DOI:
10.1039/C8RA01891J
(Paper)
RSC Adv., 2018,
8, 18364-18371
Synthesis of MoS2 nanosheets for mercury speciation analysis by HPLC-UV-HG-AFS
Received
4th March 2018
, Accepted 11th May 2018
First published on 18th May 2018
Abstract
Mercury species have aroused wide concern in the past several decades due to their high toxicity. However, it is still difficult to detect ultra-trace mercury species due to their biochemical transformation in complex samples. To establish a simpler and more sensitive method for pre-concentration and determination of trace mercury species, molybdenum disulfide (MoS2) nanosheets with sulfur-rich characteristics and enlarged interlayer spacing were prepared by a hydrothermal method coupled with a sonication-assisted liquid exfoliation method and acted as solid-phase extraction adsorbent. The nano-MoS2 had high adsorption capacity, fast adsorption rate and excellent selectivity towards mercury ions (Hg2+), methyl mercury (MeHg+) and ethyl mercury (EtHg+) in a wide pH range and complex matrices. And it could be easily regenerated by 4 mol L−1 HCl and reused several times. After optimizing HPLC-UV-HG-AFS conditions, a great linearity (1.0–10.0 μg L−1, R2 = 0.999 for Hg2+, MeHg+ and EtHg+), lower detection limits (0.017, 0.037 and 0.021 ng mL−1 for Hg2+, MeHg+ and EtHg+, respectively), relative standard deviations (<5%) and addition recoveries of the samples within 82.75–113.38% were observed. In summary, trace inorganic and organic mercury species in environmental and biological samples could be selectively enriched by the prepared nano-MoS2 and efficiently seperated and detected by HPLC-UV-HG-AFS. The present study will help provide a better strategy for environmental monitoring and health assessment of mercury pollutants.
1. Introduction
Mercury compounds are dangerous pollutants due to their bio-accumulation and persistence in the environment and ecosystems.1 Different mercury species have dissimilar toxicity, and different transport pathways and biogeochemical behaviours. Inorganic mercury can be transformed into more toxic organic mercury, which is easier to accumulate in the human body by the respiratory and gastrointestinal tract, and then damage the central nervous system.2 Therefore, it is urgent to develop sensitive methods for speciation analysis of mercury in environmental and biological samples to determine their different risks to public health.
During the past few decades, atomic adsorption spectrometry (AAS),3 atomic emission spectrometry (AES),4 atomic fluorescence spectrometry (AFS)5 and inductively coupled plasma mass spectrometry (ICP-MS)6 are usually used to detect total mercury species but not a particular form. To distinguish different mercury species, hyphenated methods were developed. High performance liquid chromatography (HPLC)-AFS was one of the most widely used methods due to its simple operation, high sensitivity, strong anti-interference ability and wide accessibility to instruments.7 However, the concentrations of organic mercury in environmental and biological samples are too low to reach the detection limits of the HPLC-AFS system. Thus, pre-concentration plays a vital role in analysis of ultra-trace mercury species.
Previous studies have shown that solid phase extraction (SPE) had higher selectivity and good reproducibility compared with liquid–liquid extraction.8 Various materials9,10 have been used as adsorbents for mercury species, Cd(II), Zn(II), Ni(II) and many other metal species, including thiol-functionalized mesoporous silica (FMMS),11,12 thiol-functionalized graphene oxide/Fe–Mn composite (SGO/Fe–Mn),13 two-dimensional transition-metal dichalcogenides (TMDs),14,15 ion-imprinted magnetic nanoparticles (IIMN)16 and graphene/biochar composite (G/BC).17 As a typical TMD, molybdenum disulfide (MoS2) has been used in the preconditioning of environmental samples due its few-layered structure, high special surface area, abundant binding sites (sulfur atoms) and strong affinity towards Hg0 and Hg2+.18,19 However, nano-MoS2 for simultaneously enriching inorganic and organic mercury species in environmental and biological samples has not been reported. In the present study, a hydrothermal method in combination with sonication-assisted liquid exfoliation was used to obtain nano-MoS2 with fewer-layers, expanded inter-layer distance and abundant active sites. High adsorption and desorption efficiency were also achieved by adjusting physicochemical parameters. Finally, a method of HPLC-UV irradiation (UV)-hydride generation (HG)-AFS was developed for detecting trace mercury species in real samples.
2. Experimental
2.1 Reagents and materials
Thiourea and sodium molybdate dehydrate (Na2MoO4·2H2O, 99.0%), acetic ammonium (CH3COONH4, 98.0%), potassium hydroxide (KOH, 85.0%), sodium hydroxide (NaOH, 98.0%), potassium persulfate (K2S2O8, 99.0%), hydrochloric acid (HCl, 36–38%) and sodium borohydride (NaBH4, 97.0%) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Beijing, China) and they were analytical reagent grade. L-cysteine (98.5%) and methanol (chroma-pure, 99.9%) were obtained from Sigma-Aldrich reagents Co., Ltd. (MO, USA). The standard solution of mercury ion (Hg2+, GBW08617), methylmercury (MeHg+, GBW08675) and ethyl mercury (EtHg+, GBW(E)081524) were purchased from the National Research Center for certified reference materials (Beijing, China). All reagents used in the experiments were prepared by ultrapure water (18.2 MΩ cm).
2.2 Apparatus and HPLC-UV-HG-AFS analysis procedure
Different mercury species were measured by HPLC-AFS (SA-20 and AFS-922, Beijing JiTian Apparatus Co., Ltd, China). The HPLC system was equipped with quaternary pump, degasser, manual stainless sampler injector and 100 μL sample loop. Ultrasonic apparatus (KH-500DB, China) was used for nano-MoS2 synthesis; field emission scanning electron microscope (FESEM, Zeiss merlin, Germany), JEM-2100 High resolution transmission electron microscopy (HRTEM, JEOL, Japan), atomic force microscopy (AFM, NT-MDT Prima, RUS), X-ray diffraction (XRD, Bruker AXS, DEU), X-ray photoelectron spectroscopy ESCALAB 250Xi Instrument (XPS, Thermo, USA) and Raman HORIBA evolution microscopy (HORIBA, FRA) were employed to make MoS2 characterization.
Procedure of HPLC-UV-HG-AFS: Step 1: separation, mercury species (inorganic and organic mercury) were separated through an Athena C18 column (250 × 4.6 mm, 5 μm, CNW, Germany), an isocratic elution profile was used for chromatographic separation of mercury species. Step 2: oxidization, all mercury species were converted to inorganic mercury after oxidization and the irradiation of UV lamp (150 W). Step 3: detection, elemental mercury vapour was purged into the gas–liquid separator and dragged into the detector by argon stream and the detection limits were 0.165, 0.190, 0.192 μg L−1 for Hg2+, MeHg+ and EtHg+, respectively. Other instrument conditions can be seen in Table 1.
Table 1 Experimental conditions of HPLC-HG-UV-AFS
| Instrument conditions |
Parameters |
| Column and sample injection volume |
Athena-C18 column (250 × 4.6 mm, 5 μm), 100 μL |
| Mobile phase composition and flow rate |
CH3OH 10%/90% H2O – 0.46% NH4Ac – 0.12% L-cys, 1.0 mL min−1 |
| Photomultiplier tube negative high pressure |
270 V |
| High performance hollow cathode lamp current |
30 mA |
| Atomic heater height |
8 mm |
| Carrier and shielded argon flow |
300 and 600 mL min−1 |
| Carrying current |
2% HNO3 (v/v) |
| Reducing agent |
2% NaBH4 (m/v)–0.5% NaOH (m/v) |
| Oxidizing agent |
0.5% K2S2O4 (m/v)–0.25% KOH (m/v) |
2.3 Preparation and characterization of nano-MoS2
2.3.1 Preparation of adsorbent. After comparing the simplicity and effectiveness of hydrothermal,20,21 chemical vapour deposition,22–24 ion-intercalated exfoliation25,26 and sonication or microwave-assisted exfoliation methods,27,28 hydrothermal method in combination with sonication-assisted liquid exfoliation method was used to synthesize MoS2 with expanded interlayer spacing. 4.959 g thiourea and 3.678 g Na2MoO4·2H2O were dissolved into 75 mL ultrapure water and heated at 180 °C for 24 h. After cooling to room temperature, the obtained products were placed under sonication at 150 W for 12 h. Finally, the materials were washed with ultrapure water for several times and heated at 60 °C overnight.
2.3.2 Adsorption and desorption. 100 μg MoS2 was added into 5 mL ultrapure water containing 200 μg mercury species after the pH was adjusted to 5.0 ± 0.1 by 0.1 mol L−1 NaOH and HNO3. Adsorption was conducted by shaking at 500 rpm and 25 ± 1 °C for 10 min. After reaching to equilibrium, MoS2 was separated by centrifugation and the supernatant was used to measure mercury species by HPLC-UV-HG-AFS. In terms of desorption process, 5 mL HCl solution as eluent was added into the precipitation and then vortexed at 1000 rpm and 25 ± 1 °C for 10 min. Then the eluent was obtained by centrifugation for subsequent HPLC-UV-HG-AFS analysis. The blank test was also conducted in the same process.The adsorption kinetic and isotherm researches were designed according to previous study.29 In the adsorption kinetic experiments, 1.0 mg nano-MoS2 was added into 5 mL 200 mg L−1 Hg2+, 2 mg L−1 MeHg+ and 2 mg L−1 EtHg+ solution with pH values of 5.0 ± 0.1 and adsorbed for different time intervals (0, 1, 5, 10, 20, 30, 40, 50, 60 min). Then, MoS2 was separated and the concentrations of mercury species in supernatant liquid were measured. In the adsorption isotherm experiments, 1.0 mg MoS2 was added into 5 mL Hg2+ and alkyl mercury aqueous solution (0, 50, 100, 200, 250, 300, 350, 400 mg L−1 for Hg2+ and 0, 10, 20, 30, 40, 50 mg L−1 for MeHg+, MeHg+ was selected to represent alkyl mercury in this experiment). Higher concentration of Hg2+ should be avoided to form Hg(OH)2, which may result in overestimating the adsorption capacity of MoS2. After enough adsorption time, the concentrations of mercury species in supernatant liquid were also measured.
2.3.3 Effects of pH and co-existing metal ions. 1.0 mg MoS2 was added into 10 mL 100 mg L−1 Hg2+, 1 mg L−1 MeHg+ and 1 mg L−1 EtHg+ solution and adsorbed for 10 min at the pH values ranged from 3.0 to 10.0. Some several metal ions, which usually present together with inorganic and organic mercury species in environmental and biological samples, were selected to add into the mercury species solutions, including alkali metal ions and heavy metal ions (0–20 mg L−1 K+, Na+, Ca2+, Mg2+, Cu2+, Fe3+, Pb2+, Ag+, Cd2+, As3+). After saturated adsorption time, the concentrations of mercury species in the supernatant liquid were measured.
2.4 HPLC-UV-HG-AFS analysis and quality control
Tap water, Tianyuan Lake water and fish tissue were collected from local laboratory and market (Nanjing, China) as real samples. The water samples were collected with glass containers, which were soaked with 10% nitric acid for 24 h and washed with ultrapure water for several times before sampling. The sampler was immersed in a certain depth underwater to avoid pollutant on sediment and lake surface for the reason that mercury species are easily adsorbed on suspended solids. 5% nitric acid and 0.5% potassium permanganate as stabilizer were added into water samples, and the solution was filtered through 0.45 μm microporous membrane before test. Solid samples were washed with ultrapure water, cut and immersed in 5 mol L−1 hydrochloric acid for 24 h and then shaking at 500 rpm for 1 h, finally the extracting solution was filtered through 0.45 μm membrane and pH was adjusted to 5.0 ± 0.1 by NaOH. Subsequently, the mercury species in these samples were enriched by prepared nano-MoS2 and then injected into HPLC-UV-HG-AFS for separation and detection. The blank experiments were also performed at the same time.
According to IUPAC, the detection limit was defined to be LOD = 3 × SD/K, where SD is the standard deviation of signal values obtained by measuring blank sample for six times, K is the slope of the standard curve. The relative standard deviations (RSDs) of this method for Hg2+, MeHg+ and EtHg+ were evaluated at concentration of 2 μg L−1, 6 μg L−1 and 10 μg L−1, respectively. Low (0.5 μg L−1), medium (1.0 μg L−1) and high (1.5 μg L−1) mercury species standard solution were added into the samples to calculate their recoveries, which could be used to estimate the accuracy.
3. Results and discussion
3.1 Characterization of MoS2 nanomaterials
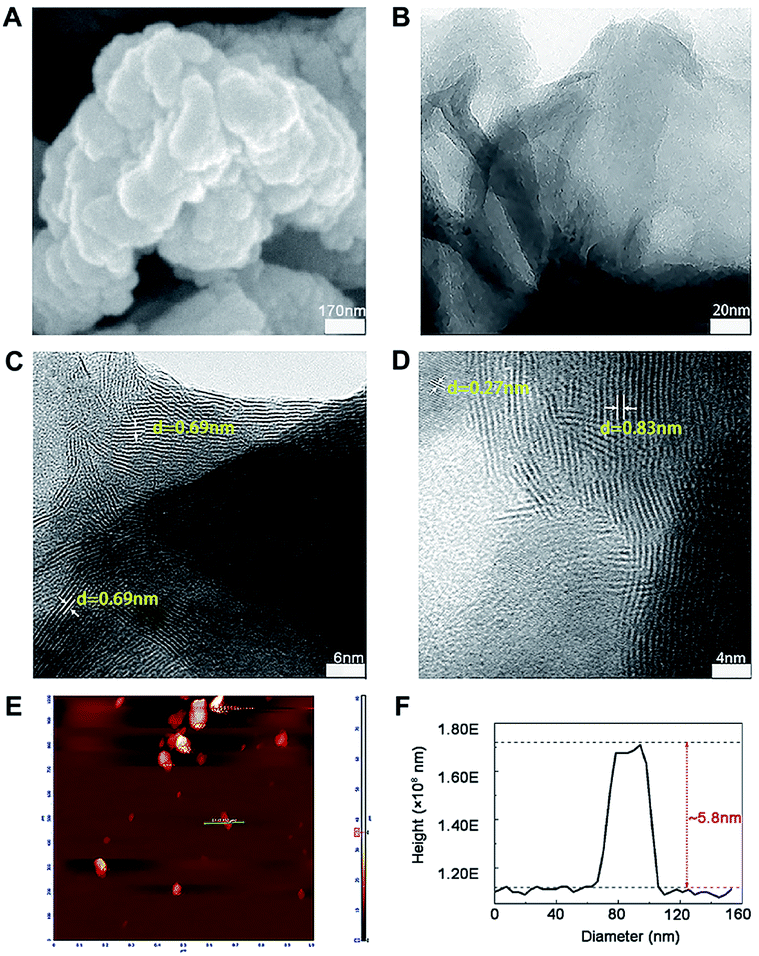
The morphology and microstructure of prepared MoS2 were achieved from the SEM, TEM and HRTEM images (Fig. 1A–D). The diameter of MoS2 ranged from hundreds of nano-meters to micro-meters (Fig. 1A and B). The HRTEM images clearly showed the S–Mo–S layer had a curved stripe-like feature and the distance between layers was 0.69–0.83 nm (Fig. 1C and D), which was in good agreement with the typical thickness of monolayer MoS2,30 indicating these nano-MoS2 had expanded interlayer spacing. The cross section analysis along with the line was conducted based on the AFM image according to previous study.31 As shown in Fig. 1E, MoS2 had thin thickness at around several nano-meters, the step height between the substrate and the nanosheets was around 5.8 nm (Fig. 1F), indicating the nano-MoS2 might contain eight-layers for the monolayer thickness was 0.69 nm.
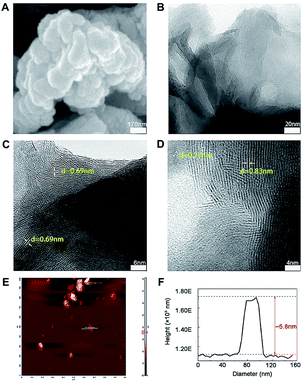 |
| | Fig. 1 Representative images of nano-MoS2 from (A) scanning electron microscope, (B) transmission electron microscope, (C and D) different versions of high resolution transmission electron microscope and (E) atomic force microscope. (F) The thickness of nano-MoS2 was obtained from atomic force microscope. | |
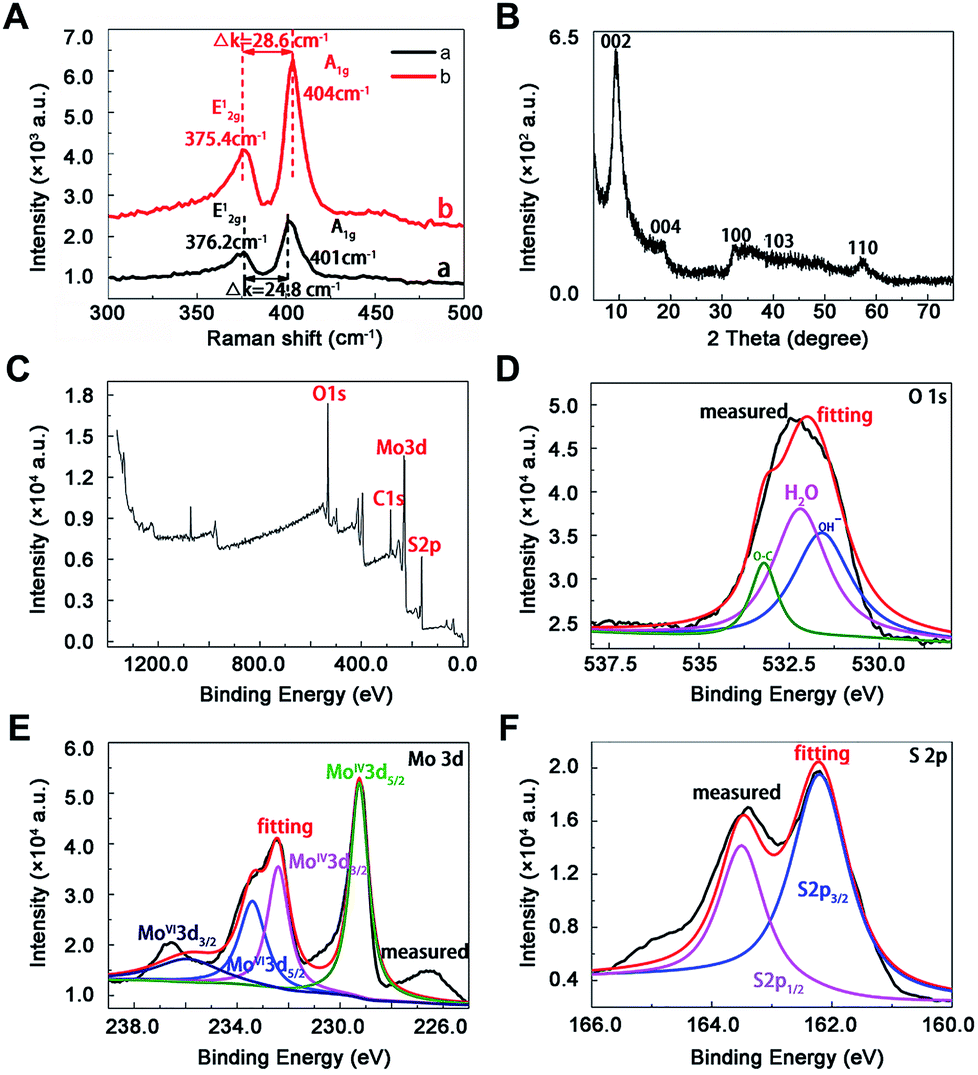
As shown in Raman spectra (Fig. 2A), there were two characteristic peaks at the wavenumber of 376.2 and 401 cm−1, which were consistent with the E12g and A1g mode of typical MoS2 layered structure, where E12g mode represents in-plane motion and A1g mode represents out-of-plane motion.32 The distance between the E12g and A1g peaks could indicate the variation in thickness. The frequency of A1g modes had a bit red shift after sonication for 12 h (curve a and b in Fig. 2A), indicating the thickness of MoS2 decreased with liquid exfoliation but in-plane structure had no change.33
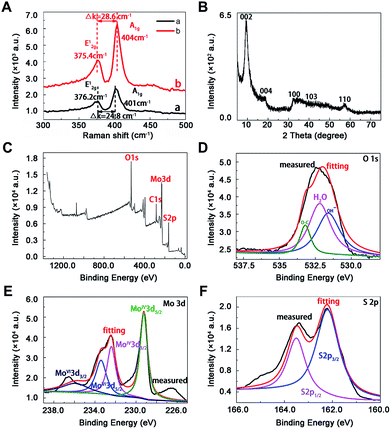 |
| | Fig. 2 Characterization of MoS2 by spectra. (A) Raman spectra of nano-MoS2 with (curve a) and without (curve b) sonication, (B) X-ray diffraction pattern of nan0-MoS2, X-ray photoelectron spectroscopy spectra of nano-MoS2 in the (C) survey, (D) O 1s, (E) Mo 3d and (F) S 2p region. | |
Towards the XRD pattern (Fig. 2B), there was a significant difference between prepared nanomaterials and commercial bulk 2H-MoS2 (JCPDS 77-1716) and 2H-MoS2 crystal (PDF 01-087-2416), one peak appeared at low-angle region (10°) corresponding to (002) reflection with d-spacing values of 8.3 Å, which could be ascribed to oxygen incorporation and indicated an enlarged interlayer spacing of MoS2. This result agreed well with above HRTEM image. The distance value was estimated to be near 5.13 Å according to the interlayer spacing value (8.3 Å) and the thickness (3.17 Å) of the S–Mo–S structure,30 which was larger than the values of bulk MoS2 (2.98 Å) and other nano-MoS2 with lithium ion intercalated (3.18–3.73 Å),21 thus the interlayer spacing of the prepared materials was enough to embed mercury ions. In this way, highly crystalline nano-MoS2 with expanded inter-layer spacing had been synthesized successfully.
The chemical states of O, Mo and S atoms were obtained in XPS spectra (Fig. 2C–F). The O1s results might derive from the minor oxidation in the natural formation process (Fig. 2D), and the binding energies of Mo and S were in good agreement with those of commercial MoS2, where 232.5 eV and 229.4 eV belonged to Mo3d3/2 and Mo3d5/2 (Fig. 2E), 163.5 eV and 162.2 eV belonged to S2p1/2 and S2p3/2 in nano-MoS2 (Fig. 2F). Meanwhile, some newly appeared peaks at 233.5 eV and 236 eV belonged to Mo3d5/2 and Mo3d3/2 in MoS3, probably due to partial surface oxidation upon exposure to air in the process of exfoliation.34
3.2 Pre-concentration conditions of mercury species
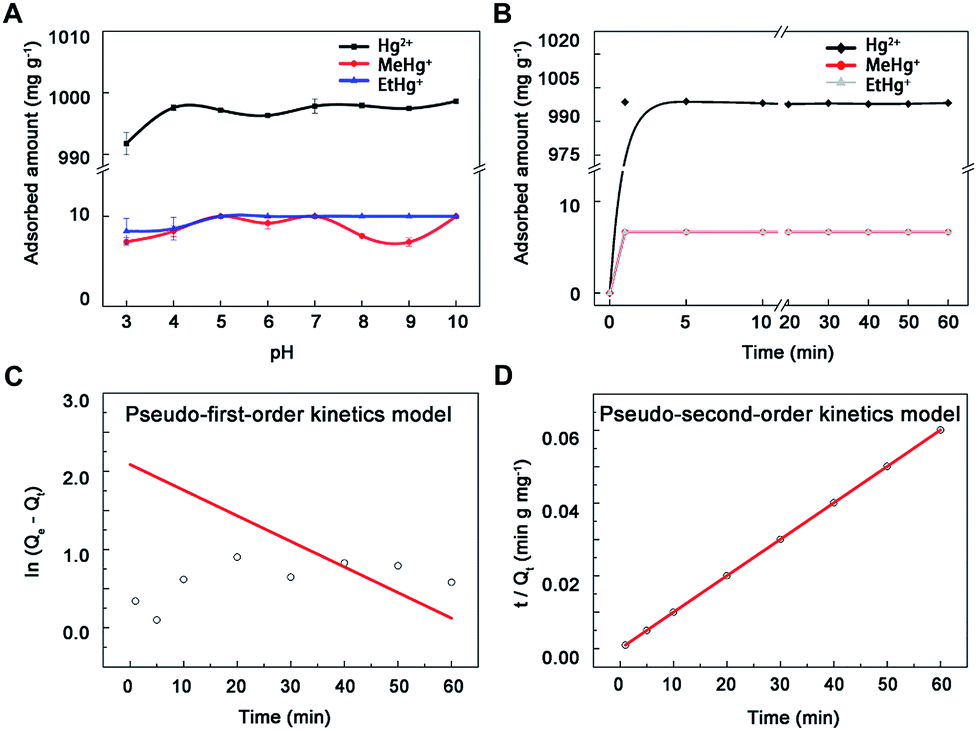
3.2.1 Experiment of pH. Nano-MoS2 exhibited high adsorption capacity to Hg2+, MeHg+ and EtHg+ in the whole pH range (Fig. 3A). The adsorption amount of MoS2 increased slightly within 3.0–5.0 and then kept at a volatile balance with pH increasing to 7.0. Previous study had shown that the surface of MoS2 was negative and the zeta potential decreased along with the increases of pH values.29 Thus, these results could be ascribed to two hypotheses: one is electrostatic attraction between the negatively charged MoS2 and positively charged mercury species, another is competitive binding between hydrogen ions and mercury species to active sites on the surface of MoS2. The electrostatic interaction will increase along with the increasing of pH before 5.0. However, low pH values provided excess H+ to compete with Hg2+ for active sites, leading to a lower adsorption capacity. In terms of higher pH values, mercury would be exist in the less positive form of HgOH+ (5.0–7.0) and Hg(OH)2 (7.0–10.0),35 resulting in the balance of adsorption in pH range of 5.0–10.0. Therefore, the pH value only needed a little adjustment to simplify the experimental process and avoid interference.
