
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel blended catalysts consisting of a TiO2 photocatalyst and an Al2O3 supported Pd–Au bimetallic catalyst for direct dehydrogenative cross-coupling between arenes and tetrahydrofuran†
Akanksha Tyagi a,
Akira Yamamotoab and
Hisao Yoshida*ab
a,
Akira Yamamotoab and
Hisao Yoshida*ab
aGraduate School of Human and Environmental Studies, Kyoto University, Yoshida Nihonmatsu-cho, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: yoshida.hisao.2a@kyoto-u.ac.jp
bElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Kyotodaigaku-Katsura, Nishikyo-ku, Kyoto 615-8520, Japan. E-mail: yoshida.hisao.2a@kyoto-u.ac.jp
First published on 2nd July 2018
Abstract
Dehydrogenative cross-coupling (DCC) is a clean methodology to make C–C bonds by using abundant C–H bonds. The blended catalyst, developed in this study, consists of a TiO2 photocatalyst and an Al2O3 supported Pd–Au bimetallic catalyst and shows superior activity to the conventional TiO2 photocatalyst loaded with the corresponding metal co-catalyst for the direct DCC between various arenes and tetrahydrofuran, with concomitant evolution of hydrogen gas. The reactions were done under mild conditions without consuming any oxidising agent or other additional chemicals. This new approach of separating the photocatalyst and the metal catalyst parts allows their independent modification to improve the overall catalytic performance.
Introduction
Dehydrogenative cross-coupling (DCC) is an efficient methodology to make carbon–carbon (C–C) bonds between two different organic compounds.1 This direct route to make C–C bonds yields hydrogen as a by-product, which can be used as a fuel or for hydrogenation reactions. Different catalysts have been developed to carry out the thermal dehydrogenation reactions,2 but the scope for further improvement remains.Recently, the use of an abundant, safe, and heterogeneous TiO2 photocatalyst for DCC has become more attractive as it works under mild reaction conditions.3 The photogenerated holes on TiO2 can activate the C–H bonds in various organic molecules to generate the corresponding radical species and protons. The generated radicals can further react with other radical species or molecules to give the coupling products by making new C–C bonds, while the protons are reduced by the photoexcited electrons to form hydrogen molecules. Thus, the TiO2 photocatalyst efficiently brings out DCC with concomitant hydrogen gas evolution, without any external oxidising agent. In many cases, metal nanoparticles are loaded on TiO2 (M/TiO2), that can accept the excited electrons from the TiO2 photocatalyst, to decrease the electron–hole recombination and increase the photocatalytic efficiency.4 In these cases, the metal nanoparticles function as an electron receiver.
On the other hand, it is notable that the metal nanoparticles loaded on TiO2 sometimes can also participate catalytically in some photocatalytic reactions, thus acting as co-catalysts.3b–3e,5 For example, Pd nanoparticles catalyse a reaction between an aromatic molecule and different carbon centred radicals,3b–3d and Pt nanoparticles catalyse reactions like photocatalytic hydrogenation,5a dehydrogenation,5c and C–H bond activation.3e,5b To develop such a hybrid catalyst system consisting of a photocatalyst and a metal co-catalyst, one may plan some modifications to improve the catalytic activity, e.g., modifying the metal co-catalyst by increasing its loading amount and dispersion, making an alloy with other metal, varying its oxidation state and so on. However, it is difficult to realise these alternations of the metal co-catalyst part on the TiO2 surface without changing the photocatalytic performance of the TiO2 part. It is because TiO2 itself is susceptible to chemical or thermal treatments and its physical properties like surface area, crystal phase, and crystal defects, may be varied.6
Here, a novel blended catalyst is proposed as one solution to this problem, which is a physical mixture of a TiO2 photocatalyst and a supported metal catalyst. Since the two catalysts can be prepared separately without affecting each other, they can be modified independently to achieve high activity entirely. Although several reports mentioned the use of a kind of the blended catalyst including a photocatalyst,7 they were employed just for investigating the origin of the catalytic activity. Employing a blended catalyst to improve the activity is uncommon, except for a few examples, such as a combination of two kinds photocatalysts to build a Z-scheme for water splitting,8a–c and a combination of a photocatalyst and an acid catalyst for C–O coupling.8d Thus, this is the first report for the blended catalyst consisting of a photocatalyst and a metal catalyst for the enhancement of the catalytic activity. We have developed it for the sp2C–sp3C photocatalytic direct DCC between various arenes and ethers like tetrahydrofuran (THF). In this study, we found that a blended catalyst consisting of a TiO2 photocatalyst and an Al2O3 supported Pd–Au bimetallic catalyst can convert various arenes and THF to their corresponding DCC products and hydrogen, with higher activity than the conventional metal loaded TiO2 photocatalyst, under mild conditions without consuming any oxidising agent or other chemicals.
