
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLatest perspectives of orally bioavailable 2,4-diarylaminopyrimidine analogues (DAAPalogues) as anaplastic lymphoma kinase inhibitors: discovery and clinical developments
Muhammad Latif
 *a,
Zaman Ashraf
b,
Sulman Basit
a,
Abdul Ghaffar
c,
Muhammad Sohail Zafar
de,
Aamer Saeed
f and
Sultan Ayoub Meo
g
*a,
Zaman Ashraf
b,
Sulman Basit
a,
Abdul Ghaffar
c,
Muhammad Sohail Zafar
de,
Aamer Saeed
f and
Sultan Ayoub Meo
g
aCollege of Medicine, Centre for Genetics and Inherited Diseases (CGID), Taibah University, Al-Madinah Al-Munawwarah, Kingdom of Saudi Arabia. E-mail: latifmayo@kaist.ac.kr
bDepartment of Chemistry, Allama Iqbal Open University, Islamabad 44000, Pakistan
cDepartment of Chemistry, University of Engineering and Technology, Lahore, Pakistan
dDepartment of Restorative Dentistry, College of Dentistry, Taibah University, Al-Madinah Al-Munawwarah, Kingdom of Saudi Arabia
eDepartment of Dental Materials, Islamic International Dental College, Riphah International University, Islamabad 44000, Pakistan
fDepartment of Chemistry, Quaid-e-Azam University, Islamabad, Pakistan
gDepartment of Physiology, College of Medicine, King Saud University, Riyadh, Kingdom of Saudi Arabia
First published on 4th May 2018
Abstract
The course of anaplastic lymphoma kinase (ALK)-rearranged non-small-cell lung cancer (NSCLC) therapy has improved impressively. The Food and Drug Administration (FDA) has approved crizotinib (Xalkori, Pfizer) as a first-in-class tyrosine kinase inhibitor (TKI) that demonstrated a substantial objective response rate (ORR) and remarkable progression-free survival (PFS). However, acquired resistance to crizotinib is still a major concern especially as the central nervous system (CNS) remains the most common sites of relapse. To combat disease resistance, limited PFS and poor CNS exposure exhibited by crizotinib (Xalkori, Pfizer) led to the discovery of numerous next generation ALK-TKIs and surprisingly most of them are 2,4-Diarylaminopyrimidine Analogues (DAAPalogues). To date, DAAPalogues have been investigated extensively to display their superior potency against numerous kinase targets especially ALK/ROS1. This review describes hit-to-drug evolution strategies, activity spectra, milestones related to medicinal chemistry discovery efforts and scalable synthetic pathways of clinically emerging DAAPalouges which are either progressing as investigational or preclinical candidates. In addition, the significance of DAAPalogues to treat the patients with ALK+-NSCLC in clinical settings has been detailed. This review is beneficial for medicinal chemists and researchers contributing to discovering ALK-TKIs to overcome existing issues related to DAAPalouges in the drug discovery process.
 Muhammad Latif | Muhammad Latif was born in 1975 in Lahore. He received his M.Phil degree from Quaid-i-Azam University, Islamabad, Pakistan. He worked as a researcher in the drug discovery division and in 2014, he obtained his PhD in medicinal chemistry from Korea University of Science and Technology, under the supervision of Professor Kwangho Lee. He also worked with Professor Jongkook Lee and developed novel synthetic methodologies. He did post-doctorate studies with Professor Sungwoo Hong at KAIST, Korea. Currently, he is working as an assistant professor at Taibah University, Madinah. His research interests include designing of druggable molecules, identification of biomarkers, synthetic methodologies and asymmetric synthesis. |
 Zaman Ashraf | Zaman Ashraf was born in Gujranwala, Pakistan, in 1977. He obtained his PhD degree under the supervision of Professor Aamer Saeed in chemistry from Quaid-i-Azam University, Islamabad, in 2011. He was awarded the prestigious BK-21 PLUS post-doctoral fellowship by Kongju National University, South Korea, in 2014. At present he is working as an assistant professor and research group leader at Allama Iqbal Open University, Islamabad, Pakistan. His research interests include designing of potent biomolecules, isocoumarin synthesis, prodrugs synthesis, liquid crystal synthesis, and drug development through enzyme-based assay. |
 Sulman Basit | Sulman Basit was born in Rawalpindi, Pakistan in 1982. He earned his M.Phil and PhD degrees in biochemistry from Quaid-i-Azam University, Islamabad. Previously, he worked as a research associate at the Department of Human Genetics, UMC Radboud Nijmegen, The Netherlands, Department of Biochemistry, Islamabad and King Saud University, Saudi Arabia. Currently he is working as an assistant professor in the College of Medicine, Taibah University, Saudi Arabia. His research interests include genetic delineation of human inherited disorders. Dr Basit has published in prestigious journals including Nature Genetics, American Journal of Human Genetics, Neurological Sciences, Clinical Genetics and Human Genetics. |
 Abdul Ghaffar | Abdul Ghaffar was born in Lahore, Pakistan. He obtained his master's degree in chemistry from the University of Siegen, Germany. In 2011, he earned his PhD from the University of Amsterdam, The Netherlands, in the area of degradation and analysis of degradable synthetic polymeric biomaterials for biomedical applications. Currently, he is holding the position of assistant professor at the Department of Chemistry, University of Engineering and Technology Lahore, Pakistan. His research interests include synthesis of biomolecules, synthesis of druggable molecules, development of synthetic methodologies, organic synthesis, and polymer synthesis. |
 Muhammad Sohail Zafar | Dr. Muhammad Sohail Zafar obtained his master's degree in Dental Materials from Queen Mary University of London, UK and PhD from the Nottingham Trent University, UK. Currently, Dr Zafar has been working as an associate professor of dental biomaterials at College of Dentistry, Taibah University, Saudi Arabia. He holds fellowships of International College of Dentists, USA (FICD) and Academy of Dentistry international, USA (FADI). He is an editorial board member of “Fluoride” (Official publication of International Society for Fluoride Research, New Zealand) and European Dental Journal. Dr Zafar's research interests include biomolecules, oral antimicrobial peptides and natural biomolecules for therapeutic applications. |
 Aamer Saeed | Aamer Saeed was born in Jhelum, Pakistan in 1964. He had been awarded the Gold Medal by Pakistan Academy of Sciences, Dr M. Razziuddin Siddiqi Prize in chemistry, an awardee of Research Productivity Award in A-1 category by Pakistan Council for Science and Technology (PCST), and a Fellow of the Chemical Society of Pakistan. He has been an Alexander von Humboldt Post-Doctoral Fellow (Germany) and also received TWAS visiting expert award. Currently, he is working as a tenured professor at the Quaid-i-Azam University, Islamabad, Pakistan. His fields of interest include medicinal chemistry, synthesis of natural products, heterocycles and fluorescent polymers. |
 Sultan Ayoub Meo | Sultan Ayoub Meo is a professor and consultant in clinical physiology, College of Medicine, King Saud University, Riyadh, Saudi Arabia. He is a medical graduate and obtained M.Phil and PhD in human physiology. He also obtained masters in medical education from University of Dundee, Scotland. He is credited with 10 books and 140 articles in peer reviewed national and international bio-medical journals. Prof. Meo has been invited as a speaker and key note speaker to deliver talks at more than 100 national and international conferences in different countries including Pakistan, Saudi Arabia, Bahrain, UAE, China, Turkey, Indonesia, UK and the USA. |
1 Introduction
Lung cancer is a fatal malignancy that has a higher mortality rate than colorectal, breast, and prostate malignancies.1–5 Lung cancer is associated with cancer related deaths globally with a truncated survival rate of about 15%. Smoking addiction is reported to be the predominant risk factor.6–10 Historically, various classifications of lung cancer have been reported but clinically, lung cancers are categorized into small-cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). Reports have revealed that SCLC is responsible for 10–15% while NSCLC is accountable for 85–90% of lung malignancies. NSCLC, typically includes squamous cell carcinoma (10–15%), adenocarcinoma (25–30%) and large cell carcinoma (10–15%) as its three key subtypes. Among these the adenocarcinoma accounts for about 40% of NSCLC cases whereas squamous cell carcinoma is reported to be responsible for about 25% of cases.11–14 Despite the devoted investigations to improve the diagnosis of lung carcinoma and treatment, the five year survival rate for patients detected with NSCLC remains very low (10–15%). In spite of technological advancements in diagnostics and its availability, more than 50% patients are diagnosed at an advanced stage. It is pertinent to note that the therapy demonstrates a palliative aspect instead of a curative intent.15–17 The lung cancer therapy, especially NSCLC has been revolutionized remarkably due to the emergence of molecular targeted therapy (MTT).18 The emergence of activated gene rearrangements in ALK as an oncogenic driver of NSCLC patients (3–7%) have made the ALK a potential therapeutic target in medicinal chemistry discovery campaigns around the globe. The purpose of current article is to review hit-to-drug evolution strategies, activity spectrum, milestones related to medicinal chemistry discovery efforts and scalable (pilot plant) synthetic pathways of clinically emerging DAAPalouges. In addition, the significance of DAAPalogues to treat the patients with ALK+-NSCLC in clinical settings has been detailed.2 ALK in non-small cell lung cancer
ALK is a receptor tyrosine kinase (RTK) and a key member of the insulin receptor protein-tyrosine kinase superfamily. It was first discovered in 1994 in the anaplastic large-cell lymphoma (ALCL) as a non-Hodgkin lymphoma subtype.19–21 ALK is a novel orphan RTK which is considered to play an essential role in usual growth and in function of nervous system. The reports have revealed that ALK is usually expressed during the growth of nervous tissues and also down-modulates in the adult.22–24 ALK is closely correlated to leukocyte tyrosine kinase (LTK)25–29 and has been found to implicate in numerous solid and hematologic cancers. Considering oncogenic manifestations, the ALK rearrangements were first identified as a fusion to a portion of the nucleophosmin (NPM) gene in 60% of ALCL, a small subset of non-Hodgkin lymphomas.20,30 It has been revealed that in lymphoma cells containing t(2;5), the NPM gene plays a critical role as an active promoter to initiate or facilitate the expression of the ALK catalytic domain. It has also been envisioned that the truncated ALK is deregulated and phosphorylates the intracellular molecules (substrate) to prompt further malignant transformations.31–33 Thus new NPM-ALK fusion protein was discovered while screening the patients associated with ALCL malignancy where the N-terminal portion of NPM was found to fuse with the kinase domain of ALK as a consequence of t(2;5)(p23;q35) translocation.34,35Since the identification of novel potential mutation driver observed in 5–10% NSCLC cases several other ALK fusion genes have also been identified recently. The fusion of the echinoderm microtubule-associated protein-like 4 gene (EML4) with the ALK gene resulted in the generation of a fusion protein (EML4-ALK) and in NSCLC it is considered to be the most significant discovery being an oncogenic factor in solid tumors.36 EML4-ALK fused protein was identified in 2007 in the Japanese patients with NSCLC.37–40 Surprisingly, since the preliminary reports regarding the discovery of the NPM-ALK fusion protein in clinical settings pertaining to the patients investigated with ALCL, a plethora of ALK fusion proteins have been described in NSCLC,37,41 inflammatory myofibroblastic tumors (IMTs),42,43 squamous cell carcinoma (SCC) of the oesophagus,44,45 renal cell carcinoma,46 and diffuse large B-cell lymphomas (DLBCLs)47 (Fig. 1). Furthermore, oncogenic progression via overexpression and point mutations has been investigated in various cancers.48–54
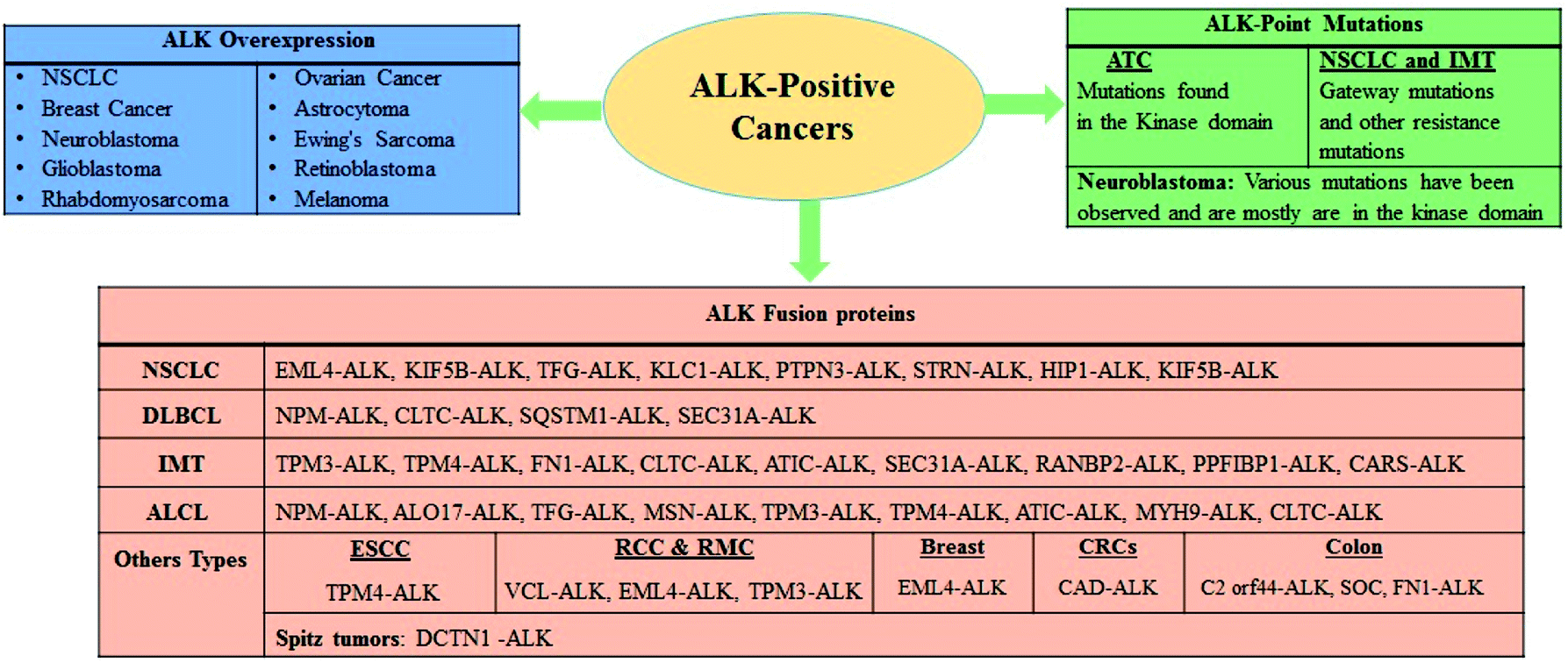 | ||
| Fig. 1 An overview of ALK in cancer and schematic summary of ALK overexpression, point mutations, and fusions in ALK positive cancer. | ||
Physiologically, the function of ALK in mammals is still under investigation and unclear. However, the significance and key role of full-length ALK-RTK in the development or onset of numerous diseases is well investigated and consequently known to be expressed in several types of tumors including NSCLC (Fig. 2). Numerous signal transduction pathways have been detected which are activated by the involvement of ALK. Initially, fusion of ALK ligand to extracellular domain occurs during the signal transduction process followed by dimerization and trans-phosphorylation which in turn prompts cellular process involved in oncogenesis. ALK signaling via several downstream pathways have been reported. Ras/Raf/MEK/ERK1/2 pathways, PI3K/Akt pathway, JAK/STAT pathway, and PLC-γ pathway have been reported to be the principle pathways activated by ALK signal transduction process. Cell survival is mediated by the JAK/STAT and PI3K/Akt pathways while PLC-γ and Ras/ERK1/2 pathways play a critical role in cell proliferation.55 To date, ample number of ALK inhibitors with promising oncolytic activity against ALK+-NSCLC tumors have been investigated and developed in the recent decade.
 | ||
| Fig. 2 The anaplastic lymphoma kinase receptor tyrosine kinase. (A) Schematic representation of pleotrophin (PTN) bound ligand activated anaplastic lymphoma kinase (ALK) receptor signalling. (B) Constitutively active ALK receptor signalling due to EML4 fusion. The fusion arises due to paracentric inversion between EML4 and ALK genes located in the short arm of chromosome 2. EML4-ALK translocation is thought to lead to the constitutive activation of oncogenic signaling through multiple pathways such as PI3K/Akt, JAK/STAT, and RAS/RAF/MEK/ERK. Aberrant activation of PI3K-Akt initiate mTOR, FOXO and GSK3β signalling which lead to survival and proliferation of tumour cells. PI3K/Akt activation also lead to MEKK2/3/MEK5/ERK5 pathway which subsequently promote the expression of the oncogene MYCN. The STAT3 signaling pathway has shown a key role in NPM-ALK mediated alteration however the significance of STAT3 activation in EML4-ALK+-NSCLC is unclear. ALK has also been shown to phosphorylate PLCγ to mediate its mitogenicity. (mTOR; mechanistic target of rapamycin, S6K; ribosomal S6 kinase, TD; trimerisation domain, TAPE; tandem atypical propeller domain, MAM; meprin, A5 protein, and protein tyrosine phosphatase Mu domain). | ||
3 ALK inhibitors in NSCLC
ALK is one of the highly competitive drug targets in oncologic drug discovery and development campaigns. ALK-fused genes such as NPM-ALK, EML4-ALK, and KIF5-ALK are putative oncogenes which are accountable for several cancers such as anaplastic large-cell lymphomas (ALCL), inflammatory myofibroblastic tumors (IMT), diffuse large B cell lymphoma (DLBCL) and a variety of solid tumor types.39,56–58 Genetic abnormalities in ALK have been investigated as key drivers of oncogene addicted lung adenocarcinoma and have boosted the researchers to develop novel and targeted therapies. Nevertheless, growing interest to develop synthetic ALK inhibitors has led to a bigger number of research reports related to new drug candidates for cancer therapy. Some of the exciting entities for ALK inhibition approved by FDA with substantial impact in the field of medical oncology are listed in Fig. 3.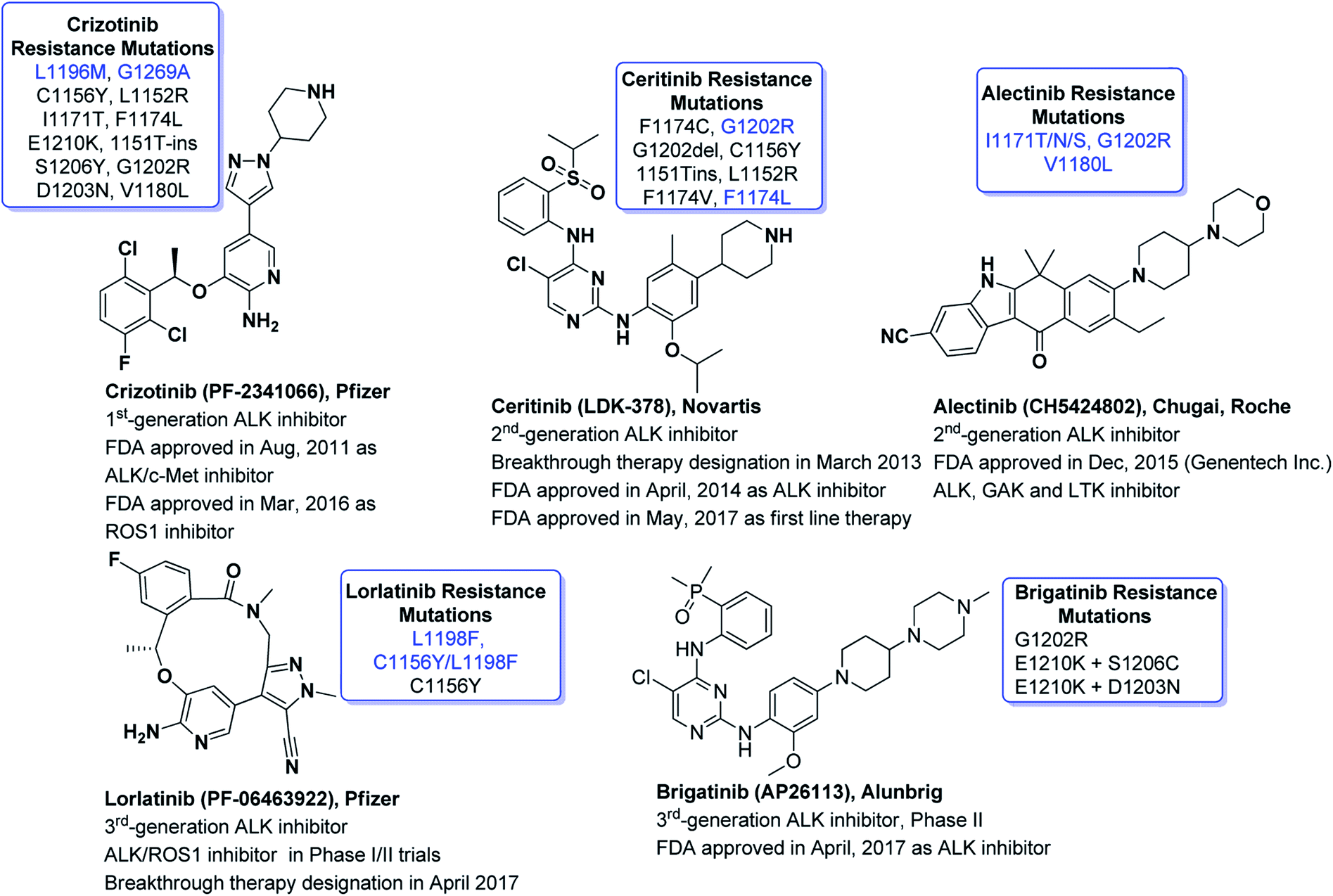 | ||
| Fig. 3 FDA approved ALK inhibitors with substantial impact in the field of medical oncology with reported point mutations to drug resistance. (Only most critical mutations are shown. The mutations written in blue are most frequent and critical.) | ||
Crizotinib (PF2341066, Xalkori), the Pfizer's first-generation dual c-Met/ALK inhibitor59 was given approval by FDA in 2011 as first line treatment for ALK+-NSCLC.60 About 1% of ROS1 rearrangements have been reported in NSCLC patients. FDA in 2016, approved the crizotinib for the management of ROS1-positive metastatic NSCLC.61 Although crizotinib demonstrated substantial objective response rate (ORR = 74%, N = 343, phase III) and remarkable progression-free survival (PFS = 10.9 months). Unfortunately, resistance to crizotinib has been reported as one of the critical issues to date.62–66 The clinical investigations have revealed that the most common identified secondary mutations are L1196M (gatekeeper mutation), G1202R, L1152R, G1269A, 1151T-ins, C1156Y, S1206Y, and F1174L.67–72 To overcome the limitations exhibited by crizotinib (Xalkori, Pfizer), extensive drug discovery campaigns have led to the innovation of numerous next generation ALK inhibitors to combat disease resistance including alectinib, ceritinib and lorlatinib. It is noteworthy, once crizotinib therapy is failed and then lorlatinib is reported to overcome entire secondary point mutations. Reportedly, point mutations such as L1196M, G1269A, S1206Y and I1171T can be overcome by ceritinib whereas L1196M, G1269A, F1174L and C1156Y can be overcome by alectinib. Although crizotinib (Xalkori, Pfizer), alectinib (CH5424802, AF-001JP, Alecensa)73 and lorlatinib (PF-06463922, 3rd-generation ALK/ROS1 macrocyclic inhibitor, Pfizer)74 are not 2,4-diarylaminopyrimidines (DAAPalogues) yet they show outstanding potential in clinical settings.
Crizotinib (acyclic) and lorlatinib (macrocyclic) are among the FDA approved ALK inhibitors (Fig. 3) which comprised of similar central core such as aminopyridine. Crizotinib (approved by FDA in 2016) is an ALK/ROS1/c-MET tyrosine kinase inhibitor that has revealed a high activity in ALK+-NSCLC. Lorlatinib is a 3rd-generation chemotype that was given breakthrough therapy designation (BTD) by FDA in 2017 and has been used for the treatment of patients with ALK-positive metastatic NSCLC which were treated earlier with one or more ALK inhibitors.75–77 We recently documented the strategic development of lorlatinib from crizotinib.78 It is noteworthy that both crizotinib and lorlatinib have aminopyridine core and are non-DAAPalogues. Notably, ceritinib (Zykadia®) is the first FDA approved DAAPalogue for ALK/ROS1 inhibition79–81 which has been described comprehensively elsewhere.82–87 Interestingly, ceritinib being a member of DAAPalogues showed potency to overcome the gatekeeper mutation of L1196M which is still considered a resistance to crizotinib (non-DAAPalogue agent). In crizotinib, the existence of the polar-NH2 (aromatic amine) substituent at C2-position of pyridine core is considered not to provide favorable interactions with a large lipophilic residue at the gatekeeper position, such as methionine. On the contrary, in ceritinib the chloro (Cl) substituent at C5-position of pyrimidine core could interact more favorably with a methionine gatekeeper and has revealed a remarkable interaction to gatekeeper mutation. On the other hand, alectinib is another FDA approved, highly selective, orally active, potent, and CNS penetrant ALK inhibitor which has shown a promising therapeutic window for the patients treated with crizotinib harboring ALK secondary mutations.88–93 Alectinib is non-DAAPalogue drug which comprised of benzo[b]carbazolone structural motif94–97 that is not ubiquitous template in second or even third generation TKIs. In medicinal chemistry campaign limited structure–activity relationship (SAR) investigation has been reported for benzo[b]carbazolone core possibly due to difficulty in chemical synthesis.97–101 However, recently reported JH-VIII-157-02 has been potent against the G1202R mutant and comprised of benzo[b]carbazolone core.102 In the designing and development of next-generation ALK-TKIs, the 2,4-diarylaminopyrimidine (DAAP) structural moiety has been incorporated extensively. The literature survey has revealed that majority of the next-generation TKIs being evaluated clinically or currently at preclinical phases have been derived from DAAP scaffold and referred as DAAPalogues. Such ALK-TKIs were developed to potently inhibit all acquired ALK mutants reported previously for first and second generation ALK-TKIs. In recent decade, rapid progress of pharmacological strategies targeting ALK has shown marvelous achievements in clinical settings. Continued research efforts of researchers have made it possible to advance several DAAPalogues to clinical trials. To date, numerous ALK inhibitors have been discovered and diversified chemical classes have been evaluated at pre-clinical or clinical phases and most of them are DAAPalogues.60,83,103–107 Some novel, potent and highly selective DAAPalogues for ALK inhibition are shown in Fig. 4.
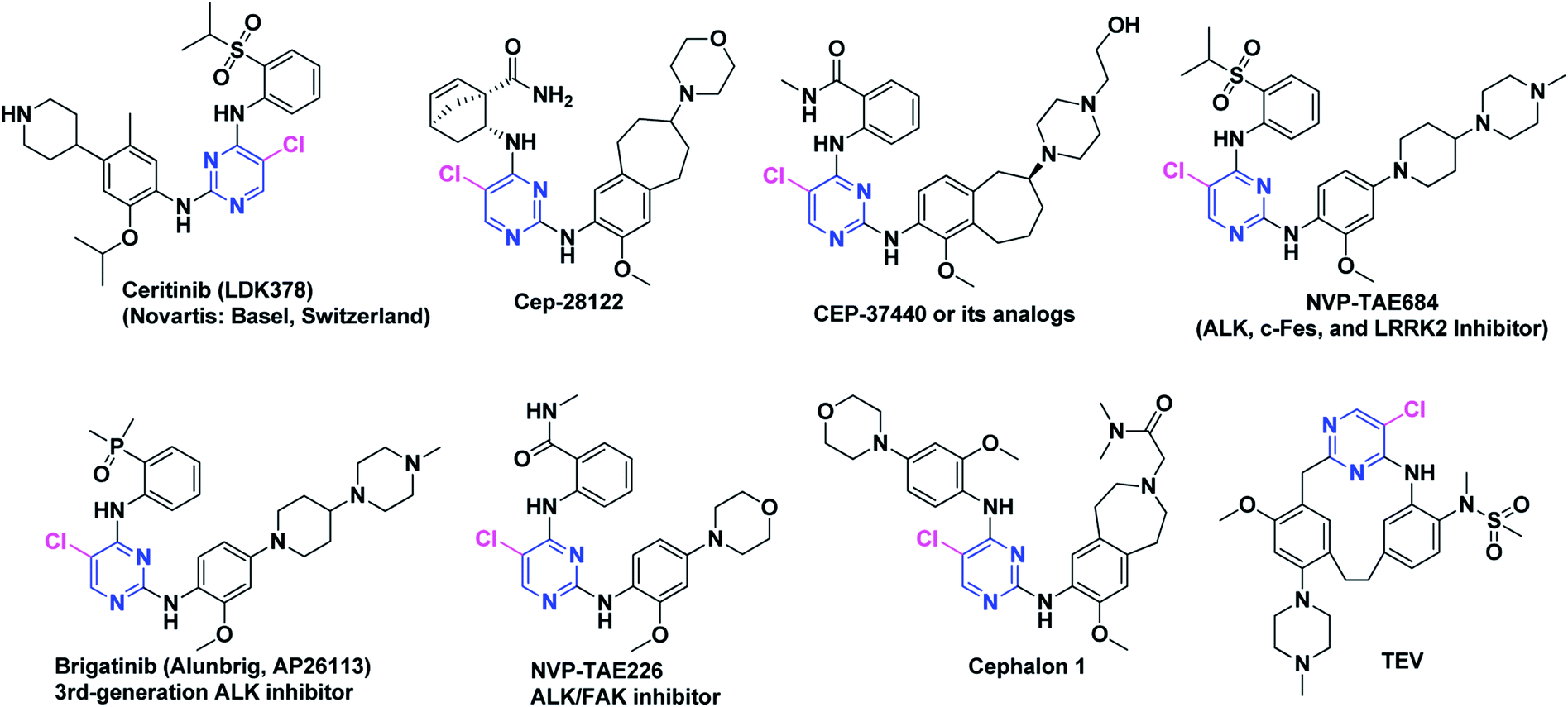 | ||
| Fig. 4 Novel, potent and selective DAAPalogues as ALK inhibitors. | ||
The DAAP skeleton as a conventional kinase inhibitor core is well recognized and such DAAPalogues has demonstrated high potency against many kinase targets, particularly ALK, c-Met and ROS1. To this end, the drug designing strategy stemmed from our literature survey has demonstrated that ceritinib (LDK-378, 2nd-generation ALK/ROS1 inhibitor Zykadia®, Novartis), brigatinib (Alunbrig™, AP26113), NVP-TAE684, Cephalon 2812, Cephalon 1, and CEP-37440 comprised of DAAP-motif are clinically novel ALK-TKIs.87,108–113 It is significant to note that on DAAP template chloro (Cl) substituent have been installed as an optimal substituent at C-5 position of pyrimidine which in most of the DAAPlouges interacts with ALKL1196 gatekeeper residue. The designing strategies, biological characteristics, therapeutic exploration, and clinical data of DAAPalogues as ALK inhibitors is well-detailed in literature. In the following sections, we have summarized hit-to-drug evolution strategies, activity spectrum, milestones related to medicinal chemistry discovery efforts, scalable synthetic pathway, and development of some potent ALK-TKIs comprised of DAAP template which are either progressing as investigational or preclinical candidates. Some key features of DAAPalogues as ALK inhibitor advancing to clinical study are shown in Table 1.
| Crizotiniba | Ceritinibb | Alectinibc | Lorlatinibd | Brigatinibe (Alunbrig) | CEP-28122f | CEP-37440g | NVP-TAE684h | CEP-1 | |
|---|---|---|---|---|---|---|---|---|---|
| Topological polar surface area (TPSA), hydrogen bond donors (HBD), breakthrough therapy designation (BTD), cellular system = (EML4-ALK), discontinued (Disc), the letter S symbolizes mutations which are “sensitive” (clinical and/or preclinical data) to a given drug entity, and “R” symbolizes resistance, data not available (NA), not determined (ND).a In 2011, FDA granted accelerated approval to crizotinib to treat patients with ALK-positive cancer but traditional approval was given in 2013. However on March 11, 2016, crizotinib got FDA approval as ROS1 inhibitor as well.b In April 2014, ceritinib obtained accelerated FDA approval for patients with ALK-positive metastatic NSCLC but on May 26, 2017, FDA granted regular approval to ceritinib.c In June 2013, FDA granted alectinib BTD based on preliminary evidence of clinical activity but got FDA approval in 2014. On Dec 11, 2015, he U.S. FDA granted accelerated approval to alecensa (alectinib) capsules for the treatment of patients with advanced (metastatic) ALK-positive NSCLC.d In 2015, lorlatinib (Pfizer) received FDA orphan drug status for therapy NSCLC patients. But in April, 2017, BTD was granted by FDA to lorlatinib for therapy of patients with ALK+-NSCLC who previously received one or more ALK inhibitors.e In 2016, FDA granted BTD to brigatinib. In April, 2017, the U.S FDA granted accelerated approval to brigatinib (ALUNBRIG) for the treatment of patients confirmed with metastatic ALK+-NSCLC who progressed on or were intolerant to crizotinib.f Further optimized to CEP-28122.g Further optimized to discover CEP-37440.h Further optimized to CEP-28122.i Further optimized to give other CEP analogs (Scheme 5).j The shown potency was against the two EML4-ALK+-NSCLC cancer cell lines such as NCI-H2228 and NCI-H3122.k The shown potency was against the two EML4-ALK+-NSCLC cancer cell lines such as NCI-H2228 and NCI-H3122. | |||||||||
| Mol. wt | 450 | 558 | 483 | 406 | 584 | 539 | 580 | 614 | 596 |
c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP logP |
4.2 | 6.5 | 5.2 | 2.7 | 5.9 | 4.7 | 4.7 | 3.9 | 5.2 |
| TPSA | 77 | 105 | 72 | 110 | 82 | — | — | — | — |
| HBD | 3 | 3 | 1 | 2 | 2 | 2 | 2 | 3 | |
| Phase | I, II, III | I, II, III | I/II | I/II | I, II, III | Pre-clinical (discontinued) | I | Pre-clinical (discontinued) | (Disc) |
| FDA approval status | b | c | d | e | f | g | h | i | j |
| Targets other than ALK, [(IC50 nM)] | ROS1, c-Met | IGF-R1, InsR, ROS1 | RET, LTK, GAK | ROS1 | ROS (1.9 nM) IGF1R (24.9 nM), FLT3 (2.1 nM) | FLT4 (IC50 = 46 nM) | RSK2, SK3, RSK4, FAK | ALK, c-Fes, LRRK2 | |
| ALK(Enzymatic) (nM) | 24 | — | — | — | 0.37 | 2.0 | 3.1 | NA | 4 |
| IC50 (nM) | |||||||||
| ALK(cellular), (nM) | 3 | 0.15 | 1.9 | 2 | 0.62 | 40 | 22 | 3 | 30 |
| EML4-ALKwt | 56 | 41 | 2 | 1.3 | 5 | — | — | 16j | |
| IC50 (nM) | 44k | ||||||||
| EML4-ALKL1196M | 549 (R) | 64 (S) | 90 (S) | 21 (S) | 3 (S) | NA | NA | NA | NA |
| EML4-ALKG1202R | 328 | 444 | 207 | 77 | 56 | ND | ND | ND | ND |
| EML4-ALKC1156Y | 144 | 164 | 2 | 1.6 | 3 | ND | ND | ND | ND |
| EML4-ALKF1174L | 81 | 101 | 3 | 0.2 | 12 | ND | ND | ND | ND |
| EML4-ALKL1152R | 645 | 2747 | 169 | 9.0 | 2300 | ND | ND | ND | ND |
| EML4-ALK1151Tins | 857 | 668 | 72 | 38 | 142 | ND | ND | ND | ND |
| EML4-ALKG1269A | 512 | 57 | 9 | 15 | 2 | ND | ND | ND | ND |
| EML4-ALKS1206Y | 65 | 33 | 2 | 4.2 | 12 | ND | ND | ND | ND |
| Ref | 65, 114 and 115 | 80, 81, 103, 113 and 115 | 89 and 115 | 74, 75, 77, 115 and 116 | 115 and 117–119 | 115 and 120 | 98 and 121–123 | 115 | |
4 Brigatinib (Alunbrig™, AP26113)- a second-generation DAAPalogue
Brigatinib (Alunbrig™, AP26113), developed by ARIAD Pharmaceuticals as antineoplastic ALK inhibitor is a 2nd-generation ALK inhibitor with DAAP structural motif. It is a potent DAAPalogue with imperious in vitro potential against ALK (IC50 = 0.6 nM L−1), ROS1 (IC50 = 1.9 nM L−1), FLT3 (IC50 = 2.1 nM L−1) and EGFR (IC50 = 1.9 nM L−1). Being a potent DAAPalogue, brigatinib received an accelerated FDA approval for therapy of patients suffering from ALK+-NSCLC who either progressed on or were intolerant to crizotinib therapy. The occurrence of resistance to crizotinib posed a therapeutic task and has led to the advancement of 2nd-generation ALK-TKIs such as brigatinib. Brigatinib have exhibited activity against treatment-resistant ALK mutants (Table 1). Brigatinib showed favorable clinical potential and satisfactory safety profile in the patients treated with crizotinib and crizotinib-naïve ALK-rearranged NSCLC patients.117,124 The structure–activity relationship reported by Katayama et al., have revealed that brigatinib also demonstrated potency against the triple-mutant EGFR (IC50 < 100 nM). Brigatinib suppresses EGFR phosphorylation and its downstream signaling pathway in cells which express several types of EGFR mutations such as EGFRC797S/del19 (IC50 = 28.4 nM), IGF1R (IC50 = 32 nM), and EGFRdel19 (IC50 = 36.8 nM).125,126 This feature has shown that brigatinib will be a promising candidate to overcome the acquired resistance of 3rd-generation EGFR-TKIs if it is used as combination therapy with anti-EGFR antibody. In the following section, description related to hit-to-drug evolution design and the milestones achieved during the development of brigatinib are summarized. These advancements ultimately led the brigatinib to its first global approval for the therapy of patients with ALK+-NSCLC who either progressed on or remained intolerant to crizotinib.126 The synthetic strategy of brigatinib (Alunbrig™) from one of the potent ALK inhibitor (AP23464, IC50 = 0.45 nM) discovered in early discovery efforts is described first.4.1 Synthetic strategy of brigatinib (Alunbrig™, AP26113) from (1)
Although phosphorous is an abundant element in human body but rarely employed in drug discovery strategies. Poor cell penetration, biological instability or low oral bioavailability have narrowed down the clinical utilization of monophosphonic or bisphosphonic acid-based drugs, phosphonates, phosphinates, and phosphate-containing prodrugs in drug discovery.Ridaforolimus (mTOR inhibitor), Src-kinase inhibitors (anti-resorptive agents) have been reported in recent decade having phosphine oxide (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) functionality. The PO bond has been investigated as a H-bond acceptor in designing of kinase inhibitors. To this end, analogous efforts related to the discovery of AP23464 (IC50 = 0.45 nM), a potent ATP-based oncogenic protein kinase inhibitor,127–129 further stimulated the discovery of potent DAAPalogue (1) where dimethylphosphine oxide (DMPO) onto the 2-anilinopyrimidine template was surveyed.111 Aforementioned investigations provoked to incorporate DMPO motif in drug designing which led to the discovery of several DAAPalouges (small-molecules) as ALK inhibitor from a known SRC inhibitor (AP23464) (Scheme 1). The DMPO installation at C-4′ enhanced the ALK activity by approximately 7-fold compared to the unsubstituted derivatives in a small focused library closely related to (1). The H-bonding ability of the DMPO functional group with the Lys-NH (K1150) on the protein was explored near the hinge region by synthesizing double-phosphorylated analog (2) which did not exhibit substantial increase in ALK activity (ALK IC50 = 1.06 nM) but demonstrated remarkably improved selectivity over both IGF1R and InsR (IGF1R IC50 = 139 nM, InsR IC50 = 726 nM).111
O) functionality. The PO bond has been investigated as a H-bond acceptor in designing of kinase inhibitors. To this end, analogous efforts related to the discovery of AP23464 (IC50 = 0.45 nM), a potent ATP-based oncogenic protein kinase inhibitor,127–129 further stimulated the discovery of potent DAAPalogue (1) where dimethylphosphine oxide (DMPO) onto the 2-anilinopyrimidine template was surveyed.111 Aforementioned investigations provoked to incorporate DMPO motif in drug designing which led to the discovery of several DAAPalouges (small-molecules) as ALK inhibitor from a known SRC inhibitor (AP23464) (Scheme 1). The DMPO installation at C-4′ enhanced the ALK activity by approximately 7-fold compared to the unsubstituted derivatives in a small focused library closely related to (1). The H-bonding ability of the DMPO functional group with the Lys-NH (K1150) on the protein was explored near the hinge region by synthesizing double-phosphorylated analog (2) which did not exhibit substantial increase in ALK activity (ALK IC50 = 1.06 nM) but demonstrated remarkably improved selectivity over both IGF1R and InsR (IGF1R IC50 = 139 nM, InsR IC50 = 726 nM).111
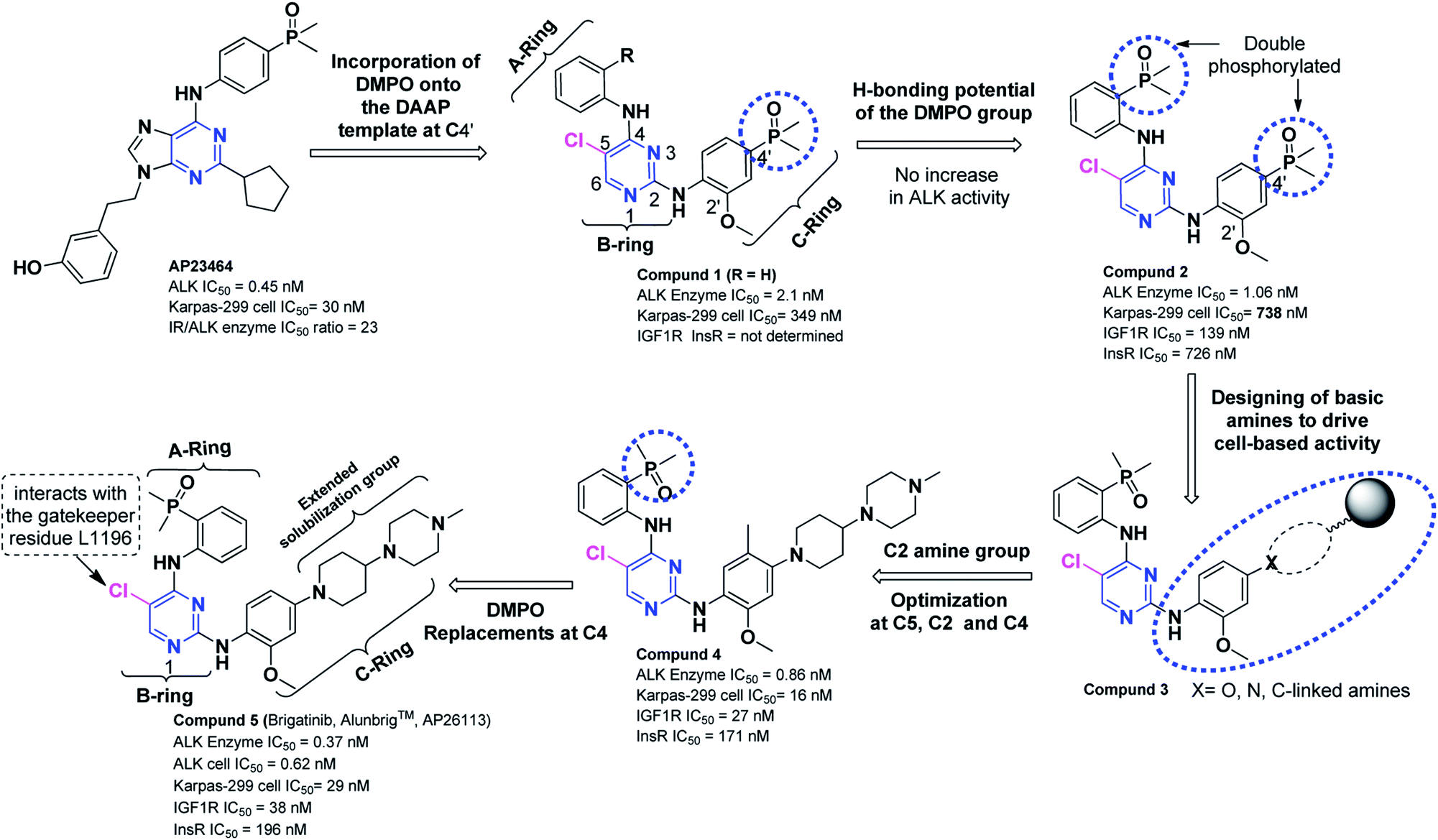 | ||
| Scheme 1 The strategic development of brigatinib (Alunbrig™, AP26113) (5) from a SRC inhibitor (AP23464). | ||
In subsequent investigations, the influence on ALK activity and cellular potency with variety of basic amines containing solubilizing groups as C-ring was surveyed by keeping DMPO motif at ortho-position on A-ring. To this end, solubilizing group screening investigation efforts led to the identification of numerous equipotent amines similar to (3). Among them DAAP (5) was most promising analog with promising potency and selectivity profile (ALK IC50 = 0.37 nM, IGF1R IC50 = 24.9 nM, InsR IC50 = 196 nM, Karpas-299 (ALK+) = 29 nM, U937 (ALK−) = 3194 nM). Subsequent lead optimization explored at other positions on the inhibitor core by maintaining the C-ring resulted in (4) bearing a methyl (Me) group at C-5′ position and it was equipotent to (5) in cellular assays but exhibited moderate enzymatic potency loss. 2′-Methoxy (ortho-MeO) at C-ring and 5-chloro (Cl) on pyrimidine core was optimal in terms of potency and selectivity. Finally to confirm the critical role DMPO functionality at C-4 of pyrimidine core, a systematic modification of DMPO was carried out with numerous H-bond acceptors (nitrile, amides, t-amides, sulfonamides, methylsulfone, isopropylsulfone acetyl etc.). The DMPO at C-4 revealed as an crucial structural unit indispensable to attain the appropriate balance between potency and selectivity of DAAPalouge (5).
Final DAAPalouge (5) (brigatinib) emerged as most the advanced clinical candidate loaded with phosphine oxide (PO) warhead and to date it is being investigated in a global phase II registration trial in patients detected with advanced malignancies and has showed activity in crizotinib-resistant patients. Brigatinib revealed almost 10-fold higher potency and selectivity compared to crizotinib.130 Brigatinib has advanced to phase I/II clinical trials which for this unique inhibitor were initiated131 in September 2011.
Brigatinib, a pyrimidine-based molecule, is a potent and orally bioavailable 2nd-generation DAAPalogues (an ALK inhibitor) which occupies the ATP-binding site of ALK preferably in U-shaped conformation.111,132 Brigatinib incorporates many chemical characteristics that enhance its affinity towards ALK inhibition that were lacked in crizotinib. These features include ortho-methoxy (OMe) group on C-ring, an extended solubilization group, an optimal chloro (Cl) substituent at C-5 and a distinctive ortho-DMPO functionality on the C-4 aniline (Scheme 1). Brigatinib demonstrated potent inhibition with a kinase activity of ALK (IC50 = 0.62 nmol L−1), IGF-1R (IC50 = 38 nmol L−1), insulin receptor kinase (IC50 = 262 nmol L−1) and for other five tested mutant variants (C1156Y IC50 = 0.6, F1174L IC50 = 1.4, L1196M IC50 = 1.7, G1202R IC50 = 4.9, R1275Q IC50 = 6.6 nmol L−1). Brigatinib also revealed a higher selectivity and only inhibited eleven additional mutants or native kinases (out of 289 screened) with IC50 < 10 nmol L−1. These included ROS1 (IC50 = 1.9 nmol L−1), FLT3 (IC50 = 2.1 nmol L−1), and mutant variants of FLT3 (D835Y) and EGFR (L858R; IC50, 1.5–2.1 nmol L−1). Brigatinib exhibited more modest activity against EGFR with a T790M resistance mutation (L858R/T790M), native EGFR, IGF1R, and INSR with (IC50, 29–160 nmol L−1) but did not inhibit c-MET (IC50 > 1000 nmol L−1). This DAAPalouge has exhibited dual inhibition of ALK and EGFR T790M. Due to this inhibitory mechanism brigatinib could be a promising therapeutic option for patients progressing on crizotinib for the activation of the EGFR bypass track. In vitro and in cellular assays against select kinases, the brigatinib has also showed a consistent data for ALK (IC50 = 0.62 nm L−1 in kinase assay, IC50 = 14 nmol L−1 in EML4-ALK as a cellular system) and ROS1 (IC50 = 1.9 nm L−1 in kinase assay, IC50 = 18 nmol L−1 in CD74-ROS1 cellular system).117
4.2 Medicinal chemistry discovery pathway of brigatinib
Pd-catalyzed phosphorous arylation (P-arylation) methodology was employed to synthesize phosphorus-containing aniline (6) directly from iodoaniline and dimethylphosphine oxide without competitive N-arylation though reactive anilino NH2 substitutent was there and such kind of transformations were reported recently.133,134 To this end, SN-Ar reaction of phosphorus-containing aniline (6) with 2,4,5-trichloropyrimidine using base mediated conditions delivered a known intermediate (7) in the following step. In another pathway, SN-Ar displacement of fluoro (F) group of 2-nitro-5-fluoroanisole (8) with secondary amine (9) using potassium carbonate as basic media yielded nitro-intermediate (10), followed by common hydrogenation provided aniline (11) in high yield. Finally, subsequent substitution at C-2 of (7) was achieved via an acid-promoted SN-Ar reaction in good yield to provide brigatinib. A summarized drug discovery synthetic pathway of brigatinib recently reported by Huang et al., is depicted in Scheme 2. | ||
| Scheme 2 Medicinal chemistry discovery pathway of brigatinib. | ||
4.3 Therapeutic trial of brigatinib (Alunbrig™)
The promising clinical activity of brigatinib in the patients pre-treated with crizotinib and crizotinib-naïve ALK rearranged NSCLC has been reported. During the advancement in phase I/II trial, single-arm study, an open label and multicenter study, the brigatinib has revealed significant clinical activity against brain metastases (NCT01449461). In order to establish the recommended dose in phase II trials, dose escalation evaluation in phase I revealed total doses of 30–300 mg per day. Phase II evaluation demonstrated three once daily dosage regimens; 90 mg, 180 mg, and 180 mg with a 7 days lead in at 90 mg. A cohort of 137 patients (n = 79, 58% were ALK-rearranged NSCLC) enrolled from academic hospitals or cancer centres in the USA and Spain in phase I/II during the period from September 2011 to July 2014 and all were treated. In phase II clinical study, among the total patients established with ALK-rearranged NSCLC (n = 79), 52 patients (66%) showed confirmed objective response. In the patients pre-treated with crizotinib (n = 71), a population of patients (n = 51) showed an objective response of 72% and objective response of 100% has been reported for crizotinib naïve patients (n = 8) (NCT01449461).
In phase I/II clinical findings, brigatinib demonstrated an objective response rate (ORR = 70%, n = 70, the patient were on pretested with crizotinib), and median progression free survival (PFS = 13.5, n = 8, previously on crizotinib). The one-year overall survival rate (OS = 80%, patients previously on crizotinib) and (OS = 100%, in crizotinib-naïve).136
In a phase II single arm trial (ALTA) study, patients (n = 222) resistant to crizotinib received brigatinib, the ORR (45%) at 90 mg day−1 and ORR (55%) at 180 mg day−1 was observed in two cohorts. In first dose-finding trial (in ALTA investigation), within the first 7 days pulmonary toxicity occurred in the 6% of patients (3% were grade 3–4) and it was correlated to the higher dose of the drug (180 mg day−1).137 In phase I/II trial and ALTA investigations in patients (n = 32), the relationship between brigatinib efficacy and ALK mutational status has also been demonstrated.138 In the samples obtained at baseline (before treatment with brigatinib), the secondary ALK mutations were detected in the 30% of patients, and the ORR was >60% both in patients with or without secondary ALK mutations which demonstrated the activity of brigatinib regardless of the presence of a secondary ALK mutations conditioning resistance to crizotinib. Although two specific mutations (F1174L and G1202R) have been detected after long-term treatment with brigatinib (in two out of the five post-brigatinib samples), however the responses were also observed when these mutations were present at baseline.139
Brigatinib is currently under evaluation in both first-line setting versus crizotinib in a phase III trial (NCT02737501) and in phase II study (NCT02706626) after unfruitful outcome of other second-generation ALK inhibitors (ceritinib and alectinib). This last study aims to explore the possibility of brigatinib to overcome the resistance to ALK inhibitors mediated by secondary ALK mutations such as G1202R and I117N/S/T.140,141 The clinical activity of brigatinib may further inspire application of phosphorus oxide group as a unique functionality more broadly in the field of medicinal chemistry. In patients with measurable CNS disease, 53% (8 out of 15) demonstrated an intracranial objective response with an intracranial disease control rate of 87%, and median duration of response was 18.9 months. Therapy emergent side effects were fatigue, nausea, diarrhea, cough and hypertension. ALTA, a phase II trial of brigatinib in patients with ALK+-NSCLC who progressed on crizotinib, has completed recruitment of patients (NCT02094573). In this trial preliminary data include 67% patient with CNS disease and 74% with prior treatment who were treated at 180 mg daily. The reported PFS was 12.9 months (95% CI: 11.1-NR), ORR 54% (97.5% CI: 43–65) and CNS response of 67% provides a favorable comparision with previously reported outcomes with ceritinib and alectinib.141,142
5 Discovery and clinical development of NVP-TAE 684 DAAPalouge
This DAAPalouge was discovered by Novartis and had 2,4-diarylaminopyrimidine (DAAP) core decorated with chloro (Cl) at C-5 position and exhibits anticancer chemotherapeutic activity. It was characterized as a highly potent and specific inhibitor of NPM-ALK. The DAAPalouge (NVP-TAE684) is an inhibitor of ALK and exhibited Ba/F3-NPM-ALK IC50 = 3.0 nM, Ba/F3-Tel-InsR IC50 = 44 nM, Ba/F3-WT IC50 = 1336 nM and Karpas-299 IC50 = 2.2 nM. Leucine-rich repeat kinase 2 (LRRK2) is another highly attractive therapeutic target and its association with Parkinson's disease has been reported. NVP-TAE684 is also a highly potent inhibitor of LRRK2 and has demonstrated promising LRRK2 kinase activity (LRRK2wt IC50 = 7.8 nM, G2019S mutant IC50 = 6.1 nM). NVP-TAE684 remarkably inhibited Ser935 and Ser910 phosphorylation of both LRRK2wt and G2019S mutant at a concentration ranging from 0.1 to 0.3 μM in cells, and in mouse spleen and kidney, but not in brain, at an oral doses of 10 mg kg−1.144 In patients who demonstrated anti-VEGF refractory cancers, c-Fes plays central role in angiogenic cellular responses of endothelial cell and has been reported another potential target. Beside this bone marrow-derived monocytic lineage regulates angiogenesis and c-Fes has been responsible to express in these cells. Investigations pertaining to c-Fes has made TAE-684 a potent inhibitor of c-Fes.145–147 NVP-TAE684 induces apoptosis and inhibited growth of diffuse large B-cell lymphoma cells in animal models and induces tumor regression. NVP-TAE684 also induces cell cycle arrest and apoptosis in animal models of anaplastic large-cell lymphoma, preventing lymphomagenesis and inducing regression.NVP-TAE684 was discovered through high throughput screening (HTS) of a kinase-directed small-molecule library aimed to explore potent entity selectively cytotoxic to BaF3 NPM-ALK but should not be toxic to non-transformed parental BaF3 cells.56 An inhibition for proliferation of BaF3 NPM-ALK cells (IC50 = 3 nM) and for parental BaF3 cells (IC50 > 1 μM) was observed which facilitated the initiation of discovery process of NVP-TAE684. Interestingly, NVP-TAE684 inhibited the human Karpas-299 ALCL cell line (IC50 = 3 nM) and showed an improvement on crizotinib. Additionally, NVP-TAE684 exhibited from 100–1000 times higher IC50 values for non-ALK kinases. The assay was comprised of a panel of thirty five (35) BaF3 cells which were transformed with several tyrosine kinases constitutively activated by fusion to the TEL oncogene. This specificity was associated as a consequence of the bulky hinge region residue at the 1198 position of the non-ALK kinases (e.g. Tyr or Phe compared to Lys existed in ALK).
In another investigation, screening of more than six hundred cancer cell lines was performed for sensitivity to obtain selective ALK inhibitor. As an outcome of the aforesaid findings, NVP-NVP-TAE684 was reported to be superior ALK-TKI over Xalkori (crizotinib) in Karpas-299 lymphoma and in the SU-DHL-1 cell lines.16 NVP-TAE684 has also emerged as selective for oncogenic ALK containing tumors.148 The evolution of NVP-TAE684 from the HTS cellular screening is summarized in Scheme 3.
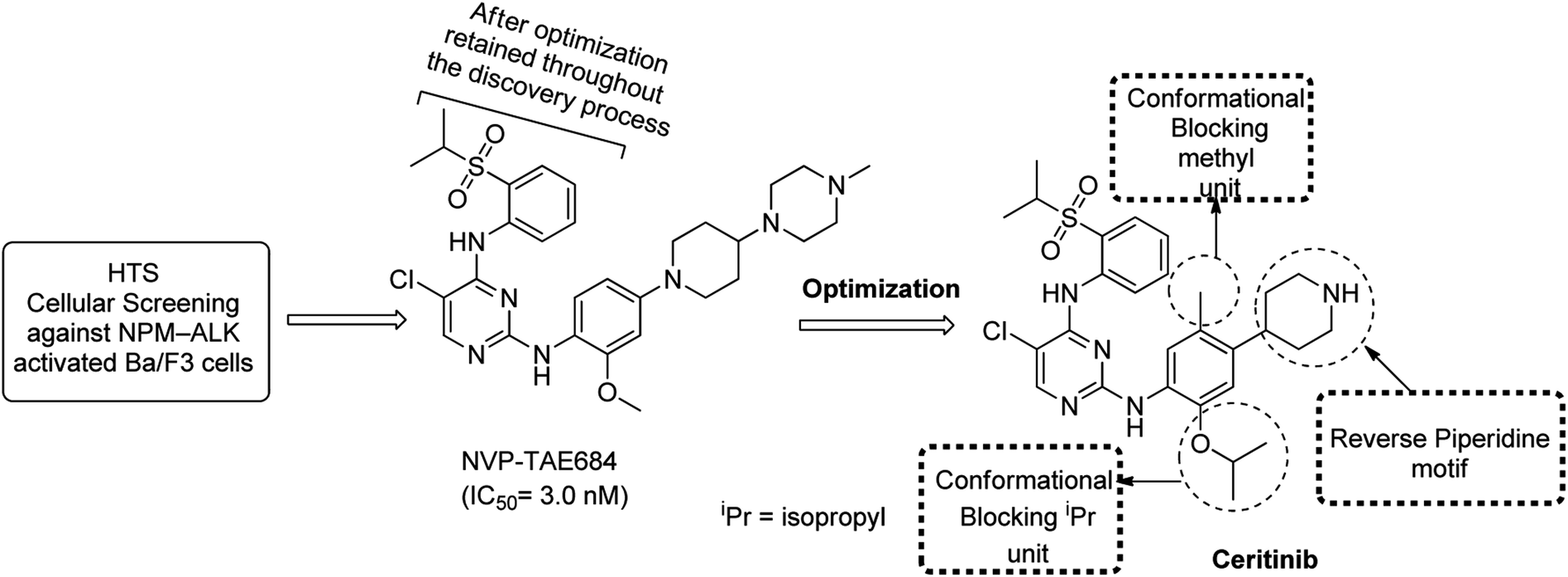 | ||
| Scheme 3 Summarized layout of NVP-TAE684 DAAPlouge and evolution of ceritinib. | ||
Although the DAAPlouge TAE-684 is a potent ALK inhibitor but later findings revealed that NVP-TAE684 upon metabolic oxidation could produce large number of reactive adducts. Metabolic oxidation was reported to be a potential threat for significant toxicological liabilities. Approximately 20% of NVP-TAE684 was found to be converted into reactive products upon incubation in liver microsomes as determined by semi-quantitative LC-MS analysis. The reactive species were determined using glutathione (GSH).149 No relationship was established between hepatotoxic drugs and the development of GSH adducts.150 However, it was hypothesized that reactive metabolites could have a critical role in idiosyncratic151 and/or in other toxicities.152 To address the further developmental deficiencies of NVP-TAE684 (as a first generation ALK inhibitor), modeling information, designing strategy, and focused investigation to achieve enhanced kinase selectivity of NVP-TAE684 was further surveyed extensively. The installation of iso-propoxy (i-Pr) instead of methoxy (MeO) revealed as well tolerated moiety to address selectivity issues. Further optimization of NVP-TAE684 and potential idiosyncratic toxicity issues led to the eventual discovery of ceritinib which got accelerated FDA approval for the patients with ALK-positive metastatic NSCLC in April 2014. However, on May 26, 2017, FDA declared regular approval to ceritinib for the patients suffering from ALK-positive metastatic NSCLC.153
6 Discovery and clinical development of Cephalon DAAPalouges
In the following sections, medicinal chemistry efforts, early discovery description and clinical developments of novel Cephalon DAAPalouges such Cephalon 1, CEP-28122 and CEP-37440 is discussed. Based on reported investigation regarding the involvement of ALK gene rearrangements as an oncogenic driver of ALK-positive cancers, the inhibition of ALK with highly selective small molecular entities has emerged as an attractive area of research and this has led the multiple pharmaceutical companies to discover novel ALK inhibitors. To this end, plethora of unique chemical scaffolds and interesting small-molecules for ALK inhibition were explored. Surprisingly, at that time the DAAPlouges were not well investigated rather tetrahydropyrido[2,3-b]pyrazine,154 indolocarbazole,155 pyridone,156 1H-pyrrolo[2,3-d]pyrimidine,157 dianilinopyrimidine,108 and aminopyridine158 were among the well probed pharmacophores.159–161Cephalon 1 (15) (Scheme 5) is a DAAPalogue derivative developed by Cephalon. It is a potent, selective, and orally bioavailable ALK inhibitor but this analog exhibited potential hERG liability. The 2,3,4,5-tetrahydro-1H-benzo[d]azepine on B-ring motif in Cephalon 1 (15) provided a suitable site in subsequent efforts for functional motif exploitation to balance out potency with promising physicochemical properties.162 This approach resulted in another potent diarylaminopyrimidine ALK inhibitor (DAAPalogue) known as CEP-28122. Interestingly, CEP-28122 was another DAAPlouge developed by Cephalon. The synthesis of CEP-28122 (17) was conducted at the department of chemistry at Cephalon, Inc., and its synthetic routes have been reported elsewhere.163 CEP-28122 is a potent, selective, and orally bioavailable ALK inhibitor. CEP-28122 (17) demonstrated ALK kinase activity (IC50 = 1.9 nM) in an enzyme-based TRF assay. It has shown concentration-dependent growth inhibition/cytotoxicity of ALK+-ALCL, NSCLC and neuroblastoma cells. CEP-28122 (17) also exhibited dose-dependent inhibition of ALK tyrosine phosphorylation in tumor xenografts in mice.164 The strategic development of Cephalon 1 (15) and CEP-28122 (17) is shown (Scheme 5). Medicinal chemistry route as an initial strategy for the synthesis of CEP-28122 (17) exploited the coupling of three key structural motifs such as a morpholine substituted on benzocycloheptane fragment as B-ring, a diaminopyrimidine central core as A-ring, and a bicyclic amino amide portion as C-ring.163 However, to meet preclinical and clinical supplies, a unique eight-step (longest linear), convergent, and scalable manufacturing route discovered later. The development of scalable new route to CEP-28122 (17) was reported in 2012 which employed several distinctive synthetic approaches such as a selective nitration using para-blocking group methodology, a one-pot amination-transfer hydrogenation to provide four reductions approximately in simultaneous fashion, resolution based on an enzymatic reaction, and the leveraging of an in situ generated mixed mesylate hydrochloride salt of CEP-28122.165 The final pilot plant synthetic pathway to CEP-28122 is shown in Scheme 4.
 | ||
| Scheme 4 Pilot plant and scalable synthesis of CEP-28122 (17). | ||
Interestingly, subsequent medicinal chemistry efforts lead to the discovery of CEP-37440 which is another emerging DAAPalouge.121 CEP-37440 highly kinase selective, potent ATP-competitive, and orally active ALK/FAK inhibitor (ALK(enzymatic) IC50 = 3.1 nM, ALK(cellular) IC50 = 22 nM) and (FAK1(enzymatic) IC50 = 2.3 nM, FAK1(cellular) IC50 = 82 nM). CEP-37440 is structurally related to CEP-28122 (an ALK inhibitor). Recently reported investigations related to CEP-37440 has led to enhanced metabolic stability, imperious pharmacokinetic features, and adequate in vitro activity in clinically described resistance mutations.112 In the following section, medicinal chemistry efforts related to early discovery of CEP-37440 (12) and clinical developments are discussed.
6.1 Synthetic strategy of emerging DAAPalouge CEP-37440 from 12
In a quest to discover potent ALK inhibitors, 7-amino-1,3,4,5-tetrahydrobenzo-[b]azepin-2-one (12) (Scheme 5) was identified as a novel DAAPalouge (a small molecule) which exhibited potent activity (ALK IC50 = 15 nM, IR IC50 = 23 nM, Karpas-299 cell IC50 = 30 nM) but the margin against IR was merely 1.5-fold. Benzamide functional group was a key potency determinant for ALK in (12). Due to the involvement of insulin receptor (IR) in the regulation of glucose transport, glycogen, and fat biosynthesis, selectivity towards IR were of particular interest. Based on promising SAR, potent enzymatic/cellular ALK inhibition, and modulation of IR activity, improvement in modest oral bioavailability and optimization of IR selectivity was surveyed on azepinone core (B-ring) (Scheme 5). Ethyl substitution on nitrogen atom of azepinone for improved pharmacokinetic properties (PK) parameters was envisioned as essential group. For reasonable selectivity, ortho-MeO group either at 6-position or 8-position of the benzazepinone was proposed to be superior. Furthermore, focused modification on A- and C-rings, a bicyclo[2.2.1]hept-5-ene ring system was identified as an alternative C-ring partner for improved IR selectivity. Further optimization of potency, off-target selectivity, and PK parameters led to the discovery of a novel and potent bridged bicyclic derivative (13) (Scheme 5). Gratifyingly, (13) demonstrated impressive activity in an ALK assay (enzymatic or cellular) measuring oncogenic NPM-ALK autophosphorylation (ALK IC50 = 14 nM, IR IC50 = 597 nM, Karpas-299 cell IC50 = 45 nM) with modestly improved IR selectivity (IR/ALK = 43). Moreover, oral administration of lead inhibitor (13) to mice induced with ALK positive ALCL SCID xenografts resulted in near-complete/complete regressions.166,167 Although (13) presented a balanced profile of ALK potency and exhibited global kinase selectivity but further efforts were needed to improve IR selectivity (enzymatic IR/ALK IC50 ratio of >100). Despite of the promising features, (13) still lacked the capability of dose escalation in preclinical toxicological models which precluded its further improvement. | ||
| Scheme 5 The strategic development of CEP-37440 from (12). | ||
Cephalon researcher during the course of their endeavors synthesized and profiled a diversity of analogs closely related to NVP-TAE226 (ref. 168) but bis-adduct (16) (Scheme 5) was discovered accidentally.169 This bis-adduct (16) demonstrated ALK inhibition equivalent to NVP-TAE226 and tremendous escalation in IR selectivity but lower (2.4-fold) cellular inhibition. The serendipitous findings were capitalized and a morpholine-phenyl moiety was incorporated as C-ring fragment which led to emergence of novel compound (14) with benzazepinone as B-ring. Compound (14) exhibited drop-off in both ALK enzymatic activity and cellular potency was observed but improvement in potency while maintaining this high degree of IR selectivity was sought. Rather than benzazepinone moiety, a structural motif such as 2,3,4,5-tetrahydro-1H-benzo[d]azepine was perceived to be a synthetically tractable functionality with potential handles to modulate both potency and physiochemical properties and this was incorporated into the pharmacophore. Extensive SAR investigation and substitutions at benzazepine nitrogen of (15) and subtle variations to the nature of groups impacted on both enzyme and cell activity prominently. Numerous analogs were designed wherein basicity of benzazepine nitrogen emerged to be critical contributing factor. Gratifyingly, novel amide (15) was identified as orally bioavailable, most potent and highly selective ALK inhibitor (ALK enzyme IC50 = 4 nM, ALK cell IC50 = 30 nM, IR enzyme IC50 = 1380 nM, IR/ALK IC50 ratio of 345) and displayed promising in vivo efficacy in ALCL tumor models.169 Candidate (15) demonstrated tumor regressions at 30 mg kg−1 and 55 mg kg−1 b.i.d., po doses, at which (15) was found to be tolerated fairly. Neither evident toxicity nor body weight loss were observed. Although compound (15) exhibited potent in vitro ALK activity but potential hERG liability was main concern (IC50 = 3.9 μM for hERG, patch clamp assay) and against the most important CYP isoforms (15) demonstrated minimal inhibition for CYP1A2 and 2D6.170
In subsequent investigations, the basic nitrogen atom was shifted outside the 7-membered ring in (17) to further control the potency and selectivity variances that were associated with the analogs of azepine (15). The new B-ring was aminobenzocycloheptene motif with (S)-configuration at the morpholino carbon juncture. To generate potent ALK inhibition, enhanced IR selectivity, and dose proportional escalation in preclinical toxicology models, a dedicated medicinal chemistry efforts were therefore initiated to evaluate the amendments on aminobenzocycloheptene ring system. With this strategy Cephalon researchers identified (17) wherein at C-2 position of azepine or azepinone fragments were replaced with aminobenzocycloheptene motif as B-ring. Furthermore, earlier established bicyclo[2.2.1]hept-5-ene ring moiety was incorporated at C-4 position of pyrimidine as C-ring to afford the fully assembled HPLC separable (S)-enantiomer DAAPalouge scaffold. Compound (17) revealed as exceedingly potent and highly selective DAAPalouge (an ALK inhibitor). The PK of compound (17) was investigated in numerous species as shown in Table 2.
| Comp. | ALK enzyme IC50 (nM) | ALK cell IC50 (nM) | INSR enzyme IC50 (nM) | INSR cell IC50 (nM) | IR/ALK IC50 | FAK enzyme IC50 (nM) | FAK cell IC50 (nM) | Karpas-299 cell IC50 (nM) | hERG IC50 (μM) | CYP IC50 (μM) | Liver microsome LM (t1/2 min) | F (%) rat | Oral AUC0−∞ (ng h mL−1) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M | R | Mo | H | |||||||||||||
| 12 | 15 | 23 | 30 | — | — | — | — | — | — | — | — | |||||
| 13 | 14 | — | — | — | 43 | — | — | 45 | — | — | — | — | — | — | — | — |
| 14 | 34 | 175 | >10000 |
— | — | — | — | — | — | — | — | — | — | — | — | — |
| 16 | 4 | — | 1860 | — | — | — | — | — | — | — | — | — | — | — | — | — |
| 15 | 4 | 30 | 1380 | — | 345 | — | — | 190 | — | — | — | — | — | — | 30 | 1019 |
| 17 | 1.9 | 42 | 662 | >10000 |
662 | 30 | 944 | 20 | >10 | >10 | 8 | 16 | 17 | 11 | 25 | 1203 |
| 18 | 9 | 200 | 508 | — | — | 6.5 | — | — | 7.9 | >10 | 40 | 39 | 7 | 11 | — | 5526 |
| 19 | 3.1 | 22 | 65 | 2000 | — | 2.0 | 80 | — | >10 | >5.6 | 40 | 40 | 21 | 40 | 42 | 8360 |
6.2 Pilot plant synthesis of CEP-37440 DAAPalouge
To provide early-phase clinical supplies of CEP-37440, Allwein et al., reviewed the previously described medicinal chemistry route of CEP-28122 and presented a scalable route to TEV-CEP-37440 (ref. 171) to obtain 22 kilograms of (19) as trihydrochloride dihydrate. The pilot plant synthesis involve the use of several steps such as a unique ring expansion approach, para-blocking group strategy followed by selective nitration, amination-hydrogenation in a single-pot, resolution of diastereomeric salt, a step to avoid a dangerous intermediates, and a large-scale production of trihydrochloride dihydrate salt were the key highlighted features. The optimized pilot plant synthesis provided TEV-37440 as trihydrochloride dihydrate in >99.5% purity and in 10.7% overall yield (Scheme 6).172 | ||
| Scheme 6 Pilot plant synthesis of CEP-37440 DAApalouge to provide clinical supplies.171 | ||
6.3 Therapeutic trial of CEP-37440 (19)
An oral dose escalation investigation for (17) was conducted in rats using variable concentrations of 10, 30, 55, and 100 mg kg−1. Compound (17) showed a proportional increase in both AUC (∼15-fold increase) and Cmax (∼9-fold increase) at 10 and 100 mg kg−1 doses. Interestingly, using dose escalation from 10 to 100 mg kg−1 (∼10-fold increment), previously discovered benzazepinone (13) displayed Cmax (only ∼2-fold increase) and AUC (only ∼2-fold increase). To this end, based on dose escalation comparative data, an enhanced PK exposure was noticed for (17) and further efforts for preclinical advancement of (13) were terminated in favor of (17).A single dose of novel CEP-28122 (17) given orally at 30 mg kg−1 revealed more than 90% inhibition of NPM-ALK phosphorylation in the tested NPM-ALK positive ALCL Karpas-299 tumor xenograft in SCID mouse. Antitumor efficacy of (17) was evaluated in a Karpas-299 tumor xenograft model in SCID mouse and dose-dependent antitumor activity was measured at both oral doses of 10 mg kg−1 and 30 mg kg−1, b.i.d. CEP-28122 (17) demonstrated complete tumor regressions at the 30 mg kg−1 oral b.i.d. dose. Administration of CEP-28122 (17) was well tolerated and at both dosing regimens neither evident toxicity nor a significant body weight loss was observed.112
Dose-dependent antitumor activity potential was also assessed in NPM-ALK-positive ALCL and EML4-ALK-positive NSCLC tumor xenografts in mice wherein CEP-28122 (17) was administered orally. It showed complete or near to complete tumor regression by following a therapy at a dose of 30 mg kg−1 b.i.d. or higher. Furthermore, CEP-28122 (17) induced growth inhibition and cytotoxicity of neuroblastoma cell lines harboring ALK activating mutants (F1174L in NB-1643 cells and R1275Q in SH-SY5Y cells, respectively). However, CEP-28122 (17) did not induce growth inhibition and cytotoxicity in those which contained ALK wild type (WT) without gene amplification (SKNAS cells). This indicated that (17) was active against the two ALK activating mutants usually found in human neuroblastoma. From the combination of entire favorable biological and pharmaceutical properties, the CEP-28122 (17) was discovered as a preclinical candidate.112 The PK characteristics of novel analogs (13–19) discovered during the strategic development of CEP-37440 from (12) are presented in Table 3.
| PK parameters | Compound | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (13) | (15) | CEP-28122 (17) | CEP-37440 (19) | |||||||||
| SCID mouse | Rat | Dog | S-D rat | CD-1 mouse | CD-1 mouse | Rat | Dog | Mo | CD-1 mouse | SD rat | Mo | |
| iv | ||||||||||||
| Dose (mg kg−1) | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| t1/2 (h) | 0.3 | 0.8 | 1.1 | 0.80 ± 0.05 | 0.7 | 0.7 | 0.9 ± 0.2 | 2.4 ± 0.2 | 1.2 ± 0.05 | 3.0 | 2 ± 0.4 | 5.4 ± 0.6 |
| Cl (mL min−1 kg−1) | 37 | 13 | 22 | 16 ± 1.5 | 20 | 29 | 17 ± 2 | 40 ± 1 | 28 ± 4 | 10 | 4 ± 0.2 | 30.±0.5 |
| % Cl/hepatic blood flow | 41 | 20 | 58 | — | — | — | — | — | — | — | — | — |
| Vd (L kg−1) | 0.9 | 0.9 | 2.1 | 1.1 ± 0.1 | 1.2 | 1.8 | 1.3 ± 0.2 | 8.2 ± 0.5 | 2.9 ± 0.3 | 2.7 | 0.8 ± 0.2 | 13.2 ± 1.9 |
| AUC0−∞ (ng h mL−1) | 446 | 1276 | 773 | 1019 ± 91 | 830 | 584 | 989 ± 127 | 409 ± 16 | 615 ± 89 | 1612 | 4005 ± 237 | 554 ± 11 |
|
||||||||||||
| Po | ||||||||||||
| Dose (mg kg−1) | 10.0 | 5.0 | 10.0 | 5.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 5.0 | 10.0 |
| Cmax (ng mL−1) | 306 ± 53 | 1192 | 880 | 599 ± 3 | 427 | 170 ± 51 | 1533 | 1340 ± 107 | 239 ± 6 | |||
| tmax (h) | 0.25 | 0.8 | 1.1 | 3.3 ± 0.7 | 0.5 | 0.25 | 1.3 ± 0.3 | 0.8 | 4.7 ± 1.3 | 2 | 3.3 ± 0.7 | 6 |
| t1/2 (h) | 2.4 | 2.2 | 1.6 | n.d | 3.1 | 2.3 | 6.6 ± 1.6 | 3.3 | 1.2 ± 0.05 | — | — | — |
| AUC0−∞ (ng h mL−1) | 1406 | 4088 | 2482 | 1395 ± 281 | 3895 | 2993 | 4833 ± 797 | 2126 | 954 ± 209 | 16429 |
8360 ± 540 | 2757 ± 114 |
| F (%) | 32 | 64 | 31 | 30 ± 6 | 64 | 51 | 50 ± 8 | 52 | 16 ± 3 | 102 | 42 ± 3 | 50 ± 3 |
Since the discovery of CEP-28122 DAAPalouge (17), the landscape of small molecule inhibitor had changed to broader extent and four distinct molecular entities had been approved by FDA to treat EML4-ALK positive NSCLC. Although (17) demonstrated overall moderate in vivo clearance potential [CD-1 mouse, Cl = 29 mL min−1 kg−1; rat, Cl = 17 mL min−1 kg−1; monkey, Cl = 28 mL min−1 kg−1], and the in vitro liver microsomal stability values [liver microsome t1/2 (min): CD-1 mouse, 13; rat, 7; human, 10] which prompted the further possibility for improvement. Innovative chemical architecture which could constrain the small-molecule ligands into the putative active conformation was executed which resulted in generation of architecturally distinctive template to overcome associated pharmaceutical liabilities. Recently focal adhesion kinase (FAK) has been investigated as a cytoplasmic protein tyrosine kinase and its overexpression and activation has been characterized in several advanced-stage solid cancers. FAK activation and overexpression are suggestive of numerous solid tumors, mainly those have susceptibility for bone metastasis especially NSCLC, ovarian, breast, prostate, and HNSCC carcinomas.173–175 Due to the growing interest in focal adhesion kinase (FAK) inhibitors and their clinical development,176 extensive SAR was sought on (17) which resulted in superior kinase selectivity profile, favorable brain penetration, and promising activity in ALK+-NSCLC tumor xenografts. Compound (18) also exhibited imperious biochemical and pharmacodynamic activity both in vitro and in vivo, and enhanced antitumor effectiveness in FAK and FAK-dependent tumors.112
In further exploration, nature of substituents (morpholine, N-methyl piperazine and methoxyethylamine) on benzocycloheptane ring system and position refinement of morpholine itself was surveyed. Compared to (19) (ALK IC50 = 3.1 nM, cell IC50 = 22 nM), both diastereomers of (18) were almost 4-fold lesser potent in enzymatic assays, and 10-fold lesser potent in cells based assays but maintained plausible hERG selectivity (7.9–9.1 μM) and CYP inhibition (>10 μM). Furthermore, an enhancement in liver microsomal stability in mouse and rat was observed but human and monkey liver microsomal stability was lesser than anticipated. Pharmacokinetics of (18) in rat demonstrated high iv AUCs but lower Cl and bioavailability (F = 11%). Encouraging results compelled to improve ALK potency by maintaining the additional FAK effectiveness (potency) and human liver microsomal metabolic stability as next envisioned agenda. Subsequent investigation led to the optimization of substituent at piperazine and C-2 position of pyrimidine. Previously reported bicyclo[2.2.1]heptene motif at C-4 of pyrimidine was main metabolic hotspot in (17). To this end, to expedite discovery efforts the racemic mixtures of aromatic sulfones, sulfonamides, N-alkylsulfonamides, imidazoles, pyrazoles, and cyclic sultams were surveyed as novel H-bond acceptors on C-4 position of pyrimidine which resulted in N-methylbenzamide as optimal C-ring partner. On the other hand, variation of N-methyl substituents on the piperazine ring to hydroxyethyl functionality was identified as a noteworthy group on B-ring.
Racemic mixture of (19) revealed as a novel lead candidate with promising ALK enzymatic activity (IC50 = 4 nM), cellular potency (IC50 = 40 nM), and acceptable dual ALK and FAK kinome selectivity. Candidate (19) proved adequate in vitro liver microsomal stability (rat and human t1/2 > 40 min) across the tested species, adequate oral bioavailability (47%), and issues of hERG activity (IC50 ≥ 10 μM) were also circumvented relative other analogs. Furthermore, (R) and (S)-enantiomers of B-ring were installed and individual enantiomers of (19) were synthesized and profiled. Findings revealed both of the enantiomers fairly comparable across off-target selectivity, on-target potency, and in vitro ADME/toxicity parameters but in spite of comparable in vitro metabolic stability of both enantiomers, (S)-enantiomer (19) showed 2-3-fold enhancement in oral bioavailability and considerable higher area under the curve (AUC). The structural basis for this observed potency was recognized using molecular docking. For clinically-derived resistance mutations against small-molecule, the ALK inhibitors compound (19) was tested for ALK mutants and showed equipotent potencies (ALK(1151Tins) Kd = 2.6 nM, ALK(C1156Y) Kd = 1.6 nM, ALK(F1174L) Kd = 1.1 nM, and ALK(L1196M) Kd = 3.3 nM) to that of FDA approved first and 2nd-generation ALK inhibitors.112
Compound (19) exhibited promising in vitro ADME properties and appropriate oral bioavailability in three investigated species. CEP-37440 (19) showed promising brain exposure which may influence physiologically related escape pathways, for example brain metastases. Moreover, dose-dependent antitumor efficacy has also been reported in several animal models of anaplastic lymphoma kinase- positive and FAK-driven cancers. CEP-37440 (19) being a dual FAK and ALK active DAAPalouge is of distinctive interest in terms of the recent observations in inflammatory breast carcinoma where the subsets are comprised of ALK or FAK oncogenic units. CEP-37440 (19) is currently undergoing human clinical trials. The effect of CEP-37440 (19) using human IBC cells and preclinical models of IBC has demonstrated that CEP-37440 inhibits the proliferation of some IBC cells by reducing the level of phospho-FAK1 (Tyr 397) but none of the cells showed expression of ALK. The investigations using IBC xenografts models revealed that compound (19) also efficiently reduces the growth of the primary tumor xenografts and inhibits the development of brain metastases in mice.177 In preclinical profiling, CEP-37440 (19) has demonstrated advantageous metabolic stability, pharmacokinetic and proven the CNS brain penetration.121
7 Conclusion and future prospectives
The trend and rapidity pertaining to drug discovery of anti-cancer agents has substantially changed the understanding of pathogenesis and cancer therapy. The extensive inclusion of computer-aided approaches has proven important involvement and has eliminated the requirement of expensive and time-consuming tasks frequently used in experimental drug discovery processes.178–180 To this end, widespread use of the techniques such as HTS, virtual screening (VS), pharmacophore modeling, and three-dimensional quantitative structure–activity relationships (3D QSAR) have made the lead compound discovery process much faster. Although the HTS is costly technique yet it has shown remarkable utilization in drug discovery campaigns since 1980. However, VS approach applied in the private sector and academia is a cost-saving technology, and has emerged as a versatile tool in the hit to lead identification and eventually becoming an integral part of drug discovery war, whereas the pharmacophore modeling has also been effectively used in recent years to identify a common pharmacophore hypothesis. In addition, the 3D QSAR model unfolds the statistical correlation between structure of designed analog and predictive functions. The 3D QSAR models are also playing significant role to accomplish greater success rate of experimentation, cost reduction of development, and short cycle of research in drug discovery.181–183Due to inclusion of above said technologies in drug discovery campaigns, the current decade (2007–2017) has dragged prodigious progress globally in NSCLC pertaining to drug development and in clinical settings.183–187 Novel targeted antineoplastic agents loaded with anticancer warhead to demonstrate imperious efficacy have been synthesized and introduced for clinical use. Second generation DAAPlouges including alectinib are known to be effective in greater than 50% NSCLC patients who were refractory to crizotinib. Unfortunately, the emergence of resistance to 2nd-generation ALK-TKIs is a major concern to date. Among the identified multiple resistance mechanisms, the G1202R mutation of ALK is emerging as resistance to all currently available ALK-TKIs including DAAPalouges. Lorlatinib (non-DAAPalouge) is a first macrocyclic 3rd-generation ALK-TKI which have demonstrated effectiveness against the G1202R mutation of ALK but again compound mutation such as C1156Y/L1198F in the ALK kinase domain has been revealed as a major concern. Brigatinib is a DAAPalouge which has shown the potential to overcome the resistance mechanism to first and second generation ALK inhibitors including the G1202R mutation of ALK in xenograft models.117 However, many other newly discovered molecular entities have demonstrated effectiveness against the G1202R mutation.188–190 Furthermore, the current median overall survival of patients with ALK-rearranged lung cancer is above 4 years only. This over survival is achieved through the treatment protocols of using sequential ALK-TKIs in addition to other FDA approved chemotherapeutics. Finally, despite the discovery the development of several potent ALK TKIs including DAAPalouges have led to the substantial increase of drugs but still it is believed that currently available therapies are palliative rather curative. Thus further discoveries are indispensable to develop superior therapeutic strategies.
Conflicts of interest
The authors also declare no conflicts of interest.Abbreviations
| ADME | Absorption, distribution, metabolism, and excretion |
| ALCL | Anaplastic large cell lymphoma |
| ALK | Anaplastic lymphoma kinase |
| ATP | Adenosine triphosphate |
| BTD | Breakthrough therapy designation |
| c-MET | Mesenchymal to epithelial transition |
| CNS | Central nervous system |
| DAAP | 2,4-Diarylaminopyrimidine |
| DAAPalogues | 2,4-Diarylaminopyrimidine analogues |
| DLBCL | Diffuse large B-cell lymphomas |
| DMPO | Dimethylphosphine oxide |
| EGFR | Endothelial growth factor receptor |
| EML4 | Echinoderm microtubule-associated protein-like 4 gene |
| FDA | Food and Drug Administration |
| GSH | Glutathione |
| HBD | Hydrogen bond donors |
| HGF/SF | Hepatocyte growth factor/scatter factor |
| HTS | Throughput screening |
| IGF1R | Insulin-like growth factor 1 receptor |
| IMT | Inflammatory myofibroblastic tumours |
| INSR | Insulin receptor |
| IR | Insulin receptor |
| JAK | Janus activated kinase |
| LRRK2 | Leucine-rich repeat kinase 2 |
| LTK | Leukocyte tyrosine kinase |
| MTT | Molecular targeted therapy |
| MW | Molecular weight |
| SN-Ar | Nucleophilic aromatic substitution |
| NPM | Nucleophosmin |
| NSCLC | Non-small-cell lung cancer |
| ORR | Objective response rate |
| P-arylation | Phosphorous arylation |
| PFS | Progression-free survival |
| PI3K | Phosphatidylinositol 3-kinase |
| PK | Pharmacokinetic properties |
| PLC | Phospholipase C |
| PTN | Pleotrophin |
| pY | Phosphotyrosine |
| ROS1 | ROS Proto-Oncogene 1 |
| SAR | Structure–activity relationship |
| SCC | Squamous cell carcinoma |
| SCLC | Small-cell-lung cancer |
| TKI | Tyrosine kinase inhibitor |
| TPSA | Topological polar surface area |
| VEGFR | Vascular endothelial growth factor receptor |
| VS | Virtual screening |
| 3D-QSAR | Three-dimensional quantitative structure–activity relationships |
Acknowledgements
The authors would like to acknowledge and grateful to Dr Farooq Ahmad Kiani research scientist at Department of Physiology and Biophysics, Boston University School of Medicine, Boston, Massachusetts, USA, for critical reading and language editing of the manuscript.References
- S. Rebecca, N. Deepa and J. Ahmedin, Ca-Cancer J. Clin., 2013, 63, 11–30 CrossRef PubMed.
- R. Siegel, J. Ma, Z. Zou and A. Jemal, Ca-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7–30 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 7–30 CrossRef PubMed.
- A. D. Lopez, N. E. Collishaw and T. Piha, Tob. Control, 1994, 3, 242 CrossRef.
- L. Collins, C. Haines, R. Perkel and R. Enck, Am. Fam. Physician, 2007, 75, 56–63 Search PubMed.
- P. National Center for Chronic Disease and S. Health Promotion Office on and Health, in The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General, Centers for Disease Control and Prevention (US), Atlanta (GA), 2014 Search PubMed.
- R. Courtney, Drug Alcohol Rev., 2015, 34, 694–695 CrossRef.
- S. McGuire, Adv. Nutr., 2016, 7, 418–419 CrossRef PubMed.
- J. Subramanian and R. Govindan, J. Clin. Oncol., 2007, 25, 561–570 CrossRef PubMed.
- W. Travis, Clin. Chest Med., 2011, 32, 669–692 CrossRef PubMed.
- M. Latif, A. Saeed and S. Kim, Arch. Pharmacal Res., 2013, 36, 1051–1054 CrossRef CAS PubMed.
- R. Clay, F. Maldonado and E. S. Edell, Interventions in Pulmonary Medicine, ed. J. P. Díaz-Jimenez and A. N. Rodriguez, Springer International Publishing, Cham, 2nd edn, 2018, pp. 313–322, DOI:10.1007/978-3-319-58036-4_19.
- G. Scagliotti, Ann. Oncol., 2007, 18(suppl. 10), 41 Search PubMed.
- U. McDermott, A. Iafrate, N. Gray, T. Shioda, M. Classon, S. Maheswaran, W. Zhou, H. Choi, S. Smith, L. Dowell, L. Ulkus, G. Kuhlmann, P. Greninger, J. Christensen, D. Haber and J. Settleman, Cancer Res., 2008, 68, 3389–3395 CrossRef CAS PubMed.
- P. Ong and D. Ost, Interventions in Pulmonary Medicine, ed. J. P. Díaz-Jimenez and A. N. Rodriguez, Springer International Publishing, Cham, 2nd edn, 2018, pp. 323–332, DOI:10.1007/978-3-319-58036-4_20.
- S. O. Dolly, D. C. Collins, R. Sundar, S. Popat and T. A. Yap, Drugs, 2017, 77, 813–827 CrossRef CAS PubMed.
- S. W. Morris, M. N. Kirstein, M. B. Valentine, K. G. Dittmer, D. N. Shapiro, D. L. Saltman and A. T. Look, Science, 1994, 263, 1281 CAS.
- M. Shiota, J. Fujimoto, T. Semba, H. Satoh, T. Yamamoto and S. Mori, Oncogene, 1994, 9, 1567–1574 CAS.
- S. W. Morris, M. N. Kirstein, M. B. Valentine, K. Dittmer, D. N. Shapiro, A. T. Look and D. L. Saltman, Science, 1995, 267, 316–317 CrossRef CAS PubMed.
- Y. S. Li, P. G. Milner, A. K. Chauhan, M. A. Watson, R. M. Hoffman, C. M. Kodner, J. Milbrandt and T. F. Deuel, Science, 1990, 250, 1690 CAS.
- T. Iwahara, J. Fujimoto, D. Wen, R. Cupples, N. Bucay, T. Arakawa, S. Mori, B. Ratzkin and T. Yamamoto, Oncogene, 1997, 14, 439 CrossRef CAS PubMed.
- S. W. Morris, C. Naeve, P. Mathew, P. L. James, M. N. Kirstein, X. Cui and D. P. Witte, Oncogene, 1997, 14, 2175–2188 CrossRef CAS PubMed.
- Y. Ben-Neriah and A. R. Bauskin, Nature, 1988, 333, 672–676 CrossRef CAS PubMed.
- A. Bernards and S. M. de la Monte, EMBO J., 1990, 9, 2279–2287 CAS.
- Y. Maru, H. Hirai and F. Takaku, Oncog. Res., 1990, 5, 199–204 CAS.
- H. Kozutsumi, H. Toyoshima, K. Hagiwara, Y. Yazaki and H. Hirai, Oncogene, 1994, 9, 2991–2998 CAS.
- S. W. Morris, C. Naeve, P. Mathew, P. L. James, M. N. Kirstein, X. Cui and D. P. Witte, Oncogene, 1997, 14, 2175–2188 CrossRef CAS PubMed.
- K. Pulford, L. Lamant, S. W. Morris, L. H. Butler, K. M. Wood, D. Stroud, G. Delsol and D. Y. Mason, Blood, 1997, 89, 1394–1404 CAS.
- R. Chiarle, C. Voena, C. Ambrogio, R. Piva and G. Inghirami, Nat. Rev. Cancer, 2008, 8, 11–23 CrossRef CAS PubMed.
- R. H. Palmer, E. Vernersson, C. Grabbe and B. Hallberg, Biochem. J., 2009, 420, 345–361 CrossRef CAS PubMed.
- A. Barreca, E. Lasorsa, L. Riera, R. Machiorlatti, R. Piva, M. Ponzoni, I. Kwee, F. Bertoni, P. P. Piccaluga, S. A. Pileri and G. Inghirami, J. Mol. Endocrinol., 2011, 47, R11–R23 CrossRef CAS PubMed.
- S. Morris, M. Kirstein, M. Valentine, K. Dittmer, D. Shapiro, D. Saltman and A. Look, Science, 1994, 263, 1281–1284 CAS.
- B. Hallberg and R. Palmer, Nat. Rev. Cancer, 2013, 13, 685–700 CrossRef CAS PubMed.
- H. Mano, Cancer Sci., 2008, 99, 2349–2355 CrossRef CAS PubMed.
- M. Soda, Y. Choi, M. Enomoto, S. Takada, Y. Yamashita, S. Ishikawa, S.-i. Fujiwara, H. Watanabe, K. Kurashina, H. Hatanaka, M. Bando, S. Ohno, Y. Ishikawa, H. Aburatani, T. Niki, Y. Sohara, Y. Sugiyama and H. Mano, Nature, 2007, 448, 561–566 CrossRef CAS PubMed.
- A. T. Shaw, B. Y. Yeap, M. Mino-Kenudson, S. R. Digumarthy, D. B. Costa, R. S. Heist, B. Solomon, H. Stubbs, S. Admane, U. McDermott, J. Settleman, S. Kobayashi, E. J. Mark, S. J. Rodig, L. R. Chirieac, E. L. Kwak, T. J. Lynch and A. J. Iafrate, J. Clin. Oncol., 2009, 27, 4247–4253 CrossRef CAS PubMed.
- T. Sasaki, S. Rodig, L. Chirieac and P. Jänne, Eur. J. Cancer, 2010, 46, 1773–1780 CrossRef CAS PubMed.
- J. Tanizaki, I. Okamoto, K. Takezawa, K. Sakai, K. Azuma, K. Kuwata, H. Yamaguchi, E. Hatashita, K. Nishio, P. A. Janne and K. Nakagawa, Br. J. Cancer, 2012, 106, 763–767 CrossRef CAS PubMed.
- K. Rikova, A. Guo, Q. Zeng, A. Possemato, J. Yu, H. Haack, J. Nardone, K. Lee, C. Reeves, Y. Li, Y. Hu, Z. Tan, M. Stokes, L. Sullivan, J. Mitchell, R. Wetzel, J. Macneill, J. M. Ren, J. Yuan, C. E. Bakalarski, J. Villen, J. M. Kornhauser, B. Smith, D. Li, X. Zhou, S. P. Gygi, T. L. Gu, R. D. Polakiewicz, J. Rush and M. J. Comb, Cell, 2007, 131, 1190–1203 CrossRef CAS PubMed.
- C. Griffin, A. Hawkins, C. Dvorak, C. Henkle, T. Ellingham and E. Perlman, Cancer Res., 1999, 59, 2776–2780 CAS.
- B. Hallberg and R. H. Palmer, Nat. Rev. Cancer, 2013, 13, 685–700 CrossRef CAS PubMed.
- F.-R. Jazii, Z. Najafi, R. Malekzadeh, T.-P. Conrads, A.-A. Ziaee, C. Abnet, M. Yazdznbod, A.-A. Karkhane and G.-H. Salekdeh, World J. Gastroenterol., 2006, 12, 7104–7112 CrossRef CAS PubMed.
- X.-L. Du, H. Hu, D.-C. Lin, S.-H. Xia, X.-M. Shen, Y. Zhang, M.-L. Luo, Y.-B. Feng, Y. Cai, X. Xu, Y.-L. Han, Q.-M. Zhan and M.-R. Wang, J. Mol. Med., 2007, 85, 863–875 CrossRef CAS PubMed.
- L. Debelenko, S. Raimondi, N. Daw, B. Shivakumar, D. Huang, M. Nelson and J. Bridge, Mod. Pathol., 2011, 24, 430–442 CrossRef CAS PubMed.
- J. Cook, L. Dehner, M. Collins, Z. Ma, S. Morris, C. Coffin and D. Hill, Am. J. Surg. Pathol., 2001, 25, 1364–1371 CrossRef CAS PubMed.
- Y. Chen, J. Takita, Y. Choi, M. Kato, M. Ohira, M. Sanada, L. Wang, M. Soda, A. Kikuchi, T. Igarashi, A. Nakagawara, Y. Hayashi, H. Mano and S. Ogawa, Nature, 2008, 455, 971–974 CrossRef CAS PubMed.
- H. Carén, F. Abel, P. Kogner and T. Martinsson, Biochem. J., 2008, 416, 153–159 CrossRef.
- R. George, T. Sanda, M. Hanna, S. Fröhling, W. Luther, J. Zhang, Y. Ahn, W. Zhou, W. London, P. McGrady, L. Xue, S. Zozulya, V. Gregor, T. Webb, N. Gray, D. Gilliland, L. Diller, H. Greulich, S. Morris, M. Meyerson and A. Look, Nature, 2008, 455, 975–978 CrossRef CAS PubMed.
- I. Janoueix-Lerosey, D. Lequin, L. Brugières, A. Ribeiro, L. de Pontual, V. Combaret, V. Raynal, A. Puisieux, G. Schleiermacher, G. Pierron, D. Valteau-Couanet, T. Frebourg, J. Michon, S. Lyonnet, J. Amiel and O. Delattre, Nature, 2008, 455, 967–970 CrossRef CAS PubMed.
- Y. Mossé, M. Laudenslager, L. Longo, K. Cole, A. Wood, E. Attiyeh, M. Laquaglia, R. Sennett, J. Lynch, P. Perri, G. Laureys, F. Speleman, C. Kim, C. Hou, H. Hakonarson, A. Torkamani, N. Schork, G. Brodeur, G. Tonini, E. Rappaport, M. Devoto and J. Maris, Nature, 2008, 455, 930–935 CrossRef PubMed.
- R. Palmer, E. Vernersson, C. Grabbe and B. Hallberg, Biochem. J., 2009, 420, 345–361 CrossRef CAS PubMed.
- S. Ogawa, J. Takita, M. Sanada and Y. Hayashi, Cancer Sci., 2011, 102, 302–308 CrossRef CAS PubMed.
- R. Roskoski Jr, Pharmacol. Res., 2013, 68, 68–94 CrossRef PubMed.
- A. Galkin, J. Melnick, S. Kim, T. Hood, N. Li, L. Li, G. Xia, R. Steensma, G. Chopiuk, J. Jiang, Y. Wan, P. Ding, Y. Liu, F. Sun, P. Schultz, N. Gray and M. Warmuth, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 270–275 CrossRef CAS PubMed.
- J. P. Koivunen, C. Mermel, K. Zejnullahu, C. Murphy, E. Lifshits, A. J. Holmes, H. G. Choi, J. Kim, D. Chiang, R. Thomas, J. Lee, W. G. Richards, D. J. Sugarbaker, C. Ducko, N. Lindeman, J. P. Marcoux, J. A. Engelman, N. S. Gray, C. Lee, M. Meyerson and P. A. Jänne, Clin. Cancer Res., 2008, 14, 4275–4283 CrossRef CAS PubMed.
- P. Minoo and H. Y. Wang, Int. J. Clin. Exp. Pathol., 2012, 5, 397–410 CAS.
- J. J. Cui, M. Tran-Dube, H. Shen, M. Nambu, P. P. Kung, M. Pairish, L. Jia, J. Meng, L. Funk, I. Botrous, M. McTigue, N. Grodsky, K. Ryan, E. Padrique, G. Alton, S. Timofeevski, S. Yamazaki, Q. Li, H. Zou, J. Christensen, B. Mroczkowski, S. Bender, R. S. Kania and M. P. Edwards, J. Med. Chem., 2011, 54, 6342–6363 CrossRef CAS PubMed.
- A. T. Shaw, U. Yasothan and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2011, 10, 897–898 CrossRef CAS PubMed.
- FDA News Release “FDA expands use of Xalkori to treat rare form of advanced non-small cell lung cancer”, https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm490329.htm, (accessed on April 2018).
- J. F. Gainor and A. T. Shaw, J. Clin. Oncol., 2013, 31, 3987–3996 CrossRef CAS PubMed.
- A. T. Shaw and J. A. Engelman, J. Clin. Oncol., 2013, 31, 1105–1111 CrossRef CAS PubMed.
- L. Friboulet, N. Li, R. Katayama, C. C. Lee, J. F. Gainor, A. S. Crystal, P. Y. Michellys, M. M. Awad, N. Yanagitani, S. Kim, A. C. Pferdekamper, J. Li, S. Kasibhatla, F. Sun, X. Sun, S. Hua, P. McNamara, S. Mahmood, E. L. Lockerman, N. Fujita, M. Nishio, J. L. Harris, A. T. Shaw and J. A. Engelman, Cancer Discovery, 2014, 4, 662–673 CrossRef CAS PubMed.
- A. T. Shaw, S. H. Ou, Y. J. Bang, D. R. Camidge, B. J. Solomon, R. Salgia, G. J. Riely, M. Varella-Garcia, G. I. Shapiro, D. B. Costa, R. C. Doebele, L. P. Le, Z. Zheng, W. Tan, P. Stephenson, S. M. Shreeve, L. M. Tye, J. G. Christensen, K. D. Wilner, J. W. Clark and A. J. Iafrate, N. Engl. J. Med., 2014, 371, 1963–1971 CrossRef PubMed.
- M. Caccese, R. Ferrara, S. Pilotto, L. Carbognin, G. Grizzi, A. Calio, M. Brunelli, F. Cuppone, S. Petraglia, A. Scarpa, G. Tortora and E. Bria, Expert Opin. Pharmacother., 2016, 17, 2253–2266 CrossRef CAS PubMed.
- Y. L. Choi, M. Soda, Y. Yamashita, T. Ueno, J. Takashima, T. Nakajima, Y. Yatabe, K. Takeuchi, T. Hamada, H. Haruta, Y. Ishikawa, H. Kimura, T. Mitsudomi, Y. Tanio and H. Mano, N. Engl. J. Med., 2010, 363, 1734–1739 CrossRef CAS PubMed.
- T. Sasaki, K. Okuda, W. Zheng, J. Butrynski, M. Capelletti, L. Wang, N. S. Gray, K. Wilner, J. G. Christensen, G. Demetri, G. I. Shapiro, S. J. Rodig, M. J. Eck and P. A. Janne, Cancer Res., 2010, 70, 10038–10043 CrossRef CAS PubMed.
- T. Sasaki, J. Koivunen, A. Ogino, M. Yanagita, S. Nikiforow, W. Zheng, C. Lathan, J. P. Marcoux, J. Du, K. Okuda, M. Capelletti, T. Shimamura, D. Ercan, M. Stumpfova, Y. Xiao, S. Weremowicz, M. Butaney, S. Heon, K. Wilner, J. G. Christensen, M. J. Eck, K. K. Wong, N. Lindeman, N. S. Gray, S. J. Rodig and P. A. Janne, Cancer Res., 2011, 71, 6051–6060 CrossRef CAS PubMed.
- R. C. Doebele, A. B. Pilling, D. L. Aisner, T. G. Kutateladze, A. T. Le, A. J. Weickhardt, K. L. Kondo, D. J. Linderman, L. E. Heasley, W. A. Franklin, M. Varella-Garcia and D. R. Camidge, Clin. Cancer Res., 2012, 18, 1472–1482 CrossRef CAS PubMed.
- C. M. Lovly and W. Pao, Sci. Transl. Med., 2012, 4, 120–122 Search PubMed.
- J. F. Gainor and A. T. Shaw, J. Clin. Oncol., 2013, 31, 3987–3996 CrossRef CAS PubMed.
- K. Kinoshita, K. Asoh, N. Furuichi, T. Ito, H. Kawada, S. Hara, J. Ohwada, T. Miyagi, T. Kobayashi, K. Takanashi, T. Tsukaguchi, H. Sakamoto, T. Tsukuda and N. Oikawa, Bioorg. Med. Chem., 2012, 20, 1271–1280 CrossRef CAS PubMed.
- T. W. Johnson, P. F. Richardson, S. Bailey, A. Brooun, B. J. Burke, M. R. Collins, J. J. Cui, J. G. Deal, Y.-L. Deng, D. Dinh, L. D. Engstrom, M. He, J. Hoffman, R. L. Hoffman, Q. Huang, R. S. Kania, J. C. Kath, H. Lam, J. L. Lam, P. T. Le, L. Lingardo, W. Liu, M. McTigue, C. L. Palmer, N. W. Sach, T. Smeal, G. L. Smith, A. E. Stewart, S. Timofeevski, H. Zhu, J. Zhu, H. Y. Zou and M. P. Edwards, J. Med. Chem., 2014, 57, 4720–4744 CrossRef CAS PubMed.
- B. J. Solomon, T. M. Bauer, E. Felip, B. Besse, L. P. James, J. S. Clancy, K. J. Klamerus, J.-F. Martini, A. Abbattista and A. T. Shaw, J. Clin. Oncol., 2016, 34, 9009 CrossRef PubMed.
- E. Felip, T. Bauer, B. Solomon, B. Besse, L. James, J. Clancy, K. Klamerus, J. F. Martini, A. Abbattista and A. Shaw, J. Thorac. Oncol., 2016, 12, S383–S384 CrossRef.
- S. Basit, Z. Ashraf, K. Lee and M. Latif, Eur. J. Med. Chem., 2017, 134, 348–356 CrossRef CAS PubMed.
- S. Basit, Z. Ashraf, K. Lee and M. Latif, Eur. J. Med. Chem., 2017, 134, 348–356 CrossRef CAS PubMed.
- V. Subbiah, D. S. Hong and F. Meric-Bernstam, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E1419–E1420 CrossRef CAS PubMed.
- I. Dagogo-Jack and A. T. Shaw, J. Clin. Oncol., 2017, 35, 2595–2597 CrossRef PubMed.
- S. M. Lim, H. R. Kim, J.-S. Lee, K. H. Lee, Y.-G. Lee, Y. J. Min, E. K. Cho, S. S. Lee, B.-S. Kim, M. Y. Choi, H. S. Shim, J.-H. Chung, Y. L. Choi, M. J. Lee, M. Kim, J.-H. Kim, S. M. Ali, M.-J. Ahn and B. C. Cho, J. Clin. Oncol., 2017, 35, 2613–2618 CrossRef PubMed.
- S. Dhillon and M. Clark, Drugs, 2014, 74, 1285–1291 CrossRef CAS PubMed.
- S. Khozin, G. M. Blumenthal, L. Zhang, S. Tang, M. Brower, E. Fox, W. Helms, R. Leong, P. Song, Y. Pan, Q. Liu, P. Zhao, H. Zhao, D. Lu, Z. Tang, A. Al Hakim, K. Boyd, P. Keegan, R. Justice and R. Pazdur, Clin. Cancer Res., 2015, 21, 2436 CrossRef CAS PubMed.
- M. R. Cooper, H. Chim, H. Chan and C. Durand, Ann. Pharmacother., 2015, 49, 107–112 CrossRef CAS PubMed.
- I. B. Muller, A. J. De Langen, R. J. Honeywell, E. Giovannetti and G. J. Peters, Expert Rev. Anticancer Ther., 2016, 16, 147–157 CrossRef CAS PubMed.
- T. S. K. Mok, L. Crino, E. Felip, R. Salgia, T. De Pas, D. S. W. Tan and L. Q. M. Chow, Cancer Treat. Rev., 2017, 55, 181–189 CrossRef CAS PubMed.
- A. T. Shaw, T. M. Kim, L. Crino, C. Gridelli, K. Kiura, G. Liu, S. Novello, A. Bearz, O. Gautschi, T. Mok, M. Nishio, G. Scagliotti, D. R. Spigel, S. Deudon, C. Zheng, S. Pantano, P. Urban, C. Massacesi, K. Viraswami-Appanna and E. Felip, Lancet Oncol., 2017, 18, 874–886 CrossRef CAS PubMed.
- T. Kodama, T. Tsukaguchi, M. Yoshida, O. Kondoh and H. Sakamoto, Cancer Lett., 2014, 351, 215–221 CrossRef CAS PubMed.
- Y. Ohe, M. Nishio, K. Kiura, T. Seto, K. Nakagawa, M. Maemondo, A. Inoue, T. Hida, H. Yoshioka, M. Harada, N. Nogami, H. Murakami, K. Takeuchi, T. Shimada, H. Kuriki, T. Tanaka and T. Tamura, J. Clin. Oncol., 2015, 33, 8061 Search PubMed.
- L. Gandhi, S. I. Ou, A. T. Shaw, F. Barlesi, A. C. Dingemans, D. W. Kim, D. R. Camidge, B. G. M. Hughes, J. C. Yang, J. de Castro, L. Crino, H. Lena, P. Do, S. Golding, W. Bordogna, A. Zeaiter, A. Kotb and S. Gadgeel, Eur. J. Cancer, 2017, 82, 27–33 CrossRef CAS PubMed.
- T. Tamura, T. Seto, K. Nakagawa, M. Maemondo, A. Inoue, T. Hida, H. Yoshioka, M. Harada, Y. Ohe, N. Nogami, H. Murakami, K. Takeuchi, T. Asakawa, K. Kikuchi, T. Tanaka and M. Nishio, Int. J. Radiat. Oncol., Biol., Phys., 2014, 90, S6 CrossRef.
- S. Peters, D. R. Camidge, A. T. Shaw, S. Gadgeel, J. S. Ahn, D. W. Kim, S. I. Ou, M. Perol, R. Dziadziuszko, R. Rosell, A. Zeaiter, E. Mitry, S. Golding, B. Balas, J. Noe, P. N. Morcos and T. Mok, N. Engl. J. Med., 2017, 377, 829–838 CrossRef CAS PubMed.
- T. Seto, K. Kiura, M. Nishio, K. Nakagawa, M. Maemondo, A. Inoue, T. Hida, N. Yamamoto, H. Yoshioka, M. Harada, Y. Ohe, N. Nogami, K. Takeuchi, T. Shimada, T. Tanaka and T. Tamura, Lancet Oncol., 2013, 14, 590–598 CrossRef CAS PubMed.
- H. Sakamoto, T. Tsukaguchi, S. Hiroshima, T. Kodama, T. Kobayashi, T. A. Fukami, N. Oikawa, T. Tsukuda, N. Ishii and Y. Aoki, Cancer Cell, 2011, 19, 679–690 CrossRef CAS PubMed.
- K. Kinoshita, T. Kobayashi, K. Asoh, N. Furuichi, T. Ito, H. Kawada, S. Hara, J. Ohwada, K. Hattori, T. Miyagi, W.-S. Hong, M.-J. Park, K. Takanashi, T. Tsukaguchi, H. Sakamoto, T. Tsukuda and N. Oikawa, J. Med. Chem., 2011, 54, 6286–6294 CrossRef CAS PubMed.
- K. Kinoshita, Y. Ono, T. Emura, K. Asoh, N. Furuichi, T. Ito, H. Kawada, S. Tanaka, K. Morikami, T. Tsukaguchi, H. Sakamoto, T. Tsukuda and N. Oikawa, Bioorg. Med. Chem. Lett., 2011, 21, 3788–3793 CrossRef CAS PubMed.
- X. Jiang, J. Zhou, J. Ai, Z. Song, X. Peng, L. Xing, Y. Xi, J. Guo, Q. Yao, J. Ding, M. Geng and A. Zhang, Eur. J. Med. Chem., 2015, 105, 39–56 CrossRef CAS PubMed.
- X. Jiang, J. Zhou, J. Ai, Z. Song, X. Peng, L. Xing, Y. Xi, J. Guo, Q. Yao, J. Ding, M. Geng and A. Zhang, Eur. J. Med. Chem., 2015, 105, 39–56 CrossRef CAS PubMed.
- Z. Song, Z. Xia, Y. Ji, L. Xing, Y. Gao, J. Ai, M. Geng and A. Zhang, Eur. J. Med. Chem., 2016, 118, 244–249 CrossRef CAS PubMed.
- M. Han, C. Wang, Y. Ji, Z. Song, L. Xing, Y. Su, X. Wang, A. Zhang, J. Ai and M. Geng, Bioorg. Med. Chem. Lett., 2016, 26, 5399–5402 CrossRef CAS PubMed.
- S. R. Peddi, S. K. Sivan and V. Manga, J. Recept. Signal Transduction, 2016, 36, 488–504 CrossRef CAS PubMed.
- J. M. Hatcher, M. Bahcall, H. G. Choi, Y. Gao, T. Sim, R. George, P. A. Janne and N. S. Gray, J. Med. Chem., 2015, 58, 9296–9308 CrossRef CAS PubMed.
- S. Dhillon and M. Clark, Drugs, 2014, 74, 1285–1291 CrossRef CAS PubMed.
- T. W. Johnson, P. F. Richardson, S. Bailey, A. Brooun, B. J. Burke, M. R. Collins, J. J. Cui, J. G. Deal, Y. L. Deng, D. Dinh, L. D. Engstrom, M. He, J. Hoffman, R. L. Hoffman, Q. Huang, R. S. Kania, J. C. Kath, H. Lam, J. L. Lam, P. T. Le, L. Lingardo, W. Liu, M. McTigue, C. L. Palmer, N. W. Sach, T. Smeal, G. L. Smith, A. E. Stewart, S. Timofeevski, H. Zhu, J. Zhu, H. Y. Zou and M. P. Edwards, J. Med. Chem., 2014, 57, 4720–4744 CrossRef CAS PubMed.
- S. Khozin, G. M. Blumenthal, L. Zhang, S. Tang, M. Brower, E. Fox, W. Helms, R. Leong, P. Song, Y. Pan, Q. Liu, P. Zhao, H. Zhao, D. Lu, Z. Tang, A. Al Hakim, K. Boyd, P. Keegan, R. Justice and R. Pazdur, Clin. Cancer Res., 2015, 21, 2436–2439 CrossRef CAS PubMed.
- E. Larkins, G. M. Blumenthal, H. Chen, K. He, R. Agarwal, G. Gieser, O. Stephens, E. Zahalka, K. Ringgold, W. Helms, S. Shord, J. Yu, H. Zhao, G. Davis, A. E. McKee, P. Keegan and R. Pazdur, Clin. Cancer Res., 2016, 22, 5171–5176 CrossRef PubMed.
- https://clinicaltrials.gov/ct2/show/NCT01970865, https://clinicaltrials.gov/show/NCT02584634, https://clinicaltrials.gov/ct2/show/NCT02927340.
- A. V. Galkin, J. S. Melnick, S. Kim, T. L. Hood, N. Li, L. Li, G. Xia, R. Steensma, G. Chopiuk, J. Jiang, Y. Wan, P. Ding, Y. Liu, F. Sun, P. G. Schultz, N. S. Gray and M. Warmuth, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 270–275 CrossRef CAS PubMed.
- C. H. Park, H. Choe, I.-Y. Jang, S. Y. Kwon, M. Latif, H. K. Lee, H. J. Lee, E. H. Yang, J. I. Yun, C. H. Chae, S. Y. Cho, S. U. Choi, J. D. Ha, H. Jung, H. R. Kim, P. Kim, C. O. Lee, C.-S. Yun and K. Lee, Bioorg. Med. Chem. Lett., 2013, 23, 6192–6196 CrossRef CAS PubMed.
- T. H. Marsilje, W. Pei, B. Chen, W. Lu, T. Uno, Y. Jin, T. Jiang, S. Kim, N. Li, M. Warmuth, Y. Sarkisova, F. Sun, A. Steffy, A. C. Pferdekamper, A. G. Li, S. B. Joseph, Y. Kim, B. Liu, T. Tuntland, X. Cui, N. S. Gray, R. Steensma, Y. Wan, J. Jiang, G. Chopiuk, J. Li, W. P. Gordon, W. Richmond, K. Johnson, J. Chang, T. Groessl, Y. Q. He, A. Phimister, A. Aycinena, C. C. Lee, B. Bursulaya, D. S. Karanewsky, H. M. Seidel, J. L. Harris and P. Y. Michellys, J. Med. Chem., 2013, 56, 5675–5690 CrossRef CAS PubMed.
- W.-S. Huang, S. Liu, D. Zou, M. Thomas, Y. Wang, T. Zhou, J. Romero, A. Kohlmann, F. Li, J. Qi, L. Cai, T. A. Dwight, Y. Xu, R. Xu, R. Dodd, A. Toms, L. Parillon, X. Lu, R. Anjum, S. Zhang, F. Wang, J. Keats, S. D. Wardwell, Y. Ning, Q. Xu, L. E. Moran, Q. K. Mohemmad, H. G. Jang, T. Clackson, N. I. Narasimhan, V. M. Rivera, X. Zhu, D. Dalgarno and W. C. Shakespeare, J. Med. Chem., 2016, 59, 4948–4964 CrossRef CAS PubMed.
- G. R. Ott, M. Cheng, K. S. Learn, J. Wagner, D. E. Gingrich, J. G. Lisko, M. Curry, E. F. Mesaros, A. K. Ghose, M. R. Quail, W. Wan, L. Lu, P. Dobrzanski, M. S. Albom, T. S. Angeles, K. Wells-Knecht, Z. Huang, L. D. Aimone, E. Bruckheimer, N. Anderson, J. Friedman, S. V. Fernandez, M. A. Ator, B. A. Ruggeri and B. D. Dorsey, J. Med. Chem., 2016, 59, 7478–7496 CrossRef CAS PubMed.
- J. C. Soria, D. S. W. Tan, R. Chiari, Y. L. Wu, L. Paz-Ares, J. Wolf, S. L. Geater, S. Orlov, D. Cortinovis, C. J. Yu, M. Hochmair, A. B. Cortot, C. M. Tsai, D. Moro-Sibilot, R. G. Campelo, T. McCulloch, P. Sen, M. Dugan, S. Pantano, F. Branle, C. Massacesi and G. de Castro Jr, Lancet, 2017, 389, 917–929 CrossRef CAS.
- J. Mazieres, G. Zalcman, L. Crino, P. Biondani, F. Barlesi, T. Filleron, A. M. Dingemans, H. Lena, I. Monnet, S. I. Rothschild, F. Cappuzzo, B. Besse, L. Thiberville, D. Rouviere, R. Dziadziuszko, E. F. Smit, J. Wolf, C. Spirig, N. Pecuchet, F. Leenders, J. M. Heuckmann, J. Diebold, J. D. Milia, R. K. Thomas and O. Gautschi, J. Clin. Oncol., 2015, 33, 992–999 CrossRef CAS PubMed.
- T. P. Heffron, J. Med. Chem., 2016, 59, 10030–10066 CrossRef CAS PubMed.
- H. Y. Zou, Q. Li, L. D. Engstrom, M. West, V. Appleman, K. A. Wong, M. McTigue, Y.-L. Deng, W. Liu, A. Brooun, S. Timofeevski, S. R. P. McDonnell, P. Jiang, M. D. Falk, P. B. Lappin, T. Affolter, T. Nichols, W. Hu, J. Lam, T. W. Johnson, T. Smeal, A. Charest and V. R. Fantin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3493–3498 CrossRef CAS PubMed.
- S. Zhang, R. Anjum, R. Squillace, S. Nadworny, T. Zhou, J. Keats, Y. Ning, S. D. Wardwell, D. Miller, Y. Song, L. Eichinger, L. Moran, W. S. Huang, S. Liu, D. Zou, Y. Wang, Q. Mohemmad, H. G. Jang, E. Ye, N. Narasimhan, F. Wang, J. Miret, X. Zhu, T. Clackson, D. Dalgarno, W. C. Shakespeare and V. M. Rivera, Clin. Cancer Res., 2016, 22, 5527–5538 CrossRef CAS PubMed.
- D.-W. Kim, M. Tiseo and M.-J. Ahn, et al., Brigatinib (BRG) in patients (pts) with crizotinib (CRZ)-refractory ALK+ non-small cell lung cancer (NSCLC): First report of efficacy and safety from a pivotal randomized phase (ph) 2 trial (ALTA), J. Clin. Oncol., 2016, 34, 9007 Search PubMed.
- https://clinicaltrials.gov/show/NCT02737501, (accessed on May 13, 2017).
- M. Cheng, M. R. Quail, D. E. Gingrich, G. R. Ott, L. Lu, W. Wan, M. S. Albom, T. S. Angeles, L. D. Aimone, F. Cristofani, R. Machiorlatti, C. Abele, M. A. Ator, B. D. Dorsey, G. Inghirami and B. A. Ruggeri, Mol. Cancer Ther., 2012, 11, 670–679 CrossRef CAS PubMed.
- M. J. Jacobs, G. R. Ott and L. Courvoisier, WO 2013134353 A1, 2013.
- G. R. Ott, M. Cheng, K. S. Learn, J. Wagner, D. E. Gingrich, J. G. Lisko, M. Curry, E. F. Mesaros, A. K. Ghose, M. R. Quail, W. Wan, L. Lu, P. Dobrzanski, M. S. Albom, T. S. Angeles, K. Wells-Knecht, Z. Huang, L. D. Aimone, E. Bruckheimer, N. Anderson, J. Friedman, S. V. Fernandez, M. A. Ator, B. A. Ruggeri and B. D. Dorsey, J. Med. Chem., 2016, 59, 7478–7496 CrossRef CAS PubMed.
- Information from the National Library of Medicine, “To determine the maximum tolerated dose of oral CEP-37440 in patients with advanced or metastatic solid tumors”, https://clinicaltrials.gov/ct2/show/NCT01922752, (accessed on March 2018).
- S. N. Gettinger, L. A. Bazhenova, C. J. Langer, R. Salgia, K. A. Gold, R. Rosell, A. T. Shaw, G. J. Weiss, M. Tugnait, N. I. Narasimhan, D. J. Dorer, D. Kerstein, V. M. Rivera, T. Clackson, F. G. Haluska and D. R. Camidge, Lancet Oncol., 2016, 17, 1683–1696 CrossRef CAS PubMed.
- K. Uchibori, N. Inase, M. Araki, M. Kamada, S. Sato, Y. Okuno, N. Fujita and R. Katayama, Nat. Commun., 2017, 8, 14768 CrossRef CAS PubMed.
- A. Markham, Drugs, 2017, 77, 1131–1135 CrossRef CAS PubMed.
- T. O'Hare, R. Pollock, E. P. Stoffregen, J. A. Keats, O. M. Abdullah, E. M. Moseson, V. M. Rivera, H. Tang, C. A. Metcalf 3rd, R. S. Bohacek, Y. Wang, R. Sundaramoorthi, W. C. Shakespeare, D. Dalgarno, T. Clackson, T. K. Sawyer, M. W. Deininger and B. J. Druker, Blood, 2004, 104, 2532–2539 CrossRef PubMed.
- J. Bain, L. Plater, M. Elliott, N. Shpiro, C. J. Hastie, H. McLauchlan, I. Klevernic, J. Simon, C. Arthur, D. R. Alessi and P. Cohen, Biochem. J., 2007, 408, 297–315 CrossRef CAS PubMed.
- A. S. Corbin, S. Demehri, I. J. Griswold, Y. Wang, C. A. Metcalf 3rd, R. Sundaramoorthi, W. C. Shakespeare, J. Snodgrass, S. Wardwell, D. Dalgarno, J. Iuliucci, T. K. Sawyer, M. C. Heinrich, B. J. Druker and M. W. Deininger, Blood, 2005, 106, 227–234 CrossRef CAS PubMed.
- V. Rivera, R. Anjum, F. Wang, S. Zhang, J. Keats, Y. Ning, S. Wardwell, L. Moran, E. Ye and D. Chun, Cancer Res., 2010, 70, 3623 Search PubMed.
- S. Zhang, F. Wang, J. Keats, Y. Ning, S. D. Wardwell, L. Moran, Q. K. Mohemmad, E. Ye, R. Anjum, Y. Wang, X. Zhu, J. J. Miret, D. Dalgarno, N. I. Narasimhan, T. Clackson, W. C. Shakespeare and V. M. Rivera, 101st AACR Annual Meeting, Washington, DC, USA, 2010, pp. 17–21 Search PubMed.
- Q. Huang, T. W. Johnson, S. Bailey, A. Brooun, K. D. Bunker, B. J. Burke, M. R. Collins, A. S. Cook, J. J. Cui, K. N. Dack, J. G. Deal, Y. L. Deng, D. Dinh, L. D. Engstrom, M. He, J. Hoffman, R. L. Hoffman, P. S. Johnson, R. S. Kania, H. Lam, J. L. Lam, P. T. Le, Q. Li, L. Lingardo, W. Liu, M. W. Lu, M. McTigue, C. L. Palmer, P. F. Richardson, N. W. Sach, H. Shen, T. Smeal, G. L. Smith, A. E. Stewart, S. Timofeevski, K. Tsaparikos, H. Wang, H. Zhu, J. Zhu, H. Y. Zou and M. P. Edwards, J. Med. Chem., 2014, 57, 1170–1187 CrossRef CAS PubMed.
- Y.-C. Kim, S. G. Brown, T. K. Harden, J. L. Boyer, G. Dubyak, B. F. King, G. Burnstock and K. A. Jacobson, J. Med. Chem., 2001, 44, 340–349 CrossRef CAS PubMed.
- J. Xuan, T.-T. Zeng, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem.–Eur. J., 2015, 21, 4962–4965 CrossRef CAS PubMed.
- The U.S. Food and Drug Administration, “Drug Safety and Availability”, https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208772lbl.pdf, accessed on Dec 12, 2017.
- R. Rosell, S. N. Gettinger, L. A. Bazhenova, C. J. Langer, R. Salgia, A. T. Shaw, N. I. Narasimhan, D. J. Dorer, D. Kerstein and D. R. Camidge, J. Thorac. Oncol., 2016, 11, S114 CrossRef CAS PubMed.
- D.-W. Kim, M. Tiseo and M.-J. Ahn, et al., Brigatinib (BRG) in patients (pts) with crizotinib (CRZ)-refractory ALK+ non-small cell lung cancer (NSCLC): first report of efficacy and safety from a pivotal randomized phase (ph) 2 trial (ALTA), ASCO Meet Abstr., 2016, vol. 34(suppl. 15), p. 9060 Search PubMed.
- S. N. Gettinger, S. Zhang, J. G. Hodgson, et al., Activity of brigatinib (BRG) in crizotinib (CRZ) resistant patients (pts) according to ALK mutation status, ASCO Meet Abstr., 2016 May 31, vol. 34(suppl. 15), p. 9060 Search PubMed.
- S. N. Gettinger, S. Zhang and J. G. Hodgson, et al., Activity of brigatinib (BRG) in crizotinib (CRZ) resistant patients (pts) according to ALK mutation status, ASCO Meet Abstr., 2016 May 31, vol. 34(suppl. 15), p. 9060 Search PubMed.
- S. H. Ou, S. J. Klempner, J. R. Greenbowe, M. Azada, A. B. Schrock, S. M. Ali, J. S. Ross, P. J. Stephens and V. A. Miller, J. Thorac. Oncol., 2014, 9, 1821–1825 CrossRef CAS PubMed.
- J. K. Sabari, F. C. Santini, A. M. Schram, I. Bergagnini, R. Chen, C. Mrad, W. V. Lai, K. C. Arbour and A. Drilon, OncoTargets Ther., 2017, 10, 1983 CrossRef PubMed.
- D.-W. Kim, M. Tiseo, M.-J. Ahn, K. L. Reckamp, K. H. Hansen, S.-W. Kim, R. M. Huber, H. J. West, H. J. M. Groen, M. J. Hochmair, N. B. Leighl, S. N. Gettinger, C. J. Langer, L. G. Paz-Ares, E. F. Smit, E. S. Kim, W. G. Reichmann, D. Kerstein, F. G. Haluska and D. R. Camidge, J. Clin. Oncol., 2016, 34, 9007 Search PubMed.
- C. J. Langer, S. N. Gettinger, L. Bazhenova, R. Salgia, K. A. Gold, R. Rosell, A. T. Shaw, G. J. Weiss, D. J. Dorer, V. M. Rivera, F. G. Haluska, D. Kerstein and D. R. Camidge, J. Clin. Oncol., 2016, 34, 9057 Search PubMed.
- J. Zhang, X. Deng, H. G. Choi, D. R. Alessi and N. S. Gray, Bioorg. Med. Chem. Lett., 2012, 22, 1864–1869 CrossRef CAS PubMed.
- S. Kanda and Y. Miyata, Frontiers in Bioscience, 2011, 16, 1024–1035 CrossRef CAS.
- S. Hellwig, C. V. Miduturu, S. Kanda, J. Zhang, P. Filippakopoulos, E. Salah, X. Deng, H. G. Choi, W. Zhou, W. Hur, S. Knapp, N. S. Gray and T. E. Smithgall, Chem. Biol., 2012, 19, 529–540 CrossRef CAS PubMed.
- S. Hellwig, N. S. Gray and T. E. Smithgall, Mol. Cancer Ther., 2014, 10, A234 Search PubMed.
- U. McDermott, S. Sharma, L. Dowell, P. Greninger, C. Montagut, J. Lamb, H. Archibald, R. Raudales, A. Tam, D. Lee, S. Rothenberg, J. Supko, R. Sordella, L. Ulkus, A. Iafrate, S. Maheswaran, C. Njauw, H. Tsao, L. Drew, J. Hanke, X.-J. Ma, M. Erlander, N. Gray, D. Haber and J. Settleman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19936–19941 CrossRef CAS PubMed.
- D. P. Walker, F. C. Bi, A. S. Kalgutkar, J. N. Bauman, S. X. Zhao, J. R. Soglia, G. E. Aspnes, D. W. Kung, J. Klug-McLeod, M. P. Zawistoski, M. A. McGlynn, R. Oliver, M. Dunn, J. C. Li, D. T. Richter, B. A. Cooper, J. C. Kath, C. A. Hulford, C. L. Autry, M. J. Luzzio, E. J. Ung, W. G. Roberts, P. C. Bonnette, L. Buckbinder, A. Mistry, M. C. Griffor, S. Han and A. Guzman-Perez, Bioorg. Med. Chem. Lett., 2008, 18, 6071–6077 CrossRef CAS PubMed.
- R. S. Obach, A. S. Kalgutkar, J. R. Soglia and S. X. Zhao, Chem. Res. Toxicol., 2008, 21, 1814–1822 CrossRef CAS PubMed.
- H. Orhan and N. P. Vermeulen, Curr. Drug Metab., 2011, 12, 383–394 CrossRef CAS PubMed.
- T. A. Baillie, Chem. Res. Toxicol., 2008, 21, 129–137 CrossRef PubMed.
- FDA approved drugs, “FDA broadens ceritinib indication to previously untreated ALK-positive metastatic NSCLC”, https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm560873.htm, accessed on March, 2018.
- K. L. Milkiewicz, L. R. Weinberg, M. S. Albom, T. S. Angeles, M. Cheng, A. K. Ghose, R. C. Roemmele, J. P. Theroff, T. L. Underiner, C. A. Zificsak and B. D. Dorsey, Bioorg. Med. Chem., 2010, 18, 4351–4362 CrossRef CAS PubMed.
- W. Wan, M. S. Albom, L. Lu, M. R. Quail, N. C. Becknell, L. R. Weinberg, D. R. Reddy, B. P. Holskin, T. S. Angeles, T. L. Underiner, S. L. Meyer, R. L. Hudkins, B. D. Dorsey, M. A. Ator, B. A. Ruggeri and M. Cheng, Blood, 2006, 107, 1617–1623 CrossRef CAS PubMed.
- R. Li, L. Xue, T. Zhu, Q. Jiang, X. Cui, Z. Yan, D. McGee, J. Wang, V. R. Gantla, J. C. Pickens, D. McGrath, A. Chucholowski, S. W. Morris and T. R. Webb, J. Med. Chem., 2006, 49, 1006–1015 CrossRef CAS PubMed.
- P. Sabbatini, S. Korenchuk, J. L. Rowand, A. Groy, Q. Liu, D. Leperi, C. Atkins, M. Dumble, J. Yang, K. Anderson, R. G. Kruger, R. R. Gontarek, K. R. Maksimchuk, S. Suravajjala, R. R. Lapierre, J. B. Shotwell, J. W. Wilson, S. D. Chamberlain, S. K. Rabindran and R. Kumar, Mol. Cancer Ther., 2009, 8, 2811–2820 CrossRef CAS PubMed.
- J. G. Christensen, H. Y. Zou, M. E. Arango, Q. Li, J. H. Lee, S. R. McDonnell, S. Yamazaki, G. R. Alton, B. Mroczkowski and G. Los, Mol. Cancer Ther., 2007, 6, 3314–3322 CrossRef CAS PubMed.
- R. Li and S. W. Morris, Med. Res. Rev., 2008, 28, 372–412 CrossRef CAS PubMed.
- C. C. Lee, Y. Jia, N. Li, X. Sun, K. Ng, E. Ambing, M. Y. Gao, S. Hua, C. Chen, S. Kim, P. Y. Michellys, S. A. Lesley, J. L. Harris and G. Spraggon, Biochem. J., 2010, 430, 425–437 CrossRef CAS PubMed.
- R. T. Bossi, M. B. Saccardo, E. Ardini, M. Menichincheri, L. Rusconi, P. Magnaghi, P. Orsini, N. Avanzi, A. L. Borgia, M. Nesi, T. Bandiera, G. Fogliatto and J. A. Bertrand, Biochemistry, 2010, 49, 6813–6825 CrossRef CAS PubMed.
- D. E. Gingrich, J. G. Lisko, M. A. Curry, M. Cheng, M. Quail, L. Lu, W. Wan, M. S. Albom, T. S. Angeles, L. D. Aimone, R. C. Haltiwanger, K. Wells-Knecht, G. R. Ott, A. K. Ghose, M. A. Ator, B. Ruggeri and B. D. Dorsey, J. Med. Chem., 2012, 55, 4580–4593 CrossRef CAS PubMed.
- D. E. Gingrich, J. G. Lisko, M. A. Curry, M. Cheng, M. Quail, L. Lu, W. Wan, M. S. Albom, T. S. Angeles, L. D. Aimone, R. C. Haltiwanger, K. Wells-Knecht, G. R. Ott, A. K. Ghose, M. A. Ator, B. Ruggeri and B. D. Dorsey, J. Med. Chem., 2012, 55, 4580–4593 CrossRef CAS PubMed.
- M. Cheng, M. Quail, D. Gingrich, G. Ott, L. Lu, W. Wan, M. Albom, T. Angeles, L. Aimone, F. Cristofani, R. Machiorlatti, C. Abele, M. Ator, B. Dorsey, G. Inghirami and B. Ruggeri, Mol. Cancer Ther., 2012, 11, 670–679 CrossRef CAS PubMed.
- S. P. Allwein, R. C. Roemmele, J. J. Haley, D. R. Mowrey, D. E. Petrillo, J. J. Reif, D. E. Gingrich and R. P. Bakale, Org. Process Res. Dev., 2012, 16, 148–155 CrossRef CAS.
- G. R. Ott, R. Tripathy, M. Cheng, R. McHugh, A. V. Anzalone, T. L. Underiner, M. A. Curry, M. R. Quail, L. Lu, W. Wan, T. S. Angeles, M. S. Albom, L. D. Aimone, M. A. Ator, B. A. Ruggeri and B. D. Dorsey, ACS Med. Chem. Lett., 2010, 1, 493–498 CrossRef CAS PubMed.
- M. Cheng and G. R. Ott, Anti-Cancer Agents Med. Chem., 2010, 10, 236–249 CrossRef CAS PubMed.
- T. J. Liu, T. LaFortune, T. Honda, O. Ohmori, S. Hatakeyama, T. Meyer, D. Jackson, J. de Groot and W. K. Yung, Mol. Cancer Ther., 2007, 6, 1357–1367 CrossRef CAS PubMed.
- E. F. Mesaros, J. P. Burke, J. D. Parrish, B. J. Dugan, A. V. Anzalone, T. S. Angeles, M. S. Albom, L. D. Aimone, M. R. Quail, W. Wan, L. Lu, Z. Huang, M. A. Ator, B. A. Ruggeri, M. Cheng, G. R. Ott and B. D. Dorsey, Bioorg. Med. Chem. Lett., 2011, 21, 463–466 CrossRef CAS PubMed.
- E. F. Mesaros, J. P. Burke, J. D. Parrish, B. J. Dugan, A. V. Anzalone, T. S. Angeles, M. S. Albom, L. D. Aimone, M. R. Quail, W. Wan, L. Lu, Z. Huang, M. A. Ator, B. A. Ruggeri, M. Cheng, G. R. Ott and B. D. Dorsey, Bioorg. Med. Chem. Lett., 2011, 21, 463–466 CrossRef CAS PubMed.
- S. P. Allwein, D. R. Mowrey, D. E. Petrillo, J. J. Reif, V. C. Purohit, K. L. Milkiewicz, R. P. Bakale, M. A. Christie, M. A. Olsen, C. J. Neville and G. J. Gilmartin, Org. Process Res. Dev., 2017, 21, 740–747 CrossRef CAS.
- S. P. Allwein, R. P. Bakale, D. R. Mowrey, D. E. Petrillo and S. Kluwer, PCT Int. Appl., WO 2016105529 A1 20160630, 2016.
- J. Zhao and J. L. Guan, Cancer Metastasis Rev., 2009, 28, 35–49 CrossRef PubMed.
- J. Schwock, N. Dhani and D. W. Hedley, Expert Opin. Ther. Targets, 2010, 14, 77–94 CrossRef CAS PubMed.
- F. J. Sulzmaier, C. Jean and D. D. Schlaepfer, Nat. Rev. Cancer, 2014, 14, 598–610 CrossRef CAS PubMed.
- B. Y. Lee, P. Timpson, L. G. Horvath and R. J. Daly, Pharmacol. Ther., 2015, 146, 132–149 CrossRef CAS PubMed.
- I. Salem, M. Alsalahi, I. Chervoneva, L. D. Aburto, S. Addya, G. R. Ott, B. A. Ruggeri, M. Cristofanilli and S. V. Fernandez, Breast Cancer Res., 2016, 18, 37 CrossRef PubMed.
- G. Sliwoski, S. Kothiwale, J. Meiler and E. W. Lowe, Pharmacol. Rev., 2014, 66, 334 CrossRef PubMed.
- T. Usha, D. Shanmugarajan, A. K. Goyal, C. S. Kumar and S. K. Middha, Curr. Top. Med. Chem., 2017, 17, 3296–3307 CrossRef CAS PubMed.
- M. Zheng, J. Zhao, C. Cui, Z. Fu, X. Li, X. Liu, X. Ding, X. Tan, F. Li, X. Luo, K. Chen and H. Jiang, Med. Res. Rev., 2018, 38, 914–950, DOI:10.1002/med.21483.
- N. James, V. Shanthi and K. Ramanathan, Appl. Biochem. Biotechnol., 2017 DOI:10.1007/s12010-017-2650-x.
- N. James and K. Ramanathan, Cell Biochem. Biophys., 2017, 1–27, DOI:10.1007/s12013-017-0800-y.
- W. Fulong, L. Qianqian, W. Zhonghua, L. Dandan, P. Peng, Y. Yan, C. Siheng and W. Fanhong, Lett. Drug Des. Discovery, 2017, 14, 270–286 CrossRef.
- H. Z. Xie, H. Lan, Y. L. Pan, J. Zou, Z. R. Wang, L. L. Li, Q. Huang, H. Zhang and S. Y. Yang, Chem. Biol. Drug Des., 2013, 81, 175–184 CAS.
- S. J. Macalino, V. Gosu, S. Hong and S. Choi, Arch. Pharmacal Res., 2015, 38, 1686–1701 CrossRef CAS PubMed.
- W. Lu, R. Zhang, H. Jiang, H. Zhang and C. Luo, Front. Chem., 2018, 6, 57 CrossRef PubMed.
- N. James and K. Ramanathan, Appl. Biochem. Biotechnol., 2018, 1–24, DOI:10.1007/s12010-018-2724-4.
- C. H. Kang, J. I. Yun, K. Lee, C. O. Lee, H. K. Lee, C. S. Yun, J. Y. Hwang, S. Y. Cho, H. Jung, P. Kim, J. D. Ha, J. H. Jeon, S. U. Choi, H. G. Jeong, H. R. Kim and C. H. Park, Biochem. Biophys. Res. Commun., 2015, 464, 762–767 CrossRef CAS PubMed.
- G. R. Mathi, C. H. Kang, H. K. Lee, R. Achary, H. Y. Lee, J. Y. Lee, J. D. Ha, S. Ahn, C. H. Park, C. O. Lee, J. Y. Hwang, C. S. Yun, H. J. Jung, S. Y. Cho, H. R. Kim and P. Kim, Eur. J. Med. Chem., 2017, 126, 536–549 CrossRef CAS PubMed.
- Y. Wang, S. Chen, G. Hu, J. Wang, W. Gou, D. Zuo, Y. Gu, P. Gong and X. Zhai, Eur. J. Med. Chem., 2018, 143, 123–136 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |