
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A 3D bioinspired highly porous polymeric scaffolding system for in vitro simulation of pancreatic ductal adenocarcinoma
Stella Totti a,
Mark C. Allenbyb,
Susana Brito Dos Santosb,
Athanasios Mantalarisb and
Eirini G. Velliou*a
a,
Mark C. Allenbyb,
Susana Brito Dos Santosb,
Athanasios Mantalarisb and
Eirini G. Velliou*a
aBioprocess and Biochemical Engineering Group (BioProChem), Department of Chemical and Process Engineering, University of Surrey, Guildford, GU2 7XH, UK. E-mail: e.velliou@surrey.ac.uk; Fax: 0044-(0)-1483686577
bBiological Systems Engineering Laboratory (BSEL), Department of Chemical Engineering, Imperial College London, London, SW7 2AZ, UK
First published on 7th June 2018
Abstract
Pancreatic ductal adenocarcinoma is an aggressive disease with an extremely low survival rate. This is due to the (i) poor prognosis and (ii) high resistance of the disease to current treatment options. The latter is partly due to the very complex and dense tissue/tumour microenvironment of pancreatic cancer, which contributes to the disease's progression and the inhibition of apoptotic pathways. Over the last years, advances in tissue engineering and the development of three-dimensional (3D) culture systems have shed more light into cancer research by enabling a more realistic recapitulation of the niches and structure of the tumour microenvironment. Herein, for the first time, 3D porous polyurethane scaffolds were fabricated and coated with fibronectin to mimic features of the structure and extracellular matrix present in the pancreatic cancer tumour microenvironment. The developed 3D scaffold could support the proliferation of the pancreatic tumour cells, which was enhanced with the presence of fibronectin, for a month, which is a significantly prolonged in vitro culturing duration. Furthermore, in situ imaging of cellular and biomarker distribution showed the formation of dense cellular masses, the production of collagen-I by the cells and the formation of environmental stress gradients (e.g. HIF-1α) with similar heterogeneity trends to the ones reported in in vivo studies. The results obtained in this study suggest that this bioinspired porous polyurethane based scaffold has great potential for in vitro high throughput studies of pancreatic cancer including drug and treatment screening.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive malignancy, being the fourth leading cause of cancer deaths in the USA, the fifth in the United Kingdom and the seventh worldwide.1,2 The 5 year survival rate, which has barely improved over the last 4 decades, is 8%.2 The high disease mortality is due to (i) the lack of symptoms which leads to late stage diagnosis,3 (ii) the high metastatic likelihood4 and (iii) the high resistance to currently available treatment options.5 The latter can be partly attributed to the disease's complex tumour microenvironment (TME),6–9 which consists of intense extracellular matrix (ECM) fibrosis (desmoplasia)6,10–12 that can contribute to the disease's progression and inhibition of apoptotic pathways, both increasing treatment resistance.13–15 Classically used systems for studying pancreatic cancer are either 2D in vitro systems or animals. While 2D in vitro systems are easy to use and responsive to radiation and drugs8,16,17 they cannot accurately simulate important 3D in vivo TME aspects such as structure, porosity, presence of ECM proteins, realistic cell–cell and cell–ECM interactions, environmental gradients (nutrient, oxygen) and vascularisation.8,18–21 Several animal model systems have been developed for pancreatic cancer,22,23 the best of which involve genetic modification, that have provided considerable insights into the pathogenesis of the disease. However, they are time consuming, expensive to generate and maintain and they are not always reproducible.23–29 Advances in tissue engineering enable the development of 3D constructs that are generally less expensive than animals, more reproducible and easy to develop while they provide a more realistic structure, cell–cell, cell–ECM spatial interactions and a more realistic distribution of environmental parameters within the system, such as nutrients and oxygen, chemotherapeutic diffusion, and irradiation deposition as compared to 2D systems.8,30–36Current 3D in vitro systems of pancreatic tumours include (i) spheroid/organoid systems,37–45 (ii) hydrogel scaffolds42,46–48 and (iii) polymeric scaffolds.49–51 For example, Ware et al. have developed pancreatic cancer spheroids with a distinct cohesiveness of the cellular masses.43 Additionally, Longati et al. showed that pancreatic cancer cell lines in spheroids exhibited decreased cellular proliferation, increased ECM production and resistance to chemotherapeutic reagents as compared to a 2D monolayer culture.37 The increased chemo-resistance of pancreatic tumour spheroids in comparison with conventional 2D systems was also reported by Wen et al.44 Some features of the complex pancreatic cancer TME have been shown within spheroids. For example, Ware et al. developed a stroma rich spheroid matrix that exhibited regions of increased collagen expression compared to the non-stromal rich control system.41 Furthermore, Boj et al. constructed 3D matrigel based organoids of primary PDAC cells in order to model in vitro some essential histological and genetic characteristics of this malignancy.39,40 Raza et al. developed polymeric hydrogels of various stiffness levels. It was observed that the stiffness of the polymeric matrix affected the cellular proliferation.48 Similar results were reported by Ki et al. who observed increased pancreatic cancer cell growth after 7 days of culture for softer thiol-ene based hydrogels.47 Additionally, Chiellini et al. created two types of microstructured hydrogels, fabricated either with chitosan (CS) or with a polyelectrolyte complex formed between CS and poly(γ-glutamic acid) (γ-PGA). BxPC-3 pancreatic tumour cells proliferated in those hydrogels for 28 days, they formed aggregates and they retained cancer typical features, i.e. loss of polarity and duct-like structures. Additionally, fascin (a marker of tumour invasiveness), was expressed in the hydrogels, but not in 2D monolayer systems.46
There are a few limited studies investigating the potential of 3D polymeric scaffolds as a pancreatic cancer model. More specifically, Wang et al. showed that a fibrous polyglyconate/gelatine scaffold (4 mm inner diameter) supported the proliferation of pancreatic cancer stem cells (CSCs) in vitro for 7 days as well tumour formation and metastasis, when it was transplanted in mice.50 Furthermore, He et al. created a fibrous polymeric scaffolding system based on poly (glycolide-co-trimethylene carbonate) and gelatine for pancreatic tumour growth. This polymeric scaffold model demonstrated better neoplastic formation and accelerated tumour evolution as compared with the 2D system.51 Ricci et al. developed three different biocompatible scaffolds based on two polymers [poly (ethylene oxide terephthalate)/poly(butylene terephthalate) and poly(vinyl alcohol)/gelatine] and two polymeric formulations (fibre mesh and sponge like). The type of polymer and the formulation technique altered the internal architecture of the scaffold, which affected the cell growth and morphology as well as the tumour-specific metalloproteinases (MMPs) synthesis of PDAC.49
Among existing tissue engineering systems, polymeric scaffolds provide a more robust control of the internal structure (pore shape, size and interconnectivity) enabling the TME structural recapitulation.8,21,30,49,52–56 Additionally, surface modification of the scaffolds with proteins enables the ECM mimicry which is crucial in tissue and/or tumour development.21,55,57,58 Furthermore, the high mechanical strength of polymeric scaffolds allows the introduction of perfusion in the system for vascularization mimicry with tuneable flow rates.52,54 Overall, the above features enable the recapitulation of realistic cell–cell interactions and distributions, cell–matrix interactions as well as the formation of realistic oxygen and nutrient gradients. These gradients can be more controlled, tuned and less acute/extreme in highly porous polymeric scaffolds as compared to cell aggregate based 3D systems.8,35 Therefore, polymeric scaffolds are a promising platform for pancreatic cancer in vitro studies.
Natural and synthetic are the two main categories of polymers that are used for scaffold fabrication in tissue engineering. However, natural polymers are not preferred for load-bearing tissue applications due to their limited mechanical and physical stability and therefore synthetic polymers are currently the dominant scaffolding matrices in tissue engineering and regenerative medicine.59–61 Among the synthetic polymers, polyurethanes (PUs) are considered very promising candidates in tissue engineering due to their unique segmented structure, their wide range of mechanical and physical properties, their assortment from stable to degradable materials and their biocompatibility.61,62 PUs have been used as scaffolding materials for soft,63 cartilage,64 bone tissue engineering,65 and to recapitulate normal and abnormal haematopoiesis.21,52,55 Nevertheless, they have not been used to date for pancreatic cancer tissue engineering.
In this study, we report for the first time the use of a highly porous 3D polyurethane (PU) scaffolding system as a platform for pancreatic cancer modelling. Surface modification of the scaffold took place with fibronectin (FN) for enhancement of cellular adhesion. The cellular evolution including proliferation, morphology and cell mass formation in the scaffold was monitored for 29 days. Furthermore, sectioning of the scaffolds, fluorescent staining and imaging with confocal laser scanning microscopy (CLSM) enabled the spatial 3D determination of environmental (stress) gradients, i.e., oxidative, starvation, and ECM production within the 3D scaffolding system.