Experimental section
Catalyst preparation
The Pd loaded TiO2 sample (Pd(3.0)/TiO2) (the number in parenthesis refers to the loading amount of the metal in weight%), and the bimetallic Pd–Au loaded TiO2 sample (Pd(2.0)Au(1.0)/TiO2) was prepared by a photodeposition method as follows. For the Pd(3.0)/TiO2 sample, 4 g of TiO2 powder (JRC-TIO-8 provided by Catalysis Society of Japan, anatase phase, surface area 335 m2 g−1) was dispersed in ion-exchanged water (300 mL) and irradiated for 30 min from a ceramic xenon lamp (PE300BUV). Then, 100 mL methanol and 5.8 mL of an aqueous solution of PdCl2 (10.1 mg mL−1 of Pd) was added to the suspension and the contents were stirred for 15 min without irradiation, followed by 1 h stirring under the light irradiation. It was then filtered off with suction, washed with ion-exchanged water, and dried at 323 K for 12 h to get the Pd(3.0)/TiO2 sample. The bimetallic Pd(2.0)Au(1.0)/TiO2 sample was prepared in a similar manner by using 2 g TiO2, 175 mL water, 50 mL methanol, 6.5 mL of an aqueous solution of PdCl2 (6.12 mg mL−1 of Pd), and 4.1 mL of an aqueous solution of HAuCl4 (4.8 mg mL−1 of Au).The various Al2O3 supported samples (monometallic Pd(3.0)/Al2O3 and Au(3.0)/Al2O3 and bimetallic Pd(x)Au(y)/Al2O3) were prepared in a similar manner to the reported method9 with some modifications, as mentioned below. 2 g of the Al2O3 powder (JRC-ALO-7 provided by the Catalysis Society of Japan, γ-phase, 180 m2 g−1) and the desired volume of the aqueous solutions of PdCl2 and HAuCl4 were dispersed in 60 mL of ion-exchanged water. The suspension was vigorously stirred at room temperature for 15 min. The pH of the suspension was adjusted to 10 by using an aqueous solution of NaOH (1 M). The resultant slurry was stirred for 24 h at room temperature. Then, the contents were filtered and dried overnight in an electric oven at 323 K. Finally, the powder was reduced at 423 K under pure H2 for 30 min to get the Pd(x)Au(y)/Al2O3 samples. The total loading amount of the metals on the support was fixed to 3 weight%. For comparison, the monometallic Al2O3-supported samples, Pd(3.0)/Al2O3 and Au(3.0)/Al2O3, were also prepared by a similar procedure.
The prepared samples were characterised by various techniques like TEM, XRD, XAFS, and UV-DRS. The transmission electron microscopy (TEM) images of the various samples were taken by a JEM-2200FS Field Electron Microscope. X-ray diffraction (XRD) measurements of the Al2O3 support and various Al2O3 supported samples were carried out on a Lab X XRD-6000 Shimadzu. X-ray absorption fine structure (XAFS) was used to study the electronic state of the Pd and Au species in the Pd(x)Au(y)/Al2O3 samples. The Pd K-edge and Au LIII-edge XAFS measurements were carried out at the BL01B1 beam line of the synchrotron facility at Spring-8 in the RIKEN Harima institute (Hyogo, Japan). The measurements were done in a transmission mode. The samples were mixed with a calculated amount of boron nitride, and the mixture was shaped into a pellet which was used for measurements. The spectra were analysed by Athena software.10 The ultraviolet-visible diffuse reflectance spectroscopy (UV-DRS) measurements of the monometallic Pd(3.0)/Al2O3 and Au(3.0)/Al2O3 samples, and the bimetallic Pd(x)Au(y)/Al2O3 samples were carried out on a JASCO V-570 instrument.
Procedure for the photocatalytic activity tests
All chemicals were of analytical grade and used without further purification; THF (Wako Pure Chemicals, 99%), benzene (Nacalai Tesque, 99%), benzaldehyde (Wako Pure Chemicals, 99%), benzonitrile (Kishida Chemicals, 99%), toluene (Nacalai Tesque, 99%), and aniline (Kishida Chemicals, 99.5%). The cross-coupling product 4-(tetrahydrofuran-2-yl)benzonitrile was synthesised according to the literature11 (details are mentioned in the ESI†).The sp2C–sp3C photocatalytic direct DCC reactions between different arenes and THF were carried out in a closed reactor of a Pyrex test tube (volume = 80 mL) and the xenon lamp (PE300BUV) as the light source. The wavelength of the irradiated light was restricted to ≥350 nm, by using a long pass optical filter. The light intensity was maintained at 40 mW cm−2 (measured at 360 nm ± 60 nm). The reaction test involved the pre-treatment of the catalyst (photocatalyst or the blended catalyst) by light irradiation from the xenon lamp for 1 h. Next, the test tube was sealed with a silicon septum and after a 10 min argon purge, the reactants (arene and THF) were added and the resultant suspension was magnetically stirred under the light irradiation for the desired time. After the reaction, a part of the gaseous phase was collected in an air-tight syringe and analysed by GC-TCD (Shimadzu, GC-8A). The liquid phase was first diluted by ethanol (2 mL), filtered, and analysed by GC-MS (Shimadzu, GCMS-QP5050A) by using decane as an internal standard. The amount of all DCC products was approximately determined from the calibration curve of 4-(tetrahydrofuran-2-yl)benzonitrile. The GC-MS yield of the products (% Y) was calculated as % Y = 100 × [total amount of the DCC products (μmol)/initial amount of the arene (μmol)]. The selectivity to the DCC products, based on the arene, (% S) was calculated as % S = 100 × [total amount of DCC product (μmol)/total amount of the products from arene (μmol)].
