DOI:
10.1039/C8RA02271B
(Paper)
RSC Adv., 2018,
8, 16032-16042
Mechanistic formation of drug-encapsulated Janus particles through emulsion solvent evaporation†
Received
15th March 2018
, Accepted 24th April 2018
First published on 30th April 2018
Abstract
Janus particles are emerging as structurally unique drug carriers with the potential to deliver multiple drugs and agents. Although synthesis methods have been extensively explored to fabricate Janus particles, it remains a challenge to generate drug-loaded Janus particles through an economical, high throughput technique. Here, we report the formation of the first drug-loaded, micro-scale Janus particles prepared using a single-step emulsion solvent evaporation approach. Our results revealed that both the net charge of drug molecules (i.e. glibenclamide, tolbutamine, rapamycin and lidocaine) and polymer weight ratio (i.e. poly(lactic-co-glycolic) and polycaprolactone) were critical in determining the formation of Janus particles. The formation of drug-loaded Janus particles was proven to be thermodynamically-driven in accordance to the classical equilibrium spreading coefficient theory, which is strongly governed by interfacial tensions. Specifically, comparable interfacial tensions between the two interacting polymers with the water phase were identified to be key criteria to achieve the Janus particles hemispheric structure. Such interfacial tensions were amenable, and were found to be highly dependent on the interfacial charge density attributed to both drug and polymer ratio. Hereby, this study provides a mechanistic insight into the fabrication of drug-loaded Janus particles and paves an important path towards large-scale production of Janus particles using a simplified, single-step emulsion solvent evaporation strategy.
1. Introduction
Biomedical research has progressed rapidly over the last two decades, attributed to advances in materials and nano-science to generate drug delivery technologies for controlled, sustained and targeted delivery. Drug delivery systems are a promising pharmaceutical technology that assures better drug bioavailability, and improves treatment efficacy and safety compared to conventional medicine.1 Such encapsulation systems are especially useful for highly potent drugs that suffer from low solubility and stability. Numerous small-scale carriers, i.e. solid lipid particles, liposomes, micelles, dendrimers, microcapsules and microparticles have thus been developed for this purpose.2,3 Examples of commercially successful delivery systems are the liposome-based doxorubicin-loaded carriers, e.g. Doxil® and Myocet®, and the polymer-based microspheres, e.g. Lupron Depot® and Nutropin Depot®. From an economics perspective, the drug delivery technology market is estimated to be going to be worth USD 1.669 trillion by the year 2021.4
The microparticle, in particular, is one of the most versatile and robust delivery systems capable of encapsulating a wide spectrum of small molecules, including proteins and nucleic acids, with high efficiency, while maintaining drug stability for long-term release.5–9 Its functionality can also be broadened through the design of structurally unique particles, i.e. multi-layered and multi-compartmentalized, as a means to manipulate the pharmacokinetics of drugs.1,5–7 The Janus Particle (JP) is one example of a structurally unique, yet promising multi-drug carrier. Such particles feature segregated, anisotropic compartments on two sides of an individual particle. JP offers several distinct advantages over other particulate delivery systems, because architecturally it allows for a compartmentalized encapsulation of drugs. In addition, it can provide other value-adding features such as bio-imaging, by incorporating imaging agents into a separate compartment for combinatorial theranostic application.10
The concept of JP was first proposed by the Nobel laureate de Gennes in the 1990s.11 Over the decades, tremendous effort has been channeled to optimize the synthesis process for large-scale production of JP with specific functionalities.12–14 Common approaches such as toposelective surface modification,15–17 template-directed self-assembly18,19 and controlled surface nucleation20–22 can provide precise morphological and structural control of the JP, but they suffer in scalability.13,14 More recent techniques are thus developed to synthesize polymeric JP using microfluidic devices23,24 and through the electro-hydrodynamic jetting strategy.25 The former produces particles of limited size range and may require an additional step for polymer crosslinking, while the latter is applicable only to conductive polymers. In contrast, the phase separation method,26 e.g. emulsion solvent evaporation, is generally recognized as the most feasible method for scalable production of JP owing to its economical set-up and relatively simple process.27 The key challenge of this method, however, is the ability to control simultaneous phase separation of polymers in the dynamic colloidal system to consistently yield JP of the desired architectural design. Although several recent studies have demonstrated the possibility of generating multi-dimensional JP using biodegradable polymers through the emulsion solvent evaporation approach, there has been no report where drugs were included into the fabrication process.28,29 In fact, attempts to encapsulate drug molecules into JP through this technique have not met with much success.30 In most cases, the addition of a drug molecule appeared to alter the initial structure of the JP, of reasons yet unknown.30 This suggests a complex relationship between the drug and polymers during particle fabrication that interfered with the formation of JP. There is therefore a need to determine the relationship among drug, polymer and JP formation, in order to devise an empirical strategy to consistently generate structurally intact drug-loaded JP through the emulsion technique.
In this study, we approached the question by scoping the work using two different FDA-approved biodegradable polymers, poly(lactic-co-glycolic) (PLGA) and polycaprolactone (PCL), as well as several drugs as our model system. Instead of encapsulating the drug using a blank JP formulation, we synthesized the JP in the presence of the drug using the emulsion solvent evaporation method and systematically investigated the factors that affect JP formation in this oil-in-water system. It was found that both the weight ratio of PLGA to PCL and the net charge of the drug molecules (i.e. glibenclamide, tolbutamine, rapamycin and lidocaine) were critical in governing the switch between anisotropic (i.e. JP) and core–shell (i.e. non-JP) structure, highlighting the importance of drug–polymer interaction in the fabrication process. Specifically, different drug molecules appeared to modulate the interfacial tensions, based on the net charge/charge density, of the interacting polymers with the water phase that primes the formation/de-formation of JP. These formulation principles can therefore serve as a theoretical framework for the generation of nano- to micro-scale, drug-loaded JP in a high throughput manner.
2. Experimental section
2.1 Materials
Glibenclamide and tolbutamide were purchased from Tocris Bioscience (UK). Rapamycin was obtained from Apollo Scientific (UK). Lidocaine was obtained from Sigma Aldrich (US). Poly(lactic-co-glycolic) (PLGA, lactide/glycolide ratio = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, IV = 0.2) was obtained from Corbion Purac (NL). Polycaprolactone (PCL, MW: 10 kDa) and poly(vinyl alcohol) (PVA, 87–90% hydrolyzed, MW: 30–70 kDa) were purchased from Sigma Aldrich (US). Dulbecco's PBS (without Ca2+ and Mg2+) was bought from GE Healthcare (UK). Dichloromethane (DCM) was purchased from Aik Moh Paints & Chemicals Pte Ltd (SG).
:50, IV = 0.2) was obtained from Corbion Purac (NL). Polycaprolactone (PCL, MW: 10 kDa) and poly(vinyl alcohol) (PVA, 87–90% hydrolyzed, MW: 30–70 kDa) were purchased from Sigma Aldrich (US). Dulbecco's PBS (without Ca2+ and Mg2+) was bought from GE Healthcare (UK). Dichloromethane (DCM) was purchased from Aik Moh Paints & Chemicals Pte Ltd (SG).
2.2 Fabrication of Janus particles
One-step oil-in-water emulsion solvent evaporation method was used to prepare the Janus particles. A total of 300 mg PLGA and PCL, in different weight ratios, were dissolved in three milliliters of dichloromethane (DCM). Drugs including, glibenclamide, tolbutamide, rapamycin and lidocaine as well as trypan blue were loaded at 2% or 10% of the total polymer weight (w/w). Both drug and polymers were mixed in DCM till completely dissolution. The drug–polymer solvent was then emulsified in water phase by stirring at 400 rpm for three hours at room temperature, in the absence of light. The water phase was prepared by dissolving PVA in 250 mL deionized water at 0.5% (v/v). Hydrochloric solution (1 M) was used for pH adjustment of water phase prior to emulsion step. The microparticles generated were harvested after washing with deionized water for five times. The microparticles were lyophilized and stored at −20 °C prior to further analysis.
