of TiO2 and influence the PEC activity of the TiO2 NTAs.
 | ||
| Fig. 2 SEM observation and the corresponding EDX pattern for the pristine TiO2 and black TiO2 NTAs samples. | ||
The capacitance of the TiO2 NTA and black TiO2 NTA electrodes was determined using the Mott–Schottky equation:
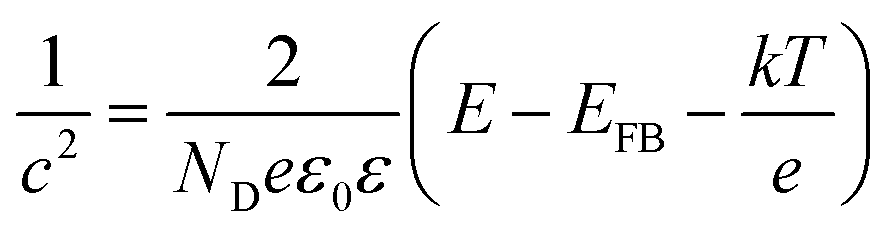
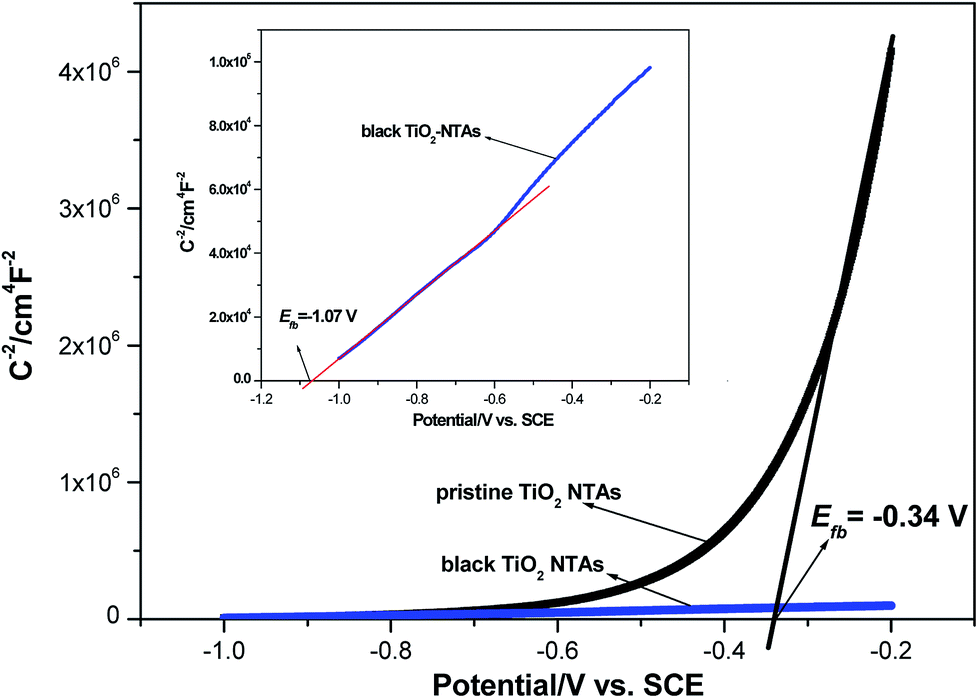 | ||
| Fig. 3 Mott–Schottky plots of TiO2 NTAs and black TiO2 NTAs electrodes. | ||
Additionally, the ND value was calculated from the plots in Fig. 3 to evaluate the semiconductor characteristic behavior using the following equation:
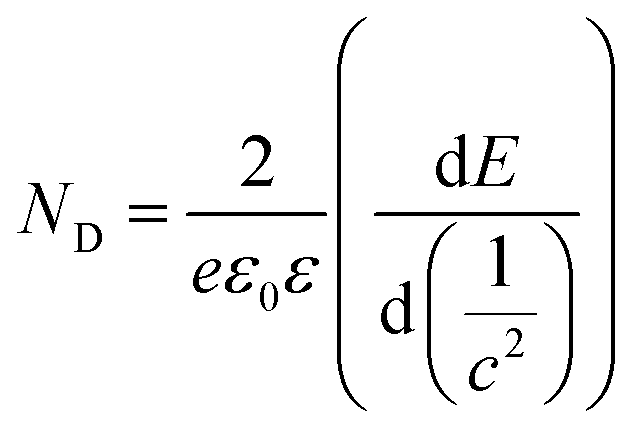
To investigate the photoelectrochemical behavior of the black TiO2 NTA electrodes prepared using various reduction potentials and times, current–voltage measurements were carried out in a 0.2 M Na2SO4 solution under solar-light illumination. The LSV photocurrent responses of the different photoelectrodes (see Fig. 4) indicated that the dark-current response of the black TiO2 NTA electrodes was negligible, which indicated that no electrochemical oxidation occurred. However, a notable enhancement of the photocurrent was observed for all photoelectrodes under illumination, and the photocurrent was potential-dependent. The high photocurrent of the semiconductor reflected the high separation and transfer efficiency of photogenerated charge carriers.52 As shown in Fig. 4, the black TiO2 NTA electrodes showed higher photocurrent density than the TiO2 NTA reference. This result indicated that the black TiO2 NTA electrodes have higher separation and transfer efficiency of photogenerated carriers than the TiO2 NTA reference.
