
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trifluoromethylated proline analogues as efficient tools to enhance the hydrophobicity and to promote passive diffusion transport of the L-prolyl-L-leucyl glycinamide (PLG) tripeptide†
Martin Olivera,
Charlène Gadaisa,
Júlia García-Pindadob,
Meritxell Teixidób,
Nathalie Lensen *a,
Grégory Chaume*a and
Thierry Brigauda
*a,
Grégory Chaume*a and
Thierry Brigauda
aLaboratoire de Chimie Biologique (LCB), Université de Cergy-Pontoise, 5 mail Gay-Lussac, Neuville-sur-Oise, 95031 Cergy-Pontoise, France. E-mail: nathalie.lensen@u-cergy.fr; gregory.chaume@u-cergy.fr
bInstitute for Research in Biomedicine (IRB Barcelona), Barcelona Institute of Science and Technology (BIST), C/ Baldiri Reixac 10, 08028 Barcelona, Spain
First published on 18th April 2018
Abstract
The synthesis of four CF3-proline analogues of the PLG peptide is reported. Our results show that the incorporation of trifluoromethylated amino acids (Tfm-AAs) at the N-terminal position of a peptide significantly increases its hydrophobicity. In addition, depending on the relative configuration and the position of the CF3 group, Tfm-AAs can also promote passive diffusion transport.
Introduction
The use of peptides as therapeutic agents are gaining interest and the number of peptides entering into clinical trials is in steady increase.1 Nonetheless their development is hampered by the lack of metabolic stability, poor transport properties, low oral bioavailability as well as poor target specificity. Therefore, efforts are still needed to circumvent these drawbacks and the incorporation of constrained unnatural amino acids into the peptide chains is one of the common strategies to improve their biological profile. In this context, fluorinated amino acids are very attractive compounds.2 The introduction of fluorine atoms and fluoroalkyl groups into peptides and peptidomimetics presents the following advantages: (i) it can greatly increase the local hydrophobicity,3 it also modulates the peptide conformation and assembly,4 (ii) it confers a better resistance to proteolysis5 and (iii) fluorine atoms can be used as highly sensitive labels for 19F-NMR/MRI/MRS spectroscopy, because of 19F 100% natural abundance and the absence of background signals in natural and biological media.6 Unfortunately, the use of fluorinated peptides remains limited, especially for amino acids bearing a trifluoromethyl group adjacent to the amino group such as trifluoromethylated amino acids (Tfm-AAs). Indeed, only few synthetic methods are available for their synthesis in enantiopure form resulting in a small panel of fluorinated amino acids. Moreover, their incorporation into peptides is still challenging due to the stereoelectronic effects imparted by the CF3 group which strongly decrease the nitrogen atom nucleophilicity7 and limits the coupling reactions at their N-termini.Our group develops for several years efficient methods for the synthesis of Tfm-AAs8 and their incorporation into peptides.9 We have reported that, unlike the non-fluorinated series, the coupling reactions at the C-termini of Tfm-AAs can be achieved with unprotected amino group because of its strong deactivation by the electron-withdrawing effect of the neighbouring CF3 group.9a In contrast, the decrease of the nucleophilicity of the amino group together with the steric hindrance of the CF3 group significantly impedes the coupling reaction of Tfm-AAs at their N-terminus and specific activations such as mixed anhydrides or amino acid chlorides have to be used.9b,c,10 We applied our methodologies to the synthesis of several Tfm-AAs containing peptides in order to investigate their specific physicochemical and/or biological properties.11
Among the targeted peptides, we are interested in the endogenous brain peptide L-prolyl-L-leucyl glycinamide 1 (PLG). Because of its structural simplicity, and its relevance in a wide range of pharmacological activities in the central nervous system (CNS),12 several conformationally constrained analogues of PLG have been reported in literature.13 From our side, we have previously described the synthesis of the (S)-α-CF3-proline analogue (S,S)-2 of PLG. In vivo biological activity studies on rat models revealed that (S,S)-2 displays, after intraperitoneal injection, higher analgesic effects in paw pressure test during acute pain and superior anti-opioid effects in stress-induced analgesia compared to PLG 1 (Fig. 1).11a,b Because PLG 1 is known to cross the blood–brain barrier (BBB),14 we hypothesized that the superior analgesic effect of (S,S)-2 could be probably due to the presence of the CF3 group. Indeed, the significant decrease of the proline basicity7 and its low protonation ability should lead to an increase in the hydrophobicity parameter enabling the fluorinated PLG analogue (S,S)-2 to better cross the BBB and enter the CNS. In order to confirm our hypothesis, we report here the synthesis of four CF3-proline analogues of PLG. Their hydrophobicity as well as their ability to cross the BBB were assessed and compared to PLG 1.
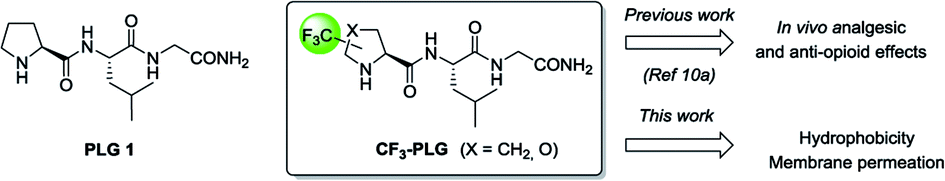 | ||
| Fig. 1 Chemical structure of PLG and CF3-PLG analogues. | ||
Results and discussion
Synthesis of the CF3-proline analogues of PLG
Four fluorinated analogues of PLG, corresponding to two pairs of diastereomers, have been considered to evaluate the effect of the CF3 group in α position of the N-terminal amino group on hydrophobicity and membrane permeation. The collected data should also provide valuable information related to the influence of the configuration of the chiral center bearing the CF3 group and its position along the 5-membered ring. In addition to the (S)-α-CF3-proline (S)-4 used for the synthesis of peptide (S,S)-2, three other fluorinated amino acids, namely the (R)-α-CF3-proline (R)-4, the (S,S)-CF3-pseudoproline (S,S)-5 and the (R,S)-CF3-pseudoproline (R,S)-5, were selected to substitute the proline residue into the PLG sequence. The preparation of these trifluoromethylated amino acids in enantiopure form has been previously reported by our group.8a,dThe synthesis of the corresponding peptides [(R,S)-2, (S,S,S)-3 and (R,S,S)-3] has been performed following the protocol described for the synthesis of (S,S)-2.11a As mentioned before, the incorporation of the fluorinated proline surrogates into the peptide chain does not required preliminary protection of the amino group. Nevertheless, the reverse addition of the unprotected CF3-amino acids to the preformed hydrochloride salt of (S)-Leu-Gly-NH2 dipeptide using the classical 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) and 1-hydroxybenzotriazole (HOBt) coupling reagents is required to prevent the formation of diketopiperazine side products. While the synthesis of the (S)-α-CF3-proline containing peptide (S,S)-2 occurred in very good yield (89%), the coupling reaction starting from (R)-α-CF3-proline (R)-4 gave the corresponding peptide (R,S)-2 in low yield (22%) (Scheme 1). This result may be explained by a mismatched double stereodifferentiation between the (R)-α-CF3-proline residue and the (S)-Leu-Gly-NH2 dipeptide. The peptide 3 diastereomers [(S,S,S)-3 and (R,S,S)-3] were obtained by coupling (S,S)- and (R,S)-CF3-pseudoproline 5 with the (S)-Leu-Gly-NH2 dipeptide (53% and 55% respectively).
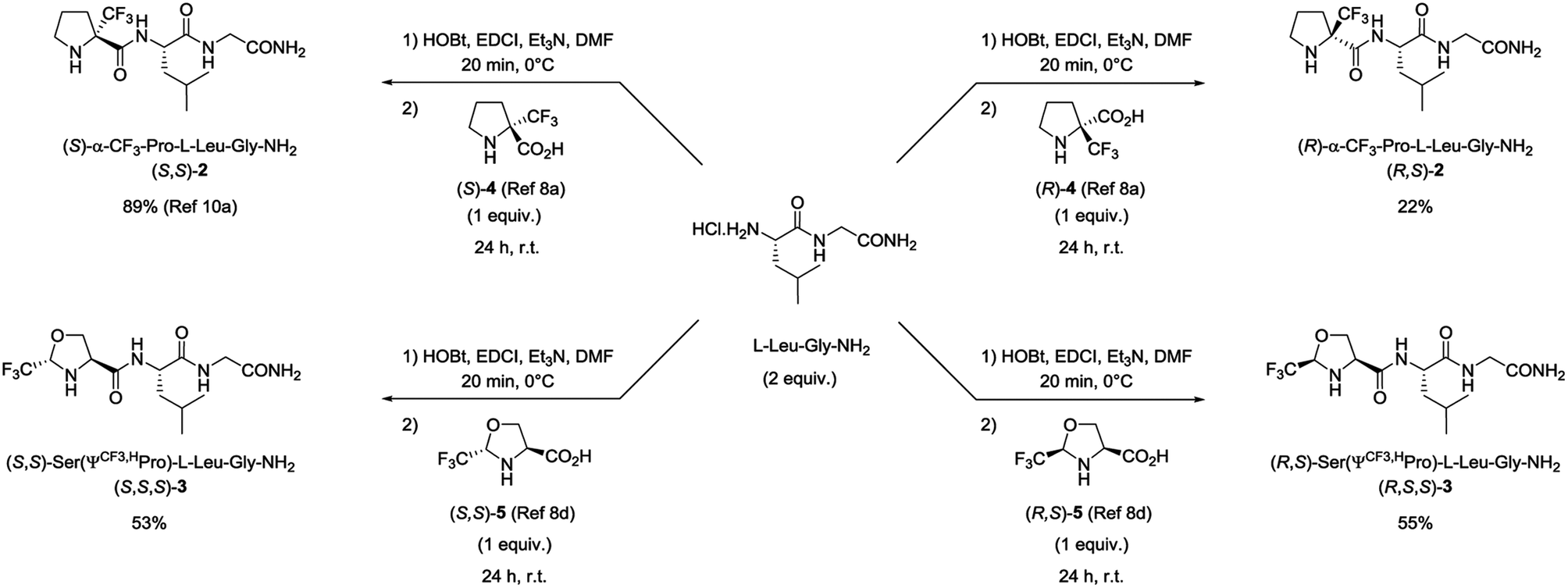 | ||
| Scheme 1 Synthesis of CF3-proline and CF3-pseudoproline containing PLG analogues. | ||
Measurement of the isocratic hydrophobicity index φ0
The hydrophobicity of the fluorinated analogues of PLG has then been assessed and compared to the native PLG 1 in order to investigate the specific effect imparted by the CF3 group. For this purpose, we applied a chromatographic method, first developed by Valko et al.,15 which proved to be very convenient for evaluating the hydrophobicity index of fluorinated peptides.16 It is based on the measurement of the isocratic hydrophobicity index (φ0) which is defined as the percentage (by volume) of organic solvent required to achieve an equal distribution of compound between the polar mobile and apolar stationary phases. Thus, higher is the φ0 value, more hydrophobic is the compound. The method involves the measurement of the logarithm of retention factor k (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k) values by reversed-phase high-performance liquid chromatography (RP-HPLC) using various organic-phase concentrations in the mobile phase (in our case, the acetonitrile is used). By plotting the logk values as a function of the acetonitrile concentration, the φ0 value can be obtained from the slope and the intercept of the linear regression (φ0 = −intercept/slope). A good correlation between the φ0 and the logP values was shown.15 The hydrophobicity index of our peptides was first measured at pH 7. The fluorinated PLG analogues 2 and 3 (entries 2–5, Table 1) display