
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic extractant with an Fe3O4@SiO2 core and aqueous ammonia coating for microextraction of petroleum acids†
Gang-Tian Zhu *a,
Fei Liua,
Sheng Hea,
Xiao-Mei Heb,
Shu-Kui Zhuc and
Yu-Qi Fengb
*a,
Fei Liua,
Sheng Hea,
Xiao-Mei Heb,
Shu-Kui Zhuc and
Yu-Qi Fengb
aKey Laboratory of Tectonics and Petroleum Resources (Ministry of Education), China University of Geosciences, Wuhan 430074, P. R. China. E-mail: zhugangtian@163.com
bKey Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), Department of Chemistry, Wuhan University, Wuhan 430072, P. R. China
cState Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences, Wuhan 430074, P. R. China
First published on 29th May 2018
Abstract
Magnetic aqueous ammonia (MAA) was prepared as a magnetic extractant for dispersive microextraction of petroleum acids (PAs). The amount of extractant in MAA was custom-made by a simple approach. In the MAA composed of an aqueous ammonia coating and Fe3O4@SiO2 core, the coating is a base extractant that can selectively extract acids, while the magnetic core serves as a support to achieve dispersion as well as rapid magnetic retrieval of the extractant during the extraction processes. This is the first use of reusable, stable and modifiable Fe3O4@SiO2 as a support instead of bare Fe3O4 in a magnetic particle assisted dispersive liquid–liquid microextraction technique. The parameters that affect extraction efficiency were investigated. The sampling step as well as the desorption step can be completed in 2 min. The linear ranges are 5–5000 ng g−1, while the limits of quantification range from 2.5 to 6.2 ng g−1. The recoveries in spiked crude oil samples are in the range of 79.1% to 112.1% with relative standard deviations less than 11.3% (intra-day) and 13.4% (inter-day). Finally, the proposed method was applied to the analysis of PAs in diluted crude oils with different maturities. In comparison to the existing methods for extraction of PAs, the proposed method provides superior performances including high throughput (12-well plate), high degree of sample clean-up, and low consumption of separation material, solvent and time.
1. Introduction
The analysis of petroleum acids (PAs) in crude oils is of great significance for studies on corrosion issues in oil refinery,1 petroleum geochemistry2 and oil degradation.3 In addition, the types and content of PAs are important values in environmental risk assessment in oil production, storage and transportation.4 Hence, there is a continuing demand for the determination of PAs in crude oils. Due to the extreme complexity of crude oil, selective extraction of PAs from crude oil is an indispensable step prior to instrumental analysis.To date, liquid–liquid extraction (LLE)2,5 and solid phase extraction (SPE)6,7 are the two main methods for extraction of PAs in crude oil samples. SPE has advantages of solvent-saving (green) and high-throughput. However, compared with SPE, LLE is a more popular technique that can provide a high degree of sample clean-up without the need for the preparation of solid sorbents or specialized devices.8 Besides, to some extent, LLE has better reproducibility than SPE, because the preparation of a solution (liquid extractant) is often more reproducible than the preparation of a functionalized solid sorbent in different labs. Nevertheless, there are some drawbacks of the LLE method including the issues caused by emulsion formation and the large consumptions of time and solvent.9 Liquid phase microextraction (LPME), a relatively environment friendly (green) technique that employs a minimal amount of solvent,10 has been proved to be a good solution to overcome the above problems in various samples,11–13 although it has not yet been applied to the extraction of PAs in crude oils.
Dispersive liquid–liquid microextraction (DLLME) is a relatively new mode of LPME developed in 2006.14 In DLLME, fine droplets composed of disperser solvent and extractant solvent are dispersed in sample solution.15 High extraction efficiency and short extraction equilibrium time can be achieved because of the high contact area between the sample and the dispersive extractant.16,17 However, in traditional DLLME, the retrieval of extractant involves tedious centrifugation steps, and the requirement of the use of extractant with density higher than sample solution restricts the application.18 To overcome these issues, various techniques, including solidification of floating organic droplet,19 solvent de-emulsification,20 in-syringe21 and nanoparticle assisted DLLME have been developed.22 Among them, magnetic nanoparticles assisted DLLME has some unique merits, although it needs external magnet and elution step with additional solution.18,23,24 Using nanoparticles as support to disperse extractant can enhance the mass transfer rate, while the extractant could be retrieved easily under the help of an external magnet. This technique combines the advantages of DLLME and magnetic SPE. However, there are still some drawbacks. The bare Fe3O4, which was used in almost all reported magnetic nanoparticles assisted DLLME methods,18,23–25 is unstable in acidic solution and may lead to irreversible adsorption of acid organic analytes or nonspecific adsorption of interferents, which will restrict the application. Besides, the Fe3O4 support, prepared by using appreciable consumptions of time and reagents, was disposable, which may due to its poor stability and the difficulty of completely desorption of compounds adsorbed on Fe3O4. Moreover, study on preparation of magnetic extractant with custom-made amount of extractant is still lacking. The use of insufficient extractant would decrease the maximum extraction capacity, while excess extractant may lead to low recovery because the extractant free from magnetic support (also contains analytes after extraction) would be discarded.
In this study, attempts were made to improve the magnetic nanoparticles assisted DLLME technique and develop a method based on the improved technique for extraction of PAs. A novel magnetic extractant (magnetic aqueous ammonia, MAA) was prepared for dispersive microextraction of PAs. The amount of the extractant coating was custom-made by a simple method. Fe3O4@SiO2 with good stability and modifiable surface was utilized as the support instead of bare Fe3O4 for the first time. Aqueous ammonia with excellent specificity for acid compounds was selected as the extractant. The MAA was in the form of nanoscale dispersive units during extraction, leading to fast equilibrium and high recovery. Compared with the existing LLE methods for PA extraction,2,5 this first proposed DLLME-based method has the unique properties of easy operation, rapidness, solvent-saving and high throughput while remaining high degree of sample clean-up and good reproducibility.
2. Experimental section
2.1 Chemicals and materials
Ethylene glycol, ethylene diamine, ferric trichloride hexahydrate (FeCl3·6H2O), sodium acetate, ethanol, tetraethyl orthosilicate (TEOS), methanol and aqueous ammonia (NH3·H2O, 25 wt% in H2O) were purchased from Shanghai General Chemical Reagent Factory (Shanghai, China). Trifluoroacetic acid (TFA), dichlorodimethylsilane, trans-4-(trifluoromethyl)cinnamic acid (TCA), decanoic acid (DA), lauric acid (LA), cyclohexanecarboxylic acid (CHA), 1-adamantanecarboxylic acid (ACA), 1-adamantaneacetic acid (AAA) and N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) were purchased from Aladdin Chemical Reagent (Shanghai, China). Bicyclo[2,2,1]heptane-2-carboxylic acid (BCHCA) and 3-noradamantanecarboxylic acid (NACA) were supplied by Thermo Fisher Scientific (MA, USA). Trans-4-ethylcyclohexanecarboxylic acid (t-ECHA), trans-4-isopropylcyclohexanecarboxylic acid (t-iPCHA), trans-4-butylcyclohexanecarboxylic acid (t-BCHA) and trans-4-pentylcyclohexanecarboxylic acid (t-PCHA) were purchased from Adamas-beta (Shanghai, China). HPLC grade n-hexane and acetone were obtained from Fisher Scientific (PA, USA). Crude oils were supported by our cooperative oil company.2.2 Synthesis of Fe3O4@SiO2 nanoparticles
Fe3O4 nanoparticles were synthesized through solvothermal reaction according to the previous works.26,27 Typically, 5 g of FeCl3·6H2O was dissolved in 100 mL of ethylene glycol, then 15 g of sodium acetate and 50 mL of ethylene diamine were added to the solution. After vigorously stirring for 0.5 h, the homogenous mixture was transferred into a Teflon-lined autoclave (200 mL). The autoclave was heated to 200 °C and held for 8 h. Finally, the product was washed by ethanol and deionized water for several times, then dried at 60 °C in a vacuum oven for 6 h.Fe3O4@SiO2 nanoparticles were prepared by the Stöber method28 with some modifications.29 Briefly, 0.6 g of Fe3O4 was homogeneously dispersed in a solution composed of 467 mL of ethanol, 139 mL of deionized water and 15 mL of aqueous ammonia. The dispersion was vigorously stirred for 0.5 h. Then, under continuous stirring, a solution composed of 30 mL of TEOS and 70 mL of ethanol was slowly added to the dispersion. After stirring for 8 h at room temperature, silica was coated onto Fe3O4 to form Fe3O4@SiO2. The product was repeatedly washed by ethanol and deionized water, and then dried at 60 °C for 8 h.
Transmission electron microscopy (TEM) image of the as-prepared Fe3O4@SiO2 was obtained from JEM-2100F transmission electron microscope (JEOL, Tokyo, Japan).
2.3 Hydrophobic modification of glass vial surface
To prevent the attachment and agglomeration of MAA on the wall of glass vial during extraction, the vial was pretreated by silylation to change its hydrophilic surface into hydrophobic surface. Briefly, glass vials were immersed into dichlorodimethylsilane/n-hexane (5/95, v/v) for 12 h. Then the silylated glass vials were taken out from the solution, and washed by methanol and n-hexane for several times, and dried at 60 °C.2.4 Preparation of samples
Crude oils were dissolved in n-hexane (100 mg mL−1) and spiked with 11 PA standards (Table 1) at a certain concentration (100 ng mL−1) to study the extraction efficiency under different conditions. To obtain a blank sample for calibration and validation purposes, a crude oil was fractionated into saturates, aromatics, resins and asphaltenes (SARA fractionation method),30 and then the saturates, aromatics and asphaltenes fractions were remixed together as the matrix free of PAs.| Analytes | Chemical structure | Mw | tR | Quantifier (m/z) |
|---|---|---|---|---|
| CHA | 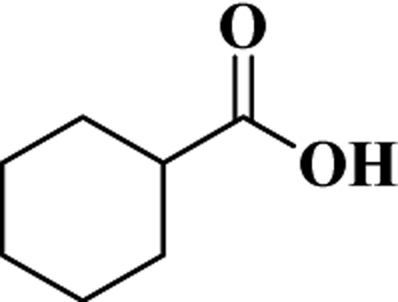 |
128 | 9.2 | 185 |
| t-ECHA | 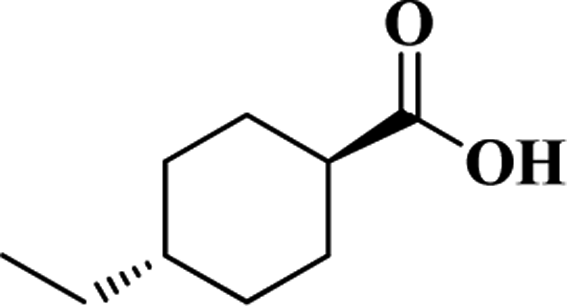 |
156 | 12.6 | 213 |
| t-iPCHA | 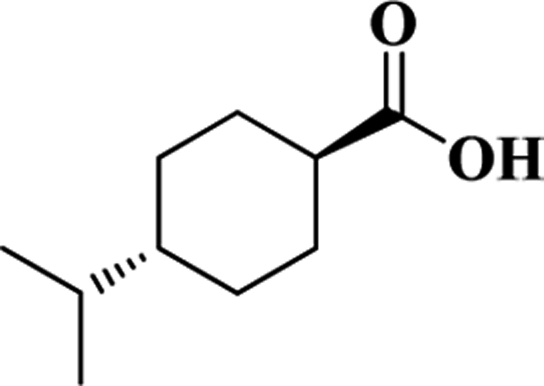 |
170 | 14.4 | 227 |
| t-BCHA |  |
184 | 16.2 | 241 |
| t-PCHA |  |
198 | 17.9 | 255 |
| DA | 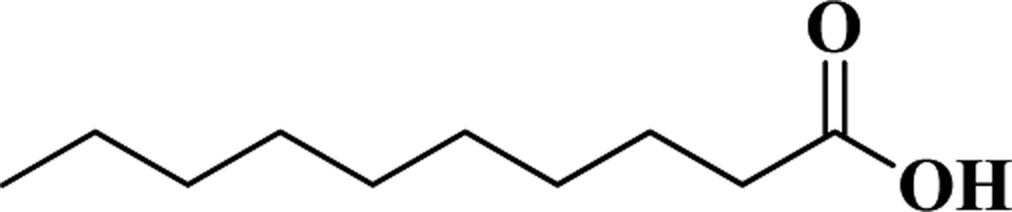 |
172 | 13.9 | 229 |
| LA |  |
200 | 17.4 | 257 |
| BCHCA |  |
140 | 10.8 | 197 |
| NACA |  |
166 | 14.0 | 223 |
| ACA | 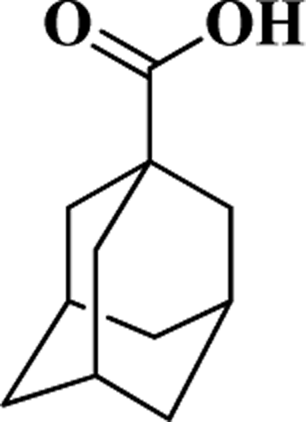 |
180 | 16.5 | 237 |
| AAA | 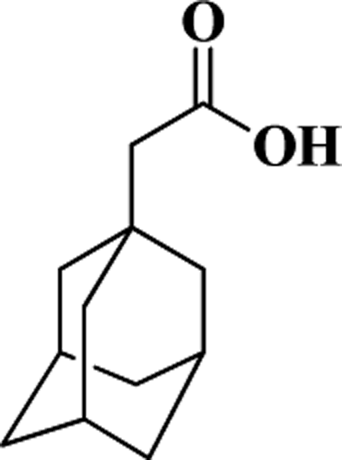 |
194 | 18.1 | 251 |
| TCA (IS) |  |
216 | 15.3 | 273 |
2.5 Dispersive microextraction
The dispersive microextraction procedure is shown in Scheme 1. Fe3O4@SiO2 nanoparticles (3 mg) were put into a silylated glass vial, then 100 μL of aqueous ammonia containing 10 wt% NH3·H2O was added. After vortex for 1 min, the redundant aqueous ammonia was removed to form MAA with custom-made amount of extractant. Then 1 mL of 100 mg mL−1 crude oil sample was added and the mixture was shaken under vortex for 2 min. Under the help of an external magnet, the analytes-trapped MAA was collected in a few seconds, and the solution after extraction was removed and discarded. After washing twice with n-hexane (1 mL) under vortex for 0.5 min, the PAs trapped extractant was desorbed by 200 μL of acetone under vortex for 2 min. The desorption solution was evaporated to dryness under a gentle nitrogen stream at room temperature. The residue was redissolved in 50 μL of n-hexane, and then derivatized with 10 μL of MTBSTFA under vortex at room temperature for 5 min prior to GC-MS analysis.31 | ||
| Scheme 1 (a) The preparation of MAA. (b) The work flow for the dispersive microextraction of PAs. | ||
2.6 GC-MS analysis
Analysis of PAs was carried out on an Agilent GC-MS (CA, USA) consisting of a 7890 GC, a 7683B autosampler and a 5975C MS. The conditions are the same with those in our previous work.31 The GC separation was achieved on an Agilent DB-5MS column (30 m × 0.25 mm × 0.25 μm). The oven temperature was programmed from 100 °C to 210 °C at a rate of 6 °C min−1, then to 300 °C at a rate of 12 °C min−1 and held for 5 min. Helium (99.9995%) was used as the carrier gas at a flow rate of 1.0 mL min−1. The injection volume was 1.0 μL in splitless mode. The temperature of injection port, transfer-line, ion source and quadrupole were held at 300, 300, 230 and 150 °C, respectively. The solvent delay was 8.5 min. The detection was performed in full scan mode with a m/z range from 50 to 550.3. Results and discussion
3.1 Preparations for dispersive microextraction
The whole procedure for the proposed method includes hydrophobic modification of glass vial, preparation of MAA and dispersive microextraction.First, the surface of the glass vial was hydrophobically modified by silylation of the hydroxyl groups on the vial surface. This is an indispensable process, otherwise all the magnetic aqueous extractant in nonpolar or low-polar solution would agglomerate and attach on the vial surface. Compared with the previous works,23,24 the modification method in this work is much simpler without heating and adjustment of pH, and the silane solution can be reused.
Second, as shown in Scheme 1a, Fe3O4 nanoparticles were prepared by a solvothermal reaction according to the previous works.26,27 Then, SiO2 was coated on the Fe3O4 to form Fe3O4@SiO2 by the Stöber method28 with some modifications.29 Subsequently, the Fe3O4@SiO2 and excess amount of extractant were added into a hydrophobically modified glass vial, and some amount of the extractant would be adsorbed on the hydrophilic surface of Fe3O4@SiO2; after removing the redundant extractant free from Fe3O4@SiO2 by a pipette under the help of an external magnet, MAA was obtained. This “overfeed first, then get rid of the redundant” strategy, which is a common method in preparation of saturated solution, ensures the amount of extractant matches perfectly with the adsorption capacity of the support. In other words, the amount of extractant in MAA is custom-made. By using 10 wt% NH3·H2O as extractant, the relation between the weights of the adsorbed extractant and the Fe3O4@SiO2 support is shown in Fig. S1.† It can be seen that the amount of the extractant is proportional to that of Fe3O4@SiO2; the mass ratio of extractant to support is about 4.1.
According to the TEM image of Fe3O4@SiO2 (Fig. S2†), it can be seen that the average diameter of Fe3O4 core is about 130 nm, and the average thickness of SiO2 shell is about 30 nm. When MAA was dispersed in nonpolar or low polar solution under vortex, the aqueous ammonia would aggregate on the hydrophilic surface of dispersive Fe3O4@SiO2 particles and the MAA would be in the form of “nano-droplets”.23 The diameter of the MAA, d, can be expressed as follows:

When the extractant is 10 wt% NH3·H2O, the calculated result indicates that the size of the dispersive MAA is about 430 nm, and the thickness of the extractant coating is about 120 nm. This is the first report on the evaluation of the size of a magnetic extractant.
Third, the as-prepared MAA was directly used for dispersive microextraction of PAs (Scheme 1b). Under vortex, the MAA was in the form of nanoscale dispersive units during extraction, which permits a fast mass transfer between phases because of the extremely high contact area between the sample and the extractant. In the sampling process, PAs were selectively trapped into the base extractant. Then the extractant was retrieved together with the Fe3O4@SiO2 support under the help of an external magnet. In the desorption process, the trapped analytes as well as the extractant were desorbed from the Fe3O4@SiO2 support, and the support could be reused. Note that during the extraction processes, a 12-well plate was utilized to improve the throughput of the method.
3.2 Condition optimization
To investigate the influence of the amount of extractant on extraction efficiency, MAA with excess amount of extractant was also prepared and used as a comparison (Fig. 1a). As shown in Fig. 1b, the excess amount of extractant resulted in low recovery, because the redundant extractant free from Fe3O4@SiO2, which also contained analytes, could not be retrieved and would be discarded together with sample solution after extraction. This result indicates the necessity of controlling the amount of extractant in preparation of magnetic extractant. | ||
| Fig. 1 (a) Schematic of the dispersive microextraction with MAA containing excess amount of extractant. (b) Comparison of the extraction efficiencies of MAA with custom-made amount of extractant and excess amount of extractant. | ||
A series of parameters including type of extractant, amount of Fe3O4@SiO2, extraction time and desorption time were investigated to achieve the best performance.
Different types of aqueous solutions, including acidic solution (TFA/H2O, 1/99, v/v), pure water and basic solution (1 wt%, 5 wt% and 10 wt% NH3·H2O in H2O) were used for coating Fe3O4@SiO2 to prepare magnetic extractants. Additionally, Fe3O4@SiO2 without any extractant was used as a comparison. The results are shown in Fig. 2 and S3.† It can be seen that Fe3O4@SiO2 without any extractant can also adsorb PAs, although with poor recovery, which may result from the hydrogen bonding interaction between PAs and hydroxyl groups on the surface of Fe3O4@SiO2.31 The acidic extractant possesses the worst extraction efficiency due to the exclusion effect between TFA and PAs. The pure water extractant shows acceptable recovery to some PAs; however, interestingly, the water extractant has poor recoveries to the PAs with relatively longer aliphatic chains. The basic extractants display good extraction efficiency to all the PAs, because base can promote the ionization of PAs and facilitate the mass transfer of PAs from sample solution to extractant. Among the three basic extractants with different contents of NH3·H2O, 10 wt% NH3·H2O in H2O possesses the superior performance (Fig. S3†). Therefore, 10 wt% NH3·H2O in H2O was selected to prepare magnetic extractant in the further experiments.
 | ||
| Fig. 2 Effect of the type of extractant on extraction efficiency. The amount of Fe3O4@SiO2 was 10 mg. | ||
Various amounts of Fe3O4@SiO2 ranging from 1 to 20 mg were utilized to prepare MAA for dispersive microextraction. The results are shown in Fig. S4.† When only 1 mg of Fe3O4@SiO2 was used, the recoveries can also higher than 80% to all PAs. This may profit from the extremely abundant contact area between the analytes and the nanoscale magnetic extractant. The recoveries remain almost unchanged from 2 to 20 mg of Fe3O4@SiO2. Considering the stability and the effect of aqueous solution in the subsequent evaporation step, 3 mg of Fe3O4@SiO2 was used in the further experiments.
The effect of extraction time was examined from 1 to 30 min. As shown in Fig. S5,† the extraction equilibrium can be reached in 2 min. Such a fast equilibrium comes from the rapid mass transfer between the analytes and the dispersive nanoscale magnetic extractant. In the further experiments, the extraction time for sampling was kept at 2 min.
Acetone was chosen as the desorption solution because of its mutual solubility with water, good solubility to PAs and low boiling point. As shown in Scheme 1b, in the desorption process, the aqueous extractant as well as the analytes was dissolved from Fe3O4@SiO2 support by acetone. The desorption time was optimized. Fig. S6† shows that, with the increase of desorption time from 2 to 30 min, there are no obvious variations for the recoveries of PAs. Therefore, 2 min of desorption time was selected for the further experiments.
Under these optimized conditions, the sample clean-up of the proposed method was studied by analysis of crude oil samples spiked with PAs before and after extraction. The results are shown in Fig. 3. In the direct analysis, high abundances of hydrocarbons dominate the chromatogram, and it is impossible to identify the signals of PAs from the chromatogram (Fig. 3a). In contrast, after the microextraction, all the 11 PAs were easily detected with few interference signals, indicating high degree of sample clean-up (Fig. 3b). Additionally, to further investigate the sample clean-up, the same crude oil without added PAs (a real sample) was also extracted by MAA. As shown in Fig. S7,† most of the detected signals can be assigned to native acids in the real sample, and some are polydimethylsiloxanes from column bleeding or injection port septum; no obvious hydrocarbon signal was detected. This result further demonstrates the high degree of sample clean-up in the proposed method.
 | ||
| Fig. 3 The total ion chromatograms of crude oil samples spiked with PAs before (a) and after (b) extraction with MAA followed by analysis with GC-MS in full-scan mode. To protect GC-MS from overload or pollution, sample (a) used 50 μL of 10 mg mL−1 crude oil in hexane spiked with 2 μg mL−1 PAs for derivatization and analysis. Sample (b) was prepared by using 1 mL of 100 mg mL−1 crude oil in hexane spiked with 100 ng mL−1 PAs for dispersive extraction with MAA. | ||
3.3 Reusability
The reusability of the Fe3O4@SiO2 support was studied. Fig. S8† shows that there is no obvious difference in the extraction efficiency for the same Fe3O4@SiO2 after used for 0, 10, 20 and 30 times, demonstrating that the Fe3O4@SiO2 is stable and can be reused at least 30 times. This is the first time to achieve the reuse of the magnetic support in the magnetic particles assisted DLLME technique. It can significantly increase the practical value of the technique, because the major consumption (time and reagent) in preparation of a magnetic extractant is the preparation of the magnetic particles. One of the reasons for the feasibility of reuse is that the Fe3O4@SiO2 can hardly contact oil sample directly under protection by the aqueous coating. The other one reason is that the SiO2 surface has poor or no retention for most organics in the mixture of acetone and aqueous ammonia. Another benefit of the SiO2 shell is its modifiable surface, which endows the feasibility to design magnetic particles with specific surface property for given requirements in magnetic particles assisted DLLME.3.4 Background contamination
To check the background levels of contamination in the whole procedure, equipment blank (EB), laboratory reagent blank (LRB), procedural blank (PB) and matrix blank (MB) were analyzed.32 The EB was reagent hexane objected into GC-MS without any extraction or purification step. The LRB was hexane injected between samples to confirm that there was no carry-over of PAs between injections. The PB was hexane that was processed similarly to oil samples by passing through the whole analytical procedure. The MB was the mixture of saturates, aromatics and asphaltenes fractions obtained from SARA fractionation of crude oils, which was analyzed after passing through the whole analytical procedure. All these blanks did not contain assignable targeted PA signal. This indicates that there was no background contamination in the laboratory supplies and the devices used in the whole analytical procedure, and the mixture of saturates, aromatics and asphaltenes fractions can be used as a blank sample for calibration and validation purposes.3.5 Method validation
To validate the dispersive microextraction method for analysis of PAs, calibration curves were generated by analysis of blank samples (the mixture of saturates, aromatics and asphaltenes fractions) spiked with PA standards and IS. Table S1† shows that good linear correlations were obtained with correlation coefficients (R) higher than 0.9954. The linear ranges are 5–5000 ng g−1. The limits of detection (LODs) and limits of quantification (LOQs), which were calculated as the signal to noise ratios of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 10:1, respectively, range from 0.7 to 1.9 ng g−1 and 2.5 to 6.2 ng g−1, respectively (Table S2†). Recoveries were obtained by using crude oil samples spiked with PAs at 10, 100 and 1000 ng g−1. First, the concentrations (C0) of the 11 PAs in the original crude oil sample (without added PAs) were measured and calculated based on the calibration curves. Second, the concentrations (C1) of PAs in the crude oil samples spiked with PAs were measured by the same method. Then the measured amounts (C2) of the spiked PAs were obtained by subtracting C0 from C1. Finally, the recoveries were obtained by comparing the measured value (C2) with the corresponding spiked value (10, 100 or 1000 ng g−1). Table S2† displays that the mean recoveries are between 79.1% and 112.1%, and the intra-day and inter-day RSDs are less than 11.3% and 13.4%, respectively. These results demonstrate that the accuracy and the precision of the proposed method are acceptable for the analysis of PAs.
:1 and 10:1, respectively, range from 0.7 to 1.9 ng g−1 and 2.5 to 6.2 ng g−1, respectively (Table S2†). Recoveries were obtained by using crude oil samples spiked with PAs at 10, 100 and 1000 ng g−1. First, the concentrations (C0) of the 11 PAs in the original crude oil sample (without added PAs) were measured and calculated based on the calibration curves. Second, the concentrations (C1) of PAs in the crude oil samples spiked with PAs were measured by the same method. Then the measured amounts (C2) of the spiked PAs were obtained by subtracting C0 from C1. Finally, the recoveries were obtained by comparing the measured value (C2) with the corresponding spiked value (10, 100 or 1000 ng g−1). Table S2† displays that the mean recoveries are between 79.1% and 112.1%, and the intra-day and inter-day RSDs are less than 11.3% and 13.4%, respectively. These results demonstrate that the accuracy and the precision of the proposed method are acceptable for the analysis of PAs.
3.6 Applications in real samples
In general, PAs (also known as naphthenic acids) are more likely to be found in immature or biodegraded crude oils.2,33 Here, the proposed method was applied to determine the 11 PAs in two immature and two mature crude oils. As listed in Table 2, 8, 3, 2 and 2 PAs were detected in the four crude oil samples, respectively. These results also preliminary indicate that the varieties or the concentrations of PAs in the two immature oils are more than those in the two mature oils. The expanded uncertainties of the determined contents of the PAs in Table 2 were calculated according to the Guide to the Expression of Uncertainty in Measurement.34,35 The calculation details are shown in ESI.†| Crude oil (maturity) | Contents ± expanded uncertainties (ng g−1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CHA | t-ECHA | t-iPCHA | t-BCHA | t-PCHA | DA | LA | BCHCA | NACA | ACA | AAA | |
| a Not detected. | |||||||||||
| 1 (immature) | —a | 24.6 ± 2.7 | 34.2 ± 3.1 | 48.4 ± 4.3 | 55.7 ± 4.7 | 182.5 ± 19.0 | 290.2 ± 24.4 | — | — | 41.3 ± 3.0 | 38.1 ± 3.4 |
| 2 (immature) | 19.9 ± 4.1 | — | — | — | — | 369.0 ± 30.9 | 808.2 ± 58.7 | — | — | — | — |
| 3 (mature) | — | — | — | — | — | 35.8 ± 3.7 | 75.7 ± 7.4 | — | — | — | — |
| 4 (mature) | — | — | — | — | — | 37.9 ± 3.5 | 79.6 ± 7.1 | — | — | — | — |
3.7 Comparison with the existing methods
To assess the analytical performance of this proposed method, a comparison with the existing methods for analysis of PAs is shown in Table 3. Among these methods, the proposed method needs low consumptions of sample, solvent and time; moreover, because of the reusability of the Fe3O4@SiO2 support, the actual consumption of the separation material in the proposed method is the lowest. These benefits make the consumptions of the proposed method lower compared with the existing methods.| Method | Sample | Consumption | Throughputa | Instrument | LOQ (ng g−1) | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| Separation material | Solvent (mL) | Time (min) | ||||||
| a The number of samples for simultaneously handling.b Not provided.c Alumina modified with 1,4-bis-(n-propyl)diazoniabicyclo[2.2.2]octane chloride silsesquioxane.d Amino-functionalized silica.e 0.25 M solution of sodium hydroxide in 30% methanol. | ||||||||
| SPE | Crude oil (1–2 g) | SAX (10 g) | >260 | >60 | —b | GC-MS | — | 6 |
| SPE | Crude oil (-) | Dab-Al2O3c (0.5 g) | 55 | >30 | — | GC-MS | — | 7 |
| Micro-SPE | Crude oil (0.1 g) | AFSd (5 mg) | 3 | <5 | 1 | GC-MS | 6–20 | 31 |
| LLE | Crude oil (15 g) | NaOH/MeOHe (50 mL) and hexane (10 mL) | 190 | >60 | 1 | GC × GC-MS | — | 2 |
| LLE | Tailings water (100 mL) | HAc/EtAc (75 mL) | >85 | >40 | 1 | LC-MS | 0.3–6 | 39 |
| Centrifugation | River water (10 mL) | — | 0 | >5 | — | LC-MS | 100 | 40 |
| Dispersive microextraction | Crude oil (0.1 g) | NH3·H2O/H2O (100 μL) | 3.2 | 6 | 12 | GC-MS | 2.5–6.2 | This work |
As for green aspects,36 the proposed microextraction method has some definite advantages. A green LPME-based approach is often described in literature as the three R's, which stands for replace, reduce and recycle (replacement of toxic solvents with green solvents, reduction of solvent consumption and waste production, and solvent recycling).10,37,38 In this work, organic solvents were still needed. Hexane was used to dilute or dissolve crude oil sample, and acetone was used in the desorption step. However, profited by the small scale of the extraction, only a small amount of organic solvent was employed. In addition, different from the existing LLE methods that used large amount of organic solvent as extractant (Table 3), the proposed dispersive microextraction method employed small amount of aqueous solution as extractant. Besides, the Fe3O4@SiO2 support could be reused. Hence, compared with the previous related works (Table 3), the proposed method is relatively more in line with a green approach.
Another merit of the proposed method is that the throughput can be easily scaled up by a multi-well plate. Additionally, due to the extremely high contact area between the analytes and the dispersive nanoscale magnetic extractant, the proposed microextraction method possesses high sensitivity.
4. Conclusions
In summary, a novel magnetic extractant (MAA) was successfully prepared for the dispersive microextraction of petroleum acids (PAs) in crude oils. In the MAA, the Fe3O4 core endows the magnetic retrieval property, the SiO2 shell protects Fe3O4 from acid and provides hydrophilic surface to adsorb extractant, while the aqueous ammonia extractant offers selective adsorption of PAs. The amount of extractant was custom-made by a simple approach, which can be applied to prepare other magnetic extractants for specific applications. Profited by the high contact area between the analytes and the dispersive nanoscale magnetic extractant, the method possesses fast extraction equilibrium and high recovery. The throughput can be easily scaled up by a multi-well plate. The reusability of the magnetic silica support makes the method low-cost. Additionally, the high degree of sample clean-up and the good reproducibility further suggest the great potential of the proposed method in the analysis of PAs in crude oils. We would like to combine this technique with comprehensive two-dimensional gas chromatography/mass spectrometry for comprehensive profiling PAs in crude oils. We also believe magnetic silica coated with polar extractant (aqueous solution, methanol) will be a promising material in extraction of polar analytes from edible oil and petroleum samples; and the modifiability of the magnetic silica might derive more types of magnetic extractants such as nonpolar or low polar extractant coated hydrophobically modified magnetic silica.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (21607137), Fundamental Research Funds for the Central Universities, China University of Geosciences (Wuhan) (CUG160606), and China National Science and Technology Major Project (2016ZX05006003-001).Notes and references
- M. P. Barrow, L. A. McDonnell, X. D. Feng, J. Walker and P. J. Derrick, Anal. Chem., 2003, 75, 860–866 CrossRef PubMed.
- C. E. West, J. Pureveen, A. G. Scarlett, S. K. Lengger, M. J. Wilde, F. Korndorffer, E. W. Tegelaar and S. J. Rowland, Rapid Commun. Mass Spectrom., 2014, 28, 1023–1032 CrossRef PubMed.
- X. Fu, Z. Dai, S. Tian, J. Long, S. Hou and X. Wang, Energy Fuels, 2008, 22, 1923–1929 CrossRef.
- I. Dzidic, A. C. Somerville, J. C. Raia and H. V. Hart, Anal. Chem., 1988, 60, 1318–1323 CrossRef.
- W. K. Seifert and W. G. Howells, Anal. Chem., 1969, 41, 554–562 CrossRef.
- D. M. Jones, J. S. Watson, W. Meredith, M. Chen and B. Bennett, Anal. Chem., 2001, 73, 703–707 CrossRef PubMed.
- J. F. de Conto, S. Nascimento Jdos, D. M. de Souza, L. P. da Costa, S. M. Egues, S. Freitas Ldos and E. V. Benvenutti, J. Sep. Sci., 2012, 35, 1044–1049 CrossRef PubMed.
- J. Płotka-Wasylka, K. Owczarek and J. Namieśnik, TrAC, Trends Anal. Chem., 2016, 85, 46–64 CrossRef.
- Y. Yan, X. Chen, S. Hu and X. Bai, J. Chromatogr. A, 2014, 1368, 1–17 CrossRef PubMed.
- A. Spietelun, L. Marcinkowski, M. de la Guardia and J. Namiesnik, Talanta, 2014, 119, 34–45 CrossRef PubMed.
- C. Huang, K. F. Seip, A. Gjelstad, X. Shen and S. Pedersen-Bjergaard, Anal. Chem., 2015, 87, 6951–6957 CrossRef PubMed.
- J. Lee and H. K. Lee, Anal. Chem., 2011, 83, 6856–6861 CrossRef PubMed.
- L. Guo, N. Binte Nawi and H. K. Lee, Anal. Chem., 2016, 88, 8409–8414 CrossRef PubMed.
- M. Rezaee, Y. Assadi, M. R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef PubMed.
- M. Rezaee, Y. Yamini and M. Faraji, J. Chromatogr. A, 2010, 1217, 2342–2357 CrossRef PubMed.
- M. I. Leong, M. R. Fuh and S. D. Huang, J. Chromatogr. A, 2014, 1335, 2–14 CrossRef PubMed.
- L. Guo, S. H. Chia and H. K. Lee, Anal. Chem., 2016, 88, 2548–2552 CrossRef PubMed.
- Z. G. Shi and H. K. Lee, Anal. Chem., 2010, 82, 1540–1545 CrossRef PubMed.
- F. R. Mansour and N. D. Danielson, Talanta, 2017, 170, 22–35 CrossRef PubMed.
- H. Chen, R. Chen and S. Li, J. Chromatogr. A, 2010, 1217, 1244–1248 CrossRef PubMed.
- F. Maya, B. Horstkotte, J. M. Estela and V. Cerda, Anal. Bioanal. Chem., 2012, 404, 909–917 CrossRef PubMed.
- N. Campillo, P. Viñas, J. Šandrejová and V. Andruch, Appl. Spectrosc. Rev., 2016, 52, 267–415 CrossRef.
- Q. Zhao, F. Wei, N. Xiao, Q. W. Yu, B. F. Yuan and Y. Q. Feng, J. Chromatogr. A, 2012, 1240, 45–51 CrossRef PubMed.
- Q. Zhao, Q. Lu, Q. W. Yu and Y. Q. Feng, J. Agric. Food Chem., 2013, 61, 5397–5403 CrossRef PubMed.
- R. A. Perez, B. Albero, J. L. Tadeo and C. Sanchez-Brunete, Anal. Bioanal. Chem., 2016, 408, 8013–8023 CrossRef PubMed.
- Q. Gao, D. Luo, M. Bai, Z. W. Chen and Y. Q. Feng, J. Agric. Food Chem., 2011, 59, 8543–8549 CrossRef PubMed.
- S. N. Xu, Q. Zhao, H. B. He, B. F. Yuan, Y. Q. Feng and Q. W. Yu, Anal. Methods, 2014, 6, 7046–7053 RSC.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- G. T. Zhu, X. S. Li, Q. Gao, N. W. Zhao, B. F. Yuan and Y. Q. Feng, J. Chromatogr. A, 2012, 1224, 11–18 CrossRef PubMed.
- W. Zhang, S. Zhu, L. Pang, X. Gao, G. T. Zhu and D. Li, J. Chromatogr. A, 2016, 1478, 75–83 CrossRef PubMed.
- G. T. Zhu, S. He, X. M. He, S. K. Zhu and Y. Q. Feng, RSC Adv., 2017, 7, 40608–40614 RSC.
- U. J. Kim and K. Kannan, Anal. Chem., 2018, 90, 3291–3298 CrossRef PubMed.
- J. A. Valencia-Dávila, C. Blanco-Tirado and M. Y. Combariza, Fuel, 2017, 193, 168–177 CrossRef.
- P. Konieczka and J. Namiesnik, Anal. Chem., 2008, 53, 785–796 Search PubMed.
- P. Konieczka and J. Namiesnik, J. Chromatogr. A, 2010, 1217, 882–891 CrossRef PubMed.
- J. Płotka-Wasylka, Talanta, 2018, 181, 204–209 CrossRef PubMed.
- C. J. Welch, N. Wu, M. Biba, R. Hartman, T. Brkovic, X. Gong, R. Helmy, W. Schafer, J. Cuff and Z. Pirzada, TrAC, Trends Anal. Chem., 2010, 29, 667–680 CrossRef.
- J. Plotka, M. Tobiszewski, A. M. Sulej, M. Kupska, T. Gorecki and J. Namiesnik, J. Chromatogr. A, 2013, 1307, 1–20 CrossRef PubMed.
- M. Bataineh, A. C. Scott, P. M. Fedorak and J. W. Martin, Anal. Chem., 2006, 78, 8354–8361 CrossRef PubMed.
- D. Shang, M. Kim, M. Haberl and A. Legzdins, J. Chromatogr. A, 2013, 1278, 98–107 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02262c |
| This journal is © The Royal Society of Chemistry 2018 |