DOI:
10.1039/C8RA02483A
(Paper)
RSC Adv., 2018,
8, 21871-21878
Efficient solar-driven conversion of nitrogen to ammonia in pure water via hydrogenated bismuth oxybromide†
Received
21st March 2018
, Accepted 25th May 2018
First published on 13th June 2018
Abstract
Solar-driven reduction of dinitrogen to ammonia under mild conditions has attracted widespread interest in recent years. In this study, we first report low-temperature hydrogenated BiOBr for the direct synthesis of ammonia from dinitrogen with high efficiency under solar-light irradiation. In a proof of concept, the hydrogenation treatment can lead to surface disorder due to the strong reducing capacity of hydrogen. Oxygen atoms can be activated, and they can escape from the surface structure to form oxygen vacancies. Then, defect engineering can broaden the photoelectricity absorption window and effectively trigger interfacial electron transfer from the semiconductor to the combined nitrogen. This method exhibits a satisfactory result for photocatalytic nitrogen fixation, yielding about 2.6 times more NH3 than that obtained from the original sample. The corresponding apparent quantum efficiency can reach a significant value of 2.1% under 380 nm monochromatic light irradiation. These results may pave a new way for the synthesis of highly active photocatalysts for efficient nitrogen fixation under solar light irradiation.
1. Introduction
To the best of our knowledge, 60% atmospheric nitrogen can be fixed by molybdenum-dependent nitrogenase, which is an excellent enzyme for the reduction of nitrogen to ammonia in biological species.1,2 The basic concept is as follows: adenosine triphosphate (ATP) must be combined with magnesium (Mg) to form the Mg–ATP complex. The typical (Fe-) protein and (MoFe-) protein act as electronic carriers to deliver electrons in the entire biological nitrogen fixation process (eqn (1)).3–5 Compared to biological nitrogen fixation, the photocatalytic reduction of atmospheric nitrogen to ammonia under solar light has received significant attention;6–11 it has significant potential due to which it can be explored in the future for nitrogen fixation due to a fast and convenient pathway. Semiconductor photocatalysis is a green reaction that requires mild conditions, and it is driven by abundant solar energy; in contrast, the industrial process (classical Haber–Bosch process, 25 MPa 723–773 K) requires large quantities of chemical fuels and causes environmental damage.12 Furthermore, in the photocatalytic nitrogen fixation process, water is utilized as the reducing agent to react with valence band holes (VB h+) (eqn (2)), and nitrogen is reduced by the conduction band electrons (CB e−) to form ammonia (eqn (3)).13,14| | |
N2 + 6H+ + nMg–ATP + 6e− → 2NH3 + nMg–ADP + nPi
| (1) |
| | |
2H2O + 4h+ → O2 + 4H+
| (2) |
| | |
N2 + 6H+ + 6e− → 2NH3
| (3) |
However, in the past several decades, the results for photocatalytic nitrogen fixation have been far from satisfactory. The nitrogen molecule is highly stable on account of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond. The decomposition of nitrogen molecules into atoms requires absorption energy of 941.69 kJ mol−1. Additionally, a crucial yet unsolved issue is the inefficient steering of electrons from the bulk to the surface. Therefore, such significant challenges must be overcome to achieve efficient photocatalytic nitrogen fixation.
N triple bond. The decomposition of nitrogen molecules into atoms requires absorption energy of 941.69 kJ mol−1. Additionally, a crucial yet unsolved issue is the inefficient steering of electrons from the bulk to the surface. Therefore, such significant challenges must be overcome to achieve efficient photocatalytic nitrogen fixation.
Nowadays, a variety of photocatalysts have been utilized in ammonia formation such as graphitic carbon nitride (g-C3N4),15–17 TiO2,18,19 MoS2,20 and WO3·H2O.21 Bismuth oxybromide (BiOBr), a favorable layered ternary oxide semiconductor, has low-cost and is non-toxic and thus, it can be a good starting material for photocatalytic nitrogen fixation. BiOBr possesses a sandwich-like structure consisting of [Bi2O2]2+ layers and two bromine ion layers. Compared to anatase TiO2, BiOBr is advantageous due to its open crystalline structure. BiOBr has attracted extensive attention due to its narrow band gap, more positive conduction band minimum (CBM), wide photoabsorption region and consistent surface structure.22–26 However, due to its poor combining capacity with nitrogen, only a small amount of ammonia can be produced.
Herein, we report a low-temperature hydrogen annealing method to successfully synthesize BiOBr hollow microspheres having abundant vacancies. The BiOBr hollow microspheres show excellent photocatalytic nitrogen fixation, with only water as the reactant. The hydrogenation process can result in disordered nanoparticle states owing to the reduction properties of H2, causing the activation of oxygen atoms and their escape from the surface structure. This is a facile strategy for generating oxygen vacancies (OVs) and controlling the amount of OVs. Then, the low-temperature hydrogenation reaction can prevent structural collapse. Simultaneously, the structure of the hollow microspheres has a high H2 storage capacity. It is possible for the photocatalysts to be in a hydrogen-rich environment during the reaction process. This process is simple, and it promotes excellent absorbance under solar light. In addition, the efficient energy coupling among excitons, photons and dinitrogen in the photocatalytic system enables photocatalytic solar-driven nitrogen fixation to ammonia with distinguished performance.
2. Experimental section
2.1 Preparation of BiOBr hollow microspheres
In a typical procedure, 1.92 g Bi(NO3)3·5H2O and 0.416 g NaBr were dissolved in 40 ml of heated ethylene glycol (EG) under magnetic stirring. After complete dissolution to form a transparent mixture, the above-mentioned solution was added dropwise into 120 ml isopropanol (IPA). After stirring for 30 min, the final solution was transferred into a 200 ml Teflon-stainless steel autoclave, which was then sealed and heated at 433 K for 12 h. After the autoclave had cooled to room temperature, the resulting product was collected by centrifugation and washed with distilled water and anhydrous ethanol. The final product was dried overnight in the drying bin.
2.2 Preparation of hydrogenated BiOBr hollow microspheres (H-BiOBr)
BiOBr powder was calcined in a tube furnace at 473 K for 4 h under H2 atmosphere, with a heating rate of 10°C min−1. For control experiments, deficient OVs of BiOBr hollow microspheres (A-BiOBr) were formed by following the same calcination method except for the use of an air atmosphere.
2.3 Characterization
The crystalline structure and the purity of the samples were characterized by the powder X-ray diffraction (XRD) analysis on a Shimadzu XRD-6100 diffractometer at 40 kV and 40 mA with Cu Kα radiation; the scan rate was 8° min−1, and the 2 theta range was 10–80°. The chemical composition and distribution of the elements were determined by energy dispersive spectroscopy (EDS). The morphology of the samples was observed via field-emission scanning electron microscopy (JSM-7800F, JEOL). For TEM and high resolution TEM (HRTEM) analyses, a JEOL JEM-2100F transmission electron microscope was used. Optical absorption property was tested by UV-vis diffuse reflectance spectra (UV-vis DRS, CARY 100 & 300, VARIAN); BaSO4 was used as a reflecting basement. The PL spectra were analysed via a Hitachi F-7000 fluorescence spectrophotometer. X-ray photoelectron spectroscopy (XPS) measurements were carried out with a Thermo VG ESCALAB-250 system with Al-K and Mg-K sources operating at 15 kV. Room-temperature ESR spectra were measured using a JEOL JES-FA200 ESR spectrometer (300 K, 9.062 GHz). Chemical bonds and functional groups were tested by Fourier transform infrared spectroscopy (FT-IR, Nicolet 380). Nitrogen adsorption–desorption isotherms were measured using Micromeritics Tristar 3000 at 77 K; the pore size distribution and the specific surface area were tested using the Barrett–Joyner–Halenda (BJH) and the Brunauer–Emmett–Teller (BET) methods.
2.4 Photocatalytic tests
All photocatalytic fixation nitrogen experiments were carried out at room temperature with a 300 W xenon lamp (PLS-SXE 300, Beijing Perfect Light Co., Ltd., China) to simulate sunlight. For the photocatalytic nitrogen fixation process, 0.02 g of catalyst was added into 100 ml distilled water in the nitrogen fixation reactor, and the reactor was equipped with water circulation to maintain a constant room temperature. The mixture solution was constantly agitated in the dark with pure nitrogen bubbled at a flow rate of 65 ml min−1 for 30 min. The concentration of the solution was measured by Nessler's colorimetry with a UV-vis spectrometer. The apparent quantum efficiency (AQE) was measured in the same nitrogen fixation condition with different monochromatic light irradiations (using different band-pass filters at 380 nm, 420 nm, 460 nm, 520 nm, and 600 nm). The light intensity was measured by an irradiatometer (Beijing Normal University photoelectric instrument factory, FZ-A), and the AQE values at different wavelengths were calculated using the following equation:27
here, Na is the Avogadro's constant, M is the number of moles of generated ammonia, P is the optical power density, S is the irradiated area, t is the irradiation time, λ is the wavelength of monochromatic light, h is the Planck's constant, and c is the velocity of light.
2.5 NH3 concentration detection
The concentration of the solution was measured by Nessler's colorimetry with a UV-vis spectrometer (Shimadzu UV-2550).28 At first, 5 ml of solution was removed every 30 min under simulated sunlight. Then, the catalyst was removed with a 0.22 μm membrane filter and placed in a 20 ml sample tube. Next, 0.2 ml of Nessler's reagent was added to the sample tube and mixed fully. The mixture was left to stand for 10 min for full color processing. Finally, the concentration of NH3 was tested with a UV-vis spectrometer at 420 nm. The standard curve of NH3 with Nessler's regent is shown in Fig. S1.†
2.6 Electrochemical and photoelectrochemical measurements
Electrochemical and photoelectrochemical measurements were carried out in a three electrode system with saturated 0.1 M Na2SO4 solution. The glassy carbon electrode was used as the working electrode, the platinum electrode was used as the counter electrode, and the saturated calomel electrode was used as the reference electrode. For the photoelectrochemical process, 4 mg catalyst and 10 μl of Nafion solution (5 wt%) were dispersed in 400 μl distilled water and 200 μl ethanol mixture solution; after ultrasonication for 20 min, 10 μl solution was pipetted onto the working electrode surface and then finally dried at room temperature. An electrochemical workstation (CHI 660E) was used in the photoelectrochemical measurements: Electrochemical impedance spectroscopy (EIS) was carried out with 10 mV current at 0.1–105 Hz. Photocurrent responses were obtained under turn on–off light irradiation (300 W Xe lamp; PLS-SXE 300, Beijing Perfect Light Co., Ltd.). The Mott–Schottky plot was measured under the dark condition, with 1000 Hz and 1 mV amplitude at potentials from −0.5 V to 1 V.
3. Results and discussion
3.1 Morphology and structure
The morphology of samples was analyzed by SEM (Fig. 1). It was easy to identify many flower-like microspheres, whereas the collapsed part showed a hollow structure (Fig. 1a and b). In the preparation process, EG and bismuth nitrate produced a slow nucleation; the surface energy of nanoparticles was reduced to assemble the microspheres.29 Then, the microcrystallites on the surface of the substance were assembled and dissolved; [Bi2O2]2+ and Br− ions were generated via solvothermal conditions.30,31 The process of dissolution and crystallization was carried out repeatedly by Ostwald ripening. Finally, the flower-like hollow microspheres were formed via aggregation of sheets.32,33 The morphology of H-BiOBr was not noticeably different from that of BiOBr, as visualized by SEM (Fig. 1d and e). The structure was further analyzed by TEM (Fig. S1†); the HRTEM image of BiOBr showed clearer lattice fringes (Fig. 1c). H-BiOBr displayed nanoscale fine crystalline structure with interplanar spacing of 0.277 nm, corresponding to the (110) spacing of BiOBr. It was easy for the oxygen atoms to escape at the edge of the (001) spacing via hydrogenation treatment.34 The lattice edge is indistinctive in the dotted portion, and the lattice fringe is distorted in the dashed circles (Fig. 1f). X-ray diffraction (XRD) is an effective tool to prove the crystalline structure of samples. The XRD patterns of the two samples are shown in Fig. 2a. Several clear diffraction peaks for BiOBr and H-BiOBr were observed at 10.9°, 25.3°, 32.3°, 39.4°, 46.3°, 57.3°, 67.6° and 76.9°, which were indexed to (001), (011), (110), (112), (020), (212), (220) and (130) crystal planes (JCPDS 73-2061), respectively, of the tetragonal phase of BiOBr. The hydrogenation process did not change the crystalline structure of the catalysts. However, the crystallinity of H-BiOBr clearly decreased, because the hydrogenation operation can produce more defects to inhibit the crystal growth of BiOBr.35,36 The surface area of the particles and the pore size distribution were analyzed via nitrogen adsorption–desorption isotherms. Both of these nitrogen adsorption–desorption isotherms revealed a type IV classification, with conventional hysteresis loops in the relative pressure range of 0.4–1.0, which corresponded with mesoporous characteristics. The hydrogenated sample exhibiting the H3 hysteresis loop had a larger adsorbing capacity at the high pressure site than the untreated sample with the H4 hysteresis loop (Fig. 2b). The surface areas before and after hydrogenation were significantly reduced depending on nanocrystal growth through calcination in the BiOBr shell (the data on surface area and average pore diameter are presented in Table S1 in the ESI†). The chemical bonds and functional groups of the catalysts were tested by FT-IR spectra. Fig. S2† shows the peak for the conventional Bi-O functional group at 510 cm−1. In the FT-IR spectra, the relative intensity of H-BiOBr decreased because of the reduction in surface area, corresponding to active hydroxyl vibrational modes at 1600 and 3448 cm−1; it could lose the internal hydroxyl inside the BiOBr shell, leading to pore collapse through hydrogenation treatment. The results were consistent with the results of nitrogen adsorption–desorption isotherms.37
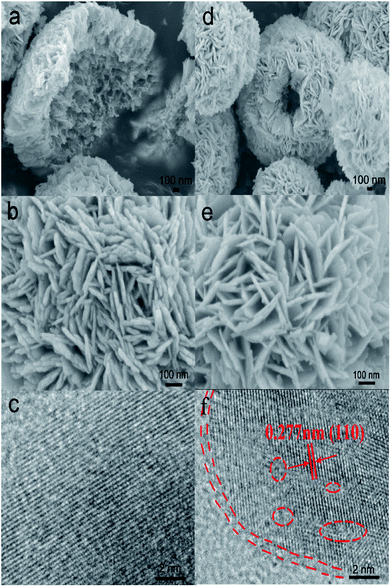 |
| | Fig. 1 SEM images of BiOBr at different magnifications (a and b) and H-BiOBr (d and e). The HRTEM image of BiOBr (c) and H-BiOBr (f); the edge location and lattice disorder via hydrogenation treatment are marked with red dashed lines and circles. | |
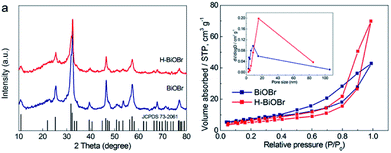 |
| | Fig. 2 XRD patterns of BiOBr and H-BiOBr (a). Nitrogen adsorption–desorption isotherms of BiOBr and H-BiOBr; inset: the corresponding pore-size distribution. | |
3.2 Chemical composition
The elemental compositions of the samples were verified by energy dispersive spectroscopy (EDS). It was easy to identify bromine, oxygen, and bismuth (Fig. S3†). The corresponding elemental information is shown in Table S2.† The ratio of the atomic numbers of Br, O, and Bi was about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, demonstrating the successful synthesis of the BiOBr catalyst. X-ray photoelectron spectroscopy (XPS) was further used to analyze the elemental composition, atom content, valence state, and surface energy state. The XPS survey spectra of the two samples revealed that the elemental compositions were in accordance with the results of the EDS images. Both of these samples exhibited no clear changes in their binding energies. The corresponding surface energy states and element valence states were studied (Fig. 3a). The high-resolution XPS spectrum of Bi 4f of the original sample revealed two main peaks corresponding to Bi 4f5/2 and Bi 4f7/2 at 164.5 and 159.2 eV, respectively, which illustrated the presence of the +3 oxidation state. However, in the hydrogenated sample, two main peaks appeared at 164.3 and 159 eV (Fig. 3b). The hydrogenated sample exhibited a shift toward lower binding energy, which illustrated the presence of low-charge bismuth ions in H-BiOBr. This conclusion verified that Bi3+ ions were reduced to low-charge Bi3−x ions (oxygen vacancies) through the simple hydrogenation method.38 The XPS spectrum of Br 3d further proved the influence of OVs (Fig. 3c). The corresponding peaks of the two samples appeared at 68.5 and 68.2 eV, which indicated the presence of Br−1. The high-resolution XPS spectrum of O 1s is shown in Fig. 3d. The peak at 530.3 eV for both samples can be assigned to the main Bi–O peak (lattice oxygen). This peak at 532 eV derived from the oxygen species chemisorbed (adsorbed oxygen) via OVs, appeared in the spectrum of H-BiOBr; this is a typical characteristic of hydrogenation, which results in the formation of stabilized OVs via adsorbed oxygen species.39–41
:1:1, demonstrating the successful synthesis of the BiOBr catalyst. X-ray photoelectron spectroscopy (XPS) was further used to analyze the elemental composition, atom content, valence state, and surface energy state. The XPS survey spectra of the two samples revealed that the elemental compositions were in accordance with the results of the EDS images. Both of these samples exhibited no clear changes in their binding energies. The corresponding surface energy states and element valence states were studied (Fig. 3a). The high-resolution XPS spectrum of Bi 4f of the original sample revealed two main peaks corresponding to Bi 4f5/2 and Bi 4f7/2 at 164.5 and 159.2 eV, respectively, which illustrated the presence of the +3 oxidation state. However, in the hydrogenated sample, two main peaks appeared at 164.3 and 159 eV (Fig. 3b). The hydrogenated sample exhibited a shift toward lower binding energy, which illustrated the presence of low-charge bismuth ions in H-BiOBr. This conclusion verified that Bi3+ ions were reduced to low-charge Bi3−x ions (oxygen vacancies) through the simple hydrogenation method.38 The XPS spectrum of Br 3d further proved the influence of OVs (Fig. 3c). The corresponding peaks of the two samples appeared at 68.5 and 68.2 eV, which indicated the presence of Br−1. The high-resolution XPS spectrum of O 1s is shown in Fig. 3d. The peak at 530.3 eV for both samples can be assigned to the main Bi–O peak (lattice oxygen). This peak at 532 eV derived from the oxygen species chemisorbed (adsorbed oxygen) via OVs, appeared in the spectrum of H-BiOBr; this is a typical characteristic of hydrogenation, which results in the formation of stabilized OVs via adsorbed oxygen species.39–41
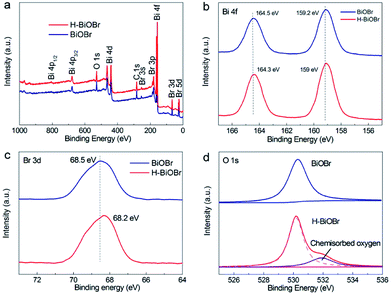 |
| | Fig. 3 XPS spectra of BiOBr and H-BiOBr. Survey (a), Bi 4f (b), Br 3d (c), and O 1s (d). | |
3.3 Broadening photoelectricity window
The UV/vis diffuse reflectance spectra of BiOBr and H-BiOBr are shown in Fig. 4a. Remarkably, H-BiOBr showed intense absorption in the visible light region compared to the original sample, indicating that the low-temperature hydrogen treatment enabled H-BiOBr to form a narrow band gap with enhanced light-harvesting performance. The corresponding Tauc plot is shown in Fig. 4b; the band gaps of BiOBr and H-BiOBr were about 2.88 and 2.76 eV, respectively. The energy band gap was evaluated via the following formula (eqn (4)):here, A, hv, Eg and α are the proportional constant, photon energy, energy gap and absorption index, respectively. A relatively narrow band gap can shorten the distance of the electron transition from the valence band to the conduction band, accelerating the separation of photogenerated electron–hole pairs, which reveals the superior sunlight-driven activities. Correspondingly, the conduction band (CB) and valence band (VB) potentials of the photocatalysts are important for photocatalytic activity. Mott–Schottky plots were adopted to indirectly calculate the CB potentials via flat band potentials (EFB). As shown in Fig. 4c, the positive slope of the Mott–Schottky plots indicated that both samples were n-type semiconductors. Then, the flat band potentials of BiOBr and H-BiOBr were obtained by extrapolating the tangent lines to 1/C2 = 0; they were found to be 0.12 V and 0.22 V, respectively (versus the saturated calomel electrode at pH = 7), which corresponded to 0.36 V and 0.46 V, respectively, versus the normal hydrogen electrode (NHE).42,43 According to previous reports, the flat band potential of an n-type semiconductor equals the Fermi level. Thus, the EF values of BiOBr and H-BiOBr were 0.36 V and 0.46 V, respectively.44–48 VB-XPS spectra were measured to establish the energy band structure of the semiconductors (Fig. 4d). The energy band structure of BiOBr was effectively modified via hydrogenation treatment. The data revealed that the spacings between EF and the valence band maximum (VBM) were 1.58 and 1.40 eV for BiOBr and H-BiOBr, respectively. The calculated VB potentials of the two samples were 1.94 and 1.86 V, respectively.49 The CB potentials of the two semiconductors were calculated on the basis of the equation ECB = Eg − EVB. It was worth noting that the distance from EF to CB was closer for the two semiconductors, implying that both samples were n-type semiconductors. These results were consistent with the results of Mott–Schottky plots. The energy band structure diagram is provided in Fig. 4e.50 It can be seen that the CB positions of the BiOBr samples were higher than E0 (N2/NH3 = −0.09 V vs. NHE). This indicated that BiOBr can reduce N2 to NH3 based on thermodynamics.51 Photoluminescence (PL) spectroscopy can further test the photon-generated carrier recombination rate of the catalysts (Fig. 4f). High PL intensity indicates high recombination rate of free charge carriers and low efficiency of charge carrier transfer and separation. The low recombination rate can enhance quantum yield and photocatalytic activities. H-BiOBr displayed lower fluorescence intensity at the 340 nm excitation wavelength compared to BiOBr; this was possibly due to vacancy defects, which resulted in the formation of an imperfect structure. Thereby, these defects captured electrons to reduce the recombination rate. For photoelectrochemical measurements, transient photocurrent (I–t curve) and electrochemical impedance spectroscopy (EIS) are effective methods to further investigate surface charge recombination. From Fig. 5a, we can deduce that the photocurrent density of H-BiOBr showed good quality relative to that of BiOBr under on/off cycles with simulated sunlight illumination. Thus, the hydrogenation method produced higher separation efficiency of holes and electrons compared to the results of primary samples. The photocurrent density of the sample was slightly reduced owing to slight light corrosion. Furthermore, EIS Nyquist plots are shown in Fig. 5b, which adopted Randles circuit as a fitting circuit. As depicted in the figure, the hydrogenated sample revealed a smaller arc radius relative to the primary sample. All these results indicated that the introduced OVs serve as donor densities, which greatly promotes the charge transportation and separation efficiency of the photogenerated carriers while suppressing the recombination of electron–hole pairs. To further verify the condition of OVs, both samples were analyzed via electron spin resonance (ESR) spectra (Fig. 5c). All the samples exhibited uniform ESR signal at g = 2.004, which is a typical signal for OVs.52–54 The signal intensity of H-BiOBr was higher than that of BiOBr, which could be ascribed to the numerous OVs. The relatively weak signal of BiOBr suggesting few OVs is due to the reducibility of EG. The A-BiOBr sample exhibited the weakest signal intensity than the other two samples (Fig. S5†). The corresponding photocatalytic nitrogen fixation performance was also the poorest (Fig. S6†). It can be inferred that the amount of oxygen vacancies is a key factor in improving photocatalytic nitrogen fixation performance.
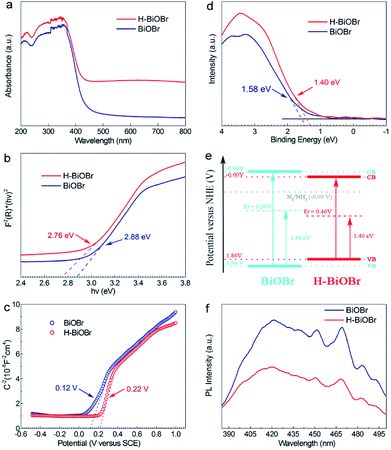 |
| | Fig. 4 UV-vis absorption spectra of BiOBr and H-BiOBr (a), the corresponding Tauc plot (b), Mott–Schottky plots of as-prepared two samples (c), valence band XPS spectra of BiOBr and H-BiOBr (d), proposed band structure schematic diagram (e), the PL spectra of BiOBr and H-BiOBr (f). | |
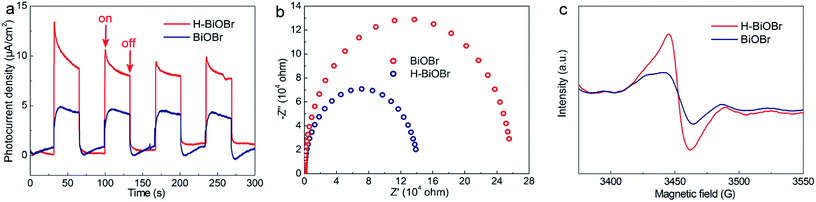 |
| | Fig. 5 Amperometric I–t curves for BiOBr and H-BiOBr under simulated sunlight illumination (a), EIS spectrum of two samples with no illumination (i.e., dark) (b), ESR spectra (c). | |
3.4 Photocatalytic performance and mechanism
The photocatalytic performance of nitrogen fixation of H-BiOBr was verified using only water as the solvent under simulated sunlight irradiation (Fig. 6a). Surprisingly, H-BiOBr exhibited strong nitrogen reduction ability in the above situation; it produced about 2.6 times more NH3 than BiOBr, which could be due to more photoinduced surface OVs via the low-temperature hydrogenation method. Compared to commercial P25 and carbonaceous materials such as g-C3N4 nanosheets, the H-BiOBr sample produced about 4.8 times and 3.6 times higher NH3, respectively, under the same condition (Fig. S7†). For comparison, photocatalytic nitrogen fixation tests were processed in argon gas. As expected, almost no NH3 was produced in the two samples. This further proved that the produced ammonia was converted through nitrogen fixation. Furthermore, we conducted dark nitrogen fixation experiments for the two kinds of catalysts (Fig. S8†). Clearly, under dark reactions, the catalysts exhibited poor NH3 production. It was determined that light irradiation could affect the activation of OVs on the catalyst surface. The apparent quantum efficiency (AQE) is a precise quantification for photocatalytic nitrogen fixation performance. The AQE values of the H-BiOBr photoharvester at different wavelengths along with the UV-vis DRS spectrum is shown in Fig. 6b. The AQE value decreased with the increase in the wavelength of monochromatic light, which was in accordance with the absorption edge of the UV-vis DRS spectrum. Under 380 nm monochromatic light irradiation, the AQE value was calculated to be about 2.1%. The performances of photocatalytic nitrogen fixation for other photocatalysts are exhibited in Table S3.† For controlling the reaction time, we conducted photocatalytic nitrogen fixation experiments via calcined BiOBr at different temperatures (Fig. 6c). The color of the sample changed from yellow-white (100 °C) to grey (200 °C), which was due to the loss of oxygen atoms via hydrogen reduction. The color of the sample became black when the temperature rose to 300 °C; the sample thus exhibited a new XRD pattern as its crystalline structure had changed (Fig. S9 and S10†). This new XRD pattern could be indexed to the presence of both Bi (JCPDS 44-1246) and BiOBr (JCPDS 73-2061) phases. This result was obtained due to the high temperature hydrogenation treatment, which caused more oxygen atoms to escape and the bismuth atoms to be exposed on the surface. Furthermore, high stability of photoreactivity is one of the important factors for catalysts. Hence, we verified the H-BiOBr nitrogen fixation performance via cycling experiments (Fig. 7a). After performing the cycling test four times, a slight decrease in photoreactivity for NH3 evolution was observed, which indicated that H-BiOBr possessed excellent photocatalytic stability for photocatalytic nitrogen fixation. The XRD pattern of H-BiOBr after four cycles is shown in Fig. 7b. It can be seen that the crystalline structure of the sample did not significantly change during the photocatalytic reaction process, which further confirmed the photocatalytic stability. Based on the as-prepared H-BiOBr, the photocatalytic nitrogen fixation model is exhibited in Fig. 7c, which only used water as the reactant under solar light. With the above discussion, we can infer that the OV-induced defect states might act as the initial charge carrier acceptors to inhibit photon-generated electron and hole recombination and promote interfacial charge transfer from the excited H-BiOBr microsphere to pre-activated N2 on OVs. These defects corresponded to the numerous active sites in the process of the reaction. The possible explanation shown in Fig. 7d demonstrates that these active sites were introduced as a sub-band in the band structure.55,56 The conventional excitation path of the electrons was considered to be from VB to CB (Path III); nevertheless, for H-BiOBr, the electrons were first excited from VB to the defect level (Path I) and then, the electrons were excited from the defect level to CB (Path II). This defect level was equivalent to a springboard in the band structure, which could capture more electrons to decrease the recombination rate for the photogenerated carriers and accelerate the transfer rate to CB. Moreover, the photogenerated electrons could be quickly transferred to the surface of the catalysts, or they were captured via vacancies in a proper band gap. This triggered photocatalytic nitrogen fixation via effective transfer from the semiconductor to combine nitrogen.
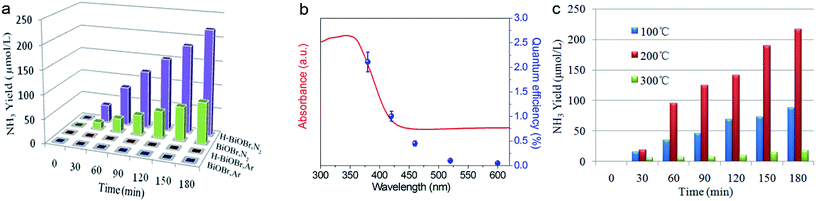 |
| | Fig. 6 Quantitative determination of photocatalytic NH3 generated under simulated sunlight illumination. Light source: 300 W Xe lamp; catalyst: 20 mg; solution: deionized water 100 ml (a), wavelength-dependence of the AQE of H-BiOBr under different monochromatic light irradiations (b), quantitative determination of photocatalytic NH3 generated at different temperatures (c). | |
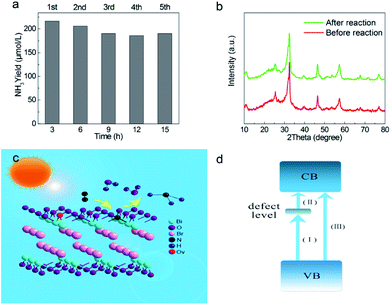 |
| | Fig. 7 Multicycle photocatalytic NH3 generation under simulated sunlight illumination (a), XRD pattern of H-BiOBr before and after multicycle (b), proposed photocatalytic nitrogen fixation mechanism (c), illustration of defect engineering possible electrons excitation pathways in band structure (d). | |
4. Conclusions
In summary, we have successfully synthesized H-BiOBr hollow microspheres via a low-temperature hydrogen annealing method. We obtained remarkable photocatalytic nitrogen fixation performance under sunlight without using any organic scavengers and noble metal co-catalysts. H-BiOBr microspheres produced about 2.6 times more NH3 than the unmodified sample. Furthermore, the corresponding AQE value reached a satisfactory value of 2.1% under 380 nm monochromatic light irradiation. This simple and innovative approach could generate and control surface defects. The oxygen vacancies in this disorder engineering revealed a superior effect in many aspects including proper band gap, intense absorbance, excellent electron capture ability, effective interfacial electron transfer, and reduced recombination rate of electrons–hole pairs. Thus, this study clearly demonstrated that hydrogenation treatment might serve as a versatile approach to modify catalysts, which is applicable to the development of low-cost and green photocatalysts. The mechanism of photocatalytic nitrogen fixation emphasized the advantage of defect engineering in material chemistry. Finally, we believe that this study represents a new step in the manufacture of innovative materials for photocatalytic nitrogen fixation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The research was supported by the National Natural Science Foundation of China (Grant no. 21476033).
Notes and references
- R. H. Burris, Annu. Rev. Plant Physiol., 1966, 17, 155–184 CrossRef
 .
. - W. F. Hardy and R. C. Burns, Annu. Rev. Biochem., 1968, 37, 331–358 CrossRef .
- R. Dixon and D. Kahn, Nat. Rev. Microbiol., 2004, 2, 621 CrossRef PubMed .
- J. Kim and D. C. Rees, Biochemistry, 1994, 33, 389–397 CrossRef PubMed .
- T. A. Bazhenova and A. E. Shilov, Coord. Chem. Rev., 1995, 144, 69–145 CrossRef .
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef .
- X. Chen, N. Li, Z. Kong, W.-J. Ong and X. Zhao, Mater. Horiz., 2018, 5, 9–27 RSC .
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef .
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836 CrossRef PubMed .
- Y. Zhao, Y. Zhao, G. I. N. Waterhouse, L. Zheng, X. Cao, F. Teng, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Mater., 2017, 29, 1703828 CrossRef PubMed .
- S. Sun, Q. An, W. Wang, L. Zhang, J. Liu and W. A. Goddard III, J. Mater. Chem. A, 2017, 5, 201–209 Search PubMed .
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef PubMed .
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef PubMed .
- S. Wang, X. Hai, X. Ding, K. Chang, Y. Xiang, X. Meng, Z. Yang, H. Chen and J. Ye, Adv. Mater., 2017, 29, 1701774 CrossRef PubMed .
- G. Dong, W. Ho and C. Wang, J. Mater. Chem. A, 2015, 3, 23435–23441 Search PubMed .
- S. Cao, H. Chen, F. Jiang and X. Wang, Appl. Catal., B, 2018, 224, 222–229 CrossRef .
- Q. Zhang, S. Hu, Z. Fan, D. Liu, Y. Zhao, H. Ma and F. Li, Dalton Trans., 2016, 45, 3497–3505 RSC .
- W. Zhao, J. Zhang, X. Zhu, M. Zhang, J. Tang, M. Tan and Y. Wang, Appl. Catal., B, 2014, 144, 468–477 CrossRef .
- K. Hoshino, R. Kuchii and T. Ogawa, Appl. Catal., B, 2008, 79, 81–88 CrossRef .
- S. Sun, X. Li, W. Wang, L. Zhang and X. Sun, Appl. Catal., B, 2017, 200, 323–329 CrossRef .
- X. Li, W. Wang, D. Jiang, S. Sun, L. Zhang and X. Sun, Chem.–Eur. J., 2016, 22, 13819–13822 CrossRef PubMed .
- J. Zhang, F. Shi, J. Lin, D. Chen, J. Gao, Z. Huang, X. Ding and C. Tang, Chem. Mater., 2008, 20, 2937–2941 CrossRef .
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef PubMed .
- Y. Huo, J. Zhang, M. Miao and Y. Jin, Appl. Catal., B, 2012, 111–112, 334–341 CrossRef .
- D. Wu, L. Ye, H. Y. Yip and P. K. Wong, Catal. Sci. Technol., 2017, 7, 265–271 Search PubMed .
- J. Xia, S. Yin, H. Li, H. Xu, L. Xu and Y. Xu, Dalton Trans., 2011, 40, 5249–5258 RSC .
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2018, 140, 526 CrossRef PubMed .
- R. H. Leonard, Clin. Chem., 1963, 9, 417–422 Search PubMed .
- M. Shang, W. Wang and H. Xu, Cryst. Growth Des., 2009, 9, 991–996 Search PubMed .
- L. Zhang, T. Xu, X. Zhao and Y. Zhu, Appl. Catal., B, 2010, 98, 138–146 CrossRef .
- X. Haolan, W. Wenzhong, Z. Wei and Z. Lin, Nanotechnology, 2006, 17, 3649 CrossRef .
- G. Xi, K. Xiong, Q. Zhao, R. Zhang, H. Zhang and Y. Qian, Cryst. Growth Des., 2006, 6, 577–582 Search PubMed .
- L. Zhang, W. Wang, Z. Chen, L. Zhou, H. Xu and W. Zhu, J. Mater. Chem., 2007, 17, 2526–2532 RSC .
- D. Wu, B. Wang, W. Wang, T. An, G. Li, T. W. Ng, H. Y. Yip, C. Xiong, H. K. Lee and P. K. Wong, J. Mater. Chem. A, 2015, 3, 15148–15155 Search PubMed .
- T.-D. Nguyen-Phan, S. Luo, Z. Liu, A. D. Gamalski, J. Tao, W. Xu, E. A. Stach, D. E. Polyansky, S. D. Senanayake, E. Fujita and J. A. Rodriguez, Chem. Mater., 2015, 27, 6282–6296 CrossRef .
- L. Han, Z. Ma, Z. Luo, G. Liu, J. Ma and X. An, RSC Adv., 2016, 6, 6643–6650 RSC .
- T. Leshuk, R. Parviz, P. Everett, H. Krishnakumar, R. A. Varin and F. Gu, ACS Appl. Mater. Interfaces, 2013, 5, 1892–1895 Search PubMed .
- X. Y. Kong, W. P. C. Lee, W.-J. Ong, S.-P. Chai and A. R. Mohamed, ChemCatChem, 2016, 8, 3074–3081 CrossRef .
- C. Shi, X. Dong, J. Wang, X. Wang, H. Ma and X. Zhang, RSC Adv., 2017, 7, 26717–26721 RSC .
- K. Zhao, L. Zhang, J. Wang, Q. Li, W. He and J. J. Yin, J. Am. Chem. Soc., 2013, 135, 15750–15753 CrossRef PubMed .
- N. Zhang, X. Li, H. Ye, S. Chen, H. Ju, D. Liu, Y. Lin, W. Ye, C. Wang, Q. Xu, J. Zhu, L. Song, J. Jiang and Y. Xiong, J. Am. Chem. Soc., 2016, 138, 8928–8935 CrossRef PubMed .
- S. J. Hong, S. Lee, J. S. Jang and J. S. Lee, Energy Environ. Sci., 2011, 4, 1781–1787 Search PubMed .
- K. Sayama, A. Nomura, T. Arai, T. Sugita, R. Abe, M. Yanagida, T. Oi, Y. Iwasaki, Y. Abe and H. Sugihara, J. Phys. Chem. B, 2006, 110, 11352–11360 CrossRef PubMed .
- D. E. Scaife, Sol. Energy, 1980, 25, 41–54 CrossRef .
- E. Gao, W. Wang, M. Shang and J. Xu, Phys. Chem. Chem. Phys., 2011, 13, 2887–2893 RSC .
- X. Wu, Y. H. Ng, L. Wang, Y. Du, S. X. Dou, R. Amal and J. Scott, J. Mater. Chem. A, 2017, 5, 8117–8124 Search PubMed .
- X. Tu, S. Luo, G. Chen and J. Li, Chem. - Eur. J., 2012, 18, 14359–14366 CrossRef PubMed .
- H. Li, H. Yu, X. Quan, S. Chen and Y. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 2111–2119 Search PubMed .
- H. Li, T. Hu, N. Du, R. Zhang, J. Liu and W. Hou, Appl. Catal., B, 2016, 187, 342–349 CrossRef .
- Y. Bai, L. Ye, T. Chen, L. Wang, X. Shi, X. Zhang and D. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 27661–27668 Search PubMed .
- H. Katsuyoshi, Chem.–Eur. J., 2001, 7, 2727–2731 CrossRef .
- F. Lei, Y. Sun, K. Liu, S. Gao, L. Liang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2014, 136, 6826–6829 CrossRef PubMed .
- Y. Hao, X. Dong, X. Wang, H. Ma and X. Zhang, J. Mater. Chem. A, 2017, 5, 5426–5435 Search PubMed .
- Y. Hao, X. Dong, S. Zhai, H. Ma, X. Wang and X. Zhang, Chem.–Eur. J., 2016, 22, 18722–18728 CrossRef PubMed .
- X. Pan, M.-Q. Yang, X. Fu, N. Zhang and Y.-J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC .
- X. Y. Kong, Y. Y. Choo, S.-P. Chai, A. K. Soh and A. R. Mohamed, Chem. Commun., 2016, 52, 14242–14245 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02483a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Nan Zheng,
Hongchao Ma and
Xiufang Zhang
*,
Nan Zheng,
Hongchao Ma and
Xiufang Zhang