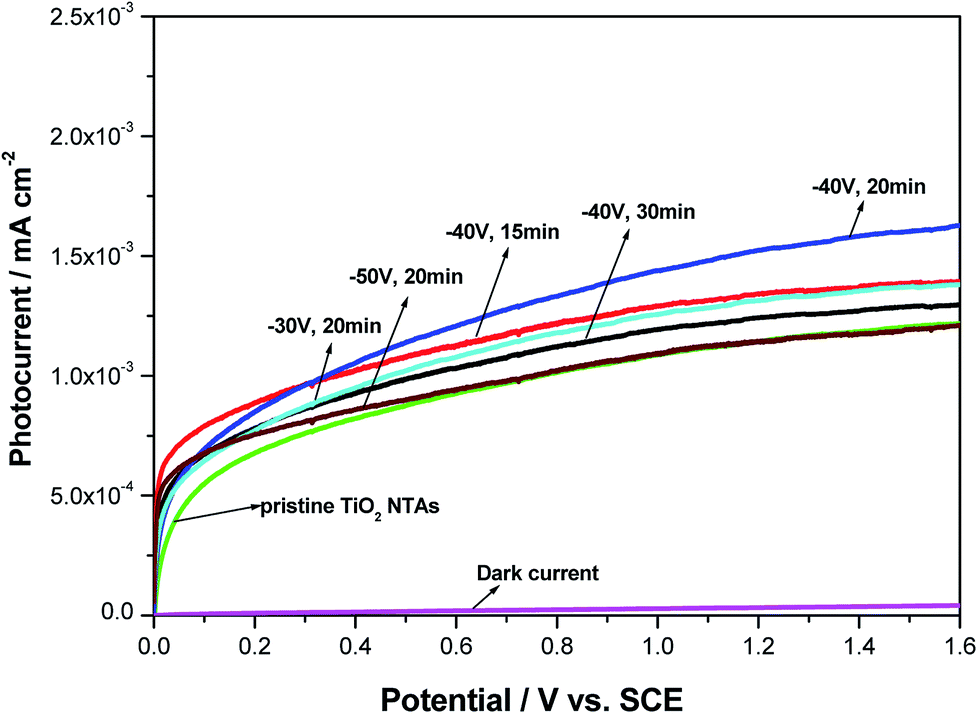 | ||
| Fig. 4 Photocurrent–potential curves of the pristine TiO2 and black TiO2 NTAs electrodes prepared using different electrochemical reduction potentials and times. | ||
To evaluate the electrochemical properties of the black TiO2 NTAs, cyclic voltammograms were recorded in a 0.5 M Na2SO4 aqueous solution. As shown in Fig. 5, the TiO2 NTA curve showed a strong correlation between the current and potential due to their n-type semiconductor properties. The high resistances of the TiO2 NTAs at higher potentials led to a decrease in the current, resulting in the pinched shape of the curve. In contrast with the TiO2 NTAs, the black TiO2 NTA samples exhibited capacitive curves with rectangular shapes.28,53 A large, rectangular CV curve is clearly observed for the black TiO2 NTA electrodes at higher current, which indicated that the black TiO2 NTA samples had high conductivity.27 The enhanced conductivity of the black TiO2 NTA can be attributed to the trapped electrons at the Ti3+ sites generated by cathodic polarization.39,40
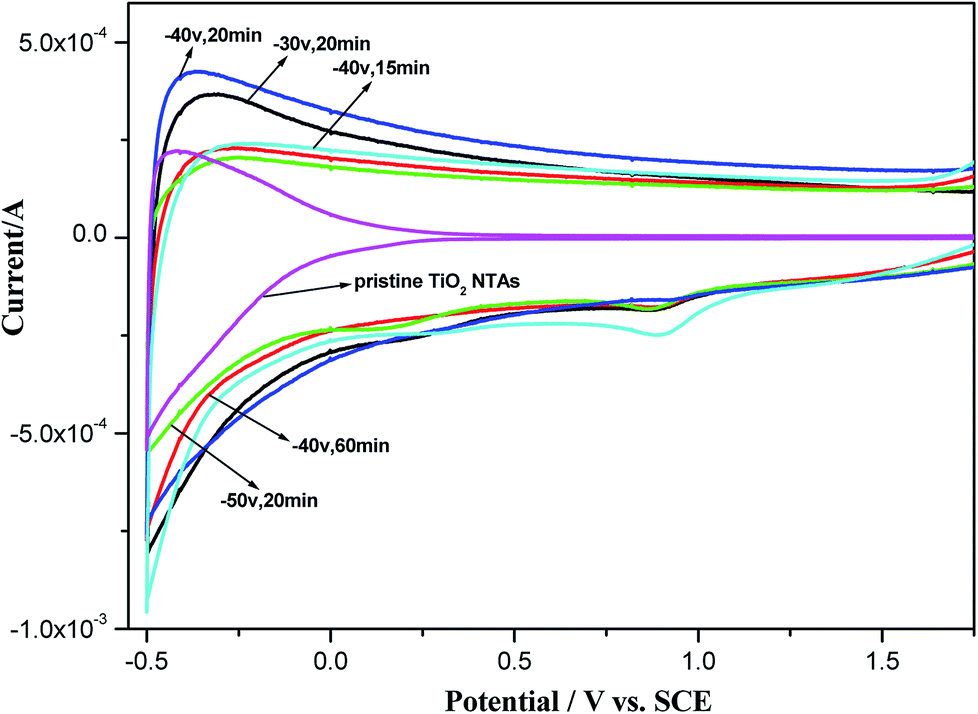 | ||
| Fig. 5 Cyclic voltammograms of the pristine TiO2 and black TiO2 NTA electrodes prepared using different electrochemical reduction potentials and times. | ||
As reported in the previous studies,32–34 amount of ˙OH (from the electro-catalytic decomposition of water on the electrode surface) could be detected using the analysis of the fluorescence spectra (benzoic acid had a known reaction with ˙OH in aqueous media and the product is p-hydroxybenzoic acid (p-HBA). The p-HBA could be detected using fluorescence spectroscopy). To investigate amount of hydroxyl radical (˙OH), the electrolysis of benzoic acid solution was performed in present work. Initial