2.3 Characterization of Janus particles
Optical images of microparticles were acquired using a light microscope equipped with a camera (Olympus CX21, JP). A total of 100–200 microparticles for each fabrication was imaged and analyzed using ImageJ (1.51f) to determine the size distribution of microparticles. The surface and cross-section of microparticle was visualized using a scanning electron microscope (SEM JEOL5410/6360, JP) with thermionic tungsten filament at an operating voltage of 5 kV. Briefly, the microparticles were first mounted on a carbon tape and dissected using a razor blade after rapid freezing in liquid nitrogen for 60 s in order to provide a cross-sectional view. To differentiate the PLGA compartment from the PCL compartment, intact microparticles were treated with acetone to dissolve PLGA but leaving the PCL compartment of a JP intact. After prior treatment finish (cross-section dissection or acetone treatment), the microparticle samples were coated with gold using a sputtering machine (JEOL Auto Fine Coater JFC-1600, JP) for 70–90 s prior to SEM imaging. The drug encapsulation efficiency (EE) for each individual drug was determined using the HPLC method as described previously.31 Briefly, the encapsulated drug was first extracted from the JP and the drug concentration was determined according to an earlier established calibration curve for each individual drug type. For example, rapamycin was analysed using Agilent Poroshell 120 column at a flow rate of 0.7 mL min−1 (80% methanol and 20% water). The drug peak was detected at a retention time of 7 min (278 nm). The EE was determined to be 92.89% ± 6.37%. Tolbutamide, glibenclamide and lidocaine were analysed using Agilent zorbax eclipse XDB-C18 column (250 × 4.6 mm, 5 μm). Tolbutamide peak was detected at 6.5 min (262 nm) at a flow rate of 1.0 mL min−1 (40% acetonitrile and 60% water, 0.05% trimethylamine), while glibenclamide peak was detected at 8 min at a flow rate (275 nm) of 1.0 mL min−1 (with 50% acetonitrile and 50% water). On the other hand, lidocaine peak was detected at 5–6 min (263 nm) at flow rate of 1 mL min−1 (30% methanol and 70% 0.01 mol L−1 NaH2PO4 (pH = 2)). The EE were determined to be 95.77% ± 1.2%, 97.94% ± 5.46% and 80.68% ± 2.6% for tolbutamide, glibenclamide and lidocaine, respectively. For non-drug molecule, i.e. trypan blue, visual observation of blue color was used to ascertain its successful encapsulation.
2.4 Distribution of glibenclamide in a Janus particle using confocal Raman microscopy
The spatial distribution of the drug glibenclamide in a JP was mapped using a confocal Raman microscope (WITec Raman Imaging, Savoy, IL), consisting of a high power NIR diode laser 785 nm excitation, a single monochromator (Acton) employing a 300 groove mm−1 grating and a thermoelectrically cooled 1024 by 128 pixel array CCD camera (Andor Technology). Using the “Raman Spectral Imaging” mode of the instrument, Raman spectrums were obtained by scanning the sample at each polymer compartment. Microparticles were mounted on a double side tape secured on a glass slide. The samples were excited using 785 nm laser at 100 mW. All Raman spectra were collected using a plan achromatic objective at 20×, with numerical aperture 0.4. A static Raman spectra window from 0–3000 cm−1 was applied in all measurements, with an acquisition time of 180 scans each time. The collective Raman spectrum was processed using the instrument default software to generate the average spectrum for polymer and drug component based on their unique Raman shift.
2.5 Interfacial tension measurement
The interfacial tensions between PLGA/DCM and PVA/water (i.e. γ12) as well as PCL/DCM with PVA/water (i.e. γ23) were determined with an optical contact angle measuring and contour system (OCA 15Pro, DataPhysics, US) using the pendant drop method.32 Briefly, a droplet of PLGA/DCM or PCL/DCM immersed in PVA/water hanging on a dosing needle was imaged using a camera. The dimension/shape of the droplet was subsequently analyzed with the DataPhysics SCA 22 software module based on the Young–Laplace's equation33 to determine the interfacial tension. On the other hand, the interfacial tension between PLGA and PCL (i.e. γ13) was estimated based on the surface energy of PLGA and PCL according to the Owens–Wendt method (Table S1†). Briefly, two standard solutions cyclohexane and water were used to measure their contact angles with PLGA and PCL films using the optical contact angle measuring and contour system (OCA 15Pro, DataPhysics, US). The interfacial energy and thus the interfacial tension were calculated based on the contact angles determined. All measurements were done in triplicates.
3. Results
3.1 Glibenclamide drives Janus particle formation at a specific polymer weight ratio
Microparticles consisting of biodegradable polymers, i.e. PLGA and PCL, at different weight ratios, as well as the model drug glibenclamide, were first synthesized using a one-step oil-in-water emulsion solvent evaporation technique. Without glibenclamide, microparticles with core–shell structures, were consistently generated regardless of the polymer weight ratios (Fig. 1(A1–A3)). In contrast, with the addition of glibenclamide, a bi-compartmental JP structure readily appeared at the polymer weight ratio 20:10, but not at 15:15 and 10:20 ratios (Fig. 1(A4–A4)). These bi-compartmental microparticles were generally characterized by a distinct, hemispheric Janus structure where both PLGA and PCL polymers appeared to occupy almost an equal volume of the microparticle (Fig. 1B1), which is different from the JP morphology commonly synthesized in the absence of drugs.34 The anisotropic characteristic of JP was evidenced by acetone treatment,35 which resulted in the full dissolution of the PLGA component, leaving the PCL compartment with pox-like surfaces (Fig. 1B2). Glibenclamide was found to be distributed in both compartments of the JP as indicated by the confocal Raman microscopic mapping (Fig. S1†). Consistent with this, the release profile of glibenclamide from JP appeared to be modulated according to the combined drug release characteristics of monolayer PLGA and PCL microparticles (Fig. S2†). Importantly, JP demonstrated greater control over burst release of glibenclamide compared to the core–shell microparticles, highlighting the unique property of Janus structure for drug delivery. Quantitative microscopy image analysis further revealed that more than 90% of the particles yielded were JP with mean diameter of 104.5 μm and standard deviation of 37.9 μm (Fig. S3†).
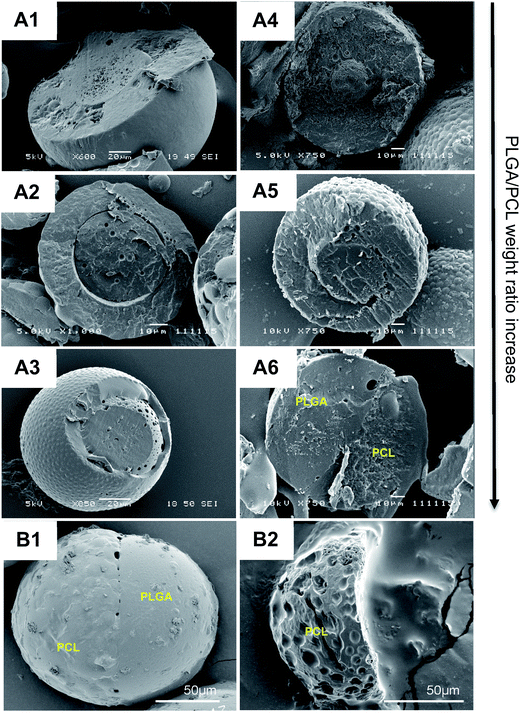 |
| | Fig. 1 Microparticles fabricated at different PLGA/PCL weight ratios in the presence or absence of glibenclamide. Cross-sectional views of blank (A1–A3) and glibenclamide-loaded (A4–A6) microparticles under scanning electron microscope (SEM). Microparticles were fabricated at different PLGA/PCL weight ratios (w/w), i.e. 10:20 (A1 and A4), 15:15 (A2 and A5), and 20:10 (A3 and A6). Surface morphology of an intact, glibenclamide-loaded microparticle characterized with Janus structure (B1) and SEM image of that Janus particle after acetone treatment (B2). Scale bar: (A1 and A3), 20 μm; (A2 and A4–A6), 10 μm; (B1–B2), 50 μm. | |
3.2 The negative charge state of glibenclamide determines the particle morphology and yield at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :10 polymer weight ratio
:10 polymer weight ratio
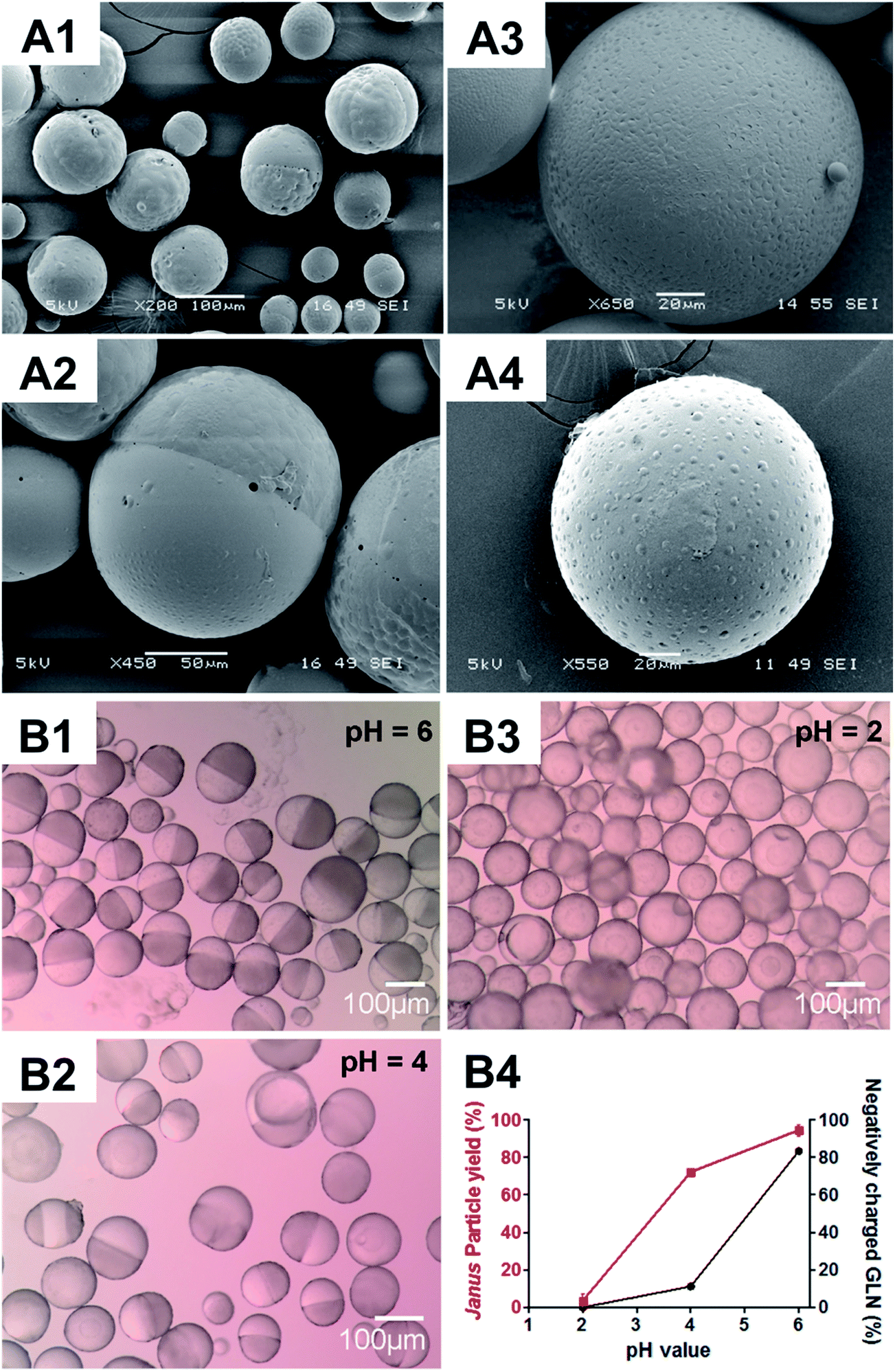
To gain a further insight into the relationship between glibenclamide and microparticle formation, different concentrations of glibenclamide were supplemented in the fabrication process whilst the PLGA/PCL polymer weight ratio was maintained at 20:10 (Fig. S4†). While core–shell (i.e. non-JP) particles were dominant in the absence of glibenclamide (Fig. S4A†), more than 90% of the microparticles were JP when glibenclamide at 2% (w/w) was included (Fig. S4B†). A higher concentration of glibenclamide, i.e. 10% (w/w), however, neither altered the Janus structure nor significantly increased the JP yield (96.6 ± 0.9% for 10% glibenclamide vs. 92.5 ± 0.5% for 2% glibenclamide, P > 0.05) (Fig. S4C†). Interestingly, it was also shown that the negatively charged glibenclamide could be functionally substituted with other negatively charged drugs such as tolbutamide (Fig. 2A1) or a negatively charged agent like trypan blue (Fig. 2A2). However, addition of a neutral drug, e.g. rapamycin (Fig. 2A3) or a positively charged drug, e.g. lidocaine (Fig. 2A4), did not yield any JP at the same PLGA/PCL weight ratio 20:10. This suggests a charge-dependent selectivity in JP formation. To further confirm the impact of negative charge on JP formation, the relationship between the charge density of glibenclamide at different pH and the JP yield was quantified (Fig. 2B). The amount of JP formed was found to be positively correlated with the negative charge density of glibenclamide (Fig. S5† and 2B4). For example, at pH 6, 83.4% of glibenclamide were expected to carry a negative charge and that resulted in more than 92% of microparticles with a Janus structure (Fig. 2B1). However, the JP yield decreased to 71% at pH 4 and JP disappeared entirely at pH 2, where the negative charge densities of glibenclamide were reduced to 11.4% and 0.1%, respectively (Fig. 2B2 and B3).
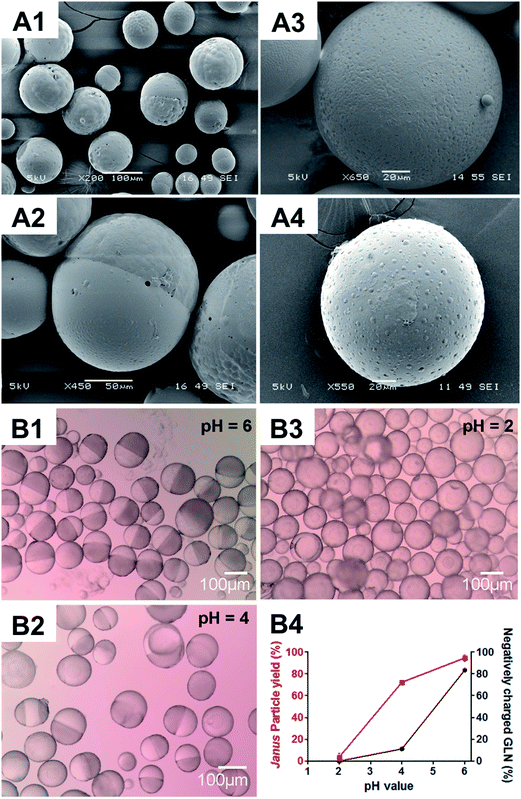 |
| | Fig. 2 Microparticles fabricated at the PLGA/PCL weight ratio 20:10 in the presence of drugs with different charges (A) or charge densities (B). Scanning electron microscopy (SEM) images of PLGA/PCL microparticles fabricated in the presence of drugs with different charges, i.e. tolbutamine (negatively charged) (A1), trypan blue (negatively charged) (A2), rapamycin (non-charged) (A3) and lidocaine (positively charged) (A4). Light microscopy images of PLGA/PLCA microparticles fabricated with glibenclamide at different, negatively charged densities, i.e. 83.4%, 11.2% and 0.1%, generated at pH 6.0 (B1), pH 4.0 (B2) and pH 2.0 (B3), respectively, according to the Henderson–Hasselbalch equation. The relationship between pH, negatively charged density of glibenclamide (GLN) and the yield of microparticles with Janus structure (B4). Scale bar: (A1 and B1–B3), 100 μm; (A2), 50 μm; (A3–A4), 20 μm. | |
3.3 Glibenclamide alters the interfacial tensions in favor of JP formation at 20:10 polymer weight ratio
In an emulsion system, the interfacial tensions (γ) between different phases determine the spreading coefficients (S), which can be defined thermodynamically based on the eqn (1).36–38 This plays a key role in shaping the particle morphology, as different combinations of spreading coefficients can influence the degree of polymer engulfment during phase separation, according to the spreading coefficient theory.37,38 By Harkin's definition, a positive spreading coefficient means the material will spread, while a negative value means the material will contract.38 In this study, the emulsion system consisted of three different phases namely, the oil phase PLGA/DCM (Phase 1), the water phase PVA (Phase 2) and the second oil phase PCL/DCM (Phase 3). Correspondingly, there could be three different polymer engulfment configurations at the equilibrium state, including the full (2), the partial (3) and no engulfment (4).| | |
Harkin's equation: Si = γjk − (γij + γik)
| (1) |
| | |
Full engulfment (i.e. core–shell): S1 < 0, S2 < 0, S3 > 0 or S1 > 0, S2 < 0, S3 < 0
| (2) |
| | |
Partial engulfment (i.e. Janus): S1 < 0, S2 < 0, S3 < 0
| (3) |
| | |
No engulfment (i.e. individual): S1 < 0, S2 > 0, S3 < 0
| (4) |
Here, γ12 and γ23 are the interfacial tensions of the water phase PVA with the polymer PLGA or PCL, respectively, and γ13 is the interfacial tension between PLGA and PCL. The latter was determined based on the solid state of PLGA and PCL as it gave more accurate morphology prediction.39 It is assumed that the interfacial tension at the solid state remained constant regardless of the drug added (i.e. γ13 = 0.65). On the other hand, it is hypothesized the negatively charged glibenclamide modulates the interfacial tensions of the polymers with the water phase (i.e. γ12 and γ23), favoring the JP formation, specifically at 20:10 polymer weight ratio. Without drug, at PLGA/PCL weight ratio of 20:10, the interfacial tensions differed markedly from each other and the spreading coefficients, i.e. S1 > 0, S2 < 0, S3 < 0, met the full engulfment criteria, resulting in core–shell microparticles (Table 1). In contrast, the interfacial tension between PLGA–PVA (γ12 = 7.04) was found comparable to that of PCL–PVA (γ23 = 7.05) when glibenclamide was included. Accordingly, all the spreading coefficients were estimated to be negative, with S1 (−0.64) nearly equivalent to S3 (−0.67) but significantly less negative than S2 (−13.43). This satisfied the partial engulfment requirement for JP formation, i.e. S1 < 0, S2 < 0, S3 < 0. Such drug modulation effect, however, was different when positively charged lidocaine and the neutral rapamycin were supplemented at the same 20:10 polymer weight ratio. Although both interfacial tensions, i.e. γ12 and γ23, were reduced with addition of lidocaine or rapamycin, the full engulfment configuration remained unchanged (Table 1). Similarly, at PLGA/PCL weight ratio of 15:15, addition of the negatively charged glibenclamide also affected the interfacial tensions (as compared to the same formulation without drug), but this time, the partial engulfment configuration was not attainable. These observations were consistent with the spreading coefficient theory, in which the thermodynamic requirements for JP had to be achieved.
Table 1 Interfacial tensions and spreading coefficients at different PLGA/PCL weight ratios
| PLGA/PCL ratioa |
Drug chargeb |
Interfacial tensionc |
Spreading coefficiente |
Predictedf |
Observed |
| γ12 |
γ23 |
γ13d |
S1 |
S2 |
S3 |
| Each emulsion system consists of three phases, i.e. Phase 1: PLGA/DCM; Phase 2: PVA (water) and Phase 3: PCL/DCM. The weight proportion of each polymer used in the fabrication is indicated. PVA: poly(vinyl alcohol); DCM: dichloromethane. Drugs with different charges were included in the fabrication, i.e. glibenclamide (negatively charged), rapamycin (neutral) and lidocaine (positively charged). n.a.: not applicable. Interfacial tensions between PLGA/DCM and PVA/water (γ12) as well as PCL/DCM and PVA/water (γ23) were determined using the pendant drop method. Meanwhile, the interfacial tension between PLGA/DCM and PCL/DCM (γ13) was estimated based on the surface energy of PLGA and PCL according to the Owens–Wendt method (Table S1). The interfacial tension is expressed in mN m−1. Phase 1 and 3 are miscible at the beginning of the emulsion. Therefore the interfacial tension between the solid state of PLGA and PCL (i.e. γ13) was used to represent the final form as it gave more accurate morphology prediction.39 It is assumed that the interfacial tension at the solid state remained the same with drug addition.40 The spreading coefficient (S) is calculated based on Harkin's equation, Si = γjk − (γij + γik). The spreading coefficient is expressed in mN m−1. According to the classic spreading coefficient theory37 – core–shell: S1 < 0, S2 < 0, S3 > 0 or S1 > 0, S2 < 0, S3 < 0; Janus: S1 < 0, S2 < 0, S3 < 0; individual particles: S1 < 0, S2 > 0, S3 < 0. |
| 20:10 |
n.a. |
6.69 |
10.56 |
0.65 |
3.22 |
−16.60 |
−4.53 |
Core–shell |
Core–shell |
| 20:10 |
Negative |
7.04 |
7.05 |
0.65 |
−0.64 |
−13.43 |
−0.67 |
Janus |
Janus |
| 20:10 |
Neutral |
5.78 |
6.62 |
0.65 |
0.19 |
−11.74 |
−1.50 |
Core–shell |
Core–shell |
| 20:10 |
Positive |
3.75 |
8.60 |
0.65 |
4.20 |
−11.70 |
−5.50 |
Core–shell |
Core–shell |
| 15:15 |
n.a. |
6.72 |
5.93 |
0.65 |
−1.44 |
−12.00 |
0.14 |
Core–shell |
Core–shell |
| 15:15 |
Negative |
4.38 |
6.43 |
0.65 |
1.40 |
−10.16 |
−2.70 |
Core–shell |
Core–shell |
3.4 Modulation of interfacial tensions by polymer weight ratio allows the formation of lidocaine- and rapamycin-loaded Janus particles
Besides net charge of the drug, interfacial tensions also changed with polymer weight ratios, e.g. 20:10 vs. 15:15 (Table 1). It was found that at a weight ratio of 19:11, JP were obtained even in the absence of drugs (Fig. 3A). At this weight ratio, addition of rapamycin or lidocaine also yielded JP (Fig. 3C and D). However, this time, encapsulation of glibenclamide resulted in core–shell microparticles (Fig. 3B), likely due to more positive spreading coefficients that shift away from partial engulfment. The subsequent interfacial tension and spreading coefficient measurements agreed with this hypothesis (Table 2). With a newly adjusted weight ratio of 19:11, the interfacial tensions of both PLGA–PVA (γ12 = 5.40) and PCL–PVA (γ23 = 5.64), without any drug, now favor the partial engulfment condition. Interestingly, addition of the neutral rapamycin increased the interfacial tensions at both interfaces (i.e. γ12 = 6.50 and γ23 = 6.04) while supplementation of the positive lidocaine decreased those measurements (i.e. γ12 = 4.85 and γ23 = 4.28). Despite the differences, the resulting spreading coefficients fulfilled the partial engulfment criteria in both cases and hence, the formation of lidocaine- and rapamycin-loaded JP. These findings clearly suggest that interactions between the drug and the polymers quantitatively dictate the interfacial tensions of the emulsion system, and determines the degree of polymer engulfment.
 |
| | Fig. 3 Microparticles fabricated in the absence or presence of drugs with different charges at the PLGA/PCL weight ratio 19:11. Scanning electron microscopy (SEM) images of PLGA/PCL microparticles fabricated, at the specific polymer weight ratio 19:11, without drug (A) or with drugs carrying different charges, i.e. glibenclamide (negatively charged) (B), rapamycin (non-charged) (C) and lidocaine (positively charged) (D). Scale bar: (A and D), 20 μm; (B), 10 μm; (C), 50 μm. | |
Table 2 Interfacial tensions and spreading coefficients at PLGA/PCL weight ratio 19:11
| PLGA/PCL ratioa |
Drug chargeb |
Interfacial tensionc |
Spreading coefficiente |
Predictedf |
Observed |
| γ12 |
γ23 |
γ13d |
S1 |
S2 |
S3 |
| Each emulsion system consists of three phases, i.e. Phase 1: PLGA/DCM; Phase 2: PVA (water) and Phase 3: PCL/DCM. The weight proportion of each polymer used in the fabrication is indicated. PVA: poly(vinyl alcohol); DCM: dichloromethane. Drugs with different charges were included in the fabrication, i.e. glibenclamide (negatively charged), rapamycin (neutral) and lidocaine (positively charged). n.a.: not applicable. Interfacial tensions PLGA/DCM and PVA/water (γ12) as well as PCL/DCM and PVA/water (γ23) were determined using the pendant drop method. Meanwhile, the interfacial tension between PLGA and PCL (i.e. γ13) was estimated based on the surface energy of PLGA and PCL according to the Owens–Wendt method (Table S1). The interfacial tension is expressed in mN m−1. Phase 1 and 3 are miscible at the beginning of the emulsion. Therefore the interfacial tension between the solid state of PLGA and PCL (i.e. γ13) was used to represent the final form as it gave more accurate morphology prediction.39 It is assumed that the interfacial tension at the solid state remained the same with drug addition.40 The spreading coefficient (S) is calculated based on Harkin's equation, Si = γjk − (γij + γik). The spreading coefficient is expressed in mN m−1. According to the classic spreading coefficient theory37 – core–shell: S1 < 0, S2 < 0, S3 > 0 or S1 > 0, S2 < 0, S3 < 0; Janus: S1 < 0, S2 < 0, S3 < 0 and separate particles: S1 < 0, S2 > 0, S3 < 0. |
| 19:11 |
n.a. |
5.40 |
5.64 |
0.65 |
−0.41 |
−10.39 |
−0.89 |
Janus |
Janus |
| 19:11 |
Neutral |
6.50 |
6.04 |
0.65 |
−1.11 |
−11.89 |
−0.19 |
Janus |
Janus |
| 19:11 |
Negative |
5.82 |
5.06 |
0.65 |
−1.41 |
−10.23 |
0.11 |
Core–shell |
Core–shell |
| 19:11 |
Positive |
4.85 |
4.28 |
0.65 |
−1.22 |
−8.48 |
−0.08 |
Janus |
Janus |
4. Discussion
The emulsion solvent evaporation is one of the most cost-effective and highly scalable technologies for microparticle synthesis.5,41 Fabrication of drug-loaded microparticles especially the anisotropic Janus particle (JP) via this approach, however, has yet to be established. In this study, we have methodically examined the mechanism controlling the formation of drug-loaded JP in an oil-in-water emulsion system. We found that these bi-compartmental microparticles could be fabricated explicitly at the partial engulfment condition according to the classic spreading coefficient theory. Critical determinants, including the polymer weight ratio and the net charge of drug molecules, were found to be strongly linked with the alteration of the interfacial tensions, a key component of the classic theory. Understanding the dynamic relationship between the drug nature and the polymer may now enable the formulation of an empirical strategy to synthesize drug-loaded JP via the emulsion approach.
JP formation based on the phase separation method, without any drug encapsulation, has been well described according to the spreading coefficient theory.37,38,42 Here, we extended the application of this thermodynamic rule in synthesizing JP with different drug loads. Regardless of the type of drug molecules incorporated or the polymer weight ratio used, the Janus structure (i.e. the partial engulfment configuration) was achieved only when all three spreading coefficients in an emulsion system were negative or whenever |γ12 − γ23| < γ13 (Tables 1 and 2). However, unlike the acorn-, snowman- or the dumbbell-shape JP commonly synthesized in the absence of drug molecules,14 microparticles fabricated in this study were uniquely characterized with two equal hemispheres, which is designated here as the JP hemisphere (Fig. 1–3). The different JP morphology is likely due to the varying degrees of partial engulfment during phase separation (Fig. S6†).37,43 Such JP hemisphere morphology is also in line with the prediction by the extended theory of Torza and Mason, where the contact angle θ between two phases at the three-phase equilibrium point of JP can be calculated based on Torza–Mason's equation (Fig. S6†).37 The contact angle between the two interacting polymers (i.e. θ2) of a JP hemisphere is approximately 180°, which could only occur when the interfacial tensions between the PLGA–PVA (i.e. γ12) and PCL–PVA (i.e. γ23) are almost equivalent. Comparable interfacial tensions (i.e. γ12 ≈ γ23) with an absolute difference less than the interfacial tension between the polymers (γ13) are therefore not only critical in driving the JP formation but also in governing the JP morphology. Given that JP will be used for drug delivery applications, a consistent particle surface to area ratio, as exemplified by the hemisphere morphology, is thus highly desirable.
The interfacial tension is defined as the energy cost per unit area associated with creation of an interface between two adjacent phases.44 Our data revealed that the interfacial tensions between the polymers and the water phase (i.e. γ12 and γ23) were highly dynamic, and susceptible to changes induced by drug molecules (Tables 1 and 2). However, it remains unclear how a drug molecule may alter the interfacial tension and what is the mechanism underscoring the transformation event. Further study indicated that the interfacial tension change was strongly linked with the net charge of a drug molecule, and this might be further influenced by the type and the amount of polymer used in the emulsion system (Table 3). For example, addition of the positively charged lidocaine consistently decreased the interfacial tension regardless of the type (i.e. PLGA or PCL) or the amount of polymer (100 to 200 mg) used. In contrast, the negatively charged glibenclamide reduced the interfacial tension only when the polymer (i.e. PLGA or PCL) was in a lower weight proportion (e.g. 100–150 mg). When a higher weight range of polymer was involved (e.g. 150–200 mg), glibenclamide increased the interfacial tension of the respective polymer with the water phase. The neutral drug rapamycin was the only exception that either increased or decreased the interfacial tensions concurrently irrespective to the type and the amount of polymer.
Table 3 Alteration of interfacial tensions by drugs at different polymer weightsa
| PLGA/DCM-PVA/water (γ12) |
PCL/DCM-PVA/water (γ23) |
| |
Drug |
|
Drug |
| Each emulsion system consists of three phases, i.e. Phase 1: PLGA/DCM; Phase 2: PVA (water) and Phase 3: PCL/DCM. Interfacial tensions between PLGA and PVA (i.e. γ12) as well as PCL and PVA (i.e. γ23) were determined using the pendant drop method. The increase or decrease of the interfacial tensions (i.e. γ12 and γ23), in the presence of drugs, was determined with reference to that of the blank particle (without drug addition) at the respective polymer weight. Drugs used including, glibenclamide (negatively charged), rapamycin (neutral) and lidocaine (positively charged). The result shown here is compiled from the data presented in Tables 1 and 2. |
| PLGA (mg) |
Negative |
Positive |
Neutral |
PCL (mg) |
Negative |
Positive |
Neutral |
| 200 |
↑ |
↓ |
↓ |
100 |
↓ |
↓ |
↓ |
| 190 |
↑ |
↓ |
↑ |
110 |
↓ |
↓ |
↑ |
| 150 |
↓ |
— |
— |
150 |
↑ |
— |
— |
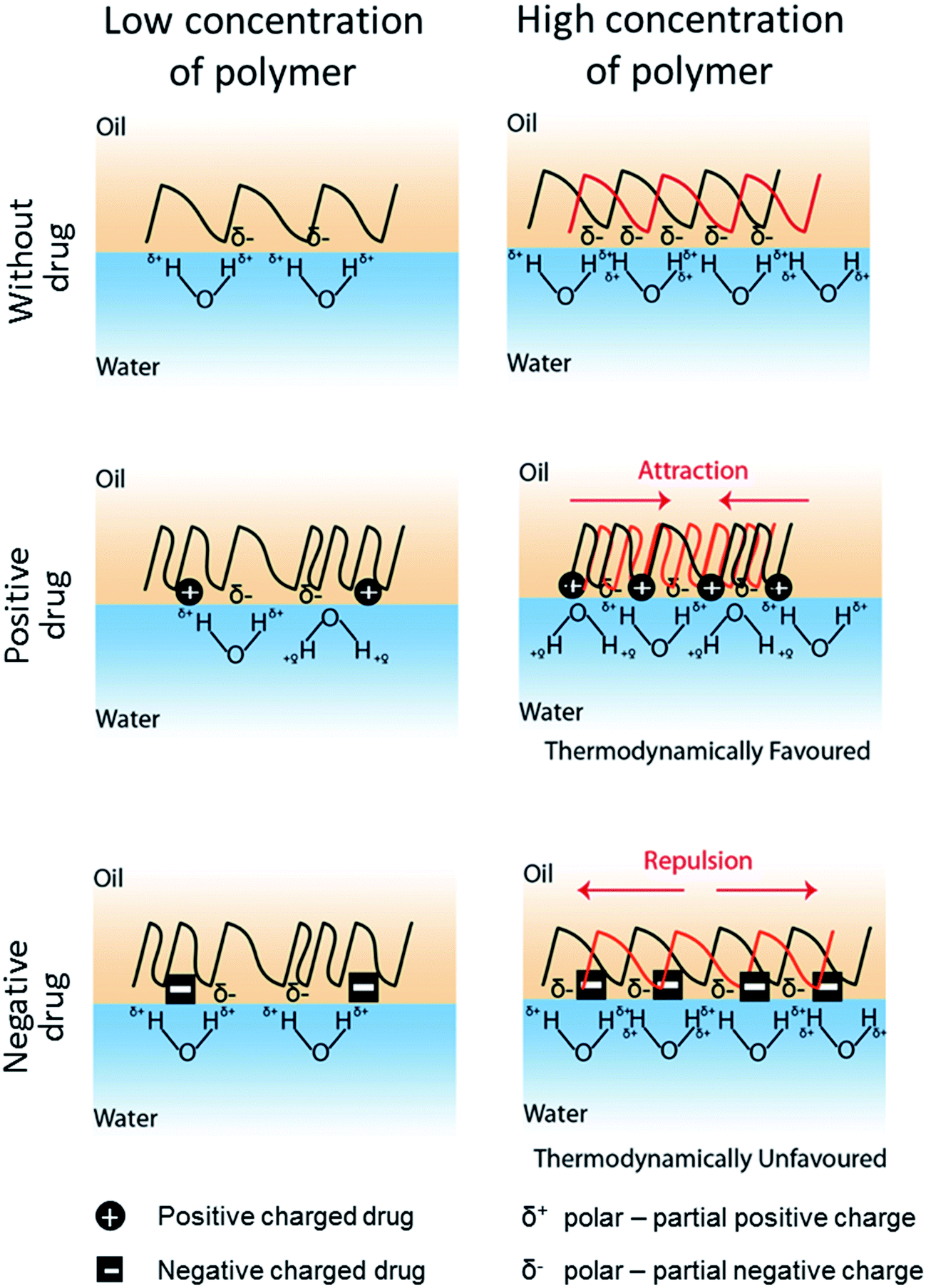
In conjunction with the classical theory described above, we propose here a model to define the drug-induced interfacial tension change based on the interfacial charge density attributed by drug-polymer interaction (Fig. 4). In this model, it is hypothesized that a drug molecule interacts with a polymer at a specific molar ratio that alters the surface hydrophobicity of the polymer. Similar to prior work,45,46 the electrical charge of the drug molecule causes the polymer to become more polar or hydrophilic and thus, reduces the interfacial tension of the polymer with the water phase in the emulsion system. In fact, a similar role by a positively charged surfactant DMBA in lowering the oil-in-water interfacial tension has also been reported.47 The charge density of a polymer, as conferred by the drug molecule, will be proportional to the amount of polymer used in the emulsion. While a higher charge density may reduce the hydrophobicity of a polymer, accumulation of like charges (e.g. negatively charge drug added to partial negatively charged polymer – PLGA or PCL) will promote charge repulsion, especially during phase separation when the drug–polymer concentrates. Prior study of PMMA/PS polymer difference phase separation behavior in corresponding to SDS surfactant concentration indicates similar phenomenon.48 Hence, the PLGA/PCL scenario here becomes thermodynamically unfavorable. A consequence of such is the rise of the interfacial tension as in the case when a high proportion of PLGA (i.e. 190 and 200 mg) or PCL (i.e. 150 mg) was mixed with negatively charged glibenclamide (Table 3). In line with our hypothesis, a lower amount of PLGA (i.e. 150 mg) or PCL (i.e. 100 and 110 mg) may therefore chelate less glibenclamide, thus reducing the amount of like charges at the interface that is below the repulsive threshold (Fig. 4). Since both PLGA and PCL are slightly polar with multiple carbonyl functional groups that display a partial negative charge (δ−), interaction between the positively charged lidocaine with either polymer is therefore thermodynamically favorable. This may also explain for the constant reduction in the interfacial tension when lidocaine was added to either polymer, regardless of the polymer weight (Table 3). For a non-charged, neutral drug like rapamycin, its effect on interfacial tension is less conspicuous. It is therefore likely that a highly hydrophobic rapamycin may have altered the polymer property and behavior differently from the concept of charge density proposed here. Although this work appears to be preliminary and the charge density-driven model remains to be attested by other polymer and drug combinations, it provides the first molecular insight into the possibilities of how a drug molecule may influence the behavior of a polymer in an emulsion system and drive the formation of different microparticles, including core–shell and Janus particles. The findings in this study clearly suggest that it is possible to encapsulate almost any drug molecule into a JP by modulating the interfacial tensions between the polymers and the water phase. Our proposed model further submits that this can be achieved by adjusting the interfacial charge density, through the tuning of the net charge of the drug molecule (e.g. by altering the emulsion pH) or the amount of polymer. It is important to next determine the extent to which a drug molecule of interest may influence the interfacial tension for different types of polymers at various polymer concentrations. Such outcomes will be critical to provide a quantitative measurement or a standard for the formation of drug-encapsulating JP. Although micron JP (10 s–100 s μm) were synthesized in this study as a model, it is highly possible that sub-micron, drug-loaded JP can also be generated via the same emulsion solvent evaporation approach.49 In fact, the flexibility for particle size tuning will be a great advantage for different drug delivery applications. For example, micron JP can be used for intraperitoneal delivery where the peritoneal cavity is used as a reservoir for the slow release of drugs from larger particles that cannot escape from the peritoneal region.50,51 In addition, the use of micron JP can also be used to deliver drugs into the anterior chamber of the eye for islet transplantation.52 Sub-micron JP, on the other hand, can be used for other routes of drug delivery, including oral and intravenous administrations.53,54
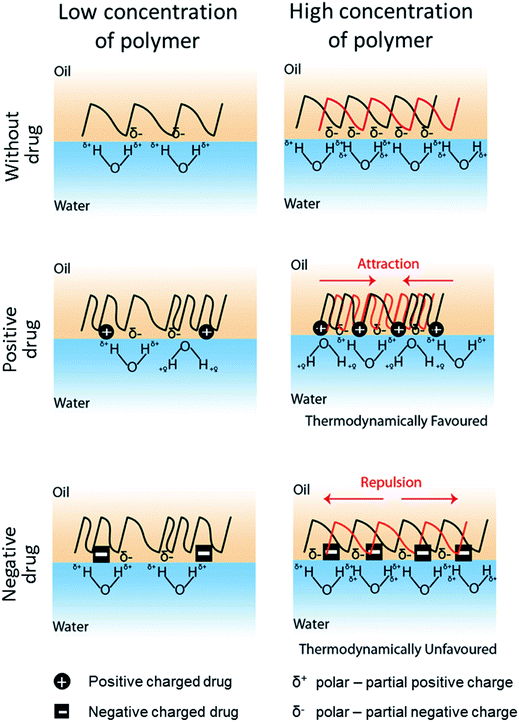 |
| | Fig. 4 The proposed model for drug-induced interfacial tension change. The interfacial tension is referred to the energy cost per unit area associated with creation of an interphase between the polymer (i.e. PLGA or PCL dissolved in DCM) and the water phase. Both PLGA and PCLs are slightly polar with multiple carbonyl groups displaying partial negative charge (δ−). When exposed to water, hydrogen bonding between water molecules and polymer dominates. The less hydrophobic the polymer surface is, there will be greater interaction between polymer and water, and hence the lower the interfacial tension. Drug molecules, either positively ( ) or negatively ( ) or negatively ( ) charged, interact with the polymer at specific molar ratios (presumably via hydrophobic or van der Waals interactions), causing the polymer to become more hydrophilic and thus, reducing the interfacial tension at low polymer weight. At high polymer concentration, more drug molecules are expected to interact with the polymer chains, resulting in higher interfacial charge density. This is thermodynamically favorable for interaction between positively charged drug and the polymer as opposite attracts. However, it is unfavorable for negatively charged drug interacting with the polymer when the charge density reaches a repulsive threshold, as similar charges repel. Consequently, addition of a positively charged drug reduces the interfacial tension, while a negatively charged drug increases the interfacial tension at high polymer concentration. ) charged, interact with the polymer at specific molar ratios (presumably via hydrophobic or van der Waals interactions), causing the polymer to become more hydrophilic and thus, reducing the interfacial tension at low polymer weight. At high polymer concentration, more drug molecules are expected to interact with the polymer chains, resulting in higher interfacial charge density. This is thermodynamically favorable for interaction between positively charged drug and the polymer as opposite attracts. However, it is unfavorable for negatively charged drug interacting with the polymer when the charge density reaches a repulsive threshold, as similar charges repel. Consequently, addition of a positively charged drug reduces the interfacial tension, while a negatively charged drug increases the interfacial tension at high polymer concentration. | |
5. Conclusions
Janus particles (JP), of anisotropic structure, can be fabricated through a one-step emulsion solvent evaporation technique, with high yield and a precise control of particle structure. The addition of a drug into this emulsion however changes the way JP are formed. Both the net charge of the drug and the polymer amount used (i.e. weight ratio of PLGA/PCL) in the emulsion play important roles in altering polymer interaction, and thus the final structure of the particles, i.e. core–shell or JP. These findings offer a new perspective in which the presence of a drug could alter the particle morphology by influencing the interfacial tension in the emulsion system. JP formation via phase separation, with or without drug encapsulation, is a thermodynamic process that is solely determined by the emulsion interfacial tensions, that is according to the spreading coefficient theory. This mechanistic insight would therefore allow for the production of drug-encapsulating JP that could be used for various drug delivery applications.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
These authors declare no conflict of interest.
Abbreviations
| JP | Janus particles |
| SEM | Scanning electron microscope |
| PLGA | Poly(lactic-co-glycolic) |
| PCL | Polycaprolactone |
| GLN | Glibenclamide |
| DCM | Dichloromethane |
Acknowledgements
The authors would like to acknowledge the financial support from the Singapore Centre for Environmental Life Sciences Engineering (SCELSE) (M4330001.C70), the School of Materials Science and Engineering (M020070110), the NTU-National Healthcare Group (NTU-NHG) grant (ARG/14012), the Ministry of Education Tier 1 grant (RG11/16), and the NTU-HSPH grant (NTU-HSPH 17002).
References
- C. L. Ventola, Pharm. Ther., 2012, 37, 512–525 Search PubMed.
- A. Fahr and X. Liu, Expert Opin. Drug Delivery, 2007, 4, 403–416 CrossRef CAS PubMed.
- J. R. Baker Jr, Hematology/the Education Program of the American Society of Hematology. American Society of Hematology. Education Program, 2009, pp. 708–719, DOI:10.1182/asheducation-2009.1.708.
- Drug Delivery Technology Market by Route of Administration (Oral (Solid), Pulmonary (Nebulizer), Injectable (Device), Ocular (Liquid), Topical (Solid), Implantable (Active), Transmucosal (Oral)), Patient Care Setting (Hospital, ASC) – Global Forecast to 2021, http://www.marketsandmarkets.com/Market-Reports/drug-delivery-technologies-market-1085.html, (accessed Feb, 2017).
- W. L. Lee and S. C. J. Loo, J. Drug Targeting, 2012, 20, 633–647 CrossRef CAS PubMed.
- W. L. Lee, E. Widjaja and S. C. J. Loo, Small, 2010, 6, 1003–1011 CrossRef CAS PubMed.
- M. P. A. Lim, W. L. Lee, E. Widjaja and S. C. J. Loo, Biomater. Sci., 2013, 1, 486–493 RSC.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- K. K. Kim and D. W. Pack, in BioMEMS and Biomedical Nanotechnology: Volume I Biological and Biomedical Nanotechnology, ed. M. Ferrari, A. P. Lee and L. J. Lee, Springer, US, Boston, MA, 2006, vol. 1, pp. 19–50 Search PubMed.
- C. Kaewsaneha, P. Tangboriboonrat, D. Polpanich, M. Eissa and A. Elaissari, ACS Appl. Mater. Interfaces, 2013, 5, 1857–1869 CAS.
- P.-G. de Gennes, Angew. Chem., Int. Ed., 1992, 31, 842–845 CrossRef.
- S. Jiang, Q. Chen, M. Tripathy, E. Luijten, K. S. Schweizer and S. Granick, Adv. Mater., 2010, 22, 1060–1071 CrossRef CAS PubMed.
- A. Perro, S. Reculusa, S. Ravaine, E. Bourgeat-Lami and E. Duguet, J. Mater. Chem., 2005, 15, 3745 RSC.
- A. Walther and A. H. Muller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed.
- L. Carbone and P. D. Cozzoli, Nano Today, 2010, 5, 449–493 CrossRef CAS.
- J. Choi, Y. Zhao, D. Zhang, S. Chien and Y. H. Lo, Nano Lett., 2003, 3, 995–1000 CrossRef CAS.
- Z. Bao, L. Chen, M. Weldon, E. Chandross, O. Cherniavskaya, Y. Dai and J. B. H. Tok, Chem. Mater., 2002, 14, 24–26 CrossRef CAS.
- Y. Xia, Y. Yin, Y. Lu and J. McLellan, Adv. Funct. Mater., 2003, 13, 907–918 CrossRef CAS.
- D. Wang and H. Mohwald, J. Mater. Chem., 2004, 14, 459–468 RSC.
- S. Reculusa, C. Poncet-Legrand, A. Perro, E. Duguet, E. Bourgeat-Lami, C. Mingotaud and S. Ravaine, Chem. Mater., 2005, 17, 3338–3344 CrossRef CAS.
- T. Teranishi, Y. Inoue, M. Nakaya, Y. Oumi and T. Sano, J. Am. Chem. Soc., 2004, 126, 9914–9915 CrossRef CAS PubMed.
- A. Pfau, R. Sander and S. Kirsch, Langmuir, 2002, 18, 2880–2887 CrossRef CAS.
- I. U. Khan, C. A. Serra, N. Anton, X. Li, R. Akasov, N. Messaddeq, I. Kraus and T. F. Vandamme, Int. J. Pharm., 2014, 473, 239–249 CrossRef PubMed.
- S. Yang, F. Guo, B. Kiraly, X. Mao, M. Lu, K. W. Leong and T. J. Huang, Lab Chip, 2012, 12, 2097–2102 RSC.
- A. C. Misra, S. Bhaskar, N. Clay and J. Lahann, Adv. Mater., 2012, 24, 3850–3856 CrossRef CAS PubMed.
- S. Han, H. An, H. Tao, L. Li, Y. Qi, Y. Ma, X. Li, R. Wang and J. Zhang, Chem. Commun., 2018, 54, 3174–3177 RSC.
- J. Hu, S. Zhou, Y. Sun, X. Fang and L. Wu, Chem. Soc. Rev., 2012, 41, 4356–4378 RSC.
- X. Huang, Q. Qian and Y. Wang, Small, 2014, 10, 1412–1420 CrossRef CAS PubMed.
- X. Cao, W. Li, T. Ma and H. Dong, RSC Adv., 2015, 5, 79969–79975 RSC.
- F. S. Romanski, J. S. Winkler, R. C. Riccobene and M. S. Tomassone, Langmuir, 2012, 28, 3756–3765 CrossRef CAS PubMed.
- Y. L. Fan, H. W. Hou, H. M. Tay, W. M. Guo, P. O. Berggren and S. C. Loo, AAPS PharmSciTech, 2017, 18, 2648–2657 CrossRef CAS PubMed.
- J. D. Berry, M. J. Neeson, R. R. Dagastine, D. Y. Chan and R. F. Tabor, J. Colloid Interface Sci., 2015, 454, 226–237 CrossRef CAS PubMed.
- C. E. Stauffer, J. Phys. Chem., 1965, 69, 1933–1938 CrossRef CAS.
- W. Li, H. Dong, G. Tang, T. Ma and X. Cao, RSC Adv., 2015, 5, 23181–23188 RSC.
- D. Cheng, X. Cao, H. Gao, X. Ye, W. Li and Y. Wang, RSC Adv., 2014, 4, 9031 RSC.
- A. Loxley and B. Vincent, J. Colloid Interface Sci., 1998, 208, 49–62 CrossRef CAS PubMed.
- S. Torza and S. G. Mason, J. Colloid Interface Sci., 1970, 33, 67–83 CrossRef CAS.
- W. D. Harkins and A. Feldman, J. Am. Chem. Soc., 1922, 44, 2665–2685 CrossRef CAS.
- A. L. Tasker, J. P. Hitchcock, L. He, E. A. Baxter, S. Biggs and O. J. Cayre, J. Colloid Interface Sci., 2016, 484, 10–16 CrossRef CAS PubMed.
- E. J. Pollauf and D. W. Pack, Biomaterials, 2006, 27, 2898–2906 CrossRef CAS PubMed.
- K. J. Pekarek, J. S. Jacob and E. Mathiowitz, Adv. Mater., 1994, 6, 684–687 CrossRef CAS.
- Y. C. Chen, V. Dimonie and M. S. El-Aassei, Pure Appl. Chem., 1992, 64, 1691–1696 CrossRef CAS.
- Y. Wang, C. Zhang, C. Tang, J. Li, K. Shen, J. Liu, X. Qu, J. Li, Q. Wang and Z. Yang, Macromolecules, 2011, 44, 3787–3794 CrossRef CAS.
- A. Marchand, J. H. Weijs, J. H. Snoeijer and B. Andreotti, Am. J. Phys., 2011, 79, 999–1008 CrossRef CAS.
- V. G. Babak, F. Baros, O. Boulanouar, F. Boury, M. Fromm, N. R. Kildeeva, N. Ubrich and P. Maincent, Colloids Surf., B, 2007, 59, 194–207 CrossRef CAS PubMed.
- Y. V. Chernysheva, V. G. Babak, N. R. Kildeeva, F. Boury, J. P. Benoit, N. Ubrich and P. Maincent, Mendeleev Commun., 2003, 13, 65–67 CrossRef.
- H.-Y. Kwon, J.-Y. Lee, S.-W. Choi, Y. Jang and J.-H. Kim, Colloids Surf., A, 2001, 182, 123–130 CrossRef CAS.
- N. Saito, Y. Kagari and M. Okubo, Langmuir, 2007, 23, 5914–5919 CrossRef CAS PubMed.
- R. Deng, H. Li, J. Zhu, B. Li, F. Liang, F. Jia, X. Qu and Z. Yang, Macromolecules, 2016, 49, 1362–1368 CrossRef CAS.
- W. L. Lee, W. M. Guo, V. H. B. Ho, A. Saha, H. C. Chong, N. S. Tan, E. Y. Tan and S. C. J. Loo, Acta Biomater., 2015, 27, 53–65 CrossRef CAS PubMed.
- S. Ohta, S. Hiramoto, Y. Amano, S. Emoto, H. Yamaguchi, H. Ishigami, J. Kitayama and T. Ito, Mol. Pharm., 2017, 14, 3105–3113 CrossRef CAS PubMed.
- Y. L. Fan, H. W. Hou, H. M. Tay, W. M. Guo, P.-O. Berggren and S. C. J. Loo, AAPS PharmSciTech, 2017, 18, 2648–2657 CrossRef CAS PubMed.
- W. H. De Jong and P. J. A. Borm, Int. J. Mol. Med., 2008, 3, 133–149 CAS.
- Z. Lu, J. Wang, M. G. Wientjes and J. L. S. Au, Future Oncol., 2010, 6, 1625–1641 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Raman spectra, drug release profiles, JP size distribution, microparticles fabricated at different glibenclamide concentrations, predicted charge status of glibenclamide at different pH, JP morphology according to Torza–Mason's equation, calculation of interfacial tensions. See DOI: 10.1039/c8ra02271b |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Yuansiang Luia,
Dionaldo Zudhistiraa and
Say Chye Joachim Loo
ab,
Yuansiang Luia,
Dionaldo Zudhistiraa and
Say Chye Joachim Loo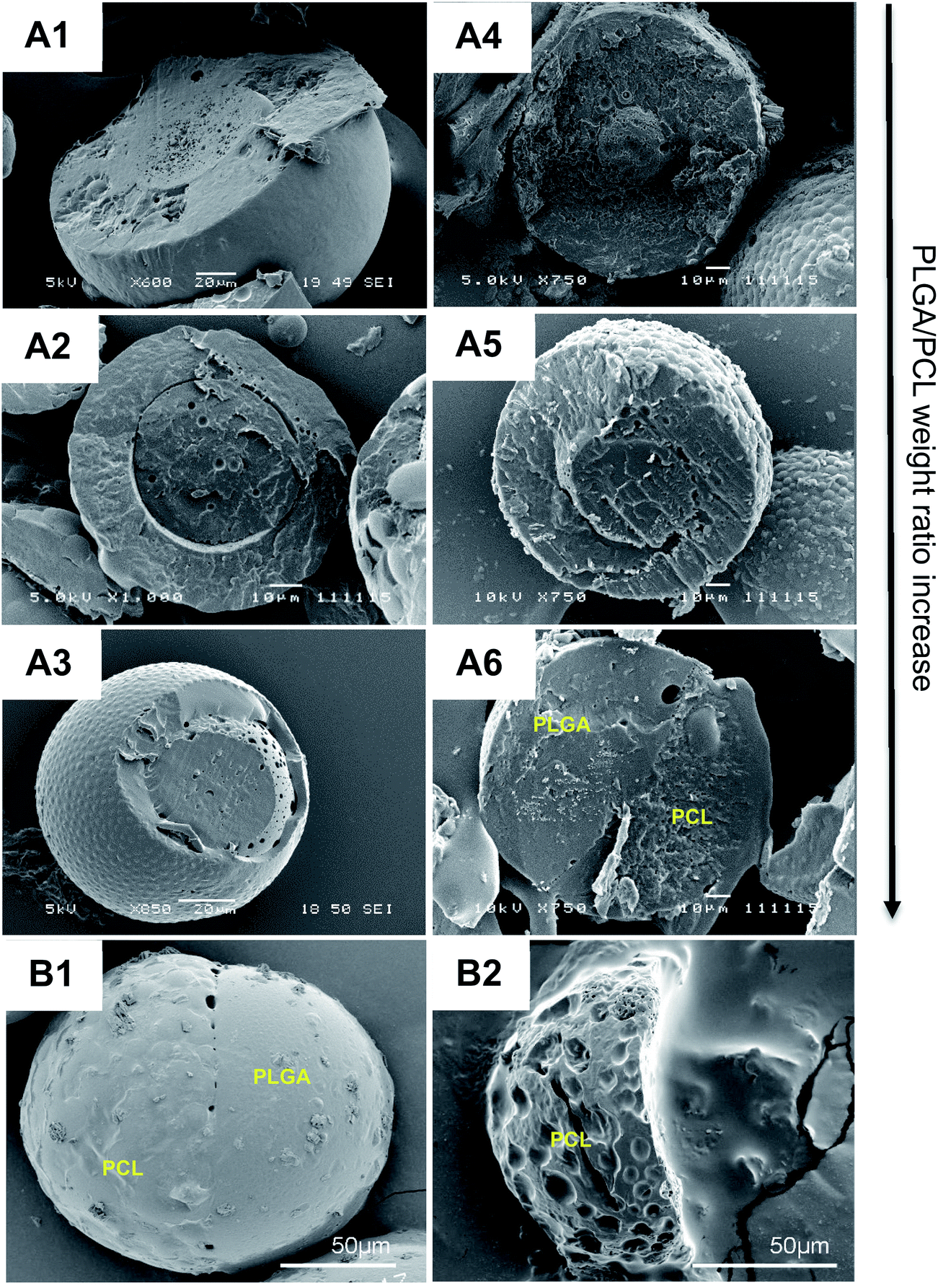

 ) or negatively (
) or negatively ( ) charged, interact with the polymer at specific molar ratios (presumably via hydrophobic or van der Waals interactions), causing the polymer to become more hydrophilic and thus, reducing the interfacial tension at low polymer weight. At high polymer concentration, more drug molecules are expected to interact with the polymer chains, resulting in higher interfacial charge density. This is thermodynamically favorable for interaction between positively charged drug and the polymer as opposite attracts. However, it is unfavorable for negatively charged drug interacting with the polymer when the charge density reaches a repulsive threshold, as similar charges repel. Consequently, addition of a positively charged drug reduces the interfacial tension, while a negatively charged drug increases the interfacial tension at high polymer concentration.
) charged, interact with the polymer at specific molar ratios (presumably via hydrophobic or van der Waals interactions), causing the polymer to become more hydrophilic and thus, reducing the interfacial tension at low polymer weight. At high polymer concentration, more drug molecules are expected to interact with the polymer chains, resulting in higher interfacial charge density. This is thermodynamically favorable for interaction between positively charged drug and the polymer as opposite attracts. However, it is unfavorable for negatively charged drug interacting with the polymer when the charge density reaches a repulsive threshold, as similar charges repel. Consequently, addition of a positively charged drug reduces the interfacial tension, while a negatively charged drug increases the interfacial tension at high polymer concentration.