
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA convenient approach to difluoromethylated all-carbon quaternary centers via Ni(II)-catalyzed enantioselective Michael addition†
Xuan Yu,
Hui Bai,
Dong Wang,
Zhaohai Qin,
Jia-Qi Li * and
Bin Fu*
* and
Bin Fu*
Department of Applied Chemistry, China Agricultural University, West Yuanmingyuan Rd. 2, Beijing 100193, People's Republic of China. E-mail: jiaqili@cau.edu.cn; fubinchem@cau.edu.cn
First published on 25th May 2018
Abstract
A Ni(II)-catalyzed enantioselective Michael addition of 2-acetyl azarenes with β-difluoromethyl substituted nitroalkenes was successfully realized, which afforded chiral CF2H-containing compounds in good enantioselectivities (up to 93% ee). This protocol provides a new convenient approach to all-carbon quaternary stereogenic centers featuring a CF2H group.
1 Introduction
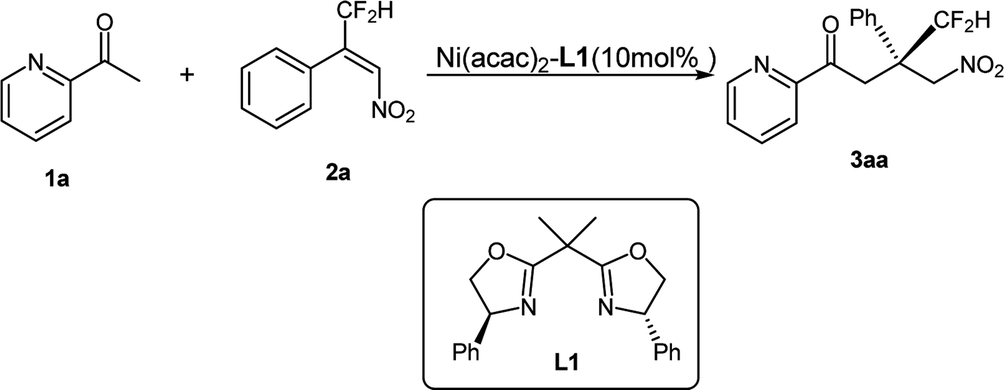
The introduction of a CF2H group into organic compounds modulates the metabolic ability, lipophilicity, hydrogen-bonding potency and bioactivity. Therefore, CF2H-containing compounds have found widespread application in pharmaceuticals, agrochemicals and enzyme inhibitors.1 During the past few years, methods for incorporating CF2H into organic molecules have increased rapidly.2 In the meantime, some successful examples on the synthesis of chiral CF2H containing compounds have also been explored by means of catalytic asymmetric methods.3 In 2012, Zhou et al. reported that nitrogen-based Lewis bases could effectively activate difluoroenoxysilanes for a highly enantioselective synthesis of 3-difluoroalkyl 3-hydroxyoxindoles.4a In 2015, Zhou's group employed a chiral secondary amine phosphoramide to catalyze efficiently the Mukaiyama–Michael addition of fluorinated enol silyl ethers to tetrasubstituted olefins, giving the oxindole products featuring an all-carbon quaternary stereocenter with either a difluoroalkyl or monofluoroalkyl group.4b Subsequently they explored another Mukaiyama–Mannich reaction of fluorinated enol silyl ethers and cyclic N-sulfonyl ketimines by employing a hydroquinine-derived urea catalyst, affording benzosultam-based α-amino acid derivatives featuring a fluoroalkyl group.4c In 2015, Mikami et al. reported that the highly enantioselective ene reactions with difluoropyruvate by using a dicationic palladium catalyst, giving α-CF2H tertiary alcohols.5 In 2016, Jacobsen reported one exciting reaction of β-substituted styrenes with commercially available reagents (m-chloroperbenzoic acid and hydrogen fluoride pyridine) to access a variety of products bearing difluoromethylated tertiary or quaternary stereocenters in the presence of simple chiral aryl iodide catalyst.6 Very recently, Hoveyda reported an efficient enantioselective addition of readily accessible Z-γ-substituted boronic acid pinacol ester compounds to fluoroalkyl-substituted ketones.7 Despite these notable advances in the introduction of CF2H-moiety to chiral products, general and convenient methods remain limited.It is well-known that the construction of all-carbon quaternary stereocenter is one challenging and even formidable topic in organic synthesis.8 In our recent research work, we have demonstrated that the construction of all-carbon quaternary stereocenters bearing a CF3-group or –CO2R could be realized by a Ni-bis(oxazoline) catalyst.9 Thus, we envision that whether the corresponding optical CF2H-containing compounds could be achieved by the same catalytic system. To the best of our knowledge, only two successful examples involving chiral all-carbon quaternary center bearing a difluoroalkyl group have been reported by Zhou and Jacobsen.4b,6 Therefore, the development of efficient and concise method for the construction of all-carbon quaternary stereocenter featuring CF2H group is of great significance and highly desirable for medicinal research. As a continuation of our ongoing research to explore efficient and economical asymmetric methodology,10 we herein report our recent findings on the addition of β-difluoromethyl nitroalkene with 2-acetyl azarenes.
2 Results and discussion
Based on our previous result,9 the complex of Ni(acac)2-Ph-BOX L1 was selected as the optimal catalyst. The reaction in the presence of 10 mol% of Ni(acac)2 and 11 mol% of L1 proceeded for 5 h in iso-propanol (i-PrOH) at room temperature, and afforded the addition product 3aa in 97% yield with 74% ee (Table 1, entry 1), lowering the reaction temperature to 0 °C and −20 °C led to an obvious improvement of the enantioselectivity (79% and 91% ee, respectively, entries 2–3), albeit along with a longer reaction time. Comparing with the substrate of β-CF3 substituted nitroalkene, this reaction exhibited more higher reactivity and somewhat lower enantioselectivity under the same reaction condition, which is presumably attributed to the low steric hindrance of –CF2H group. Moreover, other conditions including solvent and the additives were also investigated, but no more yields or ee values were obtained. Then the optimal condition was identified as the following: Ni(acac)2-L1 (10 mol%), i-PrOH, −20 °C.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | T | t (h) | Additive | Yield (%)b | ee (%)c |
a Unless otherwise noted, reactions were conducted with Ligand–metal (1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mol%), 1a (0.1 mmol), and 2a (0.15 mmol) in solvent (1.5 mL).b Isolated yields.c Determined by chiral HPLC. :1, 10 mol%), 1a (0.1 mmol), and 2a (0.15 mmol) in solvent (1.5 mL).b Isolated yields.c Determined by chiral HPLC. |
||||||
| 1 | i-PrOH | rt | 5 | — | 97 | 74 |
| 2 | i-PrOH | 0 °C | 12 | — | 95 | 79 |
| 3 | i-PrOH | −20 °C | 60 | — | 92 | 91 |
| 4 | MeOH | −20 °C | 60 | — | 50 | 75 |
| 5 | DCM | −20 °C | 60 | — | 73 | 50 |
| 6 | THF | −20 °C | 60 | — | 77 | 70 |
| 7 | n-Pentanol | −20 °C | 60 | — | 61 | 80 |
| 8 | EtOH | −20 °C | 60 | — | 80 | 81 |
| 9 | i-BuOH | −20 °C | 60 | — | 95 | 83 |
| 10 | n-BuOH | −20 °C | 60 | — | 95 | 83 |
| 11 | i-PrOH | rt | 5 | K2CO3 | 83 | 2 |
| 12 | i-PrOH | rt | 5 | CH3ONa | 72 | 21 |
| 13 | i-PrOH | rt | 5 | 4 Å | Trace | — |
Under the optimized reaction conditions, a variety of aromatic heterocycle of 2-acetyl azarenes were further investigated, as outlined in Table 2. For various azarenes containing a five-membered N-heterocycle, the reaction demonstrated good enantioselectivities and moderate reactivities (67–93% ee and 40–68% yields, entries 1–5). Among them 2-acetyl N-methyl 2-imidazole furnished the best catalytic results (93% ee, entry 3). Moreover, the substrates 3ag–3ai containing six-membered N-heterocycles such as pyrazinyl, quinolinyl, 2-benzopyrazinyl and quinoxalinyl were also suitable reaction partners. A range of 71–80% ee values were obtained, although a relatively low reactivity was displayed in the case of 2-quinolinyl and 2-benzopyrazinyl groups (entries 7 and 8). In addition, when using the bulky 2-acetyi 6,7-dihydro-5H-quinolin-8-one as Michael donor, no reaction was observed (entry 9).
|
||||
|---|---|---|---|---|
| Entry | 1 | Product | Yield (%)b | ee (%)c |
| a Unless otherwise noted, reactions were performed with L1–Ni(acac)2 (1.1:1, 10 mol%), 1 (0.1 mmol), and 2a (0.15 mmol) in i-PrOH (1.5 mL) at −20 °C for 60 h.b Yields of isolated products.c The ee value was determined by chiral HPLC analysis.d n.r = no reaction. | ||||
| 1 | 2-Oxazolyl | 3ab | 58 | 71 |
| 2 | 2-Thiazolyl | 3ac | 48 | 81 |
| 3 | N-methyl 2-imidazolyl | 3ad | 68 | 93 |
| 4 | 2-Benzoxazolyl | 3ae | 40 | 67 |
| 5 | 2-Benzothiazolyl | 3af | 48 | 75 |
| 6 | 2-Pyrazinyl | 3ag | 76 | 79 |
| 7 | 2-Quinolinyl | 3ah | 45 | 71 |
| 8 | 2-Benzopyrazinyl | 3ai | 38 | 80 |
| 9 | 6,7-Dihydro-5H-quinolin-8-one | 3aj | n.rd | — |
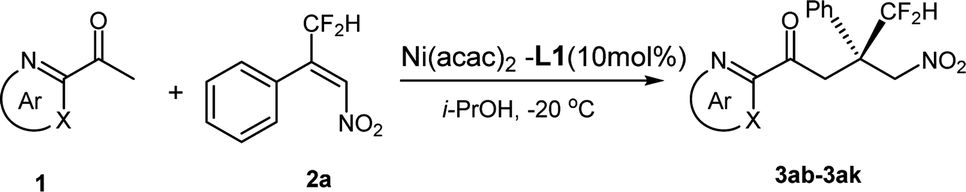
We next evaluated the scope of nitroalkenes. The results are summarized in Table 3. First, whatever an electron-rich or electron-deficient substituent at the para- or meta-position of the phenyl ring, the reaction proceeded smoothly to afford the dducts 3ba–3ga in both good yields and enantioselectivities (74–86% yields and 71–83% ees, entries 1–6). These results demonstrated that the electronic properties of the substituent on the phenyl ring of the nitroalkene has little influence on the enantioselectivity and reactivity of the reaction. However an ortho-substituent on the phenyl ring of the nitroalkene has a remarkable detrimental effect on the reaction. No reaction took place with the substrate bearing an ortho-methoxy or ortho-chloro on the phenyl ring, which is probably attributed to the bulky steric hindrance at the ortho-position of the phenyl ring (entries 7 and 8). Unfortunately, for heptyl substituted nitroalkene the poor enantioselectivity was obtained (entry 9, 20% ee), which is remarkably different from that of CF3-substituted nitroalkene. These results exhibited that, although only a fluoro-atom disparity between –CF2H and –CF3 substituted nitroalkenes, the enantioselectivity showed obvious differences under the same catalytic system. Generally β-difluoromethyl substituted nitroalkenes gave somewhat lower enantio-selectivity than corresponding β-trifluoromethyl substrates.
|
||||
|---|---|---|---|---|
| Entry | 2 | Product | Yield | ee (%)c |
| a Unless otherwise noted, reactions were performed with L1–Ni(acac)2 (1.1:1, 10 mol%), 1a (0.1 mmol), and 2 (0.15 mmol) in i-PrOH (1.5 mL) at −20 °C for 60 h.b Yields of isolated products.c The ee value was determined by chiral HPLC analysis.d n.r = no reaction. | ||||
| 1 | 4-Methylphenyl | 3ba | 84 | 76 |
| 2 | 4-Methoxyphenyl | 3ca | 81 | 83 |
| 3 | 4-Chlorophenyl | 3da | 86 | 77 |
| 4 | 4-Trifluoromethylphenyl | 3ea | 74 | 81 |
| 5 | 3-Methoxyphenyl | 3fa | 76 | 71 |
| 6 | 3-Chlorophenyl | 3ga | 85 | 77 |
| 7 | 2- Methoxyphenyl | 3ha | n.rd | — |
| 8 | 2-Chlorophenyl | 3ia | n.r | — |
| 9 | n-Heptyl | 3ja | 77 | 20 |
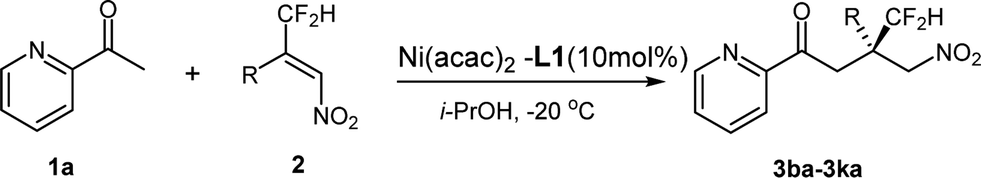
To further demonstrate the applicability of the current method in the synthesis of these types of molecules, a scale–up reaction was performed. Similar excellent yield and enantioselectivity were obtained (Scheme 1).
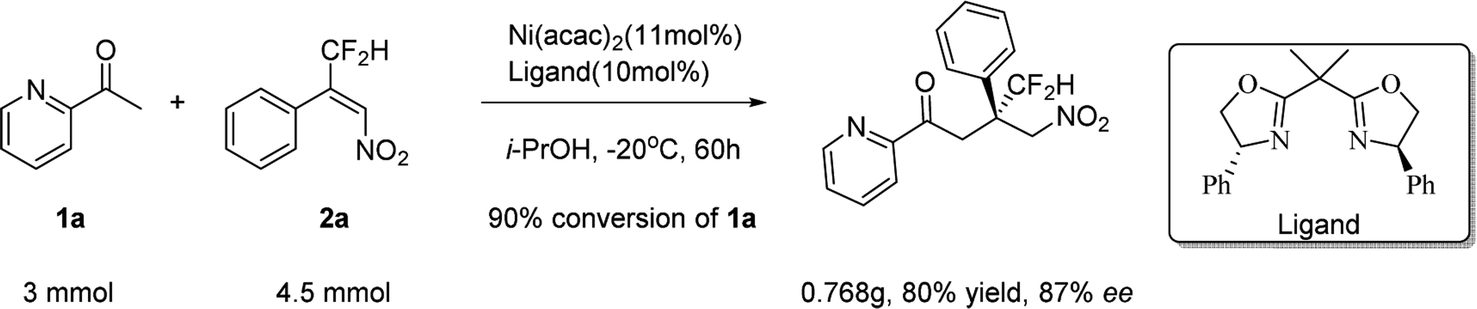 | ||
| Scheme 1 Scale up reaction. | ||
On the basis of X-ray diffraction analysis, the absolute configuration of compound 3ad was determined to be (S) (Fig. 1),11 and the configuration of other products was also assigned by analogy. Considering the observed stereochemistry, a plausible asymmetric induction model was proposed (Fig. 1). The coordination of BOX ligand L1 to Ni(acac)2 gave rise to a Ni complex. Subsequently, an enolate was resulted by the interaction of 2-acetyl azaarene to this Ni complex. Meanwhile, nitroalkene was also activated through coordination to Ni. The Si face attack of the enolate was disfavoured due to the steric hindrance between the NO2 group of nitroalkene and the phenyl substituent of the oxazoline ring, leading to the predominant Re-face addition.
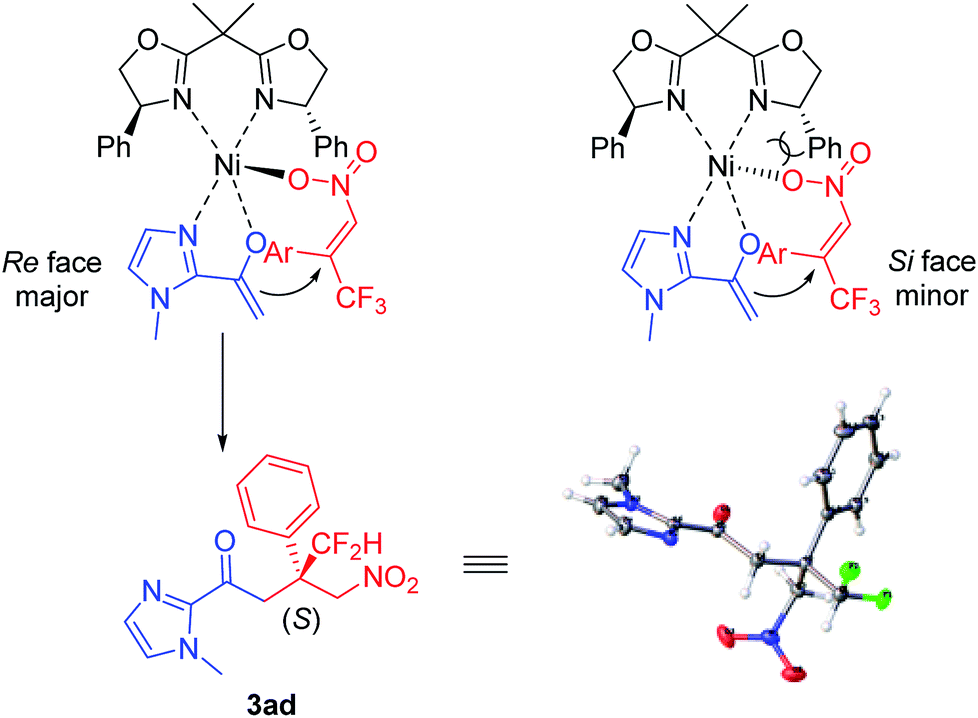 | ||
| Fig. 1 The proposed stereochemical model. | ||
However, in view of somewhat higher reactivity and lower enantioselectivity of difluoromethyl nitroalkenes in contrast to trifluoromethyl substrates under the same condition (Table 1, entries 1–3), it is believed that both electronic and steric effects of substituents on the nitroalkene have an impact on the efficiency and enantioselectivity of the reaction, but the steric effect could play a much more important role, which is basically in agreement with our previous report.9
3 Conclusions
In summary, we have developed a Ni(II)-catalyzed asymmetric Michael addition reaction of β-difluoromethyl nitroalkene and 2-acetyl azarene by employing an easily available catalyst. The reaction proceeded smoothly under mild conditions to afford the adducts in moderate to high yields and enantioselectivities (up to >93% ee). The reaction mechanism was discussed by comparison with that of β-CF3 substituted nitroalkenes. These difluoromethylated compounds bearing an all-carbon quaternary stereocenter are interesting targets for medicinal research. Further applications of this methodology to the synthesis of functional chiral molecules are in progress in our laboratory.4 Experimental section
4.1 General methods
The 1H, 13C, and 19F NMR spectra were recorded on a Bruker Avance DPX300 instrument with TMS as internal standard. Mass spectra were obtained on Bruker APEX II FT-ICRMS mass spectrometer. Optical rotations were measured on a PerkineElmer341 LC polarimeter. The enantiomeric excesses of (R)- and (S)-enantiomer were determined by Agilent 1260 HPLC analysis over a chiral column (Daicel Chiralcel OD-H, AD-H, AS-H or OJ-H; eluted with hexane/iso-propanol; UV detector). Solvents were purified and dried by standard procedures.4.2 General procedure for the asymmetric Michael addition
Under nitrogen to a solution of ligand L1 (0.011 mmol) in i-PrOH (1.5 mL) was added Ni(acac)2 (0.01 mmol). The reaction mixture was stirred for 1 h at room temperature before 2-acetyl azaarene 1 (0.1 mmol) was added. After stirring for 15 min. The resulting mixture was cooled to −20 °C, and then a solution of nitroalkene 2 (0.15 mmol) was added. The reaction proceeded to completion at −20 °C (monitored by TLC). Subsequently, water (10 mL) was added and extracted with ethyl acetate (10 mL × 3). The organic layer was combined, washed with brine, dried over Na2SO4 and concentrated. The residue was purified by flash column chromatography on silica gel (eluted with ethyl acetate/petroleum ether (1/10, v/v) to afford the desired product 3.4.3 (S)-4,4-difluoro-3-(nitromethyl)-3-phenyl-1-(pyridin-2-yl)butan-1-one (3aa)
Yellow oil, 92% yield. [α]20D = –47.1 (c = 1.32, CH2Cl2); 91% ee, determined by HPLC analysis [Daicel Chiralcel AD-H column, n-hexane/i-PrOH = 90:10, 1.0 mL min−1, 254 nm; t (major) = 14.650 min, t (minor) = 13.159 min];1H NMR (300 MHz, CDCl3) δ 8.70 (d, J = 4.2 Hz, 1H), 7.96 (d, J = 7.8 Hz, 1H), 7.81 (dt, J = 7.6, 1.1 Hz, 1H), 7.50 (dd, J = 4.9, 7.4 Hz, 1H), 7.44–7.27 (m, 5H), 6.48 (t, J = 56.0 Hz, 1H), 5.38 (s, 2H), 4.37, 4.15 (ABq, J = 18.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 197.66 (s), 152.48 (s), 148.73 (s), 136.73 (s), 134.31 (s), 128.56 (s), 128.06 (s), 127.43 (s), 126.54 (s), 121.51 (s), 116.13 (t, J = 249.1 Hz), 76.22 (s), 48.41 (t, J = 19.0 Hz), 37.04 (dd, J = 4.2, 2.9 Hz). 19F NMR (282 MHz, CDCl3) δ −125.26 (q, J = 280.3 Hz). ESI-HRMS: Calcd for C16H15F2N2O3+ ([M + H+]): 321.1040; found: 321.1038.
4.4 (S)-4,4-Difluoro-3-(nitromethyl)-1-(oxazol-2-yl)-3-phenylbutan-1-one (3ab)
Whitesolid, 58% yield. Mp. 96–110 °C. [α]20D = −17.2 (c = 0.56, CH2Cl2); 75% ee, determined by HPLC analysis [Daicel Chiralcel AD-H column, n-hexane/i-PrOH = 95:5, 1.0 mL min−1, 254 nm; t (major) = 37.009 min, t (minor) = 33.169 min]; 1H NMR (300 MHz, CDCl3) δ 7.82 (s, 1H), 7.36 (s, 6H), 6.44 (t, J = 55.8 Hz, 1H), 5.37, 5.31 (ABq, J = 12.0 Hz, 2H), 4.14, 4.02 (ABq, J = 18.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 184.05 (s), 157.52 (s), 142.29 (s), 133.92 (s), 129.33 (s), 129.06 (s), 128.64 (s), 126.58 (s), 116.14 (t, J = 249.6 Hz), 76.13 (dd, J = 5.6, 3.1 Hz), 48.77 (t, J = 19.1 Hz), 38.75 (dd, J = 4.5, 3.0 Hz). 19F NMR (282 MHz, CDCl3) δ −124.97 (q, J = 281.3 Hz). ESI-HRMS: Calcd for C14H13F2N2O4+ ([M + H+]): 311.0838; found: 311.0835.
4.5 (S)-4,4-Difluoro-3-(nitromethyl)-3-phenyl-1-(thiazol-2-yl)butan-1-one (3ac)
Orange oil, 48% yield. [α]20D = –27.5 (c = 0.78, CH2Cl2); 81% ee, determined by HPLC analysis [Daicel ChiralcelOD-H column, n-hexane/i-PrOH = 80:20, 1.0 mL min−1, 254 nm; t (major) = 14.168 min, t (minor) = 12.471 min];1H NMR (300 MHz, CDCl3) δ 8.02 (d, J = 2.8 Hz, 1H), 7.70 (d, J = 2.8 Hz, 1H), 7.50–7.27 (m, 5H), 6.46 (t, J = 55.8 Hz, 1H), 5.39, 5.33 (ABq, J = 12.0 Hz, 2H), 4.25, 4.11 (ABq, J = 18.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 189.86 (s), 166.12 (s), 144.90 (s), 134.18 (s), 128.97 (s), 128.53 (s), 127.15 (s), 126.66 (s), 116.23 (t, J = 249.5 Hz), 76.32 (dd, J = 5.6, 3.0 Hz), 48.76 (t, J = 19.1 Hz). 19F NMR (282 MHz, CDCl3) δ −125.02 (q, J = 280.9 Hz). ESI-HRMS: Calcd forC14H13F2N2O3S+ ([M + H+]): 327.0609; found: 327.0604.
4.6 (S)-4,4-Difluoro-1-(1-methyl-1H-imidazol-2-yl)-3-(nitromethyl)-3-phenylbutan-1-one (3ad)
White solid, 68% yield. Mp. 72–74 °C, [α]20D = –110.4 (c = 2.14, CH2Cl2); 93% ee, determined by HPLC analysis [Daicel Chiralcel AD-H column, n-hexane/i-PrOH = 90:10, 1.0 mL min−1, 254 nm; t (major) = 21.285 min, t (minor) = 16.802 min]; 1H NMR (300 MHz, CDCl3) δ 7.37 (s, 5H), 7.15 (s, 1H), 7.03 (s, 1H), 6.43 (t, J = 55.8 Hz, 1H), 5.36 (s, 2H), 4.23, 4.03 (ABq, J = 18.0 Hz, 2H), 3.88 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 188.24 (s), 134.56 (s), 129.36 (s), 128.83 (s), 128.31 (s), 127.62 (s), 126.74 (s), 116.37 (t, J = 249.3 Hz), 76.46 (s), 48.74 (t, J = 19.0 Hz), 38.4 (dd, J = 3.8, 3.0 Hz), 36.03 (s). 19F NMR (282 MHz, CDCl3) δ −125.21 (q, J = 280.0 Hz). ESI-HRMS: calcd for C15H16F2N3O3+ ([M + H+]): 324.1154; found: 324.1149.
4.7 (S)-1-(benzo[d]oxazol-2-yl)-4,4-difluoro-3-(nitromethyl)-3-phenylbutan-1-one (3ae)
Orange oil, 40% yield. [α]20D = –18.9 (c = 0.68, CH2Cl2); 67% ee, determined by HPLC analysis [Daicel ChiralcelOD-H column, n-hexane/i-PrOH = 80:20, 1.0 mL min−1, 254 nm; t (major) = 24.226 min, t (minor) = 19.349 min]; 1H NMR (300 MHz, CDCl3) δ 7.92 (d, J = 7.9 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.52 (dt, J = 15.0, 7.3 Hz, 2H), 7.44–7.29 (m, 5H), 6.47 (t, J = 55.7 Hz, 1H), 5.40, 5.34 (ABq, J = 12.0 Hz, 2H), 4.29, 4.19 (ABq, J = 18.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 185.96 (s), 156.69 (s), 150.92 (s), 140.32 (s), 135.32–130.96 (m), 129.10 (s), 129.01 (s), 128.69 (s), 126.60 (s), 126.02 (s), 122.44 (s), 116.15 (t, J = 249.8 Hz), 111.97 (s), 76.12 (dd, J = 5.3, 2.8 Hz), 48.85 (t, J = 19.1 Hz), 39.10 (dd, J = 4.1, 2.7 Hz). 19F NMR (282 MHz, CDCl3) δ −124.81 (q, J = 281.3 Hz). ESI-HRMS: calcd for C18H17F2N2O4+ ([M + H+]): 361.0994; found: 361.0990.
4.8 (S)-1-(benzo[d]thiazol-2-yl)-4,4-difluoro-3-(nitromethyl)-3-phenylbutan-1-one (3af)
Yellow oil, 48% yield. [α]20D = –13.8 (c = 0.45, CH2Cl2); 75% ee, determined by HPLC analysis [Daicel ChiralcelOD-H column, n-hexane/i-PrOH = 90:10, 1.0 mL min−1, 254 nm; t (major) = 26.581 min, t (minor) = 22.790 min]; 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 7.7 Hz, 1H), 7.96 (d, J = 7.4 Hz, 1H), 7.68–7.49 (m, 2H), 7.47–7.26 (m, 5H), 6.48 (t, J = 55.8 Hz, 1H), 5.40, 5.35 (ABq, J = 12.0 Hz, 2H), 4.37, 4.23 (ABq, J = 18.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 191.45 (s), 165.37 (s), 153.33 (s), 137.50 (s), 134.15 (s), 129.05 (s), 128.61 (s), 128.09 (s), 127.24 (s), 126.70 (s), 125.67 (s), 122.43 (s), 116.26 (t, J = 249.6 Hz), 76.35 (dd, J = 5.8, 3.0 Hz), 48.86 (t, J = 19.1 Hz), 38.34 (dd, J = 4.3, 3.1 Hz). 19F NMR (282 MHz, CDCl3) δ −124.89 (q, J = 280.9 Hz). ESI-HRMS: calcd for C18H15F2N2O3S+ ([M + H+]): 377.0766; found: 377.0763.
4.9 (S)-4,4-Difluoro-3-(nitromethyl)-3-phenyl-1-(pyrazin-2-yl)butan-1-one (3ag)
White solid, 76% yield. Mp. 104–106 °C, [α]20D = –40.1 (c = 1.24, CH2Cl2); 79% ee, determined by HPLC analysis [Daicel ChiralcelAS-H column, n-hexane/i-PrOH = 80:20, 1.0 mL min−1, 254 nm; t (major) = 23.778 min, t (minor) = 17.251 min]; 1H NMR (300 MHz, CDCl3) δ 9.17 (s, 1H), 8.79 (s, 1H), 8.67 (s, 1H), 7.36 (s, 5H), 6.46 (t, J = 55.8 Hz, 1H), 5.37 (s, 2H), 4.29, 4.13 (ABq, J = 18.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 197.42 (s), 148.38 (s), 146.99 (s), 143.58 (s), 143.49 (s), 134.28 (s), 128.97 (s), 128.53 (s), 126.66 (s), 116.28 (t, J = 249.4 Hz), 76.36 (dd, J = 5.8, 2.9 Hz), 48.65 (t, J = 19.1 Hz), 37.38 (dd, J = 4.5, 2.9 Hz). 19F NMR (282 MHz, CDCl3) δ −125.13 (q, J = 280.9 Hz). ESI-HRMS: calcd for C15H14F2N3O3+ ([M + H+]): 322.0998; found: 322.0995.
4.10 (S)-4,4-Difluoro-3-(nitromethyl)-3-phenyl-1-(quinolin-2-yl)butan-1-one (3ah)
Orange solid, 45% yield. Mp. 85–92 °C, [α]20D = –64.4 (c = 1.46, CH2Cl2); 70% ee, determined by HPLC analysis [Daicel Chiralcel AD-H column, n-hexane/i-PrOH = 90:10, 1.0 mL min−1, 254 nm; t (major) = 15.068 min, t (minor) = 13.060 min]; 1H NMR (300 MHz, CDCl3) δ 8.26 (dd, J = 8.3, 2.6 Hz, 2H), 8.04 (d, J = 8.5 Hz, 1H), 7.91–7.86 (m, 1H), 7.82 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H), 7.68 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 7.50–7.29 (m, 5H), 6.55 (t, J = 56.0 Hz, 1H), 5.43 (s, 2H), 4.57, 4.32 (ABq, J = 18.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 198.33 (s), 152.38 (s), 147.08 (s), 137.17 (s), 134.71 (s), 130.70 (s), 130.26 (s), 129.85 (s), 128.97 (s), 128.90 (s), 128.38 (s), 127.66 (s), 126.91 (s), δ 116.52 (t, J = 249.1 Hz), 117.86 (s), 76.75 (dd, J = 6.0, 3.1 Hz), 48.88 (t, J = 19.0 Hz), 37.15 (dd, J = 4.3, 2.8 Hz). 19F NMR (282 MHz, CDCl3) δ −125.24 (q, J = 280.2 Hz). ESI-HRMS: calcd for C20H17F2N2O3+ ([M + H+]): 371.1202; found: 371.1198.
4.11 (S)-4,4-difluoro-3-(nitromethyl)-3-phenyl-1-(quinoxalin-2-l)butan-1-one (3ai)
Yellow solid, 38% yield. Mp. 108–114 °C, [α]20D = –35.1 (c = 0.42, CH2Cl2); 80% ee, determined by HPLC analysis [Daicel ChiralcelAD-H column, n-hexane/i-PrOH = 90:10, 1.0 mL min−1, 254 nm; t (major) = 15.516 min, t (minor) = 17.459 min]; 1H NMR (300 MHz, CDCl3) δ 9.42 (s, 1H), 8.23 (d, J = 20.5 Hz, 2H), 7.92 (s, 2H), 7.40 (s, 5H), 6.53 (t, J = 55.4 Hz, 1H), 5.42 (s, 2H), 4.48, 4.29 (ABq, J = 18.0 Hz, 2H)·13C NMR (75 MHz, CDCl3) δ 197.69 (s), 145.81 (s), 144.24 (s), 142.82 (s), 140.82 (s), 134.39 (s), 132.67 (s), 131.03 (s), 130.56 (s), 129.49 (s), 129.03 (s), 128.57 (s), 126.75 (s), 116.39 (t, J = 249.3 Hz), 76.53 (m), 48.78 (t, J = 19.1 Hz), 37.19 (dd, J = 4.2, 3.0 Hz).19F NMR (282 MHz, CDCl3) δ −125.09 (q, J = 280.8 Hz). ESI-HRMS: calcd for C19H16F2N3O3+ ([M + H+]): 372.1154; found: 372.1150.
4.12 (S)-4,4-difluoro-3-(nitromethyl)-1-(pyridin-2-yl)-3-(p-tolyl)butan-1-one (3ba)
Colorless oil, 84% yield. [α]20D = –53.1 (c = 1.26, CH2Cl2); 76% ee, determined by HPLC analysis [Daicel Chiralcel OD-H column, n-hexane/i-PrOH = 90:10, 1.0 mL min−1, 254 nm; t (major) = 12.882 min, t (minor) = 14.746 min]; 1H NMR (300 MHz, CDCl3) δ 8.70 (d, J = 4.2 Hz, 1H), 7.97 (d, J = 7.8 Hz, 1H), 7.82 (dt, J = 7.6, 1.4 Hz, 1H), 7.50 (dd, J = 7.6, 4.9 Hz, 1H), 7.21 (dd, J = 27.4, 8.1 Hz, 4H), 6.45 (t, J = 56.1 Hz, 1H), 5.36 (s, 2H), 4.14, 4.34 (ABq, J = 18.0 Hz, 2H), 2.31 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 198.02 (s), 152.85 (s), 149.00 (s), 138.18 (s), 136.99 (s), 131.85–131.34 (m), 129.58 (s), 127.65 (s), 126.67 (d, J = 1.4 Hz), 121.80 (s), 116.47 (t, J = 249.1 Hz), 76.72–76.61 (m), 48.46 (t, J = 18.9 Hz), 37.31 (dd, J = 4.5, 2.9 Hz), 20.85 (s). 19F NMR (282 MHz, CDCl3) δ −125.36 (q, J = 280.0 Hz). ESI-HRMS: calcd for C17H17F2N2O3+ ([M + H+]): 335.1202; found: 335.1197.
4.13 (S)-4,4-Difluoro-3-(4-methoxyphenyl)-3-(nitromethyl)-1-(pyridin-2-yl)butan-1-one (3ca)
Yellow oil, 81% yield. [α]20D = –68.9 (c = 1.44, CH2Cl2); 84% ee, determined by HPLC analysis [Daicel Chiralcel AS-H column, n-hexane/i-PrOH = 90:10, 1.0 mL min−1, 254 nm; t (major) = 33.111 min, t (minor) = 31.418 min]; 1H NMR (300 MHz, CDCl3) δ 8.70 (d, J = 4.4 Hz, 1H), 7.97 (d, J = 7.8 Hz, 1H), 7.81 (dt, J = 7.8, 1.0 Hz, 1H), 7.50 (dd, J = 6.9, 5.1 Hz 1H), 7.28 (t, J = 7.7 Hz, 2H), 6.87 (d, J = 8.8 Hz, 2H), 6.45 (t, J = 56.2 Hz, 1H), 5.34 (s, 2H), 4.34, 4.13 (ABq, J = 18.0 Hz, 2H), 3.76 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 198.07 (s), 159.31 (s), 152.82 (s), 148.99 (s), 136.99 (s), 128.08 (s), 127.66 (s), 126.24 (s), 121.77 (s), δ 116.44 (t, J = 248.8 Hz), 114.19 (s), 76.74–76.57 (m), 55.12 (s), 48.22 (t, J = 19.0 Hz), 38.94–35.93 (m). 19F NMR (282 MHz, CDCl3) δ −125.42 (q, J = 280.0 Hz). ESI-HRMS: calcd for C17H17F2N2O4+ ([M + H+]): 351.1151; found: 351.1147.
4.14 (S)-3-(4-chlorophenyl)-4,4-difluoro-3-(nitromethyl)-1-(pyridin-2-yl)butan-1-one (3da)
Colorless oil, 86% yield. [α]20D = –55.8 (c = 1.13, CH2Cl2); 77% ee, determined by HPLC analysis [Daicel ChiralcelAS-H column, n-hexane/i-PrOH = 80:20, 1.0 mL min−1, 254 nm; t (major) = 16.024 min, t (minor) = 13.599 min]; 1H NMR (300 MHz, CDCl3) δ 8.70 (d, J = 4.6 Hz, 1H), 7.96 (d, J = 7.8 Hz, 1H), 7.83 (dt, J = 7.9, 1.1 Hz, 1H), 7.51 (dd, J = 7.4, 4.8 Hz, 1H), 7.45–7.27 (m, 4H), 6.47 (t, J = 56.0 Hz, 1H), 5.37, 5.32 (ABq, J = 12.0 Hz, 2H), 4.35, 4.11 (ABq, J = 18.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 197.78 (s), 152.63 (s), 149.05 (s), 137.07 (s), 134.55 (s), 133.06 (s), 129.00 (s), 128.40 (s), 127.83 (s), 121.83 (s), 116.00 (t, J = 249.2 Hz), 76.41 (dd, J = 5.9, 3.1 Hz), 48.42 (t, J = 19.1 Hz), 37.37 (dd, J = 4.4, 2.7 Hz). 19F NMR (282 MHz, CDCl3) δ −125.51 (q, J = 281.5 Hz). ESI-HRMS: calcd for C16H14ClF2N2O3+ ([M + H+]): 355.0656; found: 355.0653.
4.15 (S)-4,4-Difluoro-3-(nitromethyl)-1-(pyridin-2-yl)-3-(4-(trifluoromethyl)phenyl)butan-1-one (3ea)
White solid, 74% yield. Mp. 82–83 °C, [α]20D = –49.5 (c = 1.50, CH2Cl2); 81% ee, determined by HPLC analysis [Daicel ChiralcelAS-H column, n-hexane/i-PrOH = 80:20, 1.0 mL min−1, 254 nm; t (major) = 10.203 min, t (minor) = 9.375 min]; 1H NMR (300 MHz, CDCl3) δ 8.71 (s, 1H), 7.97 (s, 1H), 7.85 (s, 1H), 7.64 (s, 2H), 7.53 (s, 3H), 6.52 (t, J = 56.3 Hz, 1H), 5.40 (s, 2H), 4.41, 4.15 (ABq, J = 18.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 197.68 (s), 152.56 (s), 149.10 (s), 138.67 (s), 137.12 (s), 130.60 (q, J = 32.9 Hz), 127.92 (s), 127.55 (s), 125.73 (q, J = 3.6 Hz), 125.52 (s), 121.87 (s), 115.90 (t, J = 249.4 Hz), 76.33 (dd, J = 5.8, 3.2 Hz), 48.76 (t, J = 19.1 Hz), 37.48 (dd, J = 4.4, 2.7 Hz). 19F NMR (282 MHz, CDCl3) δ −62.88 (s), −125.52 (q, J = 282.3 Hz). ESI-HRMS: calcd for C17H14F5N2O3+ ([M + H+]): 389.0919; found: 389.0915.
4.16 (S)-4,4-Difluoro-3-(3-methoxyphenyl)-3-(nitromethyl)-1-(pyridin-2-yl)butan-1-one (3fa)
Colorless oil, 76% yield. [α]20D = –46.1 (c = 1.16, CH2Cl2); 71% ee, determined by HPLC analysis [Daicel Chiralcel OD-H column, n-hexane/i-PrOH = 90:10, 1.0 mL min−1, 254 nm; t (major) = 27.608 min, t (minor) = 20.249 min]; 1H NMR (300 MHz, CDCl3) δ 8.70 (d, J = 4.4 Hz, 1H), 7.96 (d, J = 7.9 Hz, 1H), 7.81 (dt, J = 7.7, 1.6 Hz, 1H), 7.56–7.42 (m, 1H), 7.27 (dd, J = 10.0, 6.4 Hz, 1H), 6.94 (d, J = 7.6 Hz, 2H), 6.85 (dd, J = 7.5, 1.8 Hz, 1H), 6.47 (t, J = 56.0 Hz, 1H), 5.36 (s, 2H), 4.34, 4.14 (ABq, J = 18.0 Hz, 2H), 3.74 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 197.94 (s), 159.78 (s), 152.79 (s), 149.00 (s), 137.00 (s), 136.25 (s), 129.81 (s), 127.67 (s), 121.80 (s), 119.03 (s), δ 116.44 (t, J = 249.3 Hz), 113.81 (s), 112.97 (s), 76.62 (m), 55.16 (s), 48.74 (t, J = 18.9 Hz), 37.33 (dd, J = 4.4, 2.8 Hz). 19F NMR (282 MHz, CDCl3) δ −125.10 (q, J = 280.0 Hz). ESI-HRMS: calcd for C17H17F2N2O4+ ([M + H+]): 351.1151; found: 351.1148.
4.17 (S)-3-(3-chlorophenyl)-4, 4-difluoro-3-(nitromethyl)-1-(pyridin-2-yl)butan-1-one (3ga)
Colorless oil, 85% yield. [α]20D = –51.0 (c = 1.24, CH2Cl2); 78% ee, determined by HPLC analysis [Daicel Chiralcel OD-H column, n-hexane/i-PrOH = 90:10, 1.0 mL min−1, 254 nm; t (major) = 16.516 min, t (minor) = 14.898 min]; 1H NMR (300 MHz, CDCl3) δ 8.71 (d, J = 4.5 Hz, 1H), 7.97 (d, J = 7.8 Hz, 1H), 7.83 (dt, J = 7.8, 1.6 Hz, 1H), 7.51 (dd, J = 6.8, 5.2 Hz, 1H), 7.39 (s, 1H), 7.35–7.23 (m, 3H), 6.48 (t, J = 55.9 Hz, 1H), 5.38, 5.33 (ABq, J = 12.0 Hz, 2H), 4.34, 4.11 (ABq, J = 18.0 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 197.70 (s), 152.61 (s), 149.05 (s), 137.06 (s), 136.67 (s), 134.91 (s), 129.96 (s), 128.63 (s), 127.82 (s), 127.39 (s), 125.15 (s), 121.83 (s), 115.96 (t, J = 249.4 Hz), 76.29 (dd, J = 5.8, 3.1 Hz), 48.58 (t, J = 19.2 Hz), 37.34 (dd, J = 4.2, 2.9 Hz). 19F NMR (282 MHz, CDCl3) δ −125.41 (q, J = 281.6 Hz). ESI-HRMS: calcd for C16H14ClF2N2O3+ ([M + H+]): 355.0656; found: 355.0654.
4.18 (R)-3-(difluoromethyl)-3-(nitromethyl)-1-(pyridin-2-yl)decan-1-one (3ja)
Colorless oil, 77% yield. [α]20D = +2.8 (c = 1.62, CH2Cl2); 20% ee, determined by HPLC analysis [Daicel ChiralcelOJ-H column, n-hexane/i-PrOH = 95:5, 1.0 mL min−1, 254 nm; t (major) = 10.455 min, t (minor) = 10.999 min]; 1H NMR (300 MHz, CDCl3) δ 8.68 (d, J = 4.2 Hz, 1H), 8.04 (d, J = 7.8 Hz, 1H), 7.87 (dt, J = 7.7, 1.4 Hz, 1H), 7.51 (dd, J = 7.3, 4.9 Hz, 1H), 6.24 (t, J = 55.9 Hz, 1H), 4.92, 4.86 (ABq, J = 12.0 Hz, 2H), 3.67 (s, 2H), 1.75 (dd, J = 15.2, 8.0 Hz, 2H), 1.53–1.33 (m, 2H), 1.26 (s, 8H), 0.86 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 198.79 (s), 152.96 (s), 148.95 (s), 137.00 (s), 127.58 (s), 121.70 (s), 117.43 (t, J = 247.9 Hz), 76.25 (t, J = 4.2 Hz), 44.79 (t, J = 18.6 Hz), 36.33 (t, J = 3.6 Hz), 31.62 (s), 31.07 (t, J = 2.7 Hz), 30.00 (s), 28.81 (s), 23.42 (s), 22.50 (s), 13.96 (s). 19F NMR (282 MHz, CDCl3) δ −127.79 (q, J = 282.0 Hz). ESI-HRMS: calcd for C17H25F2N2O3+ ([M + H+]): 343.1828; found: 343.1824.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from NSFC program (21172255) and the Ministry of Science and Technology of China (No. 2015BAK45B01, 2009BAK61B04) is appreciated.References
- (a) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef PubMed; (b) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef PubMed; (c) B. J. Elisabeth, M. M. M. Woodhead, Patent WO 2007096576, 2007; (d) L. David, D. Robert, S. Dean, A. Alexander, Patent WO 2007111904, 2007; (e) G. W. Rewcastle, S. A. Gamage, J. U. Flanagan, R. Frederick, W. A. Denny, B. C. Baguley, P. Kestell, R. Singh, J. D. Kendall, E. S. Marshall, C. L. Lill, W.-J. Lee, S. Kolekar, C. M. Buchanan, S. M. F. Jamieson and P. R. Shepherd, J. Med. Chem., 2011, 54, 7105–7126 CrossRef PubMed; (f) Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 CrossRef PubMed; (g) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef PubMed.
- For selected literatures on the introduction of CF2H-moiety to organic compounds: (a) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465–7478 RSC; (b) J. Liu and J. Hu, Chem.–Eur. J., 2010, 16, 11443–11454 CrossRef PubMed; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef PubMed; (d) A. G. O'Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868–11871 CrossRef PubMed; (e) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765–825 CrossRef PubMed; (f) X.-J. Tang and W. R. Dolbier Jr, Angew. Chem., Int. Ed., 2015, 54, 4246–4249 CrossRef PubMed; (g) C. S. Thomoson, X.-J. Tang and W. R. Dolbier Jr, J. Org. Chem., 2015, 80, 1264–1268 CrossRef PubMed; (h) X.-J. Tang, Z. Zhang and W. R. Dolbier Jr, Chem.–Eur. J., 2015, 21, 18961–18965 CrossRef PubMed; (i) Z. He, P. Tan, C. Ni and J. Hu, Org. Lett., 2015, 17, 1838–1841 CrossRef PubMed; (j) Y. Arai, R. Tomita, G. Ando, T. Koike and M. Akita, Chem.–Eur. J., 2016, 22, 1262–1265 CrossRef PubMed; (k) I. G. Molnár and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 5004–5007 CrossRef PubMed; (l) A. Tarui, S. Shinohara, K. Sato, M. Omote and A. Ando, Org. Lett., 2016, 18, 1128–1131 CrossRef PubMed.
- (a) W. Kashikura, K. Mori and T. Akiyama, Org. Lett., 2011, 13, 1860–1863 CrossRef PubMed; (b) Y.-L. Liu, J.-S. Yu and J. Zhou, Asian J. Org. Chem., 2013, 2, 194–206 CrossRef; (c) J.-H. Lin and J.-C. Xiao, Tetrahedron Lett., 2014, 55, 6147–6155 CrossRef.
- (a) Y.-L. Liu and J. Zhou, Chem. Commun, 2012, 1919–1921 RSC; (b) J.-S. Yu, F.-M. Liao, W.-M. Gao, K. Liao, R.-L. Zuo and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 7381–7385 CrossRef PubMed; (c) J.-S. Yu and J. Zhou, Org. Chem. Front., 2016, 3, 298–303 RSC.
- K. Aikawa, S. Yoshida, D. Kondo, Y. Asai and K. Mikami, Org. Lett., 2015, 17, 5108–5111 CrossRef PubMed.
- S. M. Banik, J. W. Medley and E. N. Jacobsen, Science, 2016, 353(6294), 51–54 CrossRef PubMed.
- F. W. van der Mei., C.-M. Qin, R. J. Morrison and A. H. Hoveyda, J. Am. Chem. Soc., 2017, 139, 9053–9065 CrossRef PubMed.
- (a) J. T. Mohr, M. R. Krout and B. M. Stoltz, Nature, 2008, 455, 323–332 CrossRef PubMed; (b) M. Bella and T. Gasperi, Synthesis, 2009, 2009, 1583–1614 CrossRef; (c) Y.-L. Liu, X. Wang, Y.-L. Zhao, F. Zhu, X.-P. Zeng, L. Chen, C.-H. Wang, X.-L. Zhao and J. Zhou, Angew. Chem., Int. Ed., 2013, 52, 13735–13739 CrossRef PubMed; (d) X.-P. Yin, X.-P. Zeng, Y.-L. Liu, F.-M. Liao, J.-S. Yu, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13740–13745 CrossRef PubMed; (e) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef PubMed.
- (a) X.-H. Hou, H.-L. Ma, Z.-H. Zhang, L. Xie, Z.-H. Qin and B. Fu, Chem. Commun., 2016, 52, 1470–1473 RSC; (b) H.-L. Ma, L. Xie, Z.-H. Zhang, L.-G. Wu, B. Fu and Z.-H. Qin, J. Org. Chem., 2017, 82, 7353–7362 CrossRef PubMed.
- (a) L. Liu, Q.-Y. Zhao, F.-P. Du, H. L. Chen, Z.-H. Qin and B. Fu, Tetrahedron: Asymmetry, 2011, 22, 1874–1878 CrossRef; (b) H. L. Ma, L. Xie, Z.-H. Zhang, J.-Q. Li, Z.-H. Qin and B. Fu, Adv. Synth. Catal., 2016, 358, 1011–1016 CrossRef; (c) L. Xie, X. Yu, J.-Q. Li, Z.-H. Zhang, Z.-H. Qin and B. Fu, Eur. J. Org. Chem., 2017, 2017, 657–661 CrossRef; (d) L. Xie, H. Bai, J.-Q. Li, X. Yu, Z.-H. Zhang and B. Fu, Tetrahedron, 2017, 73, 2923–2930 CrossRef; (e) L. Xie, H.-L. Ma, J.-Q. Li, X. Yu, Z.-H. Qin and B. Fu, Org. Chem. Front., 2017, 4, 1858–1862 RSC.
- Crystallographic Data Centre as supplementary publication number CCDC 1575270. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK. Fax: t44 1223 336033 or e-mail: E-mail: deposit@ccdc.cam.ac.uk.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization and spectra. CCDC 1575270. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra02853b |
| This journal is © The Royal Society of Chemistry 2018 |