Results and discussion
Characterisation of Pd(x)Au(y)/Al2O3 samples
Fig. 1 shows the TEM images and particle distribution histograms of three Al2O3 supported samples, monometallic Pd(3.0)/Al2O3 (Fig. 1a), monometallic Au(3.0)/Al2O3 (Fig. 1b), and bimetallic Pd(2.0)Au(1.0)/Al2O3 (Fig. 1c) that exhibited the highest catalytic activity as mentioned later (Pd/Au molar ratio: 3.5). Clearly, all samples contained small and well-dispersed nanoparticles of 3–4 nm in size.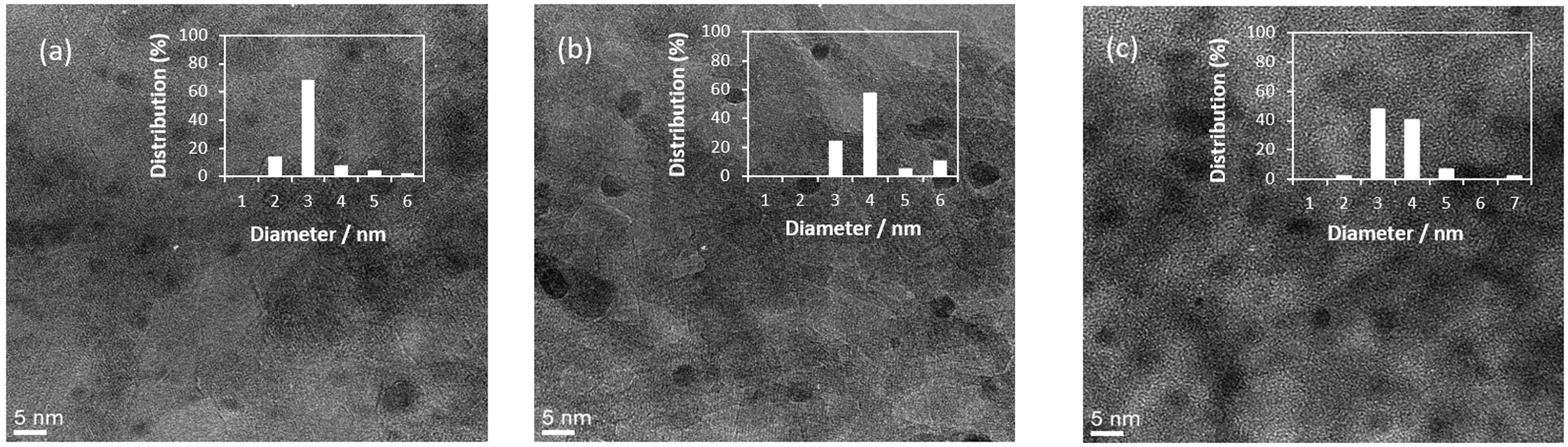 | ||
| Fig. 1 TEM images and particle distribution histograms of (a) monometallic Pd(3.0)/Al2O3, (b) Au(3.0)/Al2O3, and (c) bimetallic Pd(2.0)Au(1.0)/Al2O3 samples. | ||
Fig. 2 shows the XRD patterns of the Al2O3 support (Fig. 2a), the monometallic Pd(3.0)/Al2O3 (Fig. 2b), monometallic Au(3.0)/Al2O3 sample (Fig. 2f), and the bimetallic Pd(x)Au(y)/Al2O3 samples (Fig. 2c–e). According to literature, the Pd (111), (200), and (220) diffractions should appear at 2θ = 40.1°, 46.5°, and 68.1°, respectively (ICSD coll. code 41517) while the Au (111), (220), and (311) diffractions should be at 2θ = 38.1°, 64.5°, and 77.1°, respectively (ICSD coll. Code 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 249). However, none of these diffractions were observed in the XRD patterns of the prepared monometallic Pd(3.0)/Al2O3 and Au(3.0)/Al2O3, and bimetallic Pd(x)Au(y)/Al2O3 samples. This could be due to the small size and high dispersion of the metal nanoparticles, as revealed by TEM. Another reason could be the broad diffractions from the Al2O3 support around 2θ = 38°, 46°, and 67°,12 which may have interfered with the very small and broad diffractions from the metal nanoparticles in the prepared samples.
249). However, none of these diffractions were observed in the XRD patterns of the prepared monometallic Pd(3.0)/Al2O3 and Au(3.0)/Al2O3, and bimetallic Pd(x)Au(y)/Al2O3 samples. This could be due to the small size and high dispersion of the metal nanoparticles, as revealed by TEM. Another reason could be the broad diffractions from the Al2O3 support around 2θ = 38°, 46°, and 67°,12 which may have interfered with the very small and broad diffractions from the metal nanoparticles in the prepared samples.
 | ||
| Fig. 2 XRD patterns of the support, the monometallic and the bimetallic samples: (a) Al2O3, (b) Pd(3.0)/Al2O3, (c) Pd(2.5)Au(0.5)/Al2O3, (d) Pd(1.5)Au(1.5)/Al2O3, (e) Pd(0.5)Au(2.5)/Al2O3, and (f) Au(3.0)/Al2O3. | ||
Fig. 3 shows the normalised Pd K-edge X-ray absorption near edge structures (XANES) of the reference Pd foil (Fig. 3a), the monometallic Pd(3.0)/Al2O3 sample (Fig. 3b), and the Pd(x)Au(y)/Al2O3 samples (Fig. 3c–f). The spectral features in the prepared samples were clearly different from those of the foil, which would mainly originate from the property of the nanoparticles. The samples containing a large amount of Au exhibited slightly different shape from others (Fig. 3f), suggesting the Pd atoms are affected by the Au atoms and have local structure or electronic state different from those in the Pd metal. Also, a clear shift in the absorption edge towards the higher energy was observed in the order: the reference Pd foil (Fig. 4a), the monometallic Pd(3.0)/Al2O3 sample (Fig. 4b), the bimetallic Pd(2.5)Au(0.5)/Al2O3 (Fig. 4c) and Pd(2.0)Au(1.0)/Al2O3 samples (Fig. 4d). The shift from the Pd foil to the monometallic Pd(3.0)/Al2O3 sample is often observed in the supported metal nanoparticles, and can be attributed to the acidic property of the Al2O3 support and nanosize of the particles. The further shifts from the monometallic Pd to the bimetallic Pd–Au samples suggest that the Pd species became electron deficient upon the introduction of Au.13 This would arise if the Pd and Au atoms in nanoparticles are in intimate contact, and a transfer of electron density occurs from Pd atoms to Au atoms, i.e., Pdδ+–Auδ−. Fig. 5 shows the normalised XANES of Au LIII-edge. The XANES features of the prepared Al2O3 supported samples, especially at the higher energy region, were similar to that of Au foil indicating that the Au species in these samples was almost metallic. However, the Au absorption edge was clearly shifted in the prepared supported samples at 11919.7 eV (Fig. 5b–e) from the Au foil at 11920.2 eV (Fig. 5a), suggesting that the Au atoms became electron rich on the support. A clear absorption shoulder peak was observed around 11923 eV in the Au foil and the monometallic Au(3.0)/Al2O3 sample, which corresponds to the transition from the filled core 2p3/2 level to the vacant d orbitals.14 The intensity of this shoulder peak gradually decreased with increasing the Pd content in the bimetallic Pd(x)Au(y)/Al2O3 samples, indicating the filling of the d orbitals in Au, which would arise due to the transfer of electron density from Pd to Au atoms, i.e., Pdδ+–Auδ−. Thus, the XANES results revealed that the Pd and Au atoms had an intimate contact in the bimetallic Pd(x)Au(y)/Al2O3 samples, which affected their local structure and facilitated the electron transfer. As a result, the Pd atom are proposed to be electron deficient in the bimetallic Pd(x)Au(y)/Al2O3 samples.
 | ||
| Fig. 3 Normalised Pd K-edge XANES of metallic Pd as a reference, and the monometallic and bimetallic catalyst samples: (a) a Pd foil, (b) Pd(3.0)/Al2O3, (c) Pd(2.5)Au(0.5)/Al2O3, (d) Pd(2.0)Au(1.0)/Al2O3, (e) Pd(1.5)Au(1.5)/Al2O3, and (f) Pd(0.5)Au(2.5)/Al2O3. | ||
 | ||
| Fig. 4 Expanded normalised Pd K-edge XANES of (a) a Pd foil reference foil, (b) the monometallic Pd(3.0)/Al2O3 sample, and the bimetallic (c) Pd(2.5)Au(0.5)/Al2O3 and (d) Pd(2.0)Au(1.0)/Al2O3 samples. | ||
 | ||
| Fig. 5 Normalised Au LIII-edge XANES of a metallic Au as a reference, and the monometallic and bimetallic catalyst samples: (a) an Au foil, (b) the monometallic Au(3.0)/Al2O3 sample, and the bimetallic (c) Pd(0.5)Au(2.5)/Al2O3, (d) Pd(1.5)Au(1.5)/Al2O3, and (e) Pd(2.0)Au(1.0)/Al2O3 samples. | ||
Fig. 6 shows the UV-DR spectra of different Al2O3 supported samples. The monometallic Pd(3.0)/Al2O3 sample showed a very broad band over almost the entire range without any clear obvious peak (Fig. 6a). With the introduction of Au to the samples, the spectra gradually changed (Fig. 6b–e) and the plasmonic peak of Au nanoparticles became clearly visible around 520 nm in wavelength for the Pd(0.5)Au(2.5)/Al2O3 sample and the monometallic Au(3.0)/Al2O3 sample (Fig. 6e and f). However, the spectra of the bimetallic samples could not be simulated by that of the two monometallic Pd(3.0)/Al2O3 and Au(3.0)/Al2O3 samples, which suggests that the Pd and Au species are not independently present on the surface of the support but in close contact, and thus they affect the electronic properties of each other, which would provide unique catalytic property.
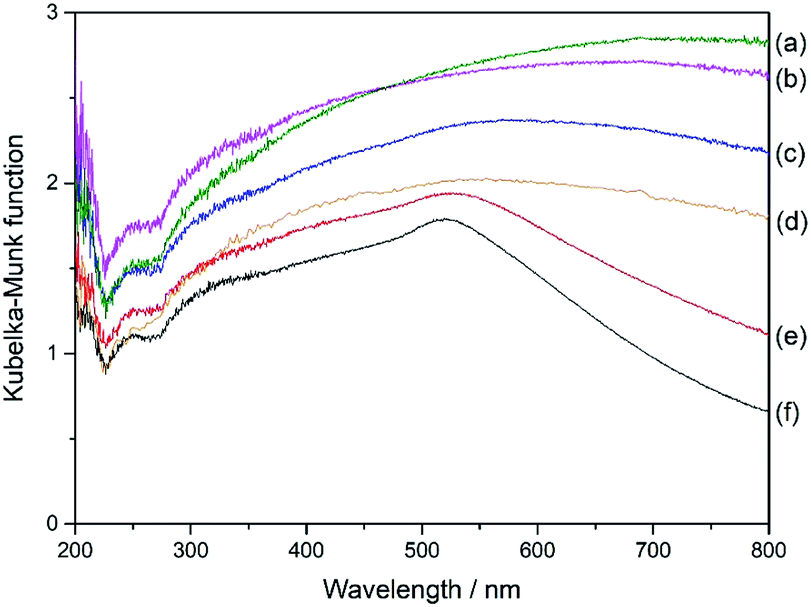 | ||
| Fig. 6 UV-DRS of the Al2O3-supported samples; (a) Pd(3.0)/Al2O3, (b) Pd(2.5)Au(0.5)/Al2O3, (c) Pd(2.0)Au(1.0)/Al2O3, (d) Pd(1.5)Au(1.5)/Al2O3, (e) Pd(0.5)Au(2.5)/Al2O3, and (f) Au(3.0)/Al2O3. | ||
Based on these results we conclude that the Pd and Au atoms in the bimetallic Pd(x)Au(y)/Al2O3 samples have a strong interaction, making the Pd atoms to be electron deficient.
Photocatalytic direct DCC between benzene and THF with different catalysts
Various photocatalysts like TiO2, Pd(3.0)/TiO2 and Pd(2.0)Au(1.0)/TiO2, and Al2O3 supported metal catalysts like Pd(3.0)/Al2O3, Au(3.0)/Al2O3 and Pd(x)Au(y)/Al2O3 were used for the photocatalytic direct DCC between arenes and THF.To begin with, the DCC of benzene (1) and THF was examined with different catalysts (Table 1). The reaction with the Pd(3.0)/TiO2 sample gave 2-phenyltetrahydrofuran (1a) as the only DCC product (Table 1 entry 1), along with small amounts of side products like octahydro-2,2′-bifuran, a homocoupling product of THF, and some unknown products (amounts not shown). The gas phase contained hydrogen (Fig. S1†), but, due to these side reactions, the amount of hydrogen and the DCC products was not balanced. Hence, the amount of hydrogen couldn't be determined precisely. The reaction done with a pristine TiO2 sample did not produce 1a (Table 1, entry 2), indicating that Pd loading was necessary for the DCC reaction.
|
|||
|---|---|---|---|
| Entry | Catalyst | 1a (μmol) | % S |
| a Reaction conditions: 2 mL (22.4 mmol) of benzene, 2 mL (24 mmol) of THF, 50 mg of photocatalyst or 100 mg of the blended catalyst (50 mg of each the photocatalyst and the metal catalyst) was used, the reaction time was 1 h. The amount of 1a was approximately determined from the calibration curve of 4-(tetrahydrofuran-2-yl)benzonitrile. Other conditions and selectivity calculations are mentioned in the Experimental section.b Not applicable.c The reaction was done without the TiO2 sample. | |||
| 1 | Pd(3.0)/TiO2 | 8.9 | >99 |
| 2 | TiO2 | 0.0 | n.a.b |
| 3 | Pd(2.0)Au(1.0)/TiO2 | 1.4 | >99 |
| 4 | TiO2 + Pd(3.0)/Al2O3 | 11.9 | >99 |
| 5c | Pd(3.0)/Al2O3 | 0.0 | n.a. |

Attempts were made to modify the Pd metal part by simultaneous loading Au on the TiO2 photocatalyst so as to make bimetallic Pd–Au nanoparticles (Pd(2.0)Au(1.0)/TiO2). However, this sample gave much lower yield of 1a (Table 1, entry 3) than the monometallic Pd(3.0)/TiO2 sample (Table 1, entry 1). Since this modification was not helpful for the enhancement of catalytic activity, we decided to separate the metal part from the TiO2 part and designed a blended catalyst consisting of a TiO2 photocatalyst and an Al2O3 supported metal catalyst.
At the beginning, the reaction was done with a blended catalyst consisting of a pristine TiO2 sample and a monometallic Pd(3.0)/Al2O3 sample (Table 1, entry 4). The reaction gave a much larger amount of 1a and a slightly smaller amount of hydrogen (not shown) than that with the Pd(3.0)/TiO2 sample (Table 1, entries 1 and 4). This means that the Pd nanoparticles can contribute to the formation of 1a even deposited on a photo-inactive support like Al2O3 instead of TiO2, or rather work more efficiently on Al2O3. This result showed that, in this reaction system, the Pd nanoparticles function not just as an electron receiver on the TiO2 photocatalyst but also as a catalyst. Moreover, the reaction done with the Pd(3.0)/Al2O3 sample alone, under light irradiation did not yield 1a (Table 1, entry 5), ruling out the possibility of plasmonic photocatalysis by the Pd metal nanoparticles as reported in some studies,15 and confirming that the coexistent TiO2 photocatalyst is essential. These results suggest that the formation of 1a is a hybrid of TiO2 photocatalysis and Pd metal catalysis. This revelation means that we can modify the property of the two catalytic components independently to improve the product yield. This time we have focused on the modification of the Pd catalyst part, and examined the bimetallic Pd(x)Au(y)/Al2O3 catalysts.
Photocatalytic direct DCC between benzene and THF with a TiO2 photocatalyst blended with Pd(x)Au(y)/Al2O3 catalysts
Table 2 shows the results of the DCC between benzene and THF with blended catalysts consisting of the TiO2 photocatalyst and various bimetallic Pd(x)Au(y)/Al2O3 catalysts. When compared to the result with the monometallic Pd(3.0)/Al2O3 catalyst (Table 1, entry 4), an introduction of a small amount of Au to Pd increased the yield of 1a (Table 2, entries 1 and 2). However, a further increase in the Au content had negative effect on the yield (Table 2, entries 3 and 4) and the monometallic Au(3.0)/Al2O3 catalyst was inactive for the formation of 1a (Table 2, entry 5). These results indicate that Pd was the catalytically active component of these bimetallic samples. Also, an optimum ratio of Pd and Au is needed for the high activity of the bimetallic samples. This suggests that the increase of the Au content would withdraw the electron density of the active Pd atoms to enhance the reaction, and then the decreasing number of the active Pd sites would decrease the activity. Furthermore, the bimetallic Pd(2.0)Au(1.0)/Al2O3 catalyst did not yield 1a or H2 gas in the absence of the TiO2 photocatalyst (Table 2, entry 6), confirming that they could not catalyse the DCC reaction alone and required TiO2 photocatalysis. This result also ruled out the possibility of plasmonic catalysis by the bimetallic samples for the production of 1a and H2.| Entry | Photocatalyst | Al2O3 supported catalyst | 1a (μmol) | % S |
|---|---|---|---|---|
| a All reaction conditions and abbreviations were the same as those described in Table 1.b Not applicable.c The reaction was done without the TiO2 sample. | ||||
| 1 | TiO2 | Pd(2.5)Au(0.5) | 13.1 | >99 |
| 2 | TiO2 | Pd(2.0)Au(1.0) | 15.2 | >99 |
| 3 | TiO2 | Pd(1.5)Au(1.5) | 5.8 | >99 |
| 4 | TiO2 | Pd(0.5)Au(2.5) | 2.2 | >99 |
| 5 | TiO2 | Au(3.0) | 0.0 | n.a.b |
| 6c | None | Pd(2.0)Au(1.0) | 0.0 | n.a. |
Scope of arene molecule for the photocatalytic direct DCC with THF
The scope of the substituted arenes was examined for the photocatalytic direct DCC reaction with THF by using different catalysts. The results are shown in Tables S1–S4 in the ESI.† Fig. 7 shows the representative results of photocatalytic direct DCC between different arenes (1–5) and THF with the blended catalyst consisting of a TiO2 photocatalyst and an Al2O3 supported Pd–Au bimetallic catalyst. Substituted benzenes with electron withdrawing (2 and 3) and electron releasing (4 and 5) substituents successfully underwent the DCC reaction with THF. Benzaldehyde (2) and a benzonitrile (3) noticeably gave appreciable yields of the DCC products with high selectivity. The details such as the regioselectivity of the DCC products have not been fully clarified yet since the syntheses of the reference products are not easy by the conventional methods. In other words, these results show the importance of the further development of these new catalyses for the photocatalytic DCC reactions. | ||
| Fig. 7 Photocatalytic direct DCC between various arenes and THF with a blended catalyst consisting of a TiO2 photocatalyst and a bimetallic Pd(2.0)Au(1.0)/Al2O3 catalyst. The general reaction conditions and % S calculation were the same as Table 1. The definition and calculation of the yield (% Y) is mentioned in the Experimental section. Briefly, 50 μL of an arene, 2 mL (24 mmol) of THF, and 25 mg of each catalyst (totally 50 mg of the blended catalyst) was used for 12 h reaction. 50 μL of each arene (1–5) corresponds to 541, 490, 485, 470, and 548 μmol, respectively. | ||
Benzaldehyde (2) and THF reacted to give a mixture of ortho, meta, and para substituted products (collectively shown as 2a) along with the oxidation and reduction products of 2, i.e., benzoic acid (2b) and benzyl alcohol (2c), respectively, as undesired by-products (Table S1†). As mentioned in the ESI,† benzoic acid (2b) was the major product of this reaction, which resulted in a low selectivity to 2a, the DCC products. The autoxidation of benzaldehyde (2) to benzoic acid (2a) is well known and the presence of adsorbed water on the TiO2 photocatalyst might promote this reaction.16 Although both the pristine TiO2 and Pd(3.0)/TiO2 samples showed low selectivity to 2a (Table S1,† entries 1 and 2), the blended catalyst consisting of the TiO2 photocatalyst and the bimetallic Pd(2.0)Au(1.0)/Al2O3 catalyst slightly increased the formation of the DCC products and suppressed the formation of the side products, which improved the selectivity to 2a (Table S1,† entry 5). Change in the reaction conditions resulted in a high selectivity of 75% (Fig. 7, 2a).
The reaction between benzonitrile (3) and THF also gave a mixture of three DCC products (collectively shown as 3a), along with benzamide (3b) (Table S2†). The blended catalyst with the bimetallic catalyst again exhibited the highest activity for the formation of the DCC products (Table S2,† entry 5), although the selectivity was lower than the photocatalyst alone due to the increased production of 3b (Table S2,† entry 3). It is notable that, however, the improved reaction condition resulted in almost complete DCC selectivity (% S > 99, Fig. 7, 3a).
The photocatalytic direct DCC reaction of toluene (4) and aniline (5) with THF also proceeded (Tables S3 and S4†). Both reactions gave a mixture of the corresponding DCC product isomers, 4a and 5a, along with some side products. Photocatalytic oxidation of toluene (4) by adsorbed water to benzaldehyde (4b) and benzyl alcohol (4c) was a competitive reaction to the DCC to give 4a. The blended catalyst with the bimetallic catalyst drastically improved the selectivity to the DCC products (Table S3,† entry 5). Under the optimised conditions shown in Fig. 7, 90% selectivity to the DCC products 4a was obtained for toluene. The reaction between aniline (5) and THF, on the contrary, yielded DCC products with poor selectivity. Due to the high reactivity of the NH2 group of aniline (5), the homocoupling of aniline to give di(phenyl)diazine (5b) was the main reaction rather than the DCC between aniline and THF (Table S4†).17 NH2 group would be readily oxidised by the photogenerated holes on TiO2, which is suggested from the fact that ammonia can be easily oxidised to become amide radicals.18 This resulted in the very low selectivity to the DCC products, 5a, even under the optimised conditions (Fig. 7).
Table 3 shows the comparison of the total yield (%) of the DCC products (μmol) and selectivity (%, in the parenthesis) for the reaction using above-mentioned substrates (1–5). For the arene without any substituent (1) and the ones with electron withdrawing substituents like CHO (2) and CN (3), the blended catalyst consisting of the TiO2 photocatalyst and the bimetallic Pd(2.0)Au(1.0)/Al2O3 catalyst exhibited a higher activity to form the corresponding DCC products than the metal loaded TiO2 photocatalysts (Pd(3.0)/TiO2 and Pd(2.0)Au(1.0)/TiO2) and the blended catalyst with the monometallic catalyst (TiO2 + Pd(3.0)/Al2O3) (Table 3, entries 1–3). So, the new blended catalyst, with the bimetallic catalyst, developed in this study is the most selective and active catalyst for the photocatalytic direct DCC reactions between these arenes and THF. Also, the lower reactivity of the arenes with electron releasing substituents (4 and 5) (Table 3, entries 4 and 5) indicates that an electron deficient arene is more reactive towards the cross-coupling reaction than an electron rich arene. These results suggest that the photogenerated THF radical species is nucleophilic, so that it would prefer to react with an electron deficient aromatic ring. This property of THF radical is opposite to a photocatalytically generated electrophilic OH radical observed in our previous study.17a These results are consistent with the fact that the electron deficient Pd species in the Pd–Au bimetallic catalyst is more active than pure Pd metal catalyst for the DCC reactions. This electron deficient Pd species can withdraw the electron density from an adsorbed arene molecule to accelerate its reaction with the nucleophilic THF radical.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Arene | DCC products | Yield of DCC products, Y (%)b | |||
| Pd(3.0)/TiO2 | Pd(2.0)Au(1.0)/TiO2 | TiO2 + Pd(3.0)/Al2O3 | TiO2 + Pd(2.0)Au(1.0)/Al2O3 | |||
| a 2 mL (19–24 mmol) of arene, 2 mL (24 mmol) of THF, and 50 mg of the photocatalyst or 100 mg of the blended catalyst were used, other conditions were the same as those described in Table 1.b The total yield, Y (%), of DCC products. The value in parenthesis shows % S. The definition of % Y and % S is mentioned in the Experimental section.c 2 mL (22.4 mmol) of benzene (1).d 2 mL (19.9 mmol) of benzaldehyde (2).e 2 mL (19.4 mmol) of benzonitrile (3).f 2 mL (19.01 mmol) of toluene (4).g 2 mL (21.9 mmol) of aniline (5). | ||||||
| 1c | 1 | 1a | 0.04(>99) | 0.006(>99) | 0.05(>99) | 0.06(>99) |
| 2d | 2 | 2a | 1.24(36) | 0.42(32) | 1.06(43) | 1.73(46) |
| 3e | 3 | 3a | 0.10(80) | 0.08(82) | 0.06(68) | 0.27(57) |
| 4f | 4 | 4a | 0.05(64) | 0.02(74) | 0.01(50) | 0.03(90) |
| 5g | 5 | 5a | 0.02(19) | 0.003(6) | 0.02(14) | 0.02(23) |

Proposed reaction mechanism for the photocatalytic direct DCC between arenes and THF
Based on the mechanism proposed in the previous study3c and the above results, the following reaction mechanism can be proposed for the photocatalytic direct DCC reaction between arenes and THF with the blended catalyst, which is a hybrid of TiO2 photocatalysis and metal catalysis by Pd–Au bimetallic nanoparticles (Fig. 8). The photo-excitation of TiO2 generates electrons and holes in the conduction band and the valence band, respectively (i). The photogenerated hole oxidizes the THF molecule to form a radical species and proton (ii). The proton is reduced to hydrogen radical by the photoexcited electron on TiO2 (iii). The THF radical can migrate to the bimetallic catalyst and attack to the carbon in the aromatic ring of the activated arene molecule, adsorbed on the Pd–Au bimetallic catalyst (iv), which gives the DCC product with the elimination of a hydrogen radical (v). The two hydrogen radicals can combine to give a hydrogen molecule either on the TiO2 surface or on the bimetallic catalyst (vi). | ||
| Fig. 8 Proposed mechanism for the photocatalytic direct DCC between arenes and THF by the blended catalyst consisting of a TiO2 photocatalyst and a supported Pd–Au bimetallic catalyst. | ||
As mentioned before, electron deficient arenes exhibited the highest activity for this reaction. So, the nature of Pd catalysis in this photocatalytic reaction is proposed to be the activation of the aromatic ring by withdrawing its electron density, which facilitates its reaction with a photogenerated THF radical. The presence of an electron deficient Pd species, formed by the introduction of Au, would promote this reaction.
According to this mechanism for the DCC, it is also proposed that the THF radical species, photocatalytically formed on the TiO2 surface, can migrate to the separated metal catalyst. This suggests that the life of the THF radical species is not so short in these condition. Additionally, the coupling of two THF radical species can give the homocoupling products like octahydro-2,2′-bifuran.3e But, as results show, this was a very minor reaction, meaning that the THF radical would selectively react with the activated arene molecule to give the DCC products.
Conclusions
In the present work, we found that the photocatalytic direct DCC between various arenes and THF was a hybrid of TiO2 photocatalysis and Pd metal catalysis. The Pd metal catalyst can work even if they were on a photo-inactive support like Al2O3. Based on these findings, we developed a novel blended catalyst consisting of a TiO2 photocatalyst and an Al2O3 supported Pd–Au bimetallic catalyst that exhibited higher activity and selectivity for the photocatalytic direct DCC between arenes and THF than the conventional metal loaded TiO2 photocatalyst alone. Although the reported yields are still low, this work has provided some new insights about the mechanisms of the photocatalytic organic synthesis reactions and opened new prospects for the catalyst design. This blended catalyst has wide flexibility for modification, like, the photocatalyst part by using a co-catalyst or by changing the photocatalyst itself, the metal catalyst part by changing the loading amount, dispersion, or the metal component, and also the ratio of the two catalytic components. Thus, the blended catalyst can be further improved and become available for other photocatalytic direct DCC reactions and also other reaction systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. Ken-ichi Fujita and his laboratory members at Kyoto University for their help to synthesize the specific compound. We thank Dr Tsutomu Kiyomura, Kyoto University, for TEM measurements which was supported by Kyoto University Nano Technology Hub in "Nanotechnology Platform Project" sponsored by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. We acknowledge the kind help of the laboratory members for XAFS measurements (Spring-8, Hyogo, Japan, proposal no.: 2017A1477, 2016A1170). A. Tyagi would like to thank JICA for providing the financial assistance under IITH–JICA ‘Friendship’ project.Notes and references
- (a) C. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef PubMed.
- (a) J. J. H. B. Sattler, J. R. Martinez, E. S. Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613 CrossRef PubMed; (b) M. R. Nabid, Y. Bide and B. Etemadi, New J. Chem., 2017, 41, 10773 RSC; (c) K. A. Goulas, Y. Song, G. R. Johnson, J. P. Chen, A. A. Gokhale, L. C. Grabow and F. D. Toste, Catal. Sci. Technol., 2018, 8, 314 RSC.
- (a) C. Vila and M. Rueping, Green Chem., 2013, 15, 2056 RSC; (b) H. Yoshida, Y. Fujimura, H. Yuzawa, J. Kumagai and T. Yoshida, Chem. Commun., 2013, 49, 3793 RSC; (c) A. Tyagi, T. Matsumoto, T. Kato and H. Yoshida, Catal. Sci. Technol., 2016, 6, 4577 RSC; (d) E. Wada, T. Takeuchi, Y. Fujimura, A. Tyagi and H. Yoshida, Catal. Sci. Technol., 2017, 7, 2457 RSC; (e) A. Tyagi, A. Yamamoto, T. Kato and H. Yoshida, Catal. Sci. Technol., 2017, 7, 2616 RSC.
- (a) A. J. Bard, Science, 1980, 207, 139 CrossRef PubMed; (b) C. M. Wang, A. Heller and H. Gerischer, J. Am. Chem. Soc., 1992, 114, 5230 CrossRef; (c) S. Vaidyanathan, W. E. Eduardo and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943 CrossRef PubMed; (d) J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900 CrossRef PubMed.
- (a) B. Ohtani, K. Iwai, S. Nishimoto and S. Sato, J. Phys. Chem. B, 1997, 17, 3349 CrossRef; (b) J. M. Herrmann, Top. Catal., 2006, 39, 3 CrossRef; (c) Y. Shiraishi, Y. Sugano, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2012, 49, 1656 CrossRef PubMed.
- (a) J. G. Yu, H. G. Yu, B. Cheng, X. J. Zhao, J. C. Yu and W. K. Ho, J. Phys. Chem. B, 2003, 107, 13871 CrossRef; (b) J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766 CrossRef PubMed; (c) C. H. Lin, J. H. Chao, C. H. Liu, J. C. Chang and F. C. Wang, Langmuir, 2008, 24, 9970 CrossRef PubMed.
- (a) W. Zhang, Y. Wang, Z. Wang, Z. Zhong and R. Xu, Chem. Commun., 2010, 46, 7631 RSC; (b) H. Cheng, X. J. Lv, S. Cao, Z. Y. Zhao, Y. Chen and W. F. Fu, Sci. Rep., 2016, 6, 19846 CrossRef PubMed; (c) X. Xu, G. Liu, C. Randorn and J. T. S. Irvine, Int. J. Hydrogen Energy, 2011, 36, 13501 CrossRef.
- (a) K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2001, 2416 RSC; (b) Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536 CrossRef; (c) K. Maeda, ACS Catal., 2013, 3, 1486 CrossRef; (d) E. Wada, A. Tyagi, A. Yamamoto and H. Yoshida, Photochem. Photobiol. Sci., 2017, 16, 1744 RSC.
- K. Taniguchi, X. Jin, K. Yamaguchi and N. Mizuno, Chem. Commun., 2015, 51, 14969 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef PubMed.
- D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715 CrossRef PubMed.
- L. Samain, A. Jaworski, M. Edén, D. M. Ladd, D. K. Seo, F. J. G. Gracia and U. Häussermann, J. Solid State Chem., 2014, 217, 1 CrossRef.
- A. F. Lee, C. J. Baddeley, C. Hardcare, R. M. Ormerod and R. M. Lambert, J. Phys. Chem., 1995, 99, 6096 CrossRef.
- H. Miura, K. Endo, R. Ogawa and T. Shishido, ACS Catal., 2017, 7, 1543 CrossRef.
- (a) T. T. Trinh, R. Sato, M. Sakamoto, Y. Fujiyoshi, M. Haruta, H. Kurata and T. Teranishi, Nanoscale, 2015, 7, 12435 RSC; (b) W. Yang, Y. Xiong, L. Zou, Z. Zou, D. Li, Q. Mi, Y. Wang and H. Yang, Nanoscale Res. Lett., 2016, 11, 283 CrossRef PubMed.
- (a) M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, Chem. Commun., 2010, 46, 145 RSC; (b) M. Sankar, E. Nowicka, E. Carter, D. M. Murphy, D. W. Knight, D. Bethell and G. J. Hutchings, Nat. Commun., 2014, 5, 3332 CrossRef PubMed.
- (a) H. Yuzawa, M. Aoki, K. Otake, T. Hattori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116, 25376 CrossRef; (b) H. Yuzawa, J. Kumagai and H. Yoshida, J. Phys. Chem. C, 2013, 117, 11047 CrossRef.
- H. Yuzawa, T. Mori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116, 4126 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 4-(tetrahydrofuran-2-yl)benzonitrile and the results of photocatalytic direct DCC between various arenes and THF are mentioned in the ESI. See DOI: 10.1039/c8ra02948b |
| This journal is © The Royal Society of Chemistry 2018 |