
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA facile and general approach for the direct fabrication of N-rGO–metal(metal oxides)–Pt composites as electrocatalyst for oxygen reduction reactions†
Xiaohong Chen a,
Zhaoyang Ningc,
Zongshang Zhoub,
Xundao Liua,
Jianlong Leia,
Supeng Pei*ab and
Yongming Zhang*a
a,
Zhaoyang Ningc,
Zongshang Zhoub,
Xundao Liua,
Jianlong Leia,
Supeng Pei*ab and
Yongming Zhang*a
aSchool of Chemistry and Chemical Engineering, Shanghai Key Lab of Electrical Insulation and Thermal Aging, Shanghai Electrochemical Energy Devices Research Center, Shanghai Jiao Tong University, No. 800 Dongchuan Rd., Minhang District, Shanghai 200240, China. E-mail: peisupeng@126.com; ymzsjtu@gmail.com
bSchool of Chemical and Environmental Engineering, Shanghai Institute of Technology, Shanghai 201418, China
cShandong Fuyuan Energy Conservation and Environmental Protection Engineering Co., Ltd., Jinan, 250000, China
First published on 31st July 2018
Abstract
Nitrogen-doped reduced graphene oxide–metal(metal oxides) nanoparticle (N-rGO–M(MO) NPs, M = Fe, MO: M = Co, Mn) composites were prepared through a facile and general method at high temperature (800 °C). M(MO) were well-dispersed and tightly anchored on graphene sheets, which were doped with nitrogen simultaneously and further loaded with Pt nanoparticles. Those results showed a more positive onset potential, higher cathodic density, and higher electron transfer number for the ORR in alkaline media. Furthermore, N-rGO–metal(metal oxides)–Pt (N-rGO–M(MO)–Pt) nanoparticles show better durability than the commercial Pt/C catalyst, and can be used as promising potential materials in practical applications.
1 Introduction
The electrocatalyst for the oxygen reduction reaction (ORR) has recently attracted a tremendous amount of attention, due to the significance in various renewable energy applications, such as fuel cells and metal–air batteries.1–8 Recently, ORR catalyzed by Pt-based catalysts has demonstrated a high onset potential and fast kinetics and a four-electron transfer process.9–14 Meanwhile, heterogeneous atom doping could also potentially produce localized highly active regions of graphene, while the electric conductivity of the functionalized graphene is improved.15–23According to the previous reports,24–27 N-rGO–M(MO) composites were synthesized by different methods, such as precursor pyrolysis, the hydrothermal method, the template method and others. Shihe Yang reported cobalt-embedded nitrogen doped carbon nanotubes for OER/ORR bifunctional catalysts, designed and prepared using CoPt as the Co and nitrogen sources, which is particularly suitable for synthesizing novel catalysts.28 Hongjie Dai reported a Co3O4/N-doped graphene composite by a two-step method as one of the rare and highest performance bi-functional catalysts for ORR and water oxidation/OER.29 Fu et al. established a soft template F-127 and hard template tellurium nanowires to produce a one-dimensional hierarchical porous PtCu alloy nanostructure with large mesopores, which exhibits enhanced electrocatalytic performance for the ORR in acid media.30 Therefore, a more efficient and convenient procedure would be desirable for improving efficiency. It was believed that combining platinum, transition metal oxides and conductive nanocarbon materials such as graphene and carbon nanotubes is an effective approach for enhancing the catalytic activity of oxides. This is attributed to an increased number of available active sites and providing efficient charge transport channels.
Herein, we reported a facile, yet general approach to prepare N-rGO–M(MO) nanoparticles, which was coreduced from metal salt and melamine at a high temperature (800 °C). Those results indicated that the M(MO) NPs were strongly bound to uniformly distributed on the N-rGO, while Pt nanoparticles could be easily loaded on the N-rGO–M(MO) materials (Scheme 1). Remarkably, the N-rGO–M(MO)–Pt composites showed a much more positive ORR onset potential (66, 140 and 253 mV) than that of 20% Pt/C, suggesting synergistic ORR catalytic activity of Pt, M(MO) and N-rGO in the composites. Therefore, the half-wave potential, Jk and durability of N-rGO–M(MO)–Pt composites are demonstrated.
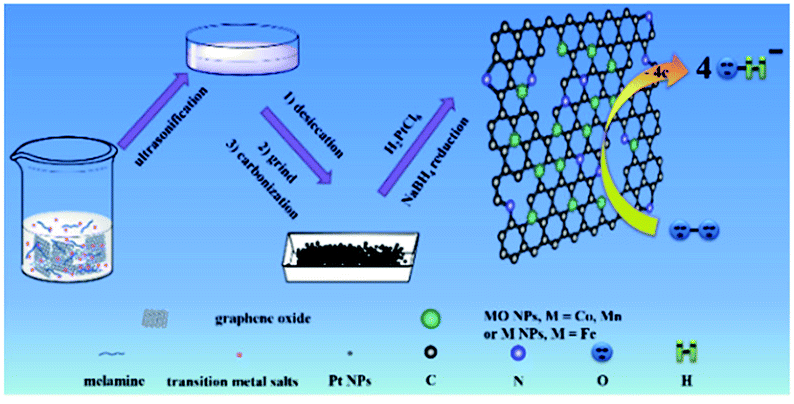 | ||
| Scheme 1 Fabrication procedure for N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt composites. | ||
2 Experimental
2.1 Synthesis of nitrogen-doped reduced graphene (N-rGO), N-rGO–Co/CoO, N-rGO–Fe and N-rGO–Mn oxides
The main text of the article should appear here with headings as appropriate. We used high-purity argon as protective ambient to anneal a mixture of GO, melamine and Co(Ac)2. Detailed procedure is as follows: GO powder, melamine and Co(Ac)2 were ground together in an agate mortar for about 5 min, and the mixture was then placed in the center of a quartz tube with a flow of argon. Before the furnace was heated to 800 °C, Ar gas was flowed for about 20 min. When the center of the furnace reached the designed reaction temperature (e.g., 800 °C), the mixture was annealed for 1 h at this temperature. After that, the sample was cooled to room temperature under Ar ambient. Finally, the products were taken out of the quartz tube. N-rGO, N-rGO–Co/CoO, N-rGO–Fe and N-rGO–Mn oxides was synthesized from GO using a similar procedure.2.2 Synthesis of N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt composites
20 mL of N-rGO–Co/CoO (1 mg mL−1) aqueous dispersion was prepared by ultrasonification, into which H2PtCl6 aqueous solution (9.2 mL, 4 mg mL−1) was dropped under nitrogen. Then 100 mL of NaBH4 (2 mg mL−1) was added dropwise. After stirring for 2 h and standing overnight, the resulting N-rGO–Co/CoO–Pt catalyst was filtrated and washed with DIW several times. The resulting product was dried in vacuum at 40 °C for 24 h. For comparison, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt was prepared according to the similar procedures. Note: the Pt loading of the catalysts is 10 wt% unless specified.2.3 Preparation of Pt/C, N-rGO–Co/CoO, N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt modified electrode
Prior to use, glassy carbon electrode (GCE, φ = 3 mm) was polished with 0.05 μm gamma alumina powders, then rinsed thoroughly with ethanol and water in an ultrasonic bath to remove any alumina residues, and finally dried at room temperature. 4 μL of 1 mg mL−1 sonicated 20% commercial Pt–C, N-rGO–Co/CoO, N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt with ethanol and water suspension was dropped on the pretreated bare GCE or GC disk using a micropipet tip and dried in air.2.4 Cyclic voltammograms (CV) measurement
A conventional cell with a three-electrode configuration was served throughout this work. CV measurement was conducted at 25 °C using an Autolab PGSTAT302 (Metrohm) electrochemical test system using saturated Ag/AgCl as the reference electrode, a Pt wire as the counter electrode and the sample modified glassy carbon electrode as the working electrode. 0.1 M KOH aqueous solution was used as the electrolyte, which was saturated with O2 by bubbling it prior to the start of each experiment. A flow of O2 was maintained over the electrolyte during the recording of CVs in order to ensure continuous O2 saturation. The working electrode was cycled at least 5 times before data recorded at a scan rate of 10 mV s−1. In control experiments, CV measurements were also performed under N2 atmosphere. In all electrochemical measurements, 0.1 M KOH aqueous solution saturated with nitrogen or oxygen was used as the electrolyte.2.5 Rotating disk electrode (RDE) measurement
For the RDE measurement, catalyst inks were prepared by the same method as that of CV analysis described above. 4 μL of 5 mg mL−1 ink was loaded on a glassy carbon RDE (φ = 3 mm). The working electrode was cathodically scanned at a rate of 10 mV s−1 with varying rotating speed from 400 to 2000 rpm. The ORR current was determined by subtracting the N2 current from the O2 current. Koutecky–Levich plots (J−1 vs. ω−1/2) were analyzed at various electrode potentials. The slopes of their best linear fit lines were used to calculate the number of electrons transferred (n) on the basis of the Koutecky–Levich equation: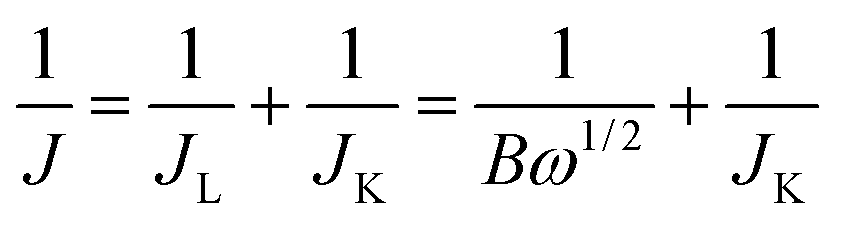
| B = 0.62nFC0(D0)2/3v−1/6Jk = nFkC0 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−3 mol cm−3), D0 is the diffusion coefficient of O2 (1.9 × 10−5), v is the kinematic viscosity of the electrolyte (0.01 m2 s−1), and k is the electron-transfer rate constant.
485 C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−3 mol cm−3), D0 is the diffusion coefficient of O2 (1.9 × 10−5), v is the kinematic viscosity of the electrolyte (0.01 m2 s−1), and k is the electron-transfer rate constant.
2.6 Materials and methods
The main text of the article should appear here with headings as appropriate. Melamine, Co(Ac)2, FeCl3 and MnCl2 were bought from Sinopharm Chemical Reagent CO. Ltd. (China). Other chemicals such as ethanol, KOH and N,N-dimethylformamide (DMF) bought from Shanghai Lingfeng Chemical Reagent CO. Ltd., were all of analytical grade and used as received. All solutions used in the electrochemical experiments were freshly prepared with Millipore water (resistivity ∼ 18.2 MΩ). The morphology of N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt was characterized by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) using JEM-2010HT and JEOL2100F (both from Electron Optics Laboratory Co., Ltd., Japan), respectively. Powder X-ray diffraction (XRD) analyses were performed on a diffractometer (Bruker, German, APLX-DUO) with Cu Kα radiation. Raman spectra were recorded using a Thermo Fisher H31XYZE-US with an excitation wavelength of 532 nm. X-ray photoelectron spectroscopy (XPS, Kα) analyses were carried out on an AXIS UltraDLD X-ray photoelectron spectrometer system equipped with Al radiation as a probe, and the analysis spot size was 400 μm in diameter.All electrochemical measurements were performed on an Autolab PGSTAT302N (Metrohm, Switzerland) electrochemical test system. Cyclic voltammograms were collected in a three-electrode system (GCE or modified GCE as the working electrode, a Pt wire as counter electrode, and an Ag/AgCl as the reference) at room temperature. For RDE measurements, a glassy carbon rotating disk electrode was used as the working electrode. Linear sweep voltammetry was performed at the GC disk electrode. In all of the electrochemical measurements, 0.1 M KOH aqueous solution saturated with nitrogen or oxygen was used as the electrolyte.
3 Results and discussion
The composition and crystal structure of N-rGO–Co/CoO, N-rGO–Co/CoO–Pt, N-rGO–Fe, N-rGO–Fe–Pt, N-rGO–Mn oxides and N-rGO–Mn oxides–Pt composite nanofibers were firstly characterized by XRD and Raman (Fig. 1 and 2). The XRD pattern confirmed the formation of Co (JCPDS no. 15-0806) and CoO (JCPDS no. 65-5474) and N-rGO in the composites (Fig. 1A). The C (002) diffraction peak in N-rGO–Co/CoO seems to be broader and faint, indicating the relatively less dense packing of graphene sheets due to N-induced structural defects. Meanwhile, the XRD pattern confirmed the formation of Fe5C2 (JCPDS no. 35-0772), MnOOH (JPCDS no. 41-1379) and Mn2O3 (JPCDS no. 24-0508) in the composites (Fig. 1C and E). The characteristic diffraction peaks indicated that Fe nanoparticles and Mn oxides were efficiently deposited on the graphene surface.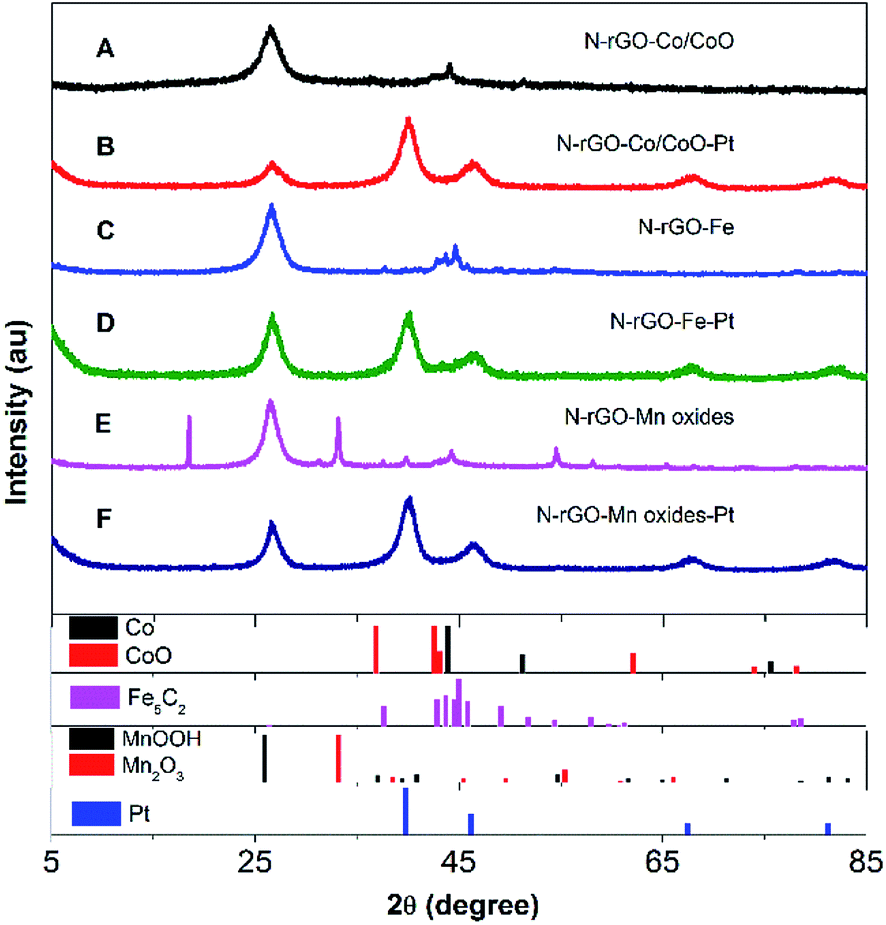 | ||
| Fig. 1 XRD patterns of (A) N-rGO–Co/CoO, (B) N-rGO–Co/CoO/Pt, (C) N-rGO–Fe, (D) N-rGO–Fe–Pt, (E) N-rGO–Mn oxides and (F) N-rGO–Mn oxides–Pt. | ||
 | ||
| Fig. 2 TEM images of (A) N-rGO-Co/CoO–Pt, (B) N-rGO–Fe–Pt and (C) N-rGO–Mn oxides–Pt and EDS spectra of (D) N-rGO–Co/CoO–Pt, (E) N-rGO–Fe–Pt and (F) N-rGO–Mn oxides–Pt. | ||
Furthermore, after Pt deposition on the three composites, XRD patterns of the composites showed strong diffraction peaks at 2θ = 40.2°, 46.3°, 67.8° and 81.2°, which are assignable to typical face-centered-cubic (111), (200), (220) and (311) crystalline planes of Pt, respectively (Fig. 1B, D and E). This fact indicates the absence of cobalt ions, iron oxide and manganese oxides diffractions was most likely due to the low crystallinity of the transition metal oxides and the heavy atom effect from platinum. Good dispersion, few aggregates and proper size of Pt NPs on N-rGO–Co/CoO, N-rGO–Fe and N-rGO–Mn oxides would bestow them with outstanding catalytic performance.
To gain more morphology information about N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt, the TEM images and EDS spectra were characterized (Fig. 2). Fig. 2A shows spherical Co/CoO–Pt NPs are uniformly decorated on N-rGO surfaces, and they are fairly monodispersed, with an average diameter of 4–6 nm. The relatively even distribution and size of Pt nanoparticles may be ascribed to a large number of Co/CoO NPs and defects of N-rGO, which not only provide many anchor sites for the uniform deposition of Pt nanoparticles but also enhance the interaction among Pt nanoparticles, Co/CoO NPs and N-rGO by coordination interaction as discussed above,25 indicating N, Co, O and Pt components, which is further confirmed by EDS and HRTEM measurements (Fig. 2D and S1, ESI†). Besides, the phenomena of N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt are the same as that of N-rGO–Co/CoO–Pt. The structure has shown good electrical conductivities due to the electron hopping between different valence states of metals in O-sites and should also provide necessary surface redox active metal centers for O2 adsorption and activation.
Raman spectroscopy is a useful tool for the characterization of carbon structure of N-rGO–Co/CoO, N-rGO–Fe and N-rGO–Mn oxides materials. As depicted in Fig. 3, the two prominent peaks at 1355 and 1585 cm−1 are the characteristic peaks of D and G bands from N-rGO. The intensity ratio (r = ID/IG) of the well-documented D band and G band of N-rGO–Co/CoO (2.98), N-rGO–Fe (3.66) and N-rGO–Mn oxides (4.40) was enhanced, compared with that of N-rGO (r = 1.27), (i) defects due to nitrogen doping onto the conjugated carbon framework and (ii) disorder of rGO layers. The G band of N-rGO–Co/CoO, N-rGO–Fe and N-rGO–Mn oxides shifted to 1590 cm−1, due to nitrogen doped or strain effect. Furthermore, compared to the N-rGO, the peaks can be found for both D and G bands, hinting a significant charge transfer between N-rGO sheets and M(MO) nanoparticles, which is helpful for improving their electrochemical performance. In the case of N-rGO–Co/CoO, N-rGO–Fe and N-rGO–Mn oxides, a 4–6 cm−1 red shift of both D and G bands was observed. It is generally accepted that the relative intensity ratio of these bands is a measure of the disorder and extent of defects in the graphitic materials, which is favorable for the further electrochemical processes.
 | ||
| Fig. 3 Raman spectra of N-rGO–Co/CoO, N-rGO–Fe and N-rGO–Mn oxides. | ||
In order to obtain more information of the nitrogen, cobalt and oxygen functionalities after codoping processes, N-rGO–Co/CoO–Pt composites were analyzed by XPS in the region of 0–1300 eV (Fig. 4A). The N 1s spectra of N-rGO–Co/CoO–Pt can be fitted into three peaks at 398.9, 399.9, and 401.4 eV (Fig. 4B, Table 1). The peaks with lower binding energy located at about 398.9 and 399.9 eV, respectively, correspond to pyridine-like and pyrrole-like nitrogen, which can contribute to the π-conjugated system with a pair of p-electrons in the graphene layers. When carbon atoms within the graphene layers are substituted by nitrogen atoms in the form of “graphitic” nitrogen, the corresponding peak in the high-resolution N 1s spectra is located at 401.4 eV. The results demonstrate the quantity of nitrogen precursor melamine and high temperature were the key factor to modulate the chemical state of nitrogen in doped graphene. The Co 2p XPS spectra of the composite exhibit two peaks at 779.8 and 795.4 eV, corresponding to the Co 2p1/2 and Co 2p3/2 spin orbit peaks of Co/CoO (Fig. 4C). Co and CoO spectra are presented in Fig. 4 with spectral fitting parameters given in Table 1, which suggests the fractions of 38.6% and 61.4% for Co and CoO, respectively. Additionally, the higher binding energy 2p3/2 multiplet or satellite structures of the CoO with the Co 2p1/2 peak at 795.4 eV maybe effectively cause a slight overestimation of the metal, compared to the oxide component. While the peak at 531.7 eV is corresponded to the oxygen species in the CoO phase, that at 533.0 eV are assignable to the residual oxygen containing groups (such as OH and COOH) on the surface of graphene (Fig. 4D). The XPS spectra of N-rGO–Mn oxides–Pt also confirmed the nitrogen, manganese and oxygen functionalities (Fig. S2, ESI†). The XPS spectra of N-rGO–Fe–Pt were not characterized, due to the magnetic properties of Fe.
 | ||
| Fig. 4 XPS spectra of the (A) survey scan, (B) N 1s region, (C) Co 2p region and (D) O 1s region of the N-rGO–Co/CoO–Pt. | ||
| Compound | Peak 1 (eV) | % | Peak 2 (eV) | % | Peak 3 (eV) | % | Peak 4 (eV) | % |
|---|---|---|---|---|---|---|---|---|
| Co | 778.2 | 38.5 | 781.1 | 38.5 | 783.1 | 23.0 | — | — |
| CoO | 779.8 | 70.0 | 782.1 | 11.3 | 785.5 | 9.0 | 786.5 | 9.6 |
To assess the catalytic activity of N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt catalysts for ORR, CV and measurements on a RDE were conducted. Apparently, N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt exhibited a pronounced electrocatalytic ORR activity associated with a more positive ORR onset potential (−0.15 V, −0.17 V and −0.13 V relative to Ag/AgCl), similar to the 20% Pt/C (−0.12 V) (Fig. 5A), suggesting synergistic ORR catalytic activity of N-rGO, M(MO) and Pt nanoparticles in the composites.31,32
 | ||
| Fig. 5 (A) CV curves of the N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt on glassy carbon electrodes in O2-saturated (solid line) and N2-saturated (dot line) 0.1 M KOH with scan rate of 10 mV s−1. (B) Linear sweep voltammetry (LSV) for the N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt, N-rGO–Mn oxides–Pt and 20% Pt/C in O2-saturated 0.1 M KOH with a sweep rate of 10 mV s−1 at 1600 rpm. (C–E) LSV curves of N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt in O2-saturated electrolytes at different rotation rates (in rpm), the inset shows the corresponding Koutecky–Levich plot of N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt derived from the RDE curves at −0.4–0.8 V with the electron transport number (n). | ||
To examine the reaction kinetics for N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt, linear sweep voltammograms (LSVs) were further recorded in an O2-saturated 0.1 M KOH electrolyte at a scan rate of 10 mV s−1 using a rotating disk electrode (RDE) (Fig. 5B). Among these prepared freestanding catalysts, N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt showed an enhanced ORR performance (Table 2) with more positive onset potential (−0.014, −0.040 and 0.073 V) and large current density (Jk = 8.00, 1.49 and 9.11 mA cm−2, respectively). The half-wave potential at 1600 rpm was −0.17, −0.39 and −0.22 V, respectively, almost more positive than of Pt/C (−0.27 V). This is contributed to the synergistic effects of three components and the interpenetrated network structure in combination Pt, M(MO) nanoparticles with N-rGO, effectively increasing basal spacing and electrical conductivity. Furthermore, RDE curves of N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt at various rotation speeds were measured to determine its ORR kinetic performance in 0.1 M KOH (Fig. 5C–E).33 The linearity and parallelism of the K–L plots suggested reaction kinetics toward the concentration of dissolved oxygen and similar electron transfer numbers for ORR at different potentials (Fig. 5C–E inset). The electron transfer numbers (n) of N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt were calculated to be 3.83, 3.98 and 4, respectively, at −0.4 V from the slopes of Koutecky–Levich plots, indicated those materials can favour ∼4e oxygen reduction process, similar to that of 20% Pt/C catalyst (n ∼ 4 for Pt/C).34 The electrochemical impedance spectra of the electrode prepared with N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt, N-rGO–Mn oxides–Pt and 20% Pt/C was shown in Fig. S3.† The low resistance of N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt, N-rGO–Mn oxides–Pt and 20% Pt/C (Table S1, ESI†) indicates its better charge conductivity, which may arise from heteroatom-doping induced charge transfer in carbon and the high electrical conductivity of those metal materials.
| On set potential (V) | Half-wave potential (V) | Jk (mA cm−2) | n | |
|---|---|---|---|---|
| N-rGo–Co/CoO–Pt | −0.014 | −0.17 | 8.00 | 3.83 |
| N-rGo–Fe–Pt | −0.040 | −0.39 | 1.49 | 3.98 |
| N-rGo–Mn oxides–Pt | 0.073 | −0.22 | 9.11 | 4 |
| 20% Pt/C | 0.030 | −0.27 | 6.4 | 4 |
In addition to, the long-term stability of catalysts is one of the major concerns in microbial fuel cell technology. The stability of N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt, N-rGO–Mn oxides–Pt and Pt/C catalysts was tested at a constant voltage of −0.35 V (vs. Ag/AgCl) for 10000 s in O2-saturated 0.1 M KOH (Fig. 6). The chronoamperometric response of N-rGO–Co/CoO–Pt and N-rGO–Fe–Pt under alkaline conditions, retained a higher relative current of ∼82.74% and 84.08%, respectively, compared to 20% Pt/C (81.23%). The better durability of N-rGO–Co/CoO–Pt and N-rGO–Fe–Pt can be ascribed to the unique confined structure of Co/CoO and Fe NPs within the graphene layers, which can enhance their interfacial contact, suppress the dissolution/agglomeration of Co/CoO and FeNPs, and facilitate the transport of electrolyte ions. However, the chronoamperometric response of N-rGO–Mn oxides–Pt retained a lower relative current of ∼72.43% which could be attributed to the instability of the Mn oxides in alkaline solution.
 | ||
| Fig. 6 Chronoamperometric responses of N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt, N-rGO–Mn oxides–Pt and 20% commercial Pt/C modified GC electrodes at −0.35 V with the speed at 1600 rpm in O2-saturated 0.1 M KOH. | ||
4 Conclusions
We developed a simple high-temperature method to synthesize a N-rGO–M(MO) composite on N-doped reduced graphene oxide, which was further loaded with Pt nanoparticles. Those results indicated that N-rGO–Co/CoO–Pt, N-rGO–Fe–Pt and N-rGO–Mn oxides–Pt showed excellent electrocatalytic activity for the ORR in alkaline electrolytes, such as a higher current density, higher electron transfer number (∼4), and better durability. This resulted from synergistic effects of three components and the interpenetrated network structure in combinations of Pt, M(MO) nanoparticles with N-rGO. Therefore, the synthetic strategy of composites may be further extended to develop other metal or metal oxides/graphene-based monolithic materials for various applications, such as sensors, batteries, and supercapacitors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge National Natural Science Foundation of China (Project No. 21203118).Notes and references
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- K. Kamiya, R. Kamai, K. Hashimoto and S. Nakanishi, Nat. Commun., 2014, 5, 5040 CrossRef PubMed.
- H. Zeng, W. Wang, J. Li, J. Luo and S. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 8721–8729 CrossRef PubMed.
- T. Li, Y. Lu, S. Zhao, Z.-D. Gao and Y.-Y. Song, J. Mater. Chem. A, 2018, 6, 3730–3737 RSC.
- W. Wang, L. Kuai, W. Cao, M. Huttula, S. Ollikkala, T. Ahopelto, A.-P. Honkanen, S. Huotari, M. Yu and B. Geng, Angew. Chem., Int. Ed., 2017, 56, 14977–14981 CrossRef PubMed.
- S. Zhang, Y. Zhang, W. Jiang, X. Liu, S. Xu, R. Huo, F. Zhang and J.-S. Hu, Carbon, 2016, 107, 162–170 CrossRef.
- R. Huo, W.-J. Jiang, S. Xu, F. Zhang and J.-S. Hu, Nanoscale, 2014, 6, 203–206 RSC.
- M. Luo, Y. Sun, X. Zhang, Y. Qin, M. Li, Y. Li, C. Li, Y. Yang, L. Wang, P. Gao, G. Lu and S. Guo, Adv. Mater., 2018, 30, 1705515 CrossRef PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef PubMed.
- P. Hernandez-Fernandez, F. Masini, D. N. McCarthy, C. E. Strebel, D. Friebel, D. Deiana, P. Malacrida, A. Nierhoff, A. Bodin, A. M. Wise, J. H. Nielsen, T. W. Hansen, A. Nilsson, I. E. Stephens and I. Chorkendorff, Nat. Chem., 2014, 6, 732–738 CrossRef PubMed.
- C.-T. Hsieh, Y.-Y. Liu, D.-Y. Tzou and W.-Y. Chen, J. Phys. Chem. C, 2012, 116, 26735–26743 CrossRef.
- S. Pedireddy, H. K. Lee, W. K. Tjiu, I. Y. Phang, R. H. Tan, S. Q. Chua, C. Troadec and X. Y. Ling, Nat. Commun., 2014, 5, 4957 CrossRef PubMed.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–69 CrossRef PubMed.
- J. B. Luo, L. Wang, D. Mott, P. N. Njoki, Y. Lin, T. He, Z. Xu, B. N. Wanjana, I.-I. S. Lim and C.-J. Zhong, Adv. Mater., 2008, 20, 4342–4347 CrossRef.
- A. Holewinski, J.-C. Idrobo and S. Linic, Nat. Chem., 2014, 6, 828–834 CrossRef PubMed.
- Y. Ito, H. J. Qiu, T. Fujita, Y. Tanabe, K. Tanigaki and M. W. Chen, Adv. Mater., 2014, 26, 4145–4150 CrossRef PubMed.
- J. H. Lee, N. Park, B. G. Kim, D. S. Jung, K. Im, J. Hur and J. W. Choi, ACS Nano, 2013, 7, 9366–9374 CrossRef PubMed.
- H.-W. Liang, X. Zhuang, S. Brüller, X. L. Feng and K. Müllen, Nat. Commun., 2014, 5, 3312 CrossRef PubMed.
- T. Horita, K. Yamaji, M. Ishikawa, N. Sakai, H. Yokokawa, T. Kawada and T. Kato, J. Electrochem. Soc., 1998, 145, 3196–3202 CrossRef.
- S.-S. Li, H.-P. Cong, P. Wang and S.-H. Yu, Nanoscale, 2014, 6, 7534–7541 RSC.
- J. Liang, R. F. Zhou, X. M. Chen, Y. H. Tang and S. Z. Qiao, Adv. Mater., 2014, 26, 6074–6079 CrossRef PubMed.
- Y. Meng, X. Zou, X. Huang, A. Goswami, Z. Liu and T. Asefa, Adv. Mater., 2014, 26, 6510–6516 CrossRef PubMed.
- X. C. Dong, H. Xu, X. W. Wang, Y. X. Huang, M. B. Chan-Park, H. Zhang, L. H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6, 3206–3213 CrossRef PubMed.
- D. A. C. Brownson, L. C. S. Figueiredo-Filho, X. Ji, M. Gómez-Mingot, J. Iniesta, O. Fatibello-Filho, D. K. Kampouris and C. E. Banks, J. Mater. Chem. A, 2013, 1, 5962–5972 RSC.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef PubMed.
- Y. Liang, H. Wang, P. Diao, W. Chang, G. Hong, Y. Li, M. Gong, L. Xie, J. Zhou and J. Wang, J. Am. Chem. Soc., 2012, 134, 15849–15857 CrossRef PubMed.
- Z. Yang, X. Zhou, Z. Jin, Z. Liu, H. Nie, X. Chen and S. Huang, Adv. Mater., 2014, 26, 3156–3161 CrossRef PubMed.
- Z. Wang, S. Xiao, Z. Zhu, X. Long, X. Zheng, X. Lu and S. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 4048–4055 CrossRef PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef PubMed.
- S. Fu, C. Zhu, J. Song, M. Engelhard, H. Xia, D. Du and Y. Lin, ACS Appl. Mater. Interfaces, 2016, 8, 22196–22200 CrossRef PubMed.
- W. Wei, Y. Tao, W. Lv, F. Y. Su, L. Ke, J. Li, D. W. Wang, B. Li, F. Kang and Q. H. Yang, Sci. Rep., 2014, 4, 6289 CrossRef PubMed.
- Z. Wu, K. Parvez, A. Winter, H. Vieker, X. Liu, S. Han, A. Turchanin, X. Feng and K. Müllen, Adv. Mater., 2014, 26, 4552–4558 CrossRef PubMed.
- Z. Wang, R. Jia, J. Zheng, J. Zhao, L. Li, J. Song and Z. Zhu, ACS Nano, 2011, 5, 1677–1684 CrossRef PubMed.
- K. Yamamoto, T. Imaoka, W. J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397–402 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02448k |
| This journal is © The Royal Society of Chemistry 2018 |