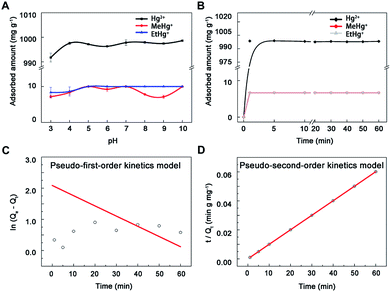 |
| | Fig. 3 The adsorption of mercury species onto nano-MoS2. (A) Effect of pH on mercury species adsorption onto nano-MoS2. (B) Adsorption kinetics spectra of MoS2 towards mercury species, (C) linear fitting results of pseudo-first-order kinetics model and (D) pseudo-second-order kinetics model toward mercury ions. | |
3.2.2 Adsorption kinetics experiment. The adsorption proceeded at a high rate during the first 2 min and reached equilibrium at around 5 min, meanwhile the adsorption percentages were clearly 100% (Fig. 3B), which probably resulted from large surface area and short diffusion path of MoS2.21,36 To further explore the mechanism of the adsorption process, pseudo-first-order (eqn (1)) and pseudo-second-order kinetics models (eqn (2)) were used (mercury ion as a model ion in this study), k1 (min−1) and k2 (mg μg−1 min−1) were the equilibrium rate constants of pseudo-first-order and pseudo-second-order, Qt (μg mg−1) and Qe (μg mg−1) were the adsorbed concentration at time t (min) and equilibrium time. The adsorption kinetics data (R2 = 1) fitted better with the pseudo-second-order model than the other (Fig. 3C and D), suggesting the adsorbent had uniform surface and there was a chemical interaction between adsorbent and adsorbate.14 Additionally, the calculated data (1000 mg g−1 for Hg2+) from the pseudo-second-order curve were consistent with results in this study, indicating the adsorption processes were under chemical process control. Thus, 5 min was selected as adsorption time to ensure complete reaction.| |
ln(Qe − Qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qe − K1t Qe − K1t
| (1) |
| | |
t/Qt = 1/(K2Qe2) + t/Qe
| (2) |
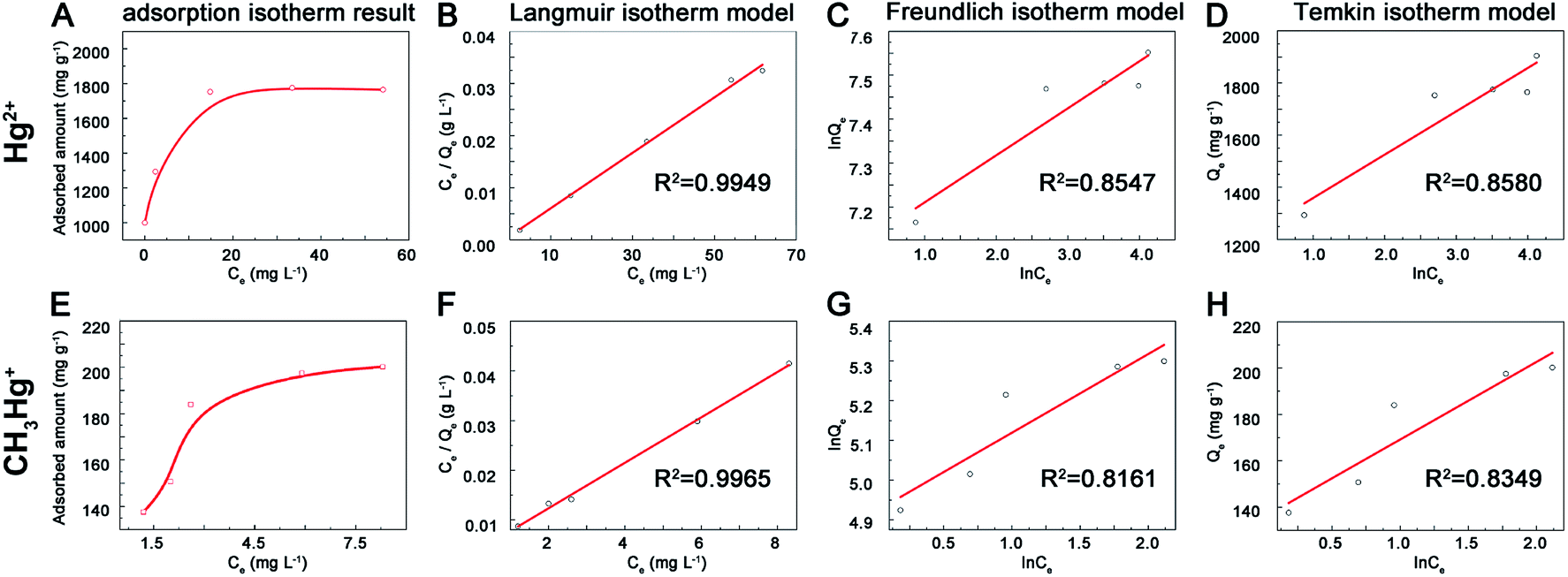
3.2.3 Adsorption isotherms experiment. The adsorption capacity increased along with the initial Hg2+ concentration and then reached to equilibrium (Fig. 4). The maximum adsorption capacities of prepared MoS2 were 1.77 g g−1 for Hg2+ (Fig. 4A) and 200 mg g−1 for MeHg+ (Fig. 4E), which were higher than other adsorbents reported previously (Table 1) and these effects may result from the rich exposure of active sites on the surface of MoS2. To further explore the mechanism of adsorption process, the Langmuir (eqn (3)), Freundlich (eqn (4)) and Temkin (eqn (5)) isotherm models were used. Qe (mg g−1) was the adsorbed concentration at equilibrium time, Qm (mg g−1) was the maximum adsorption capacity of Langmuir model, Ce was the aqueous concentration of adsorption equilibrium, K1, K2 and K3 represented the Langmuir constant (L mg−1), Freundlich constant (mg g−1) and Temkin constant (L g−1), n and B represented the adsorption intensity constant and adsorption heat constant (J mol−1). The isotherms data fitted better with Langmuir model than the other two models for the correlation coefficient (R2 = 0.995, 0.996 for Hg2+ and MeHg+) (Fig. 4B–D and F–H). Furthermore, the calculated data (Qm) (1881.8 and 218.3 mg g−1 for Hg2+ and MeHg+) from the Langmuir curves were close to the results data, indicating the adsorption was monolayer. According to previous study,21 Hg2+ could spontaneously intercalate into S–Mo–S layers and then interact with S atoms to form Hg–S species. In terms of organic mercury species, previous study11 suggested that MeHg+ and EtHg+ could react with sulfhydryl group, but the sulfhydryl groups had higher affinity to divalent Hg2+ rather than monovalent MeHg+ when they were together. The interaction between MeHg+ and MoS2 might ascribe to the electrostatic attraction and binding cooperation. The adsorption capacity of MoS2 towards MeHg+ was less than Hg2+, probably because MeHg+ could only bind to active sites on the surface-layer of MoS2 but not intercalate into interlayers for space steric hindrance effects. Moreover, the actual adsorption capacities were lower than the theoretical values (the theoretical adsorption capacity is estimated to be 2587 mg Hg2+ g−1 MoS2 based on a stoichiometric S/Hg ratio of 1:1) might due to the oxidation of exposed edge sites on MoS2 surface layers.34| | |
Langmuir: Ce/Qe = Ce/Qm + 1/(K1 × Qm)
| (3) |
| | |
Freundlich: lnQe = (lnCe)/n + lnK2
| (4) |
| | |
Temkin: Qe = B × ln(K3 × Ce)
| (5) |
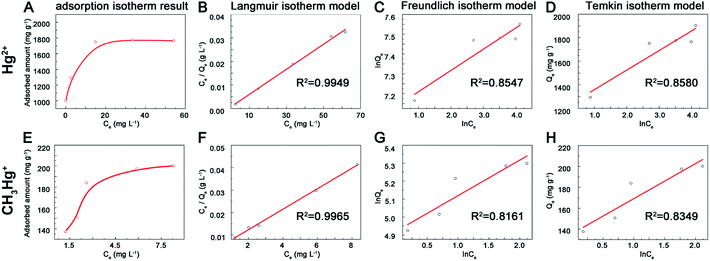 |
| | Fig. 4 Adsorption isotherm spectra of MoS2 towards (A) mercury (E) and methylmercury ions, linear fitting results of Langmuir (B and F), Freundlich (C and G), Temkin (D and H) towards mercury and methyl mercury ions. | |
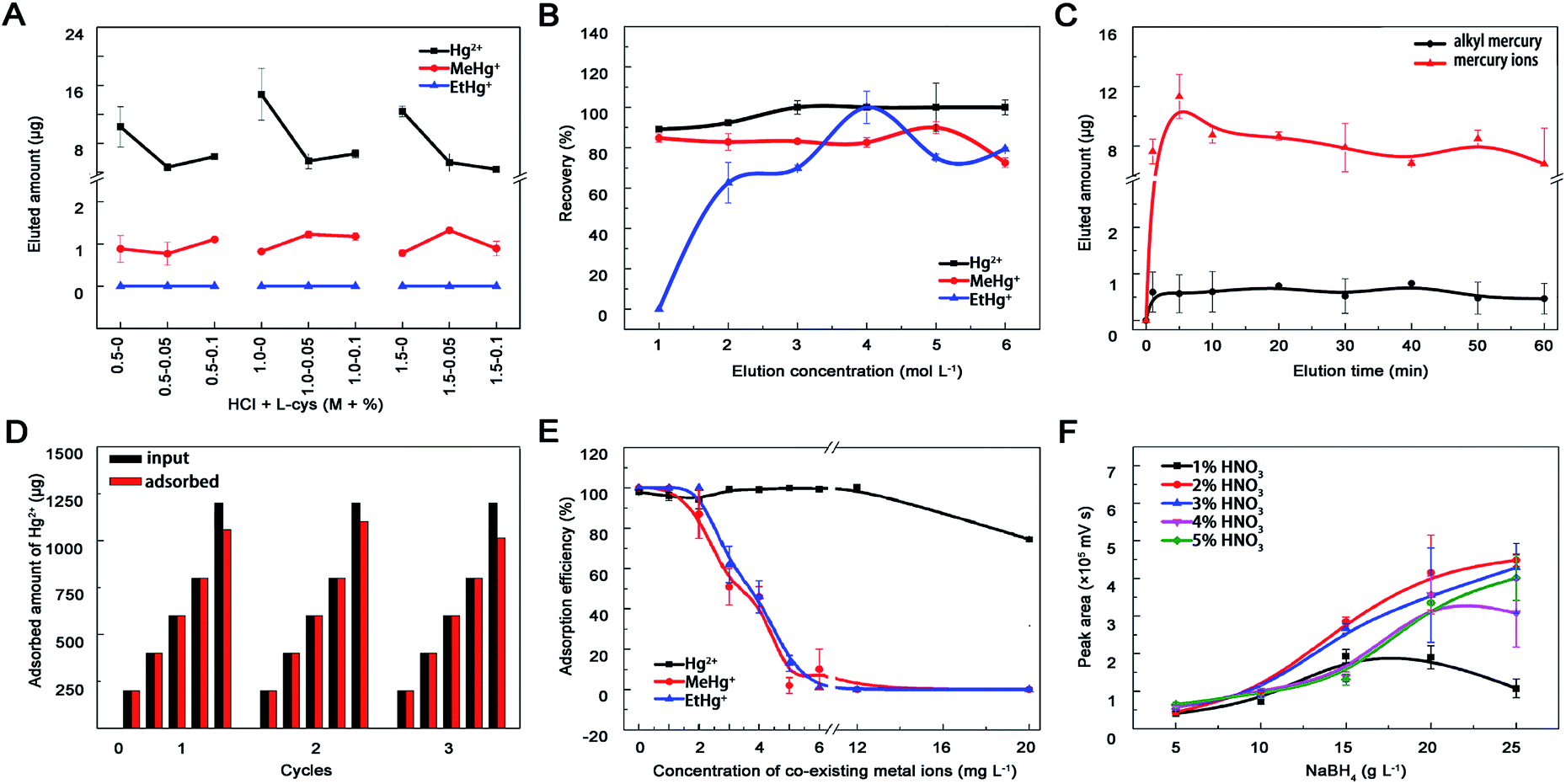
3.2.4 Eluent concentration and elution time. Acid and L-cysteine were chosen as eluents due to effects of pH and mobile phase on elution efficiency.11 Hg2+ was completely eluted by HCl, MeHg+ can quantitatively desorbed with L-cys, while EtHg+ could not be eluted absolutely (Fig. 5A). Progressively, a series concentration of HCl was set out as eluent and these mercury species were almost entirely eluted by 4 mol L−1 HCl (Fig. 5B). Except for eluent type and concentration, elution time may affect the elution efficiency either. The desorption proceeded at a high rate in the first 5 min and reached to equilibrium at around 10 min, meanwhile the recoveries could reach to 90% (Fig. 5C). Therefore, 4 mol L−1 HCl and 10 min elution time were selected for regeneration process and the adsorption capacity of MoS2 towards Hg2+ maintained relatively stable during three recycles of adsorption and regeneration (Fig. 5D).
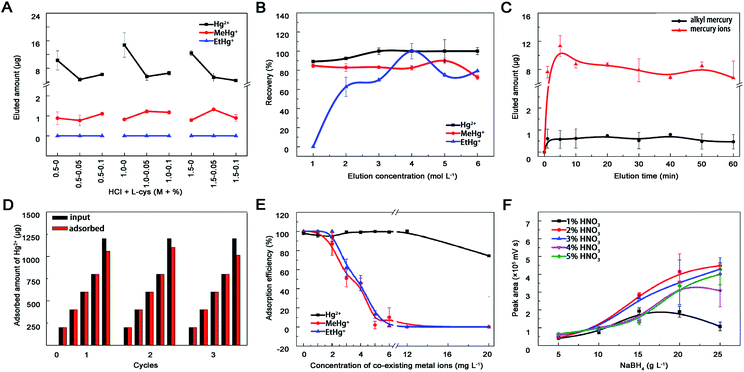 |
| | Fig. 5 Optimization of the experimental conditions. Effects of (A) HCl-L-cys concentration, (B) HCl concentration and (C) elution time on mercury species desorption from nano-MoS2. (D) Adsorption capacity of MoS2 during three cycles of regeneration and reuse. The material was regenerated by introducing 4 mol L−1 HCl. (E) Effect of alkali metal ions (K, Na, Ca, Mg) and heavy metal ions (Cu, Fe, Ag, As, Pb, Cd) on Hg2+ (6.0 mg L−1), MeHg+ (0.2 mg L−1) and EtHg+ (0.2 mg L−1) adsorption onto nano-MoS2 (6 mg L−1). (F) Effects of carrying current and reducing agent ratio on HPLC-AFS signal intensity. Nitric acid acted as carrying current and its' concentration ranged from 1–5%, sodium borohydride acted as reducing agent and its' concentration ranged from 5–25 g L−1. Once one parameter changed, the others would be under their optimal conditions. | |
3.2.5 Interference of co-existing metal ions. Previous studies showed that MoS2 had superior selectivity toward Hg2+ and negligible capture capability for various competitive ions,26 including alkali metal ions (K+, Na+, Ca2+, Mg2+) and heavy metal ions (Cu2+, Fe3+, Pb2+, Ag+, Cd2+, As3+). The adsorption efficiency of MoS2 toward MeHg+ and EtHg+ but not Hg2+ decreased significantly when the concentration of co-existing metal ions was within 2–12 mg L−1 (Fig. 5E), which may result from their similar affinity to the active sites (sulfur atoms), bigger steric hindrance and weaker electrostatic interactions between monovalent alkyl mercury and MoS2. Generally, the concentrations of co-existing heavy metal ions in environmental and biological samples were lower than our experiment settings, therefore this prepared material could be used for enriching mercury species in real samples.
3.3 HPLC-UV-HG-AFS analysis
In this study, mobile phase including 10% methyl alcohol, 0.12% L-cys and 0.46% acetic ammonium severed as organic modifier, chelating agent and buffer to optimize and separate the chromatographic peaks of different mercury species.41 Finally, good resolution (R > 1.5), higher recoveries and shorter analyses time (less than 12 min) were achieved. As shown in Fig. 5F, signal intensity of Hg in AFS increased within 1–2% HNO3 followed by a slightly decrease beyond 5%. In terms of NaBH4, the signal intensity increased with the concentration from 5 to 20 g L−1 and differed within 20–25 g L−1. This probably because the mercury species could not be reduced to Hg0 completely at lower HNO3 and NaBH4 concentrations, but at higher concentrations the mercury vapour would be diluted by the generated hydrogen gas, which could also quench mercury fluorescence. No obvious influence of oxidant concentration on the signal intensity was observed with the help of online decomposed ultraviolet light irradiation system, which could simplify the process and reduce contamination from additional chemicals. Therefore, 20 g L−1 NaBH4, 2% HNO3 and 5 g L−1 K2S2O8 were selected in this study.
The effectiveness and commonality of this established method were further evaluated by comparing LODs with other materials (Table 2), testing linearity, sensitivity, accuracy and reproducibility (Table 3). Then, mercury species in water and fish tissue samples were detected through the whole process and their concentrations were below the maximum level in drinking water (WHO and China: <1 μg L−1; EPA: <2 μg L−1), surface water (EU legislation: <0.07 μg L−1) and fish (China: total Hg < 0.3 μg g−1 wet wt). Based on the recoveries of standard addition (Table 4), the prepared nano-MoS2 may provide a new strategy to detect mercury pollutants in environmental and biological samples by HPLC-UV-HG-AFS.
Table 2 Applications of MoS2 nanoparticles as adsorbent for mercury species
| Chemical species |
Adsorbents |
Samples |
Analytical technique |
Adsorbed amount (mg g−1) |
LOD (μg L−1) |
Ref. |
| Hg2+ |
Au NPs–Al2O3 |
Water |
ICP-MS |
676 |
2.8 × 10−5 |
37 |
| MeHg+ |
215.6 |
6.5 × 10−6 |
| EtHg+ |
245.7 |
2.1 × 10−5 |
| PhHg+ |
277.6 |
1.9 × 10−5 |
| Hg2+ |
Magnetic sulfur-doped porous carbon |
Water |
ICP-MS |
343 |
5.2 × 10−4 |
38 |
| Hg2+ |
Magnetic PPy–GO |
Water |
AAS |
400.0 |
— |
39 |
| MeHg+ |
Fe3O4@SiO2–RSH |
Water |
HPLC-AFS |
14.4 |
0.2 |
11 |
| EtHg+ |
15.0 |
0.3 |
| Hg2+ |
FeCo/GC NCs@MSNs-SHs |
Water |
ICP-AES |
221.4 |
— |
12 |
| Hg2+ |
MoS2 nanosheets |
Water |
ICP-MS |
2563 |
— |
21 |
| Hg2+ |
MoS2 nanosheets |
— |
AAS |
305 |
— |
29 |
| 2D-M-500 |
800 |
| Hg2+ |
Graphene/biochar composite |
Water |
AFS |
16.3 |
2.0 |
17 |
| MeHg+ |
SGO/Fe–Mn-ne |
Water |
AFS |
28.0 |
0.043 |
13 |
| SGO/Fe–Mn-ac |
36.3 |
| SGO/Fe–Mn-am |
43.88 |
| MeHg+ |
Fe3O4@SiO2 NPs |
Aqueous water |
CE-ICP-MS |
25 |
8.4 × 10−5 |
16 |
| Hg2+ |
Fe3O4@SiO2@g-MPTS |
Sea water |
ICP-MS |
83.8 |
1.07 × 10−4 |
40 |
| Hg2+ |
MoS2 |
Water |
HPLC-UV-HG-AFS |
1770 |
0.017 |
This work |
| MeHg+ |
Fish tissue |
200 |
0.037 |
| EtHg+ |
|
|
0.021 |
Table 3 Analysis of mercury species by HPLC-UV-HG-AFS after pre-concentrated by MoS2 adsorbent
| Species |
Hg2+ |
MeHg+ |
EtHg+ |
| Linear regression |
I = 6535.5 × C + 326.7 |
I = 6675.5 × C − 886.0 |
I = 6406.8 × C − 305.2 |
| R2 |
0.999 |
0.999 |
0.999 |
| RSD (%), [6 ng mL−1, n = 6] |
3.9 |
2.5 |
3.8 |
| LOD (ng mL−1) |
0.017 |
0.037 |
0.021 |
Table 4 Determination of mercury species in real sample
| Samples |
Hg2+ |
MeHg+ |
EtHg+ |
| Added (ng mL−1) |
Found (ng mL−1) |
Recovery (%) |
Added (ng mL−1) |
Found (ng mL−1) |
Recovery (%) |
Added (ng mL−1) |
Found (ng mL−1) |
Recovery (%) |
| Not detected. |
| Fish tissue |
0 |
0.4963 |
0 |
0 |
0.0542 |
0 |
0 |
NDa |
0 |
| 0.5 |
0.9971 |
99.86 |
0.5 |
0.4411 |
88.22 |
0.5 |
0.4482 |
89.64 |
| 1.0 |
1.3448 |
84.85 |
1.0 |
0.8500 |
85.00 |
1.0 |
0.8319 |
83.19 |
| 1.5 |
1.8161 |
87.99 |
1.5 |
1.3434 |
89.56 |
1.5 |
1.3600 |
90.67 |
| Tap water |
0 |
0.9876 |
0 |
0 |
ND |
0 |
0 |
ND |
0 |
| 0.5 |
1.4928 |
101.04 |
0.5 |
0.4137 |
82.75 |
0.5 |
0.5654 |
113.08 |
| 1.0 |
1.9363 |
94.86 |
1.0 |
1.0211 |
102.11 |
1.0 |
0.9772 |
97.72 |
| 1.5 |
2.5037 |
101.07 |
1.5 |
1.7007 |
113.38 |
1.5 |
1.6442 |
109.61 |
| Lake water |
0 |
1.0189 |
0 |
0 |
ND |
0 |
0 |
ND |
0 |
| 0.5 |
1.4532 |
86.86 |
0.5 |
0.4229 |
84.58 |
0.5 |
0.4181 |
83.62 |
| 1.0 |
1.8805 |
86.16 |
1.0 |
1.0526 |
105.26 |
1.0 |
0.8451 |
84.51 |
| 1.5 |
2.5001 |
98.75 |
1.5 |
1.6250 |
108.34 |
1.5 |
1.6299 |
108.66 |
4. Conclusions
In this study, a new solid phase extraction method based on nano-MoS2 was established for the enrichment of mercury species in environmental and biological samples. The prepared nano-MoS2 with widened interlayer spacing structure and more exposed sulfur atoms can be served as an excellent adsorbent, which has extraordinary selectivity and high adsorption capacity towards mercury species. The nano-MoS2 coupled with HPLC-UV-HG-AFS offers a better strategy to enhance sensitivity and simplicity for the simultaneous speciation analysis of trace mercury species in real samples, and it has potential significant importance for environmental monitoring and health assessment.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Natural Science Foundations of China (81673228 and 81473020), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (2014).
Notes and references
- P. A. Ariya, M. Amyot, A. Dastoor, D. Deeds, A. Feinberg, G. Kos, A. Poulain, A. Ryjkov, K. Semeniuk, M. Subir and K. Toyota, Chem. Rev., 2015, 115, 3760–3802 CrossRef PubMed.
- B. Michalke, D. Willkommen, E. Drobyshev and N. Solovyev, TrAC, Trends Anal. Chem., 2017 DOI:10.1016/j.trac.2017.08.008.
- M. Enrico, G. Le Roux, L. E. Heimburger, P. Van Beek, M. Souhaut, J. Chmeleff and J. E. Sonke, Environ. Sci. Technol., 2017, 51, 5899–5906 CrossRef PubMed.
- J. Mo, Q. Li, X. Guo, G. Zhang and Z. Wang, Anal. Chem., 2017, 89, 10353–10360 CrossRef PubMed.
- Q. Chen, Y. Lin, Y. Tian, L. Wu, L. Yang, X. Hou and C. Zheng, Anal. Chem., 2017, 89, 2093–2100 CrossRef PubMed.
- M. He, L. Huang, B. Zhao, B. Chen and B. Hu, Anal. Chim. Acta, 2017, 973, 1–24 CrossRef PubMed.
- Y. M. Liu, F. P. Zhang, B. Y. Jiao, J. Y. Rao and G. Leng, J. Chromatogr. A, 2017, 1493, 1–9 CrossRef PubMed.
- R. A. Gil, P. H. Pacheco, S. Cerutti and L. D. Martinez, Anal. Chim. Acta, 2015, 875, 7–21 CrossRef PubMed.
- C. Bendicho, C. Bendicho-Lavilla and I. Lavilla, TrAC, Trends Anal. Chem., 2016, 77, 109–121 CrossRef.
- G. Giakisikli and A. N. Anthemidis, Anal. Chim. Acta, 2013, 789, 1–16 CrossRef PubMed.
- G. Li, M. Liu, Z. Zhang, C. Geng, Z. Wu and X. Zhao, J. Colloid Interface Sci., 2014, 424, 124–131 CrossRef PubMed.
- Y. Hong, D. J. Kim, I. A. Choi, M. Pal, G. Lee, K. M. Nam and W. S. Seo, RSC Adv., 2018, 8, 1089–1097 RSC.
- Y. Huang, J. Tang, L. Gai, Y. Gong, H. Guan, R. He and H. Lyu, Chem. Eng. J., 2017, 319, 229–239 CrossRef.
- H. Zhao, X. Mu, G. Yang, M. George, P. Cao, B. Fanady, S. Rong, X. Gao and T. Wu, Appl. Energy, 2017, 207, 254–264 CrossRef.
- X. Guo, X. Zhang, S. Zhao, Q. Huang and J. Xue, Phys. Chem. Chem. Phys., 2016, 18, 228–233 RSC.
- W. Jiang, X. Jin, X. Yu, W. Wu, L. Xu and F. Fu, J. Chromatogr. A, 2017, 1496, 167–173 CrossRef PubMed.
- J. Tang, H. Lv, Y. Gong and Y. Huang, Bioresour. Technol., 2015, 196, 355–363 CrossRef PubMed.
- H. Zhao, H. Fan, G. Yang, L. Lu, C. Zheng, X. Gao and T. Wu, Energy Fuels, 2018, 32, 5338–5344 CrossRef.
- J. P. Rooney, Toxicology, 2007, 234, 145–156 CrossRef PubMed.
- J. A. Darr, J. Zhang, N. M. Makwana and X. Weng, Chem. Rev., 2017, 117, 11125–11238 CrossRef PubMed.
- K. Ai, C. Ruan, M. Shen and L. Lu, Adv. Funct. Mater., 2016, 26, 5542–5549 CrossRef.
- H. Li, Y. Li, A. Aljarb, Y. Shi and L. J. Li, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.7b00212.
- S. Boandoh, S. H. Choi, J. H. Park, S. Y. Park, S. Bang, M. S. Jeong, J. S. Lee, H. J. Kim, W. Yang, J. Y. Choi, S. M. Kim and K. K. Kim, Small, 2017, 13(39), 1701306 CrossRef PubMed.
- S. Najmaei, Z. Liu, W. Zhou, X. Zou, G. Shi, S. Lei, B. I. Yakobson, J. C. Idrobo, P. M. Ajayan and J. Lou, Nat. Mater., 2013, 12, 754–759 CrossRef PubMed.
- Y. Yu, G. Li, L. Huang, A. Barrette, Y. Q. Cai, Y. Yu, K. Gundogdu, Y. W. Zhang and L. Cao, ACS Nano, 2017, 11, 9390–9396 CrossRef PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1–18 CrossRef.
- Z. Liu, Y. Wang, Z. Wang, Y. Yao, J. Dai, S. Das and L. Hu, Chem. Commun., 2016, 52, 5757–5760 RSC.
- X. Hai, K. Chang, H. Pang, M. Li, P. Li, H. Liu, L. Shi and J. Ye, J. Am. Chem. Soc., 2016, 138, 14962–14969 CrossRef PubMed.
- F. Jia, Q. Wang, J. Wu, Y. Li and S. Song, ACS Sustainable Chem. Eng., 2017, 5, 7410–7419 CrossRef.
- V. P. Pham and G. Y. Yeom, Adv. Mater., 2016, 28, 9024–9036 CrossRef PubMed.
- X. Zhang, F. Jia, B. Yang and S. Song, J. Phys. Chem. C, 2017, 121, 9938–9943 Search PubMed.
- C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone and S. Ryu, ACS Nano, 2010, 4, 2695–2700 CrossRef PubMed.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutierrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 2898–2926 CrossRef PubMed.
- J. Martincova, M. Otyepka and P. Lazar, Chem.–Eur. J., 2017, 23, 13233–13239 CrossRef PubMed.
- H. C. H. Hahne and W. Kroontje, J. Environ. Qual., 1973, 2, 444–450 CrossRef.
- H. Zhao, X. Mu, G. Yang, C. Zheng, C. Sun, X. Gao and T. Wu, Appl. Surf. Sci., 2017, 420, 439–445 CrossRef.
- S.-I. Lo, P.-C. Chen, C.-C. Huang and H.-T. Chang, Environ.
Sci. Technol., 2012, 46, 2724–2730 CrossRef PubMed.
- C. Peng, M. He, B. Chen, L. Huang and B. Hu, Analyst, 2017, 142, 4570–4579 RSC.
- C. Zhou, H. Zhu, Q. Wang, J. Wang, J. Cheng, Y. Guo, X. Zhou and R. Bai, RSC Adv., 2017, 7, 18466–18479 RSC.
- C. Huang and B. Hu, Spectrochim. Acta, Part B, 2008, 63, 437–444 CrossRef.
- Z. Yun, B. He, Z. Wang, T. Wang and G. Jiang, Talanta, 2013, 106, 60–65 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Lei Li
*a and
Lei Li