
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormal total synthesis of histrionicotoxin alkaloids via Hg(OTf)2-catalyzed cycloisomerization and SmI2-induced ring expansion†
Kunihiro Matsumura,
Keisuke Nishikawa,
Hiroaki Yoshida,
Matsumi Doe and
Yoshiki Morimoto *
*
Department of Chemistry, Graduate School of Science, Osaka City University Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: morimoto@sci.osaka-cu.ac.jp
First published on 21st March 2018
Abstract
The efficient formal total synthesis of histrionicotoxin alkaloids was achieved. In this process, two key reactions were used to construct a core 1-azaspiro[5.5]undecane framework common to histrionicotoxins: a mercuric triflate (Hg(OTf)2)-catalyzed cycloisomerization of a linear substrate, which was developed in our laboratory, and a samarium iodide (SmI2)-mediated ring expansion.
Introduction
(−)-Histrionicotoxin 283A (HTX-283A, 1, Fig. 1) is an azaspirocyclic histrionicotoxin alkaloid that was first isolated from skin extracts of the Colombian poison arrow frog Dendrobates histrionicus in 1971 by Witkop et al.1 It exhibits highly selective inhibition of nicotinic acetylcholine receptors. Since Witkop et al. separated a mixture of six alkaloids including 1 from the skin extracts of 1110 frogs, other members of this alkaloid family have also been identified. In 1992, (−)-histrionicotoxin 235A (HTX-235A, 2) was also isolated as a major constituent by Spande et al. from the other poison frog Dendrobates auratus.2 The bioactivities have led to its use as important probes in neurophysiology. Establishing an efficient synthetic pathway for histrionicotoxin alkaloids is essential to investigate the development of new biological tools and the structure–activity relationships in more detail.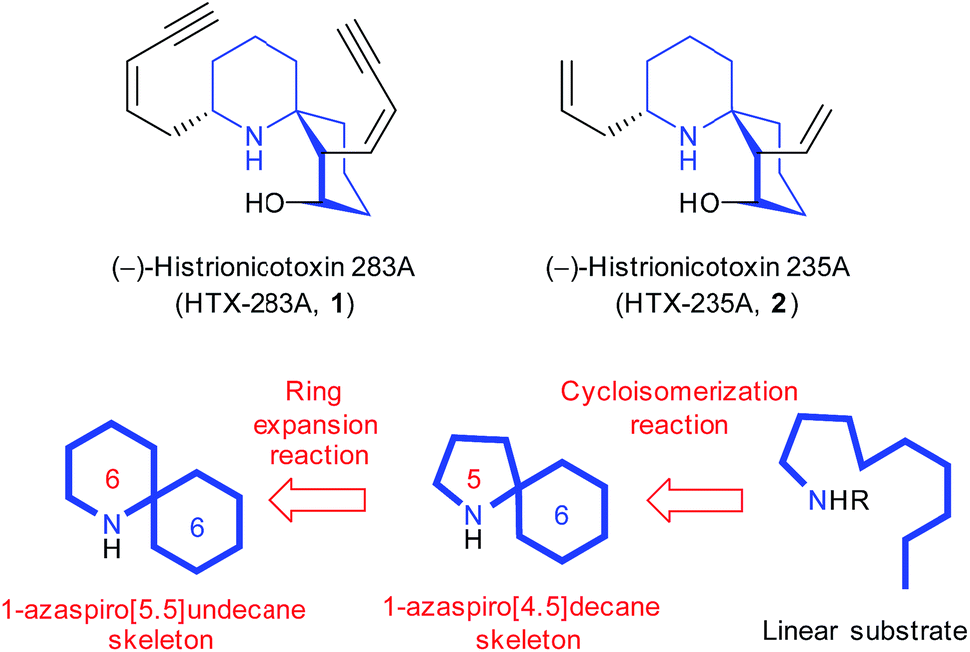 | ||
| Fig. 1 Chemical structures of (−)-HTX-283A (1) and (−)-HTX-235A (2). | ||
Regarding the structural features, compound 1 has a unique chemical structure characterized by a core spiropiperidine structure and two cis-enyne side chains. The core spirocyclic skeleton of 1 is conserved in 2, but the two side cis-enyne groups are replaced by allylic and vinylic groups. The spirocyclic skeleton structure of histrionicotoxin alkaloids has prompted many synthetic organic chemists to promote the total syntheses so far.3 In this contribution, we intended to realize the efficient construction of the core 1-azaspiro[5.5]undecane skeleton common to histrionicotoxin alkaloids based on ring expansion of a 1-azaspiro[4.5]decane one that could be formed in a stereoselective manner from a linear substrate by our original Hg(OTf)2-catalyzed cycloisomerization reaction4 (Fig. 1). In this letter, we report the formal synthesis of histrionicotoxins via two key steps: Hg(OTf)2-catalyzed cycloisomerization and SmI2-mediated ring expansion reactions.
Results and discussion
The retrosynthetic analysis of histrionicotoxin alkaloids is shown in Scheme 1. The spirocyclic ring compound 3 has been converted into histrionicotoxins via introduction of an allylic group by Tokuyama et al.3i Therefore, we planned to synthesize the 1-azaspiro[5.5]undecane skeleton 3 by applying a SmI2-mediated ring expansion reaction, reported by Honda et al.,5 to 1-azaspiro[4.5]decane skeleton 4, which would be derived from spirocyclic compound 5. Compound 5 would be constructed by the key Hg(OTf)2-catalyzed cycloisomerization reaction of linear ynone 6, which would be prepared through acylation of sulfone 8 with pyrrolidinone 7, derived from L-glutamic acid according to the known method.6 Compound 8 would be synthesized by alkylation of a lithium acetylide of alkyne 9 with commercially available 1,3-diiodopropane (10).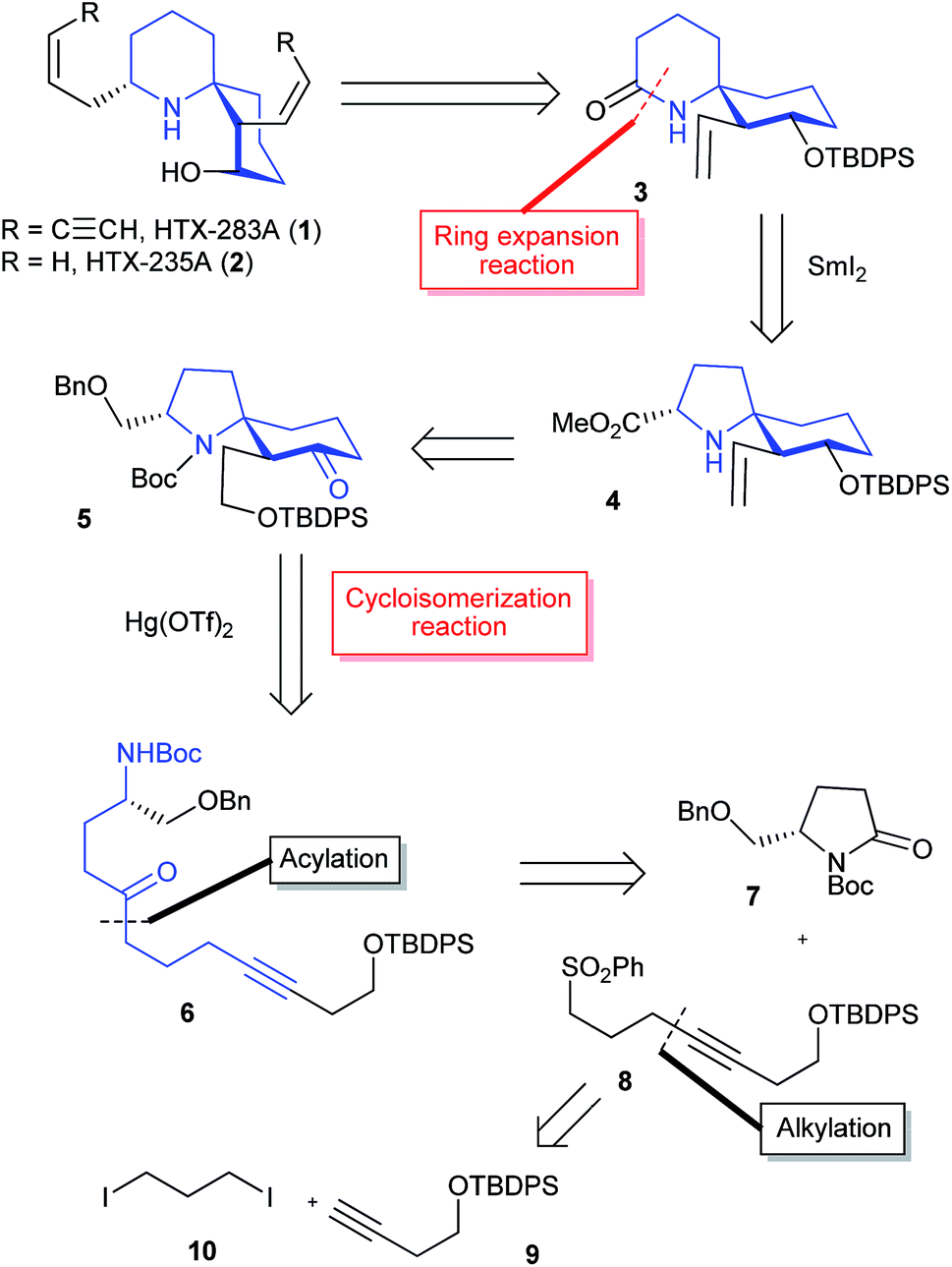 | ||
| Scheme 1 Retrosynthetic analysis of histrionicotoxins. | ||
The total synthesis commenced with the known silyl ether 9, derived from commercially available 3-butyn-1-ol (Scheme 2).7 Alkylation of a lithium acetylide of 9 with 1,3-diiodopropane (10) followed by sulfonylation of the iodo moiety gave sulfone 8. After acylation of an α-anion of 8 with the known pyrrolidinone 7, the cyclization precursor 6 was prepared through SmI2-mediated desulfonylation.8
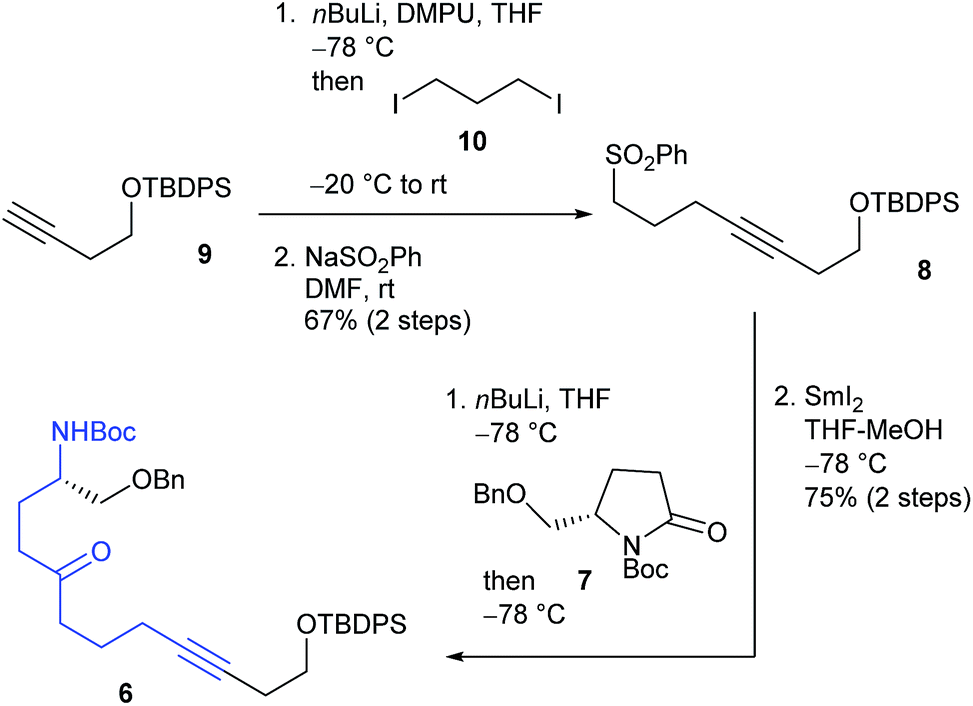 | ||
| Scheme 2 Synthesis of the cyclization precursor 6. | ||
We examined the Hg(OTf)2-catalyzed cycloisomerization reaction of precursor 6 as the first key reaction (Table 1). As expected, the reaction proceeded in a stereoselective manner to provide the desired spirocyclic product 5 in an isolated yield of 58%, along with a minor diastereomer 11 (11% and 12%), when 6 was allowed to react with Hg(OTf)2 (5 and 10 mol%) in MeCN at 0 °C (entries 1 and 2, respectively). Increasing the catalyst loading to 20 mol% afforded a better yield (67%) of 5 (entry 3). When the catalyst loading was increased to 30 and 50 mol%, the yield of 5 decreased to 52% and 38%, respectively (entries 4 and 5). The reaction at −20 °C resulted in a decrease in the yield of 11 (trace) (entry 6). When the reaction solution was gradually warmed to room temperature from −20 °C, the result was the same as that in entry 3 (entry 7). Finally, the effect of reaction temperature was examined (entries 8–10). Entry 9 showed the best conditions (cat. 20 mol%, −30 °C) in terms of the yield of 5. Lowering the temperature resulted in a gradual decrease in the yield of 11. Stereochemical assignments of 5 and 11 were achieved by their NOESY spectra; see (ESI†). The reaction mechanism, which we propose at present, is shown in Table 1.
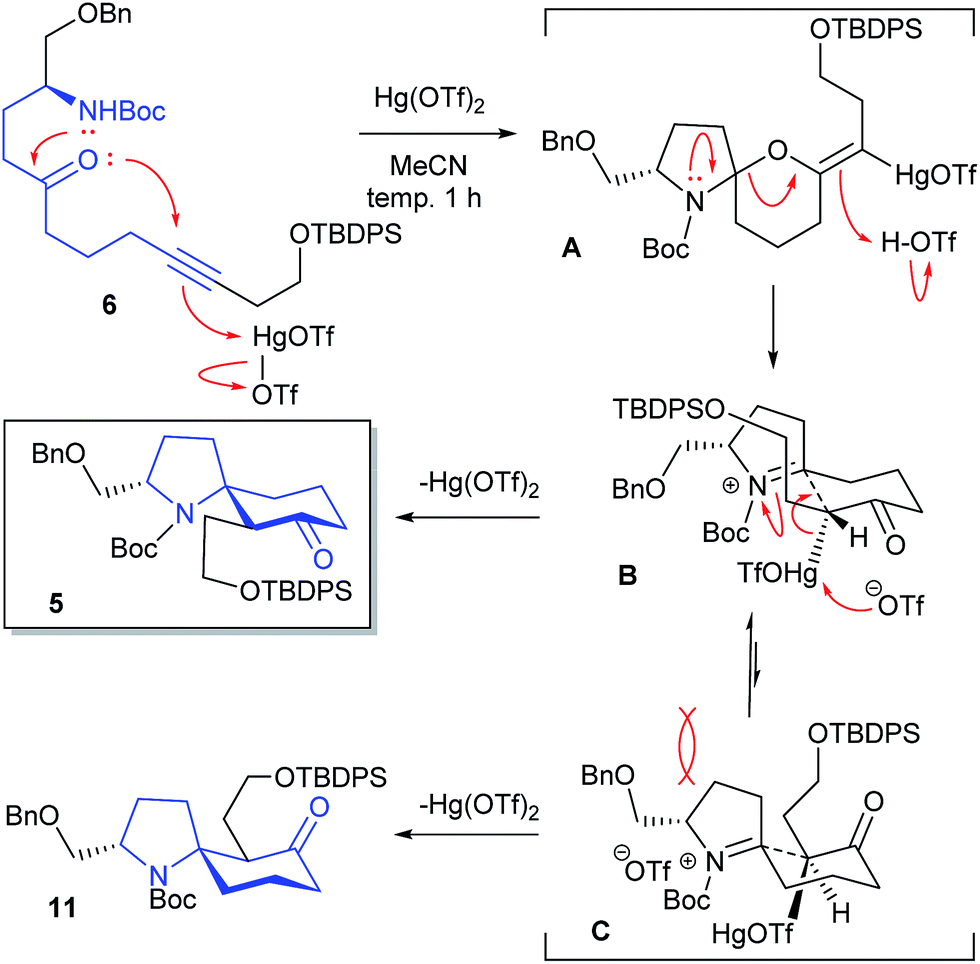
The aminoketal A would be formed through a 6-exo-dig intramolecular oxymercuration to the alkyne π-electron activated by coordination of Hg(OTf)2 followed by nucleophilic addition of the nitrogen function. The intermediate A could be cleaved by protonation with the generated TfOH to give an iminium ion intermediate B or C. The construction of a carbocycle via Ferrier-type cyclization would provide the desired spirocyclic product 5 with regeneration of the catalyst. Considering the chair-like transition states B and C, the desired 5 would be diastereoselectively obtained by way of the more stable transition state B without steric repulsion with a benzyloxymethyl group as outlined in Table 1. The by-product 11 would be produced through C.9
According to Procter's conditions,10 treatment of spirocyclic product 5 with SmI2 in the presence of H2O and triethylamine afforded the desired eq-alcohol 12 as a major diastereomer (58%), along with ax-alcohol 12 (40%) (Scheme 3).11 The undesired ax-12 was oxidized with Dess–Martin periodinane (DMP) for the recycling use.12 After MOM protection of a hydroxy group in eq-12, a benzylic group was removed by hydrogenolysis using Pd/C. After a hydroxy group was converted into a carboxylic acid through one-pot oxidation using 1,5-dimethyl-nor-AZADO (DMN-AZADO) and NaClO2,13 the esterification using MeI and Cs2CO3 afforded ester 14. Deprotection of a TBDPS group using tetrabutylammonium fluoride (TBAF) provided the desired alcohol 15.
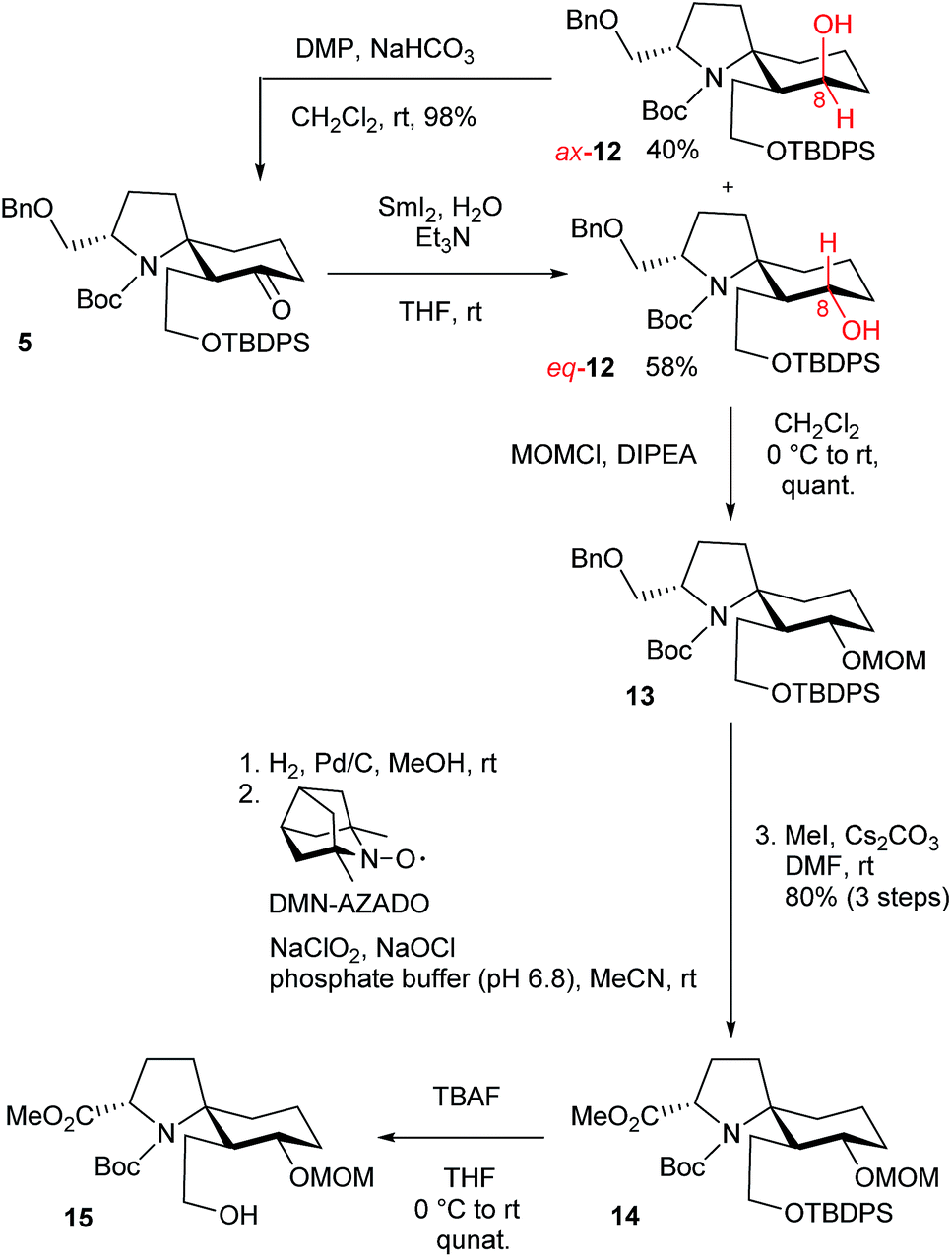 | ||
| Scheme 3 Synthesis of alcohol 15. | ||
The formal total syntheses of histrionicotoxins were completed from 15 as outlined in Scheme 4. After a vinylic group was constructed by Nishizawa–Grieco elimination,3i,14 deprotection of MOM and Boc groups afforded vinyl alcohol 16. The conformation of the spirocycle in 16 was assigned based on the NOESY spectra; see ESI.† The TBDPS protection of a hydroxy group in 16 provided silyl ether 4. Compound 4 was further converted into 1-azaspiro[5.5]undecane 3 in high yield through the key SmI2-mediated ring expansion reaction in the presence of HMPA and pivalic acid.5 It is the first example that the SmI2-mediated radical ring expansion was applied for constructing such a complex system as a 1-azaspiro[5.5]undecane skeleton of histrionicotoxins. Compound 3 is a key intermediate in the total synthesis of histrionicotoxin alkaloids, (−)-HTX-283A (1) and (−)-HTX-235A (2), by Tokuyama's group.3i The spectral data (1H- and 13C-NMR) and the optical rotation of our synthetic 3 were consistent with those reported for the previous synthetic compound.3i It has been reported that compound 3 can be transformed to (−)-1 via (−)-2.3i
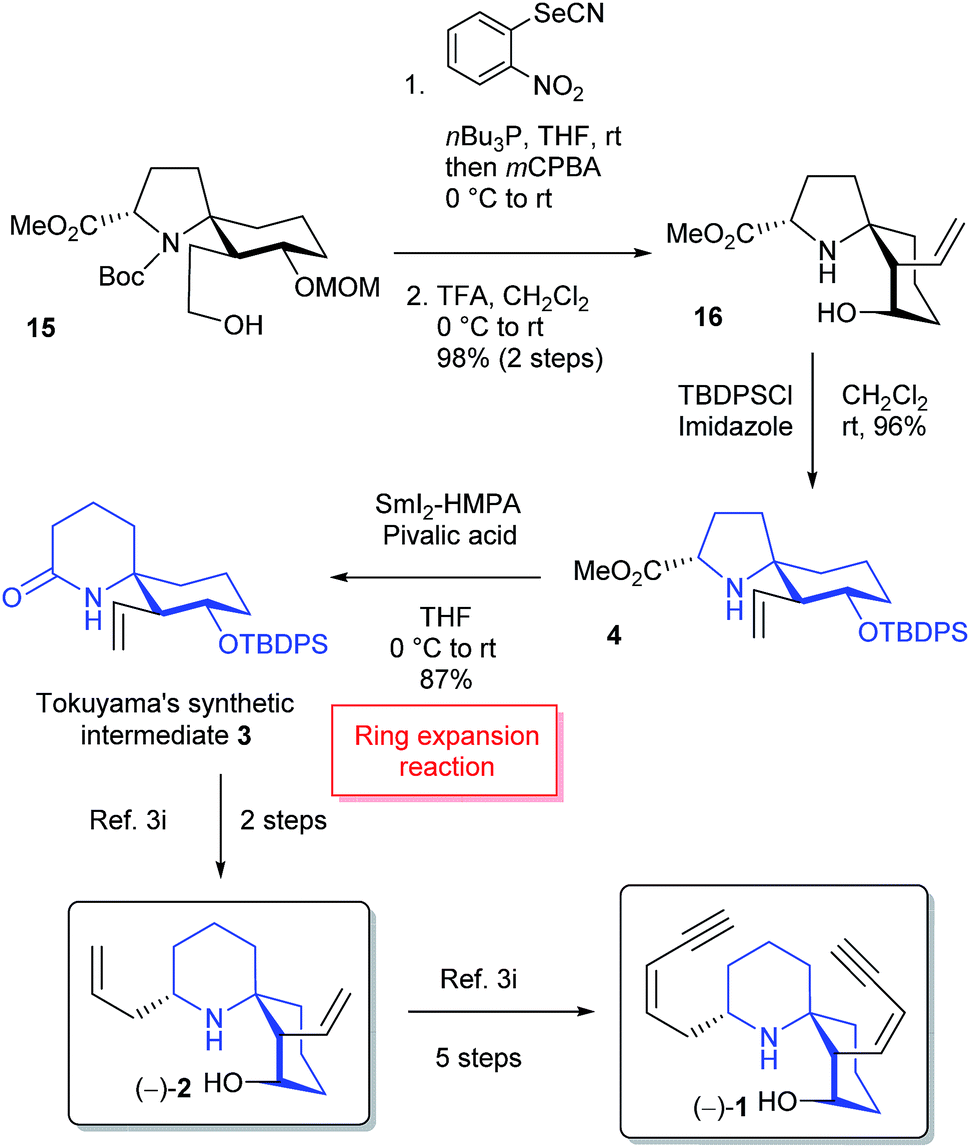 | ||
| Scheme 4 Formal synthesis of histrionicotoxin alkaloids. | ||
Conclusions
In summary, the combination of Hg(OTf)2-catalyzed cycloisomerization and SmI2-mediated ring expansion reactions made it possible to efficiently construct a 1-azaspiro[5.5]undecane framework of histrionicotoxin alkaloids. The synthetic efficiency of key intermediate 3 (15% overall yield and 15 steps based on the known silyl ether 9) was demonstrated in comparison with Tokuyama's method3i (5% overall yield and 14 steps based on the readily available ketodiester and benzylamine). Our synthetic method will be useful to synthesize HTXs and the derivatives as biological probes.Experimental
General procedures
1H-NMR spectra were recorded in deuteriochloroform on Bruker Biospin Avance 300 nanobay (300 MHz), JEOL JNM-ECZ400S or Bruker Biospin Avance III HD 400 (400 MHz), and Bruker Biospin Avance III HD 600 (600 MHz) spectrometers. 13C-NMR spectra were measured in deuteriochloroform on Bruker Biospin Avance 300 nanobay (75 MHz), JEOL JNM-ECZ400S or Bruker Biospin Avance III HD 400 (100 MHz), and Bruker Biospin Avance III HD 600 (150 MHz) spectrometers. Chemical shifts were reported in parts per million (ppm) from tetramethylsilane with the solvent resonance as the internal standard (CDCl3: 7.26 ppm for 1H-NMR, 77.0 ppm for 13C-NMR). Splitting patterns were designated as “s, d, t, q, and m” to indicate “singlet, doublet, triplet, quartet, and multiplet,” respectively. IR spectra were recorded on a JASCO FT/IR-4100 spectrophotometer by the attenuated total reflection (ATR) method, unless otherwise noted. High-resolution mass spectra were obtained on a JEOL AccuTOF LC-plus JMS-T100LP (DART). Optical rotations were determined on a JASCO DIP-370 digital polarimeter. Mp are uncorrected and were recorded on a Yanagimoto micro melting point apparatus. Analytical TLC was carried out by precoated silica gel (Merck TLC plates silica gel 60 F254). Flash column chromatography was performed with Merck silica gel 60 (particle size 63–200 μm), Wakogel® 60N (particle size 38–100 μm), and KANTO silica gel 60N (particle size 40–50 μm). All reactions were performed in oven-dried glassware. Tetrahydrofuran (THF) was distilled over sodium metal/benzophenone ketyl. Acetonitrile (MeCN), dichloromethane (CH2Cl2), and triethylamine (Et3N) were distilled over calcium hydride. Hexamethylphosphoric triamide (HMPA), N,N′-dimethylpropyleneurea (DMPU), and N,N-dimethylformamide (DMF) were distilled over calcium hydride under reduced pressure. Methanol (MeOH) was distilled from Mg(OMe)2.Experimental procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90) on silica gel to give a mixture including a desired mono-iodide, which was used in the next reaction without further purification.
:90) on silica gel to give a mixture including a desired mono-iodide, which was used in the next reaction without further purification.To a solution of the mixture including a desired mono-iodide in DMF (68 mL) was added NaSO2Ph (1.68 g, 10.2 mmol) at room temperature under a nitrogen atmosphere, and the solution was stirred for 5 h. After the reaction was quenched with H2O, the resulting mixture was extracted with EtOAc (×3). The organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/hexane, 30:70) on silica gel to give 8 (2.24 g, 4.56 mmol, 67% in 2 steps) as a colorless oil: Rf = 0.58 (EtOAc/hexane, 30:70); 1H-NMR (300 MHz, CDCl3) δ 7.91–7.85 (2H, m), 7.70–7.58 (5H, m), 7.56–7.48 (2H, m), 7.47–7.34 (6H, m), 3.69 (2H, t, J = 7.1 Hz), 3.22–3.12 (2H, m), 2.42–2.33 (2H, m), 2.28–2.19 (2H, m), 1.85 (2H, quintet, J = 7.3 Hz), 1.03 (9H, s); 13C-NMR (75 MHz, CDCl3) δ 139.0, 135.5, 133.7, 133.6, 129.7, 129.3, 128.0, 127.7, 79.0, 78.6, 62.6, 55.2, 26.7, 22.8, 22.1, 19.2, 17.6; IR (ATR) 3070, 3051, 2997, 2956, 2931, 2857, 1769, 1588, 1508, 1472, 1447, 1428, 1388, 1362, 1308, 1261, 1152, 1111, 1089, 1059, 1025, 915, 822, 800 cm−1; DART-HRMS calcd for C29H35O3SSi [(M + H)+] 491.2076, found 491.2078.
:90) on silica gel to afford a mixture including a desired sulfone, which was used in the next reaction without further purification.To a solution of the mixture including a desired sulfone in THF (6.0 mL) and MeOH (4.0 mL) was added SmI2 (15.0 mL, 1.50 mmol, 0.100 M in THF) at −78 °C under a nitrogen atmosphere, and the solution was stirred for 1 h. The reaction was quenched with a saturated aqueous solution of NaHCO3, and the mixture was extracted with EtOAc (×3). The organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (EtOAc/hexane, 30:70) on silica gel to provide 6 (404 mg, 617 μmol, 75% in 2 steps) as a colorless oil: Rf = 0.37 (EtOAc/hexane, 30:70); [α]25D −12.3 (c 2.31, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.72–7.64 (4H, m), 7.46–7.26 (11H, m), 4.74 (1H, br d, J = 11.3 Hz), 4.52 (1H, d, J = 11.9 Hz), 4.46 (1H, d, J = 12.0 Hz), 3.73 (2H, t, J = 7.1 Hz), 3.71 (1H, m), 3.44 (2H, d, J = 4.0 Hz), 2.52–2.36 (6H, m), 2.14 (2H, td, J = 7.0, 2.3 Hz), 1.92–1.61 (4H, m), 1.42 (9H, s), 1.05 (9H, s); 13C-NMR (75 MHz, CDCl3) δ 210.1, 155.7, 138.0, 135.5, 133.6, 129.6, 128.4, 127.64, 127.61, 127.57, 80.4, 79.2, 77.7, 77.2, 73.1, 72.2, 62.8, 49.9, 41.5, 39.3, 28.3, 26.7, 26.1, 22.9, 22.8, 19.2, 18.1; IR (ATR) 3371, 3070, 3049, 3031, 2956, 2932, 2894, 2858, 1712, 1589, 1499, 1473, 1453, 1428, 1389, 1365, 1246, 1221, 1171, 1111, 1059, 1028, 915, 823 cm−1; DART-HRMS calcd for C40H54NO5Si [(M + H)+] 656.3771, found 656.3789.
:90) on silica gel to give 5 (32.8 mg, 50.0 μmol, 77%) and 11 (2.6 mg, 3.97 μmol, 6%) as each colorless oil. 5: Rf = 0.58 (EtOAc/hexane, 30:70); [α]26D −15.9 (c 0.34, CHCl3); 1H-NMR (600 MHz, CDCl3, 25 °C, two rotamers in a 4:1 ratio) δ 7.68–7.61 (4H, m), 7.42–7.21 (11H, m), 4.55 (0.2H, d, J = 12.0 Hz), 4.51 (1.6H, s), 4.49 (0.2H, d, J = 12.0 Hz), 4.20 (0.2H, br s), 4.01 (0.8H, br s), 3.84 (0.8H, dd, J = 10.2, 1.8 Hz), 3.69 (1H, td, J = 9.2, 4.6 Hz), 3.55 (0.2H, d, J = 4.7 Hz), 3.53–3.44 (2H, m), 3.36 (0.8H, dd, J = 8.9, 7.5 Hz), 3.30 (0.2H, d, J = 9.8 Hz), 2.96 (0.8H, td, J = 13.6, 4.2 Hz), 2.62 (0.2H, td, J = 13.4, 4.2 Hz), 2.36–2.22 (1.8H, m), 2.21–1.99 (1.2H, m), 1.93–1.63 (5.2H, m), 1.61–1.53 (0.8H, m), 1.52–1.38 (2H, m), 1.38 and 1.36 (total 9H, each s), 1.03 (9H, s); 13C-NMR (150 MHz, CDCl3, 25 °C, two rotamers in a 4:1 ratio) δ 210.9, 210.0, 153.3, 152.9, 138.5, 138.2, 135.49, 135.45, 134.1, 133.9, 133.84, 133.82, 129.43, 129.42, 129.36, 129.3, 128.3, 128.2, 127.6, 127.5, 127.4, 127.3, 80.2, 79.4, 73.0, 71.2, 70.8, 70.3, 69.4, 63.2, 62.8, 59.7, 59.1, 54.3, 52.4, 42.0, 41.8, 37.7, 36.8, 33.7, 32.4, 28.5, 28.4, 27.0, 26.84, 26.80, 26.6, 26.3, 25.8, 21.5, 19.1; IR (ATR) 3070, 3048, 3032, 2956, 2929, 2858, 1712, 1688, 1589, 1541, 1472, 1455, 1428, 1388, 1364, 1317, 1298, 1254, 1219, 1171, 1111, 1072, 1029, 999, 973, 941, 909, 886, 866, 823, 772 cm−1; DART-HRMS calcd for C40H54NO5Si [(M + H)+] 656.3771, found 656.3773. 11: Rf = 0.53 (EtOAc/hexane, 30:70); [α]24D −8.0 (c 0.87, CHCl3); 1H-NMR (600 MHz, CDCl3, 25 °C, two rotamers in a 5:1 ratio) δ 7.68–7.60 (4H, m), 7.42–7.25 (11H, m), 4.56 (0.83H, d, J = 12.1 Hz), 4.53, 4.51 (each 0.17H, d, J = 12.1 Hz), 4.50 (0.83H, d, J = 12.1 Hz), 4.15 (0.17H, br s), 3.98 (0.83H, br t, J = 7.0 Hz), 3.91 (0.83H, d, J = 9.7 Hz), 3.87 (0.17H, dd, J = 10.1, 2.1 Hz), 3.69 (0.17H, m), 3.67–3.55 (1.83H, m), 3.55–3.44 (1H, m), 3.37 (0.17H, m), 3.25 (0.83H, t, J = 8.9 Hz), 3.02 (0.17H, td, J = 13.5, 4.3 Hz), 2.73 (0.83H, td, J = 13.3, 4.0 Hz), 2.42–2.17 (2.17H, m), 2.15–2.02 (0.83H, m), 1.97–1.33 (8H, m), 1.42 and 1.37 (total 9H, each s), 1.03 (1.53H, s), 1.02 (7.47H, s); 13C-NMR (150 MHz, CDCl3, 25 °C, two rotamers in a 5:1 ratio) δ 210.5, 209.5, 153.2, 152.9, 138.3, 138.1, 135.54, 135.52, 134.13, 134.10, 134.07, 133.9, 129.6, 129.4, 128.4, 128.3, 127.7, 127.64, 127.59, 127.53, 127.50, 80.2, 79.6, 73.2, 71.0, 70.7, 70.5, 70.4, 62.83, 62.75, 58.2, 57.8, 52.4, 52.1, 41.5, 41.4, 32.2, 30.6, 28.4, 26.9, 26.8, 25.7, 25.3, 24.5, 23.8, 21.9, 21.4, 19.18, 19.16; IR (ATR) 3069, 3049, 3030, 2959, 2931, 2857, 1712, 1688, 1588, 1473, 1455, 1428, 1366, 1306, 1256, 1169, 1110, 1026, 975, 955, 908, 854, 823, 804 cm−1; DART-HRMS calcd for C40H54NO5Si [(M + H)+] 656.3771, found 656.3771.:90) on silica gel to provide eq-12 (77.4 mg, 118 μmol, 58%) and ax-12 (54.1 mg, 82.2 μmol, 40%) as each colorless oil: eq-12: Rf = 0.54 (EtOAc/hexane, 30:70); [α]26D −15.1 (c 1.40, CHCl3); 1H-NMR (600 MHz, CDCl3, 25 °C, two rotamers in a 2:1 ratio) δ 7.71–7.63 (4H, m), 7.46–7.36 (6H, m), 7.36–7.23 (5H, m), 4.75 (0.33H, br s), 4.54 (0.67H, d, J = 13.0 Hz), 4.49 (0.67H, d, J = 11.6 Hz), 4.47 (0.33H, d, J = 12.0 Hz), 4.46 (0.33H, d, J = 12.1 Hz), 4.23 (0.67H, d, J = 3.1 Hz), 4.10 (0.33H, m), 3.91 (0.67H, m), 3.77 (0.33H, dt, J = 10.2, 4.1 Hz), 3.72–3.65 (0.67H, m), 3.61 (0.67H, td, J = 10.0, 3.7 Hz), 3.57 (0.33H, m), 3.56 (0.33H, td, J = 9.3, 2.5 Hz), 3.49 (0.67H, dd, J = 8.9, 3.1 Hz), 3.45 (0.33H, dd, J = 8.9, 7.7 Hz), 3.43–3.32 (1H, m), 3.29 (0.67H, t, J = 8.5 Hz), 2.77 (0.67H, br t, J = 7.4 Hz), 2.39 (0.67H, td, J = 13.2, 4.2 Hz), 2.37 (0.33H, m), 2.16 (0.33H, td, J = 12.9, 3.6 Hz), 2.10–1.99 (1H, m), 1.99–1.88 (1H, m), 1.88–1.53 (7H, m), 1.45–1.16 (2H, m), 1.38 (3H, s), 1.26 (6H, s), 1.07 (3H, s), 1.04 (6H, s); 13C-NMR (150 MHz, CDCl3, 25 °C, two rotamers in a 2:1 ratio) δ 153.5, 152.6, 138.6, 138.3, 135.64, 135.59, 135.56, 133.0, 132.6, 132.5, 129.94, 129.90, 129.72, 129.67, 128.4, 128.3, 127.8, 127.7, 127.6, 127.44, 127.39, 79.9, 79.0, 73.11, 73.06, 73.0, 71.5, 70.7, 68.9, 68.3, 65.1, 64.6, 59.0, 58.6, 50.0, 47.3, 37.9, 36.8, 35.1, 35.0, 33.4, 31.8, 31.4, 31.1, 28.5, 28.3, 26.8, 26.7, 26.0, 20.58, 20.56, 19.0, 18.9; IR (ATR) 3435, 3070, 3048, 3032, 2956, 2929, 2858, 1688, 1589, 1541, 1472, 1455, 1428, 1388, 1364, 1317, 1298, 1254, 1219, 1171, 1111, 1072, 1029, 999, 973, 941, 909, 886, 866, 823, 772, 736, 700 cm−1; DART-HRMS calcd for C40H56NO5Si [(M + H)+] 658.3928, found 658.3935. ax-12: Rf = 0.67 (EtOAc/hexane, 30:70); [α]27D −26.3 (c 0.82, CHCl3); 1H-NMR (600 MHz, CDCl3, 25 °C, two rotamers in a 3:1 ratio) δ 7.70–7.62 (4H, m), 7.47–7.22 (11H, m), 4.57 (0.25H, d, J = 12.0 Hz), 4.53 (0.75H, d, J = 12.1 Hz), 4.48 (0.25H, d, J = 12.0 Hz), 4.50 (0.75H, d, J = 11.9 Hz), 4.18 (1H, br d, J = 2.4 Hz), 4.12 (0.25H, m), 3.94 (0.75H, m), 3.82–3.71 (1H, m), 3.71–3.60 (1H, m), 3.60 (0.25H, dd, J = 9.0, 3.3 Hz), 3.50 (0.75H, dd, J = 8.8, 3.1 Hz), 3.39 (0.25H, t, J = 8.5 Hz), 3.24 (0.75H, t, J = 8.8 Hz), 2.81 (1H, m), 2.49 (0.75H, td, J = 13.0, 3.2 Hz), 2.44 (0.25H, m), 2.25 (0.75H, br s), 2.10 (0.25H, br s), 1.89–1.75 (3H, m), 1.75–1.53 (5H, m), 1.53–1.28 (2H, m), 1.39 (2.25H, s), 1.32 (6.75H, s), 1.05 (2.25H, s), 1.04 (6.75H, s); 13C-NMR (150 MHz, CDCl3, 25 °C, two rotamers in a 3:1 ratio) δ 153.7, 152.8, 138.7, 138.4, 135.6, 135.5, 133.3, 133.2, 129.80, 129.78, 129.74, 129.71, 128.4, 128.3, 127.8, 127.73, 127.71, 127.6, 127.5, 127.4, 127.3, 79.5, 78.8, 73.02, 72.98, 71.3, 70.7, 68.9, 68.6, 67.1, 66.7, 63.7, 63.5, 58.5, 58.1, 43.1, 41.1, 39.3, 38.4, 34.2, 33.1, 33.0, 32.6, 29.3, 29.0, 28.6, 28.5, 26.9, 26.81, 26.79, 26.2, 19.03, 19.01, 18.7, 18.6; IR (ATR) 3485, 3070, 3049, 3031, 2957, 2929, 2858, 1673, 1473, 1454, 1428, 1388, 1364, 1322, 1253, 1219, 1169, 1108, 1081, 1028, 996, 940, 908, 858, 823, 805, 772, 735, 700 cm−1; DART-HRMS calcd for C40H56NO5Si [(M + H)+] 658.3928, found 658.3942.:90) on silica gel to provide eq-12 (2.8 mg, 3.96 μmol, 16%) and ax-12 (9.8 mg, 14.9 μmol, 62%) as each colorless oil.:90) on silica gel to give 5 (74.0 mg, 113 μmol, 98%) as a colorless oil.:90) on silica gel to afford 13 (112 mg, 160 μmol, quant.) as a colorless oil: Rf = 0.62 (EtOAc/hexane, 30:70); [α]23D −27.6 (c 0.66, CHCl3); 1H-NMR (300 MHz, CDCl3, 25 °C, two rotamers in a 2:1 ratio) δ 7.69–7.60 (4H, m), 7.43–7.25 (11H, m), 4.57 (0.67H, d, J = 7.0 Hz), 4.53 (0.33H, d, J = 7.3 Hz), 4.50 (2H, s), 4.46 (0.67H, d, J = 6.8 Hz), 4.44 (0.33H, d, J = 7.3 Hz), 4.10 (0.33H, br s), 3.95–3.76 (1.67H, m), 3.66–3.51 (1.33H, m), 3.51–3.40 (1H, m), 3.30 (3H, s), 3.25 (0.67H, m), 3.16 (1H, td, J = 10.2, 4.4 Hz), 2.50 (0.67H, quintet, J = 5.3 Hz), 2.37 (0.67H, td, J = 12.7, 3.5 Hz), 2.20–2.03 (0.66H, m), 2.03–1.50 (8H, m), 1.45–1.08 (3H, m), 1.34 (3H, s), 1.23 (6H, s), 1.022 (3H, s), 1.015 (6H, s); 13C-NMR (75 MHz, CDCl3, 25 °C, two rotamers in a 2:1 ratio) δ 153.3, 152.6, 138.7, 138.4, 135.5, 134.3, 134.20, 134.18, 129.4, 129.3, 128.4, 128.3, 127.6, 127.53, 127.50, 127.4, 95.6, 95.4, 80.8, 80.5, 79.7, 78.9, 73.0, 71.5, 70.7, 68.3, 67.7, 64.82, 64.77, 59.1, 58.5, 55.5, 55.4, 44.7, 42.8, 37.8, 36.9, 33.5, 32.7, 32.10, 32.07, 32.04, 32.02, 28.5, 28.3, 26.91, 26.88, 26.6, 26.1, 20.5, 20.4, 19.2; IR (ATR) 3069, 3046, 3030, 2954, 2929, 2883, 2858, 2822, 1688, 1636, 1589, 1541, 1473, 1455, 1428, 1388, 1371, 1364, 1318, 1300, 1254, 1172, 1143, 1105, 1078, 1038, 998, 939, 915, 884, 863, 823, 805 cm−1; DART-HRMS calcd for C42H60NO6Si [(M + H)+] 702.4190, found 702.4196.DMN-AZADO (9.2 mg, 55.3 μmol), NaClO2 (81.9 mg, 906 μmol), and NaOCl (5.2 mg, 69.9 μmol) were added to a solution of the mixture including a desired alcohol in MeCN (600 μL) and phosphate buffer (pH 6.8, 190 μL) at room temperature under a nitrogen atmosphere. The mixture was stirred for 22 h, and the reaction was quenched with H2O. After the resulting mixture was extracted with CHCl3, the organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. A mixture including a desired carboxylic acid was directly used in the following reaction.
To a solution of the mixture including a desired carboxylic acid in DMF (1.2 mL) were added Cs2CO3 (13.3 mg, 40.8 μmol) and MeI (4.3 μL, 69.0 μmol) at room temperature under a nitrogen atmosphere. After the solution was stirred for 9 h, the reaction was quenched with a saturated aqueous solution of Na2CO3. After the resulting mixture was extracted with EtOAc (×3), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Purification by flash silica gel column chromatography (EtOAc/hexane, 5:95) afforded 14 (29.7 mg, 46.4 μmol, 80% in 3 steps) as a colorless oil: Rf = 0.41 (EtOAc/hexane, 30:70); [α]23D −21.7 (c 0.45, CHCl3); 1H-NMR (300 MHz, CDCl3, 25 °C, two rotamers in a 3:1 ratio) δ 7.68–7.61 (4H, m), 7.45–7.30 (6H, m), 4.55 (0.75H, d, J = 7.0 Hz), 4.51 (0.25H, d, J = 7.1 Hz), 4.44 (1H, d, J = 7.1 Hz), 4.28 (0.25H, dd, J = 8.5, 4.6 Hz), 4.11 (0.75H, dd, J = 7.9, 6.3 Hz), 3.90 (1H, m), 3.71 (0.75H, s), 3.70 (2.25H, s), 3.58 (1H, m), 3.29 (0.75H, s), 3.28 (2.25H, m), 3.12 (1H, m), 2.78–2.63 (0.5H, m), 2.49 (0.75H, ddd, J = 10.1, 6.2, 3.1 Hz), 2.27 (0.75H, td, J = 13.5, 5.0 Hz), 2.16–1.86 (2H, m), 1.83–1.45 (6H, m), 1.37–1.14 (3H, m), 1.34 (2.25H, s), 1.25 (6.75H, s), 1.03 (2.25H, s), 1.02 (6.75H, s); 13C-NMR (75 MHz, CDCl3, 25 °C, two rotamers in a 3:1 ratio) δ 174.0, 173.9, 153.6, 152.2, 135.49, 135.47, 134.5, 134.3, 134.2, 134.1, 129.44, 129.41, 129.39, 129.36, 127.6, 127.52, 127.50, 127.47, 95.5, 95.2, 80.5, 80.4, 80.1, 79.6, 69.3, 68.4, 64.82, 64.80, 61.64, 61.59, 55.5, 55.4, 52.0, 51.8, 44.9, 43.1, 35.0, 33.3, 32.6, 32.2, 32.12, 32.06, 31.9, 30.3, 28.4, 28.0, 27.4, 27.3, 26.9, 26.6, 20.6, 19.2, 19.1; IR (ATR) 3071, 3051, 2950, 2931, 2888, 2859, 2822, 1749, 1701, 1685, 1624, 1590, 1577, 1569, 1558, 1541, 1522, 1507, 1497, 1489, 1473, 1457, 1429, 1389, 1376, 1364, 1327, 1297, 1272, 1257, 1197, 1177, 1146, 1133, 1110, 1082, 1040, 1008, 999, 941, 915, 881, 857, 824, 805 cm−1; DART-HRMS calcd for C36H54NO7Si [(M + H)+] 640.3670, found 640.3657.
:30) on silica gel to afford 15 (22.6 mg, 56.3 μmol, quant.) as a colorless oil: Rf = 0.19 (EtOAc/hexane, 50:50); [α]24D −29.7 (c 0.58, CHCl3); 1H-NMR (300 MHz, CDCl3, 25 °C, two rotamers in a 4:1 ratio) δ 4.81 (0.2H, d, J = 7.1 Hz), 4.78 (0.8H, d, J = 7.1 Hz), 4.62 (0.2H, d, J = 7.2 Hz), 4.59 (0.8H, d, J = 7.1 Hz), 4.39 (0.2H, m), 4.26 (0.8H, m), 3.72 (3H, s), 3.62 (1H, br s), 3.42 (0.6H, s), 3.38 (2.4H, s), 3.23 (1H, m), 3.04 (1H, br s), 2.86 (0.8H, ddd, J = 8.4, 6.1, 2.3 Hz), 2.50 (0.2H, ddd, J = 8.8, 6.0, 2.2 Hz), 2.27 (0.8H, td, J = 13.0, 3.7 Hz), 2.21–2.03 (2.2H, m), 1.94–1.65 (6H, m), 1.65–1.45 (2H, m), 1.31 (1H, m), 1.38 (9H, s); 13C-NMR (75 MHz, CDCl3, 25 °C, two rotamers in a 4:1 ratio) δ 173.9, 173.7, 153.8, 152.8, 95.00, 94.97, 81.1, 80.3, 79.8, 79.5, 69.9, 69.0, 63.0, 62.9, 61.9, 61.8, 56.1, 55.9, 52.0, 51.9, 46.8, 45.2, 34.5, 32.8, 32.0, 31.8, 31.7, 31.6, 31.3, 29.8, 28.5, 28.2, 27.1, 26.6, 20.6; IR (ATR) 3463, 2975, 2950, 2931, 2887, 2868, 2824, 1748, 1700, 1684, 1559, 1541, 1520, 1507, 1474, 1456, 1437, 1391, 1365, 1328, 1295, 1276, 1255, 1198, 1175, 1146, 1131, 1120, 1101, 1037, 942, 916, 879, 854, 792 cm−1; DART-HRMS calcd for C20H36NO7 [(M + H)+] 402.2492, found 402.2501.To a solution of the mixture including a desired olefin in CH2Cl2 (800 μL) was slowly added TFA (200 μL) at 0 °C under a nitrogen atmosphere. After the mixture was allowed to warm to room temperature, the solution was stirred for 12 h. After the reaction was quenched with a saturated aqueous solution of NaHCO3, the resulting mixture was extracted with CHCl3 (×3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Purification by flash silica gel column chromatography (MeOH/CHCl3, 10:90) afforded 16 (9.0 mg, 37.6 μmol, 98% in 2 steps) as a colorless oil: Rf = 0.50 (MeOH/CHCl3, 10:90); [α]27D −49.3 (c 0.90, CHCl3); 1H-NMR (600 MHz, CDCl3) δ 5.73 (1H, dt, J = 16.9, 10.0 Hz), 5.17 (1H, dd, J = 17.0, 1.4 Hz), 5.16 (1H, dd, J = 10.1, 1.7 Hz), 3.85 (1H, dd, J = 8.8, 5.6 Hz), 3.80 (1H, m), 3.75 (3H, s), 2.38 (1H, dd, J = 9.1, 2.7 Hz), 2.19 (1H, m), 1.94 (1H, m), 1.88–1.78 (3H, m), 1.73–1.61 (2H, m), 1.56 (1H, dt, J = 13.5, 4.3 Hz), 1.46 (1H, dt, J = 14.0, 3.8 Hz), 1.25 (1H, m); 13C-NMR (150 MHz, CDCl3) δ 175.2, 136.7, 118.8, 72.4, 65.8, 58.7, 52.4, 51.7, 36.0, 34.4, 30.3, 28.9, 18.0; IR (ATR) 3282, 3074, 3005, 2930, 2856, 1736, 1699, 1635, 1507, 1456, 1438, 1356, 1339, 1327, 1284, 1260, 1232, 1206, 1153, 1117, 1092, 1074, 1032, 996, 968, 921, 901, 869, 856, 808 cm−1; DART-HRMS calcd for C13H22NO3 [(M + H)+] 240.1600, found 240.1560.
:90) (×3), the combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography (MeOH/CHCl3, 2:98) on silica gel to afford 4 (35.8 mg, 74.9 μmol, 96%) as a colorless oil: Rf = 0.44 (MeOH/CHCl3, 2:98); [α]26D −35.1 (c 0.40, CHCl3); 1H-NMR (600 MHz, CDCl3) δ 7.72–7.64 (4H, m), 7.46–7.33 (6H, m), 5.50 (1H, dt, J = 17.0, 10.0 Hz), 5.07 (1H, dd, J = 10.2, 2.0 Hz), 5.02 (1H, d, J = 16.8 Hz), 3.76 (1H, t, J = 7.5 Hz), 3.70 (1H, m), 3.68 (3H, s), 2.20 (1H, t, J = 8.2 Hz), 1.94 (1H, m), 1.87 (1H, m), 1.74 (1H, m), 1.60–1.43 (3H, m), 1.43–1.12 (4H, m), 1.05 (9H, s); 13C-NMR (150 MHz, CDCl3) δ 175.4, 137.3, 136.02, 135.97, 135.5, 134.5, 134.0, 129.6, 129.5, 127.5, 127.4, 118.8, 74.0, 64.5, 59.1, 51.9, 57.3, 37.4, 33.0, 32.3, 28.8, 27.0, 19.5, 19.2; IR (ATR) 3373, 3071, 3050, 2998, 2932, 2892, 2858, 1737, 1639, 1540, 1523, 1510, 1458, 1429, 1361, 1311, 1282, 1254, 1200, 1158, 1109, 1086, 1029, 1002, 919, 885, 822, 793 cm−1; DART-HRMS calcd for C29H40NO3Si [(M + H)+] 478.2777, found 478.2811.:50) on silica gel to give 3 (10.9 mg, 24.3 μmol, 87%) as a colorless oil: Rf = 0.25 (MeOH/CHCl3, 2:98); [α]28D −57.1 (c 0.55, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.74–7.64 (4H, m), 7.47–7.34 (6H, m), 5.47 (1H, dt, J = 16.9, 10.1 Hz), 4.97 (1H, dd, J = 10.2, 1.8 Hz), 4.83 (1H, dd, J = 16.8, 1.4 Hz), 3.88 (1H, br s), 2.40–2.15 (3H, m), 1.93 (1H, m), 1.82–1.56 (3H, m), 1.56–1.22 (7H, m), 1.14 (9H, s); 13C-NMR (75 MHz, CDCl3) δ 170.9, 136.0, 135.9, 135.1, 133.5, 133.3, 129.9, 129.7, 127.64, 127.60, 118.7, 74.3, 56.7, 55.7, 34.2, 33.2, 31.0, 29.1, 27.1, 19.0, 16.5, 16.0; IR (ATR) 3358, 3207, 3071, 3048, 2932, 2857, 1659, 1589, 1463, 1428, 1406, 1391, 1363, 1335, 1283, 1220, 1184, 1165, 1110, 1084, 1071, 1025, 994, 957, 936, 919, 875, 841, 822, 795 cm−1; DART-HRMS calcd for C28H38NO2Si [(M + H)+] 448.2672, found 448.2682.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Astellas Foundation and the JGC-S Scholarship Foundation (Nikki Saneyoshi Scholarship).Notes and references
- J. W. Daly, I. Karle, C. W. Myers, T. Tokuyama, J. W. Waters and B. Witkop, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1870–1875 CrossRef CAS.
- T. F. Spande, H. M. Garraffo, J. W. Daly, T. Tokuyama and A. Shimada, Tetrahedron, 1992, 48, 1823–1836 CrossRef CAS.
- (a) For a review, see: A. Sinclair and R. A. Stockman, Nat. Prod. Rep., 2007, 24, 298–326 RSC; (b) A. C. Carey, M. Aratani and Y. Kishi, Tetrahedron Lett., 1985, 26, 5887–5890 CrossRef; (c) G. Stork and K. Zhao, J. Am. Chem. Soc., 1990, 112, 5875–5876 CrossRef CAS; (d) G. M. Williams, S. D. Roughley, J. E. Davies and A. B. Holmes, J. Am. Chem. Soc., 1999, 121, 4900–4901 CrossRef CAS; (e) R. A. Stockman, Tetrahedron Lett., 2000, 41, 9163–9165 CrossRef CAS; (f) E. C. Davison, M. E. Fox, A. B. Holmes, S. D. Roughley, C. J. Smith, G. M. Williams, J. E. Davies, P. R. Raithby, J. P. Adams, I. T. Forbes, N. J. Press and M. J. Thompson, J. Chem. Soc., Perkin Trans. 1, 2002, 1494–1514 RSC; (g) M. S. Karatholuvhu, A. Sinclair, A. F. Newton, M. L. Alcaraz, R. A. Stockman and P. L. Fuchs, J. Am. Chem. Soc., 2006, 128, 12656–12657 CrossRef CAS PubMed; (h) Y. Adachi, N. Kamei, S. Yokoshima and T. Fukuyama, Org. Lett., 2011, 13, 4446–4449 CrossRef CAS PubMed; (i) M. Sato, H. Azuma, A. Daigaku, S. Sato, K. Takasu, K. Okano and H. Tokuyama, Angew. Chem., Int. Ed., 2017, 56, 1087–1091 CrossRef CAS PubMed.
- (a) K. Nishikawa, S. Kikuchi, S. Ezaki, T. Koyama, H. Nokubo, T. Kodama, Y. Tachi and Y. Morimoto, Org. Lett., 2015, 17, 5772–5775 CrossRef CAS PubMed; (b) K. Nishikawa, K. Yamauchi, S. Kikuchi, S. Ezaki, T. Koyama, H. Nokubo, K. Matsumura, T. Kodama, M. Kumagai and Y. Morimoto, Chem.–Eur. J., 2017, 23, 9535–9545 CrossRef CAS PubMed.
- (a) T. Honda and M. Kimura, Org. Lett., 2000, 2, 3925–3927 CrossRef CAS PubMed; (b) M. Katoh, R. Matsune, H. Nagase and T. Honda, Tetrahedron Lett., 2004, 45, 6221–6223 CrossRef CAS; (c) T. Honda, R. Takahashi and H. Namiki, J. Org. Chem., 2005, 70, 499–504 CrossRef CAS PubMed; (d) M. Katoh, C. Hisa and T. Honda, Tetrahedron Lett., 2007, 48, 4691–4694 CrossRef CAS.
- T. Katoh, Y. Nagata, Y. Kobayashi, K. Arai, J. Minami and S. Terashima, Tetrahedron, 1994, 50, 6221–6238 CrossRef CAS.
- R. C. Clark, S. Y. Lee and D. L. Boger, J. Am. Chem. Soc., 2008, 130, 12355–12369 CrossRef CAS PubMed.
- G. A. Molander and G. Hahn, J. Org. Chem., 1986, 51, 1135–1138 CrossRef CAS.
- For a discussion about the more detailed transition states, see ref. 4.
- (a) D. J. Procter, M. Spain and M. Szostak, Chem. Commun., 2011, 47, 10254–10256 RSC; (b) M. Szostak, M. Spain and D. J. Procter, Org. Lett., 2012, 14, 840–843 CrossRef CAS PubMed; (c) M. Szostak, M. Spain and D. J. Procter, Org. Lett., 2014, 16, 5052–5055 CrossRef CAS PubMed.
- The reduction of a carbonyl moiety in spirocyclic product 5 was performed using various reduction methods. When compound 5 was allowed to react with NaBH4, the undesired ax-12 was obtained as a major isomer (62%), along with the desired eq-12 (16%). The use of Super-Hydride® (lithium triethylborohydride) also gave ax-12 (76%). In the borohydride reduction of 5, an equatorial hydride attack could predominate as a result of avoiding the axial alkyl group. In the 1H NMR spectrum of a major rotamer of ax-12, the coupling pattern of C8-H at 4.18 ppm was observed as a doublet with J = 2.4 Hz, showing an equatorial proton. When using Bouveault–Blanc reduction, compound eq-12 was stereoselectively obtained, but the yield was very low (22%). The reduction of 5 under Birch conditions afforded a debenzylated compound of eq-12 in low yield (<30%). The Corey–Bakshi–Shibata (CBS) asymmetric reduction resulted in the recovery of the starting material. The Luche reduction gave eq-12 as a major product (59%, ax-12: 30%).
- Compound 5 could be recovered by DMP oxidation and again submitted to the SmI2-mediated reduction, and after one cycle, the desired eq-12 was obtained in 81% overall yield based on initial loading of the compound 5. Two cycles gave eq-12 with 90% overall yield.
- R. Doi, M. Shibuya, T. Murayama, Y. Yamamoto and Y. Iwabuchi, J. Org. Chem., 2015, 80, 401–413 CrossRef CAS PubMed.
- P. A. Grieco, S. Gilman and M. Nishizawa, J. Org. Chem., 1976, 41, 1485–1486 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02011f |
| This journal is © The Royal Society of Chemistry 2018 |