
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of partially oxidized Mn3O4-functionalized carbon cathodes for rechargeable Li–O2 batteries†
Juhyoung Kim ,
Inhan Kang,
Soyeon Kim and
Jungwon Kang*
,
Inhan Kang,
Soyeon Kim and
Jungwon Kang*
Department of Advanced Materials Science and Engineering, Mokpo National University, 61 dorim-ri, 1666 Yeongsan-ro, Cheonggye-myeon, Muan-gun, Jeonnam, South Korea. E-mail: jwkang17@mokpo.ac.kr
First published on 19th June 2018
Abstract
High charging overpotential (low energy efficiency) is one of the most important challenges preventing the use of current nonaqueous Li–O2 batteries. This study demonstrates direct in situ-incorporation of metal oxides on carbon during synthesis and the associated application to nonaqueous Li–O2 battery catalysts. The partially oxidized Mn3O4 (Mn3O4/Mn5O8)-incorporating carbon cathode shows an average overpotential reduction of ∼8% charge/discharge during 40 cycles in a rechargeable nonaqueous Li–O2 cell. Here, we suggested the possibility that only a small amount of the oxide species (<5%) could show catalytic effects during charge in a rechargeable Li–O2 cell.
Introduction
Lithium–oxygen (Li–O2) batteries have attracted considerable scientific attention due to their high theoretical specific energy density (∼3 kW h kg−1), which could possibly satisfy the demand for use in improved electric vehicles, if realised.1–4 However, despite this high energy density, Li–O2 batteries present many scientific challenges that have also prevented their commercialization, such as their large polarization, low round-trip efficiency and poor cyclability.5–7 Typically, a reachargeable nonaqueous Li–O2 battery is composed of a Li metal anode, a nonaqueous Li+ ion conducting electrolyte and a porous cathode and is based on a net electrochemical reaction (2Li+ + O2 + 2e− ↔ Li2O2) with a thermodynamically determined reversible potential (U0 = 2.96 V, versus Li/Li+).8–10 However, there are large differences between the theoretical and empirical potential, and this problem presents one of the greatest challenges. Early studies on Li–O2 batteries showed a modest overpotential (∼0.3 V) for discharging but a high overpotential (∼1.5 V),11,12 and the charge overpotential is particularly large due to decomposition of the insulating discharge product, Li2O2.13 This overpotential results in a reduction in energy efficiency (the energy released during discharging/the energy required during charging).Many efforts have been made to suppress this energy loss during either the discharge or recharge process by adding a catalytic substance, such as a metal and a metal oxide into the carbon.14 However, owing to the catalytic effects of certain materials, the use of precious metals (Pt, Au, Ru etc.) and the complex routes used to synthesize metal oxides are undesirable.15,16 In addition, when only mechanical mixing of catalysts containing carbon is employed,17 electronic conductivity of the cathodes and inappropriate contact between the catalyst/carbon interfaces results. Furthermore, the metal/carbon ratio needs to be carefully controlled for heavy metals during the mixing process, as it can cause a decrease in the gravimetric energy density.18,19
This study demonstrates the direct in situ-incorporation of metal oxides on carbon during synthesis and the associated application to nonaqueous Li–O2 battery catalysts. The partially oxidized Mn3O4 (Mn3O4/Mn5O8)-incorporated carbon cathode shows an average reduction of ∼8% charge/discharge overpotential during 40 cycles in a rechargeable nonaqueous Li–O2 cell.
Experimental
Preparation of materials
The detailed preparation procedure for partially oxidized Mn3O4 (Mn3O4/Mn5O8)-incorporated carbon is shown in Scheme 1. Manganese(II) acetate tetrahydrate ((CH3COO)2Mn·4H2O-98%, Daejung) was dissolved in ethanol (99.9%, Daejung). Carbon paper (P50, AvCarb®) was dipped in the manganese acetate solution and then dried, and the manganese acetate-immersed P50 was heated at 300 °C in air for 10 h. Manganese oxide-functionalized P50 was obtained, and the manganese oxide was determined as being partially oxidized Mn3O4 (Mn3O4/Mn5O8)-functionalized carbon using X-ray photoelectron spectroscopy (XPS), transmission electron microscope (TEM), and X-ray diffraction (XRD) patterns. | ||
| Scheme 1 Partially oxidized Mn3O4 (Mn3O4/Mn5O8)-incorporated carbon preparation procedure. | ||
TEGDME (tetraethylene glycol dimethyl ether, ≥99%) was purchased from Sigma-Aldrich and dried over a freshly activated molecular sieve with a 4 Å pore size. LiTFSI (bis(trifluoromethane)sulfonimide lithium salt, Sigma-Aldrich) was dried in a vacuum oven at 150 °C for 24 h. P50 carbon paper was purchased from AvCarb®. Carbon cathodes were dried in a vacuum oven (150 °C) and transferred to an Ar-filled glove box for cell fabrication.