concentration of benzoic acid is 200 mg L−1 and 0.5 mol L−1 Na2SO4 was used as a supporting electrolyte. During electrolysis, the sample was collected and analyzed at a desired time interval. The fluorescence intensity of solution was measured at 440 nm with excitation at 325 nm by a fluorescence spectrophotometer. Fig. 6 shows the photoluminescence (PL) spectra for the photoelectrolysis of the benzoic acid solution over the TiO2 NTA and black TiO2 NTA electrodes. As seen in Fig. 6, the fluorescence intensity of the black TiO2 NTA electrodes was higher than that of the TiO2 NTA electrode at all times, indicating that the black TiO2 NTA electrodes generated more ˙OH. The PL results demonstrated that the black TiO2 NTAs had better current efficiency for the generation of hydroxyl radicals. Obviously, the higher efficiency for the generation of hydroxyl radicals was favorable for the photoelectrocatalytic degradation of organic pollutants.
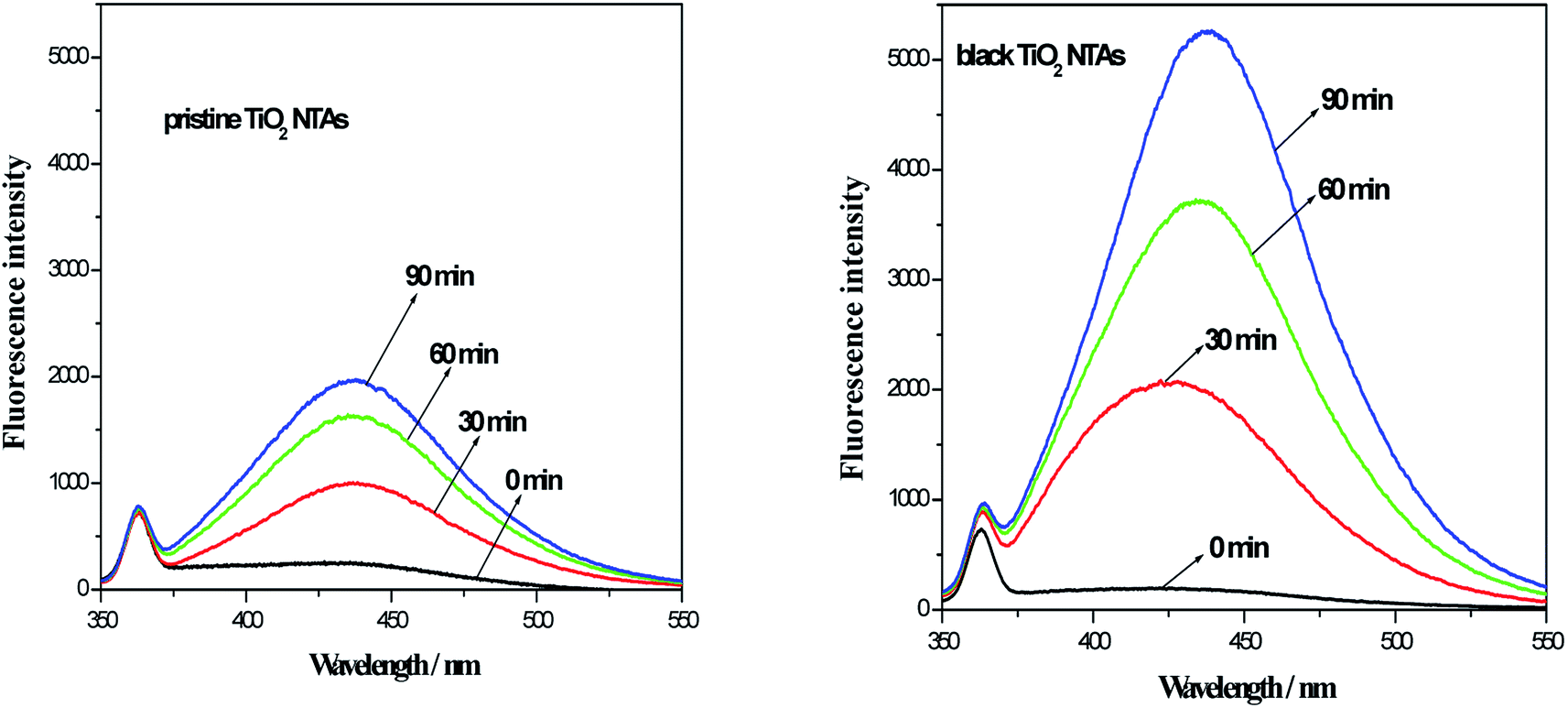 | ||
| Fig. 6 Fluorescence spectra of the TiO2 NTA and black TiO2 NTA electrodes during the photoelectrolysis of benzoic acid (200 mg L−1 benzoic acid and 0.5 mol L−1 Na2SO4 as a supporting electrolyte). | ||
The photo-electrocatalytic (PEC) degradation efficiency of KN-R over the black TiO2 NTA electrodes prepared using different reduction potentials and times was investigated, as shown in Fig. 7a and b. The degradation rate of KN-R over the black TiO2 NTA electrodes depended on the reduction voltage and time; and the black TiO2 NTA electrode prepared at −40 V for 20 min exhibited the highest KN-R removal ability. However, excessive electrochemical reduction treatment markedly damaged the pore structure of the TiO2 NTAs, which resulted in a decline in the catalytic performance of the TiO2 NTA electrodes. The effect of the anodic potential on the PEC performance of the black TiO2 NTAs for the degradation of KN-R was also investigated. As shown in Fig. 8c, the PEC degradation rate increased gradually with increasing anodic bias in the potential range of 0–6.0 V. Furthermore, to explore the effect of electro-oxidation on the KN-R removal, a set of pure electro-oxidation measurements were carried out by applying an anodic bias (0–6.0 V) in dark conditions. The electrochemical oxidation of KN-R was not observed when the anodic potential was lower than 3.0 V. However, the electrochemical oxidation of KN-R was non-negligible when the anodic potential was greater than 3.0 V, and KN-R degradation was significantly enhanced at higher potentials. A similar phenomenon was observed in previous work.54 Fig. 7d shows the degradation rates under various degradation conditions (the electrocatalytic (EC), photocatalytic (PC) and photo-electrocatalytic (PEC) processes) for the black TiO2 NTAs prepared at −40 V for 20 min. As shown in Fig. 7d, the PEC degradation rate of KN-R over the black TiO2 NTAs electrode was larger than the sum of both the EC and PC rates, which suggested that an obvious synergetic effect occurred during the PEC process for the black TiO2 NTAs electrode. The black TiO2 NTAs electrode exhibited considerable electrocatalytic oxidation of KN-R due to the excellent electrochemical characteristics of the black TiO2 NTAs. For comparison, the degradation rate of the TiO2 NTAs electrode was also measured under PEC, EC and PC conditions. Fig. 7 shows that a significant synergetic effect also existed in the PEC process for the TiO2 NTAs electrode. However, the TiO2 NTAs electrode did not demonstrate electrocatalytic degradation of KN-R at an anodic potential of 6.0 V. Based on the above results, it can be demonstrated that the black TiO2 NTAs exhibited better PEC, EC and PC performances than the TiO2 NTAs.