2. Materials and methods
2.1. Fabrication and sterilisation of the 3D scaffolds
The PU scaffolds were fabricated by the Thermally Induced Phase Separation (TIPS) method, which is described in Fig. 1.21,55,66,67 PU beads (Noveon, Belgium) were dissolved in dioxan (5% w/v) (99.8% anhydrous pure, Sigma-Aldrich, UK) and the solution was quenched at −80 °C for 2 h. Thereafter, the solvent was removed by freeze drying in a poly-ethylene glycol (PEG) bath at −15 °C under 0.01 mbar vacuum pressure. The scaffolds were then cut in 5 × 5 × 5 mm3 cubes. The average pore size was 100–150 μm, the porosity 85–90%, as determined by Mercury Intrusion Porosimetry (MIP) (PoreMaster33, Quantachrome) and the specific pore volume obtained using helium displacement pycnometry (AccuPyc, 1330 V3.00) was 2.3 ± 0.48 cm3 g−1. The compression modulus was 28 ± 3 kPa, as reported previously by Safinia et al.67 Sterilisation of the scaffolds took place by washing them with 70% v/v ethanol solution for 3 h followed by exposure to a UV/ozone generator for 10 min (BioForce Nanosciences, USA).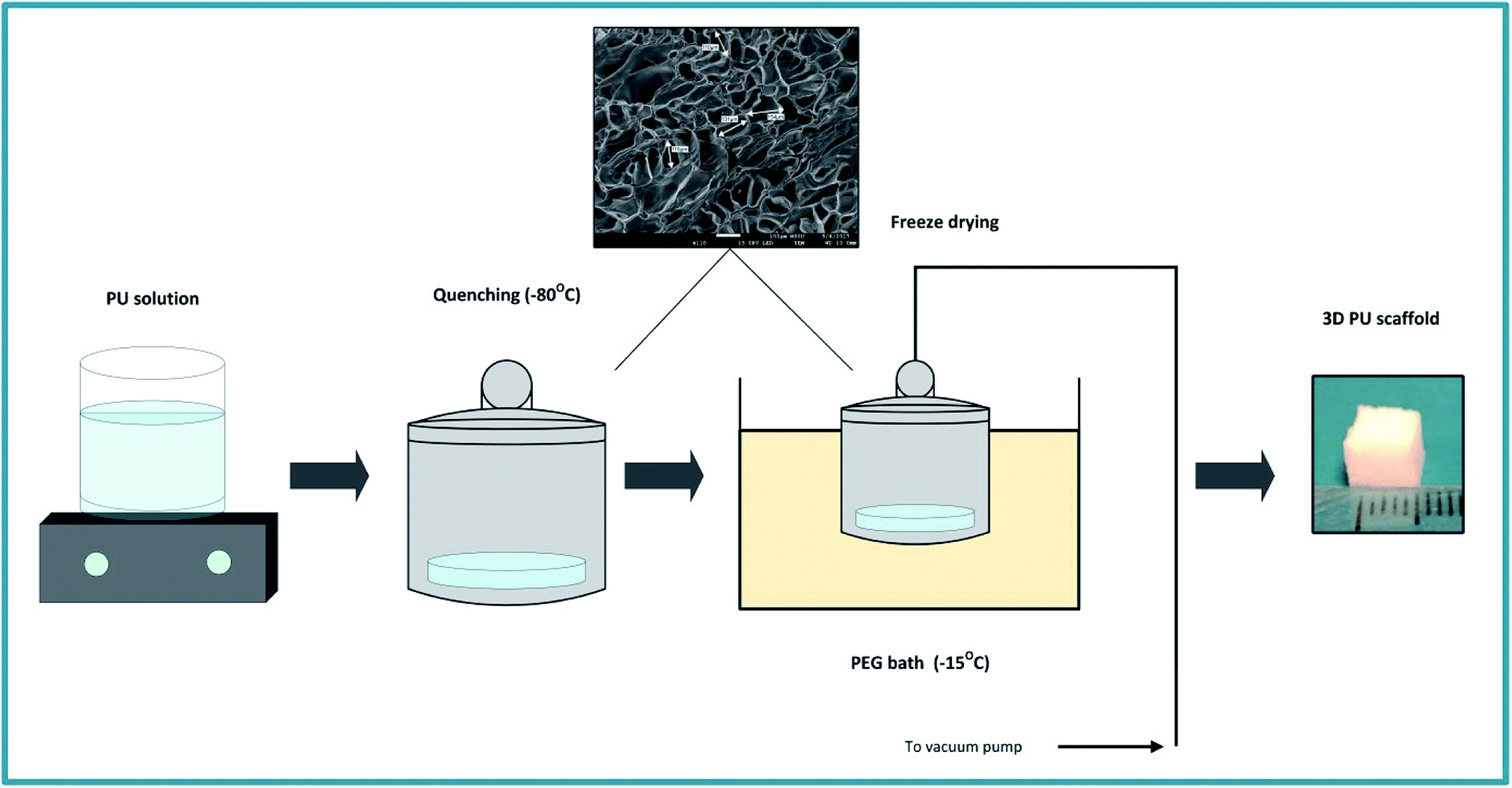 | ||
| Fig. 1 Schematic representation of the PU scaffold fabrication process with the Thermally Induced Phase Separation (TIPS). | ||
2.2. Surface modification of the 3D scaffolds
The PU scaffolds were coated with fibronectin (FN) from bovine plasma (Sigma-Aldrich, UK), to mimic a crucial ECM protein dominant in pancreatic cancer as well as to enhance the cell adhesion on the PU matrix. The protein coating took place via adsorption of the FN on the PU.68,69 More specifically, as previously described,21,55 the scaffolds were dipped in Phosphate-Buffered Saline (PBS, Sigma-Aldrich, UK) for 10 min and centrifuged (in PBS) for 10 min at 2500 rpm. Then, they were transferred to the FN solution and centrifuged for 20 min at 2000 rpm. Centrifuging ensures better penetration and uniform distribution of the protein solution in the PU matrix. Then, one more centrifugation step in PBS for 10 min at 1500 rpm is carried out to unblock the surface pores of the scaffolds. The FN coating concentration was 25 μg mL−1.21,55,70,712.3. 3D cell culture
The human pancreatic adenocarcinoma cell lines AsPC-1 (ATCC® UK, CRL-1682) and PANC-1 (Sigma-Aldrich UK, ECAAC 87092802) were expanded in tissue culture plastic flasks (Fisher Scientific, UK) in Dulbecco's modified Eagle's medium (DMEM) with high glucose (Lonza, UK), while the BxPC-3 (ATCC® UK, CRL-1687) cell line was cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Lonza, UK) with high glucose, according to the supplier's culture protocol, in a humidified incubator at 37 °C and 5% CO2 until an adequate number of cells for each experiment was achieved. Both culture media were supplemented with 10% v/v heat inactivated foetal bovine serum (Fisher Scientific, UK), 100 U mL−1 penicillin/streptomycin (Sigma-Aldrich, UK) and 2 mM L-glutamine (Sigma-Aldrich, UK) stock solutions. Mycoplasma negative cultures were ensured by regular mycoplasma screening tests (Lonza, UK).For all conditions under study, 100 μL of cell suspension (5 × 105 cells per 100 μL) was seeded in sterile scaffolds, which were placed in 24 well-plates and allowed to settle for 15 min in an incubator at 37 °C in a 5% CO2 atmosphere. Then, 1.5 mL of the appropriate cell culture medium was added in each well. Scaffolds were placed in an incubator at 37 °C, 5% CO2 and 20% O2. Cell growth was monitored for 29 days (5 weeks). Cell culture medium was replenished every two to three days. In order to avoid cell confluency at the bottom of the wells resulting from scaffold egress, scaffolds were placed in a new well-plate on a weekly basis.
2.4. MTS cell viability assay
Quantitative assessment of cell viability/proliferation in the scaffolds took place by measuring the increase of metabolically active cells using the tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] MTS reagent (Promega, CellTiter96® Aqueous Solution Cell Proliferation Assay, WI USA). The reagent was supplemented to fresh culture medium at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 according to the manufacturer's protocol and added to the 3D scaffold cultures prior to a 3 h incubation at 37 °C, according to the manufacturer's protocol. Afterwards, the absorbance was measured at 490 nm on a plate reader (Synergy HT, BioTek, VT, USA).
:5 according to the manufacturer's protocol and added to the 3D scaffold cultures prior to a 3 h incubation at 37 °C, according to the manufacturer's protocol. Afterwards, the absorbance was measured at 490 nm on a plate reader (Synergy HT, BioTek, VT, USA).
2.5. Scanning electron microscopy (SEM)
The 3D scaffolds were sectioned and the cell distribution and adhesion on/in the pores of the matrix was observed with scanning electron microscopy (SEM). More specifically, scaffolds were collected at the beginning (day 1) and at the culture endpoint (day 29), snap frozen in liquid nitrogen for 15 min and then preserved in −80 °C until fixation. At fixation, samples where sectioned approximately in the middle with a razor and then directly immersed in 4% v/v cold formaldehyde solution (Sigma-Aldrich, UK) for 2 h at room temperature. Thereafter, post fixation took place including 2 washing steps with PBS for 15 min each, followed by 4 washing steps with deionized water for 15 min each to ensure removal of the residual crystals. Afterwards, the scaffold sections were air dried overnight in an aseptic environment. The specimens were sputter coated with gold in an argon atmosphere 24 h prior the SEM imaging. The scanning electron microscopy was performed on a JOEL JMS610LA (JEOL USA, MA, USA) microscope at different magnifications.2.6. Immunofluorescence assays
In situ immunofluorescence (IF) staining of the scaffolds took place for the spatial determination of the (i) cell organisation and/or cell masses formations (4′,6-diamidino-2-phenylindole, DAPI), (ii) cell proliferation (Ki-67), (iii) ECM production (collagen-I), (iv) potential environmental stress gradients, i.e., oxygen/oxidative stress (HIF-a) and nutrient/starvation stress (LC3). More specifically, scaffolds were collected at the middle (day 15) and the end of the culturing period (day 29), snap frozen in liquid nitrogen for 15 min and then preserved at −80 °C until sectioning as previously described.52 Prior to the IF assay preparation, multiple scaffold sections of ∼1 mm were generated. Briefly, the preparation included overnight fixation in 4% w/v paraformaldehyde, then 2 h permeabilisation in 0.1% Triton X-100 (Sigma Aldrich, UK) at room temperature followed by 4 h blocking in 10% of donkey serum (Abcam, UK), overnight primary antibody staining, 6 h secondary antibody staining and overnight counterstaining all at 4 °C. Cells and scaffold structure were evaluated with a Leica SP5 inverted confocal microscope and processed with Leica LAS AF software (Leica, Milton Keynes, UK). Each step employed solvents of either 1% w/v bovine serum albumin (Sigma-Aldrich, UK), 0.5% v/v Tween-20 (Promega, UK) and 0.01% w/v NaN3 in PBS, or only 0.01% w/v NaN3 in PBS after secondary antibody staining. Each step was separated by at least two washes with above solvent buffer. Primary antibodies and isotype controls are summarised in Table 1. Secondary antibodies consisted of: donkey anti rat Alexa Fluor 488 (AF 488), donkey anti mouse Alexa Fluor 555 (AF 555) and donkey anti rabbit 647 (AF 647; all Fisher Scientific, UK) at 1:500 dilution. Counterstain consisted of: 5 μg mL−1 DAPI (Fisher Scientific, UK).
| Primary | Isotype | |
|---|---|---|
| a All products were obtained from Abcam, UK.b IgG: immunoglobulin G. | ||
| Fig. 4a | 11.17 μg mL−1 rat anti human Ki-67 | 11.17 μg mL−1 rat IgG1b |
| 16.67 μg mL−1 mouse anti human HIF-1α | 16.67 μg mL−1 mouse IgG2b | |
| 8.34 μg mL−1 rabbit anti human collagen-I | 8.34 μg mL−1 rabbit IgG | |
| Fig. 4b and c | 8.00 μg mL−1 rabbit anti human LC3 A/B | 8.00 μg mL−1 rabbit IgG |
| 16.67 μg mL−1 mouse anti human HIF-1α | 16.67 μg mL−1 mouse IgG2b | |
| Fig. 6 | 16.67 μg mL−1 mouse anti human HIF-1α | 16.67 μg mL−1 mouse IgG2b |
| 8.34 μg mL−1 rabbit anti human collagen-I | 8.34 μg mL−1 rabbit IgG | |
| Fig. 7 | 11.17 μg mL−1 rat anti human Ki-67 | 11.17 μg mL−1 rat IgG1b |
| 16.67 μg mL−1 mouse anti human HIF-1α | 16.67 μg mL−1 mouse IgG2b | |
| 8.34 μg mL−1 rabbit anti human collagen-I | 8.34 μg mL−1 rabbit IgG | |
2.7. Cell viability confocal imaging
As described in section 2.6, scaffolds were collected at day 29, snap frozen in liquid nitrogen for 15 min and then preserved at −80 °C. After sectioning, each scaffold section was incubated with Calcein-AM (2 μM; Life Technologies, Paisley, UK) and ethidium homodimer-1 (4 μM; Life Technologies) in culture medium for 1 h at 37 °C. The presence of live (green) and dead (red) cells was immediately evaluated with Leica SP5 inverted confocal microscope and processed with Leica LAS AF software (Leica, Milton Keynes, UK).2.8. Confocal laser scanning microscopy (CLSM) imaging
Immunofluorescent samples were imaged on a Leica SP5 inverted confocal microscope and processed with Leica LAS AF software (Leica, Milton Keynes, UK) using 405, 453, 488, 543, and 633 nm lasers and filters for DAPI, reflectance, AF488, AF555, and AF647 stains for 2 sequential scans. Confocal images were captured using a 10× dry objective, with a 512 × 512 pixel resolution and 5 μm Z-stack distance, as previously described.52 Those image acquisition settings were identical for the positive and the corresponding isotype control. Multiple replicates from multiple scaffold areas where imaged and representative images are presented.2.9. Statistical analysis
Statistical analyses were performed from N = 3 independent cultures where n = 2–3 replicate scaffold measurements per culture were averaged. Error bars represent standard deviation. Analysis of variance (One-way or Two-way ANOVA) and unpaired, 2-tailed Student's t-test was performed using GraphPad Prism® software with a p-value threshold 0.05 to evaluate whether there was a statistical difference between the experimental conditions under study.3. Results
3.1. Long term cultivation of pancreatic cell lines in the PU scaffolding system
AsPC-1, PANC-1 and BxPC-3 pancreatic cancer cell lines were seeded (5 × 105 cells) in uncoated PU scaffolds and cell viability/proliferation was monitored for 29 days. As shown in Fig. 2, high viability was maintained for all the cell lines in the 3D scaffolds. Furthermore, a significant increase in the proliferation took place, for the AsPC-1 and PANC-1 cell lines (Fig. 2a and b). BxPC-3, did not present any significant increase in absorbance indicating a low level of growth rate (Fig. 2c), which is consistent with previously reported 2D studies.72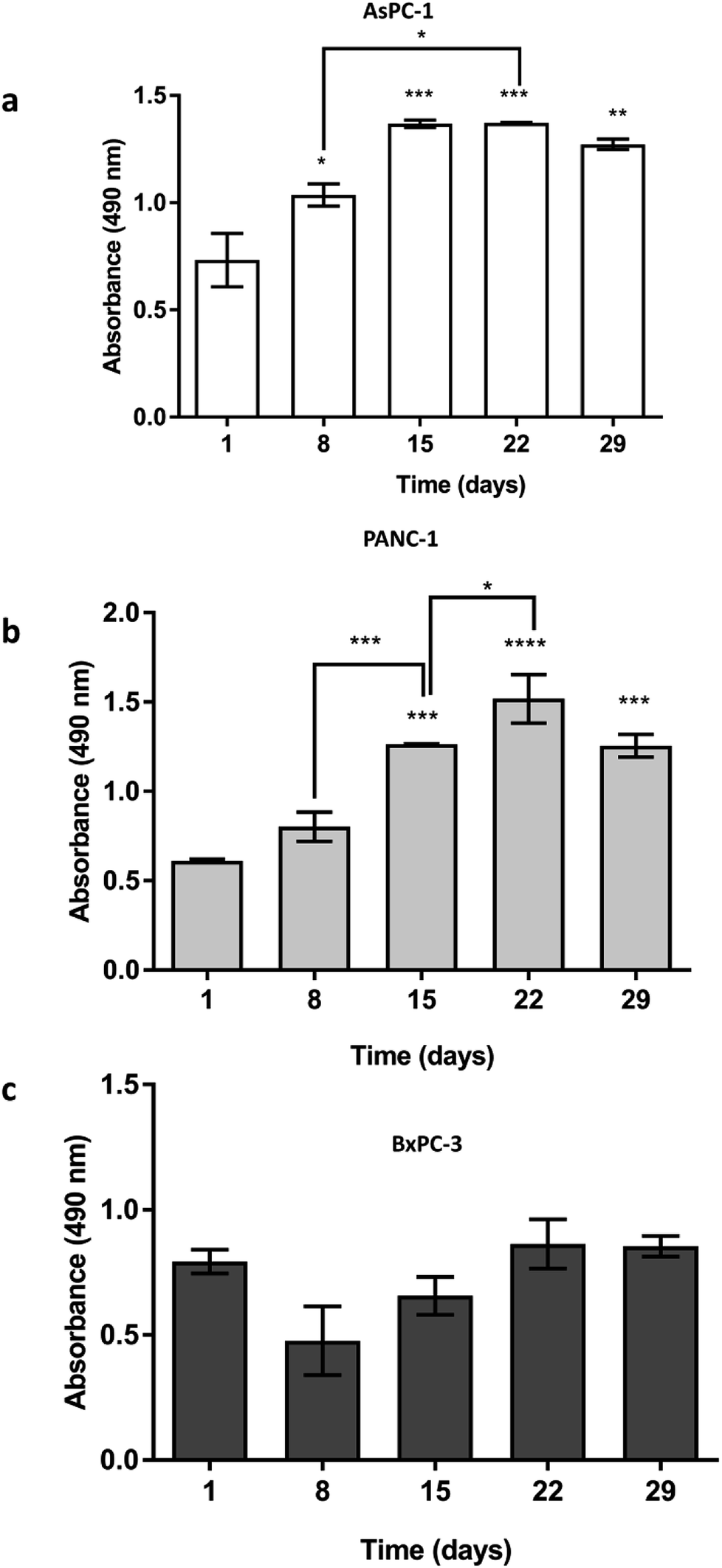 | ||
| Fig. 2 Growth of (a) AsPC-1, (b) PANC-1 and (c) BxPC-3 pancreatic cancer cell lines in uncoated PU scaffolds for 29 days. Data are presented as mean ± S.D (N = 3, n = 3). Statistical differences for the cell growth are marked by asterisks (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). N = number of independent experiments; n = number replicates. | ||
3.2. In situ cellular characterisation in the PU scaffolding system
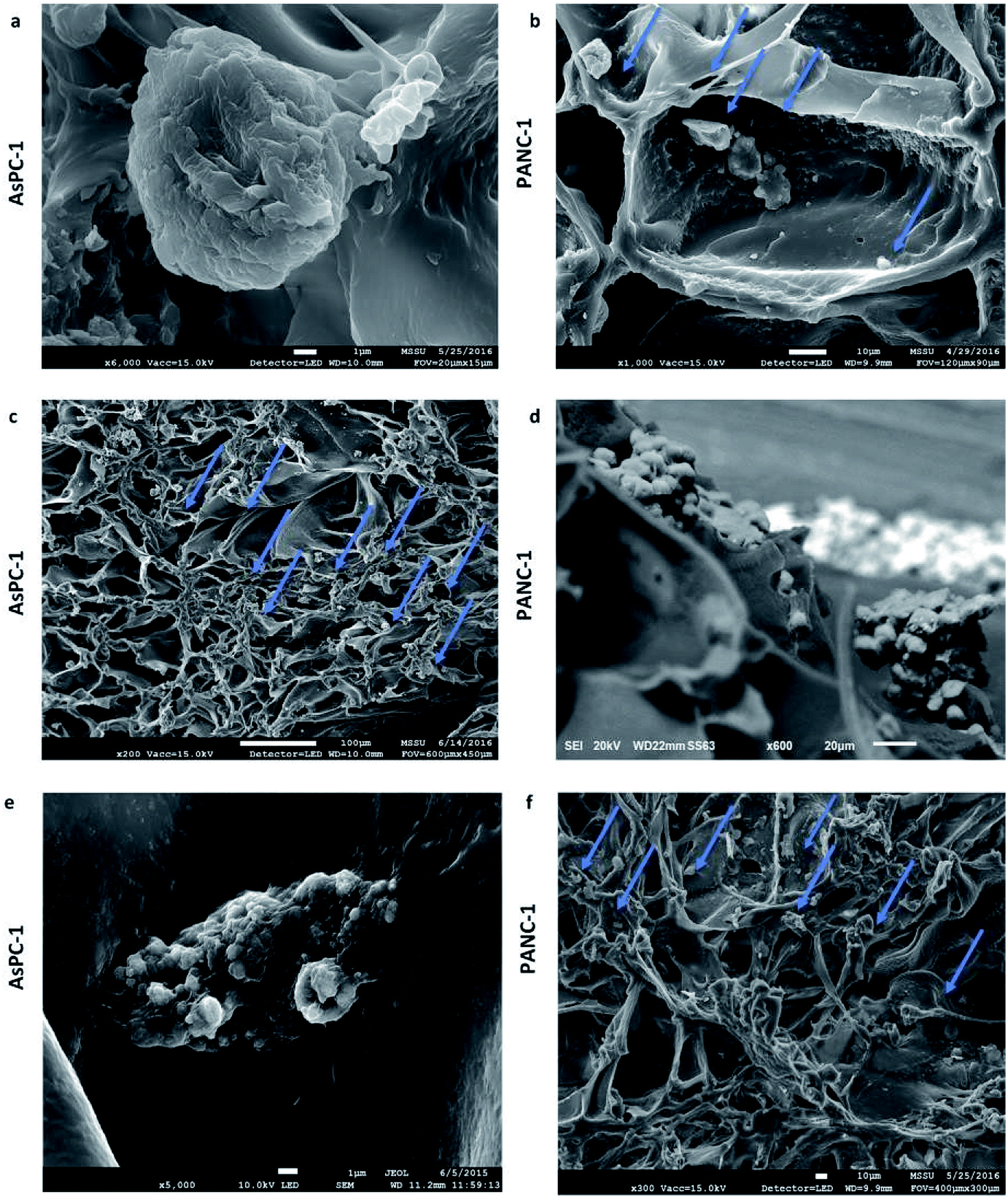 | ||
| Fig. 3 Scanning electron microscopy images of AsPC-1 and PANC-1 cells in sections of the uncoated (UN) PU scaffolds at day 1 (a and b) and day 29 (c–f) of culture. | ||
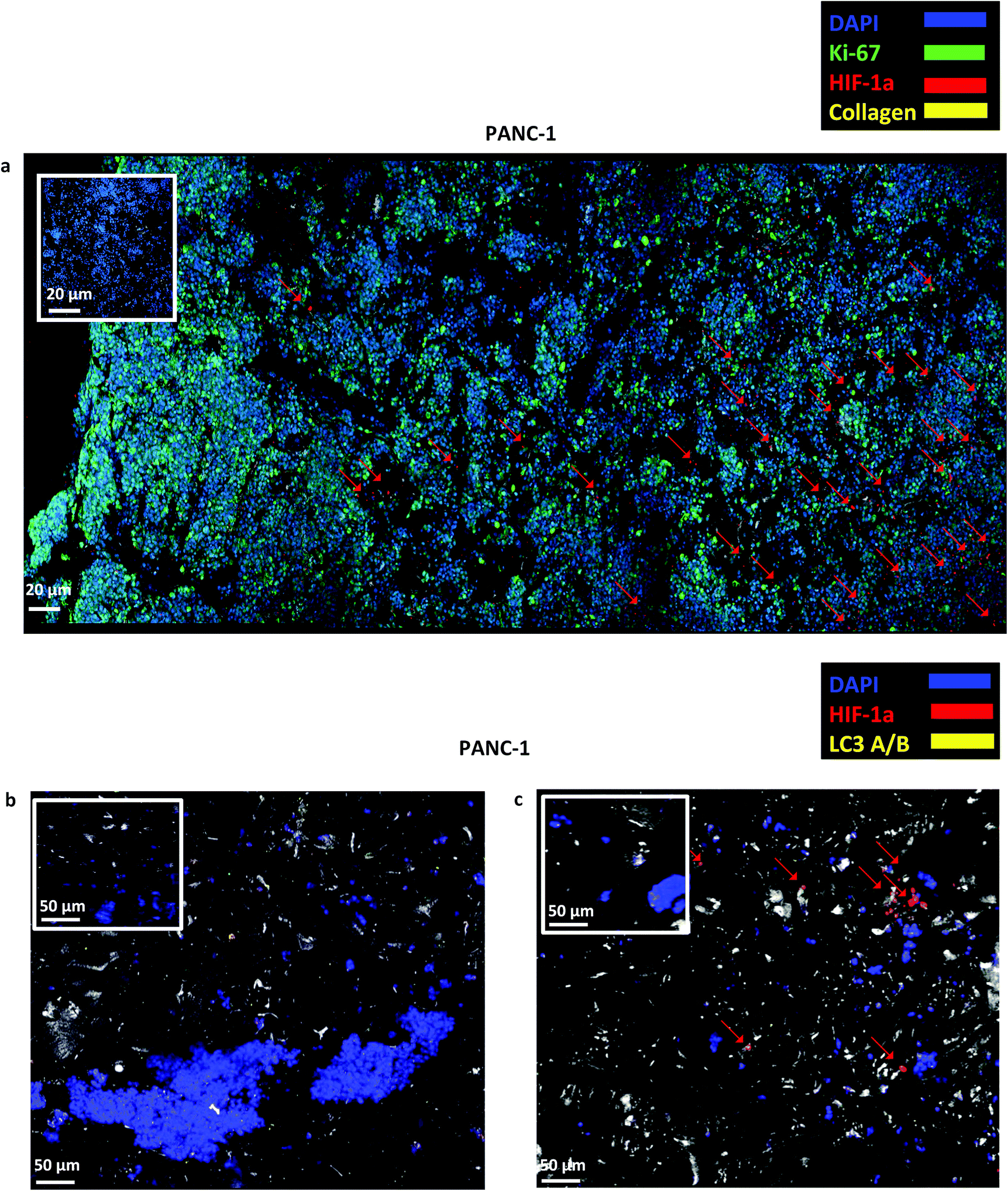 | ||
| Fig. 4 (a) Representative immunofluorescence CLSM images of Ki-67, HIF-1α and collagen-I distribution in sections of uncoated PANC-1 PU scaffolds at day 29 of culture. (b and c) Representative immunofluorescence CLSM of HIF-1α and LC3-A/B distribution in uncoated PANC-1 PU scaffolds at day 1 (b) and day 29 (c) of culture. For each image the corresponding isotype control was generated. | ||
3.3. Engineering the PU scaffolding system to recapitulate ECM features
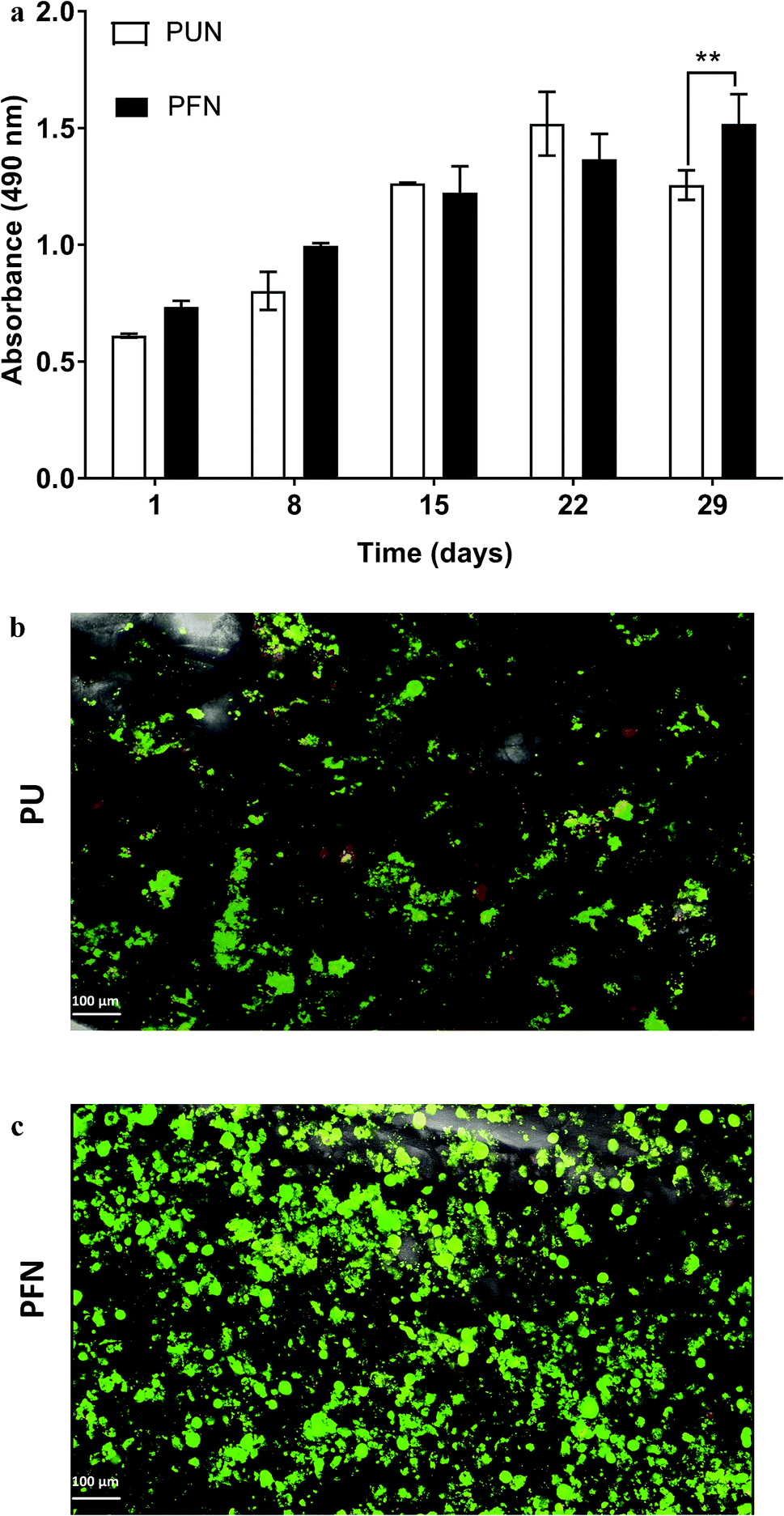 | ||
| Fig. 5 (a) Growth of PANC-1 cells in uncoated (PUN) and FN coated (PFN) PU scaffolds for 29 days. Data are presented as mean ± S.D. (N = 2, n = 3). Statistical differences are marked by asterisks (*p < 0.05; **p < 0.01). N = number of independent experiments; n = number replicates. (b and c) Visualization of PANC-1 cells in uncoated (PUN) and FN coated (PFN) scaffolds respectively with fluorescence Live (green, Calcein AM) and Dead (red, Ethidium homodimer-1) viability assay at day 29 of culture. | ||
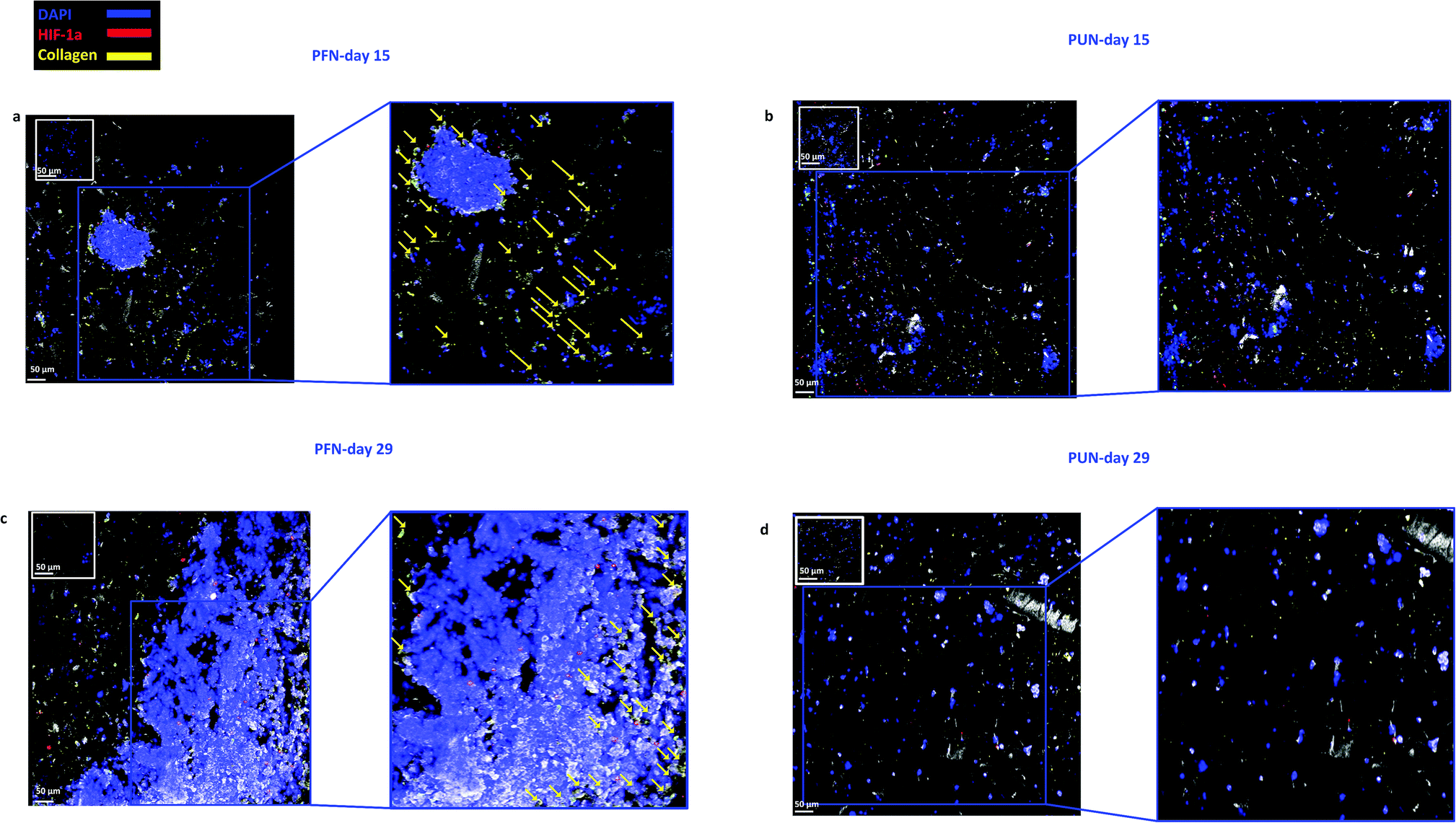 | ||
| Fig. 6 Representative immunofluorescence CLSM images of HIF-1α and collagen-I distribution in FN coated (a and c) and uncoated (b and d) PANC-1 scaffolds at day 15 and day 29 of culture. For each image the corresponding isotype control was generated. | ||
Finally, staining and imaging a wide scaffold area at the endpoint of the culture provides a map of the cellular distribution, the ECM production and the oxidative stress (HIF-1α) biomarker expression within the scaffold. Spatial variations in collagen-I, HIF-1α and Ki-67 expression as well as cell self-organisation are presented in Fig. 7. Different densities of cell masses, most of them maintaining their proliferative properties (Ki-67 positive), were formed within the PU matrix. The cell masses were surrounded by collagen-I molecules and expressed locally oxidative stress gradients (HIF-1α) (Fig. 7).
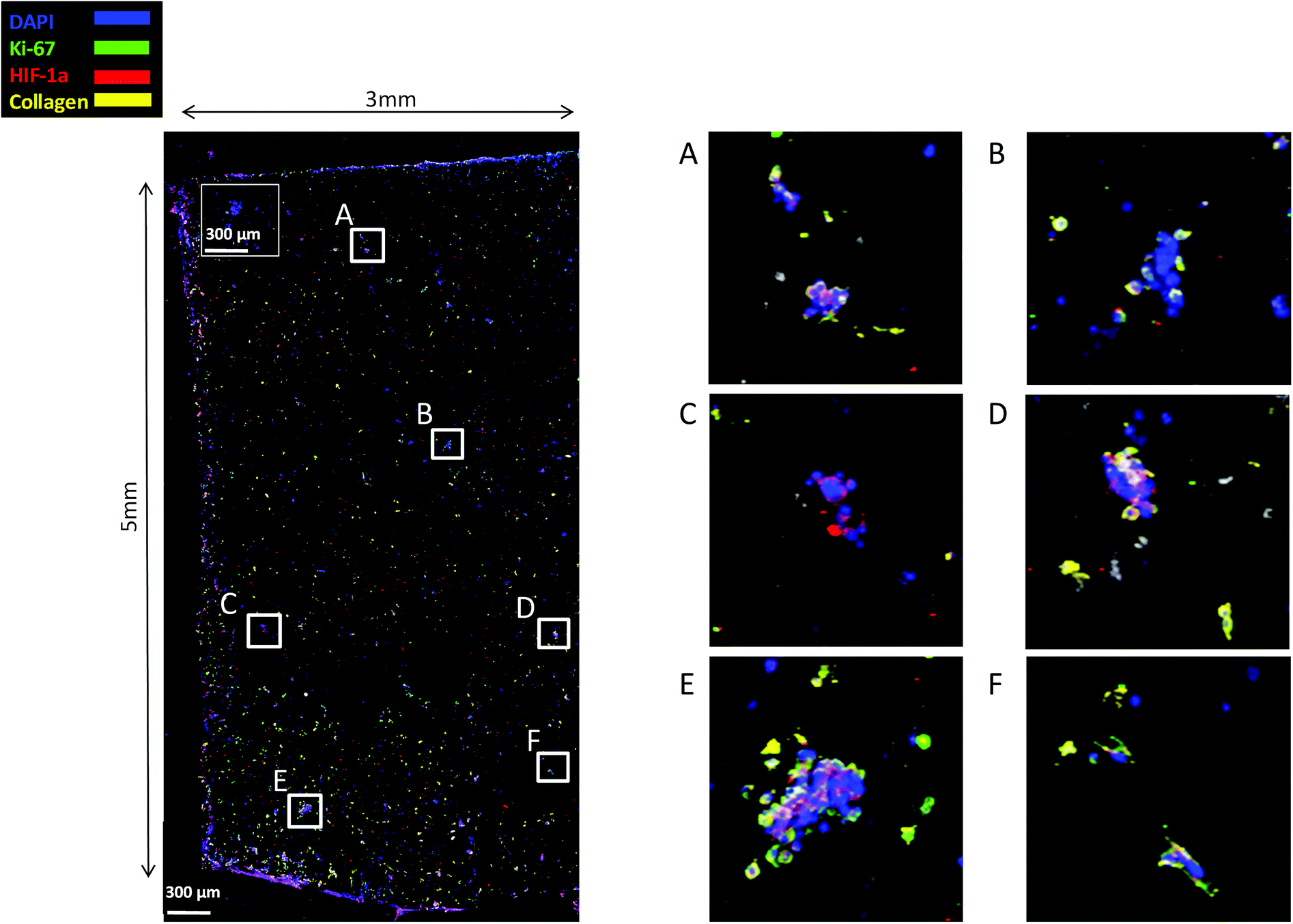 | ||
| Fig. 7 CLSM image of Ki-67, HIF-1α and collagen-I distribution in a wide PANC-1 FN coated scaffold area at day 29 of culture. For each image the corresponding isotype control was generated. | ||
4. Discussion
Overall, in this work, a 3D highly porous PU scaffold coated with fibronectin (FN) was developed and assessed as an in vitro platform for pancreatic cancer studies. The developed scaffolding system was able to support the long term cellular growth and proliferation (Fig. 2, 4a and 5–7), cell self-organisation (Fig. 3 and 5–7) and ECM protein, i.e., collagen I, production of pancreatic cancer cells (Fig. 5 and 6). Furthermore, in situ mapping of various environmental stress biomarkers, i.e., oxygen and nutrient stress, revealed the formation of some oxygen/oxidative stress gradients, i.e., formation of some hypoxic areas, but no nutrient/starvation stress gradients within the scaffold (Fig. 4, 6 and 7). This is crucial, as the accurate topological identification of stress biomarkers' signals can lead to a better understanding of the 3D tumour response and/or resistance to treatment, e.g., areas of higher or lower resistance.77,78 Furthermore, the fact that the environmental gradients are not extreme is of importance as most currently available 3D systems for pancreatic cancer induce artificially high oxygen gradients that lead to a large non-proliferative centre, which is not always present in vivo.32 To our knowledge, this is the first study that reports the long term growth and evolution of pancreatic cancer cells in a highly porous ECM coated PU scaffolding system. Some previous studies have explored pancreatic cancer cell evolution in 3D polymeric systems, however it is the first time that the PU is used as scaffolding material to generate a highly porous sponge-like matrix which, to the authors' knowledge, resulted to the longest reported cell proliferation period of pancreatic tumour cells, along the study of Chiellini et al. Most pancreatic cancer studies in 3D systems to date report an in vitro culture period of 7–14 days.46,49–51 Long term cultivation is important as it could enable the conduction of fractionated treatment followed by long term post-treatment observations.Evolution of pancreatic cancer cells in the highly porous PU scaffold