Characterization of materials
Morphology and elemental mapping were conducted using field-emission transmission electron microscopy (FETEM) (Philips Tecnai F20) at 200 kV; XPS analysis was conducted using Al Kα as the X-ray source (Thermo VG Scientific instrument, Multilab 2000); the powder X-ray diffraction pattern (XRD) of prepared samples was measured using a PANalytical X'Pert PRO Multi-Purpose X-ray Diffractometer; and thermal analysis was conducted using a Mettler Toledo. X-ray absorption spectroscopy (XAS) was performed using a Rigaku Model R-XAS.Fabrication of Li–O2 cells
Swagelok-type cells were composed of a Li metal anode (Honjo Metal Co.) with carbon (AvCarb P50) as the prepared cathode and the GF (GF/C™, Whatman™) as a separator and the cell fabricated in an Ar-filled glove box (MBraun, H2O < 1 ppm). High purity oxygen gas (>99.999%) was blown into the carbon cathode through a capillary (inlet) attached to the upper side of the Swagelok®-type cells and then out of the cathode through another capillary (outlet), to replace argon with oxygen within the cell. An oxygen environment of ∼1.5 bar was maintained during discharging and charging by opening the inlet capillary and closing the outlet. Electrochemical properties for the assembled Li–O2 cells were analysed using a VMP3 potentiostat (Biologic Science Instrument).Results and discussion
Fig. 1 shows TG-DTA curves of (a) P50 in nitrogen and (b) manganese(II) acetate tetrahydrate in air. The TG curve (Fig. 1a, green) of P50 shows a weight loss of ∼30% at ∼600 °C, and the corresponding endothermic peak is shown in the DTA curve (Fig. 1a, blue). This weight loss implies the occurrence of moisture desorption, thermal desorption of organic and inorganic matter absorbed by activated carbon, and oxidation of amorphous carbon.20 In the TG-DTA curves shown in Fig. 1b, it is evident that manganese(II) acetate tetrahydrate begins to lose water at around room temperature to produce anhydrous salts that are then decomposed to MnO at about 320 °C.21 To conduct a thermal analysis, an annealing temperature of 300 °C was used for the direct in situ-incorporation of manganese oxides on carbon during synthesis, as carbon paper (P50) is drastically oxidized at temperatures over 400 °C. Fragments of partially oxidized P50 were observable in the sample heated at 400 °C; this sample showed poor electrochemical properties, as evidenced in Fig. S1 (in the ESI†).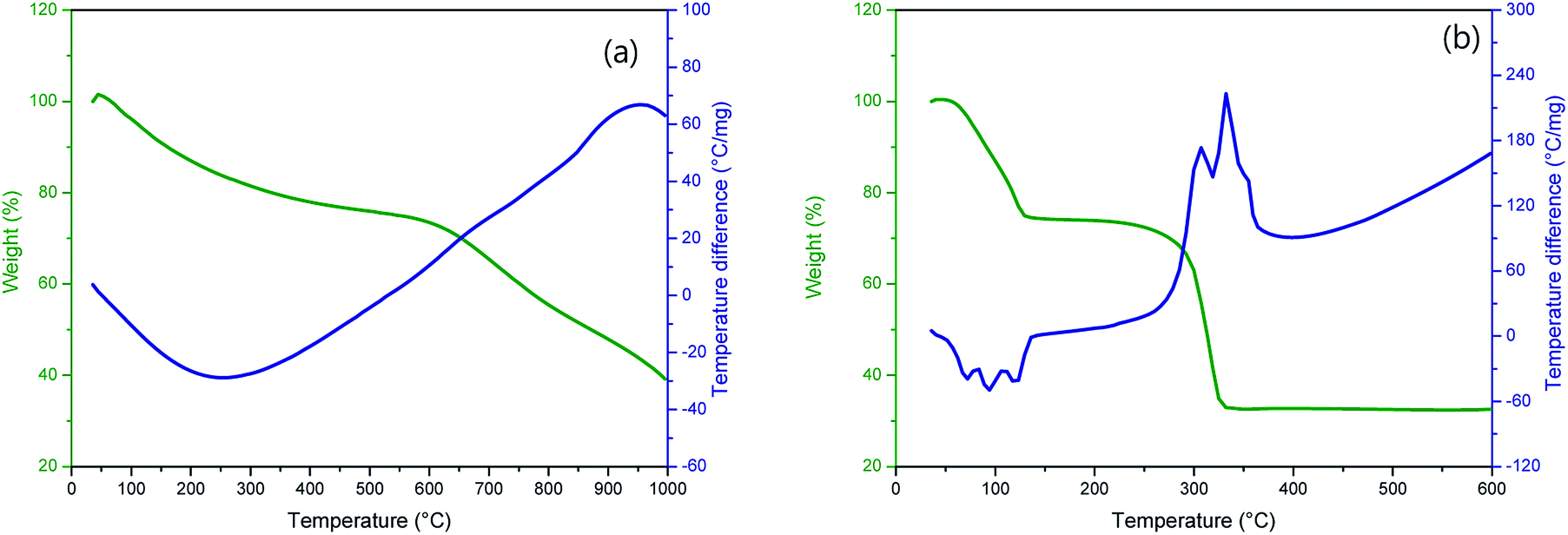 | ||
| Fig. 1 TG-DTA curves of (a) P50 in nitrogen, and (b) manganese(II) acetate tetrahydrate in air. | ||
The incorporation of manganese oxide to carbon in the final product was confirmed by X-ray diffraction (XRD); however, it was not possible to obtain a distinct diffraction pattern of manganese oxide, as shown in Fig. S2 (in the ESI†), as the manganese-oxide included only a few amount (4.33 w%, element analysis by TEM). Fig. 2 shows the XRD pattern of Mn-acetate when heated only to 300 °C; the powder is verified as a composite of major-Mn3O4/minor-Mn5O8. Based on this result, it was hypothesized that the final product of partially oxidized Mn3O4 (Mn3O4/Mn5O8)-incorporated carbon could be obtained (as shown in Scheme 1).
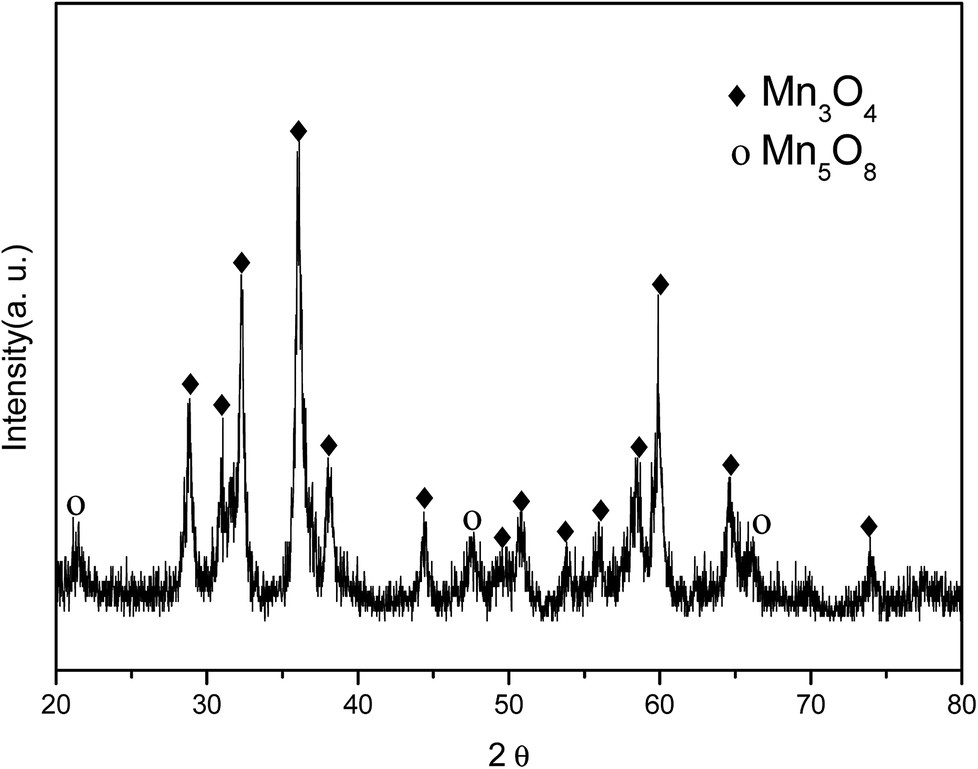 | ||
| Fig. 2 XRD pattern of Mn-acetate heated at 300 °C for 10 h in air. | ||
The precise stoichiometry of the manganese oxide-incorporated carbon obtained was then further investigated using XPS. Fig. 3 shows XPS survey scan analysis at a binding energy range between 0 and 1400 eV, where there is no evidence of Mn-related signals in the pristine P50 (Fig. 3a). However, the manganese oxide-functionalized P50 clearly shows doublet splitting of Mn 2p3/2 and Mn 2p1/2 at binding energies of 642.18 eV and 654.21 eV, respectively, as shown in Fig. 3b.22 Various Mn ionic species exist in the sample of partially oxidized manganese oxide-incorporated carbon, and the position and intensity of the XPS peaks are considered to identify a consistent oxide formula.
 | ||
| Fig. 3 XPS survey scan analysis of (a) pristine P50 and (b) partially oxidized Mn3O4-functionalized P50. | ||
Fig. 4 shows the curve-fitted XPS Mn 2p. There is no signal that indicates a series of manganese oxides in the pristine P50 sample (Fig. 4a). Fig. 4b shows the curve-fitted Mn 2p of the partially oxidized Mn3O4-functionalized P50, which was prepared as illustrated in Scheme 1 (with a heating temperature of 300 °C). Fig. 4c shows results of Mn-acetate powder heated at 300 °C for 10 h in air, which was examined by XRD and determined as a composite of major-Mn3O4/minor-Mn5O8. XRD did not confirm the presence of manganese oxide in the sample of the partially oxidized Mn3O4-functionalized P50, due to the low amount of manganese oxide present. However, the XPS signal was analogous in samples of the partially oxidized Mn3O4-functionalized P50 (Fig. 4b) and the powder of Mn-acetate heated at 300 °C for 10 h in air (Fig. 4c). The oxidation states of the Mn atom in manganese oxides were then further examined by peak deconvolution, on the assumption that the intensity ratios related to Mn2+ and Mn4+ have corresponding peak positions of 641.98 and 643.52 eV, respectively.
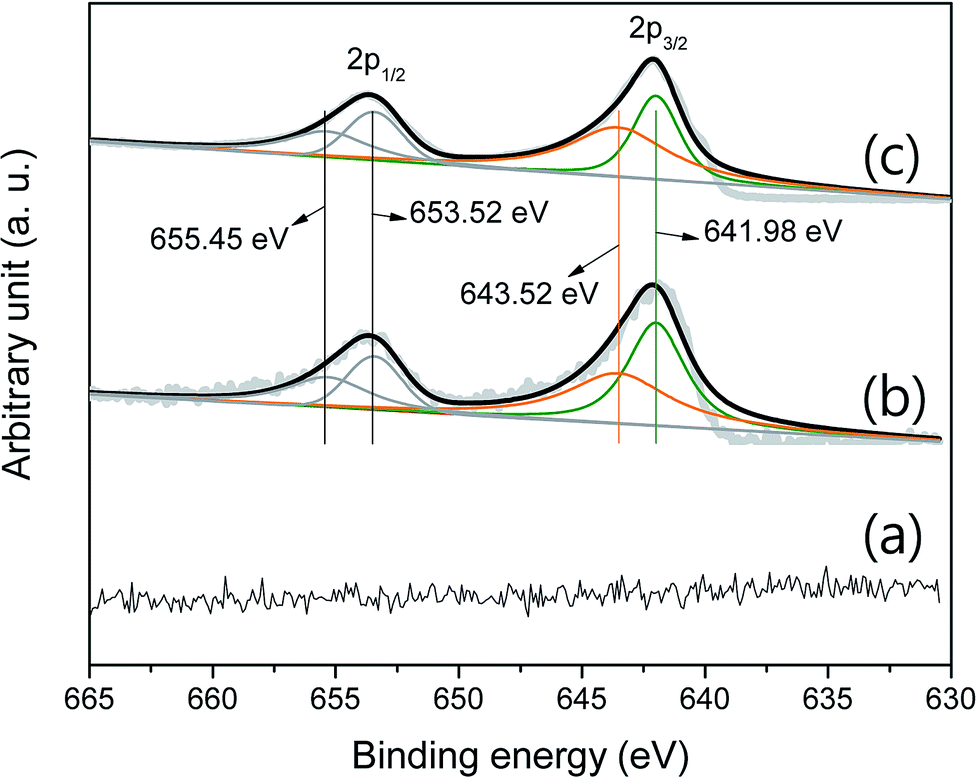 | ||
| Fig. 4 Curve-fitted XPS Mn 2p of (a) pristine P50, (b) partially oxidized Mn3O4-functionalized P50, and (c) powder of Mn-acetate heated at 300 °C for 10 h in air. | ||
Based on the similarity of the XPS signal for both samples (Fig. 4b and c), it was considered that the species of functionalized manganese oxides were a composite of major-Mn3O4/minor-Mn5O8; this was confirmed by XRD analysis.
To elucidate further on the oxidation state of the manganese oxide-incorporated carbon obtained, X-ray absorption near-edge structure (XANES) analysis was conducted. Fig. 5 shows XANES spectra of the partially oxidized Mn3O4-functionalized P50 (a), the powder of Mn-acetate heated at 300 °C for 10 h in air (b) and the references corresponding to Mn3O4, Mn2O3, MnO2 and MnO. The observed Mn valence in the partially oxidized Mn3O4-functionalized P50 (Fig. 5a) and the powder of Mn-acetate heated at 300 °C for 10 h in air (Fig. 5b) shows similarity, and the XANES spectra of both samples are analogous to the Mn3O4 reference (Fig. 5). The XANES spectra of Mn2O3 (Mn+3 state) shows slightly higher state than the partially oxidized Mn3O4-functionalized P50 of which oxidation state is clearly less than Mn+4 (MnO2), but greater than Mn+2 (MnO) as depicted in Fig. 4. This trend is clearly in accordance with the XPS characterization.
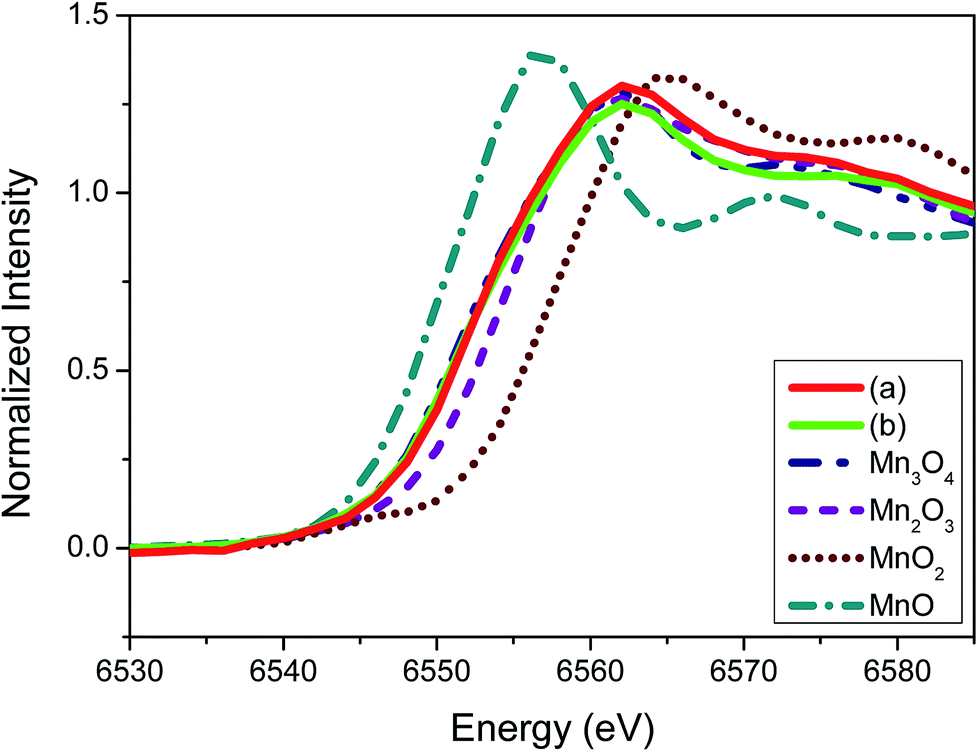 | ||
| Fig. 5 XANES spectra of (a) the partially oxidized Mn3O4-functionalized P50, (b) the powder of Mn-acetate heated at 300 °C for 10 h in air and, the references corresponding to Mn3O4, Mn2O3, MnO2 and MnO. | ||
The extended X-ray absorption fine structure (EXAFS) spectra of the partially oxidized Mn3O4-functionalized P50 (Fig. 6) provide further information about the related phases. Fig. 6 shows the EXAFS spectra of the partially oxidized Mn3O4-functionalized P50 (a), the powder of Mn-acetate heated at 300 °C for 10 h in air (b) and the references corresponding to Mn3O4, Mn2O3, MnO2 and MnO. The EXAFS results are also in the similar context for defining our prepared sample as the partially oxidized Mn3O4-functionalized P50. The EXAFS spectra of the partially oxidized Mn3O4-functionalized P50 (Fig. 6a) is analogous to the sample of Mn-acetate heated at 300 °C for 10 h in air (Fig. 6b) of which feature well matches that of Mn3O4 (Fig. 6). Unfortunately the detailed local environment of Mn in the partially oxidized Mn3O4-functionalized P50 cannot be easily investigated by EXAFS due to its little amount of manganese oxide contents. This XANES and EXAFS results clearly follow the trend observed with XPS and XRD characterization in which our prepared sample in the procedure (Scheme 1) defined the partially oxidized Mn3O4-functionalized P50.
 | ||
| Fig. 6 XANES spectra of (a) the partially oxidized Mn3O4-functionalized P50, (b) the powder of Mn-acetate heated at 300 °C for 10 h in air and, the references corresponding to Mn3O4, Mn2O3, MnO2 and MnO. | ||
The particles and shape of partially oxidized Mn3O4-functionalized P50 were confirmed using HR-TEM. Fig. 7 shows HR-TEM images of (a and b) pristine P50 and (c and d) partially oxidized Mn3O4-functionalized P50. The absence of particles is evident, and only amorphous carbon exists on the carbon nanofiber in the pristine P50 sample (Fig. 5a and b). However, Fig. 7c and d show that the nanoparticles of manganese oxides are well dispersed in amorphous carbon in the partially oxidized Mn3O4-functionalized P50. The solid contact formed during the in situ method between carbon and oxide-particles led to catalytic effects (reduction of the overpotential), despite the inclusion of a few manganese oxides in the carbon for application to rechargeable Li–O2 batteries. A dark-field TEM image (Fig. 8a) also shows amorphous carbon on carbon nanofiber features, and this was confirmed by elemental mapping for C (Fig. 8c), O (Fig. 8d) and Mn (Fig. 8e), respectively. In pristine P50 (Fig. S2 in the ESI†), there are no manganese elements, however the Mn element is seen dispersed on the assignment of the dark field image (Fig. 8a), which is also shown in Fig. 8e. This evidence also demonstrates that manganese oxide is well-functionalized on the carbon surface.
 | ||
| Fig. 7 HR-TEM images of (a and b) pristine P50 and (c and d) partially oxidized Mn3O4-functionalized P50. | ||
 | ||
| Fig. 8 (a) Dark field (b) elemental mapping images and corresponding elemental mapping images for (c) C, (d) O, and (e) Mn of partially oxidized Mn3O4-functionalized P50. | ||
Fig. 9 shows the electrochemical properties at a current of 0.1 mA for 5 h (limited capacity: 0.5 mA h); (a) first discharge/charge profiles and (b) cycle performances and energy efficiency for nonaqueous Li–O2 cells. The pristine and partially oxidized Mn3O4-functionalized P50 cathodes in the Li–O2 cells (Fig. 9a) show an average discharge potential of 2.6 V, which is similar to that determined in previous reports.22–24 However, Fig. 9a shows a reduction in the charge overpotential: during charging, the energy efficiency of ∼8% is increased in the cell where partially oxidized Mn3O4-functionalized P50 is applied to cathodes (red), compared to the pristine one (black). This increased energy efficiency is maintained during 40 cycles, although the degree decreases as the cycling proceeds, as shown in Fig. 9b.
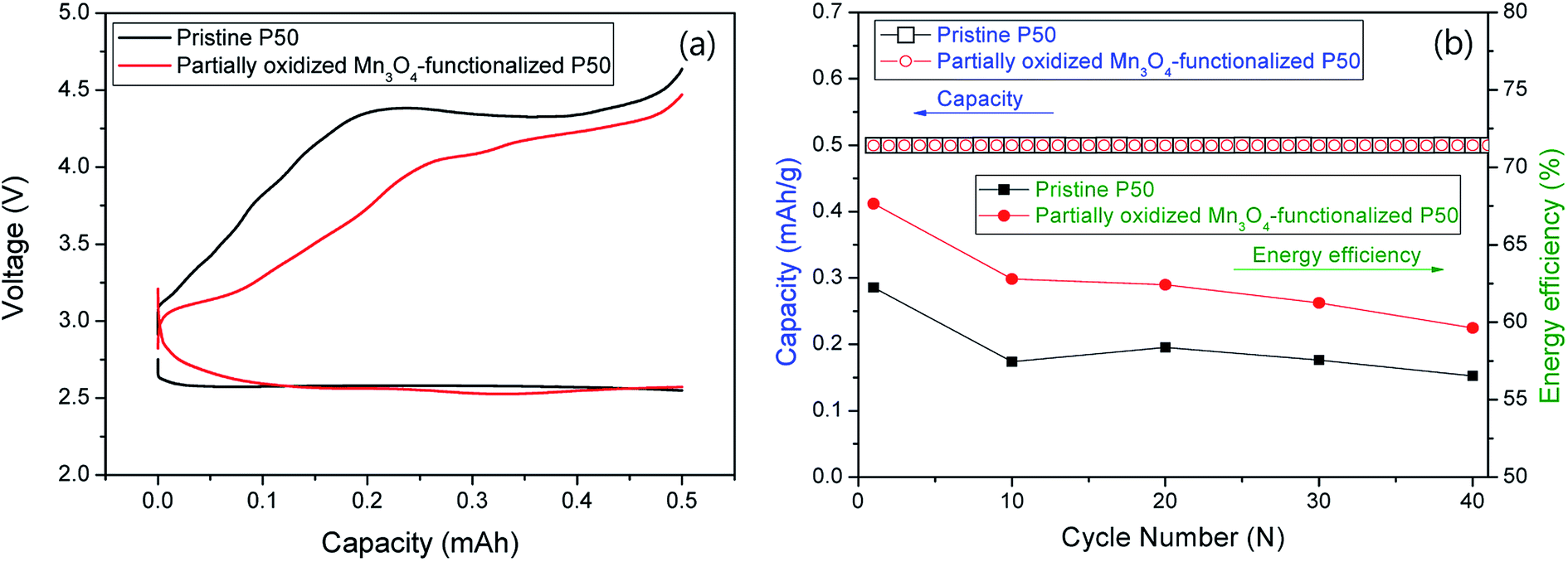 | ||
| Fig. 9 (a) First discharge/charge graph and (b) cycle performances and energy efficiency for nonaqueous Li–O2 cells. | ||
Conclusion
In this study, partially oxidized Mn3O4-functionalized P50 was prepared by dipping carbon paper (P50) in Mn-acetate solution and heating at 300 °C to apply manganese oxide directly on the carbon. Results showed an improvement in the contact between carbon and the oxide, but only a small amount of the oxide species (<5%) showed catalytic effects during charge in a rechargeable Li–O2 cell. This study proposes a simple synthetic route involving oxide application to the carbon surface. Results show that the reduced gravimetric capacity for heavy metal is relieved when using oxide catalysts in Li–O2 cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This Research was supported by Research Funds of Mokpo National University in 2017.References
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193 CrossRef.
- M. Balaish, A. Kraytsberg and Y. Ein-Eli, Phys. Chem. Chem. Phys., 2014, 16, 2801 RSC.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef PubMed.
- N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994 CrossRef PubMed.
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. S. -Horn, Energy Environ. Sci., 2013, 6, 750–768 RSC.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114(23), 11721 CrossRef PubMed.
- R. Black, B. Adams and L. F. Nazar, Adv. Energy Mater., 2012, 2, 801 CrossRef.
- F. Li, T. Zhang and H. Zhou, Energy Environ. Sci., 2013, 6, 1125 RSC.
- M. L. Thomas, K. Yamanaka, T. Ohta and H. R. Byon, Chem. Commun., 2015, 51, 3977 RSC.
- Y. Chen, F. Li, D.-M. Tang, Z. Jian, C. Liu, D. Golberg, A. Yamada and H. Zhou, J. Mater. Chem. A, 2013, 1, 13076 RSC.
- Y.-C. Lu, H. A. Gasteiger, M. C. Parent, V. Chiloyan and Y. S. -Horn, Electrochem. Solid-State Lett., 2010, 13(6), A69–A72 CrossRef.
- N. Ding, S. W. Chien, T. S. A. Hor, R. Lum, Y. Zong and Z. Liu, J. Mater. Chem. A, 2014, 2, 12433 RSC.
- J. S. Hummelshoj, J. Blomqvist, S. Datta, T. Vegge, J. Rossmeisl, K. S. Thygesen, A. C. Luntz, K. W. Jacobsen and J. K. Nørskov, J. Chem. Phys., 2010, 132, 071101 CrossRef PubMed.
- Y. Shao, S. Park, J. Xiao, J.-G. Zhang, Y. Wang and J. Liu, ACS Catal., 2012, 2, 844 CrossRef.
- R. Choi, J. Jung, G. Kim, K. Song, Y.-I. Kim, S. C. Jung, Y.-K. Han, H. Song and Y.-M. Kang, Energy Environ. Sci., 2014, 7, 1362 RSC.
- E. Yilmaz, C. Yogi, K. Yamanaka, T. Ohta and H. R. Byong, Nano Lett., 2013, 13, 4679 CrossRef PubMed.
- Z. Zhang, L. Su, M. Yang, M. Hu, J. Bao, J. Wei and Z. Zhou, Chem. Commun., 2014, 50, 776 RSC.
- X. Zhang, C. Wang, Y.-N. Chen, X.-G. Wang, Z. Xie and Z. Zhou, J. Power Sources, 2018, 377, 136 CrossRef.
- X. Zhang, X. Zhang, X.-G. Wang, Z. Xie and Z. Zhou, J. Mater. Chem. A, 2016, 4, 9390 RSC.
- D. B. Radic, M. M. Stanojevi, M. O. Obradovic and A. M. Jovovic, J. Therm. Sci., 2017, 21, 1067 CrossRef.
- E.-H. M. Diefallah, Thermochim. Acta, 1992, 202, 1 CrossRef.
- D. Sun, Y. Shen, W. Zhang, L. Yu, Z. Yi, W. Yin, D. Wang, Y. Huang, J. Wang, D. Wang and J. B. Goodenough, J. Am. Chem. Soc., 2014, 136, 8941 CrossRef PubMed.
- E. N. Nasybulin, W. Xu, B. L. Mehdi, E. Thomsen, M. H. Engelhard, R. C. Masse, P. Bhattacharya, M. Gu, W. Bennett, Z. Nie, C. Wang, N. D. Browning and J.-G. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 14141 CrossRef PubMed.
- H. Nie, Y. Zhang, J. Li, W. Zhou, Q. Lai, T. Liu and H. Zhang, RSC Adv., 2014, 4, 17141 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02920b |
| This journal is © The Royal Society of Chemistry 2018 |