
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RNA: packaged and protected by VLPs†
Po-Yu Fanga,
Jessica C. Bowman a,
Lizzette M. Gómez Ramosab,
Chiaolong Hsiaoc and
Loren Dean Williams*a
a,
Lizzette M. Gómez Ramosab,
Chiaolong Hsiaoc and
Loren Dean Williams*a
aSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: loren.williams@chemistry.gatech.edu
bSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA
cInstitute of Biochemical Sciences, National Taiwan University, Taipei 10617, Taiwan, Republic of China
First published on 12th June 2018
Abstract
Virus Like Particles (VLPs) are devices for RNA packaging, protection and delivery, with utility in fundamental research, drug discovery, and disease treatment. Using E. coli for combined expression and packaging of non-viral RNAs into Qβ VLPs, we investigated the extent of chemical protection conferred by packaging of RNA in VLPs. We also probed relationships between packaging efficiency and RNA size, sequence and intrinsic compaction. We observe that VLP packaging protects RNA against assault by small diffusible damaging agents such as hydroxyl radicals and divalent cations. By contrast, the extent of unmediated cleavage, in the absence of reactive species, is the same for RNA that is free or packaged within VLPs, and is very slow. In vivo packaging of RNA within VLPs appears to be more efficient for intrinsically compact RNAs, such as rRNA, and less efficient for unstructured, elongated RNA such as mRNA. Packaging efficiency is reduced by addition of the ribosome binding site to a target RNA. The Qβ hairpin is necessary but not sufficient for efficient packaging.
Introduction
RNA-based tools have utility in fundamental research, drug discovery, and disease treatment1–3 in part because of the programmable specificity of base-pairing interactions. Small interfering RNAs (siRNAs) and microRNAs (miRNAs) modulate gene expression by inhibiting translation or by stimulating degradation of mRNAs.4,5 RNA can be active as antibacterial or antiviral agents.6,7Impediments to broadly applicable RNA-based therapeutics remain. Cellular uptake is inhibited by the high anionic charge of RNA.8 Chemical and biological stability of RNA is low.9,10 RNA production on large scales by in vitro transcription11 or solid-phase synthesis12 is problematic.13,14
The packaging of RNA by protein(s) in vivo, in coupled RNA production and packaging systems, may be effective for overcoming at least a subset of these challenges.15 RNA expression in vivo16–18 can employ recombinant nucleic acids that encode polymerase promoters, affinity tags, functional ‘target’ sequences and genes for packaging proteins.
Virus Like Particles (VLPs) are packaging systems that can be used to deliver RNA and other cargo. Qβ and MS2 VLPs are derived from single-strand RNA bacteriophages. Their genomes are small and their protein capsids are T = 3 icosahedral nanoparticles with diameters of ∼30 nm, formed of 180 copies of the coat protein (CP).19–21 Qβ VLPs are hollow with pores 1.4 nm in diameter.20
Lau et al.22 and Fang et al.15 previously demonstrated packaging of non-viral RNA in Qβ VLPs by co-expressing target RNA and CP from distinct plasmids in E. coli. In vivo packaging of target RNA in VLPs is promoted by an RNA hairpin (hp), derived from Qβ RNA, which has high affinity for CP (Fig. 1).23,24 CP binds to hp during VLP assembly in vivo through RNA backbone, sugar (2′ hydroxyl), and sequence-specific interactions.25 RNA secondary structure also promotes viral assembly; RNA stem-loops are thought to promote capsid formation, while CP interactions with RNA promote RNA collapse.26,27
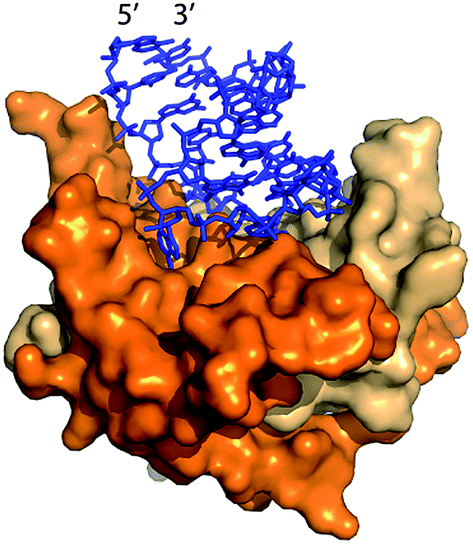 | ||
| Fig. 1 The Qβ hairpin (blue) in complex with the coat protein (two copies are shown) in the assembled Qβ VLP (PDB 4L8H).25 | ||
Biological stability of RNA can be increased by fusing target RNA to stable folds such as 5S rRNA or tRNA. These fusions allow simultaneous production and purification of active RNA in fermentation-based systems.28–33 tRNA fusions were produced and packaged in MS2 VLPs within E. coli by Ponchon et al.14 We recently demonstrated simultaneous production and packaging of active RNAi in Qβ VLPs using a novel scaffold.15
Here, we explore factors that influence the stability and quantity of target RNA that is packaged in Qβ VLPs in vivo. We report that packaging in VLPs chemically protects RNA from small diffusible chemicals that can readily penetrate the VLP. It appears that packaging chemically stabilizes RNA by mechanisms beyond direct exclusion of reactive species from proximity to the RNA. We show that intrinsic compaction of RNA may increase the efficiency of assembly. Intrinsically compact RNA, here derived from rRNA, appears to package with high efficiency in vivo.
Methods
Qβ CP and Qβ VLP–RNA expression vectors
A two-plasmid expression system was used for production of Qβ VLPs and non-viral RNA in E. coli (Fig. 2). Two origins of replication and two antibiotic resistance markers were used within the same host. The pBR322 origin of replication and kanamycin resistance gene of pET-28b (+) (Novagen) are compatible with the CloDF13 origin and streptomycin resistance gene in pCDF-1b (Novagen), allowing for co-expression in the same bacterial cell. Therefore, we chose pCDF-1b as the coat protein expression vector (pCDF-CP, Fig. 2a) and pET-28b (+) as the RNA expression vector (pET-RNA, Fig. 2b). The non-viral RNAs and Qβ CP were cloned downstream of the T7 promoter/lac operator.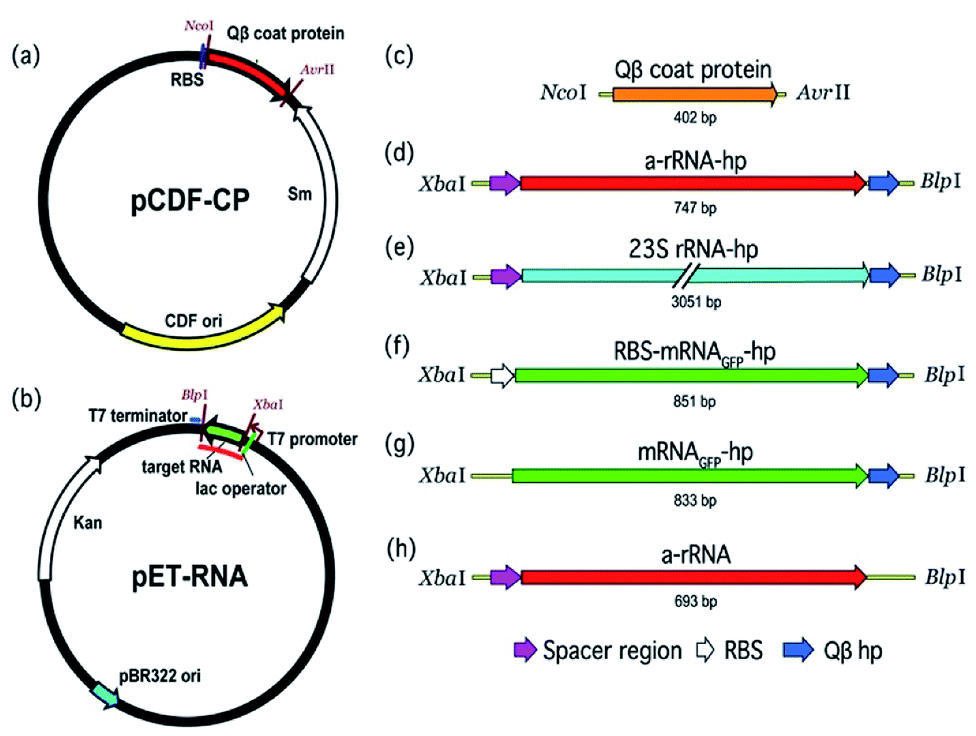 | ||
| Fig. 2 The pCDF-CP and pET-RNA plasmids used to co-express VLP protein and RNA in E. coli. (a) The protein expression vector (pCDF-CP) encoding Qβ VLP. (b) The generic RNA production vector (pET-RNA) used to express the series of RNAs in this study. The red line on pET-RNA plasmid (b) indicates the RNA transcript. (c) The gene cloned to pCDF-1b to create pCDF-CP is Qβ coat protein. (d–h) Genes cloned to pET-28b (+) to create the series of pET-RNAs are (d) a-rRNA-hp, (e) 23S rRNA-hp, (f) RBS-mRNAGFP-hp, (g) mRNAGFP-hp, and (h) a-rRNA. ‘hp’ is Qβ hairpin; ‘RBS’ is ribosomal binding site. | ||
Expression of Qβ VLP and Qβ VLP–RNA
E. coli BL21(DE3) containing pCDF-CP vector was inoculated in NZY medium (1.0% select peptone (NZ amine), 0.5% sodium chloride, 0.5% yeast extract) containing streptomycin (50 μg mL−1) and incubated overnight at 37 °C. Qβ VLP production was initiated with the addition of 1% overnight culture to ZYM-5052 auto-induction medium.35 Production culture was incubated for 24 h at 37 °C. Cells were collected by centrifugation at 4 °C and 6500 × g for 30 min. Cells were resuspended in an equal volume of Qβ buffer (10 mM MgCl2 and 20 mM Tris–HCl, pH 7.5) and lysed by sonication at 40 watts for 3 min with 10 s on/off intervals. The lysate was centrifuged for 30 minutes at 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 × g. Ammonium sulfate precipitation (2 M) was followed by 30 minute centrifugation at 23400 × g to obtain crude VLPs, which were suspended in 1 mL Qβ buffer and extracted three times with 1:1 n-butanol:chloroform. VLPs in the aqueous layer were purified by step sucrose gradient ultracentrifugation (10–40% w/v) at 40000 rpm for 2 hours. Qβ VLPs were precipitated with 20% w/v PEG8000. Resuspended Qβ VLPs were dialyzed against dialysis buffer (20 mM Tris–HCl, pH 7.5 and 50 mM NaCl) for 2 h. Qβ VLPs were characterized by SDS polyacrylamide gel electrophoresis. Qβ VLP–RNA complexes were obtained as described above but by co-expression of pCDF-CP/pET-RNA selected with streptomycin/kanamycin (50 μg mL−1).
400 × g. Ammonium sulfate precipitation (2 M) was followed by 30 minute centrifugation at 23400 × g to obtain crude VLPs, which were suspended in 1 mL Qβ buffer and extracted three times with 1:1 n-butanol:chloroform. VLPs in the aqueous layer were purified by step sucrose gradient ultracentrifugation (10–40% w/v) at 40000 rpm for 2 hours. Qβ VLPs were precipitated with 20% w/v PEG8000. Resuspended Qβ VLPs were dialyzed against dialysis buffer (20 mM Tris–HCl, pH 7.5 and 50 mM NaCl) for 2 h. Qβ VLPs were characterized by SDS polyacrylamide gel electrophoresis. Qβ VLP–RNA complexes were obtained as described above but by co-expression of pCDF-CP/pET-RNA selected with streptomycin/kanamycin (50 μg mL−1).
RNA extraction from Qβ VLPs
To extract RNA from purified VLPs, 50 μg of VLP was added to extraction buffer (5% SDS, 25 mM DTT, 20 mM Tris–HCl, pH 7.5, and 50 mM NaCl) to 250 μL. The solution was incubated at room temperature for 20 minutes. An equivalent volume of low pH phenol–chloroform was then added to the solution. RNA extracts were centrifuged for 15 minutes at 16300 × g. The RNA retained in the aqueous phase was precipitated in ammonium acetate at −20 °C for 24 h followed by 15 minute centrifugation at 16300 × g. The RNA pellet was washed three times with 80% ethanol. RNA was dissolved in nuclease free water and stored at −80 °C. In this extraction method, RNA yield is about 4–5 μg for every 50 μg VLP or VLP–RNA. RNA samples were analyzed by denaturing urea polyacrylamide gel electrophoresis or Agilent 2100 Bioanalyzer.
Transmission electron microscopy
TEM images were obtained with a Hitachi H-7500 electron microscope. Samples were prepared by fixing with 250 μL of glutaraldehyde (8%) per 500 μL of sample (1 μg mL−1). A sample volume of 15 μL was deposited on a copper grid and incubated for 5 min. Excess sample was wicked off with filter paper. The grid was then placed face down on a droplet of 2% ammonium molybdate stain for 2 min. Excess stain was wicked off with filter paper and the grid was dried for 2 h before imaging.Hydroxyl radical cleavage
Following established methods,36 VLP–RNA assemblies (250 μg) or unpackaged RNA (25 μg) were added to pre-cleavage solution (0.3% H2O2, 3.3 mM sodium ascorbate, and 20 mM Tris–HCl, pH 7.4) to a volume of 50 μL. Cleavage reactions were initiated by addition of 10 μL of freshly prepared 200 μM iron(II) EDTA with a 2:1 ratio of (NH4)2Fe(SO4)2·6H2O to EDTA. Reactions were quenched at various timepoints (0–180 minutes) by addition of 20 μL of 0.1 M thiourea. For reactions with varying concentrations of hydroxyl radical, 10 μL of iron(II) EDTA (0–1500 μM) was prepared and added to solutions of VLP–RNA complexes or unpackaged RNA in solution as described above. RNA extraction was performed according to the procedures above.
Magnesium-mediated in-line cleavage
Following established methods,37 VLP–RNA assemblies (250 μg) or unpackaged RNA (25 μg) were added to Mg2+ cleavage solution (25 mM MgCl2, 20 mM Tris–HCl, pH 7.4) to a final volume of 60 μL. Samples were incubated at 37 °C for various time periods (0–120 hours). VLP–RNA complexes or unpackaged RNA were added to solutions of varying concentration of MgCl2 (0–300 mM). Samples were incubated at 37 °C for 15 minutes. Reactions were quenched by freezing at −80 °C. RNA extraction was as described above.Unmediated cleavage
To investigate chemical stability of the RNA in the absence of reactive species, VLP–RNA complexes or unpackaged RNAs were incubated in a solution lacking divalent cations (20 mM Tris–HCl, pH 7.4). These samples were incubated at room temperature for various time periods.RNA quantitation
Reaction products were run on denaturing urea polyacrylamide gels and stained with SYBR® Green II. Band intensities were integrated with AlphaView® software for FluorChem systems. Packaging efficiencies were determined by normalization of packaged target RNA to co-packaged CP mRNA from the same co-expression/purification.Results
Here we assessed: (i) the chemical stability conferred to RNA by packaging in Qβ VLPs, and (ii) the effect of specific RNA characteristics on in vivo packaging efficiency. A series of RNAs were co-expressed with Qβ VLP CP in E. coli (Fig. 2). The RNAs are of different lengths, extents of compaction and sequence. We investigated the chemical robustness of packaged RNA in the presence of reactive small molecules, and the effect of RNA length and structure on packaging efficiency.Qβ VLP packaging protects RNA from chemical degradation
To investigate the effect of VLP packaging on the chemical robustness of RNA, VLPs containing ‘a-rRNA’38 were constructed and treated with reactive chemicals. a-rRNA is a well-characterized RNA derived from the ancestral core of the Thermus thermophilus large ribosomal subunit rRNA.38 Here, a-rRNA was fused to the Qβ hairpin (a-rRNA-hp, Fig. 2d).It was previously shown that VLPs protect RNA from degradation by protein nucleases,39 which are larger than the pore size of the VLPs. Here we investigated the ability of RNA packaged in Qβ VLPs to resist degradation by small diffusible molecules that are smaller than the VLP pore size. We compared rates of chemical degradation of a-rRNA-hp packaged in VLPs to a-rRNA-hp free in solution. The amount of uncleaved RNA was determined for a variety of timepoints.
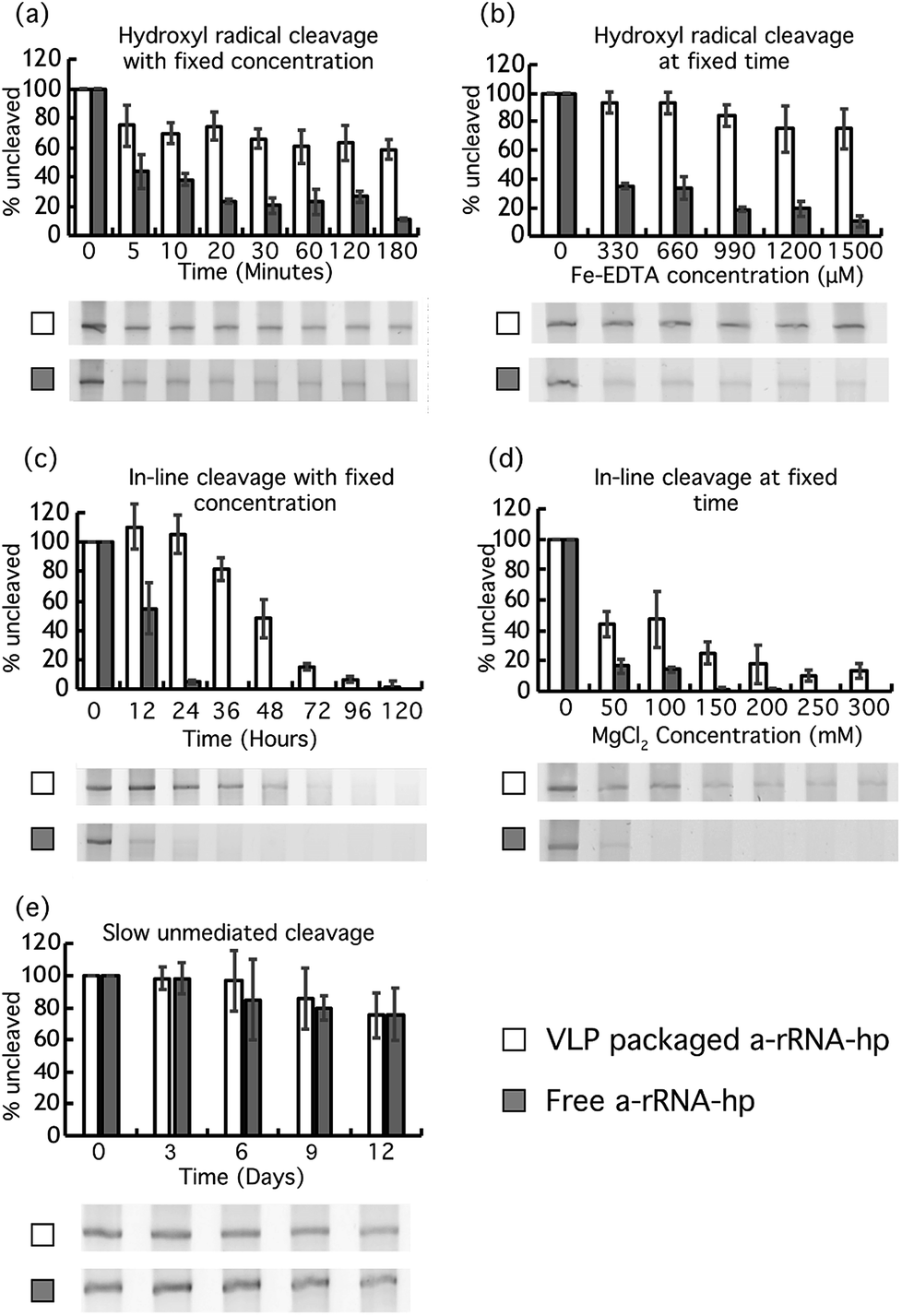 | ||
| Fig. 3 VLP packaging protects RNA. VLP–packaged a-rRNA-hp and free a-rRNA-hp were incubated under conditions (a) that promote Fenton chemistry (33 μM Fe-EDTA, H2O2 and atmospheric oxygen at 37 °C) at various timepoints, (b) that promote Fenton chemistry (varying [Fe-EDTA]) at constant time, (c) that promote inline cleavage (25 mM MgCl2 at 37 °C) at various timepoints, (d) that promote inline cleavage (varying [Mg2+] at 37 °C) at constant time, and (e) in the absence of divalent cations at neutral pH at various timepoints. For all experiments here, the uncleaved a-rRNA-hp was isolated on denaturing urea PAGE and quantitated with AlphaView® software. Error bars represent the 95% confidence interval of the quantitation. | ||
Similarly, protection of a-rRNA-hp within VLPs is observed in experiments in which the Fe-EDTA concentration is varied at fixed time (Fig. 3b). After 15 min in 1500 μM Fe-EDTA and 0.3% H2O2, about 80% of VLP–packaged a-rRNA-hp remains intact, while less than 20% of free RNA remains.
Magnesium is known to catalyze in-line cleavage of RNA.37,41 After 36 hours of incubation in 25 mM MgCl2 at 37 °C, the a-rRNA-hp within VLPs is about 80% intact. By contrast, less than 5% of free a-rRNA-hp is intact under the same conditions (Fig. 3c).
Similarly, VLP packaging increases the concentration of MgCl2 required for fixed extent of RNA cleavage at fixed time (Fig. 3d). About 45% of VLP–packaged a-rRNA-hp remains intact after 24 hours at a MgCl2 concentration of 100 mM. There is no detectable intact RNA remaining after the same incubation time for free RNA under the same conditions.
Our results show that both VLP–packaged and free a-rRNA-hp degrade slowly at 37 °C in the absence of Mg2+ and Fe2+-EDTA/H2O2. There is no significant difference in rates of degradation of VLP–packaged RNA and free RNA under the conditions of this experiment. The VLP does not appear to retard the rate of unmediated RNA degradation that occurs over extended periods of time.
Effects of RNA length and intrinsic compaction on packaging in VLPs
To investigate the impact of RNA size and structure on RNA packaging efficiency in Qβ VLPs, three representative RNAs of disparate length and structure were co-expressed with CP in E. coli. VLPs were purified and RNA packaging efficiency was estimated. SDS-PAGE analysis and transmission electron microscopy confirmed that co-expression of pCDF-CP and pET-RNA leads to expression of CP monomer (Fig. 4a) and canonical VLPs (Fig. 4b). RNA packaging was confirmed by denaturing urea PAGE of RNA extracted from purified VLPs (Fig. 4c and d).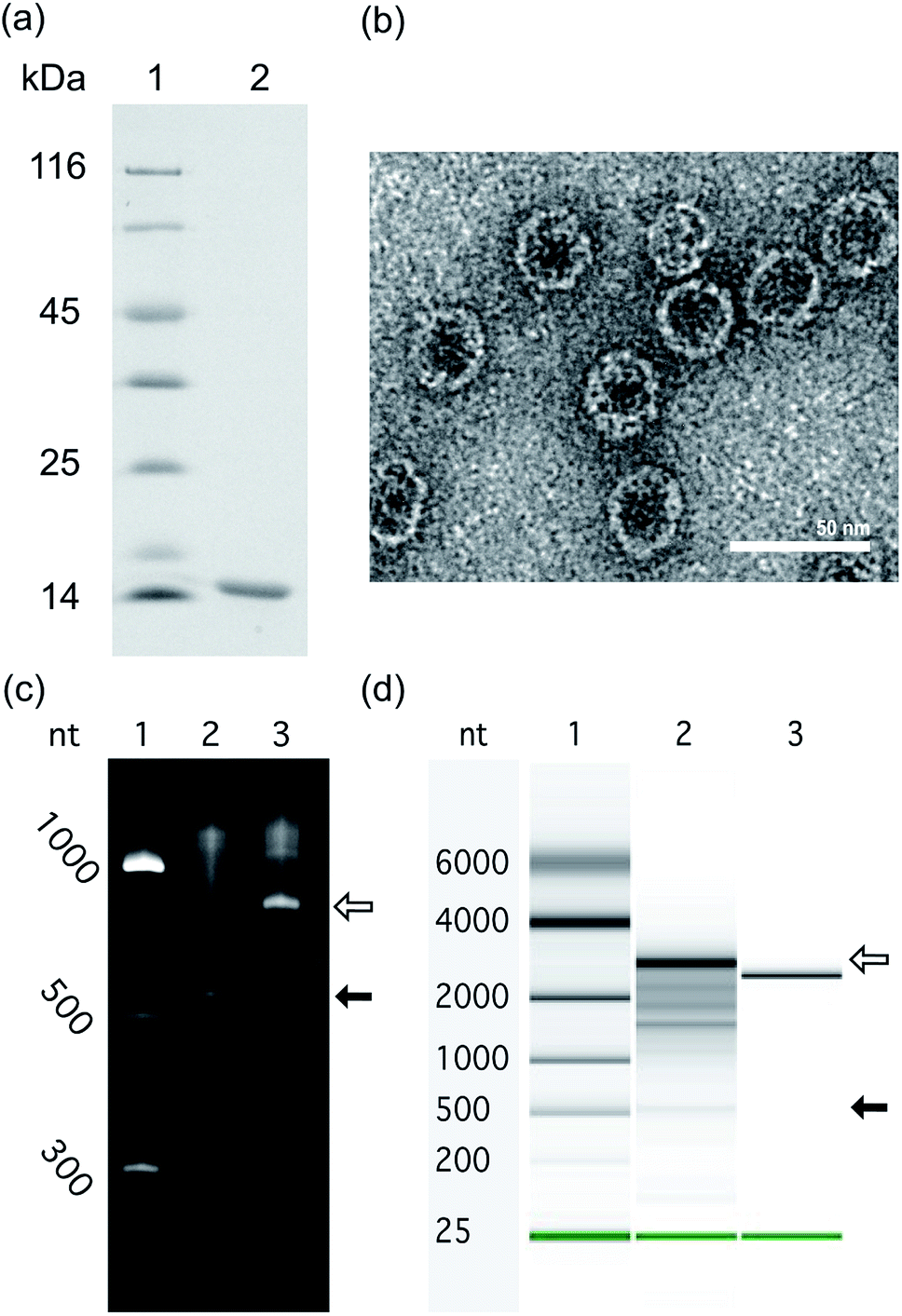 | ||
| Fig. 4 In vivo expressed RNA is packaged within VLPs to form VLP–RNA assemblies. (a) Purified Qβ VLPs analyzed by SDS-PAGE. The Qβ CP monomer is 14.5 kDa. Lane 1: protein sizing ladder. Lane 2: Qβ CP monomer from purified Qβ VLPs. (b) Transmission electron microscope images of Qβ VLP containing a-rRNA-hp. Scale bar = 50 nm. (c) In vivo expressed a-rRNA-hp, extracted from purified Qβ VLPs, subjected to electrophoresis on denaturing urea PAGE. Lane 1: RNA ladder, Lane 2: RNAs of Qβ CP expressed alone. Lane 3: RNAs of purified Qβ CP co-expressed with a-rRNA-hp. The open arrow indicates the a-rRNA-hp (747 nt). The closed arrow indicates the Qβ CP mRNA. (d) In vivo expressed 23S rRNA-hp, extracted from purified Qβ VLP, subjected to microfluidic-based chip electrophoresis. Lane 1: RNA sizing ladder, Lane 2: RNAs extracted from purified Qβ CP co-expressed with 23S rRNA-hp (3051 nt). Lane 3: 23S rRNA prepared by in vitro transcription (2966 nt). The open arrow in panel (d) indicates the 23S rRNA-hp. The closed arrow indicates the Qβ CP mRNA. | ||
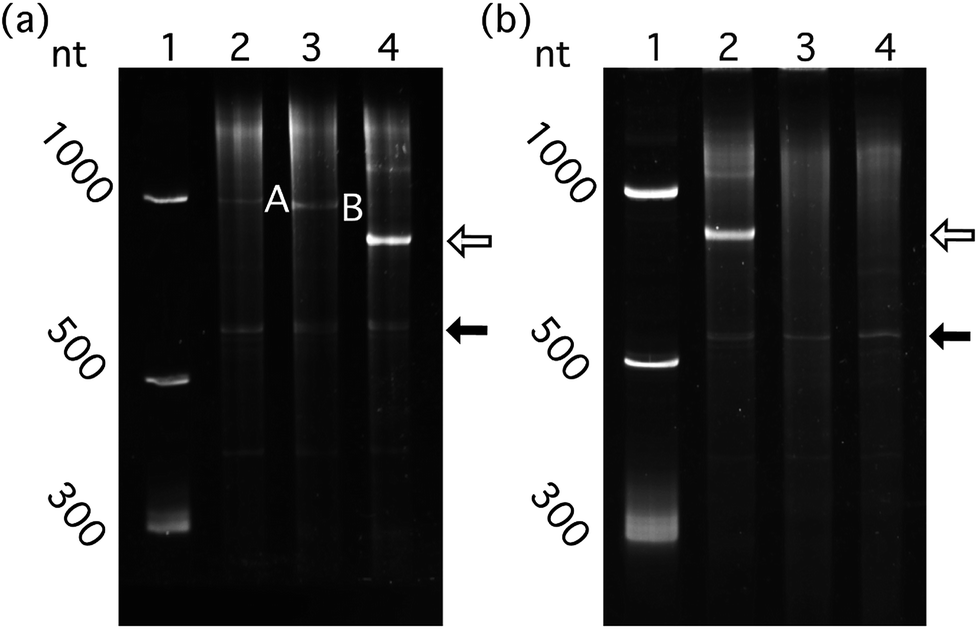 | ||
| Fig. 5 Highly structured RNAs bearing the Qβ hp and lacking the RBS package efficiently in Qβ VLP in vivo. (a) Structured and unstructured RNAs, packaged in vivo by Qβ CP. Lane 1: RNA ladder. Lane 2: RNAs extracted from purified Qβ CP co-expressed with RBS-mRNAGFP-hp. Lane 3: RNAs from purified Qβ CP co-expressed with mRNAGFP-hp. Lane 4: RNAs from purified Qβ CP co-expressed with a-rRNA-hp. (b) RNAs with and without the Qβ hp, co-expressed in vivo with Qβ CP. Lane 1: RNA ladder. Lane 2: RNAs of purified Qβ CP co-expressed with a-rRNA-hp. Lane 3: RNAs of purified Qβ CP co-expressed with a-rRNA (without Qβ hp). Lane 4: RNAs of Qβ CP expressed alone. One hundred nanograms of total VLP–extracted RNA was loaded to each well for denaturing PAGE. Band A: RBS-mRNAGFP-hp (851 nt). Band B: mRNAGFP-hp (833 nt, without the RBS). The open arrow indicates the a-rRNA-hp (747 nt). The closed arrow indicates the Qβ CP mRNA. | ||
To probe the impact of RNA length and compactness on packaging efficiency, three appropriate RNAs were co-expressed with CP (Fig. 2).
We used gel mobility to qualitatively characterize the extent of compaction of a-rRNA-hp compared to mRNAGFP-hp. The mobility of a-rRNA-hp increases relative to mRNAGFP-hp when gel conditions are converted from non-native to native (data not shown). This increase in mobility is consistent with a greater compaction of a-rRNA-hp over mRNAGFP-hp in the native state.44
RNA packaging efficiency is influenced by competing RNA binding factors
We determined whether ribosomes can compete with Qβ VLP packaging by binding target RNA. The effect of competition on packaging efficiency was assessed by co-expression of CP with pET-RNA encoding mRNAGFP-hp fused to the ribosomal binding site (RBS)45 (RBS-mRNAGFP-hp, Fig. 2b and f). Cloning at the XbaI/BlpI sites to generate all other constructs in the series of pET-RNAs used in this study eliminated the RBS of pET-28b (+). Therefore, mRNAGFP-hp represents a negative control for competition relative to RBS-mRNAGFP-hp.Quantitative comparison of packaged RBS-mRNAGFP-hp with mRNAGFP-hp shows that RNA packaging efficiency in VLPs decreases when the RNA contains the RBS (Fig. 5a, p < 0.01, n = 6). This result suggests competition between ribosomal binding and Qβ assembly in vivo.
Packaging of target RNA requires the Qβ hairpin
The Qβ hp is necessary for efficient packaging of essentially any RNA within a Qβ VLP. a-rRNA fused to the Qβ hp (a-rRNA-hp) shows the highest packaging efficiency of any RNA tested here. We investigated the importance of the Qβ hp to packaging of a highly structured RNA by removing Qβ hp from a-rRNA-hp (a-rRNA, Fig. 2b and h). Removal of the Qβ hp essentially abolishes packaging of a-rRNA (Fig. 5b).Together, these results suggest that efficient packaging of RNA within VLPs in vivo may depend upon the compactness of the target RNA structure, the absence of competing binding motifs in the RNA, and the presence of the Qβ hp.
Discussion
The potency and efficacy VLP–packaged RNA is related to the functionality, purity and quantity of the packaged RNA, and to the chemical stability of the RNA within the VLP. Using E. coli for combined expression and packaging of RNA into Qβ VLPs, we probed relationships between packaging efficiency and RNA size and intrinsic compaction. We characterized the degree of RNA protection from small diffusible agents such as hydroxyl radical and magnesium cations conferred by packaging in VLPs.RNA protection by VLP packaging
RNA packaged within MS2 VLPs is completely protected from digestion by plasma ribonucleases.39 The mechanism of this protection is steric; the enzymes are significantly larger than the pores in the VLP capsid. Here we establish that packaging in VLPs protects RNA against small, diffusible damaging agents that are smaller than the capsid pores.We determine the level of protection afforded against metal ion mediated strand cleavage by packaging in Qβ VLPs. Mechanisms and levels of protection against small reactive molecules such as metal ions are expected to be distinct from those of nucleases because Qβ VLPs contain pores20 that are significantly larger than the metal ions and their hydrates. Mg2+ has an ionic diameter of 0.13 nm and a fully hydrated diameter of just less than 1 nm.46 Pores in VLPs allow entry and exit of metal ions and species such as hydroxyl radical. Diffusion of metal ions into Qβ VLPs has been demonstrated by Finn, who showed that RNA packaged in VLPs is degraded by lead(II) acetate.47 It has been shown that RNA within MS2 VLPs is degraded by high pH.48
Our results demonstrate, as expected, that Mg2+ and Fe2+ enter VLPs and degrade RNA. The results are consistent with expectations that rates of RNA cleavage caused by these metals are highly attenuated for compact RNA packaged in the interior of VLPs compared to rates of cleavage of free RNA in solution. RNA is substantially protected against hydroxyl radical and magnesium-mediated in-line cleavage by packaging in VLPs (Fig. 3). It is known that rates of hydroxyl radical49 and magnesium-mediated in-line cleavage41 of RNA are influenced by RNA compaction and dynamics. RNA is more compact and less dynamic within VLPs than RNA that is free in solution.50
The RNA backbone is cleaved spontaneously in both the presence and in absence of magnesium.37,41 The mechanism is thought to be the same for both reactions; intramolecular in-line cleavage with a linear arrangement of the nucleophile (the 2′ oxygen), the electrophile (the P) and the leaving group (the 5′ oxygen). The reaction is faster in the presence of magnesium because it stabilizes the transition state by accepting a proton from the 2′ oxygen and/or by withdrawing electron density from the electrophilic phosphorous. Flexible regions of RNA are most labile to in-line cleavage because they more frequently occupy transient conformational states similar to the transition state.37,41 As expected, in the presence of magnesium the rate of cleavage decreases when RNA is packaged in the VLP. The reaction rate decreases because the dynamics of RNA within the VLP are suppressed by confinement and association with CP. Packaging of RNA in Qβ VLPs protects RNA not only against nucleases51 but against assault by small diffusible species.
Structured RNA may pack more efficiently than unstructured RNA
The results indicate that in vivo RNA packaging of RNA within VLPs may be most efficient for intrinsically compact RNA such as rRNA and less efficient for dynamic, elongated RNA such as mRNA. The Qβ hp is necessary but not sufficient for efficient packaging.Stockley previously suggested that for viral RNA, stem-loops dispersed through the MS2 viral genome interact with CP during assembly52–54 and tentatively identified 60 RNA stem-loops in the MS2 genome.26 RNA collapse during assembly in vitro depends initially on RNA–protein interactions, followed by CP–CP interactions.55 Packaging involves multiple weak RNA–CP interactions along the length of the viral RNA and a single strong interaction with the MS2 hp.27 MS2 and Qβ CP share about 25% amino acid sequence identity,56–58 with highly similar three-dimensional structures and RNA binding sites.
We conclude that in vivo packaging efficiency of non-viral RNA within Qβ VLPs may be high when the RNA (i) is intrinsically compact, (ii) includes the Qβ hp, (iii) lacks binding sites for assemblies that compete with CP (e.g., the RBS), and (vi) is as large as the 23S rRNA. We prepared a series of RNAs (Fig. 2) and co-expressed the RNAs with Qβ CP in E. coli. The RNAs expressed here contain (i) a-rRNA, a ∼650 nt intrinsically compact RNA stabilized by 11 GNRA tetraloops,38 (ii) 23S rRNA, a ∼2970 nt intrinsically compact RNA containing over forty stem-loops, or (iii) mRNAGFP, an unstructured ∼780 nt RNA. The expressed RNAs either contained or lacked the Qβ hp, and/or a RBS. The results show that both a-rRNA-hp and 23S rRNA-hp are efficiently packaged within Qβ VLP in vivo (Fig. 4), suggesting that, within the limits explored here, RNA length is not a strong predictor of packaging efficiency. We compared the packaging efficiency of intrinsically compact a-rRNA-hp and unstructured mRNAGFP-hp. The results of these experiments show greater packaging of intrinsically compact rRNA than unstructured mRNA (Fig. 5a). We conclude that intrinsic RNA compaction influences the efficiency of in vivo RNA packaging in Qβ VLPs.
Qβ hp is indispensable for efficient packaging of RNA
The Qβ hp is necessary but not sufficient for efficient packaging of RNA into VLPs within E. coli (Fig. 5b). This finding is consistent with previous work showing that RNA lacking the MS2 hairpin does not interact with MS2 CP in vitro.55 The MS2 hairpin is thought to be the initiation trigger for viral capsid formation.59–62 Our results confirm that numerous GNRA tetraloops found on the surface of a-rRNA,38 do not substitute for the Qβ hp during RNA packaging in vivo.Competing binding processes attenuate RNA packaging in VLPs
Finn and coworkers found that VLP packaging of CP mRNA competes with CP expression. When the Qβ hp is fused to the 3′ end of the CP mRNA, expression of Qβ CP decreases.63 Our results show that target RNA packaging efficiency in VLPs is slightly decreased when an RBS is introduced upstream of the mRNAGFP-hp (Fig. 5a). The presence of the RBS decreases RBS-mRNAGFP-hp packaging in the VLP relative to mRNAGFP-hp, suggesting that Qβ CP and the ribosome compete for binding to RBS-mRNAGFP-hp. Based on previous studies and our current findings, we expect that introduction of additional high-affinity RNA binding sites could facilitate or attenuate RNA packaging efficiency.Off-target RNA packaging
We and others observe that non-target RNAs, including Qβ CP mRNA, tRNAs, etc., are packaged within VLPs along with target RNA. Contaminating RNAs compete with target RNA during co-assembly. The absolute amount of contaminating RNA appears to depend of the extent of packaging of the target RNA. If target RNA is expressed at low levels, contains motifs (e.g., RBS) with affinity for competing species or lacks the Qβ hp, then the amount of contaminating RNA is increased. In total, it appears that sufficient RNA is packaged in the VLP to fully compensate for the cationic charge of Qβ CP.64 It is possible that RNA is required within the VLP for neutralization of positive charge.65 In our two-plasmid expression system, T7 RNA polymerase was used to induce high-level expression of both Qβ CP and target RNA. Without the simultaneous expression of RNA in vivo, high concentrations of positively charged viral CP may form non-specific RNA–CP interactions that serve to stabilize the protein structure and neutralize RNA charge.66Relevance to VLP-based therapies
Qβ VLPs are non-infectious and non-replicative platforms for delivery of various therapies with high stability and biocompatibility.67 VLPs have been shown to enhance the immunogenicity of some peptides through surface display.68 Expression of Qβ VLPs in E. coli packages host RNAs capable of inducing immune response in animal models, an incidental adjuvant.67,69 Several VLP-based therapies have already shown “good safety and tolerability” in clinical trials.67The immunogenicity of VLPs presents a challenge for their use in systematic delivery. However, efforts to attenuate immunogenicity are in progress in several laboratories, for example by “PEGylation”. This approach has been shown to limit the primary immune response of mice to cowpea mosaic virus,70 and to decrease immune response in rats administered adenovirus-based therapy targeting metastatic colonies.71 PEG is already clinically approved and is in use in pharmaceuticals.
We previously demonstrated delivery of functional RNAi to cancer cell lines using a highly structured RNAi scaffold co-expressed with VLPs in E. coli.15 The current study explored the role of RNA sequence and structure in efficient packaging by co-expression in E. coli, and the stability of RNA within VLPs once purified. Though these specific RNAs are not intended for use in VLP-based therapies, they represent a range of properties relevant to the development of RNA-based VLP therapeutics.
Conclusions
Using E. coli for combined expression and packaging of non-viral RNA in Qβ VLPs, we probed relationships between in vivo packaging efficiency and RNA size, sequence, and intrinsic compaction. We purified assembled VLPs and evaluated the extent of resistance to metal ion mediated degradation conferred on RNA by packaging in VLPs. We conclude that in vivo packaging in Qβ VLPs (i) protects target RNA against cleavage by hydroxyl radical and magnesium, and (ii) is efficient when the target RNA is intrinsically compact, includes the Qβ hairpin, and lacks the RBS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Georgia Research Alliance Grant No. GRA.VL15.B11; National Aeronautics and Space Administration grants NNX16AJ28G and NNX16AJ29G; National Science Foundation Graduate Research Fellowship Program [DGE-1650044 to L. M. G. R.]; and the Alfred P. Sloan & Georgia Tech UCEM Minority Doctoral Fellowship (to L. M. G. R). The authors thank Chieri Ito and Drs M. G. Finn and Brande Jones for helpful discussions.Notes and references
- E. Puerta-Fernández, C. Romero-López, A. Barroso-delJesus and A. Berzal-Herranz, FEMS Microbiol. Rev., 2003, 27, 75–97 CrossRef.
- R. S. Pillai, S. N. Bhattacharyya and W. Filipowicz, Trends Cell Biol., 2007, 17, 118–126 CrossRef PubMed.
- V. Pelechano and L. M. Steinmetz, Nat. Rev. Genet., 2013, 14, 880–893 CrossRef PubMed.
- B. L. Davidson and P. B. McCray, Nat. Rev. Genet., 2011, 12, 329–340 CrossRef PubMed.
- J. Goodchild, in Therapeutic Oligonucleotides, Springer, 2011, pp. 1–15 Search PubMed.
- B. L. Geller, J. Deere, L. Tilley and P. L. Iversen, J. Antimicrob. Chemother., 2005, 55, 983–988 CrossRef PubMed.
- T. K. Warren, K. L. Warfield, J. Wells, D. L. Swenson, K. S. Donner, S. A. Van Tongeren, N. L. Garza, L. Dong, D. V. Mourich and S. Crumley, Nat. Med., 2010, 16, 991–994 CrossRef PubMed.
- J. Wang, Z. Lu, M. G. Wientjes and J. L. S. Au, AAPS J., 2010, 12, 492–503 CrossRef PubMed.
- J. Ross, Microbiol. Rev., 1995, 59, 423–450 Search PubMed.
- N. Dam Mikkelsen and K. Gerdes, Mol. Microbiol., 1997, 26, 311–320 CrossRef PubMed.
- J. F. Milligan, D. R. Groebe, G. W. Witherell and O. C. Uhlenbeck, Nucleic Acids Res., 1987, 15, 8783–8798 CrossRef PubMed.
- W. S. Marshall and R. J. Kaiser, Curr. Opin. Chem. Biol., 2004, 8, 222–229 CrossRef PubMed.
- F. H. Nelissen, E. H. Leunissen, L. van de Laar, M. Tessari, H. A. Heus and S. S. Wijmenga, Nucleic Acids Res., 2012, 40, e102 CrossRef PubMed.
- L. Ponchon, M. Catala, B. Seijo, M. El Khouri, F. Dardel, S. Nonin-Lecomte and C. Tisné, Nucleic Acids Res., 2013, 41, e150 CrossRef PubMed.
- P.-Y. Fang, L. M. Gómez Ramos, Y. S. Holguin, C. Hsiao, J. C. Bowman, H.-W. Yang and L. D. Williams, Nucleic Acids Res., 2016, 45, 3519–3527 CrossRef PubMed.
- B. Lago, J. Birnbaum and A. Demain, Appl. Microbiol., 1972, 18, 430–436 Search PubMed.
- F. W. Studier and B. A. Moffatt, J. Mol. Biol., 1986, 189, 113–130 CrossRef PubMed.
- P. Lopez and M. Dreyfus, Biochimie, 1996, 78, 408–415 CrossRef PubMed.
- K. Machida and H. Imataka, Biotechnol. Lett., 2015, 37, 753–760 CrossRef PubMed.
- R. Golmohammadi, K. Fridborg, M. Bundule, K. Valegård and L. Liljas, Structure, 1996, 4, 543–554 CrossRef PubMed.
- W. Fiers, R. Contreras, F. Duerinck, G. Haegeman, D. Iserentant, J. Merregaert, W. Min Jou, F. Molemans, A. Raeymaekers and A. Van den Berghe, Nature, 1976, 260, 500–507 CrossRef PubMed.
- J. L. Lau, M. M. Baksh, J. D. Fiedler, S. D. Brown, A. Kussrow, D. J. Bornhop, P. Ordoukhanian and M. G. Finn, ACS Nano, 2011, 5, 7722–7729 CrossRef PubMed.
- A. E. Ashcroft, H. Lago, J. Macedo, W. T. Horn, N. J. Stonehouse and P. G. Stockley, J. Nanosci. Nanotechnol., 2005, 5, 2034–2041 CrossRef PubMed.
- G. W. Witherell and O. C. Uhlenbeck, Biochemistry, 1989, 28, 71–76 CrossRef PubMed.
- J. Rumnieks and K. Tars, J. Mol. Biol., 2014, 426, 1039–1049 CrossRef PubMed.
- E. C. Dykeman, P. G. Stockley and R. Twarock, J. Mol. Biol., 2013, 425, 3235–3249 CrossRef PubMed.
- A. Borodavka, R. Tuma and P. G. Stockley, RNA Biol., 2013, 10, 481–489 CrossRef PubMed.
- K. O. Hedenstierna, Y.-H. Lee, Y. Yang and G. E. Fox, Syst. Appl. Microbiol., 1993, 16, 280–286 CrossRef PubMed.
- S. Umekage and Y. Kikuchi, J. Biotechnol., 2009, 139, 265–272 CrossRef PubMed.
- J.-M. Masson and J. H. Miller, Gene, 1986, 47, 179–183 CrossRef PubMed.
- T. Meinnel, Y. Mechulam and G. Fayat, Nucleic Acids Res., 1988, 16, 8095 CrossRef PubMed.
- C. Tisné, M. Rigourd, R. Marquet, C. Ehresmann and F. Dardel, RNA, 2000, 6, 1403–1412 CrossRef.
- L. Ponchon and F. Dardel, Nat. Methods, 2007, 4, 571–576 CrossRef PubMed.
- J. C. Bowman, B. Azizi, T. K. Lenz, P. Roy and L. D. Williams, in Recombinant and In Vitro RNA Synthesis: Methods and Protocols, Methods in Molecular Biology, ed. G. L. Conn, Springer Science, LLC, 2012, vol. 941, pp. 19–41 Search PubMed.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 CrossRef PubMed.
- T. D. Tullius and B. A. Dombroski, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 5469–5473 CrossRef.
- E. E. Regulski and R. R. Breaker, Post-Transcriptional Gene Regulation, 2008, pp. 53–67 Search PubMed.
- C. Hsiao, T. K. Lenz, J. K. Peters, P. Y. Fang, D. M. Schneider, E. J. Anderson, T. Preeprem, J. C. Bowman, E. B. O'Neill, L. Lie, S. S. Athavale, J. J. Gossett, C. Trippe, J. Murray, A. S. Petrov, R. M. Wartell, S. C. Harvey, N. V. Hud and L. D. Williams, Nucleic Acids Res., 2013, 41, 3373–3385 CrossRef PubMed.
- B. L. Pasloske, C. R. Walkerpeach, R. D. Obermoeller, M. Winkler and D. B. DuBois, J. Clin. Microbiol., 1998, 36, 3590–3594 Search PubMed.
- J. Prousek, Pure Appl. Chem., 2007, 79, 2325–2338 CrossRef.
- M. Forconi and D. Herschlag, Methods Enzymol., 2009, 468, 91–106 Search PubMed.
- Q. M. Yi and K. P. Wong, Biochem. Biophys. Res. Commun., 1982, 104, 733–739 CrossRef PubMed.
- T. K. Lenz, A. M. Norris, N. V. Hud and L. D. Williams, RSC Adv., 2017, 7, 54674–54681 RSC.
- C. Cantor and P. Schimmel, Biophysical Chemistry (I-III), Academic Press, New York, 1984 Search PubMed.
- R. A. Calogero, C. L. Pon, M. A. Canonaco and C. O. Gualerzi, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 6427–6431 CrossRef.
- M. E. Maguire and J. A. Cowan, BioMetals, 2002, 15, 203–210 CrossRef PubMed.
- M. L. Hovlid, J. L. Lau, K. Breitenkamp, C. J. Higginson, B. Laufer, M. Manchester and M. G. Finn, ACS Nano, 2014, 8, 8003–8014 CrossRef PubMed.
- J. M. Hooker, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 3718–3719 CrossRef PubMed.
- J. A. Latham and T. R. Cech, Science, 1989, 245, 276–282 Search PubMed.
- K. C. Dent, R. Thompson, A. M. Barker, J. A. Hiscox, J. N. Barr, P. G. Stockley and N. A. Ranson, Structure, 2013, 21, 1225–1234 CrossRef PubMed.
- C. R. WalkerPeach, M. Winkler, D. B. DuBois and B. L. Pasloske, Clin. Chem., 1999, 45, 2079–2085 Search PubMed.
- D. Peabody, Mol. Gen. Genet., 1997, 254, 358–364 CrossRef PubMed.
- I. Hirao, M. Spingola, D. Peabody and A. D. Ellington, Mol. Diversity, 1998, 4, 75–89 CrossRef PubMed.
- D. Mills, C. Priano, P. Merz and B. Binderow, J. Virol., 1990, 64, 3872–3881 Search PubMed.
- A. Borodavka, R. Tuma and P. G. Stockley, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15769–15774 CrossRef PubMed.
- W. T. Horn, K. Tars, E. Grahn, C. Helgstrand, A. J. Baron, H. Lago, C. J. Adams, D. S. Peabody, S. E. V. Phillips, N. J. Stonehouse, L. Liljas and P. G. Stockley, Structure, 2006, 14, 487–495 CrossRef PubMed.
- D. S. Peabody, EMBO J., 1993, 12, 595 Search PubMed.
- F. Lim, M. Spingola and D. S. Peabody, J. Biol. Chem., 1996, 271, 31839–31845 CrossRef PubMed.
- D. Beckett, H.-N. Wu and O. C. Uhlenbeck, J. Mol. Biol., 1988, 204, 939–947 CrossRef PubMed.
- D. Beckett and O. C. Uhlenbeck, J. Mol. Biol., 1988, 204, 927–938 CrossRef PubMed.
- P. G. Stockley, O. Rolfsson, G. S. Thompson, G. Basnak, S. Francese, N. J. Stonehouse, S. W. Homans and A. E. Ashcroft, J. Mol. Biol., 2007, 369, 541–552 CrossRef PubMed.
- J. M. Gott, L. J. Wilhelm and O. C. Uhlenbeck, Nucleic Acids Res., 1991, 19, 6499–6503 CrossRef PubMed.
- J. D. Fiedler, C. Higginson, M. L. Hovlid, A. A. Kislukhin, A. Castillejos, F. Manzenrieder, M. G. Campbell, N. R. Voss, C. S. Potter, B. Carragher and M. G. Finn, Biomacromolecules, 2012, 13, 2339–2348 CrossRef PubMed.
- D. Muriaux, J. Mirro, D. Harvin and A. Rein, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5246–5251 CrossRef PubMed.
- V. A. Belyi and M. Muthukumar, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17174–17178 CrossRef PubMed.
- B. Devkota, A. S. Petrov, S. Lemieux, M. B. Boz, L. Tang, A. Schneemann, J. E. Johnson and S. C. Harvey, Biopolymers, 2009, 91, 530–538 CrossRef PubMed.
- J. G. H. Low, L. S. Lee, E. E. Ooi, K. Ethirajulu, P. Yeo, A. Matter, J. E. Connolly, D. A. G. Skibinski, P. Saudan, M. Bachmann, B. J. Hanson, Q. Lu, S. Maurer-Stroh, S. Lim and V. Novotny-Diermayr, Vaccine, 2014, 32, 5041–5048 CrossRef PubMed.
- A. C. Tissot, R. Renhofa, N. Schmitz, I. Cielens, E. Meijerink, V. Ose, G. T. Jennings, P. Saudan, P. Pumpens and M. F. Bachmann, PLoS One, 2010, 5, e9809 Search PubMed.
- W. Liao, Z. Hua, C. Liu, L. Lin, R. Chen and B. Hou, J. Immunol., 2017, 198, 3846 CrossRef PubMed.
- K. S. Raja, Q. Wang, M. J. Gonzalez, M. Manchester, J. E. Johnson and M. G. Finn, Biomacromolecules, 2003, 4, 472–476 CrossRef PubMed.
- Y. Eto, Y. Yoshioka, T. Ishida, X. Yao, T. Morishige, S. Narimatsu, H. Mizuguchi, Y. Mukai, N. Okada, H. Kiwada and S. Nakagawa, Biol. Pharm. Bull., 2010, 33, 1540–1544 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02084a |
| This journal is © The Royal Society of Chemistry 2018 |