
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe formation and growth of calcium sulfate crystals through oxidation of SO2 by O3 on size-resolved calcium carbonate†
Ying Zhangab,
Shengrui Tong*a,
Maofa Ge *abc,
Bo Jinga,
Siqi Houab,
Fang Tanab,
Yi Chenab,
Yucong Guoa and
Lingyan Wud
*abc,
Bo Jinga,
Siqi Houab,
Fang Tanab,
Yi Chenab,
Yucong Guoa and
Lingyan Wud
aState Key Laboratory for Structural Chemistry of Unstable and Stable Species, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: gemaofa@iccas.ac.cn; tongsr@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cCAS Research/Education Center for Excellence in Molecular Sciences, Beijing 100190, P. R. China
dState Key Laboratory of Severe Weather & Key Laboratory of Atmospheric Chemistry of CMA, Chinese Academy of Meteorological Sciences, Beijing 100081, P. R. China
First published on 1st May 2018
Abstract
Calcium sulfate is a major constituent of atmospheric sulfate, with a typical rod-like morphology ranging from several hundred nanometers to approximately two micrometers observed in field studies. However, the chemical formation mechanism is still not well known. In this study, the kinetics and mechanism for the formation and growth of rod-like calcium sulfate crystals through oxidation of SO2 by O3 on size-resolved CaCO3 at different relative humidity (RH) were investigated using diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) and scanning electron microscopy (SEM). We found that the concentration and formation rate of sulfate decreased with the increasing diameter of CaCO3 particles, and thus smaller particles could enhance the formation of sulfate due to more reactive sites on smaller particles. The rod-like calcium sulfate crystals were formed only at RH above 60% and in the presence of reactant gases through the heterogeneous pathway. The liquid-like water layer formed by promotion of high RH in the presence of reactant gases could facilitate the formation and aggregation of calcium sulfate hydrates and thus promote the formation and growth of rod-like calcium sulfate crystals. This study provides a possible mechanism for the formation and growth of rod-like calcium sulfate crystals existing in the atmosphere.
Introduction
Haze, with a high frequency of occurrence in China, exerts adverse impacts on the global climate and human health such as inducing many respiratory diseases.1,2 Atmospheric sulfate presented an explosive growth during haze episodes and contributed significantly to the formation of severe haze.3–5 Furthermore, as one of the most important constituents of aerosols in atmosphere, the sulfate can serve as a cloud condensation nuclei (CCN) and scatter solar radiation, and thus affects the climate and weather.6–9 Sulfate also can impact on reactivity, acidity and hygroscopicity of aerosols by changing the mixing state, components and morphology of particles.10–13 Sulfate originates from primary sources including sea spray aerosol and volcanic ash and from secondary sources by oxidation of SO2, carbonyl sulfur (COS) or dimethyl sulfide (DMS).14,15 SO2 with concentration up to several tens ppb in polluted days, is an important gaseous pollutant largely released by fossil fuel combustion.16,17 Sulfate is produced mainly from the oxidation of SO2 by OH radical in the gas-phase15 and by H2O2, O3 and NO2 in liquid phase.18,19 The catalyzation of transition metal ions (for instance, Fe3+ and Mn2+) on the oxidation of SO2 is another important pathway to form sulfate.20,21 Heterogeneous oxidation of SO2 by oxidants such as O3 and NO2 on mineral dust, sea salt or soot is proposed as a significant source of sulfate.22–25 These processes lead to the formation of different types of sulfate, for example, sodium sulfate, ammonium sulfate and calcium sulfate.Calcium sulfate is a main component of atmospheric sulfate.26 The strong correlation between SO42− and Ca2+ within aerosol particles has been revealed in field measurements.27,28 Also, a large fraction of calcium-containing particles is found to be calcium sulfate component,29 which is a major type of sulfate in fine particles and composes almost total sulfate in coarse particles.30 Rod-like morphology ranging from several hundred nanometers to approximately two micrometers are identified as a typical morphology of calcium sulfate in field studies.31–34 Until now, little attention has focused on the mechanism for the formation of rod-like calcium sulfate crystals. Rod-like calcium sulfate with diameters of 10 to 500 nm could be generated by the adsorption of SO2 on particles at high RH due to the role of adsorbed water proposed by field studies.35 Li et al. also found that the formation of secondary regular particles like gypsum was promoted by high humidity.36 From above, it could be speculated that the formation and growth of rod-like calcium sulfate crystals is influenced by humidity. Furthermore, Gustafsson et al. found that the amount of water adsorbed on the mineral dust aerosols decreased with the increasing particle diameters.37 Takahashi et al. suggested that the ratio of gypsum in total calcium-containing particles is larger for smaller particles, due to the more thorough reaction of SO2 in smaller particles.38 However, the key factors for formation and growth of the rod-like calcium sulfate crystals are still unclear in the atmosphere.
CaCO3 is proved to be one of the most important precursors of calcium sulfate in the atmosphere.39,40 CaCO3 comprises up to 30% of the total mineral dust,41 and could be transported to long distance and wide range,42 and thus it is often a naturally available precursor. As one of reactive components of atmospheric mineral dust,43 CaCO3 is liable to be converted into calcium sulfate through reacting with trace acidic gaseous pollutants in the atmosphere.44 Several studies have focused on the formation of calcium sulfate transformed from CaCO3. It was found that RH and temperature could influence the formation of sulfate on CaCO3 particles.45–47 Ma et al. showed that a deliquescent layer is crucial for the formation of gypsum.48 However, few studies have been conducted with respect to the evolution of morphology for the sulfate formed on the CaCO3. In this study, we aim at providing a mechanistic understanding on how rod-like calcium sulfate crystals are formed in the atmosphere and investigating whether RH and particle diameters of reactant particles could impact on the formation process. O3 is an efficient oxidant and could accelerate the formation of sulfate by oxidizing sulfite on particles,49,50 hence is chosen as a model oxidant in this study. Therefore, we have undertaken an experimental study to explore the formation and growth of calcium sulfate crystals by oxidation of SO2 by O3 on size-resolved CaCO3 at different RH. Products and kinetics were investigated by diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). The evolution of the sizes and morphology of calcium sulfate was observed by scanning electron microscopy (SEM).
Experimental section
Sources of chemicals
Three kinds of CaCO3 particles with different diameters were purchased from commercial sources (InnoChem, Beijing InnoChem Science & Technology Co., Ltd.). The shapes and diameters were observed by SEM and analyzed by image analysis software (NIH, Imagej). The smallest CaCO3 particles were approximately square, with diameter of 65 ± 10 nm determined by edge length. The other two larger particles were spindle shape, and the diameters were calculated to be 270 ± 50 nm and 1000 ± 300 nm, respectively, by the mean value of the short and long axes of the spindles.51 The three kinds of particles were denoted as 65 nm, 270 nm and 1000 nm CaCO3 particles, respectively, with the corresponding Brunauer–Emmett–Teller (BET) surface area of the samples measured to be 23.742 m2 g−1, 13.461 m2 g−1 and 5.301 m2 g−1 (Autosorb-1-MP automatic equipment (Quanta Chrome Instrument Co.)). CaCO3 particles were heated for 3 h for outgassing before the BET measurement. N2 (>99.999%, Beijing Tailong Electronics Co., Ltd.) and O2 (>99.998%, Orient Center Gas Science & Technology Co., Ltd.) were used to simulate the ambient air, both of which were desiccated by silica gel and molecular sieve to ensure that RH was less than 1%. SO2 (500 ppm, Beijing Huayuan Gas Chemical Industry Co. Ltd.) and ozone (generated by irradiating a dry flow of pure O2 with an ultraviolet lamp (Heraeus Ltd., ZSZ-8)) were used as the reactant gases. Before each experiment, pure O2 was passed through an ultraviolet lamp compartment of the ozone generator (UVP, Model SOG-2) for 1 h to achieve the steady concentration of ozone. The ozone concentration measured by an ozone analyzer (Model 49i, Thermo, USA) was controlled by varying the flux of oxygen exposed to ultraviolet light. Ultrapure water (18.2 MΩ cm) used for humidifying N2 was obtained from Water Purification System (Dura FV).Gas supply system
The gas supply system was composed of five Teflon inlet lines. The first and second line supplied SO2 and a mixture of ozone and oxygen, respectively. The concentrations of both SO2 and ozone were 4.9 × 1014 molecules cm−3 in this study. The third line was used to supply humid N2 produced by bubbling a dry flow of pure N2 through ultrapure water. The fourth and fifth line provided dry N2 and O2 for dilution to simulate the ambient air. The mixing of all gases with a total flow of 400 sccm before entering the reaction chamber resulted in a synthetic air (20% O2 and 80% N2) with additions of SO2, O3 and H2O. Mass flow controllers (Beijing Sevenstar electronics Co., Ltd.) were used to adjust the flux of the five lines to get the desired concentration of SO2 and O3, as well as the expected RH. A commercial humidity and temperature sensor (HMT330, Vaisala) was used to measure the RH and temperature at the inflow of the sample cell with a measurement accuracy of ± 1% RH and ± 0.2 K, respectively.Measurement methods
DRIFTS was employed to record the reaction process in situ on CaCO3 particles. A Nicolet FTIR spectrometer 6700 was equipped with a liquid-nitrogen-cooled narrow band mercury–cadmium–telluride (MCT) detector, Praying Mantis accessory (Model DRP, Harrick Scientific Corp.), and a high temperature reaction chamber (Model HVC-DRP-3, Harrick Scientific Corp.), used for recording the DRIFTS spectra in the spectral range of 650 to 4000 cm−1 at a resolution of 4 cm−1. 100 scans were averaged for each spectrum corresponding to a time resolution of 40 s in this study. DRIFTS connected with the gas supply system has been described in detail elsewhere.52–54 The reaction chamber was cleaned with alcohol and then evacuated under high vacuum over 1 h. Samples of about 30 mg, 37 mg and 45 mg were placed into the stainless steel cup (10 mm in diameter, 0.5 mm in depth) for 65 nm, 270 nm and 1 μm CaCO3, respectively. Afterwards, the sample cup was put into the reaction chamber, with temperature controlled by an automatic temperature controller (ATC-024-2, Harrick Scientific Corp.) combined with circulating cooling water. The sample was kept at 673 K for 1 h to remove adsorbed species from the surface and then cooled to 298 K for 1 h to ensure a stable condition.55 The spectra of unreacted particles at a specific RH were recorded as background after synthetic air was introduced into the reaction chamber for about 1 h to achieve water adsorption equilibrium on the samples. SO2 and O3 with specific concentrations were stabilized through the bypass line for about 30 min and 1 h, respectively, and then were introduced into the reaction chamber with an average residence time of 2.5 s. Spectra of products were shown as positive bands while losses of surface species as negative bands. All the spectral data was automatically collected by series program in OMINC software during a typical experiment time of 120 min. Each experiment at the same RH was repeated at least three times, and the reproducibility of results was within acceptable error probably caused by artificial operation such as packing samples in the sample cup.46S-4800 SEM (Hitachi, Japan) operated at 10 kV was employed to determine the morphology features and particle diameters of CaCO3 particles before and after the reaction. Several hundred micrograms samples were sprayed onto the surface of silicon wafers coated by gold. The silicon wafer was then put into the SEM chamber for observation of the unreacted particles after heating at 673 K for 1 h. Three silicon wafers sprayed with the three size-resolved CaCO3 particles were placed into the sample cup and reacted in the DRIFTS chamber under the same conditions as samples directly in the sample cup. After reaction, the silicon wafers coated by gold were used to determine the morphology features of particles by SEM.
Results and discussion
The surface products, the kinetics and morphology features during the heterogeneous reactions were analyzed based on the measurements by DRIFTS and SEM. Then, a mechanism was proposed based on the result analysis.Characterization of surface products
Fig. 1 represented the in situ DRIFTS spectra of surface products when the size-resolved samples were exposed to SO2 and O3 simultaneously for 120 min at different RHs. The adsorption bands were almost the same for three size-resolved CaCO3 at the same RH. The peaks in the range from 990 to 1270 cm−1 were ascribed to the stretching mode (ν) of adsorbed sulfate (SO42−).56 Under dry condition (1.5% RH), peaks at 985 cm−1 belonged to ν(SO32−) in calcium sulfite.57 The peaks at 839 cm−1 and 1689 cm−1 were assigned to the bending mode (δ) and ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of CO32− in carbonic acid adsorbed on the particle surface, respectively, indicating the formation of H2CO3.58 Hence, the products were CaSO4, CaSO3 and adsorbed H2CO3 at 1.5% RH. At 19% RH, peaks at 985 cm−1 disappeared while peaks at 839 cm−1 and 1689 cm−1 still existed. New peaks at 995 cm−1 belonged to ν(SO42−) in CaSO4,56 and new absorption bands at 1648, 3248 and 3510 cm−1 were attributed to δ(OH) and ν(OH) of adsorbed water on the particle surfaces.47 Thus, the products were CaSO4 and adsorbed H2CO3 at 19% RH. At 53% RH, peaks at 839 cm−1 and 1689 cm−1 also disappeared. Peaks at 1196 cm−1 shifted to 1190 cm−1, which indicated the formation of new sulfate products. New bands at 1629, 3552, and 3610 cm−1 were assigned to the δ(OH) and ν(OH) of water in bassanite (CaSO4·0.5H2O).59 Hence, bassanite was formed at 53% RH. At 90% RH, peaks at 995 cm−1 and 1136 cm−1 shifted to 1006 cm−1 and 1142 cm−1, respectively, which belonged to ν(SO42−) in calcium sulfate hydrates, and thus suggested that more calcium sulfate hydrates were formed on the CaCO3 particles.60 Double peaks of δ(OH) at 1629 and 1685 cm−1 and three peaks of ν(OH) at 3248, 3406, and 3552 cm−1 belonged to absorbance of water in CaSO4·2H2O (gypsum).61,62 Therefore, bassanite and gypsum were formed at 90% RH in this study.
O) of CO32− in carbonic acid adsorbed on the particle surface, respectively, indicating the formation of H2CO3.58 Hence, the products were CaSO4, CaSO3 and adsorbed H2CO3 at 1.5% RH. At 19% RH, peaks at 985 cm−1 disappeared while peaks at 839 cm−1 and 1689 cm−1 still existed. New peaks at 995 cm−1 belonged to ν(SO42−) in CaSO4,56 and new absorption bands at 1648, 3248 and 3510 cm−1 were attributed to δ(OH) and ν(OH) of adsorbed water on the particle surfaces.47 Thus, the products were CaSO4 and adsorbed H2CO3 at 19% RH. At 53% RH, peaks at 839 cm−1 and 1689 cm−1 also disappeared. Peaks at 1196 cm−1 shifted to 1190 cm−1, which indicated the formation of new sulfate products. New bands at 1629, 3552, and 3610 cm−1 were assigned to the δ(OH) and ν(OH) of water in bassanite (CaSO4·0.5H2O).59 Hence, bassanite was formed at 53% RH. At 90% RH, peaks at 995 cm−1 and 1136 cm−1 shifted to 1006 cm−1 and 1142 cm−1, respectively, which belonged to ν(SO42−) in calcium sulfate hydrates, and thus suggested that more calcium sulfate hydrates were formed on the CaCO3 particles.60 Double peaks of δ(OH) at 1629 and 1685 cm−1 and three peaks of ν(OH) at 3248, 3406, and 3552 cm−1 belonged to absorbance of water in CaSO4·2H2O (gypsum).61,62 Therefore, bassanite and gypsum were formed at 90% RH in this study.
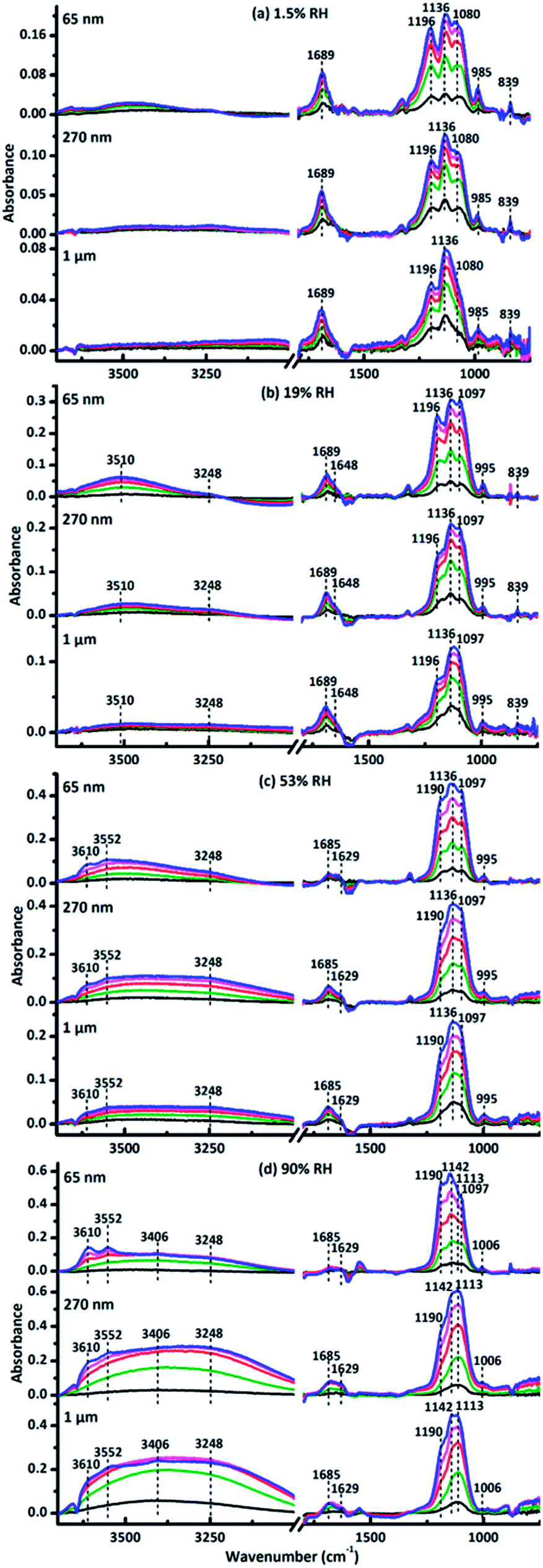 | ||
| Fig. 1 In situ DRIFTS spectra when SO2 and O3 were exposed simultaneously to 65 nm, 270 nm and 1 μm CaCO3 particles, respectively, at (a) dry condition, 1.5% RH, (b) 19% RH, (c) 53% RH, (d) 90% RH. Absorption lines are recorded at 10 (black), 30 (olive), 60 (red), 90 (magenta) and 120 min (blue), respectively. | ||
The sulfate formation kinetics on size-resolved CaCO3 particles
The concentration of the ions formed during the reactions showed non-linear dependence on integrated area calculated by Kubelka–Munk function, while showed linear relationship with integrated absorbance.54 Hence, integrated absorbance from 1020 to 1270 cm−1 as a function of reaction time was used to follow the concentration of sulfate ions during the reaction of SO2 with O3 on the surface of 65 nm, 270 nm and 1 μm CaCO3 particles at various RH, respectively. The amount of sulfate formed during the reaction was calculated by DRIFTS calibration curve (seen in Fig. S1†). The DRIFTS calibration curve was made by a series of known weight ratios of CaSO4 and CaCO3 mixtures, which showed that the amount of sulfate had a good linear relationship with the DRIFTS signal and could be expressed by:| (Integrated absorbance) × f = [SO42−] | (1) |
Here f is the conversion factor. The calculated f value was 6.83 × 1018 ions g−1 ABU−1 (ABU, absorbance unit) for three kinds of CaCO3 particles, consistent with the order of magnitude of the values in previous studies.45,46 Additional experiments proved that particle diameter and RH had no influence on f value. Fig. 2 represents sulfate ion concentrations as a function of particle diameter. The concentrations of sulfate ions decreased with increasing particle diameter of CaCO3. The differences of sulfate ion concentrations for three size-resolved CaCO3 were almost the same. It could be explained by the fact that the smaller particles with higher specific surface area and more active sites could adsorb more water and SO2, and thus concentration of sulfate decreased with increasing particle diameter of CaCO3 particles.
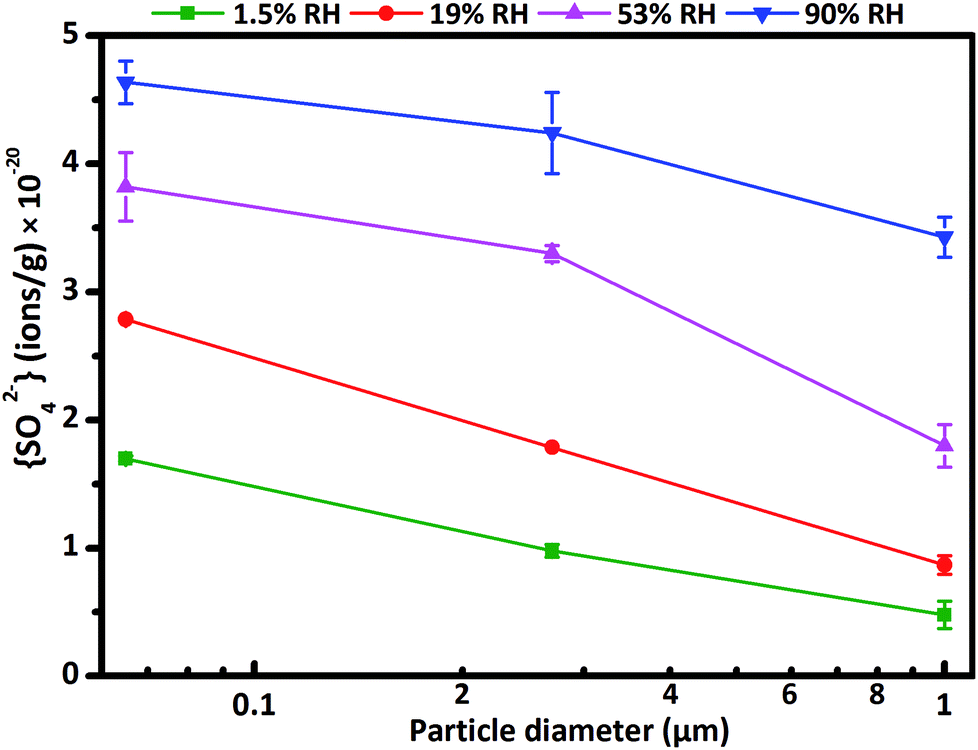 | ||
| Fig. 2 The concentration of sulfate ions formed on particle surfaces during the reaction at 1.5% RH (olive), 19% RH (red), 53% RH (magenta) and 90% RH (blue) and reaction time of 120 min as a function of CaCO3 particle diameter. | ||
The formation rate of sulfate d[SO42−]/dt can be quantified through the slope of sulfate concentration as a function of reaction time. The formation process of sulfate on CaCO3 surfaces exhibited two stages. At the beginning of the reaction, the rapid increase of sulfate concentration was denoted as the initial stage, afterwards, the formation rate of sulfate decreased gradually until reached a stable rate after a certain time, and the stable stage appeared.63 The formation rate of sulfate ions decreased with increasing particle diameter in the two stages (seen in Fig. 3), consistent with the variation of concentration of sulfate ions as a function of particle diameter. In initial stage, the differences of sulfate formation rate for three size-resolved CaCO3 were almost unchanged at 1.5% RH, 19% RH and 53% RH, and decreased at 90% RH. During the initial stage, SO2 had no time to diffuse into the CaCO3 samples, as a result, the formation rate of sulfate was mainly determined by surface reactive sites and the amount of adsorbed SO2. Since the surface adsorbed water could occupy the reactive sites, the surface reactive sites decreased with increasing RH. 65 nm CaCO3 particles with higher specific surface area could adsorb more water than 270 nm and 1 μm CaCO3 particles at the same RH and thus the surface reactive sites decreased much faster. At 90% RH, after the water adsorption equilibrium was achieved, the left surface reactive sites were not enough for the adsorption of SO2, hence, the formation rate decreased for 65 nm CaCO3 compared with that at 19% and 53% RH. Therefore, the differences of sulfate formation rate in initial stage among the three kinds of CaCO3 decreased at 90% RH. In stable stage, the differences of sulfate formation rate for three size-resolved CaCO3 were larger under humid condition than under dry condition. It has been proved that the amount of surface adsorbed water increased with enhanced RH, which could promote the adsorption of SO2 and improve the ion mobility.64 Consequently, more SO2 could diffuse into the bulk of the samples under humid condition than dry condition and thus accelerated the formation of sulfate in the stable stage. Considering more water adsorbed on the surface of smaller CaCO3 particles, it could be understood that the differences of formation rate in stable stage were larger under humid condition compared with that under dry condition. The differences of sulfate ion concentrations seemed to be determined by formation rate both in initial stage and stable stage.
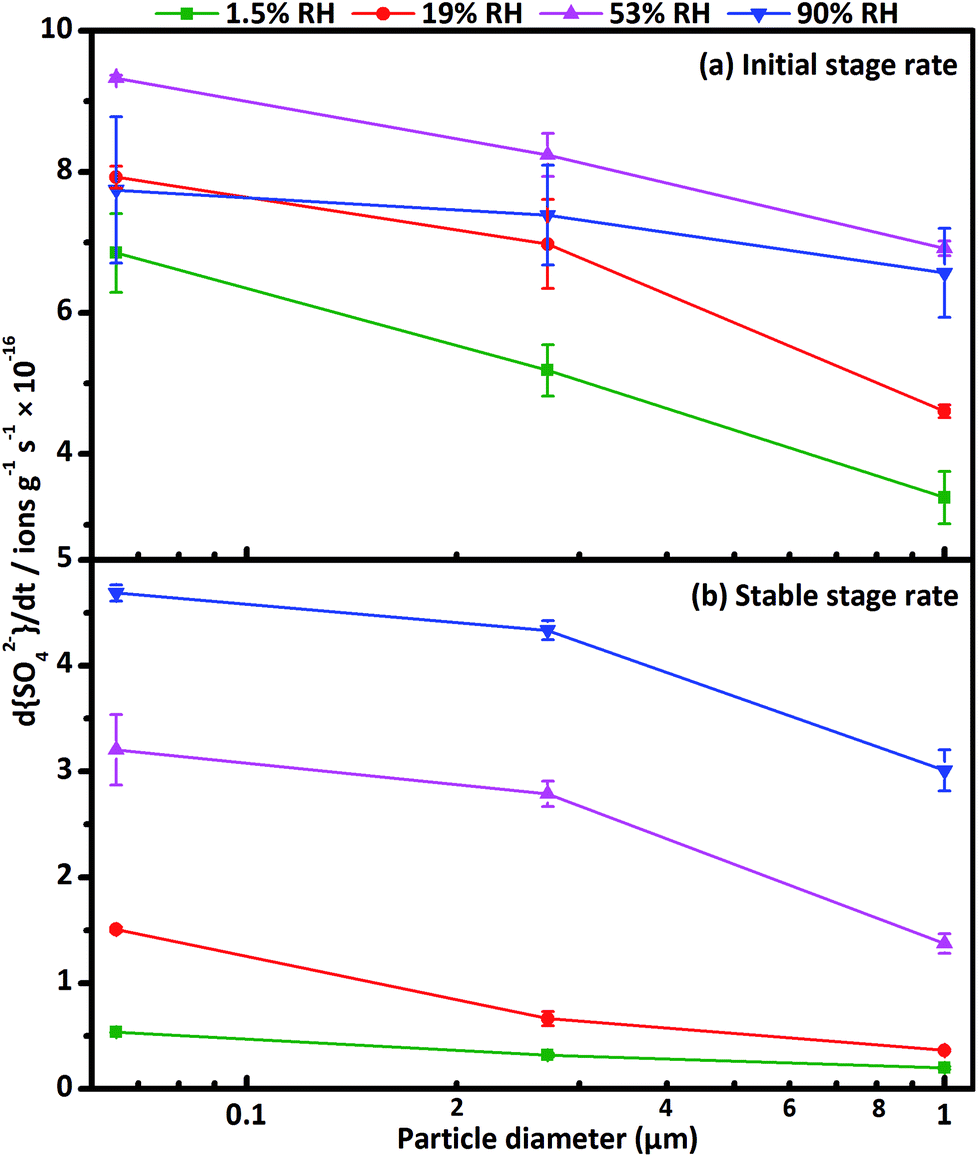 | ||
| Fig. 3 Formation rate of sulfate d[SO42−]/dt (a) in initial stage and (b) in stable stage at 1.5% RH (olive), 19% RH (red), 53% RH (magenta) and 90% RH (blue) as a function of CaCO3 particle diameter. | ||
The reactive uptake coefficient γ, was defined as the ratio of the rate of the reactive gases collisions with the surface to the rate of total gases surface collisions (Z). The γ for the heterogeneous oxidation of SO2 was deduced from DRIFTS experiments. On the basis of the assumption that the rate of reactive SO2 collision with the surface was equal to the sulfate formation rate d[SO42−]/dt, the reactive uptake coefficients of SO2 can be calculated by the following equations.
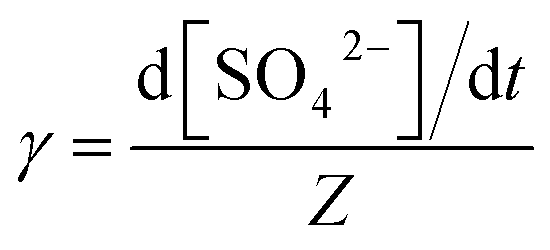 | (2) |
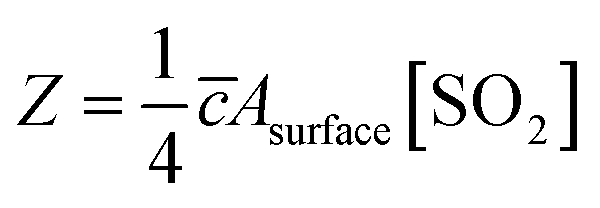 | (3) |
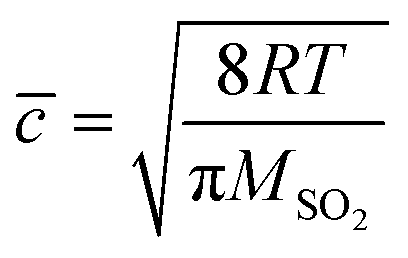 | (4) |
Here, Z is the rate of total collisions between SO2 and particles, ![[c with combining macron]](https://www.rsc.org/images/entities/i_char_0063_0304.gif) represents the mean molecular velocity of SO2, Asurface is the effective surface area of CaCO3 particles, and [SO2] is the gas-phase concentration of SO2. R is the molar gas constant, T refers to the temperature, and MSO2 represents the molecular weight of SO2. If the reaction probability is high, sulfate was formed only on the surface of the samples since SO2 has no time to diffuse into the CaCO3 samples, and thus the geometric surface area of the samples would be the effective surface area. If the reaction probability is low, sulfate formed was distributed evenly through the whole samples since SO2 could diffuse into the sample entirely, and the effective surface area would be the BET surface area.65 Hence, the calculated uptake coefficients of SO2 on CaCO3 samples based on the BET and geometric surface area were considered as the lower and upper limits, respectively.49 Uptake coefficients were denoted as γ0 and γss in initial stage and stable stage, respectively. In this study, γ0 using geometric surface areas were determined to be (6.71 ± 0.55) × 10−4, (6.41 ± 0.45) × 10−4 and (5.06 ± 0.55) × 10−4 for 65 nm, 270 nm and 1 μm CaCO3 particles, respectively. These values were in the range of values determined under the same condition by Wu et al. (2011) and Li et al. (2006). On the basis of BET surface areas, our uptake coefficients γ0 were (7.51 ± 0.61) × 10−8, (1.00 ± 0.07) × 10−7 and (1.66 ± 0.18) × 10−7 for 65 nm, 270 nm and 1 μm CaCO3 particles, respectively. The values for 65 nm and 270 nm CaCO3 particles were lower than γ0 determined under the same condition in previous studies.45,46 It could be attributed to the fact that the BET surface areas of 65 nm and 270 nm CaCO3 particles were much larger than that in literatures.45,46 γ0 and γss varied with diameters of CaCO3 particles for adsorption of SO2 at various RHs were listed in Tables 1 and 2, respectively. Due to different amount of 65 nm, 270 nm and 1 μm CaCO3 particles packed in the sample cup, the γ based on geometric surface areas did not show regular change with the diameters of CaCO3 particles. In contrast to the variation tendency of sulfate formation rate with particle diameters, the γ using BET surface areas increased with increasing diameters of CaCO3 particles, due to the great differences of the BET surface areas among three size-resolved CaCO3 particles.
represents the mean molecular velocity of SO2, Asurface is the effective surface area of CaCO3 particles, and [SO2] is the gas-phase concentration of SO2. R is the molar gas constant, T refers to the temperature, and MSO2 represents the molecular weight of SO2. If the reaction probability is high, sulfate was formed only on the surface of the samples since SO2 has no time to diffuse into the CaCO3 samples, and thus the geometric surface area of the samples would be the effective surface area. If the reaction probability is low, sulfate formed was distributed evenly through the whole samples since SO2 could diffuse into the sample entirely, and the effective surface area would be the BET surface area.65 Hence, the calculated uptake coefficients of SO2 on CaCO3 samples based on the BET and geometric surface area were considered as the lower and upper limits, respectively.49 Uptake coefficients were denoted as γ0 and γss in initial stage and stable stage, respectively. In this study, γ0 using geometric surface areas were determined to be (6.71 ± 0.55) × 10−4, (6.41 ± 0.45) × 10−4 and (5.06 ± 0.55) × 10−4 for 65 nm, 270 nm and 1 μm CaCO3 particles, respectively. These values were in the range of values determined under the same condition by Wu et al. (2011) and Li et al. (2006). On the basis of BET surface areas, our uptake coefficients γ0 were (7.51 ± 0.61) × 10−8, (1.00 ± 0.07) × 10−7 and (1.66 ± 0.18) × 10−7 for 65 nm, 270 nm and 1 μm CaCO3 particles, respectively. The values for 65 nm and 270 nm CaCO3 particles were lower than γ0 determined under the same condition in previous studies.45,46 It could be attributed to the fact that the BET surface areas of 65 nm and 270 nm CaCO3 particles were much larger than that in literatures.45,46 γ0 and γss varied with diameters of CaCO3 particles for adsorption of SO2 at various RHs were listed in Tables 1 and 2, respectively. Due to different amount of 65 nm, 270 nm and 1 μm CaCO3 particles packed in the sample cup, the γ based on geometric surface areas did not show regular change with the diameters of CaCO3 particles. In contrast to the variation tendency of sulfate formation rate with particle diameters, the γ using BET surface areas increased with increasing diameters of CaCO3 particles, due to the great differences of the BET surface areas among three size-resolved CaCO3 particles.
| CaCO3 particle diameter (nm) | 1.5% RH | 19% RH | 53% RH | 90% RH | ||||
|---|---|---|---|---|---|---|---|---|
| γ0 (geometric), (×10−4) | γ0 (BET), (×10−7) | γ0 (geometric), (×10−4) | γ0 (BET), (×10−7) | γ0 (geometric), (×10−4) | γ0 (BET), (×10−7) | γ0 (geometric), (×10−4) | γ0 (BET), (×10−7) | |
| 65 | 6.71 ± 0.55 | 0.75 ± 0.06 | 7.76 ± 0.16 | 0.87 ± 0.02 | 9.13 ± 0.04 | 1.02 ± 0.01 | 7.58 ± 1.02 | 0.85 ± 0.11 |
| 270 | 6.41 ± 0.45 | 1.00 ± 0.07 | 8.63 ± 0.78 | 1.35 ± 0.12 | 10.2 ± 0.38 | 1.59 ± 0.06 | 9.15 ± 0.88 | 1.43 ± 0.14 |
| 1000 | 5.06 ± 0.55 | 1.66 ± 0.18 | 6.90 ± 0.14 | 2.26 ± 0.04 | 10.4 ± 0.15 | 3.39 ± 0.05 | 9.84 ± 0.95 | 3.22 ± 0.31 |
| CaCO3 particle diameter (nm) | 1.5% RH | 19% RH | 53% RH | 90% RH | ||||
|---|---|---|---|---|---|---|---|---|
| γss (geometric), (×10−4) | γss (BET), (×10−8) | γss (geometric), (×10−4) | γss (BET), (×10−8) | γss (geometric), (×10−4) | γss (BET), (×10−8) | γss (geometric), (×10−4) | γss (BET), (×10−8) | |
| 65 | 0.52 ± 0.01 | 0.59 ± 0.01 | 1.48 ± 0.02 | 1.65 ± 0.02 | 3.14 ± 0.32 | 3.51 ± 0.36 | 4.59 ± 0.08 | 5.13 ± 0.08 |
| 270 | 0.40 ± 0.02 | 0.62 ± 0.04 | 0.82 ± 0.08 | 1.28 ± 0.13 | 3.45 ± 0.15 | 5.39 ± 0.23 | 5.37 ± 0.11 | 8.37 ± 0.18 |
| 1000 | 0.30 ± 0.02 | 0.98 ± 0.07 | 0.55 ± 0.01 | 1.79 ± 0.05 | 2.06 ± 0.14 | 6.74 ± 0.46 | 4.51 ± 0.29 | 14.8 ± 0.96 |
The variation in morphology features of particles
The morphology features of particles before and after the reaction observed by the SEM were shown in Fig. 4 and 5. It is interesting that the morphologies of sulfate products were almost the same for size-resolved CaCO3 particles at the same RH. At about 50% RH, some microcrystallites appeared on the surface of the samples at the reaction time of 120 min, which were consistent with the appearance of bassanite based on in situ DRIFTS spectra. At 90% RH, rod-like calcium sulfate crystals with several hundred nanometers in length and approximately 100 nm in width occurred as the reaction lasted 120 min, corresponding to the formation of bassanite and gypsum based on the IR results. Afterwards, more rods with length ranging from several hundred nanometers (reaction lasted 200 min) to over one micrometers (reaction lasted 400 min) were observed with the extended reaction time. These rods grew on and stretched out of the samples and covered the surface of particles. The length of rods was no more than 2 μm even after the reaction time of 24 h under 90% RH condition (Fig. 5(2)) accompanied with appearance of some microcrystallites. According to the results of DRIFTS and SEM, when bassanite appeared without the presence of gypsum at 50% RH, microcrystallites were observed without rods. At higher RH, when gypsum was observed with certain amount, the rods also appeared. Hence, it could be speculated that microcrystallites and rods were largely composed of bassanite and gypsum, respectively.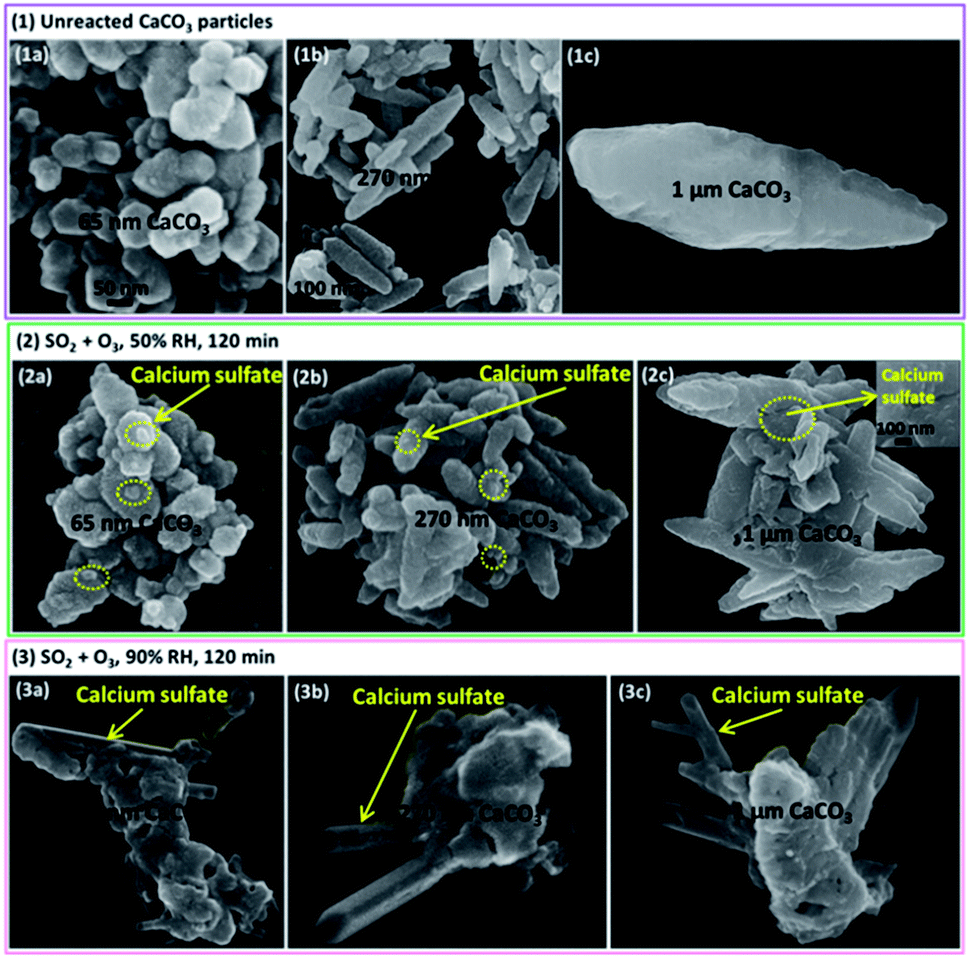 | ||
| Fig. 4 The SEM pictures of three kinds of samples (a) 65 nm CaCO3, (b) 270 nm CaCO3 and (c) 1 μm CaCO3 at 298 K. (1) Unreacted particles, (2 and 3) after SO2 (4.9 × 1014 molecules cm−3) and O3 (4.9 × 1014 molecules cm−3) were exposed simultaneously for 120 min at 50% RH and 90% RH, respectively. | ||
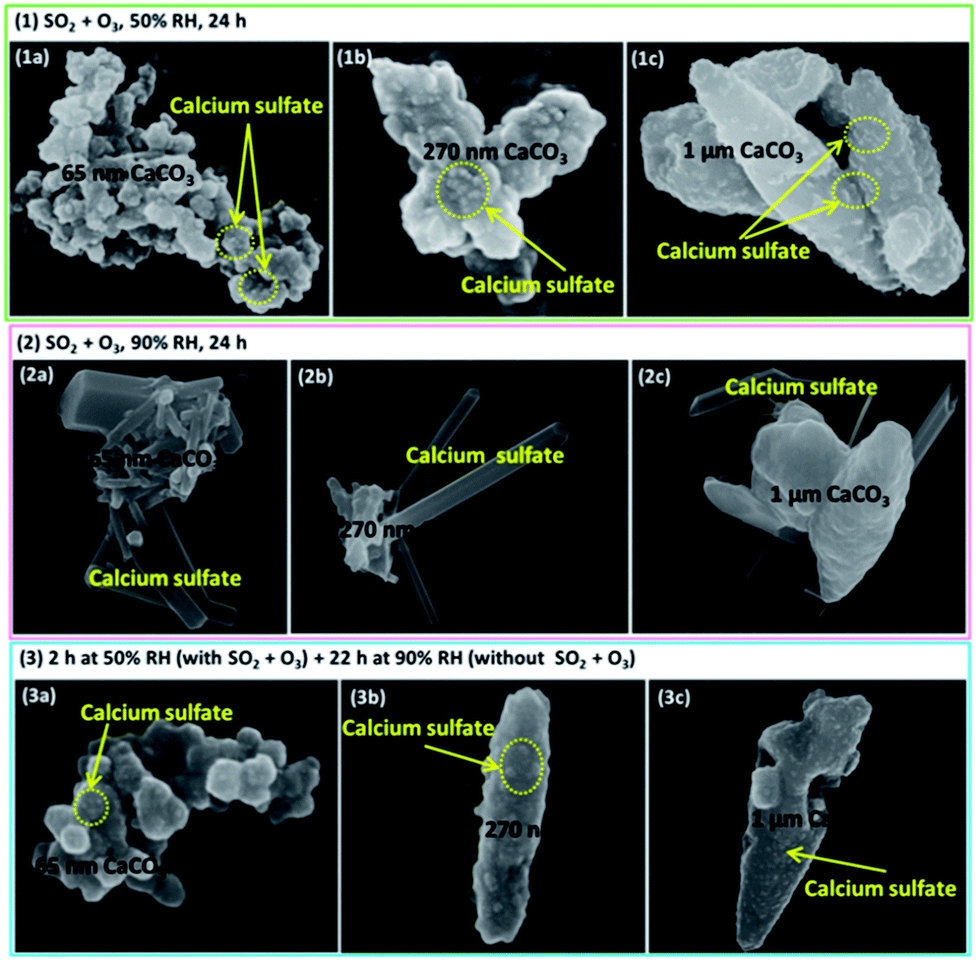 | ||
| Fig. 5 The SEM pictures of three kinds of samples (a) 65 nm CaCO3, (b) 270 nm CaCO3 and (c) 1 μm CaCO3 after SO2 and O3 were exposed simultaneously at 298 K. (1 and 2) Reaction time of 24 h at 50% RH and 90% RH, respectively, (3) reaction time of 2 h at 50% RH, and then maintained for 22 h without SO2 and O3 at 90% RH. | ||
To confirm the formation conditions of rod-like calcium sulfate crystals, further experiments were conducted. First, when the reaction lasted 24 h at 50% RH (Fig. 5(1)), only microcrystallites were observed. Second, the reaction time was 120 min at 50% RH, and then the RH was elevated to 90% for about 24 h in the absence of SO2 and O3 (Fig. 5(3)). Only microcrystallites appeared, and no rods could be observed. Third, at the reaction time of 120 min and 60% RH, rod-like calcium sulfate crystals with sizes of 50–100 nm in length and several tens nanometers in width appeared along with the microcrystallites similar to observed at 50% RH (Fig. S2†). It indicated that even more microcrystallites were formed, they could not aggregate together to form rod-like crystals at 50% RH. Furthermore, microcrystallites formed at 50% RH also could not coagulate to form rod-like crystals even the RH increased to 90%. It could be speculated that presence of reaction gases at RH above 60% was necessary for the formation of rod-like calcium sulfate crystals. The comparisons of size of calcium sulfate crystals formed at 60% and 90% RH indicated that sizes of rod-like calcium sulfate crystals increased with increasing RH above 60%.
Proposed mechanisms
A mechanism for the formation and growth of rod-like calcium sulfate crystals by the heterogeneous reactions of SO2 with O3 on size-resolved CaCO3 particles at different RHs was proposed. Under dry condition, CaSO4 was formed on the surface of CaCO3 particles upon adsorption of SO2 and oxidation by O3. Under humid condition, adsorbed water on calcium carbonate surface could facilitate the formation of Ca(OH)(HCO3), which was reactive towards SO2 and thus promoted the adsorption of SO2.64 Hence, the amount of adsorbed SO2 increased with elevated RH and thus accelerated the formation of sulfate. In contrast, when SO2 existed in the reaction systems, the particle surface became more hydrophilic upon reaction,47 and thus promoted further adsorption of water. When the amount of adsorbed SO2 and water were both enough, calcium sulfate hydrates could be formed immediately with the reaction. Hence, bassanite was formed at 50% RH and resulted in the formation of calcium sulfate microcrystallites on the surface of CaCO3 particles. At higher RH, gypsum was also formed by the heterogeneous reaction process. Above 50% RH, the liquid-like water layer was formed by multilayer coverages of surface adsorbed water accompanied with the promotion effect of SO2, which could facilitate the formation and aggregation of calcium sulfate hydrates during the reactions, and thus led to the formation of rod-like calcium sulfate crystals. Therefore, it could be understood that the high RH (above 60%) and the presence of reactant gases is requirement for the formation and growth of rod-like calcium sulfate crystals.Upon the aggregation of sulfate on the particle surface, fresh reactive sites of CaCO3 were exposed and thus further reinforced the formation of sulfate. The sizes and amount of rod-like calcium sulfate crystals increased with RH above 60% and increased with reaction time below 400 min. When the whole surface of CaCO3 have been covered by calcium sulfate crystals as shown in Fig. 5(2a), the reactive sites were prevented from exposing to reactant gases, and thus further formation of sulfate was inhibited in the deeper position of the samples. As a result, the limiting size of rod-like calcium sulfate crystals was approximately 2 μm in length during the heterogeneous reactions.
Conclusions
This study provides experimental evidence for heterogeneous formation pathway of rod-like calcium sulfate crystals in the atmosphere. The key factors for the pathway to form rod-like calcium sulfate crystals are RH above 60% and the presence of reactant gases. We show that liquid-like water layer formed by promotion of high RH in the presence of reactant gases could promote the formation and growth of rod-like calcium sulfate crystals by facilitating the formation and aggregation of calcium sulfate hydrates. Reactant particle diameters could affect the formation of sulfate. Results here imply that concentration and formation rate of sulfate decreased with increasing diameter of CaCO3 particles. Along with the longer suspending time in the air, CaCO3 particles with smaller diameters could accordingly contribute greater to sulfate formation.The rod-like shape is a typical morphology of calcium sulfate crystals observed in most field studies, which is consistent with the morphology of calcium sulfate crystals acquired by laboratory heterogeneous reactions in this study. Thus, the heterogeneous reaction pathway is a possible source of rod-like calcium sulfate crystals in the atmosphere. Calcium sulfate crystals with different morphology also existed in the atmosphere. Guo et al. found that calcium sulfate derived from coal fly ash were euhedral crystals or globular melt droplets.66 Pan et al. showed that calcium sulfate emitted from coal-fired power plant after wet flue gas desulfurization were prismatic or clintheriform shape with size distribution centered at approximately 0.2 μm.67 It could be speculated that the calcium sulfate observed in field study may not be originated from the direct emission of coal-fired power plant or coal fly ash. Therefore, it is a feasible method to recognize different origins or formation pathways of sulfate by the morphology of the particles in the atmosphere. The present study has examined only the morphology of calcium sulfate formed by heterogeneous reactions. Further laboratory studies on characterizing morphology features of sulfate formed through other formation pathways are needed to better understand the origin of sulfate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Natural Science Foundation of China (Contract No. 91544227), and the National Key Research and Development Program of China (2016YFC0202700).References
- A. Nel, Science, 2005, 308, 804–806 CrossRef CAS PubMed.
- X. Y. Zhang, Y. Q. Wang, T. Niu, X. C. Zhang, S. L. Gong, Y. M. Zhang and J. Y. Sun, Atmos. Chem. Phys., 2012, 12, 779–799 CrossRef CAS.
- X. G. Liu, K. Sun, Y. Qu, M. Hu, Y. L. Sun, F. Zhang and Y. H. Zhang, Aerosol Air Qual. Res., 2015, 15, 2246–2257 CrossRef CAS.
- Y. S. Wang, L. Yao, L. L. Wang, Z. R. Liu, D. S. Ji, G. Q. Tang, J. K. Zhang, Y. Sun, B. Hu and J. Y. Xin, Sci. China: Earth Sci., 2014, 57, 14–25 CrossRef CAS.
- J. H. Tan, J. C. Duan, D. H. Chen, X. H. Wang, S. J. Guo, X. H. Bi, G. Y. Sheng, K. B. He and J. M. Fu, Atmos. Res., 2009, 94, 238–245 CrossRef CAS.
- R. Zhang, G. Li, J. Fan, D. L. Wu and M. J. Molina, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5295–5299 CrossRef CAS PubMed.
- S. E. Bauer, M. I. Mishchenko, A. A. Lacis, S. Zhang, J. Perlwitz and S. M. Metzger, J. Geophys. Res.: Atmos., 2007, 112, D06307 CrossRef.
- G. H. Li, Y. Wang and R. Y. Zhang, J. Geophys. Res.: Atmos., 2008, 113, D15211 CrossRef.
- Y. Wang, Q. Wan, W. Meng, F. Liao, H. Tan and R. Zhang, Atmos. Chem. Phys., 2011, 11, 12421–12436 CrossRef CAS.
- F. Tan, B. Jing, S. R. Tong and M. F. Ge, Sci. Total Environ., 2017, 586, 930–938 CrossRef CAS PubMed.
- J. J. Cao, X. X. Tie, W. F. Dabberdt, T. Jie, Z. Z. Zhao, Z. S. An, Z. X. Shen and Y. C. Feng, J. Geophys. Res.: Atmos., 2013, 118, 4834–4846 Search PubMed.
- B. Jing, S. R. Tong, Q. F. Liu, K. Li, W. G. Wang, Y. H. Zhang and M. F. Ge, Atmos. Chem. Phys., 2016, 16, 4101–4118 CAS.
- Q. F. Liu, B. Jing, C. Peng, S. R. Tong, W. G. Wang and M. F. Ge, Atmos. Environ., 2016, 125, 69–77 CrossRef CAS.
- M. O. Andreae and P. J. Crutzen, Science, 1997, 276, 1052–1058 CrossRef CAS.
- J. H. P. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, Hoboken, New Jersey, 3rd edn, 2016 Search PubMed.
- C. R. Usher, A. E. Michel and V. H. Grassian, Chem. Rev., 2003, 103, 4883–4939 CrossRef CAS PubMed.
- C. F. Cullis and M. M. Hirschler, Atmos. Environ., 1980, 14, 1263–1278 CrossRef CAS.
- O. V. Rattigan, J. Boniface, E. Swartz, P. Davidovits, J. T. Jayne, C. E. Kolb and D. R. Worsnop, J. Geophys. Res.: Atmos., 2000, 105, 29065–29078 CrossRef CAS.
- Y. F. Cheng, G. J. Zheng, C. Wei, Q. Mu, B. Zheng, Z. B. Wang, M. Gao, Q. Zhang, K. B. He, G. Carmichael, U. Poschl and H. Su, Sci. Adv., 2016, 2, e1601530 Search PubMed.
- E. Harris, B. Sinha, D. van Pinxteren, A. Tilgner, K. W. Fomba, J. Schneider, A. Roth, T. Gnauk, B. Fahlbusch, S. Mertes, T. Lee, J. Collett, S. Foley, S. Borrmann, P. Hoppe and H. Herrmann, Science, 2013, 340, 727–730 CrossRef CAS PubMed.
- W. W. Yang, J. H. Zhang, Q. X. Ma, Y. Zhao, Y. C. Liu and H. He, Sci. Rep., 2017, 7, 4550 CrossRef PubMed.
- J. Y. Park and M. Jang, RSC Adv., 2016, 6, 58617–58627 RSC.
- H. He, Y. S. Wang, Q. X. Ma, J. Z. Ma, B. W. Chu, D. S. Ji, G. Q. Tang, C. Liu, H. X. Zhang and J. M. Hao, Sci. Rep., 2014, 4, 04172 CrossRef PubMed.
- C. Ye, H. Li, T. Zhu, J. Shang, Z. Zhang and D. Zhao, Sci. China: Chem., 2010, 53, 2652–2656 CrossRef CAS.
- X. He, S. F. Pang, J. B. Ma and Y. H. Zhang, Atmos. Environ., 2017, 165, 198–206 CrossRef CAS.
- Y. Wang, G. S. Zhuang, Y. L. Sun and Z. S. An, Atmos. Environ., 2006, 40, 6579–6591 CrossRef CAS.
- D. Cesari, A. Donateo, M. Conte, E. Merico, A. Giangreco, F. Giangreco and D. Contini, Atmos. Res., 2016, 174, 106–119 CrossRef.
- X. Huang, Y. Song, C. Zhao, M. M. Li, T. Zhu, Q. Zhang and X. Y. Zhang, J. Geophys. Res.: Atmos., 2014, 119, 14165–14179 CAS.
- S. Hoornaert, H. Van Malderen and R. Van Grieken, Environ. Sci. Technol., 1996, 30, 1515–1520 CrossRef CAS.
- K. Sun, X. G. Liu, J. W. Gu, Y. P. Li, Y. Qu, J. L. An, J. L. Wang, Y. H. Zhang, M. Hu and F. Zhang, J. Environ. Sci., 2015, 32, 155–167 CrossRef PubMed.
- W. J. Li, S. Z. Zhou, X. F. Wang, Z. Xu, C. Yuan, Y. C. Yu, Q. Z. Zhang and W. X. Wang, J. Geophys. Res.: Atmos., 2011, 116, D09301 Search PubMed.
- Z. B. Shi, D. Z. Zhang, M. Hayashi, H. Ogata, H. Z. Ji and W. Fujiie, Atmos. Environ., 2008, 42, 822–827 CrossRef CAS.
- W. J. Li, L. Y. Shao, D. Z. Zhang, C. U. Ro, M. Hu, X. H. Bi, H. Geng, A. Matsuki, H. Y. Niu and J. M. Chen, J. Cleaner Prod., 2016, 112, 1330–1349 CrossRef CAS.
- B. Kong, B. H. Guan, M. Z. Yates and Z. B. Wu, Langmuir, 2012, 28, 14137–14142 CrossRef CAS PubMed.
- W. J. Li and L. Y. Shao, Atmos. Chem. Phys., 2009, 9, 1863–1871 CrossRef CAS.
- W. J. Li and L. Y. Shao, Environ. Monit. Assess., 2010, 161, 565–573 CrossRef CAS PubMed.
- R. J. Gustafsson, A. Orlov, C. L. Badger, P. T. Griffiths, R. A. Cox and R. M. Lambert, Atmos. Chem. Phys., 2005, 5, 3415–3421 CrossRef CAS.
- Y. Takahashi, T. Miyoshi, M. Higashi, H. Kamioka and Y. Kanai, Environ. Sci. Technol., 2009, 43, 6535–6540 CrossRef CAS PubMed.
- D. F. Zhao, X. J. Song, T. Zhu, Z. F. Zhang, Y. J. Liu and J. Shang, Atmos. Chem. Phys., 2018, 18, 2481–2493 CAS.
- H. Boke, E. H. Gokturk and E. N. C. Saltik, Mater. Lett., 2002, 57, 935–939 CrossRef CAS.
- T. Claquin, M. Schulz and Y. J. Balkanski, J. Geophys. Res.: Atmos., 1999, 104, 22243–22256 CrossRef CAS.
- J. J. Cao, S. C. Lee, X. Y. Zhang, J. C. Chow, Z. S. An, K. F. Ho, J. G. Watson, K. Fung, Y. Q. Wang and Z. X. Shen, J. Geophys. Res.: Atmos., 2005, 110, D03203 Search PubMed.
- B. J. Krueger, V. H. Grassian, J. P. Cowin and A. Laskin, Atmos. Environ., 2004, 38, 6253–6261 CrossRef CAS.
- Y. Takahashi, T. Miyoshi, S. Yabuki, Y. Inada and H. Shimizu, Atmos. Environ., 2008, 42, 6535–6541 CrossRef CAS.
- L. Li, Z. M. Chen, Y. H. Zhang, T. Zhu, J. L. Li and J. Ding, Atmos. Chem. Phys., 2006, 6, 2453–2464 CrossRef CAS.
- L. Y. Wu, S. R. Tong, W. G. Wang and M. F. Ge, Atmos. Chem. Phys., 2011, 11, 6593–6605 CAS.
- H. A. Al-Hosney and V. H. Grassian, Phys. Chem. Chem. Phys., 2005, 7, 1266–1276 RSC.
- Q. X. Ma, H. He, Y. C. Liu, C. Liu and V. H. Grassian, Phys. Chem. Chem. Phys., 2013, 15, 19196–19204 RSC.
- M. Ullerstam, R. Vogt, S. Langer and E. Ljungstrom, Phys. Chem. Chem. Phys., 2002, 4, 4694–4699 RSC.
- W. J. Liu, X. He, S. F. Pang and Y. H. Zhang, Atmos. Environ., 2017, 167, 245–253 CrossRef CAS.
- T. Kojima, P. R. Buseck and J. M. Reeves, J. Geophys. Res.: Atmos., 2005, 110, D09203 CrossRef.
- C. Borensen, U. Kirchner, V. Scheer, R. Vogt and R. Zellner, J. Phys. Chem. A, 2000, 104, 5036–5045 CrossRef.
- H. Shaka, W. H. Robertson and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2007, 9, 1980–1990 RSC.
- R. Vogt and B. J. Finlaysonpitts, J. Phys. Chem., 1994, 98, 3747–3755 CrossRef CAS.
- C. M. Koretsky, D. A. Sverjensky, J. W. Salisbury and D. M. DAria, Geochim. Cosmochim. Acta, 1997, 61, 2193–2210 CrossRef CAS.
- D. Peak, R. G. Ford and D. L. Sparks, J. Colloid Interface Sci., 1999, 218, 289–299 CrossRef CAS PubMed.
- A. P. Prince, P. Kleiber, V. H. Grassian and M. A. Young, Phys. Chem. Chem. Phys., 2007, 9, 3432–3439 RSC.
- H. A. Al-Hosney and V. H. Grassian, J. Am. Chem. Soc., 2004, 126, 8068–8069 CrossRef CAS PubMed.
- S. E. Bogushevich and G. N. Lysenko, Russ. J. Inorg. Chem., 2009, 54, 618–623 CrossRef.
- P. S. R. Prasad, V. K. Chaitanya, K. S. Prasad and D. N. Rao, Am. Mineral., 2005, 90, 672–678 CrossRef CAS.
- J. Bensted and S. Prakash, Nature, 1968, 219, 60–61 CrossRef CAS.
- P. K. Mandal and T. K. Mandal, Cem. Concr. Res., 2002, 32, 313–316 CrossRef CAS.
- F. Tan, S. R. Tong, B. Jing, S. Q. Hou, Q. F. Liu, K. Li, Y. Zhang and M. F. Ge, Atmos. Chem. Phys., 2016, 16, 8081–8093 CAS.
- J. Baltrusaitis, C. R. Usher and V. H. Grassian, Phys. Chem. Chem. Phys., 2007, 9, 3011–3024 RSC.
- S. R. Tong, L. Y. Wu, M. F. Ge, W. G. Wang and Z. F. Pu, Atmos. Chem. Phys., 2010, 10, 7561–7574 CAS.
- Z. B. Guo, Z. Q. Li, J. Farquhar, A. J. Kaufman, N. P. Wu, C. Li, R. R. Dickerson and P. C. Wang, J. Geophys. Res.: Atmos., 2010, 115, D00K07 CrossRef.
- D. P. Pan, H. Wu and L. J. Yang, J. Environ. Sci., 2017, 55, 303–310 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02050g |
| This journal is © The Royal Society of Chemistry 2018 |