The developed highly porous PU scaffolding system supported the long term cultivation and proliferation of 3 different PDAC cell lines, i.e., AsPC-1, PANC-1, BxPC-3, for 29 days (Fig. 2). It is noteworthy that the pancreatic cancer cell lines maintained their intrinsic kinetics and typical morphological characteristics within the polymeric matrices (Fig. 2 and 3). AsPC-1 and PANC-1 cells had similar growth rates in the scaffolding system, while BxPC-3 cell line had a slower growth evolution, which is in alignment with the reported duplication times of these cell lines.72 In terms of morphological characteristics within the 3D scaffold, as verified by SEM imaging, the AsPC-1 cells retained their foci-like epithelial shape (Fig. 3a),79,80 while PANC-1 cells had an elongated epithelial morphology (Fig. 3b).81,82 This scaffolding system allowed large population of cells to grow, the majority of which were positive proliferative (Ki-67 positive) on day 29 (Fig. 4a). Similar images of cellular masses during PDAC evolution in a rat model were reported by Ignat et al.83 Likewise, another study for PDAC detection demonstrated dense cell masses in the tumour area of human pancreatic cancer xenografts.84 This suggests that this scaffolding system provides realistic in vivo recapitulation of the pancreatic tumour self-organisation facilitating high cellular densities and cell–cell interactions within the 3D structure.The lack of nutrient gradients despite the high cell densities on day 29 of PANC-1 scaffold culture was verified since the LC3 autophagic marker (Fig. 4b and c), a marker which is mainly upregulated under starvation conditions, was not expressed.85 Nevertheless, local hypoxic areas were present mainly where dense overpopulated masses exist, mimicking the low oxygen levels that naturally occur within the pancreatic tumour (Fig. 4a). Similar results of HIF-1α expression in patient derived samples were demonstrated by Salnikov et al.86 Other studies have also reported the mimicry of hypoxic areas in vitro, leading however to extreme oxygen gradients which lead to high apoptotic regions which are not always reported in vivo.87–89
Evolution of pancreatic cancer cells in FN coated highly porous PU scaffolds
FN is a major ECM constituent of PDAC that mediates cell adhesion. We therefore, modified further the PU scaffolds with FN, to promote the cell adhesion, but also to provide partially the ECM mimicry to the cells. Cellular viability/proliferation in FN coated scaffolds was significantly higher as compared to the uncoated scaffolds at day 29 of culture (Fig. 5a). However, the difference in cell growth between the uncoated and FN coated scaffolds was noticeably smaller than other reported biological systems in PU scaffolds,55 showing that pancreatic cancer cell lines grow significantly well in the PU scaffolds even without ECM coating support. Similarly to our findings, Raza et al. reported the effect of FN derived peptides on PANC-1 growth in 3D PEG-based hydrogels and showed that PANC-1 cells exhibited higher proliferation at the end of the culture in the FN enriched hydrogels compared to the FN-free gels.48 Additionally, Miyamoto et al. reported that pancreatic cancer cell lines growth was enhanced on FN coated 2D monolayers compared to the controls (uncoated surfaces) at the end of the culture.90 From the above studies in both 2D and 3D systems it is evident that FN stimuli promotes cell growth in vitro. Further to the pancreatic cancer cell growth/proliferation we observed that the FN coating affected the cellular spatial self-organisation in the PU matrix. More specifically, the cells formed larger masses in the FN coated scaffolds as compared to uncoated ones where they formed smaller masses (Fig. 6). We have previously reported this formation of PANC-1 cellular masses in carbon nanotube based films.80 FN is considered to interact with the cell surface integrin receptors, which may result in enhancing the cell–cell interactions and consequently leading to cell clustering.75,91 This evidence is further supported by the group of Da Rocha-Azevedo et al., who showed that the cell contraction in clustering requires the presence of FN.92 Furthermore, the secretion of collagen-I was monitored, as collagen-I is an important feature of PDAC microenvironment which is highly secreted within the densely-packed pancreatic tumour stroma, as proven by patient and animal studies.76,93,94 We observed a significant collagen-I secretion in the FN coated scaffolds (Fig. 6a, c and 7) and no collagen-I secretion in uncoated scaffolds (Fig. 4a, 6b and d). Expression of high collagen levels from the pancreatic cancer cells in multilayer FN–gelatin nanofilms was also reported by Matsusaki et al.95Finally, sectioning and imaging of a wide scaffold area of the FN coated scaffold (essentially ‘zooming-out’ the FN scaffold) enabled the biomarker mapping of a tissue-scale area, therefore, providing insightful information about on formed cell heterogeneity. It should be stated that, to best of our knowledge, this is the first reported image of this size (∼3 × 5 × 1 mm3), as most CSLM images of both in vivo and in vitro systems are within the range of microns.46,49,83,84,86 As can be seen in Fig. 7, the majority of the cell population that existed in the scaffold is proliferative (Ki-67) positive on day 29 of culture, revealing the long term maintenance of the cell culture within the 3D matrix. At this point it should be mentioned that there are very few 3D systems studying the pancreatic cancer cell Ki-67 expression and none after 15 days of culture, as the centre of such 3D models usually contains arrested proliferation and accumulation of apoptotic due to oxidative stress regions.41,43,89 The pore size and interconnection of this scaffolding system allows the cells to proliferate throughout the matrix area, with local hypoxic areas (HIF-1α positive) being heterogeneously distributed throughout the scaffold area (Fig. 7). This is of great importance, since hypoxia is a hallmark of all the solid tumours and due to the structural and functional abnormalities of tumour microcirculation spatial and temporal heterogeneity in the perfusion is caused.96,97 Heterogeneous HIF-1α gradient accumulation in pancreatic tumours has also been reported for patient derived tissues.86,98 Additionally, collagen-I was heterogeneously expressed by the cells within the FN coated scaffolds (Fig. 7).
5. Conclusions
Overall, in this study we developed a 3D highly porous PU scaffolding system coated with FN which shows great potential as a model for pancreatic cancer studies. The reported 3D model was able to (i) support long term growth and proliferation of pancreatic cancer cells for up to a month, (ii) allow formation of dense cellular masses, (iii) enhance the collagen-I production from the pancreatic cells and (iv) induce environmental gradients (hypoxic regions) which were not acute and therefore did not arrest cell proliferation. Furthermore, in situ mapping of a wide scaffold area revealed a high level of heterogeneity with respect to biomarker spatial distribution. Similar trends have been reported in vivo, indicating the great potential of the developed PU scaffolding system for pancreatic cancer studies. Future work will focus on (i) introducing perfusion in the system for vascularisation mimicry as well as (ii) co-culturing of pancreatic cancer cells with stromal cells in order to simulate even more accurately the pancreatic cancer tissue microenvironment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Chemical and Process Engineering Department of the University of Surrey as well as an Impact Acceleration Grant (IAA-KN9149C) of the University of Surrey, an IAA – EPSRC Grant (RN0281J) and the Royal Society. M. C. A. is grateful for the Imperial College Chemical Engineering Scholarship. M. C. A., S. B. D. S., A. M. acknowledge the support from ERC-BioBlood (no. 340719).References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 7–30 CrossRef PubMed.
- American Cancer Society, Cancer Facts & Figures 2018, Atlanta, 2018 Search PubMed.
- S. Cascinu, M. Falconi, V. Valentini and S. Jelic, Ann. Oncol., 2010, 21, 55–58 CrossRef PubMed.
- T. Kamisawa, T. Isawa, M. Koike, K. Tsuruta and A. Okamoto, Pancreas, 1995, 11, 345–349 CrossRef PubMed.
- S. Chand, K. O'Hayer, F. F. Blanco, J. M. Winter and J. R. Brody, Int. J. Biol. Sci., 2016, 12, 273–282 CrossRef PubMed.
- D. Xie and K. Xie, Genes Dis., 2015, 2, 133–143 CrossRef PubMed.
- M. W. Pickup, J. K. Mouw and V. M. Weaver, EMBO Rep., 2014, 15, 1243–1253 CrossRef PubMed.
- S. Totti, S. I. Vernardis, L. Meira, P. A. Perez-Mancera, E. Costello, W. Greenhalf, D. Palmer, J. Neoptolemos, A. Mantalaris and E. G. Velliou, Drug Discovery Today, 2017, 22, 690–701 CrossRef PubMed.
- L. G. Melstrom, M. D. Salazar and D. J. Diamond, J. Surg. Oncol., 2017, 116, 7–15 CrossRef PubMed.
- T. M. Gress, A. Menke, M. Bachem, F. Muller-Pillasch, V. Ellenrieder, H. Weidenbach, M. Wagner and G. Adler, Digestion, 1998, 59, 625–637 Search PubMed.
- S. Pandol, M. Edderkaoui, I. Gukovsky, A. Lugea and A. Gukovskaya, Clin. Gastroenterol. Hepatol., 2009, 7, S44–S47 CrossRef PubMed.
- C. J. Whatcott, C. H. Diep, P. Jiang, A. Watanabe, J. LoBello, C. Sima, G. Hostetter, H. M. Shepard, D. D. Von Hoff and H. Han, Clin. Cancer Res., 2015, 21, 3561–3568 CrossRef PubMed.
- H. E. Barker, J. T. E. Paget, A. A. Khan and K. J. Harrington, Nat. Rev. Cancer, 2015, 15, 409–425 CrossRef PubMed.
- C. J. Whatcott, R. G. Posner, D. D. Von Hoff and H. Han, in Pancreatic Cancer and Tumor Microenvironment, ed. P. J. Grippo and H. G. Munshi, Trivandrum, 2012 Search PubMed.
- N. Dumont, B. Liu, R. A. DeFilippis, H. Chang, J. T. Rabban, A. N. Karnezis, J. A. Tjoe, J. Marx, B. Parvin and T. D. Tlsty, Neoplasia, 2013, 15, 249–262 CrossRef PubMed.
- K. Oonishi, X. Cui, H. Hirakawa, A. Fujimori, T. Kamijo, S. Yamada, O. Yokosuka and T. Kamada, Radiother. Oncol., 2012, 105, 258–265 CrossRef PubMed.
- L. R. Jaidev, U. M. Krishnan and S. Sethuraman, Mater. Sci. Eng., C, 2015, 47, 40–47 CrossRef PubMed.
- C. M. Nelson and M. J. Bissell, Annu. Rev. Cell Dev. Biol., 2006, 22, 287–309 CrossRef PubMed.
- A. Birgersdotter, R. Sandberg and I. Ernberg, Semin. Cancer Biol., 2005, 15, 405–412 CrossRef PubMed.
- E. Cukierman, R. Pankov and K. M. Yamada, Curr. Opin. Cell Biol., 2002, 14, 633–639 CrossRef PubMed.
- E. G. Velliou, S. B. D. Santos, M. M. Papathanasiou, M. Fuentes-Gari, R. Misener, N. Panoskaltsis, E. N. Pistikopoulos and A. Mantalaris, Bioprocess Biosyst. Eng., 2015, 38, 1589–1600 CrossRef PubMed.
- M. Mattie, A. Christensen, M. S. Chang, W. Yeh, S. Said, Y. Shostak, L. Capo, A. Verlinsky, Z. An, I. Joseph, Y. Zhang, S. Kumar-Ganesan, K. Morrison, D. Stover and P. Challita-Eid, Neoplasia, 2013, 15, 1138–1150 CrossRef PubMed.
- P. A. Perez-Mancera, C. Guerra, M. Barbacid and D. A. Tuveson, Gastroenterology, 2012, 142, 1079–1092 CrossRef PubMed.
- K. P. Olive, M. A. Jacobetz, C. J. Davidson, A. Gopinathan, D. McIntyre, D. Honess, B. Madhu, M. A. Goldgraben, M. E. Caldwell and D. Allard, Science, 2009, 324, 1457–1461 CrossRef PubMed.
- A. Richmond and Y. Su, Dis. Models & Mech., 2008, 1, 78–82 Search PubMed.
- S. Loisel, M. Ohresser, M. Pallardy, D. Daydé, C. Berthou, G. Cartron and H. Watier, Crit. Rev. Oncol. Hematol., 2007, 62, 34–42 CrossRef PubMed.
- S. Y. Cho, W. Kang, J. Y. Han, S. Min, J. Kang, A. Lee, J. Y. Kwon, C. Lee and H. Park, Mol. Cells, 2016, 39, 77–86 CrossRef PubMed.
- E. K. Colvin and C. J. Scarlett, Semin. Cell Dev. Biol., 2014, 27, 96–105 CrossRef PubMed.
- M. Hogenes, M. Huibers, C. Kroone and R. de Weger, Transplant. Rev., 2014, 28, 103–110 CrossRef PubMed.
- F. J. O'Brien, Mater. Today, 2011, 14, 88–95 CrossRef.
- B. Dhandayuthapani, Y. Yoshida, ToruMaekawa and D. S. Kumar, Int. J. Polym. Sci., 2011, 2011 DOI:10.1155/2011/290602.
- E. Burdett, K. Kasper, A. G. Mikos and J. A. Ludwig, Tissue Eng., Part B, 2010, 16, 351–359 CrossRef PubMed.
- T. G. Fernandes, M. M. Diogo, D. S. Clark, J. S. Dordick and J. M. S. Cabral, Trends Biotechnol., 2009, 27, 342–349 CrossRef PubMed.
- D. Gaspar and D. I. Zeugolis, Drug Discovery Today, 2016, 21, 1341–1344 CrossRef PubMed.
- L. Meli, H. S. C. Barbosa, A. M. Hickey, L. Gasimli, G. Nierode, M. M. Diogo, R. J. Linhardt, J. M. S. Cabral and J. S. Dordick, Stem Cell Res., 2014, 13, 36–47 CrossRef PubMed.
- D.-D. Wang, W. Liu, J.-J. Chang, X. Cheng, X.-Z. Zhang, H. Xu, D. Feng, L.-J. Yu and X.-L. Wang, RSC Adv., 2016, 6, 24083–24090 RSC.
- P. Longati, X. Jia, J. Eimer, A. Wagman, M.-R. Witt, S. Rehnmark, C. Verbeke, R. Toftgård, M. Löhr and R. L. Heuchel, BMC Cancer, 2013, 13 DOI:10.1186/1471-2407-13-95.
- Y. Matsuda, T. Ishiwata, Y. Kawamoto, K. Kawahara, W.-X. Peng, T. Yamamoto and Z. Naito, Med. Mol. Morphol., 2010, 43, 211–217 CrossRef PubMed.
- S. F. Boj, C. I. Hwang, L. A. Baker, D. D. Engle, D. A. Tuveson and H. Clevers, Mol. Cell. Oncol., 2016, 3, e1014757 CrossRef PubMed.
- S. F. Boj, C. I. Hwang, L. A. Baker, I. I. C. Chio, D. D. Engle, V. Corbo, M. Jager, M. Ponz-Sarvise, H. Tiriac, M. S. Spector, A. Gracanin, T. Oni, K. H. Yu, R. van Boxtel, M. Huch, K. D. Rivera, J. P. Wilson, M. E. Feigin, D. Ohlund, A. Handly-Santana, C. M. Ardito-Abraham, M. Ludwig, E. Elyada, B. Alagesan, G. Biffi, G. N. Yordanov, B. Delcuze, B. Creighton, K. Wright, Y. Park, F. H. Morsink, I. Q. Molenaar, I. H. Borel Rinkes, E. Cuppen, Y. Hao, Y. Jin, I. J. Nijman, C. Iacobuzio-Donahue, S. D. Leach, D. J. Pappin, M. Hammell, D. S. Klimstra, O. Basturk, R. H. Hruban, G. J. Offerhaus, R. G. Vries, H. Clevers and D. A. Tuveson, Cell, 2015, 160, 324–338 CrossRef PubMed.
- M. J. Ware, V. Keshishian, J. J. Law, J. C. Ho, C. A. Favela, P. Rees, B. Smith, S. Mohammad, R. F. Hwang, K. Rajapakshe, C. Coarfa, S. Huang, D. P. Edwards, S. J. Corr, B. Godin and S. A. Curley, Biomaterials, 2016, 108, 129–142 CrossRef PubMed.
- V. Brancato, V. Comunanza, G. Imparato, D. Corà, F. Urciuolo, A. Noghero, F. Bussolino and P. A. Netti, Acta Biomater., 2017, 49, 152–166 CrossRef PubMed.
- M. J. Ware, K. Colbert, V. Keshishian, J. Ho, S. J. Corr, S. A. Curley and B. Godin, Tissue Eng., Part C, 2016, 22, 312–321 CrossRef PubMed.
- Z. Wen, Q. Liao, Y. Hu, L. You, L. Zhou and Y. Zhao, Braz. J. Med. Biol. Res., 2013, 46, 634–642 CrossRef PubMed.
- C. I. Hwang, S. F. Boj, H. Clevers and D. A. Tuveson, J. Pathol., 2016, 238, 197–204 CrossRef PubMed.
- F. Chiellini, D. Puppi, A. M. Piras, A. Morelli, C. Bartoli and C. Migone, RSC Adv., 2016, 6, 54226–54235 RSC.
- C. S. Ki, T.-Y. Lin, M. Korc and C.-C. Lin, Biomaterials, 2014, 35, 9668–9677 CrossRef PubMed.
- A. Raza, C. S. Ki and C.-C. Lin, Biomaterials, 2013, 24, 5117–5127 CrossRef PubMed.
- C. Ricci, C. Mota, S. Moscato, D. D'Alessandro, S. Ugel, S. Sartoris, V. Bronte, U. Boggi, D. Campani, N. Funel, L. Moroni and S. Danti, Biomatter, 2014, 4, e955386 CrossRef PubMed.
- X. Wang, X. Zhang, Z. Fu and H. Yina, J. Biotechnol., 2013, 166, 166–179 CrossRef PubMed.
- Q. He, X. Wang, X. Zhang, H. Han, B. Han, J. Xu, K. Tang, Z. Fu and H. Yin, Int. J. Nanomed., 2013, 8, 1167–1176 Search PubMed.
- M. C. Allenby, R. Misener, N. Panoskaltsis and A. Mantalaris, Tissue Eng., Part C, 2017, 23, 108–117 CrossRef PubMed.
- E. Velliou, M. Fuentes-Garí, R. Misener, E. Pefani, M. Rende, N. Panoskaltsis, A. Mantalaris and E. N. Pistikopoulos, Comput.-Aided Chem. Eng., 2014, 34, 225–236 Search PubMed.
- M. Fuentes-Garía, E. Velliou, R. Misener, E. Pefani, M. Rende, N. Panoskaltsis, A. Mantalaris and E. N. Pistikopoulos, Comput. Chem. Eng., 2015, 81, 80–93 CrossRef.
- T. M. Blanco, A. Mantalaris, A. Bismarck and N. Panoskaltsis, Biomaterials, 2010, 31, 2243–2251 CrossRef PubMed.
- C. Ricci, L. Moroni and S. Danti, OA Tissue Engineering, 2013, 1, 4–10 CrossRef.
- P. Lu, V. M. Weaver and Z. Werb, J. Cell Biol., 2012, 196, 395–406 CrossRef PubMed.
- D. I. Zeugolis, G. R. Paul and G. Attenburrow, J. Biomed. Mater. Res., Part A, 2009, 89, 895–908 CrossRef PubMed.
- R. Nigam and B. Mahanta, J. Tissue Sci. Eng., 2014, 5, 137–141 Search PubMed.
- G. BaoLin and P. X. Ma, Sci. China: Chem., 2014, 57, 490–500 CrossRef.
- H. Janik and M. Marzec, Mater. Sci. Eng., C, 2015, 48, 586–591 CrossRef PubMed.
- J. Kucinska-Lipka, I. Gubanska and H. Janik, Sci. World J., 2013, 2013, 450132 Search PubMed.
- J. Guan, K. L. Fujimoto, M. S. Sacks and W. R. Wagner, Biomaterials, 2005, 26, 3961–3971 CrossRef PubMed.
- S. Grad, L. Kupcsik, K. Gorna, S. Gogolewski and M. Alini, Biomaterials, 2003, 24, 5163–5171 CrossRef PubMed.
- I. C. Bonzani, R. Adhikari, S. Houshyar, R. Mayadunne, P. Gunatillake and M. M. Stevens, Biomaterials, 2007, 28, 423–433 CrossRef PubMed.
- L. Safinia, J. J. Blaker, V. Maquet, A. R. Boccaccini, A. Mantalaris and A. Bismarck, e-Polym., 2005, 5 DOI:10.1515/epoly.2005.5.1.92.
- L. Safinia, A. Mantalaris and A. Bismarck, Langmuir, 2006, 22, 3235–3242 CrossRef PubMed.
- M. Panos, T. Z. Sen and M. G. Ahunbay, Langmuir, 2012, 28, 12619–12628 CrossRef PubMed.
- Y. Zhang, C. Chai, X. S. Jiang, S. H. Teoh and K. W. Leong, Mater. Sci. Eng., C, 2007, 27, 213–219 CrossRef.
- D. M. Salvay, C. B. Rives, X. Zhang, F. Chen, D. B. Kaufman, W. L. Lowe, Jr. and L. D. Shea, Transplantation, 2008, 85, 1456–1464 CrossRef PubMed.
- D. Rajendran, A. Hussain, D. Yip, A. Parekh, A. Shrirao and C. H. Cho, J. Biomed. Mater. Res., Part A, 2017, 105, 2119–2128 CrossRef PubMed.
- E. L. Deer, J. Gonzalez-Hernandez, J. D. Coursen, J. E. Shea, J. Ngatia, C. L. Scaife, M. A. Firpo and S. J. Mulvihill, Pancreas, 2010, 39, 425–435 CrossRef PubMed.
- B. Sipos, S. Möser, H. Kalthoff, V. Török, M. Löhr and G. Klöppel, Virchows Arch., 2003, 442, 444–452 Search PubMed.
- C. He and D. J. Klionsky, Annu. Rev. Genet., 2009, 43, 67–93 CrossRef PubMed.
- M. Topalovski and R. A. Brekken, Cancer Lett., 2016, 381, 252–258 CrossRef PubMed.
- T. Imamura, H. Iguchi, T. Manabe, G. Ohshio, T. Yoshimura, Z. H. Wang, H. Suwa, S. Ishigami and M. Imamura, Pancreas, 1995, 11, 357–364 CrossRef PubMed.
- K. C. Wood, Cancer Res., 2015, 75, 4247–4251 CrossRef PubMed.
- J. P. O'Connor, E. O. Aboagye, J. E. Adams, H. J. Aerts, S. F. Barrington, A. J. Beer, R. Boellaard, S. E. Bohndiek, M. Brady, G. Brown, D. L. Buckley, T. L. Chenevert, L. P. Clarke, S. Collette, G. J. Cook, N. M. deSouza, J. C. Dickson, C. Dive, J. L. Evelhoch, C. Faivre-Finn, F. A. Gallagher, F. J. Gilbert, R. J. Gillies, V. Goh, J. R. Griffiths, A. M. Groves, S. Halligan, A. L. Harris, D. J. Hawkes, O. S. Hoekstra, E. P. Huang, B. F. Hutton, E. F. Jackson, G. C. Jayson, A. Jones, D. M. Koh, D. Lacombe, P. Lambin, N. Lassau, M. O. Leach, T. Y. Lee, E. L. Leen, J. S. Lewis, Y. Liu, M. F. Lythgoe, P. Manoharan, R. J. Maxwell, K. A. Miles, B. Morgan, S. Morris, T. Ng, A. R. Padhani, G. J. Parker, M. Partridge, A. P. Pathak, A. C. Peet, S. Punwani, A. R. Reynolds, S. P. Robinson, L. K. Shankar, R. A. Sharma, D. Soloviev, S. Stroobants, D. C. Sullivan, S. A. Taylor, P. S. Tofts, G. M. Tozer, M. van Herk, S. Walker-Samuel, J. Wason, K. J. Williams, P. Workman, T. E. Yankeelov, K. M. Brindle, L. M. McShane, A. Jackson and J. C. Waterton, Nat. Rev. Clin. Oncol., 2017, 14, 169–186 CrossRef PubMed.
- W. H. Chen, J. S. Horoszewicz, S. S. Leong, T. Shimano, R. Penetrante, W. H. Sanders, R. Berjian, H. O. Douglass, E. W. Martin and T. M. Chu, In Vitro, 1982, 18, 24–34 CrossRef PubMed.
- B. Matta-Domjan, A. King, S. Totti, C. Matta, G. Dover, P. Martinez, A. Zakhidov, R. La Ragione, H. Macedo, I. Jurewicz, A. Dalton and E. G. Velliou, J. Biomed. Mater. Res., Part B, 2017 DOI:10.1002/jbm.b.34012.
- M. E. Madden and M. P. Sarras Jr., Pancreas, 1988, 3, 512–528 CrossRef PubMed.
- K. Zeeberg, R. A. Cardone, M. R. Greco, M. Saccomano, A. Nohr-Nielsen, F. Alves, S. F. Pedersen and S. J. Reshkin, Int. J. Oncol., 2016, 49, 243–252 CrossRef PubMed.
- M. Ignat, M. Aprahamian, V. Lindner, A. Altmeyer, S. Perretta, B. Dallemagne, D. Mutter and J. Marescaux, Gastroenterology, 2009, 137, 1584–1592 CrossRef PubMed.
- Z. Cruz-Monserrate, C. L. Roland, D. Deng, T. Arumugam, A. Moshnikova, O. A. Andreev, Y. K. Reshetnyak and C. D. Logsdon, Sci. Rep., 2014, 4, 4410 CrossRef PubMed.
- S. E. Kim, H.-J. Park, H. K. Jeong, M.-J. Kim, M. Kim, O.-N. Bae and S.-H. Baek, Biochem. Biophys. Res. Commun., 2015, 463, 205–210 CrossRef PubMed.
- A. V. Salnikov, L. Liu, M. Platen, J. Gladkich, O. Salnikova, E. Ryschich, J. Mattern, G. Moldenhauer, J. Werner, P. Schemmer, M. W. Büchler and I. Herr, PLoS One, 2012, 7, e46391 Search PubMed.
- S. Nath and G. R. Devi, Pharmacol. Ther., 2016, 163, 94–108 CrossRef PubMed.
- E. Knight and S. Przyborski, J. Anat., 2015, 227, 746–756 CrossRef PubMed.
- I. Dufau, C. Frongia, F. Sicard, L. Dedieu, P. Cordelier, F. Ausseil, B. Ducommun and A. Valette, BMC Cancer, 2012, 12, 15 CrossRef PubMed.
- H. Miyamoto, T. Murakami, K. Tsuchida, H. Sugino, H. Miyake and S. Tashiro, Pancreas, 2004, 28, 38–44 CrossRef PubMed.
- A. G. Lowrie, D. M. Salter and J. A. Ross, Br. J. Cancer, 2004, 91, 1327–1334 CrossRef PubMed.
- B. Da Rocha-Azevedo, C.-H. Ho and F. Grinnell, Exp. Cell Res., 2013, 319, 546–555 CrossRef PubMed.
- S. Linder, E. Castaños-Velez, A. von Rosen and P. Biberfeld, Hepato-Gastroenterology, 2001, 48, 1321–1327 Search PubMed.
- M. T. Yip-Schneider, H. Wu, K. Stantz, N. Agaram, P. A. Crooks and C. M. Schmidt, BMC Cancer, 2013, 13, 194 CrossRef PubMed.
- M. Matsusaki, M. Komeda, S. Mura, H. Y. Tanaka, M. R. Kano, P. Couvreur and M. Akashi, Adv. Healthcare Mater., 2017, 6, 1700057 CrossRef PubMed.
- V. B. Orel, M. A. Zabolotny and V. E. Orel, Med. Hypotheses, 2017, 102, 82–86 CrossRef PubMed.
- N. C. Denko, Nat. Rev. Cancer, 2008, 8, 705–713 CrossRef PubMed.
- A. C. Koong, V. K. Mehta, Q. T. Le, G. A. Fisher, D. J. Terris, J. M. Brown, A. J. Bastidas and M. Vierra, Int. J. Radiat. Oncol., Biol., Phys., 2000, 48, 919–922 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |