
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a BiOCl1−xBrx@AgBr heterostructure with enhanced photocatalytic activity under visible light†
Chenghe Hua,
Xiaoli Dong *,
Yu Wang,
Nan Zheng,
Hongchao Ma and
Xiufang Zhang
*,
Yu Wang,
Nan Zheng,
Hongchao Ma and
Xiufang Zhang
School of Light Industry and Chemical Engineering, Dalian Polytechnic University, #1 Qinggongyuan, Dalian 116034, PR China. E-mail: dongxiaoli65@163.com
First published on 4th May 2018
Abstract
We present a facile approach to preparing a BiOCl1−xBrx@AgBr heterostructure using a two-step solvothermal method. Multiple characterisation techniques have been employed to investigate its morphology, structure, optical and electronic properties and photocatalytic performance. The photocatalytic activity of the BiOCl1−xBrx@AgBr heterostructure was sufficiently evaluated by adopting Reactive Blue KN-R as the target organic pollutant under visible light irradiation. The as-prepared BiOCl1−xBrx@AgBr exhibited much higher photocatalytic activity than BiOCl1−xBrx and BiOCl, which was ascribed to the movement of photogenerated electrons from AgBr to BiOCl1−xBrx, resulting in effective charge separation and transfer. Moreover, the modification of BiOCl1−xBrx with AgBr broadened the light absorption range, making the composite suitable for visible light excitation. The excellent photocatalytic performance provides potential opportunities to utilize BiOCl1−xBrx@AgBr for environmental purification and organic pollution treatment of water.
1. Introduction
Fossil fuels, as some of the most important sources of the energy, are utilised in an increasing number of fields such as energy sources, impetus transmission, machine manufacturing and chemical synthesis. However, effluent and solid waste typically contain a variety of organic and inorganic pollutants, which gives rise to a serious problem i.e. environmental pollution. Undoubtedly, semiconductor-based photocatalysis is regarded as one of the most efficient and economic techniques for solving this issue by utilizing solar energy to degrade pollutants into nontoxic products.1–5 It is now imperative to explore new and highly active photocatalysts with superior photocatalytic activity and good chemical stability. TiO2, as the model semiconductor photocatalyst, has been considered the most prominent material for widespread environmental applications, due to its excellent photocatalytic activity, low cost, nontoxicity, long-term chemical stability and photostability.6–11 However, lacking visible light absorption quite limits its practical applications. Therefore, the development of photocatalysts which are capable of a response to visible light have been important research topics. Among Bi-based materials, BiOCl has been widely researched due to its unique layered structure and optical properties. Recently, some researchers reported that the (001) facets of BiOCl might contribute to generating oxygen vacancies, and thus lead to high photocatalytic activity.12–17 However, the wide band gap and rapid recombination of electrons and holes restrict its application to some extent. Thus, it is necessary to improve the photocatalytic activity via enhancing the visible light harvest and restraining the charge combination, such as by nonmetal/metal doping or coupling with other semiconductors with suitable band gaps.18–28More recently, AgX (Cl, Br and I) has been regarded as a new photosensitive photocatalyst and has attracted much attention because of its high photocatalytic performance in organic pollutant degradation under sunlight irradiation. Due to its high absorption of visible light, AgX is the most promising candidate for coupling with other semiconductors to enhance their photocatalytic activity and stability of their composites.29–31 It has been reported that various semiconductors like InVO4,32 BiPO433 and TiO2 (ref. 34) coupled with AgX exhibits increased performance in eliminating organic pollutants. These results may be caused by the formation of heterojunctions with different semiconductor materials, which can facilitate interfacial charge transfer and decrease charge combination.
In previous reports, the AgBr/BiOCl heterostructure has been studied. Qi et al.35 prepared AgBr particles coupled with BiOCl composites with exposed (001) facets using solvothermal and chemical precipitation methods. The photocatalytic activity of the AgBr/BiOCl was much higher than that of pure AgBr and BiOCl, mainly due to the separation of photoinduced charges and greater number of oxygen vacancies. Chen et al.36 synthesized BiOCl-(001) nanosheets which were attached to AgBr colloidal spheres to form a AgBr/BiOCl-(001) hybrid. The composite exhibited excellent photocatalytic performance for degrading methyl orange and rhodamine B under LED irradiation, because of its response to full visible light and efficient separation of electron–hole pairs. Both of these reports provide an excellent theoretical basis for research into the AgBr/BiOCl photocatalyst, however the AgX agglomerates often lack a close connection with the original materials, resulting in increased recombination of electron–hole pairs. Therefore, synthesis of AgX with controlled shapes could improve the photocatalytic performance and stability of the hybrid.37
In this work, a BiOCl1−xBrx@AgBr heterostructure was successfully prepared via a solvothermal method. A series of measurements were used to obtain information on its crystalline structure, morphology, heterostructure interface, surface element composition, optical properties and charge transfer. AgBr could enhance the photocatalytic activity of BiOCl1−xBrx nanosheets under visible-light irradiation (λ > 420 nm) for degrading Reactive Blue KN-R. Furthermore, the BiOCl1−xBrx@AgBr displayed higher activity than AgBr spheres composited with the BiOCl1−xBrx hybrid (AgBr/BiOCl1−xBrx). Besides, the cycling experiment and the possible reaction mechanism of the BiOCl1−xBrx@AgBr heterostructure are discussed.
2. Experimental
2.1 Materials
Bismuth(III) chloride (AR) and silver nitrate (AR) were provided by Sinopharm Chemical Reagent Co., Ltd, cetyltrimethylammonium bromide (AR) was purchased from Tianjin Kemiou Chemical Reagent Co., Ltd, sodium hydroxide (AR) was supplied by Tianjin Guangfu Technology Development Co., Ltd and ethanol (GR) and ethylene glycol (AR) were obtained from Tianjin Fuyu Fine Chemical Co., Ltd, (GR). All reagents were used without further purification.2.2 Preparation of the BiOCl1−xBrx@AgBr heterostructure
2.3 Characterization
X-ray diffraction (XRD) patterns of the samples were recorded using a Shimadzu XRD-6100 diffractometer with Cu Kα radiation (λ = 0.145 nm), operating at 40 kV and 30 mA. X-ray photoelectron spectroscopy (XPS) was conducted on a VG Scientific ESCALAB250 XPS instrument. The diffuse reflectance UV-visible spectra (DRS) were recorded on a CARY 100 CONC spectrometer. The surface morphology and elemental composition of the samples were investigated using a scanning electron microscope (SEM, JSM-7800F, JEOL) equipped with an EDS detector. Transmission electron microscope (TEM) images were obtained using a JEOL JEM-2000EX.2.4 Photocatalytic activity measurements
The photocatalytic activity of the as-prepared samples was evaluated by degrading Reactive Blue KN-R (λ > 420 nm) under visible light irradiation, which was provided by a 300 W point light source coupled with a 420 nm cutoff filter. In each experiment, 20 mg of the catalyst was dispersed in 50 mL Reactive Blue KN-R solution (40 mg L−1). Prior to illumination, the suspension was magnetically stirred for 30 min in order to make quite sure the Reactive Blue KN-R reached adsorption and desorption equilibrium. Then, the suspension was exposed to visible light for 60 min with continuous stirring, and 5 mL samples of the aqueous suspension were withdrawn every 10 min. The mixture of Reactive Blue KN-R and catalyst powder was separated by centrifugation at 8000 rpm for 5 min and then filtrated using a Millipore filter with a diameter of 220 nm. Finally, the concentration variation of Reactive Blue KN-R was recorded using a UV-Vis spectrophotometer at 594 nm. The stability of the catalyst was measured with the following steps. After each run of the reaction, the catalyst was separated via centrifugation, washed with deionized water and ethanol and dried at 60 °C for 12 h. Each recycling experiment was carried out under the same conditions for 60 min and the concentration variation of Reactive Blue KN-R was determined using a UV-Vis spectrophotometer at 594 nm.2.5 Electrochemical tests
Photoelectrochemical measurements were carried out on a CHI660E electrochemical workstation (Shanghai Chenhua, P. R. China) operated with a three-electrode configuration and the as-prepared samples as the working electrode, Pt plate as the counter electrode and Ag/AgCl as the reference electrode. An aqueous solution of 0.5 M Na2SO4 was used as the electrolyte. To prepare the working electrode, 5 mg sample was suspended in a mixture of 10 μL Nafion solution (D520, DuPont), 1.5 mL ethanol and 0.5 mL ultrapure water to become a slurry, which was then dip-coated onto a glass carbon electrode and dried in the shade. The light source was a 300 W Xe lamp (PLS-SXE 300, Beijing Perfect Light Co., Ltd.). The photocurrent response was obtained using potentiostatic (current vs. time, I–t) measurements under intermittent illumination (30 s) under the conditions of no bias potential.3. Results and discussion
The composition, structure and morphology of BiOCl1−xBrx@AgBr and BiOCl were measured using XRD, XPS and SEM analysis. The XRD patterns of AgBr, BiOCl and BiOCl1−xBrx@AgBr are displayed in Fig. 1. The diffraction peaks occurring at 11.9°, 24.1°, 25.8°, 32.5°, 33.4°, 36.5°, 40.9°, 46.6°, 49.7°, 54.1°, 55.1°, 58.6°, and 60.5° are readily assigned to the (001), (002), (101), (110), (102), (003), (112), (200), (113), (211), (104), (212), and (114) lattice planes, respectively, being well matched with tetragonal BiOCl (JCPDS card no. 06-0249). Meanwhile, the characteristic peaks appearing at 26.7°, 31.0°, 44.3°, 55.0°, 64.5° and 73.3° are indexed to the (111), (200), (220), (222), (400) and (420) crystal planes of the cubic phase of AgBr (JCPDS card no. 06-0438), respectively. After coupling with AgBr, the XRD pattern of BiOCl1−xBrx@AgBr is similar to that of BiOCl; only two diffraction peaks occurring at 31.0° and 44.3° can be indexed to the (200) and (220) crystal planes of the cubic phase of AgBr, respectively. The results indicate that the BiOCl1−xBrx@AgBr heterostructure was successfully synthesized. Besides, the XRD patterns of BiOCl1−xBrx and AgBr/BiOCl1−xBrx are shown in Fig. S1 (ESI).† The XRD pattern of BiOCl1−xBrx is almost identical to that of BiOCl and no peaks of bromide are observed due to the low molecular weight content or high dispersity of bromide in the BiOCl crystal. The diffraction peaks of AgBr/BiOCl1−xBrx are indexed to the BiOCl (JCPDS card no. 06-0249) and AgBr (JCPDS card no. 06-0438) respectively. The diffraction peaks of the samples are sharp, which indicates that the samples have high crystallinity. No other characteristic peaks of impurities could be detected, indicating the high purity of the samples. Nevertheless, in the amplified XRD (Fig. S2†) patterns in the 2θ range from 10° to 14°, the diffraction peak at 2θ = 11.9° corresponding to the (001) crystal planes of BiOCl in BiOCl1−xBrx are slightly shifted to lower angles compared to BiOCl. The slight shift to lower angles of the (001) peak may be caused by the introduction of Br, because the ionic radius of Br (1.95 Å) is larger than that of Cl (1.81 Å).38 Compared to the standard card of BiOCl, the intensity of the peak located at 11.9° assigned to the plane (001) is obviously higher than that for other planes, which suggests that the samples have abundant (001) planes and these are preferentially oriented (or textured) parallel to the surface of the supporting substrate.39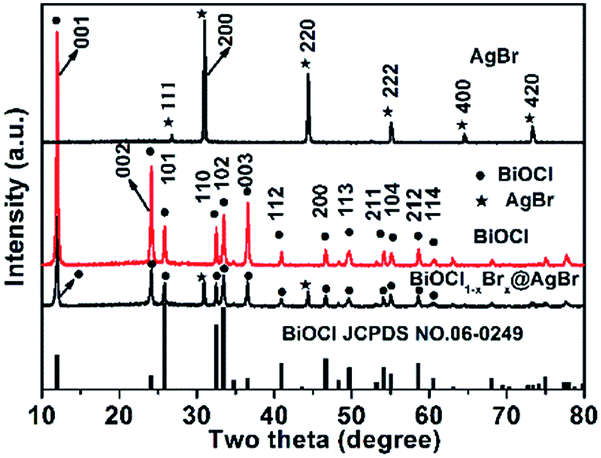 | ||
| Fig. 1 XRD diffraction patterns of AgBr, BiOCl and BiOCl1−xBrx@AgBr. | ||
The chemical compositions and valence states of the BiOCl and BiOCl1−xBrx@AgBr were characterized using XPS. The results are displayed in Fig. 2. As shown in Fig. 2a, the BiOCl sample consists of Bi, Cl and O elements. The high-resolution XPS spectra of BiOCl are shown in Fig. S3 (ESI).† The Bi 4f spectrum (Fig. S3b (ESI)†) gives two typical peaks, where the two strong peaks at 158.8 eV and 164.2 eV can be attributed to Bi 4f7/2 and Bi 4f5/2, indicating the chemical state of Bi3+. Two peaks can be observed at the binding energies of 197.1 eV and 199.0 eV corresponding to Cl 2p3/2 and Cl 2p1/2 (Fig. S3c (ESI)†), respectively. The peak of O 1s (Fig. S3d (ESI)†) can be fitted into two peaks which are located at 529.8 eV and 532.4 eV representing lattice oxygen and adsorption oxygen, respectively. The survey spectrum of BiOCl1−xBrx (Fig. S3a (ESI)†) demonstrates that Bi, Cl, O and Br are the main elements in the sample. The high-resolution XPS spectra of BiOCl1−xBrx are shown in Fig. S4.† It is worth noting that a weak peak of Br (Fig. S4c (ESI)†) from BiOCl1−xBrx can be observed at the binding energy of 68.8 eV, which is in agreement with the Br 3d3/2 peak. The weak peak may due to the low content of Br. The spectrum of O 1s (Fig. S4d (ESI)†) can be fitted into three peaks with banding energies of 529.4 eV, 530.1 eV and 532.3 eV which are assigned to lattice oxygen, defects and adsorption oxygen, respectively. The survey spectrum of BiOCl1−xBrx@AgBr reveals the existence of the elements Bi, Cl, Br, Ag and O in the composite (Fig. 2a). Furthermore, the high-resolution XPS spectra of the elements are displayed in Fig. 2b–f. The Bi spectrum can be de-convoluted into double peaks at 158.8 eV and 164.2 eV (Fig. 2b), which can be attributed to Bi 4f7/2 and Bi 4f5/2, respectively, indicating the chemical state of Bi3+. The fitted Cl 2p peaks (Fig. 2c) indicate that the peaks at 195.6 eV and 197.2 eV correspond to Cl− species. The XPS spectrum of Br is displayed in Fig. 2d; deconvolution of the Br 3d high-resolution spectrum of BiOCl1−xBrx@AgBr results in two peaks centered at 68.0 eV and 69.1 eV, corresponding to Br 3d5/2 and Br 3d3/2, respectively. The two intensive peaks located at 367.5 eV and 373.5 eV (Fig. 2e) are assigned to Ag 3d5/2 and Ag 3d3/2, respectively, indicating that the chemical state of the Ag element is +1.40 In addition, there are no peaks belonging to Ag0, suggesting that the chemical structure of AgBr is stable during the synthetic process. The spectrum of O 1s can be fitted into three peaks (Fig. 2f). The peak located at 529.8 eV originates from the lattice oxygen of BiOCl1−xBrx, whereas the peak at 530.8 eV corresponds to the defects or oxygen vacancies. The peak with higher energy (532.2 eV) is attributed to the hydroxyl group attached to the surface of the photocatalyst.
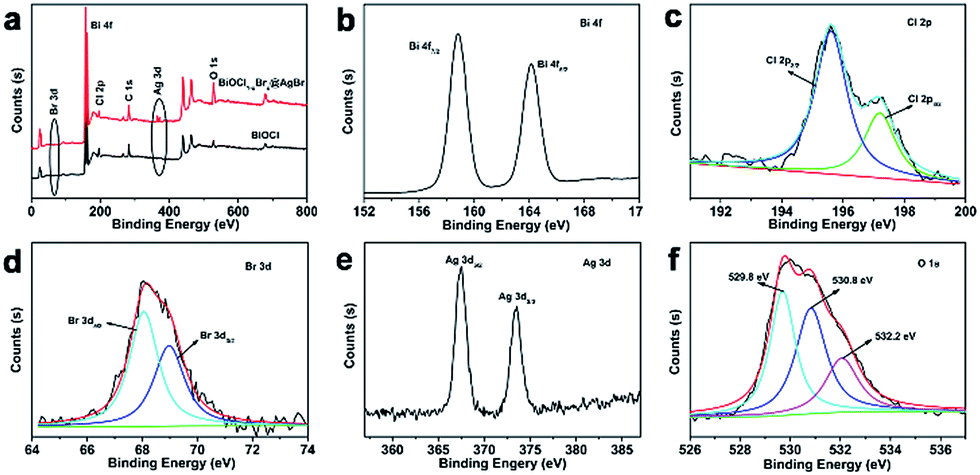 | ||
| Fig. 2 (a) XPS survey spectra of BiOCl1−xBrx@AgBr and BiOCl and (b–f) XPS spectra of the Bi, Cl, Br, Ag and O elements in BiOCl1−xBrx@AgBr. | ||
The morphology and structure of BiOCl and BiOCl1−xBrx@AgBr were observed using SEM and TEM. It is clearly seen that the BiOCl (Fig. 3a and b) has a flake-like structure with an irregular shape. The BiOCl nanosheets have a very smooth surface with a lateral width of about 400–500 nm and a thickness of approximately 50–100 nm. The HRTEM of BiOCl is displayed in Fig. 3c. The image shows an excellent crystalline structure and clear lattice fringes with an interplanar lattice spacing of 0.74 nm, which could be attributed to the (001) facet of BiOCl. The SEM and TEM images of BiOCl1−xBrx are exhibited in Fig. S5a and b (ESI).† After being decorated with CTAB, the BiOCl1−xBrx nanosheets have smoother edges and the thickness decreases to 20–60 nm. The HRTEM image of BiOCl1−xBrx shows good crystalline structure and lattice fringes with an interplanar lattice spacing of 0.28 nm, which could be attributed to the (110) facet of BiOCl. CTAB, as a kind of well-known surfactant, can self-aggregate into a variety of peculiar structures. Thus, it is widely used as a soft template to synthesize nanomaterials with controlled morphologies. The function of CTAB in this study is that of the template and Br source.41 According to previous reports,42 the release rate of Cl (from BiCl3) is higher than that of Br which is released from CTAB. During the synthesis process, a large quantity of Cl− reacts with Bi3+ to form the BiOCl nucleus and grow anisotropically throughout the solvothermal reaction. However, due to the low release rate and content, only a small quantity of Br ions exists among the BiOCl nanosheets, which further demonstrates the results obtained by XRD and XPS. Fig. S5c (ESI)† shows the SEM image of AgBr/BiOCl1−xBrx. The AgBr is in the form of colloidal spheres with a diameter of about 400–500 nm, onto which some BiOCl1−xBrx nanosheets are attached. The SEM and TEM images of BiOCl1−xBrx@AgBr are shown in Fig. 3d–f. The BiOCl1−xBrx nanosheets are covered by a thin layer of AgBr (Fig. 2d–e and S5d (ESI)†) formed from AgNO3 and CTAB. The formation was confirmed by XRD and XPS. The HRTEM image of BiOCl1−xBrx@AgBr (Fig. 3f and S6†) shows good crystalline structure and lattice fringes with an interplanar lattice spacing of 0.28 nm and 0.29 nm, which could be assigned to the (110) facet of BiOCl and (200) facet of AgBr, respectively. The SEM and TEM images demonstrate that the BiOCl1−xBrx@AgBr was successfully synthesized.
 | ||
| Fig. 3 (a) SEM images of BiOCl, (b and c) TEM and HRTEM images of BiOCl, (d) SEM image of BiOCl1−xBrx@AgBr and (e and f) TEM and HRTEM images of BiOCl1−xBrx@AgBr. | ||
EDS mapping images of the BiOCl1−xBrx@AgBr are displayed in Fig. S7 (ESI).† The results reveal that the Bi, Cl, Br, Ag and O elements are uniformly distributed throughout the BiOCl1−xBrx@AgBr sample. The high angle annular dark field scanning TEM (HAADF-STEM) and the corresponding energy-dispersive X-ray spectroscopy (EDX) elemental mapping images are displayed in Fig. 4. The images suggest that the different elements are distributed uniformly throughout the BiOCl1−xBrx nanosheets.
 | ||
| Fig. 4 HAADF-STEM image of BiOCl1−xBrx and the corresponding EDX elemental mapping analysis. | ||
The optical absorption properties of photocatalysts play an important role in their photocatalytic performance. Therefore, the DRS spectra of BiOCl, AgBr, BiOCl1−xBrx and BiOCl1−xBrx@AgBr were measured to determine their optical absorption properties and investigate the band gap energies. It can be clearly seen that the BiOCl (Fig. 5a), with a wide band gap and an absorption edge at about 359 nm, can only absorb ultraviolet light. AgBr exhibits strong optical absorption with an absorption edge at about 493 nm in the visible light region, which is in agreement with ref. 33. The light absorption edge of BiOCl1−xBrx extends to ∼400 nm. The absorption edge red-shift may be due to the presence of Br, which narrows its band-gap. The absorption edge of AgBr/BiOCl1−xBrx is at about 430 nm (Fig. S8a (ESI)†), which could be caused by coupling with AgBr spheres. As for the BiOCl1−xBrx@AgBr photocatalyst, the absorption edge shifts to longer wavelengths (about 450 nm), exhibiting an obvious red-shift compared to BiOCl. This result indicates that the presence of AgBr can enhance the optical absorption of BiOCl1−xBrx@AgBr, with the absorption edge shifting to the visible light range.
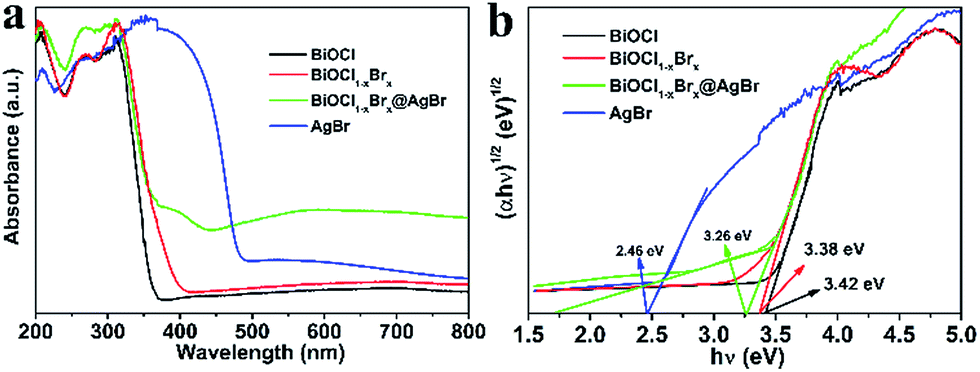 | ||
| Fig. 5 (a) UV-Vis diffuse reflectance spectra and (b) band gap energies of the samples. | ||
It is reported that eqn (1) can be used to calculate the band gap of the semiconductor on the basis of the DRS data.
| A(hν − Eg)n/2 = αhν | (1) |
| ECB = X − EC − 0.5Eg | (2) |
| EVB = ECB + Eg | (3) |
N2 adsorption/desorption measurements were conducted to characterize the specific surface area of the prepared nanomaterials. As shown in Fig. 6, the isotherms of BiOCl, BiOCl1−xBrx and BiOCl1−xBrx@AgBr are typical type IV curves with an H1 hysteresis loop. These features suggest that the obtained photocatalysts are mesoporous materials. The BET specific surface areas of BiOCl, BiOCl1−xBrx and BiOCl1−xBrx@AgBr are 4.449 m2 g−1, 6.26 m2 g−1 and 10.208 m2 g−1, respectively. The higher specific surface area could provide plentiful active reaction sites and contribute to the absorption of more pollutant molecules. Besides, the high SBET value could be attributed to diffusing photogenerated electrons and holes at the surface, which can increase the photocatalytic performance.
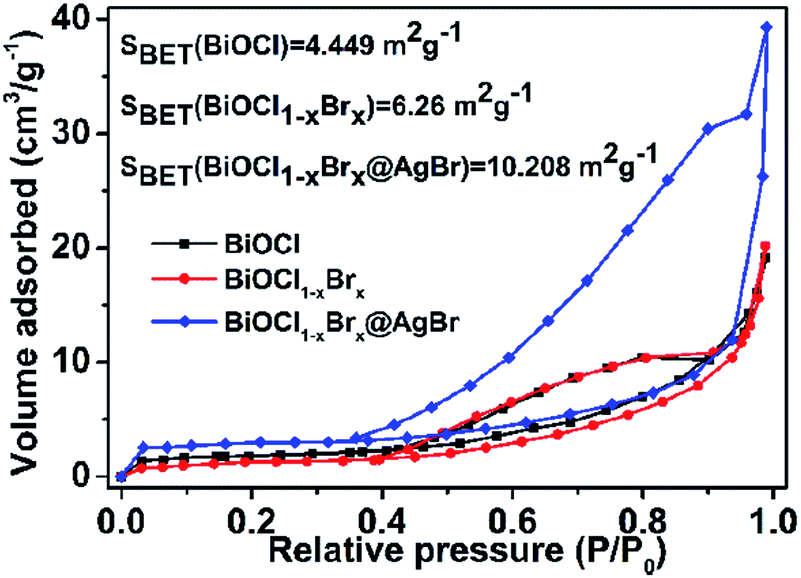 | ||
| Fig. 6 N2 adsorption–desorption isotherms of BiOCl, BiOCl1−xBrx and BiOCl1−xBrx@AgBr. | ||
The photocatalytic activity of the as-obtained samples was detected by degrading Reactive Blue KN-R under visible light (λ > 420 nm). Fig. 6a displays the relationship between the variation of Reactive Blue KN-R concentration and the reaction time of P25, BiOCl and BiOCl1−xBrx@AgBr, respectively. As seen in Fig. 7a, the blank experiment demonstrates that the Reactive Blue KN-R is inappreciably degraded in the absence of a photocatalyst, revealing that the structure of Reactive Blue KN-R is stable under visible light. The degradation rates of Reactive Blue KN-R over P25, pure BiOCl and BiOCl1−xBrx are merely around (13 ± 0.3)%, (24 ± 0.7)% and (66 ± 1.2)%, respectively, in 60 min. The BiOCl1−xBrx@AgBr heterostructure is more photoactive for Reactive Blue KN-R degradation than P25, BiOCl and BiOCl1−xBrx, and shows the best photocatalytic performance, with a degradation ratio of (92 ± 2.6)% after 60 min. Meanwhile, the degradation ratio of AgBr/BiOCl1−xBrx was also investigated using the photodegradation of Reactive Blue KN-R under visible light (λ > 420 nm). The Reactive Blue KN-R decoloration ratio reaches (82 ± 2.1)% over AgBr/BiOCl1−xBrx in 60 min (Fig. S9a†). Ofloxacin can also be used as the target antibiotic to be degraded to evaluate the photocatalytic properties of BiOCl, BiOCl1−xBrx and BiOCl1−xBrx@AgBr. The results are displayed in Fig. S10a (ESI).† The degradation efficiency of ofloxacin over BiOCl1−xBrx@AgBr is about (72 ± 3.2)% after 30 min, which is higher than when BiOCl1−xBrx (about (43 ± 1.5)%) and BiOCl (about (19 ± 0.4)%) are used as photocatalysts. Meanwhile, phenol was also chosen as an optional colourless organic substance to investigate the photocatalytic performance of BiOCl1−xBrx and BiOCl1−xBrx@AgBr. The result is displayed in Fig. S11.† After irradiation for 3.5 h, the degradation efficiencies of BiOCl1−xBrx and BiOCl1−xBrx@AgBr are (58 ± 2.5)% and (75 ± 3.7)%, respectively. As shown in Fig. 7b, the photocatalytic reaction of all the samples can be regarded as a pseudo-first order reaction. The corresponding kinetic constants (k) of Reactive Blue KN-R (OF) are listed in Table S2 (ESI).† The rate constants of P25, BiOCl, BiOCl1−xBrx and BiOCl1−xBrx@AgBr are 8.2 × 10−5 min−1, 1.6 × 10−3 min−1 (4.0 × 10−3 min−1), 1.3 × 10−2 min−1 (1.5 × 10−2 min−1) and 3.6 × 10−2 min−1 (4.1 × 10−2 min−1), respectively. AgBr/BiOCl1−xBrx has a rate constant of 2.3 × 10−2 min−1, which is about 14.4 times as high as that of BiOCl (1.6 × 10−3 min−1). It can be obviously seen from these results that the BiOCl1−xBrx@AgBr shows the best photocatalytic properties under visible light illumination, which may be caused by the efficient separation of photoinduced electron–hole pairs and the enhanced absorption of visible light.
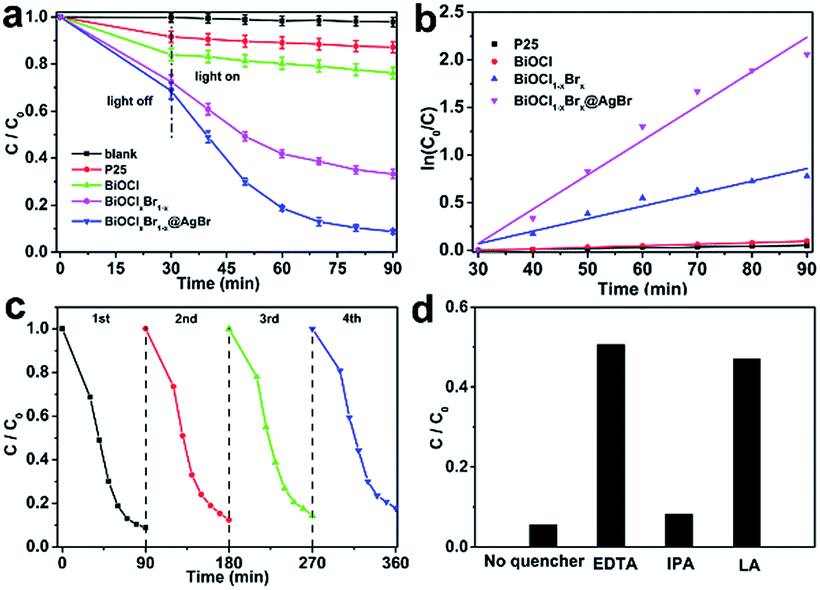 | ||
| Fig. 7 (a) Photocatalytic activities of different samples under visible light irradiation (λ > 420 nm), (b) kinetic fit for degradation of dyes with different samples, (c) cycling runs of BiOCl1−xBrx@AgBr for degradation of Reactive Blue KN-R under visible light and (d) degradation rate of Reactive Blue KN-R over BiOCl1−xBrx@AgBr after adding different scavengers. | ||
In order to assess the recyclability and stability of the BiOCl1−xBrx@AgBr photocatalyst during the degradation process, a circulation test for degradation of the Reactive Blue KN-R was carried out, and the result is displayed in Fig. 7c. After four cycles, BiOCl1−xBrx@AgBr still decomposes the Reactive Blue KN-R with only a slight decrease in photodegradation efficiency, which may be due to the loss of sample during the collection and decontamination. The stability of the sample can be further confirmed via the XRD pattern (Fig. S12†) of the BiOCl1−xBrx@AgBr heterostructure before and after the fourth Reactive Blue KN-R degradation experiment. It is worth noting that no obvious structure changes can be detected by the XRD test before and after irradiation, which can also confirm the phase stability of BiOCl1−xBrx@AgBr. Thus, the BiOCl1−xBrx@AgBr heterostructure shows extraordinary reusability and stability in photocatalytic applications.
In order to illustrate the photocatalytic mechanism, a series of sacrificial agents were introduced to detect the contributions of the active species in a photocatalytic process. The main active species can be detected through the trapping of radicals and holes during the photocatalytic process, and the results are displayed in Fig. 7d. In this study, ethylenediamine tetraacetic acid disodium salt (EDTA-2Na), isopropyl alcohol (IPA) and L-ascorbic acid (LA) are employed for quenching the holes (h+), hydroxyl radicals (˙OH) and superoxide radicals (˙O2−), respectively. As shown in Fig. 7d, the presence of EDTA decreases the degradation rate of Reactive Blue KN-R significantly, implying that the holes play a crucial role in photocatalytic systems. However, with the addition of IPA, the degradation rate of Reactive Blue KN-R has slight decrease, demonstrating that hydroxyl radicals are not the main active species during the Reactive Blue KN-R degradation process. L-Ascorbic acid significantly suppresses the degradation of Reactive Blue KN-R, so it can be concluded that superoxide radicals participate in the photocatalytic reaction. The results mentioned above suggest that the degradation reaction of Reactive Blue KN-R over the BiOCl1−xBrx@AgBr heterostructure is mainly driven by holes and superoxide radicals.
The photocurrent test is correlated with the transfer of photoinduced electrons and holes, thus the transient photocurrent–time curves can represent the separation and transfer dynamics of photogenerated charges in semiconductor photocatalysts. Fig. 8a shows the photocurrent measurements for BiOCl, BiOCl1−xBrx and BiOCl1−xBrx@AgBr. It can be observed that the photocurrent densities of BiOCl and BiOCl1−xBrx are about 0.01 μA cm−2 and 0.10 μA cm−2, respectively. Comparatively, BiOCl1−xBrx@AgBr exhibits a higher photocurrent density (0.31 μA cm−2), which is much higher than that of BiOCl and BiOCl1−xBrx, indicating the enhancement of the separation efficiency of photo-induced charges and the improvement of the transfer rate of photogenerated electrons and holes in BiOCl1−xBrx@AgBr. It is reported that the higher photocurrent density results in an enhanced charge separation efficiency and a higher interfacial transfer rate, leading to better photocatalytic properties. Thus, the photocatalytic properties of BiOCl1−xBrx@AgBr are better than that of BiOCl and BiOCl1−xBrx. The electrochemical impedances of BiOCl, BiOCl1−xBrx and BiOCl1−xBrx@AgBr were also tested to illustrate the photocatalytic enhancement mechanism. The results (Fig. 8b) show that BiOCl, BiOCl1−xBrx and BiOCl1−xBrx@AgBr have similar impedance spectra. The Nyquist circle diameter of BiOCl1−xBrx@AgBr is much smaller than that of BiOCl and BiOCl1−xBrx, indicating a lower resistance. This result suggests a faster charge transfer and slower efficiency of electron and hole recombination in the BiOCl1−xBrx@AgBr heterostructure. The impedance of BiOCl1−xBrx@AgBr was compared under light irradiation and in the dark. As seen in Fig. S13,† the semicircle diameter of BiOCl1−xBrx@AgBr under light irradiation is shorter than that in the dark, which contributes to the separation of electrons and holes.
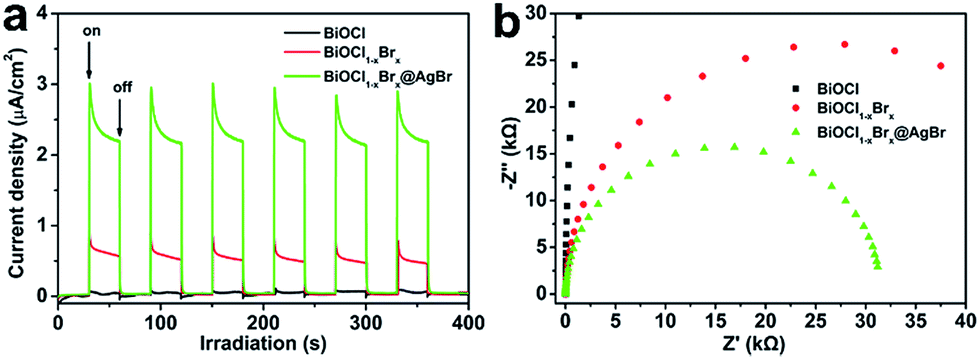 | ||
| Fig. 8 (a) Transient photocurrent responses and (b) electrochemical impedance spectroscopy of different samples. | ||
According to the results mentioned above, a general illustration for the mechanism of photocatalytic degradation over the BiOCl1−xBrx@AgBr heterostructure is given in Fig. 9. Since the redox potentials of the valence band (VB) and conduction band (CB) of BiOCl1−xBrx are both more positive than those of AgBr, AgBr and BiOCl1−xBrx possess the best interactive structure, which contributes to the effective separation of photogenerated electrons and holes. Due to the narrow band gap of AgBr, when the BiOCl1−xBrx@AgBr absorbs visible light with a certain energy, the electrons in the valence band (VB) of AgBr could be excited to the conduction band (CB), leaving holes in the valence band. Then the generated electrons transfer from the CB of AgBr to that of BiOCl1−xBrx. The electrons in the CB of BiOCl1−xBrx can react with O2 to generate ˙O2− radicals, which may react with Reactive Blue KN-R molecules to degrade them. In the meantime, the h+ in the VB of BiOCl1−xBrx can transfer to the VB of AgBr, since AgBr has a more negative VB potential than that of BiOCl1−xBrx. The h+ in the VB of AgBr can directly oxidize Reactive Blue KN-R. In this way, the generated electrons and holes in the BiOCl1−xBrx@AgBr heterostructure can be effectively separated, and eventually result in a higher photocatalytic performance.
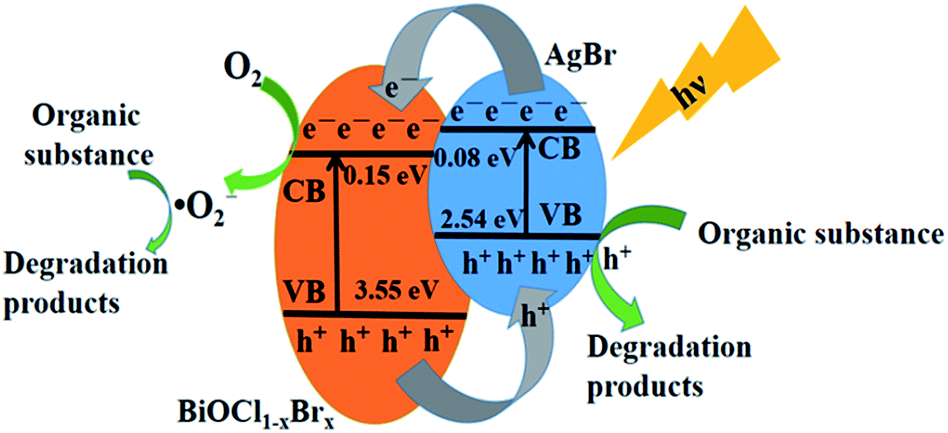 | ||
| Fig. 9 Schematic illustration of the charge transfer process in BiOCl1−xBrx@AgBr under visible light irradiation. | ||
4. Conclusions
In summary, a highly effective visible-light-response photocatalyst of BiOCl1−xBrx@AgBr was synthesized successfully via a simple two-step solvothermal method, which was based upon formation of a heterojunction interface between BiOCl1−xBrx and AgBr. The as-fabricated BiOCl1−xBrx@AgBr heterostructure exhibits superior photocatalytic activity compared with BiOCl towards the degradation of Reactive Blue KN-R under visible light illumination. The formation of the BiOCl1−xBrx@AgBr heterostructure plays a significant role in the enhancement of photocatalytic performance, which is mainly ascribed to the more efficient separation of electron–hole pairs in the two semiconductors and the enhanced absorption of visible light. Besides, the photocatalyst has good stability and reusability. Thus, the excellent photocatalytic and stability properties facilitate its practical applications in environmental purification and organic pollution treatment of water.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant no. 21476033 and Grant no. 21577008).Notes and references
- Z. Li, Y. Qu, K. Hu, M. Humayun, S. Chen and L. Jing, Appl. Catal., B, 2017, 203, 355–362 CrossRef CAS.
- X. Guo, G. Zhang, H. Cui, N. Wei, X. Song, J. Li and J. Tian, Appl. Catal., B, 2017, 217, 12–20 CrossRef CAS.
- S. Zhu, C. Yang, F. Li, T. Li, M. Zhang and W. Cao, Mol. Catal., 2017, 435, 33–48 CrossRef CAS.
- J. Hu, X. Wu, C. Huang, W. Fan and X. Qiu, Appl. Surf. Sci., 2016, 387, 45–50 CrossRef CAS.
- D. Lv, D. Zhang, X. Pu, D. Kong, Z. Lu, X. Shao, H. Ma and J. Dou, Sep. Purif. Technol., 2017, 174, 97–103 CrossRef CAS.
- J. Pan, Y. Sheng, J. Zhang, J. Wei, P. Huang, X. Zhang and B. Feng, J. Mater. Chem. A, 2014, 2, 18082–18086 CAS.
- G. Li, L. Wu, F. Li, P. Xu, D. Zhang and H. Li, Nanoscale, 2013, 5, 2118–2125 RSC.
- H. Sun, Q. He, S. Zeng, P. She, X. Zhang, J. Li and Z. Liu, New J. Chem., 2017, 41, 7244–7252 RSC.
- S. A. Bakar, G. Byzynski and C. Ribeiro, J. Alloys Compd., 2016, 666, 38–49 CrossRef CAS.
- A. K. R. Police, M. Chennaiahgari, R. Boddula, S. V. P. Vattikuti, K. K. Mandari and B. Chan, Mater. Res. Bull., 2018, 98, 314–321 CrossRef CAS.
- Y. Liu, W. Zhang, Y. Sun and W. Liang, Mater. Res. Bull., 2018, 98, 240–249 CrossRef CAS.
- L. Lei, H. Jin, Q. Zhang, J. Xu, D. Gao and Z. Fu, Dalton Trans., 2015, 44, 795–803 RSC.
- X. Jia, J. Cao, H. Lin, M. Zhang, X. Guo and S. Chen, Appl. Catal., B, 2017, 204, 505–514 CrossRef CAS.
- K. Li, Y. Liang, J. Yang, Q. Gao, Y. Zhu, S. Liu, R. Xu and X. Wu, J. Alloys Compd., 2017, 695, 238–249 CrossRef CAS.
- H. Xu, Z. Wu, M. Ding and X. Gao, Mater. Des., 2017, 114, 129–138 CrossRef CAS.
- Z. Haider, J. Y. Zheng and Y. S. Kang, Phys. Chem. Chem. Phys., 2016, 18, 19595–19604 RSC.
- F. Dong, T. Xiong, S. Yan, H. Wang, Y. Sun, Y. Zhang, H. Huang and Z. Wu, J. Catal., 2016, 344, 401–410 CrossRef CAS.
- S. Li, S. Hu, K. Xu, W. Jiang, J. Liu and Z. Wang, Nanomaterials, 2017, 7, 1–13 Search PubMed.
- S. Li, S. Hu, W. Jiang, Y. Liu, Y. Zhou, Y. Liu and L. Mo, J. Colloid Interface Sci., 2018, 521, 42–49 CrossRef CAS PubMed.
- Z. Jiang, Y. Liu, T. Jing, B. Huang, Z. Wang, X. Zhang, X. Qin and Y. Dai, RSC Adv., 2015, 5, 47261–47264 RSC.
- J. Hu, G. Xu, J. Wang, J. Lv, X. Zhang, Z. Zheng, T. Xie and Y. Wu, New J. Chem., 2014, 38, 4913–4921 RSC.
- J. Xia, L. Xu, J. Zhang, S. Yin, H. Li, H. Xu and J. Di, CrystEngComm, 2013, 15, 10132–10141 RSC.
- Y. Kan, Y. Yang, F. Teng, L. Yang, J. Xu and Y. Teng, Catal. Commun., 2016, 87, 10–13 CrossRef CAS.
- M. Palmai, E. M. Zahran, S. Angaramo, S. Balint, Z. Paszti, M. R. Knecht and L. G. Bachas, J. Mater. Chem. A, 2017, 5, 529–534 CAS.
- K. Xu, X. Fu and Z. Peng, Mater. Res. Bull., 2018, 98, 103–110 CrossRef CAS.
- S. Li, X. Shen, J. Liu and L. Zhang, Environ. Sci.: Nano, 2017, 4, 1155–1167 RSC.
- S. Li, S. Hu, W. Jiang, Y. Liu, J. Liu and Z. Wang, Mol. Catal., 2017, 435, 135–143 CrossRef CAS.
- S. Li, S. Hu, W. Jiang, Y. Liu, J. Liu and Z. Wang, J. Colloid Interface Sci., 2017, 501, 156–163 CrossRef CAS PubMed.
- Z. Xu and S.-Y. Lin, RSC Adv., 2016, 6, 84738–84747 RSC.
- D. Wei, F. Tian, Z. Lu, H. Yang and R. Chen, RSC Adv., 2016, 6, 52264–52270 RSC.
- J. Cheng, C. Wang, Y. Cui, Y. Sun, Y. Zuo and T. Wang, Mater. Lett., 2014, 127, 28–31 CrossRef CAS.
- F. Guo, W. Shi, Y. Cai, S. Shao, T. Zhang, W. Guan, H. Huang and Y. Liu, RSC Adv., 2016, 6, 93887–93893 RSC.
- D. Wang, L. Yue, L. Guo, F. Fu, X. He and H. Shen, RSC Adv., 2015, 5, 72830–72840 RSC.
- Q. Li, Y. Xing, R. Li, L. Zong, X. Wang and J. Yang, RSC Adv., 2012, 2, 9781–9785 RSC.
- Y. L. Qi, Y. F. Zheng, H. Y. Yin and X. C. Song, J. Alloys Compd., 2017, 712, 535–542 CrossRef CAS.
- G. Chen, M. Zhu and X. Wei, Mater. Lett., 2018, 212, 182–185 CrossRef CAS.
- H. Cheng, W. Wang, B. Huang, Z. Wang, J. Zhan, X. Qin, X. Zhang and Y. Dai, J. Mater. Chem. A, 2013, 1, 7131–7136 CAS.
- X. Zhang, L.-W. Wang, C.-Y. Wang, W.-K. Wang, Y.-L. Chen, Y.-X. Huang, W.-W. Li, Y.-J. Feng and H.-Q. Yu, Chem.–Eur. J., 2015, 21, 11872–11877 CrossRef CAS PubMed.
- J. Duan, G. Mou, S. Zhang, S. Wang and J. Guan, J. Mater. Chem. A, 2015, 3, 14686–14695 CAS.
- H. Zheng, P. Li, L. Gao and G. Li, RSC Adv., 2017, 7, 25725–25731 RSC.
- X. Li, C. Zhu, Y. Song, D. Du and Y. Lin, RSC Adv., 2017, 7, 10235–10241 RSC.
- M. Shang, W. Wang and L. Zhang, J. Hazard. Mater., 2009, 167, 803–809 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of AgBr/BiOCl1−xBrx, XRD patterns, XPS spectrum, SEM and TEM images, UV-Vis diffuse reflectance spectra and photocatalytic activity of BiOCl1−xBrx and AgBr/BiOCl1−xBrx; the UV-Vis absorption spectra for Reactive Brilliant Blue KN-R degradation using BiOCl1−xBrx@AgBr and BiOCl; photocatalytic activities of BiOCl and BiOCl1−xBrx@AgBr for degrading ofloxacin; PEC results of BiOCl1−xBrx; the absorption edges and band gap energies, ECB and EVB, listed in tables; the kinetic constants listed in tables. See DOI: 10.1039/c8ra02971g |
| This journal is © The Royal Society of Chemistry 2018 |