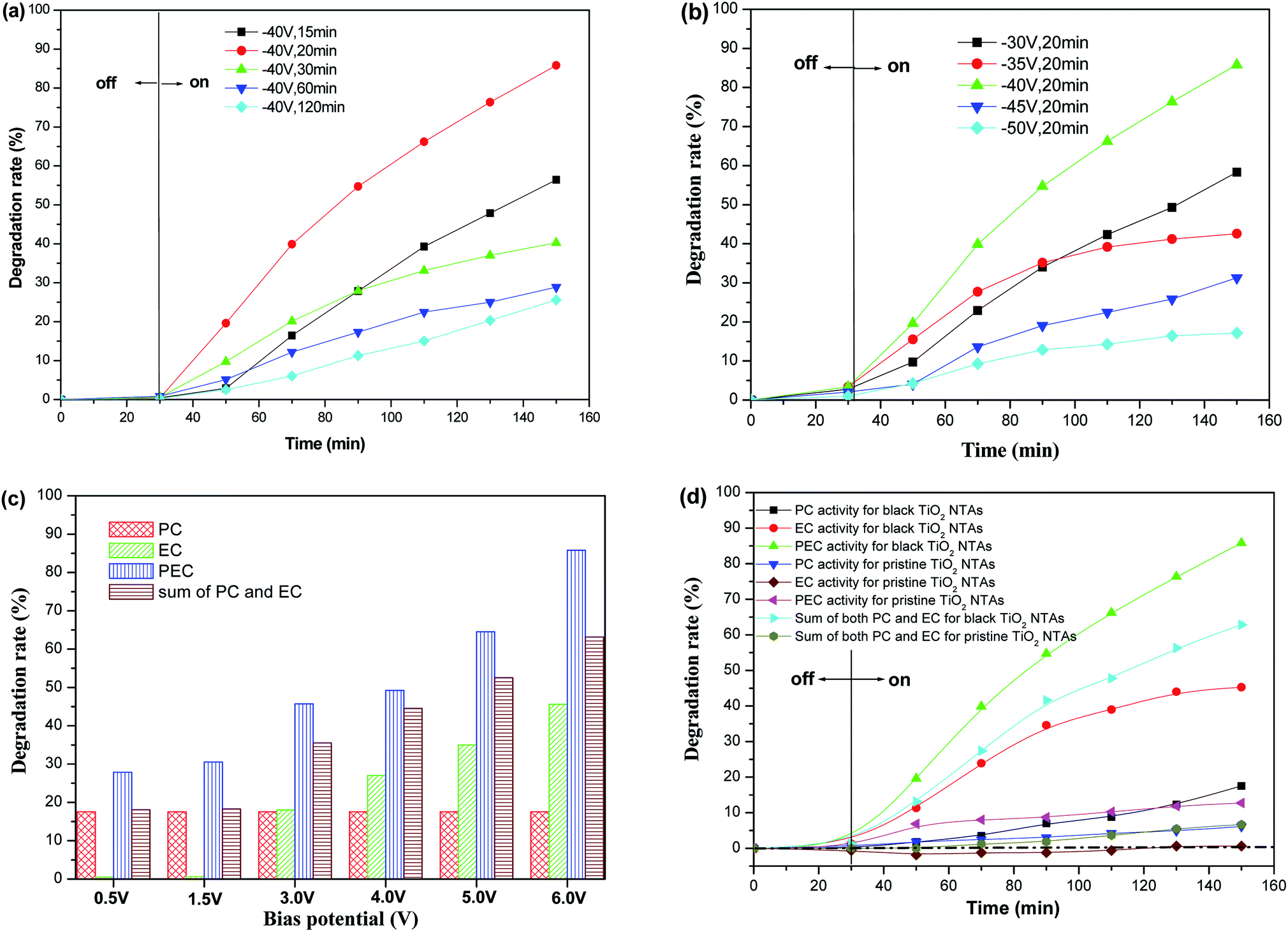 | ||
| Fig. 7 (a and b) Decolorization efficiencies of KN-R over the black TiO2 NTA electrodes prepared using different electrochemical reduction potentials and times (c) effect of the applied bias on the degradation rate of KN-R over the black TiO2 NTA electrodes (d) efficiencies of the different degradation processes of KN-R over the TiO2 NTAs and black TiO2 NTA electrodes. | ||
 | ||
| Fig. 8 Results of the recycling experiments for the black TiO2 NTA electrodes for three cycles. | ||
Based on the above results, a schematic of the PEC process on the black TiO2 NTAs is illustrated in Scheme 1. When anodic potential is lower than 3.0 V (greater than the flatband potential of the black TiO2 NTAs), only photocatalytic degradation occurred during the PEC process. Thus, the enhancement of KN-R degradation under these conditions was mainly a result of charge separation by the electric field (electro-assisted photocatalysis). When the anodic potential was increased above 3.0 V, both photocatalytic and electrochemical degradation for KN-R occurred during the PEC process. In this case, the enhancement of KN-R degradation resulted mainly from the combination of electro-oxidation and electro-photocatalysis.
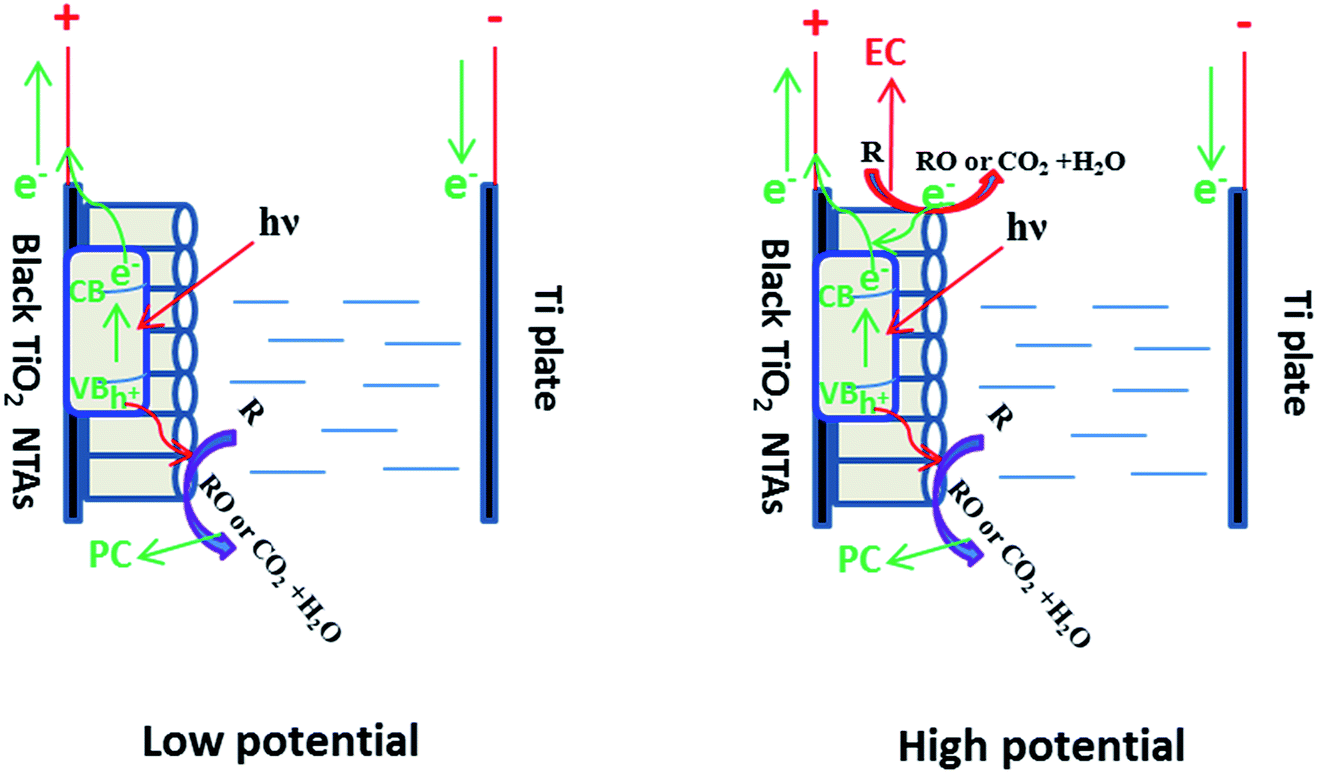 | ||
| Scheme 1 Diagram of the PEC process over the black TiO2 NTA electrode under solar light irradiation. | ||
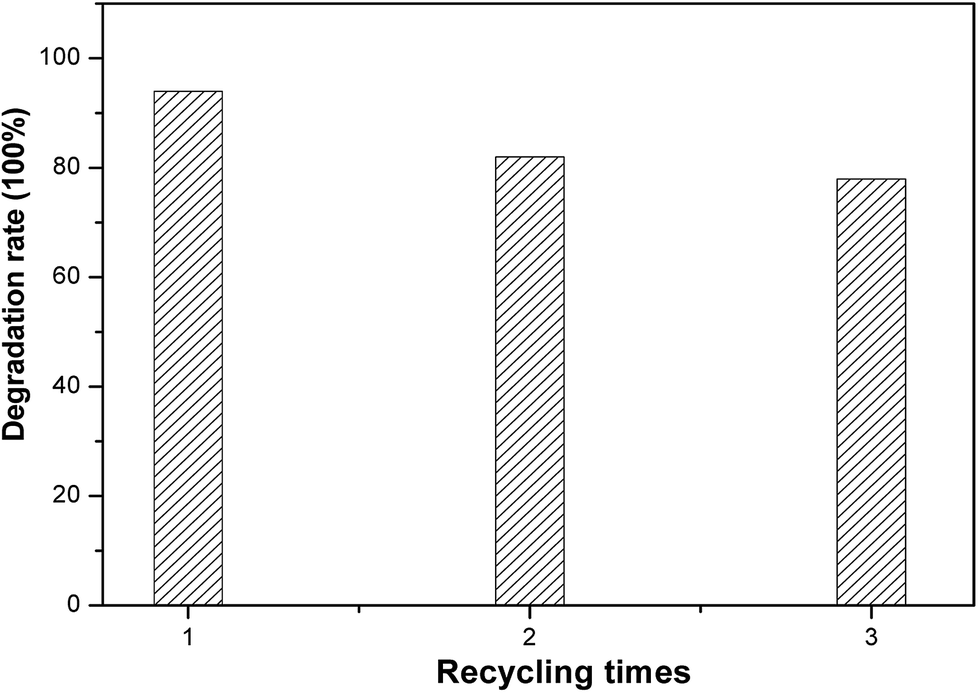
The service life of electrodes is a significant factor for their practical application. To estimate the electrochemical stability of black TiO2 NTA electrodes, recycling tests of the degradation of KN-R were carried out under solar light radiation, as shown in Fig. 8. The black TiO2 NTA electrode still exhibited a considerable removal rate (about 78%) for reactive brilliant blue KN-R after three cycles. The above results indicated that the black TiO2 NTAs can be used as an excellent PEC electrode.
4. Conclusions
In summary, highly oriented black TiO2 NTA photoanodes were fabricated using an anodization-electrochemical reduction approach. The as-obtained photoanodes showed stronger light absorption throughout the entire UV-Vis region, a more negative flatband potential, and better separation and transfer efficiency of photoinduced carriers than a conventional TiO2 NTA electrode. It was found that the photo-electrocatalytic removal of KN-R on black TiO2 NTA electrodes can be divided into two regimes, namely, electro-assisted photocatalysis at low bias and a combination of electro-assisted photocatalysis and electrochemical oxidation at high bias. The black TiO2 NTA electrode prepared by electrochemical reducing at −40 V for 20 min, exhibited a highest rate of KN-R degradation. This may be ascribed to the compromise action of conductivity and pore structure for black TiO2 NTAs under electrochemical reduction. Obviously, the black TiO2 NTAs can be suggested as a potential cost effective anode material in the field for treatment organic wastewater.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21476033), Program for Liaoning Excellent Talents in University (LR2014013), the Natural Science Foundation of Liaoning Province (20170520427) and Science and Technology Foundation of Liaoning Province (201602052).References
- X. Quan, X. L. Ruan, H. M. Zhao, S. Chen and Y. Z. Zhao, Environ. Pollut., 2007, 147, 409–414 CrossRef PubMed.
- X. Quan, S. G. Yang, X. L. Ruan and H. M. Zhao, Environ. Sci. Technol., 2005, 39, 3770–3775 CrossRef PubMed.
- J. M. Macak, M. Zlamal, J. Krysa and P. Schmuki, Small, 2007, 3, 300–304 CrossRef PubMed.
- S. P. Albu, A. Ghicov, J. M. Macak, R. Hahn and P. Schmuki, Nano Lett., 2007, 7, 1286–1289 CrossRef PubMed.
- H. F. Zhuang, C. J. Lin, Y. K. Lai, L. Sun and J. Li, Environ. Sci. Technol., 2007, 41, 4735–4740 CrossRef PubMed.
- R. Genc, M. O. Alas, E. Harputlu, S. Repp, N. Kremer, M. Castellano, S. G. Colak, K. Ocakoglu and E. Erdem, Sci. Rep., 2017, 7, 11222 CrossRef PubMed.
- S. Repp, E. Harputlu, S. Gurgen, M. Castellano, N. Kremer, N. Pompe, J. Wörner, A. Hoffmann, R. Thomann, F. M. Emen, S. Weber, K. Ocakoglu and E. Erdem, Nanoscale, 2018, 10, 1877–1884 RSC.
- L. Sun, J. Li, C. L. Wang, S. F. Li, H. B. Chen and C. J. Lin, Sol. Energy Mater. Sol. Cells, 2009, 93, 1875–1880 CrossRef.
- J. Y. Li, N. Lu, X. Quan, S. Chen and H. M. Zhao, Ind. Eng. Chem. Res., 2008, 47, 3804–3808 CrossRef.
- J. Li, H. Yun and C. J. Lin, J. Electrochem. Soc., 2007, 154, C631–C637 CrossRef.
- E. Erdem, K. Kiraz, M. Somer and R. A. Eichel, J. Eur. Ceram. Soc., 2010, 30, 289–293 CrossRef.
- H. Rumpf, H. Modrow, J. Hormes, H. J. Gläsel, E. Hartmann, E. Erdem, R. Böttcher and K. H. Hallmeier, J. Phys. Chem. B, 2001, 105, 3415–3421 CrossRef.
- Y. H. Tseng, I. G. Chang, Y. A. Tai and K. W. Wu, J. Nanosci. Nanotechnol., 2012, 12, 416–422 CrossRef PubMed.
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676–1680 CrossRef PubMed.
- T. Yamaguchi, E. Kazuma, N. Sakai and T. Tatsuma, Chem. Lett., 2012, 41, 1340–1342 CrossRef.
- M. Ye, J. Gong, Y. Lai, C. Lin and Z. Lin, J. Am. Chem. Soc., 2012, 134, 15720–15723 CrossRef PubMed.
- Z. C. Lian, W. C. Wang, S. N. Xiao, X. Li, Y. Y. Cui, D. Q. Zhang, G. S. Li and H. X. Li, Sci. Rep., 2015, 5, 10461–10471 CrossRef PubMed.
- K. P. Xie, L. Sun, C. L. Wang, Y. K. Lai, M. Y. Wang, H. B. Chen and C. J. Lin, Electrochim. Acta, 2010, 55, 7211–7218 CrossRef.
- C. L. Wang, L. Sun, H. Yun, J. Li, Y. K. Lai and C. J. Lin, Nanotechnology, 2009, 20, 295601–295606 CrossRef PubMed.
- Y. Hou, X. Y. Li, X. J. Zou, X. Q. An and G. H. Chen, Environ. Sci. Technol., 2009, 43, 858–863 CrossRef PubMed.
- G. P. Dai, J. G. Yu and G. Liu, J. Phys. Chem. C, 2011, 115, 7339–7346 Search PubMed.
- Y. B. Xie, L. M. Zhou and H. T. Huang, Mater. Lett., 2006, 60, 3558–3560 CrossRef.
- Z. H. Zhang, Y. Yuan, G. Y. Shi, Y. J. Fang, L. H. Liang, H. C. Ding and L. T. Jin, Environ. Sci. Technol., 2007, 41, 6259–6263 CrossRef PubMed.
- N. Alberto, A. Mattia, S. Saveria, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi and R. Psaro, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef PubMed.
- N. Liu, C. Schneider, D. Freitag, M. Hartmann, U. Venkatesan, J. Muller, E. Spiecker and P. Schmuki, Nano Lett., 2014, 14, 3309–3313 CrossRef PubMed.
- H. L. Cui, W. Zhao, C. Y. Yang, H. Yin, T. Q. Lin, Y. F. Shan, Y. Xie, H. Gu and F. Q. Huang, J. Mater. Chem. A, 2014, 2, 8612–8616 Search PubMed.
- C. Kim, S. Kim, J. Choi, J. Lee, J. S. Kang, Y. E. Sung, J. Lee, W. Choi and J. Yoon, Electrochim. Acta, 2014, 141, 113–119 CrossRef.
- C. Kim, S. Kim, J. Lee, J. Kim and J. Yoon, ACS Appl. Mater. Interfaces, 2015, 7, 7486–7491 Search PubMed.
- A. Q. Zhang, F. L. Gong, Y. H. Xiao, X. P. Guo, F. Li, L. Z. Wang, Y. Zhang and L. S. Zhang, J. Electrochem. Soc., 2017, 164(2), H91–H96 CrossRef.
- H. Wu, D. Li, X. Zhu, C. Yang, D. Liu, X. Chen, Y. Song and L. Lu, Electrochim. Acta, 2014, 116, 129–136 CrossRef.
- Q. Zheng, H. J. Lee, J. Lee, W. Choi, N. B. Park and C. Lee, Chem. Eng. J., 2014, 249, 285–292 CrossRef.
- M. E. Lindsey and M. A. Tarr, Chemosphere, 2000, 41, 409–417 CrossRef PubMed.
- M. L. And and M. A. Tarr, Environ. Sci. Technol., 2000, 34, 444–449 CrossRef.
- W. H. Yang, W. T. Yang and X. Y. Lin, Acta Phys.-Chim. Sin., 2012, 28, 831–836 Search PubMed.
- E. Erdem, P. Jakes, R. A. Eichel, D. C. Sinclair, M. Pasha and I. M. Reaney, Funct. Mater. Lett., 2010, 3, 65–68 CrossRef.
- J. G. Yu, H. G. Yu, B. Cheng, X. J. Zhao, J. C. Yu and W. K. Ho, J. Phys. Chem. B, 2003, 107, 13871–13879 CrossRef.
- H. M. Zhu, B. F. Yang, J. Xu, Z. P. Fu, M. W. Wen, T. Guo, S. Q. Fu, J. Zuo and S. Y. Zhang, Appl. Catal., B, 2009, 90, 463–469 CrossRef.
- X. X. Yu, J. G. Yu, B. Cheng and M. Jaroniec, J. Phys. Chem. C, 2009, 113, 17527–17535 Search PubMed.
- J. M. Macak, B. G. Gong, M. Hueppe and P. Schmuki, Adv. Mater., 2007, 19, 3027–3031 CrossRef.
- F. Fabregat-Santiago, E. M. Barea, J. Bisquert, G. K. Mor, K. Shankar and C. A. Grimes, J. Am. Chem. Soc., 2008, 130, 11312–11316 CrossRef PubMed.
- S. S. M. Lu, F. H. Pollak and P. M. Raccah, Phys. Rev. B, 1978, 17, 1970–1975 CrossRef.
- G. K. Mor, O. K. Varghese, M. Paulose and C. A. Grimes, Adv. Funct. Mater., 2005, 15, 1291–1296 CrossRef.
- Z. Wang, C. Y. Yang, T. Q. Lin, H. Yin, P. Chen, D. Y. Wan, F. F. Xu, F. Q. Huang, J. H. Lin, X. M. Xie and M. H. Jiang, Energy Environ. Sci., 2013, 6, 3007–3014 Search PubMed.
- H. Q. Tan, Z. Zhao, M. Niu, C. Y. Mao, D. P. Cao, D. J. Cheng, P. Y. Feng and Z. C. Sun, Nanoscale, 2014, 6, 10216–10223 RSC.
- S. Tominaka, Chem. Commun., 2012, 48, 7949–7951 RSC.
- R. Hahn, F. Schmidt-Stein, J. Salonen, S. Thiemann, Y. Y. Song, J. Kunze, V. P. Lehto and P. Schmuki, Angew. Chem., Int. Ed., 2009, 48, 7236–7239 CrossRef PubMed.
- H. Ge, H. Tian, Y. G. Zhou, S. Y. Wu, D. L. Liu, X. Z. Fu, X. M. Song, X. C. Shi, X. X. Wang and N. Li, ACS Appl. Mater. Interfaces, 2014, 6, 2401–2406 Search PubMed.
- H. A. Girard, N. Simon, D. Ballutaud and A. Etcheberry, C. R. Chim., 2008, 11, 1010–1015 CrossRef.
- H. Zhou and Y. R. Zhang, J. Phys. Chem. C, 2014, 118, 5626–5636 Search PubMed.
- A. I. Kontos, V. Likodimos, T. Stergiopoulos, D. S. Tsoukleris, P. Falaras, I. Rabias, G. Papavassiliou, D. Kim, J. Kunze and P. Schmuki, Chem. Mater., 2009, 21, 662–672 CrossRef.
- G. M. Wang, H. Y. Wang, Y. C. Ling, Y. C. Tang, X. Y. Yang, R. C. Fitzmorris, C. C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef PubMed.
- H. G. Kim, P. H. Borse, W. Choi and J. S. Lee, Angew. Chem., Int. Ed., 2005, 117, 4661–4665 CrossRef.
- H. Li, Z. H. Chen, C. K. Tsang, Z. Li, X. Ran, C. Lee, B. Nie, L. X. Zheng, T. Hung, J. Lu, B. C. Pan and Y. Y. Li, J. Mater. Chem. A, 2014, 2, 229–236 Search PubMed.
- C. He, X. Z. Li, N. Graham and Y. Wang, Appl. Catal., A, 2006, 305, 54–63 CrossRef.
Footnote |
| † School of Chemistry Engineering & Material, Dalian Polytechnic University, No. 1 Qinggongyuan, Ganjingzi District, Dalian 116034, P. R. China. |
| This journal is © The Royal Society of Chemistry 2018 |