similar φ0 values (ca. 30) which are significantly higher (by a factor ∼2.5) compared to the native PLG 1 (entry 1, Table 1). These results show that the incorporation of a CF3 group in α position of the N-terminal amino group enhances the hydrophobic character of the corresponding peptide. However, the configuration of the stereocenter bearing the CF3 group as well as its position along the pyrrolidine ring does not seem to play a significant role (entries 2–5, Table 1). The φ0 values were then measured at pH 2 to investigate the impact of the possible ionic form of the PLG peptides 1–3 on the hydrophobicity parameter. The lower φ0 value observed for the native PLG 1 is consistent with its expected polar ionic state (entry 1, Table 1). Surprisingly, the fluorinated PLG analogues 2 and 3 display a different behaviour at pH 2. The PLG analogues 2 bearing an α-CF3-proline exhibit significant decrease of their hydrophobicity index (entries 2 and 3, Table 1), the (R,S)-2 derivative being more sensitive than its diastereomer (S,S)-2. These results suggest that the protonation of the deactivated N-terminal amino group occurs at pH 2, increasing the polarity of the corresponding ionized peptides. In contrast, the pseudoproline containing derivatives 3 do not seem to be protonated at pH 2. Only a slight decrease in the φ0 value is observed for the (S,S,S)-3 (entry 4, Table 1), that of the (R,S,S)-3 diastereomer remaining almost unchanged (entry 5, Table 1). Nevertheless, the incorporation of a Tfm-AA residue at the N-terminal position of the PLG allows to significantly enhance the hydrophobic character.
k) values by reversed-phase high-performance liquid chromatography (RP-HPLC) using various organic-phase concentrations in the mobile phase (in our case, the acetonitrile is used). By plotting the logk values as a function of the acetonitrile concentration, the φ0 value can be obtained from the slope and the intercept of the linear regression (φ0 = −intercept/slope). A good correlation between the φ0 and the logP values was shown.15 The hydrophobicity index of our peptides was first measured at pH 7. The fluorinated PLG analogues 2 and 3 (entries 2–5, Table 1) display similar φ0 values (ca. 30) which are significantly higher (by a factor ∼2.5) compared to the native PLG 1 (entry 1, Table 1). These results show that the incorporation of a CF3 group in α position of the N-terminal amino group enhances the hydrophobic character of the corresponding peptide. However, the configuration of the stereocenter bearing the CF3 group as well as its position along the pyrrolidine ring does not seem to play a significant role (entries 2–5, Table 1). The φ0 values were then measured at pH 2 to investigate the impact of the possible ionic form of the PLG peptides 1–3 on the hydrophobicity parameter. The lower φ0 value observed for the native PLG 1 is consistent with its expected polar ionic state (entry 1, Table 1). Surprisingly, the fluorinated PLG analogues 2 and 3 display a different behaviour at pH 2. The PLG analogues 2 bearing an α-CF3-proline exhibit significant decrease of their hydrophobicity index (entries 2 and 3, Table 1), the (R,S)-2 derivative being more sensitive than its diastereomer (S,S)-2. These results suggest that the protonation of the deactivated N-terminal amino group occurs at pH 2, increasing the polarity of the corresponding ionized peptides. In contrast, the pseudoproline containing derivatives 3 do not seem to be protonated at pH 2. Only a slight decrease in the φ0 value is observed for the (S,S,S)-3 (entry 4, Table 1), that of the (R,S,S)-3 diastereomer remaining almost unchanged (entry 5, Table 1). Nevertheless, the incorporation of a Tfm-AA residue at the N-terminal position of the PLG allows to significantly enhance the hydrophobic character.
Evaluation of the passive diffusion transport by parallel artificial membrane permeability assay (PAMPA)
We then decided to evaluate the ability of our fluorinated PLG analogues 2 and 3 to cross the BBB through passive diffusion. Passive diffusion is a nonsaturable and spontaneous transport process that depends on physicochemical properties such as hydrophobicity, hydrogen bonding, pKa or molecular weight. Therefore, such evaluation should allow us to assess the relevance of the CF3 group in the passive diffusion transport and thus, demonstrate the benefit in incorporating Tfm-AAs at the N-terminus of peptides. For this purpose, we used PAMPA method, first introduced by Kansy et al.,17 and selected a mixture of porcine brain polar lipid extract as in vitro model of the BBB.18 It consists in measuring the ratio of peptide that has been transferred from a donor well into an acceptor well through the artificial membrane (see Experimental part). The rate of permeation can be determined by the effective permeability (Pe) as well as the rate of transport values. The membrane retention value can also be obtained and reflects the strength of the interaction between the lipids that mimic the membrane and the peptide. While PLG 1 is known to cross the BBB,13 its permeability was found to be very negligible and no peptide was retained in the membrane (entry 1, Table 2). Thus, PLG 1 seems to not display excellent physicochemical properties to transport through passive diffusion. This result correlates with the fact that PLG 1 has been reported to enter brain through a partially saturable transport system.14 PLG analogues 2, bearing the α-CF3-proline residue, show an ability to cross the membrane (entries 2 and 3, Table 2). According to the empirical BBB permeation prediction,19 the peptide (R,S)-2 displays a poor effective permeability (Pe < 2.0 × 106 cm s−1) while its diastereomer (S,S)-2 exhibits a moderate transport properties (2.0 × 106 < Pe < 4.0 × 106 cm s−1). Both peptides (S,S)-2 and (R,S)-2 were also found to have a strong interaction with the membrane. It is worth noting the relevant difference in terms of permeability due to the change of only one stereocenter in the molecules. On the contrary, none of the CF3-pseudoproline containing PLG analogues 3 reveal an ability to cross the membrane by passive diffusion nor a membrane affinity (entries 4 and 5, Table 2). The PAMPA assays have been performed at pH 7.4, a pH for which fluorinated PLG analogues 2 and 3 display similar hydrophobicity index values (see Table 1). Therefore, the significant difference between the peptides 2 and 3 suggests that the nature of the 5-membered ring (pyrrolidine or oxazolidine) and the position along the ring as well as the absolute configuration of the carbon bearing the CF3 group play also a key role in the passive diffusion transport.| Entry | Compound | Pe (× 106)b cm s−1 | Transport (%) (4 h)b | Membrane retentionb |
|---|---|---|---|---|
| a Data are expressed as the mean ± SD.b Parameters definitions and their calculations are provided in Experimental section.c Not determined. | ||||
| 1 | 1 | ndc | ndc | ndc |
| 2 | (S,S)-2 | 2.68 ± 0.04 | 5.34 ± 0.08 | 50% |
| 3 | (R,S)-2 | 0.31 ± 0.03 | 0.65 ± 0.06 | 30% |
| 4 | (S,S,S)-3 | ndc | ndc | ndc |
| 5 | (R,S,S)-3 | ndc | ndc | ndc |
Conclusions
We demonstrate that the incorporation of a Tfm-AA residue at the N-terminal position of a peptide can be very useful to enhance its hydrophobic character. Moreover, depending on its position along the 5-membered pyrrolidine ring, the CF3 group may promote passive diffusion transport. Thus, Tfm-AA residues could serve as potent BBB-shuttle to transport into the CNS compounds of interests that cannot cross the BBB. The use of peptide as BBB-shuttles is one of the most promising approaches to deliver safely drugs to the brain.20Experimental section
Synthesis of the CF3-Proline analogues of PLG
:10) gave pure (R,S)-2 (110 mg, 22%) as colorless oil. Rf = 0.33 (CH2Cl2/MeOH, 90:10); [α]D +20.1 (c 1.4 in MeOH). IR (neat): 3272, 2955, 1651, 1651, 1519, 1386, 1250 cm−1; 1H NMR (400 MHz, CD3OD) δ 0.93 (d, J = 6.4 Hz, 3H, Hδ Leu-H), 0.96 (d, J = 6.4 Hz, 3H, Hδ Leu-H), 1.58–1.72 (m, 3H, Hβ Leu-H, Hγ Leu-H), 1.73–1.90 (m, 2H, Hγ Pro-H), 2.12–2.28 (m, 2H, Hβ Pro), 3.02–3.14 (m, 2H, Hδ Pro), 3.78 (d, J = 17.0 Hz, 1H, Hα Gly-Ha), 3.91 (d, J = 17.0 Hz, 1H, Hα Gly-Hb), 4.41 (m, 1H, Hα Leu-H); 13C NMR (100.5 MHz, CD3OD) δ 22.0 (CH3, Cδ Leu), 23.4 (CH3, Cδ Leu), 25.9 (CH, Cγ Leu), 26.1 (CH2, Cγ Pro), 33.2 (CH2, Cβ Pro), 41.8 (CH2, Cβ Leu), 43.0 (CH2, Cα Gly), 48.2 (CH2, Cδ Pro), 53.5 (CH, Cα Leu), 72.2 (q, J = 25.9 Hz, C, Cα Pro), 127.4 (q, J = 283.7 Hz, C, CF3), 172.1 (C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 173.9 (C, CO), 174.6 (C, CO); 19F NMR (376.2 MHz, CD3OD): δ −78.9 (s, CF3); HRMS (ESI-TOF) calcd. for C14H24N4O3F3 [M + H]+ 353.1801, found 353.1801.:10) gave pure (R,S,S)-3 (212 mg, 55%) as hygroscopic white solid. Rf = 0.34 (CH2Cl2/MeOH, 90:10); [α]D −12.2 (c 1.0 in MeOH); IR (neat): 3326, 2961, 2470, 1638, 1524, 1454, 1288 cm−1; 1H NMR (400 MHz, CD3OD) δ 0.93 (d, J = 6.4 Hz, 3H, Hδ Leu-H), 0.96 (d, J = 6.0 Hz, 3H, Hδ Leu-H), 1.58–1.70 (m, 3H, Hβ Leu-H and Hγ Leu-H), 3.77 (d, J = 16.9 Hz, 1H, Hα Gly-Ha), 3.90 (d, J = 16.9 Hz, 1H, Hα Gly-Hb), 4.11–4.21 (m, 3H, Hα Ψpro-H and Hβ Ψpro-Ha), 4.37 (t, J = 6.5 Hz, 1H, Hα Leu-H), 5.10 (d, J = 6.0 Hz, 1H, Hδ Ψpro-H); 13C NMR (100.5 MHz, CD3OD) δ 21.7 (CH3, Cδ Leu), 23.5 (CH3, Cδ Leu), 25.5 (CH, Cγ Leu), 42.0 (CH2, Cβ Leu), 43.1 (CH2, Cα Gly), 53.1 (CH, Cα Leu), 60.8 (CH, Cα Ψpro), 70.6 (CH2, Cβ Ψpro), 89.1 (q, J = 33.6 Hz, CH, Cδ Ψpro), 125.1 (q, J = 282.8 Hz, C, CF3), 174.2 (C, CO), 174.6 (C, CO), 175.0 (C, CO); 19F NMR (376.2 MHz, CD3OD) δ −85.3 (bs, CF3); HRMS (ESI-TOF) calcd. for C13H22N4O4F3 [M + H]+ 355.1593, found 355.1601.:10) gave pure (S,S,S)-3 (434 mg, 53%) as white solid. Rf = 0.34 (CH2Cl2/MeOH, 90:10); mp 150 °C; [α]D −41.8 (c 0.9 in MeOH); IR (neat): 3301, 2419, 1782, 1558, 1435, 1289 cm−1; 1H NMR (400 MHz, CD3OD) δ 0.95 (d, J = 6.4 Hz, 3H, Hδ Leu-H), 0.99 (d, J = 6.4 Hz, 3H, Hδ Leu-H), 1.58–1.64 (m, 2H, Hβ Leu-H), 1.68 (m, 1H, Hγ Leu-H), 3.64 (t, J = 7.3 Hz, 1H, Hβ Ψpro-Ha), 3.74 (d, J = 17.4 Hz, 1H, Hα Gly-Ha), 3.92 (d, J = 17.4 Hz, 1H, Hα Gly-Hb), 3.98 (t, J = 7.3 Hz, 1H, Hα Ψpro-H), 4.21 (t, J = 7.3 Hz, 1H, Hβ Ψpro-Hb), 4.35 (t, J = 6.9 Hz, 1H, Hα Leu-H), 5.14 (q, J = 5.5 Hz, 1H, Hδ Ψpro-H); 13C NMR (100.5 MHz, CD3OD) δ 22.0 (CH3, Cδ Leu), 23.3 (CH3, Cδ Leu), 25.9 (CH, Cγ Leu), 41.4 (CH2, Cβ Leu), 43.1 (CH2, Cα Gly), 54.0 (CH, Cα Leu), 60.2 (CH, Cα Ψpro), 71.4 (CH2, Cβ Ψpro), 89.4 (q, J = 33.6 Hz, CH, Cδ Ψpro), 125.0 (q, J = 282.8 Hz, C, CF3), 172.3 (C, CO), 174.2 (C, CO), 175.0 (C, CO); 19F NMR (376.2 MHz, CD3OD) δ −86.1 (d, J = 5.5 Hz, CF3); HRMS (ESI-TOF) calcd. for C13H22N4O4F3 [M + H]+ 355.1593, found 355.1587.
O), 173.9 (C, CO), 174.6 (C, CO); 19F NMR (376.2 MHz, CD3OD): δ −78.9 (s, CF3); HRMS (ESI-TOF) calcd. for C14H24N4O3F3 [M + H]+ 353.1801, found 353.1801.:10) gave pure (R,S,S)-3 (212 mg, 55%) as hygroscopic white solid. Rf = 0.34 (CH2Cl2/MeOH, 90:10); [α]D −12.2 (c 1.0 in MeOH); IR (neat): 3326, 2961, 2470, 1638, 1524, 1454, 1288 cm−1; 1H NMR (400 MHz, CD3OD) δ 0.93 (d, J = 6.4 Hz, 3H, Hδ Leu-H), 0.96 (d, J = 6.0 Hz, 3H, Hδ Leu-H), 1.58–1.70 (m, 3H, Hβ Leu-H and Hγ Leu-H), 3.77 (d, J = 16.9 Hz, 1H, Hα Gly-Ha), 3.90 (d, J = 16.9 Hz, 1H, Hα Gly-Hb), 4.11–4.21 (m, 3H, Hα Ψpro-H and Hβ Ψpro-Ha), 4.37 (t, J = 6.5 Hz, 1H, Hα Leu-H), 5.10 (d, J = 6.0 Hz, 1H, Hδ Ψpro-H); 13C NMR (100.5 MHz, CD3OD) δ 21.7 (CH3, Cδ Leu), 23.5 (CH3, Cδ Leu), 25.5 (CH, Cγ Leu), 42.0 (CH2, Cβ Leu), 43.1 (CH2, Cα Gly), 53.1 (CH, Cα Leu), 60.8 (CH, Cα Ψpro), 70.6 (CH2, Cβ Ψpro), 89.1 (q, J = 33.6 Hz, CH, Cδ Ψpro), 125.1 (q, J = 282.8 Hz, C, CF3), 174.2 (C, CO), 174.6 (C, CO), 175.0 (C, CO); 19F NMR (376.2 MHz, CD3OD) δ −85.3 (bs, CF3); HRMS (ESI-TOF) calcd. for C13H22N4O4F3 [M + H]+ 355.1593, found 355.1601.:10) gave pure (S,S,S)-3 (434 mg, 53%) as white solid. Rf = 0.34 (CH2Cl2/MeOH, 90:10); mp 150 °C; [α]D −41.8 (c 0.9 in MeOH); IR (neat): 3301, 2419, 1782, 1558, 1435, 1289 cm−1; 1H NMR (400 MHz, CD3OD) δ 0.95 (d, J = 6.4 Hz, 3H, Hδ Leu-H), 0.99 (d, J = 6.4 Hz, 3H, Hδ Leu-H), 1.58–1.64 (m, 2H, Hβ Leu-H), 1.68 (m, 1H, Hγ Leu-H), 3.64 (t, J = 7.3 Hz, 1H, Hβ Ψpro-Ha), 3.74 (d, J = 17.4 Hz, 1H, Hα Gly-Ha), 3.92 (d, J = 17.4 Hz, 1H, Hα Gly-Hb), 3.98 (t, J = 7.3 Hz, 1H, Hα Ψpro-H), 4.21 (t, J = 7.3 Hz, 1H, Hβ Ψpro-Hb), 4.35 (t, J = 6.9 Hz, 1H, Hα Leu-H), 5.14 (q, J = 5.5 Hz, 1H, Hδ Ψpro-H); 13C NMR (100.5 MHz, CD3OD) δ 22.0 (CH3, Cδ Leu), 23.3 (CH3, Cδ Leu), 25.9 (CH, Cγ Leu), 41.4 (CH2, Cβ Leu), 43.1 (CH2, Cα Gly), 54.0 (CH, Cα Leu), 60.2 (CH, Cα Ψpro), 71.4 (CH2, Cβ Ψpro), 89.4 (q, J = 33.6 Hz, CH, Cδ Ψpro), 125.0 (q, J = 282.8 Hz, C, CF3), 172.3 (C, CO), 174.2 (C, CO), 175.0 (C, CO); 19F NMR (376.2 MHz, CD3OD) δ −86.1 (d, J = 5.5 Hz, CF3); HRMS (ESI-TOF) calcd. for C13H22N4O4F3 [M + H]+ 355.1593, found 355.1587.Isocratic hydrophobicity index φ0
An Agilent 1200 series high-performance liquid chromatograph coupled to an ELSD detector was used. The reversed-phase HPLC measurements were carried out on a Zorbax® RX-C18 analytical column (4.6 × 250 mm, 5 μm). The mobile phase was 50 mM ammonium acetate and HPLC grade acetonitrile as organic modifier (pH 7.0) or TFA 0.1% buffer aqueous solution and 0.1% TFA HPLC grade acetonitrile (pH 2). The mobile phase flow rate was ranging from 0.1–1.0 mL min−1. The dead time (t0) was measured by injecting sodium nitrate together with the sample at a concentration of 10 mM. The samples were dissolved at 1 mg/1 mL in water. Few drops of acetonitrile were added when the dissolution of the peptide was incomplete. The logarithm of retention factor k (logk) values were obtained according to the eqn (1):
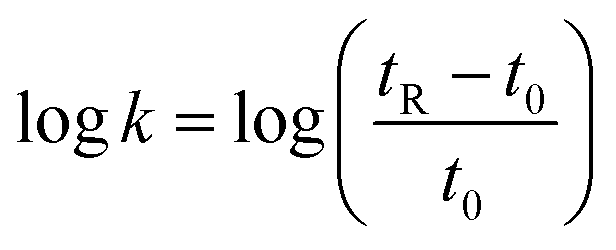 | (1) |
The logk values were plotted as a function of the acetonitrile concentration. The slope (S) and the intercept (logkw) values were calculated from at least three concentrations of acetonitrile. The correlation coefficients of the linear fit were higher than 0.99. The isocratic hydrophobicity index φ0 was calculated according to the eqn (2):
|
φ0 = −logkw/S
| (2) |
kw is the logk value extrapolated to the 0% acetonitrile concentration.
Parallel artificial membrane permeability assay (PAMPA)
The PAMPA was used to measure the capacity of the peptides 1–3 to cross the BBB by passive diffusion. The effective permeability (Pe) of the compounds was determined at an initial concentration of 5 mM. The buffer solution was prepared from a commercially concentrated one, supplied by pION. Following the manufacturer's instructions, system solution was adjusted to pH 7.4 using a 0.5 M NaOH solution. The compounds to be analyzed were dissolved in a mixture of buffer solution with 20% of 1-propanol used as co-solvent to ensure their solubility. The PAMPA sandwich was separated, and the donor wells were filled with 195 μL of each compound. A magnetic stirrer was placed in each well. The acceptor plate was put into the donor plate, ensuring that the underside of the membrane was in contact with the buffer. Then, 4 μL of a mixture of phospholipids from a porcine polar brain extract (20 mg mL−1) in dodecane (composition: 12.6% phosphatidylcholine (PC), 33.1% phosphatidylethanolamine (PE), 18.5% phosphatidylserine (PS), 4.1% phosphatidylinositol (PI), 0.8% phosphatidic acid and 30.9% of other compounds) was added to the filter of each well, followed by 200 μL of buffer solution. The plate was then covered and incubated at room temperature in a saturated humidity atmosphere for 4 h under orbital agitation at 25 μm of unstirred water layer (UWL). After this period, the solution from the donor and acceptor plates was transferred to the UPLC vials. 100 μL of each acceptor and donor samples and 100 μL of t0 samples were injected into the UPLC apparatus. Acceptor samples were analyzed by MALDI-TOF in order to study the presence of not degraded peptides and confirm their crossing. The effective permeability (Pe) after 4 h was calculated using eqn (3) and the percentage of transport with eqn (4):
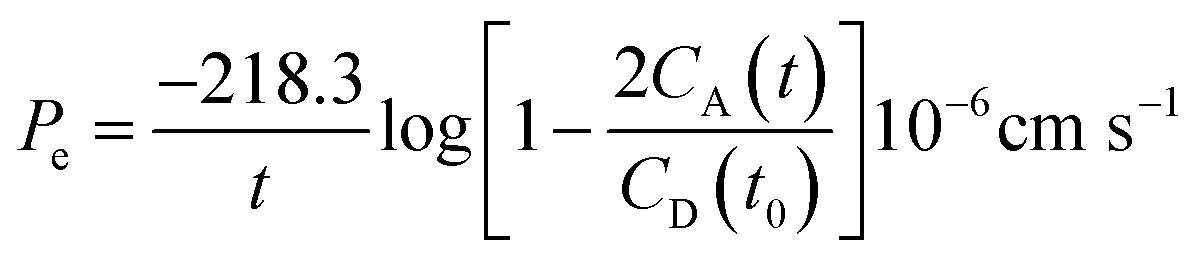 | (3) |
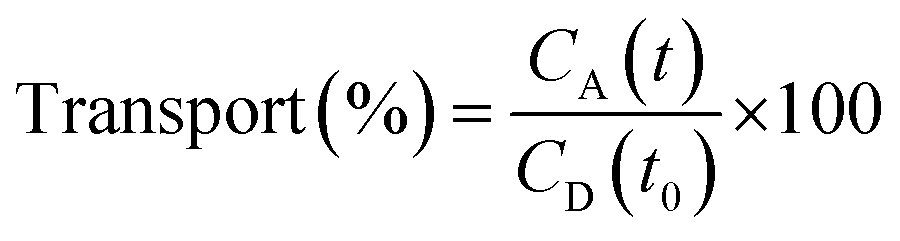 | (4) |
Membrane retention was also calculated as the difference of product at t0 and after 4 h in the donor and acceptor compartments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
French partner thanks the French Fluorine Network.References
- (a) S. Ghosh, Chem. Today, 2016, 34, 5 CAS; (b) K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122 CrossRef CAS PubMed; (c) T. Uhlig, T. Kyprianou, F. G. Martinelli, C. A. Oppici, D. Heiligers, D. Hills, X. R. Calvo and P. Verhaert, EuPa Open Proteomics, 2014, 4, 1 CrossRef.
- (a) R. Smits, C. D. Cadicamo, K. Burger and B. Koksch, Chem. Soc. Rev., 2008, 37, 1727 RSC; (b) X.-L. Qiu and F.-L. Qing, Eur. J. Org. Chem., 2011, 3261 CrossRef CAS; (c) J. L. Aceña, A. E. Sorochinsky and V. A. Soloshonok, Synthesis, 2012, 44, 1591 CrossRef.
- (a) M. Salwiczek, E. K. Nyakatura, U. I. M. Gerling, S. Ye and B. Koksch, Chem. Soc. Rev., 2012, 41, 2135 RSC; (b) V. H. Dalvi and P. J. Rossky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13603 CrossRef CAS PubMed; (c) Q. A. Huchet, B. Kuhn, B. Wagner, H. Fischer, M. Kansy, D. Zimmerli, E. M. Carreira and K. Müller, J. Fluorine Chem., 2013, 152, 119 CrossRef CAS; (d) J. R. Robalo, S. Huhmann, B. Koksch and A. V. Verde, Chem, 2017, 3, 881–897 CrossRef CAS.
- (a) U. I. M. Gerling, M. Salwiczek, C. D. Cadicamo, H. Erdbrink, C. Czekelius, S. L. Grage, P. Wadhwani, A. S. Ulrich, M. Behrends, G. Haufe and B. Koksch, Chem. Sci., 2014, 5, 819 RSC; (b) S. Huhmann, E. K. Nyakatura, H. Erdbrink, U. I. M. Gerling, C. Czekelius and B. Koksch, J. Fluorine Chem., 2015, 175, 32 CrossRef CAS.
- (a) B. Koksch, N. Sewald, H.-J. Hofmann, K. Burger and H.-D. Jakubke, J. Pept. Sci., 1997, 3, 157 CrossRef CAS PubMed; (b) V. Asante, J. Mortier, G. Wolber and B. Koksch, Amino Acids, 2014, 46, 2733 CrossRef CAS PubMed.
- For review on the use of 19F NMR, see: (a) E. N. G. Marsh and Y. Suzuki, ACS Chem. Biol., 2014, 9, 1242 CrossRef CAS PubMed; (b) A. Vulpetti and C. Dalvit, Drug Discovery Today, 2012, 17, 890 CrossRef CAS PubMed; (c) H. Chen, S. Viel, F. Ziarelli and L. Peng, Chem. Soc. Rev., 2013, 42, 7971 RSC.
- M. Schlosser, Angew. Chem., Int. Ed., 1998, 37, 1496 CrossRef CAS.
- (a) G. Chaume, M.-C. Van Severen, S. Marinkovic and T. Brigaud, Org. Lett., 2006, 8, 6123 CrossRef CAS PubMed; (b) G. Chaume, M.-C. Van Severen, L. Ricard and T. Brigaud, J. Fluorine Chem., 2008, 129, 1104 CrossRef CAS; (c) C. Caupène, G. Chaume, L. Ricard and T. Brigaud, Org. Lett., 2009, 11, 209 CrossRef PubMed; (d) G. Chaume, O. Barbeau, P. Lesot and T. Brigaud, J. Org. Chem., 2010, 75, 4135 CrossRef CAS PubMed; (e) J. Simon, T. T. Nguyen, E. Chelain, N. Lensen, J. Pytkowicz, G. Chaume and T. Brigaud, Tetrahedron: Asymmetry, 2011, 22, 309 CrossRef CAS; (f) N. Lensen, J. Marais and T. Brigaud, Org. Lett., 2015, 17, 342 CrossRef CAS PubMed; (g) H. Lubin, J. Pytkowicz, G. Chaume, G. Sizun-Thome and T. Brigaud, J. Org. Chem., 2015, 80, 2700 CrossRef CAS PubMed.
- (a) G. Chaume, N. Lensen, C. Caupène and T. Brigaud, Eur. J. Org. Chem., 2009, 5717 CrossRef CAS; (b) G. Chaume, J. Simon, C. Caupène, N. Lensen, E. Miclet and T. Brigaud, J. Org. Chem., 2013, 78, 10144 CrossRef CAS PubMed; (c) E. Devillers, J. Pytkowicz, E. Chelain and T. Brigaud, Amino Acids, 2016, 48, 1457 CrossRef CAS PubMed.
- B. Koksch, P. J. L. M. Quaedflieg, T. Michel, K. Burger, Q. B. Broxterman and H. E. Schoemaker, Tetrahedron: Asymmetry, 2004, 15, 1401 CrossRef CAS.
- (a) I. Jlalia, N. Lensen, G. Chaume, E. Dzhambazova, L. Astasidi, R. Hadjiolova, A. Bocheva and T. Brigaud, Eur. J. Med. Chem., 2013, 62, 122 CrossRef CAS PubMed; (b) A. Bocheva, H. Nocheva, I. Jlalia, N. Lensen, G. Chaume and T. Brigaud, Med. Chem., 2013, 3, 206 CAS; (c) A. Botz, V. Gasparik, E. Devillers, A. R. F. Hoffmann, L. Caillon, E. Chelain, O. Lequin, T. Brigaud and L. Khemtemourian, Biopolymers, 2015, 104, 601 CrossRef CAS PubMed; (d) J. Simon, J. Pytkowicz, N. Lensen, G. Chaume and T. Brigaud, J. Org. Chem., 2016, 81, 5381 CrossRef CAS PubMed; (e) S. L. Grage, S. Kara, A. Bordessa, V. Doan, F. Rizzolo, M. Putzu, T. Kubař, A. M. Papini, G. Chaume, T. Brigaud, S. Afonin and A. S. Ulrich, Chem. - Eur. J., 2018, 24, 4328 CrossRef CAS PubMed.
- (a) R. K. Mishra, S. Chiu, P. Chiu and C. P. Mishra, Methods Find. Exp. Clin. Pharmacol., 1983, 5, 203 CAS; (b) G. W. Reed, G. A. Olson and R. D. Olson, Neurosci. Biobehav. Rev., 1994, 18, 519 CrossRef CAS PubMed; (c) W. Pan and A. J. Kastin, Peptides, 2007, 28, 2411 CrossRef CAS PubMed.
- (a) S. Bhagwanth, R. K. Mishra and R. L. Johnson, Beilstein J. Org. Chem., 2013, 9, 204 CrossRef CAS PubMed and references cited therein;; (b) J. Ferreira da Costa, O. Caamaño, F. Fernández, X. García-Mera, I. E. Sampaio-Dias, J. M. Brea and M. I. Cadavid, Eur. J. Med. Chem., 2013, 69, 146 CrossRef CAS PubMed; (c) I. E. Sampaio-Dias, C. A. D. Sousa, X. García-Mera, J. Ferreira da Costa, O. Caamaño and J. E. Rodríguez-Borges, Org. Biomol. Chem., 2016, 14, 11065 RSC.
- L. Di, E. H. Kerns, K. Fan, O. J. McConnell and G. T. Carter, Eur. J. Med. Chem., 2003, 38, 223 CrossRef CAS PubMed.
- K. Valko and P. Slegel, J. Chromatogr., 1993, 631, 49 CrossRef CAS.
- C. Gadais, E. Devillers, V. Gasparik, E. Chelain, J. Pytkowicz and T. Brigaud, ChemBioChem, 2018 DOI:10.1002/cbic.201800088.
- M. Kansy, F. Senner and K. Gubernator, J. Med. Chem., 1998, 41, 1007 CrossRef CAS PubMed.
- W. A. Banks and A. J. Kastin, Peptides, 1994, 15, 23 CrossRef CAS.
- P. Crivori, G. Cruciani, P. Carrupt and B. Testa, J. Med. Chem., 2000, 43, 2204 CrossRef CAS PubMed.
- B. Oller-Salvia, M. Sánchez-Navarro, E. Giralt and M. Teixidó, Chem. Soc. Rev., 2016, 45, 4690 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Extended experimental details and analytical data; copies of the NMR spectra. See DOI: 10.1039/c8ra02511h |
| This journal is © The Royal Society of Chemistry 2018 |