
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlowery nickel–cobalt hydroxide via a solid–liquid sulphur ion grafting route and its application in hybrid supercapacitive storage†
Lin Ye‡
,
Zepei Bao‡,
Yuguang Zhao and
Lijun Zhao *
*
Key Laboratory of Automobile Materials, Ministry of Education and School of Materials Science and Engineering, Jilin University, Changchun, 130022, P. R. China. E-mail: lijunzhao@jlu.edu.cn
First published on 29th June 2018
Abstract
In our research, a two-step solid–liquid route was employed to fabricate flowery nickel–cobalt hydroxide with sulphur ion grafting (Ni1Co2–S). The utilization of NaOH/agar and Na2S/agar could efficiently retard the release rates of OH− or S2− ions at the solid–liquid interface due to strong bonding between agar hydrogel and these anions. Ni1Co2–S generally displays ultrathin flowery micro-frame, ultrathin internal nanosheets and expanded pore size. Besides, the introduction of suitable sulphide species into nickel–cobalt hydroxide could improve its conductivity due to the lower band gap of Ni–Co sulphide. The supercapacitive electrode Ni1Co2–S presented capacitance of 1317.8 F g−1 (at 1 A g−1) and suitable rate performance (77.9% at 10 A g−1 and 59.3% at 20 A g−1). Furthermore, a hybrid supercapacitor (HSC) was developed utilizing positive Ni1Co2–S and negative activated carbon electrodes. As expected, the HSC device exhibited excellent specific capacitance (117.1 F g−1 at 1 A g−1), considerable energy densities (46.7 W h kg−1 at 0.845 kW kg−1 and 27.5 W h kg−1 even at 9 kW kg−1) and suitable cycling performance, which further illuminated the high energy storage capacity of Ni1Co2–S.
1 Introduction
Numerous electronic equipments such as electric vehicles, consumer electronics and portable devices have facilitated our daily lives, but some adverse issues such as low recyclability and energy storage capacity remain for their extensive applications. Therefore, substantial attention is focused on achieving efficient energy storage of supercapacitors (SCs) and their successful applications in various electronic equipments due to their high power deliveries, long lifespans and rapid charge/discharge rates.1–4 To fully promote energy density and enlarge the potential window, assembling hybrid SCs is a feasible strategy, wherein a redox electrode serves as the energy source and an electrical double layer electrode serves as the power source.5,6 Meanwhile, exploring efficient functional electrode materials plays a vital role for integrated performance of HSCs. As typical battery-type materials,7–9 a class of Ni/Co hydroxides represent multiple oxidation states, high theoretical values and long discharge platforms,10–12 which endow them with high potentials in supercapacitive materials.The actual capacitance values of electrode materials are always lower than the theoretical values due to many factors such as specific surface area, porosity, conductivity, crystal structure and chemical composition.13–16 Thus, a rational structural design is highly desired to further improve electrochemical performances, especially while fabricating multi-level structures with large specific areas and optimizing components. On the one hand, versatile micro/nanoflower structures have been constructed and applied in catalysts,17 batteries,18 and supercapactors.19 Sufficient interior space and large specific surface area expose more active areas and shorten contact distance and thus, the generation of ultrathin nanosheets in flowery structures enhances electrochemical performance. On the other hand, the conductivity or stability performance of Ni/Co hydroxides needs further improvement.20–22 Recently, many studies have reported different forms of S incorporated with Ni/Co hydroxides, because Ni/Co sulphides display lower band gap values than their hydroxides.23–27 For instance, Zhou and co-workers reported a distinct core–shell Ni3S2 nanorod@ Ni(OH)2 nanosheet nanoarchitecture that presented an enhanced performance of 1037.5 F g−1 (at 5.1 A g−1) than mono-component Ni(OH)2, because the hybrid structure-integrated Ni(OH)2 nanosheets with rich active sites and crystalline Ni3S2 nanorods promoted rapid electron transfer.23 Also, core–shell NiCo2S4@Co(OH)2 nanotube arrays were prepared, which showed lower polarization and improved reactivity compared to bare NiCo2S4 structures.24 Recently, a gas–solid reaction route was selected to fabricate novel metallic nickel hydroxide nanosheets utilizing H2S gas to treat β-Ni(OH)2 nanosheets at 110 °C. The introduction of sulphur into Ni(OH)2 efficiently promoted electrical conductivity, and the material exhibited metallic property.25 Therefore, slight sulphur ion grafting route has been designed to improve the conductivities of hydroxide samples.
For the synthesis of electrode materials, various methods such as the microwave method,28 solvothermal/hydrothermal method,29,30 electro–deposition reaction31 and calcination treatment32 have been reported. In spite of great improvements in the energy conversion of SCs, high costs and unsuitable synthesis processes hinder their further applications. Up to now, few studies have been reported on the NaOH/agar and Na2S/agar hydrogel synthetic method. Herein, we used a two-step solid–liquid method to obtain Ni–Co hydroxide and Ni–Co hydroxide with S2− grating. The distinct agar hydrogel method has the following advantages: (1) the preparation of agar hydrogel only needs a short heat treatment time of 10 min, and the subsequent reactions are carried out at room temperature, which saves energy consumption; (2) there is controllable ionic diffusion in the solid–liquid interface owing to strong bonding between hydrogel and anions (OH− or S2− ions); (3) facile reaction equipments scale up sample preparation. In the hydrogel-assisted reaction, the flowery nickel–cobalt hydroxide via sulphur ions grafting not only introduces small amount of sulphide species, but also expands the pore size, which greatly stimulates charge transport. As anticipated, the Ni1Co2–S electrode and the corresponding hybrid supercapacitor (HSC) device deliver excellent capacitances, appropriate rate performances and cycling stabilities.
2 Experimental section
2.1 Preparation of NaOH/agar and Na2S/agar hydrogel
First, 12 mmol sodium hydroxide (NaOH) and 0.5 g agar powder were transferred to a 100 mL beaker. Then, 10 mL deionized water was added to the beaker. The mixture was homogeneously dispersed via consistent ultrasonication and then heated in a water bath at a temperature of 85 °C for 10 min. The obtained mixture was placed at room temperature to form NaOH/agar hydrogel. Similarly, 0.7 g agar was added to 20 mL 0.03 mol L−1 sodium sulphide (Na2S) aqueous solution, and the mixture was homogeneously dispersed, heated at 85 °C for 10 min and cooled to room temperature. The obtained hydrogel was marked as Na2S/agar hydrogel.2.2 Preparation of Ni1Co2, Ni1Co2-AN, Ni2Co1, Co3 and Ni3
First, 1.5 mmol nickel nitrate hexahydrate (Ni(NO3)2·6H2O), 3 mmol cobalt hexahydrate (Co(NO3)2·6H2O) and 1.5 mmol ammonium chloride (NH4Cl) were added to a solution containing 15 mL deionized water and 50 mL N,N-dimethyl formamide (DMF). The mixture was stirred for several minutes and gradually dropped on NaOH/agar hydrogel. After that, the beaker was placed for 24 h in a dark place without any heat treatment. The precipitate generated was collected, washed using abundant water and ethanol, and heated to dry. The above-mentioned sample was denoted as Ni1Co2. In the absence of NH4Cl, the obtained precipitate was denoted as Ni1Co2-AN. Similarly, the molar ratios of Ni(NO3)2·6H2O and Co(NO3)2·6H2O were adjusted from 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 2:1 and 0:3, and the other conditions were not changed; hence, the corresponding precipitates were marked as Ni3, Ni2Co1 and Co3.
:0, 2:1 and 0:3, and the other conditions were not changed; hence, the corresponding precipitates were marked as Ni3, Ni2Co1 and Co3.
2.3 Preparation of Ni1Co2–S, Ni1Co2-AN–S, Ni2Co1–S, Ni3–S and Co3–S
First, 50 mg Ni1Co2 precursor was dispersed in 40 mL deionized water with ultrasonication to form Ni1Co2 suspension. The above-mentioned suspension was dropped on Na2S/agar hydrogel and reacted at room temperature for 24 h. The precipitate generated was collected, washed using abundant water and ethanol, and heated to dry. The as-obtained composite was marked as Ni1Co2–S. The Ni1Co2 precursor was converted to Ni1Co2-AN, Ni2Co1, Ni3 and Co3 in turn; the samples obtained were denoted as Ni1Co2-AN–S, Ni2Co1–S, Ni3–S and Co3–S, respectively.2.4 General characterization
On the basis of frequently used characterization technologies such as field emission scanning electron microscopy (FESEM, JEOL6700F) and transmission electron microscopy (TEM, JEM-2100F), superficial microtopographies and internal structures of our samples were studied in detail. Besides, an accelerating voltage of 200 kV was employed in the selected area electron diffraction (SAED) measurement. Wide angle X-ray diffraction (XRD, D8 ADVANCE) was also employed to analyze the phase components of our samples utilizing Cu-Kα radiation with a scanning range from 10° to 80°. Furthermore, X-ray photoelectron spectroscopy (XPS, ESCAlab220i-XL, Al-Kα source) and Raman microspectrometry (Renishaw, inVia-Reflex, 532 nm) tests were performed to gain insights into the valence states of different elements and chemical bonds of our samples. Furthermore, inductively coupled plasma-optical emission spectrometry (ICP-OES, SPECTRO CIROS VISION) was used to analyze the accurate mole ratio of metal anions. Fourier transform infrared (FTIR) spectrometry was carried out to study the chemical bonds in our samples. For obtaining the Barrett–Joyner–Halenda (BJH) mode, a Micromeritics ASAP-2020M apparatus was operated.2.5 Electrode preparation and electrochemical measurements
The preparatory method of positive electrodes based on Ni–Co nanocomposites was as follows: first, we ground active materials poly(vinylidene fluoride) (PVDF) and acetylene black (AB) in a mass ratio of 85:5:10 for several minutes. The mixture was added to a suitable amount of N-methyl-2-pyrrolidone (NMP) solvent and stirred constantly to generate a uniformly distributed ropy slurry. Then, the ropy slurry was uniformly applied on a porous Ni foam (effective area of 1 cm2 was controlled) and dried at 110 °C for 6 h in a vacuum oven. A series of electrodes were prepared including Ni1Co2, Ni1Co2-AN, Ni2Co1, Ni3, Co3, Ni1Co2–S, Ni1Co2-AN–S, Ni2Co1–S, Ni3–S and Co3–S. For effective comparison, we controlled the mass loadings of the as-mentioned electrodes to 1.2 ± 0.2 mg cm−2. An active carbon (AC) electrode was also prepared utilizing a similar method except that we changed the mass ratio of the mixture (AC:PVDF:AB = 90:5:5). The mass loading of the AC electrode was controlled in the range of 2.9 ± 0.1 mg cm−2. In the Ni1Co2//AC HSC device, we utilized an Ni1Co2 electrode as the positive electrode and AC positive electrode as the negative electrode. The total mass loading of the Ni1Co2–S//AC device was controlled in the range 4.1 ± 0.3 mg.
3 Results and discussion
3.1 Characterization of morphology, microstructure and composition
The overall fabrication procedure of flowery Ni1Co2 precursor is displayed in Fig. 1. OH− ions were slowly transferred through the solid–liquid interface due to strong bonding between OH− ions and agar hydrogel. These OH− ions were slowly incorporated with Ni2+ and Co2+ ions, thus generating binary α-(Ni–Co) hydroxide. After that, the Na2S/agar hydrogel slowly provided S2− ions for partial incorporation with metal ions from nickel–cobalt hydroxide. A small amount of sulphur ions was grafted on Ni1Co2 hydroxide (Ni1Co2–S).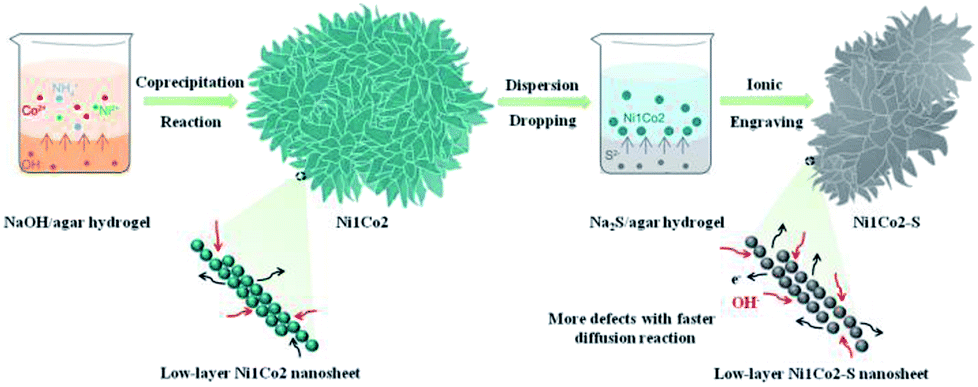 | ||
| Fig. 1 Schematic exhibition of the fabrication procedure of flowery Ni1Co2–S. | ||
As shown in Fig. 2a and S1a and b,† Ni1Co2 displayed well-proportioned flowery clusters (diameter up to several micrometers) and ultrathin internal nanosheets. In comparison, the Ni1Co2-AN sample showed irregular nanoflowers with the coexistence of small sheets and large sheets (in Fig. S2a and b†), because there was no chelation between NH4+ and OH− ions. Furthermore, the mole ratio of Ni(NO3)2:Co(NO3)2 was adjusted from 3:0, 2:1 to 0:3 to optimize the proportion. Staked nanoflowers with insufficient space (Ni2Co1 or Co3) and conglobated nanoparticles (Ni3) are exhibited in Fig. S2c–h,† indicating that various mole ratios of metal nitrates could significantly influence the ionic reaction and tune the microtopography. On the basis of the accurate ICP-OES data in Table S1,† the Ni:Co ratios of Ni1Co2, Ni1Co2-AN, Ni3, Ni2Co1 and Co3 were 1:2.27, 1:2.26, 3:0, 1.86:1 and 3:0, respectively, which matched the dosages of metal nitrates, and this indicated a uniform co-precipitation reaction.
Subsequently, we utilized Na2S/agar hydrogel to grate S2− ions on Ni1Co2 and obtain Ni1Co2–S. The lower solubilities of metal sulphides than those of hydroxides caused ion exchanges between OH− and S2− ions. Fig. S3† reveals the colour change from green (Ni1Co2) to grey green (Ni1Co2–S), which supported the occurrence of S2− ion grating processes. In Fig. 2b and S1c and d,† we can see that the pore sizes of Ni1Co2–S extended with the partial damage of pristine flowery frames. Furthermore, the BJH modes of Ni1Co2 and Ni1Co2–S were also obtained to study the details of pore size distribution. As shown in Fig. S4,† the main pore size of Ni1Co2 was concentrated in a narrow range from 1.7 to 7.5 nm, whereas that of Ni1Co2–S exhibited a wider range from 1.7 to 50 nm. This clear broadening of pore size further revealed a structural change after S2− ion grating. Fig. 2c displays the transparent structure of Ni1Co2–S, and Fig. S5† presents nanosheet thicknesses (mainly ranging from 2.2 to 5.3 nm), and these results revealed the ultrathin property of Ni1Co2–S. In the inset of Fig. 2c, the diffraction circles reveal the polycrystalline property of Ni1Co2–S. In Fig. 2d, the lattice fringes of 0.258, 0.263 and 0.266 nm resulted from the (102) planes of nickel–cobalt hydroxide. Notably, many internal pores and defects could be observed in Fig. 2d such as the red elliptical area, which probably could be ascribed to the grating effect of S2− ion. Furthermore, the elemental mapping of Ni1Co2–S is provided to illuminate element distribution. As depicted in Fig. 2e, Ni, Co, S and O uniformly existed in the flowery Ni1Co2–S architecture. Based on energy dispersive spectrometry (EDS) data shown in Fig. S6,† the mole ratio S:O was found to be 1:5.14, indicating that Ni1Co2–S was composed of Ni–Co hydroxides with slight S2− grating. Also, the mole ratio of Ni:Co reached 1:1.89, which also generally coincided with the ratio of 1:2 (dosage of metal nitrates).
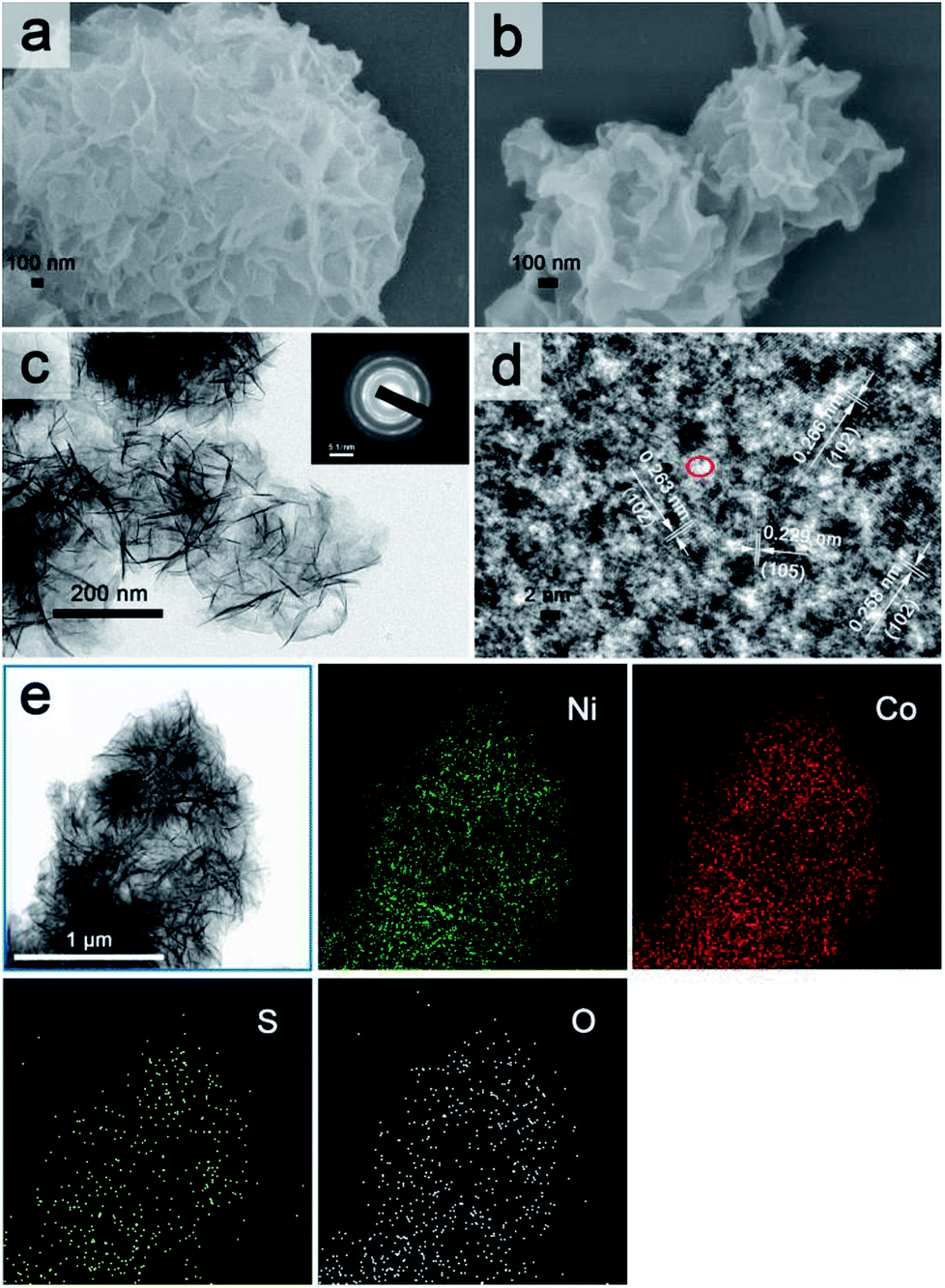 | ||
| Fig. 2 FESEM images of (a) Ni1Co2 and (b) Ni1Co2–S; (c) TEM image of Ni1Co2–S with inset of SEDA image; (d) HETEM image of Ni1Co2–S; (e) TEM figure and corresponding elemental mapping images for Ni1Co2–S. | ||
In Fig. 3, the main peaks of Ni3 are directly indexed to α-Ni hydroxide with turbostratic disorder, and the peaks of Co3 also present the property of α-Co hydroxide.33,34 Meanwhile, the peaks of Ni1Co2, Ni1Co2-AN and Ni2Co1 are analogous with those of Ni3 and Co3, indicating the formation of binary α-(Ni–Co) hydroxide. To further study the structures of these hydroxides, their FTIR patterns are displayed in Fig. S7a.† In samples of Ni1Co2, Ni1Co2-AN, Ni2Co1, Ni3 and Co3, a series of broad peaks centred at 3400–3500 cm−1 were assigned to the hydrogen bonding with intercalated water molecules. Besides, the peaks at 1000–1500 cm−1 were ascribed to intercalated NO3− ions, and the peaks at 400–700 cm−1 resulted from M (Ni/Co)–O–H bending and M–O stretching vibrations. These FTIR results demonstrated the α-hydroxide features of Ni1Co2, Ni1Co2-AN, Ni2Co1, Ni3 and Co3.33–35 Furthermore, the Raman spectra of these samples supported the chemical bonds of hydroxide precursors. In Fig. S8,† the Raman peaks of Co3 located at 184.6, 464.0, 507.6, 599.8 and 666.2 cm−1 coincided with the features of α-Co(OH)2.36–38 In the Raman pattern of Ni3, the peaks at 304.2, 400–600, and 1093.9 cm−1 corresponded to Ni–O stretching, E-type vibration of the Ni–OH lattice and hydroxyl group vibration mode of the Ni(OH)2 phase.39,40 In contrast, Ni1Co2, Ni1Co2-AN and Ni2Co1 maintained the peak locations of Ni3 and Co3 with some peak shifts, indicating the existence of binary Ni–Co hydroxide. Therefore, we successfully fabricated a series of unitary α-(Ni/Co) or binary Ni–Co hydroxides.
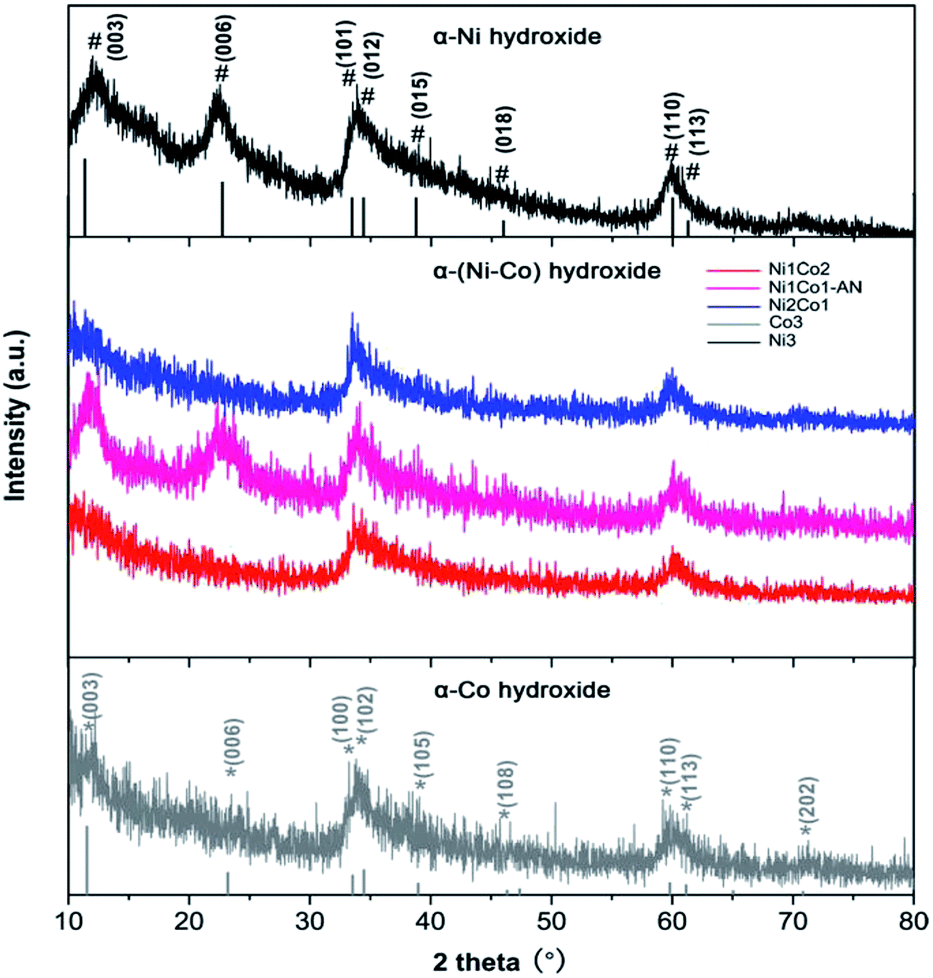 | ||
| Fig. 3 XRD patterns of Ni1Co2, Ni1Co2-AN, Ni2Co1, Co3 and Ni3 samples. | ||
In the XRD pattern shown in Fig. 4a, Ni1Co2–S displays similar peaks to Ni1Co2, demonstrating that Ni1Co2–S mainly contains α-(Ni–Co) hydroxide. Compared to peaks from Ni1Co2, a clear negative shift (ca. 0.6 to 0.9°) can be observed in the XRD pattern of Ni1Co2–S because the slight S2− grating reaction extends the internal planar spacing and pore size. No clear signals of Ni–Co sulphide can be detected because of weak crystallinity and low content of sulphide at room-temperature. By comparing the FTIR and Raman patterns of Ni1Co2 and Ni1Co2–S (in Fig. S7b and 4b†), we further ascertain the α-(Ni–Co) hydroxide component in Ni1Co2–S.
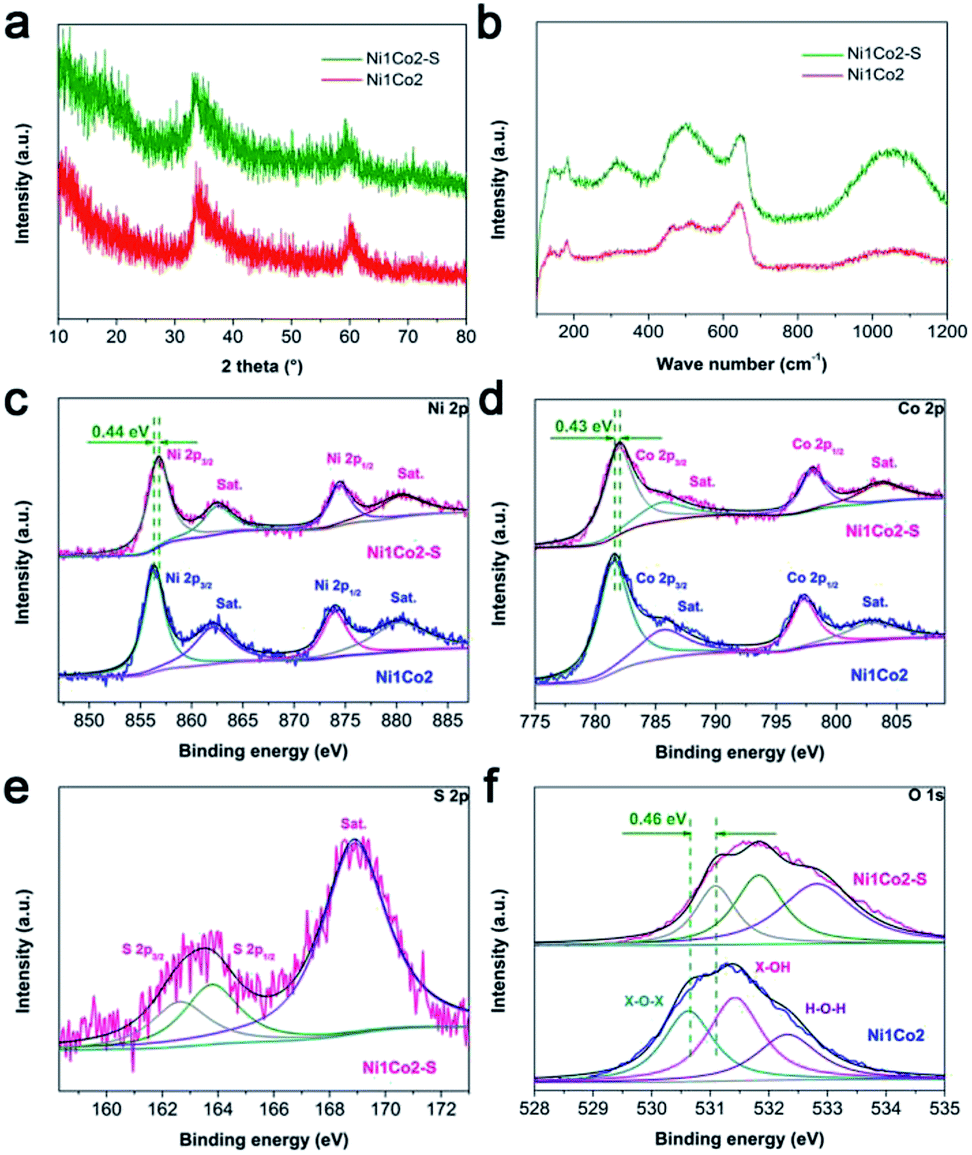 | ||
| Fig. 4 (a) XRD patterns of Ni1Co2 and Ni1Co2–S samples; (b) Raman patterns of Ni1Co2 and Ni1Co2–S samples; (c) Ni 2p, (d) Co 2p, (e) S 2p and (f) O 1s XPS spectra of Ni1Co2 and Ni1Co2–S samples. Herein, X in (f) represents Ni or Co metal element. | ||
We utilized the XPS patterns of Ni1Co2 and Ni1Co2–S to evaluate the variation before and after S2− grafting. All spectra were calibrated based on the C 1s benchmark of 284.6 eV. In the Ni 2p spectra of Ni1Co2 (in Fig. 4c), the peaks at 856.33 and 873.95 eV corresponded to Ni 2p3/2 and 2p1/2 with a separation of 17.62 eV, which was in line with the feature of the Ni(OH)2 phase.41,42 In Fig. 4d, Ni1Co2 displays Co 2p3/2 and 2p1/2 peaks (at 781.51 and 797.29 eV) with a separation of 15.78 eV, indicating the co-existence of divalent and trivalent states of Co.43,44 The divalent and trivalent states of Co co-existed in α-(Ni–Co) hydroxides. For Ni1Co2–S, the energy-bond separations of Ni 2p and Co 2p levels were 17.67 and 16.05 eV, respectively, verifying slight reduction of partial cobalt ions during the ion exchange process. In Fig. 4e, the peaks at 162.63 and 163.77 eV represented S 2p3/2 and 2p1/2 levels, respectively, and a satellite peak at 168.89 eV was ascribed to S4O62− ions with high oxidation states on the surface. Similar S 2p spectra could be found in many reported nickel/cobalt sulfides,45–47 which suggested successful grating of S2− ions in Ni1Co2–S. In contrast to the observations for Ni1Co2, Ni 2p, Co 2p and O 1s levels in the XPS spectrum of Ni1Co2–S clearly shifted to more positive levels, which was mainly due to electronic interactions involving S and Ni/Co components.25,48
3.2 Electrochemical capacitive behaviours of various electrodes
To select an efficient hydroxide precursor, we investigated the performances of Ni1Co2, Ni1Co2-AN, Ni3, Ni2Co1 and Co3 in an alkaline three-electrode system. Fig. 5a shows the CV curves of Ni1Co2, Ni1Co2-AN, Ni3, Ni2Co1 and Co3 at 5 mV s−1. The main contents of Ni1Co2, Ni1Co2-AN, Ni3, Ni2Co1 and Co3 were unitary Ni/Co or binary Ni–Co hydroxides. The energy storage mechanism could be described as follows:49–54| Ni(OH)2 + OH− ↔ NiOOH + e− | (1) |
| Co(OH)2 + OH− ↔ CoOOH + e− | (2) |
| CoOOH + OH− ↔ CoO2 + e− | (3) |
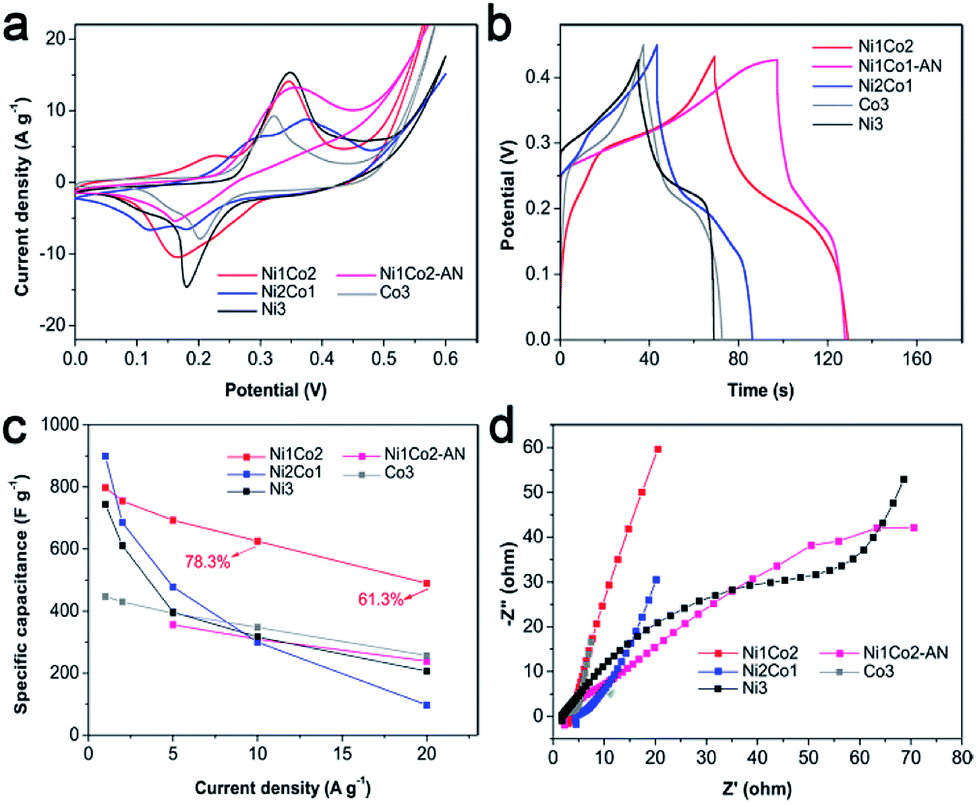 | ||
| Fig. 5 (a) CV curves at 5 mV s−1, (b) GCD curves at 5 A g−1, (c) specific capacitances at various current densities and (d) EIS spectra of Ni1Co2, Ni1Co2-AN, Ni2Co1, Co3 and Ni3 electrodes. | ||
A smaller potential disparity (ΔEa,c, the disparity between the anode and cathode peak positions) is presented in the CV curve of Ni1Co2, suggesting slight electrode polarization.55,56 Additionally, Ni1Co2 delivers longer discharge time, as shown in Fig. 5b, and better capacitance performance, as shown in Fig. 5c, thus highlighting the enhanced performance of the Ni1Co2 electrode. These results are mainly ascribed to the ultrathin flowery structure, providing more active contact sites for charge transfer. In the EIS spectra (Fig. 5d), the intersections of semicircles on the real axis, semicircle diameter at high frequency and slope of a line in the low-frequency region are employed to evaluate the equivalent series resistance (Rs), charge-transfer resistance (Rct) and diffusion resistance (Zw).57,58 The Rs values of Ni1Co2, Ni1Co2-AN, Ni2Co1, Co3 and Ni3 electrodes are 2.90, 2.05, 4.38, 1.74 and 1.65 Ω, respectively. In comparison, even though Ni1Co2 possesses higher Rs values, the smaller semicircle diameter and steeper slope indicate lower Rct and Zw values, thus revealing enhanced performance.
To further promote the performance of Ni1Co2, controllable S2− grafting was carried out to obtain Ni1Co2–S. The CV curves of Ni1Co2 and Ni1Co2–S at various scanning rates from 5 to 50 mV s−1 are presented in Fig. 6a–c. Compared to that of Ni1Co2, the extended CV area of Ni1Co2–S revealed enhanced capacitance. Interestingly, compared to the observations for Ni1Co2, there was a pair of anodic peaks that disappeared for Ni1Co2–S electrode. This peak disappearance actually resulted due to closer location of various pairs of oxidation and reduction peaks, leading to overlapping.59,60 In the CV curves of Ni2Co1–S (Fig. S9a†), there were many pairs of oxidation and reduction peaks, indicating incomplete overlapping of these peaks. Furthermore, the scatters of logIp–logν from the three pairs of oxidation peaks are fitted in Fig. 6d, wherein Ip and ν stand for scanning rate and peak current, respectively. Based on a previous study, the relationship between Ip and ν is in line with the formula I = aνb, wherein a and b represent empirical parameters; b value of 0.5 is due to the diffusion-dominated reaction, whereas b value of 1 originates from the ideal capacitive behaviour.55,61 In our study, the peak 1 and peak 2 from Ni1Co2 exhibited higher b values of 0.63 and 0.84, respectively, and the peak 3 from Ni1Co2–S exhibited a lower b value of 0.55, indicating that the capacitances of Ni1Co2 and Ni1Co2–S contained diffusion and capacitive contributions.
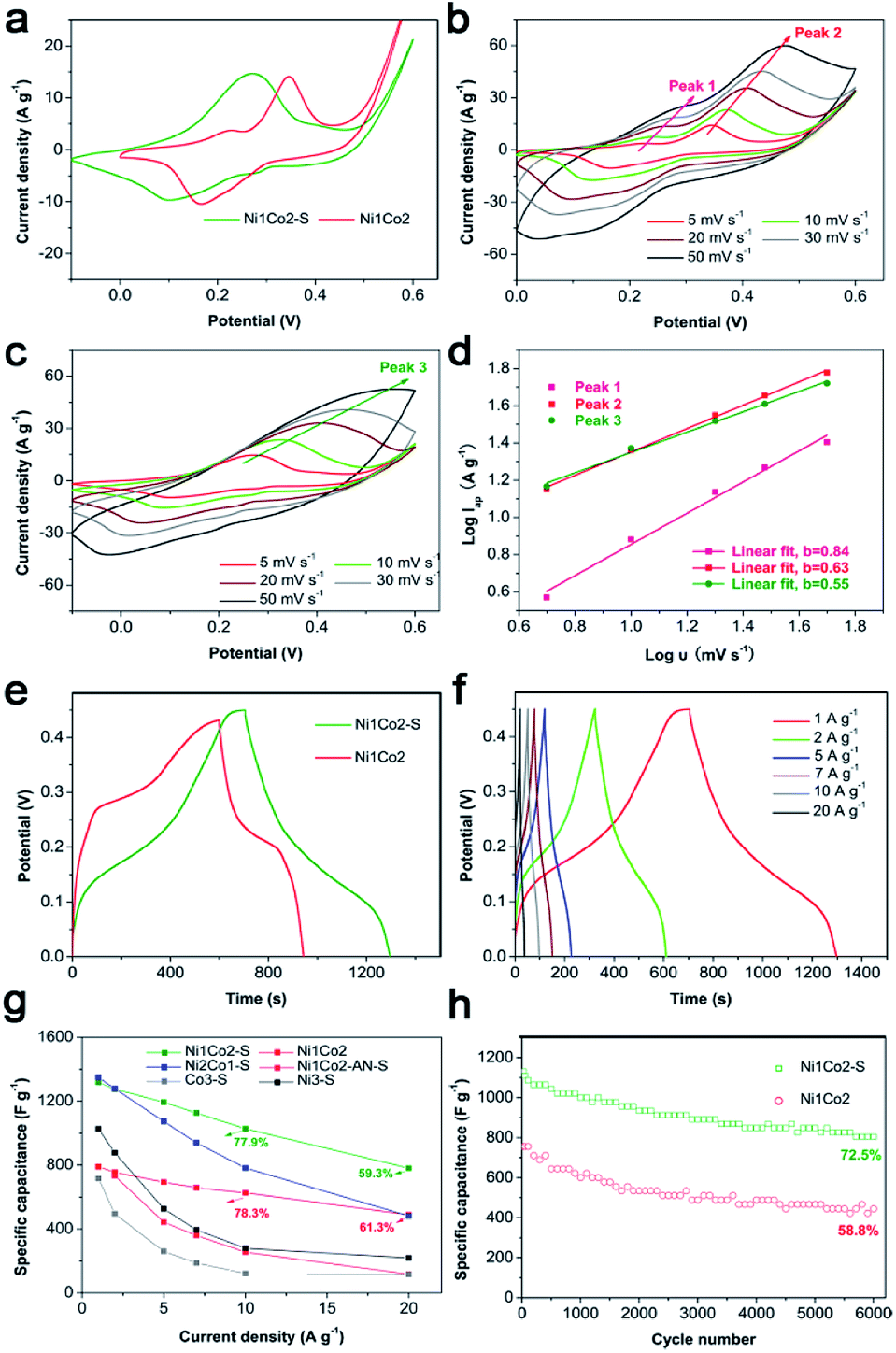 | ||
| Fig. 6 (a) CV curves of Ni1Co2 and Ni1Co2–S at 5 mV s−1; (b) CV curves of Ni1Co2 at different scanning rates; (c) CV curves of Ni1Co2–S at different scanning rates; (d) linear fit of logIap–logν scatters from three pairs of peaks in the CV curves of Ni1Co2 and Ni1Co2–S; (e) GCD curves of Ni1Co2 and Ni1Co2–S at 1 A g−1; (f) GCD curves of the as-fabricated Ni1Co2–S at different current densities; (g) capacitance performances of Ni1Co2, Ni1Co2–S, Ni1Co2-AN–S, Ni2Co1–S, Co3–S and Ni3–S electrodes at various current densities; (h) 6000 cycle duration measurements of Ni1Co2 and Ni1Co2–S at the current density of 10 A g−1. | ||
As shown in Fig. 6e and f, Ni1Co2–S displayed low discharge platform, which was in agreement with the result of CV curves. We further calculated the specific capacitances of our electrodes based on different GCD curves from 1 to 20 A g−1. As shown in Fig. 6g, Ni1Co2–S possessed enhanced specific capacitances (1317.8 F g−1 at 1 A g−1 and 781.3 F g−1 at 20 A g−1) relative to those of Ni1Co2 (789.5 F g−1 at 1 A g−1 and 489.4 F g−1 at 20 A g−1). When the Ni1Co2–S electrode was at high current density of 10 or 20 A g−1, it still displayed suitable capacitance retention of 77.9% or 59.3%, respectively. The electrochemical capacitances of Ni1Co2-AN–S, Ni2Co1–S, Ni3–S and Co3–S were also studied. Similarly, S2− ion grating enhanced the capacitance compared with that of the corresponding precursors. Ni1Co2–S displayed excellent capacitance and high rate performance. Therefore, the selection of precursor and further S2− ion grating efficiently promoted the comprehensive performances of our electrode materials.
Besides, long-term cycling duration is desired for reversible applications; hence, the cycling measurement of Ni1Co2–S was operated at a current density of 10 A g−1. In Fig. 6h, after 6000 cycles, Ni1Co2–S presented a high capacitance of 804.3 F g−1 and capacitance retention of 72.5%, which was higher than that of Ni1Co2 (58.8%). As shown in Fig. 7, Ni1Co2–S had lower semicircle diameter and steeper slope (lower Rct and Zw), further highlighting the improved resistance performance. Rct and Zw significantly increased after 6000 cycles, probably due to the damage of internal nanosheets of Ni1Co2–S (in Fig. S10†). In view of the results mentioned above, the enhancement in the electrochemical performance of Ni1Co2–S was mainly due to the following factors: (1) suitable ratio of Ni:Co could tune the nanostructure and promote electrochemical performance; (2) S was introduced into Ni–Co hydroxide, and it greatly improved the conductivity; and (3) S2− ion grafting generated many defects and extended the pore size.
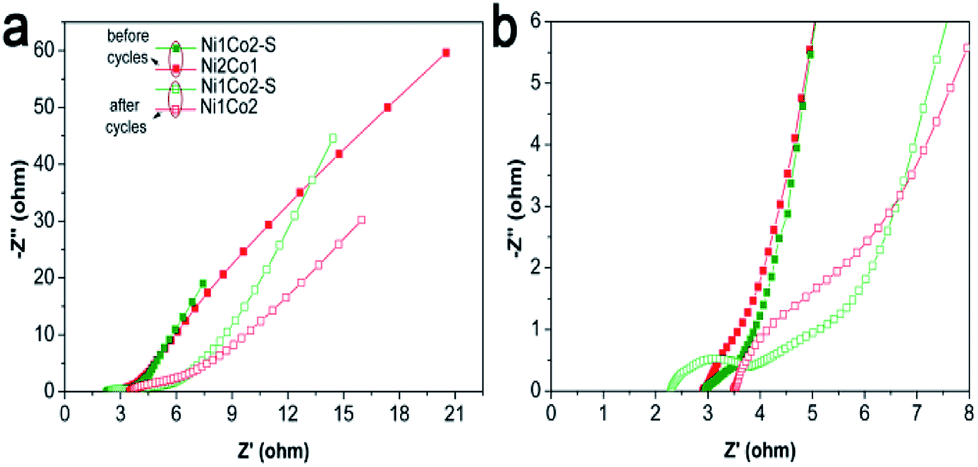 | ||
| Fig. 7 (a) EIS spectra of Ni1Co2 and Ni1Co2–S electrodes before and after 6000 cycles with frequencies from 0.01 to 100 kHz; (b) magnification of EIS spectra from 0 to 8 Ω for Z′. | ||
3.3 Electrochemical capacitive behaviour of HSC device
Considering that general three-electrode systems have narrow potential windows, we prepared an HSC device to enlarge the voltage using Ni1Co2–S and AC electrodes as positive and negative electrodes, respectively, as shown in Fig. 8a. The CV curves shown in Fig. 8b present large quasi-rectangular shapes with few weak redox peaks, which suggest the battery-type and electrical double-layer hybrid energy-storage mechanisms. Fig. 8c displays various linear GCD profiles with long discharge time, further highlighting its good capacitance performance. As depicted in Fig. 8d, Ni1Co2–S//AC delivered great specific capacitance values of 117.1, 106.3, 97.5, 82.3, 74.5 and 61.1 F g−1 at various current densities of 1, 2, 3, 5, 7 and 10 A g−1, respectively. Unfortunately, with the increase in current density, the specific capacity gradually faded, and the total voltage of the HSC also increased due to insufficient electrochemical reaction at high current density. Similar to the study of the three-electrode system, we also studied the cycling stability of Ni1Co2–S//AC with charge/discharge current density of 3 A g−1 (in Fig. 8e). The initial electrochemical measurement contributed to gradual wetting between the electrode and electrolyte. The trapped OH− ions then gradually diffused out, and the contact area between the electrode and electrolyte was enlarged, thus causing capacitance enhancement during the initial 1000 cycles.62,63 Subsequently, the capacitance of Ni1Co2–S//AC slowly faded and reached 82.4% after 7000 cycles at 3 A g−1, resulting from partial damage to the Ni1Co2–S structure.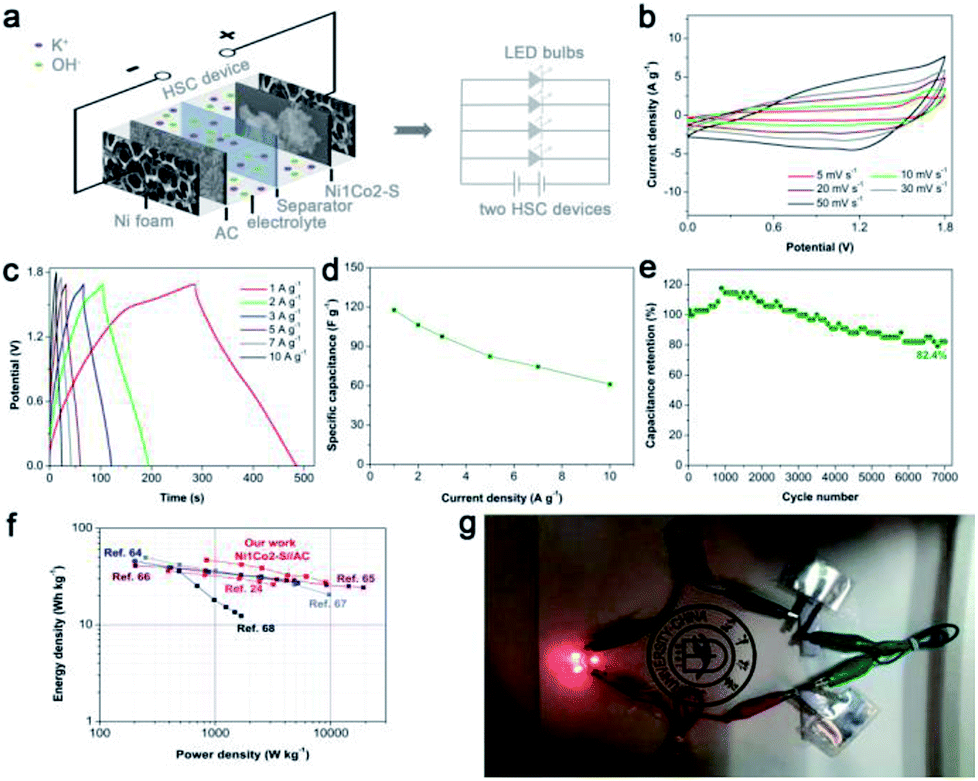 | ||
| Fig. 8 (a) Schematic diagram of Ni1Co2–S//AC and lighting LED bulb devices; (b) CV curves of the as-fabricated Ni1Co2–S//AC at different scanning rates from 5 to 50 mV s−1; (c) GCD curves of Ni1Co2–S//AC device at various current densities from 1 to 10 A g−1; (d) specific capacitance of Ni1Co2–S//AC device at various current densities from 1 to 10 A g−1; (e) cycling performance of Ni1Co2–S//AC for 7000 cycles at 3 A g−1; (f) Ragone plots of our study and other peer HSC devices; and (g) photographs of lighting LED bulbs device with fast charging current of 3 A g−1 based on total mass of active materials. | ||
The relationship between energy density and power density in Ni1Co2–S//AC is depicted in the Ragone diagram (Fig. 8f). The HSC device achieved a high energy density of 46.7 W h kg−1 at a power density of 0.845 kW kg−1 and also reached 27.5 W h kg at 9 kW kg−1, which were comparable to the results of many peer HSC devices based on core–shell NiCo2S4@Co(OH)2 nanotube arrays,24 NiCo2S4 nanosheets,64 and other cobalt/nickel hydroxide electrode materials.65–68 To trigger four light emitting diodes (LEDs, red, 5 mm), we further assembled two Ni1Co2–S//AC devices in series and charged them at 3 A g−1, as exhibited in Fig. 6g. As anticipated, the LED bulbs worked for a long duration of 2 min and displayed dim light after 5 min. These results suggested that Ni1Co2–S can serve as a promising energy storage material.
4 Conclusions
Herein, we designed a simple solid–liquid interface reaction to synthesize Ni1Co2–S using NaOH/agar and Na2S/agar hydrogels. Ni1Co2–S was mainly composed of α-(Ni–Co) hydroxide with S2− ion grating. The slight ion exchange reaction between S2− ions and Ni1Co2 introduced a small amount of sulphide to improve its conductivity and expanded the internal pore size, thus ensuring rapid charge transfer. Compared to other Ni1Co2, Ni1Co2-AN, Ni2Co1, Co3 and Ni3 electrodes, the Ni1Co2–S electrode delivered improved capacitance of 1317.8 F g−1 at 1 A g−1, suitable rate performance (77.9% at 10 A g−1 and 59.3% at 20 A g−1) and improved cycling stability. The Ni1Co2–S//AC device also exhibited a high energy density of 46.7 W h kg−1 at 0.845 W kg−1 and long cycling stability, which further demonstrated the potential advantage of Ni1Co2–S material in supercapacitive storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 51501068) and Graduate Innovation Fund of Jilin University (Grant No. 2017183).Notes and references
- K. M. Hercule, Q. Wei, A. M. Khan, Y. Zhao, X. Tian and L. Mai, Nano Lett., 2013, 13, 5685–5691 CrossRef PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef PubMed.
- X. Zhang, Y. Zhao and C. Xu, Nanoscale, 2014, 6, 3638–3646 RSC.
- T. Kim, G. Jung, S. Yoo, K. S. Suh and R. S. Ruoff, ACS Nano, 2013, 7, 6899–6905 CrossRef PubMed.
- B. Y. Guan, L. Yu, X. Wang, S. Song and X. W. Lou, Adv. Mater., 2017, 29, 1605051 CrossRef PubMed.
- J. H. Won, H. M. Jeong and J. K. Kang, Adv. Energy Mater., 2017, 7, 1601355 CrossRef.
- T. Brousse, D. Belanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef PubMed.
- Y. Gogotsi, ACS Nano, 2014, 8, 5369–5371 CrossRef PubMed.
- D. Zhou, X. Su, M. Boese, R. Wang and H. Zhang, Nano Energy, 2014, 5, 52–59 CrossRef.
- L. Wang, Z. H. Dong, Z. G. Wang, F. X. Zhang and J. Jin, Adv. Funct. Mater., 2013, 23, 2758–2764 CrossRef.
- J. Ji, L. L. Zhang, H. Ji, Y. Li, X. Zhao, X. Bai, X. Fan, F. Zhang and R. S. Ruoff, ACS Nano, 2013, 7, 6237–6243 CrossRef PubMed.
- R. B. Rakhi, W. Chen, D. Cha and H. N. Alshareef, Nano Lett., 2012, 12, 2559–2567 CrossRef PubMed.
- V. R. Shinde, S. B. Mahadik, T. P. Gujar and C. D. Lokhande, Appl. Surf. Sci., 2006, 252, 7487–7492 CrossRef.
- M. Liu, L. Gan, W. Xiong, Z. Xu, D. Zhu and L. Chen, J. Mater. Chem. A, 2014, 2, 2555–2562 RSC.
- K. Ramasamy, R. K. Gupta, S. Palchoudhury, S. Ivanov and A. Gupta, Chem. Mater., 2015, 27, 379–386 CrossRef.
- M. Chauhan, K. P. Reddy, C. S. Gopinath and S. Deka, ACS Catal., 2017, 7, 5871–5879 CrossRef.
- X. Chen, Y. Huang, K. Zhang, X. Feng and S. Li, J. Alloys Compd., 2016, 686, 905–913 CrossRef.
- G. C. Lau, N. A. Sather, H. Sai, E. M. Waring, E. Deiss-Yehiely, L. Barreda, E. A. Beeman, L. C. Palmer and S. I. Stupp, Adv. Funct. Mater., 2018, 28, 1702320 CrossRef.
- Y. Liu, N. Fu, G. Zhang, M. Xu, W. Lu, L. Zhou and H. Huang, Adv. Funct. Mater., 2017, 27, 1605307 CrossRef.
- J. H. Park, O. O. Park, K. H. Shin, C. S. Jin and J. H. Kim, Electrochem. Solid-State Lett., 2002, 5, H7–H10 CrossRef.
- D. Lee, Q. X. Xia, J. M. Yun and K. H. Kim, Appl. Surf. Sci., 2018, 433, 16–26 CrossRef.
- W. Zhou, X. Cao, Z. Zeng, W. Shi, Y. Zhu, Q. Yan, H. Liu, J. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2216–2221 RSC.
- R. Li, S. Wang, Z. Huang, F. Lu and T. He, J. Power Sources, 2016, 312, 156–164 CrossRef.
- X. Zhu, X. Dou, J. Dai, X. An, Y. Guo, L. Zhang, S. Tao, J. Zhao, W. Chu, X. C. Zeng, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 12465–12469 CrossRef PubMed.
- T. Yoon and K. S. Kim, Adv. Funct. Mater., 2016, 26, 7386–7393 CrossRef.
- T. Yan, R. Li, L. Zhou, C. Ma and Z. Li, Electrochim. Acta, 2015, 176, 1153–1164 CrossRef.
- Y. Lu, Y. Wang, Y. Zou, Z. Jiao, B. Zhao, Y. He and M. Wu, Electrochem. Commun., 2010, 12, 101–105 CrossRef.
- F.-X. Ma, H. Hu, H. B. Wu, C.-Y. Xu, Z. Xu, L. Zhen and X. W. Lou, Adv. Mater., 2015, 27, 4097–4101 CrossRef PubMed.
- S. Ratha, A. K. Samantara, K. K. Singha, A. S. Gangan, B. Chakraborty, B. K. Jena and C. S. Rout, ACS Appl. Mater. Interfaces, 2017, 9, 9640–9653 CrossRef PubMed.
- F. Grote, Z.-Y. Yu, J.-L. Wang, S.-H. Yu and Y. Lei, Small, 2015, 11, 4666–4672 CrossRef PubMed.
- B. Li, X. Li, J. Zai and X. Qian, Nano-Micro Lett., 2016, 8, 174–181 CrossRef.
- M. Rajamathi, P. V. Kamath and R. Seshadri, J. Mater. Chem., 2000, 10, 503–506 RSC.
- P. V. Kamath, G. H. A. Therese and J. Gopalakrishnan, J. Solid State Chem., 1997, 128, 38–41 CrossRef.
- M. Rajamathi, G. N. Subbanna and P. V. Kamath, J. Mater. Chem., 1997, 7, 2293–2296 RSC.
- T. Peng, H. Wang, H. Yi, Y. Jing, P. Sun and X. Wang, Electrochim. Acta, 2015, 176, 77–85 CrossRef.
- O. Bockman, T. Ostvold, G. A. Voyiatzis and G. N. Papatheodorou, Hydrometallurgy, 2000, 55, 93–105 CrossRef.
- Y. Wang, H. Wang and X. Wang, Electrochim. Acta, 2013, 92, 298–303 CrossRef.
- W. Tian, X. Wang, C. Zhi, T. Zhai, D. Liu, C. Zhang, D. Golberg and Y. Bando, Nano Energy, 2013, 2, 754–763 CrossRef.
- X. Xiong, D. Ding, D. Chen, G. Waller, Y. Bu, Z. Wang and M. Liu, Nano Energy, 2015, 11, 154–161 CrossRef.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef.
- C. Hou, X.-Y. Lang, Z. Wen, Y.-F. Zhu, M. Zhao, J.-C. Li, W.-T. Zheng, J.-S. Lian and Q. Jiang, J. Mater. Chem. A, 2015, 3, 23412–23419 RSC.
- J. Liang, R. Ma, N. Iyi, Y. Ebina, K. Takada and T. Sasaki, Chem. Mater., 2010, 22, 371–378 CrossRef.
- H. Chen, L. Hu, M. Chen, Y. Yan and L. Wu, Adv. Funct. Mater., 2014, 24, 934–942 CrossRef.
- W. Du, Z. Wang, Z. Zhu, S. Hu, X. Zhu, Y. Shi, H. Pang and X. Qian, J. Mater. Chem. A, 2014, 2, 9613–9619 RSC.
- L. Niu, Y. Wang, F. Ruan, C. Shen, S. Shan, M. Xu, Z. Sun, C. Li, X. Liu and Y. Gong, J. Mater. Chem. A, 2016, 4, 5669–5677 RSC.
- T. Li, Y. Zuo, X. Lei, N. Li, J. Liu and H. Han, J. Mater. Chem. A, 2016, 4, 8029–8040 RSC.
- J.-X. Feng, H. Xu, Y.-T. Dong, S.-H. Ye, Y.-X. Tong and G.-R. Li, Angew. Chem., Int. Ed., 2016, 55, 3694–3698 CrossRef PubMed.
- H. Ma, J. He, D.-B. Xiong, J. Wu, Q. Li, V. Dravid and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 1992–2000 CrossRef PubMed.
- Y. Zhao, H. Ma, S. Huang, X. Zhang, M. Xia, Y. Tang and Z.-F. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 22997–23005 CrossRef PubMed.
- M. Wang, F. Jin, X. Zhang, J. Wang, S. Huang, X. Zhang, S. Mu, Y. Zhao and Y. Zhao, ACS Sustainable Chem. Eng., 2017, 5, 5679–5685 CrossRef.
- Y. Zhao, Z. Chen, D.-B. Xiong, Y. Qiao, Y. Tang and F. Gao, Sci. Rep., 2016, 6, 17613 CrossRef PubMed.
- Y. Zhao, X. Zhang, J. He, L. Zhang, M. Xia and F. Gao, Electrochim. Acta, 2015, 174, 51–56 CrossRef.
- Z. Chen, D.-B. Xiong, X. Zhang, H. Ma, M. Xia and Y. Zhao, Nanoscale, 2016, 8, 6636–6645 RSC.
- J. Liu, M. Chen, L. Zhang, J. Jiang, J. Yan, Y. Huang, J. Lin, H. J. Fan and Z. X. Shen, Nano Lett., 2014, 14, 7180–7187 CrossRef PubMed.
- J. Liu, J. Wang, Z. Ku, H. Wang, S. Chen, L. Zhang, J. Lin and Z. X. Shen, ACS Nano, 2016, 10, 1007–1016 CrossRef PubMed.
- Y. Zeng, Y. Han, Y. Zhao, Y. Zeng, M. Yu, Y. Liu, H. Tang, Y. Tong and X. Lu, Adv. Energy Mater., 2015, 5, 1402176 CrossRef.
- L.-L. Zhang, H.-H. Li, C.-Y. Fan, K. Wang, X.-L. Wu, H.-Z. Sun and J.-P. Zhang, J. Mater. Chem. A, 2015, 3, 19077–19084 RSC.
- Q. Liao, N. Li, S. Jin, G. Yang and C. Wang, ACS Nano, 2015, 9, 5310–5317 CrossRef PubMed.
- Y. Cheng, H. Zhang, C. V. Varanasi and J. Liu, Energy Environ. Sci., 2013, 6, 3314–3321 RSC.
- X. Xu, J. Liu, Z. Liu, J. Shen, R. Hu, J.-W. Liu, L. Ouyang, L. Zhang and M. Zhu, ACS Nano, 2017, 11, 9033–9040 CrossRef PubMed.
- J. Chang, M. Jin, F. Yao, T. H. Kim, L. Viet Thong, H. Yue, F. Gunes, B. Li, A. Ghosh, S. Xie and Y. H. Lee, Adv. Funct. Mater., 2013, 23, 5074–5083 CrossRef.
- S. Saha, M. Jana, P. Khanra, P. Samanta, H. Koo, N. C. Murmu and T. Kuila, RSC Adv., 2016, 6, 1380–1387 RSC.
- L. Shen, J. Wang, G. Xu, H. Li, H. Dou and X. Zhang, Adv. Energy Mater., 2015, 5, 1400977 CrossRef.
- J.-C. Chen, C.-T. Hsu and C.-C. Hu, J. Power Sources, 2014, 253, 205–213 CrossRef.
- Y. Cheng, H. Zhang, C. V. Varanasi and J. Liu, Energy Environ. Sci., 2013, 6, 3314–3321 RSC.
- H. Jiang, C. Li, T. Sun and J. Ma, Chem. Commun., 2012, 48, 2606–2608 RSC.
- H. B. Li, M. H. Yu, F. X. Wang, P. Liu, Y. Liang, J. Xiao, C. X. Wang, Y. X. Tong and G. W. Yang, Nat. Commun., 2013, 4, 1894 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02791a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |