
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of toluene-4-sulfonic acid 2-(2-thiophen-2-yl-phenanthro[9,10-d]imidazol-1-yl)-ethyl ester and its application for sensitive determination of free fatty acids in ginkgo nut and ginkgo leaf by high performance liquid chromatography with fluorescence detection
Xiuli Dong,
Xueying Liu and
Xiangming Chen *
*
School of Pharmacy, Binzhou Medical University, Yantai, 264003, P. R. China. E-mail: xmch913@163.com; Tel: +86(535)6913406
First published on 22nd May 2018
Abstract
A novel fluorescent labeling reagent toluene-4-sulfonic acid 2-(2-thiophen-2-yl-phenanthro[9,10-d]imidazol-1-yl)-ethyl ester has been designed and synthesized. It was used to label twenty-six fatty acids (C5–C30) successfully in the presence of K2CO3 catalyst in N,N-dimethylformamide solvent. The reaction conditions were optimized by employing a three-factor, three-level Box–Behnken design. Derivatives were sufficiently stable to be efficiently analyzed by high-performance liquid chromatography with fluorescence detection. All fatty acid derivatives were separated on a hypersil BDS-C8 column in conjunction with a gradient elution with a good baseline resolution. Good linear correlations were observed for all fatty acids with correlation coefficients > 0.993. The established method exhibited high sensitivity and excellent repeatability. The limit of detection (at a signal-to-noise ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was 8.8–45.5 fmol. The method was used to quantify free fatty acids in ginkgo nut and ginkgo leaf samples with satisfactory results.
:1) was 8.8–45.5 fmol. The method was used to quantify free fatty acids in ginkgo nut and ginkgo leaf samples with satisfactory results.
1. Introduction
Fatty acids are widely distributed in all kinds of samples, such as living organisms, vegetable oils, and food products. They are essential structural elements of biological membranes, and play important roles at trace levels in the regulation of a variety of physiological and biological functions. Saturated fatty acids have been considered to be bad fatty acids that could cause hypercholesterol and cardiovascular disease if people eat too much every day. However, many research studies revealed that saturated fatty acids have a therapeutic action for some diseases such as alcoholic liver injury,1,2 intraparenchymal hemorrhage,3 metabolic syndrome4 and insulin resistance syndrome.5 Therefore, sensitive and accurate isolation and quantization of trace fatty acids is of great importance both in biology and medicine.The most commonly used method for the determination of free fatty acids is capillary gas chromatography (GC) with a flame ionization detector6,7 or GC coupled with mass spectrometry.8–10 GC provides excellent separation and quantification together with high sensitivity and selectivity for the analysis of multi-component samples. However, it is difficult to analyze fatty acids directly by GC because of their high polarity and low volatility. One method of overcoming this difficulty is to prepare volatile alkyl derivatives or silylation derivatives which can decrease the boiling point of fatty acids,11 but this procedure is usually very time-consuming, and the limited stability of the derivatives is also an obstacle for accurate quantitative analysis. There is a growing interest in the use of high performance liquid chromatography (HPLC) for analyzing fatty acids.12–15 HPLC analysis is usually performed at room temperature, a lower temperature compared with GC would reduce the risk of damage of heat-liable compounds. HPLC methods for the determination of fatty acids involving detectors such as mass spectrometer,15,16 chemiluminescence,17 evaporative light scattering detector,14,18 and contactless conductivity detector13 have been reported. The frequently-used detector for HPLC is an UV-vis and fluorescence detector, but most fatty acids show weak natural absorption in the visible or UV regions and no fluorescence, so it is of low sensitivity for direct analysis of fatty acids by HPLC.19 For these reasons, derivatization procedures allowing a chromophore or fluorophore to label fatty acids have been widely adopted, which allows fatty acids to be detected by an UV-vis or fluorescence detector and increases the sensitivity of the methods.11,20–23 Many pre-column ultraviolet and fluorescence labeling reagents have been developed for analysis of the compounds with carboxy groups in the last decades. These regents are classified into diazomethane-type reagents,24,25 coumarin-type derivatives,26,27 benzofurazan-type reagents,28,29 and alcohol reagents.30,31 Typical labeling reagents for carboxyl include 9-anthryldiazomethane (ADAM),25 4-aminomethyl-6,7-dimethoxycoumarin (ADC),27 7-acetylamino-4-mercapto-2,1,3-benzoxadiazole (AABD),29 1,2-benzo-3,4-dihydrocarbazole-9-ethanol (BCE),31 and so on. ADAM is not stable enough and thus cannot be stored in solution for a long time, although it can react with carboxylic acids under mild conditions without a catalyst. ADC is easily be decomposed when was exposed to moist environment and benzene or toluene which is highly toxic is used in derivatization procedure. Derivatization procedure is complicated and time-consuming. Other labeling reagents have many disadvantages in their applications such as low detection sensitivity, short detection wave-lengths, poor stability, tedious analytical procedures, and so on.32–34
Phenanthroimidazole is an excellent luminophore which could emit strong blue light with high efficiency when excited by light or electric. A serious of phenanthroimidazole-derivatives were designed and synthesized to study the possibility of use as photoelectric material. Inspired by this, we think that phenanthroimidazole could be used as a good pre-column derivative reagent after structure modified. Therefore, we designed and synthesized a novel fluorescence labeling reagent, toluene-4-sulfonic acid 2-(2-thiophen-2-yl-phenanthro[9,10-d]imidazol-1-yl)-ethyl ester (TSTPE) that emits strong fluorescence (λem = 398 nm) when exposed to excitation light (λex = 314 nm). TSTPE can react with carboxylic acid compounds in the presence of a basic catalyst and a highly sensitive method for the analysis of fatty acids using TSTPE as labeling reagent was developed. The optimum derivatization of fatty acids with TSTPE was obtained at 94 °C for 32.6 min in the presence of potassium carbonate (K2CO3) in N,N-dimethylformamide (DMF) solvent. A total of 26 saturated fatty acids were separated in 50 min on a Hypersil BDS-C8 column with gradient elution. The established method has been used to determine the saturated fatty acid compositions in ginkgo nut and ginkgo leaf with satisfactory results. Ginkgo nut, the seed of Ginkgo biloba, is an important nut for most Chinese. It's also a traditional Chinese medicine for treating cough, asthma, enuresis, alcohol misuse, pyogenic skin infections, and worm infections in the intestinal tract.35 Ginkgo leaf is also a traditional Chinese medicine used to lower lipid levels and prevent cardiovascular and cerebrovascular disease. In this study, the content of free fatty acids in ginkgo leaf and ginkgo nut was determined for the first time.
2. Experimental
2.1 Chemicals and materials
All fatty acids (C5–C30) standards were of analytical grade and purchased from Sigma-Aldrich (St. Louis, MO, USA). HPLC grade acetonitrile (ACN) was purchased from Yucheng (Shandong Province, China). N,N-Dimethylformamide (DMF), potassium carbonate (K2CO3), pyridine, and chloroform were of analytical grade obtained from Shanghai Chemical Reagent Co. (Shanghai, China). Toluene-4-sulfonic acid 2-(2-thiophen-2-yl-phenanthro[9,10-d]imidazol-1-yl)-ethyl ester (TSTPE) was synthesized in our lab as described in the synthesis section and Fig. 1. Water was purified on a Milli-Q system (Millipore, Bedford, MA). All other reagents used in this study were also of analytical grade unless otherwise stated. Ginkgo nut and ginkgo leaf samples were collected from Shandong province, Jiangsu province and Henan province of China.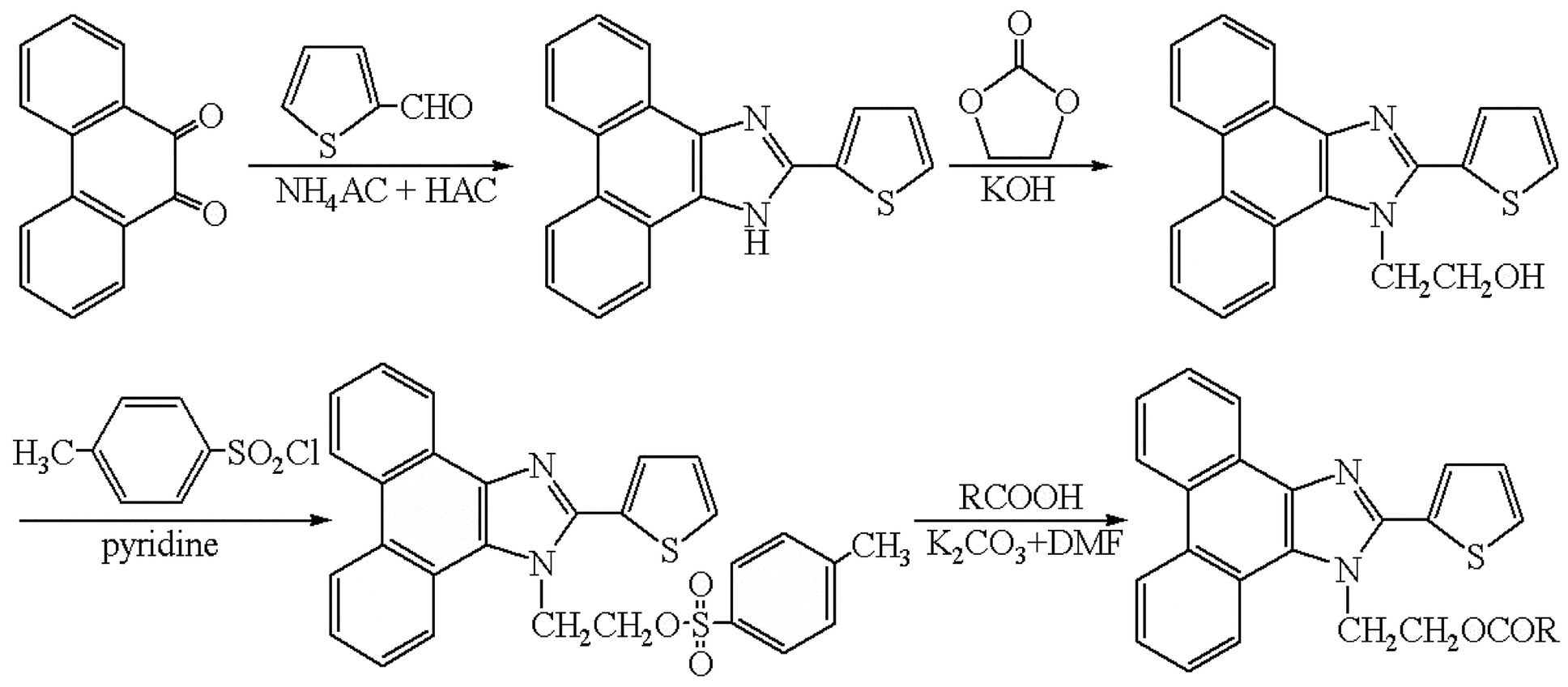 | ||
| Fig. 1 Synthesis route of TSTPE and derivatization scheme of TSTPE with fatty acids. | ||
2.2 Instruments and conditions
Agilent 1100 series HPLC (Agilent, USA) was used to perform the experiment. The HPLC system was equipped with a quaternary pump (model G1311A), an online vacuum degasser (model G1322A), an auto-sampler (model G1329A), a thermostated column compartment (model G1316A) and a fluorescence detector (FLD) (model G1321A). The HPLC system was controlled by Agilent Chemstation software (version B.01.01).Chromatographic conditions: derivatives were separated on a reversed-phase Hypersil BDS-C8 column (150 × 4.6 mm, 5 μm, Dalian Elite Analytical Instruments Co., Ltd., China) by a gradient elution. Eluent A was 5% acetonitrile in water, and B was acetonitrile. Elution conditions were as follows: initial = 60% B; 15 min = 80% B; 40 min = 100% B (kept for 15 min). The flow rate was constant at 1.0 mL min−1 and the column temperature was set at 30 °C. Before the injection of next sample, the column was equilibrated with the initial mobile phase for 5 min. The fluorescence excitation and emission wavelengths were set at 314 nm and 398 nm, respectively.
2.3 Synthesis of TSTPE
:1, v/v) to afford a brown crystal, yield 88%.:2, v/v). The crude product was dried at room temperature for 2 days and recrystallized from acetonitrile/DMF mixed solvent (acetonitrile/DMF, 4:1, v/v) to afford a yellow acicular type crystal, yield 51%.2.4 Preparation of solutions
Individual stock solutions of fatty acids (1.0 × 10−2 mol L−1) were prepared in ACN/DMF (1:1, v/v), the standard fatty acids for HPLC analysis at individual concentrations of 1.0 × 10−4 mol L−1 were prepared by diluting the corresponding stock solutions with ACN/DMF (1:1, v/v). The derivatization reagent solution (1.0 × 10−2 mol L−1) was prepared by dissolving 0.5 mg TSTPE in 10 mL DMF. When not in use, all solutions were stored at 4 °C in a refrigerator.
2.5 Extraction of fatty acids from samples
All ginkgo nut and ginkgo leaf samples were baked at 80 °C for 4 h and grinded by a Chinese medicine pulverizer. 1.0 g powdered ginkgo nut sample or ginkgo leaf sample and 10 mL chloroform were added in a round-bottom flask (50 mL) in turn. The flask was then immersed in an ultrasonic water-bath and the sample was sonicated for 60 min. The mixture was filtered and the filtrate was collected. 10 mL chloroform was added into the residue for further extraction. The twice filtrate was united and evaporated to dryness under a gentle stream of nitrogen, the residue was re-dissolved in 1.0 mL of DMF, and filtered through a 0.2 μm nylon membrane filter. The final solution was stored at 4 °C until derivatization and HPLC analysis.2.6 Derivatization procedure
20 mg K2CO3 and 400 μL DMF were added into a 2.0 mL vial, then, 100 μL fatty acids solution and 270 μL TSTPE were added successively. The vial was sealed and shaken for 1 min, and then heated at 94 °C for 32.6 min. After derivatization, 230 μL acetonitrile was added into the mixture. The final solution was directly injected into the HPLC system for analysis. The derivatization scheme of TSTPE with fatty acids is shown in Fig. 1.2.7 Experimental design for optimization of derivatization
Based on previous studies,21,40 the labeling reagent with active sulfonic ester group could easily react with fatty acids in the presence of basic catalyst and DMF. In this study, K2CO3 was used as catalyst and DMF was used as reaction medium, because TSTPE has the same sulfonic ester group as APIETS21 and TSPP.40 In order to ensure the sufficient labeling of the analyzed components by TSTPE, derivatization parameters were optimized by response surface methodology (RSM) which was based on the results obtained by single factor experiments. Compared to the classical experimental design method, RSM is more efficient and provides interaction effects on the response. The optimum conditions could be found by a small amount of tests. In this study, three factors including the amount of TSTPE (V: molar ratio of TSTPE to total free fatty acids), reaction temperature (T) and reaction time (t) were selected and optimized by employing a three-level, three-variable Box–Behnken design (BBD) from RSM. The coded and uncoded independent variables used in the RSM design and their respective levels were listed in Table 1. A total of 17 experiments were designed. The total peak area of all fatty acids was taken as the response. The surface response models were obtained by fitting the data to a polynomial model as shown below in the following equation.| Y = β0 + β1V + β2t + β3T − β12Vt − β13VT + β23tT + β11V2 + β22t2 + β33T2 |
| Run number | Independent variable | Response (total peak area) | ||
|---|---|---|---|---|
| V (molar ratio) | T (temperature, °C) | t (time, min) | ||
| 1 | 10(0) | 85(0) | 30(0) | 15591 |
| 2 | 10(0) | 100(+1) | 20(−1) | 6905 |
| 3 | 10(0) | 100(+1) | 40(+1) | 12371 |
| 4 | 10(0) | 85(0) | 30(0) | 14560 |
| 5 | 10(0) | 70(−1) | 40(+1) | 9448 |
| 6 | 8(−1) | 85(0) | 20(−1) | 4184 |
| 7 | 10(0) | 85(0) | 30(0) | 14393 |
| 8 | 8(−1) | 85(0) | 40(+1) | 9576 |
| 9 | 12(+1) | 85(0) | 20(−1) | 5365 |
| 10 | 10(0) | 85(0) | 30(0) | 14713 |
| 11 | 12(+1) | 100(+1) | 30(0) | 13901 |
| 12 | 12(+1) | 85(0) | 40(+1) | 10605 |
| 13 | 10(0) | 85(0) | 30(0) | 15350 |
| 14 | 12(+1) | 70(−1) | 30(0) | 11858 |
| 15 | 8(−1) | 70(−1) | 30(0) | 8616 |
| 16 | 10(0) | 70(−1) | 20(−1) | 5963 |
| 17 | 8(−1) | 100(+1) | 30(0) | 11707 |
A software Design-Expert 8.0.6 (State-Ease, Inc., Minneapolis MN, USA) was used to obtain the coefficients of the quadratic polynomial model. The quality of the fitted model was expressed by the determined coefficient (R2), and its statistical significance was checked by an F-test.
3. Results and discussion
3.1 Fluorescence spectra of TSTPE and fatty acid derivatives
In order to find the fluorometric detector wavelengths, the excitation and emission spectra of TSTPE (Fig. 2A) and fatty acid derivatives (Fig. 2B) were determined at the wavelength range of 200 to 550 nm. Octadecanoic acid (C18) was selected as representative fatty acid that was used to study the excitation and emission spectra of derivatives. As was shown in Fig. 2, the fluorescence spectra of TSTPE-C18 and TSTPE were very similar, but the excitation spectra of the two compounds changed a little. There was no obviously different in maximum excitation and emission wavelength for the two compounds. The fluorescence spectra of TSTPE-C18 in acetonitrile solution with different concentrations (50–100%) were also studied to evaluate the effect of mobile phase gradient on fluorescence properties. The result (Fig. 2B) showed that the maximum excitation and emission wavelengths are 314 and 398 nm, respectively. Therefore, these wavelengths were chosen as detecting wavelengths for further experiments. The fluorescence intensity of TSTPE-C18 decreased with the decrease of acetonitrile concentration, and a 40% decrease was found when acetonitrile concentration varied from 100% to 50%.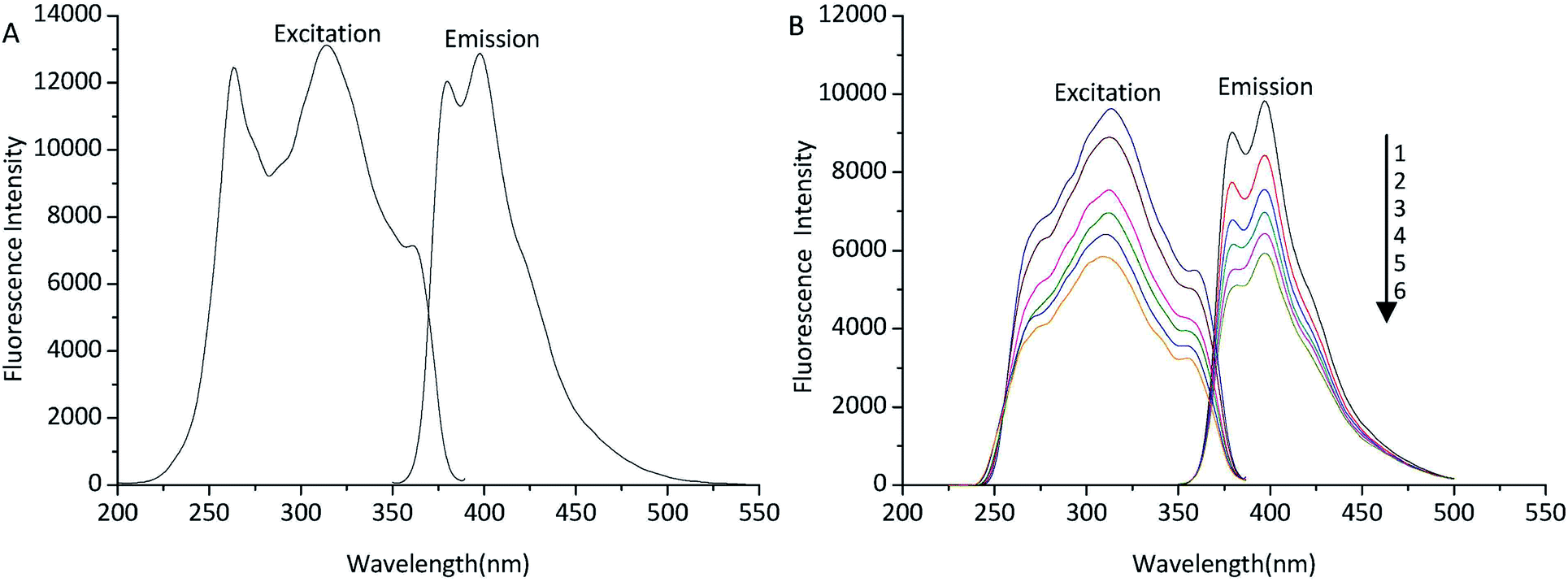 | ||
| Fig. 2 Excitation and emission spectra of TSTPE in DMF (A) and TSTPE-C18 in acetonitrile (B). 1: 100% acetonitrile, 2: 90% acetonitrile, 3: 80% acetonitrile, 4: 70% acetonitrile, 5: 60% acetonitrile, 6: 50% acetonitrile. | ||
3.2 Single factor experiments
TSTPE can react with fatty acid in the existence of basic catalyst. Several different basic catalysts including K2CO3, Na2CO3, potassium citrate, pyridine, triethylamine, 4-dimethylaminopyridine (DMAP) were tested and evaluated for their derivatization efficiency. The strongest fluorescence intensity was obtained by using K2CO3 as catalyst. The effect of the K2CO3 amount on derivatization yield was also tested and maximum response was obtained when the amount of K2CO3 was 20 mg. When the amount of K2CO3 was increased to 50 mg, no obvious increase in response was observed. Therefore, 20 mg of K2CO3 was applied in all subsequent derivatization reactions.The reaction temperature and time are always the important factors on the derivative yield. The effect of the reaction temperature was examined over the range of 60–100 °C. The fluorescence intensity increased with the rise of the temperature, and the maximum fluorescence intensity was obtained at 85 °C. When exceeding this temperature, the decreased responses and more by-product peaks were observed, while below 85 °C, the rate of reaction was decreased which led to long derivatization time. Different reaction time was tested over the range of 10–60 min to investigate the effect of time on the derivatization reaction. The maximum fluorescence intensity was obtained at 30 min, and there was little decrease for fluorescence intensity when prolonging reaction time.
The effect of TSTPE concentrations on derivatization was investigated in detail to ensure sufficient reaction of fatty acids. The result indicated that constant fluorescence intensity was obtained when ten-fold molar excess to total molar fatty acids, further increasing TSTPE concentration beyond this level had no significant effect on fluorescence intensity.
3.3 Response surface methodology
The optimum derivatization conditions including the concentration of TSTPE, derivatization temperature, and derivatization time were optimized through Box–Behnken design and response surface analysis. A total of 17 runs were tested, and the experimental results are shown in Table 1. The analysis of variance (ANOVA) for the response surface quadratic model showed that the model was significant at the level of p < 0.0001; none of the linear parameters and interaction parameters was significant; quadratic parameters were insignificant except derivatization time. The R2 value was calculated to be 0.9866, revealing that the experimental data were in good agreement with the predicted peak area values. The F-value of 2.28 for the lack of t was insignificant (p > 0.05), indicating that the model was sufficiently accurate for predicting the relevant responses. The coefficient of variation (CV%) of less than 5.95 indicates that the model was reproducible. The final estimative response model equation (based on the actual value) is as follows:| Y = 14921 + 956V + 2448t + 1125T − 38Vt − 262VT + 495tT − 2320V2 − 5169t2 − 1080T2 |
Three-dimensional response surfaces (Fig. 3) were plotted on the basis of this model equation to investigate the interaction among the variables and to determine the optimum values of each factor for the maximum derivatization yield. Fig. 3A intuitively presents the variations of peak areas with derivatization time and the molar ratio of TSTPE to fatty acids at a constant derivatization temperature of 85 °C. Fig. 3B is the response surface showing the effect of the derivatization temperature and molar ratio of TSTPE to fatty acids on the response (peak area) at a fixed derivatization time of 30 min. Fig. 3C describes the effects of different derivatization time and temperature at a fixed TSTPE-to-fatty acid molar ratio of 10:1.
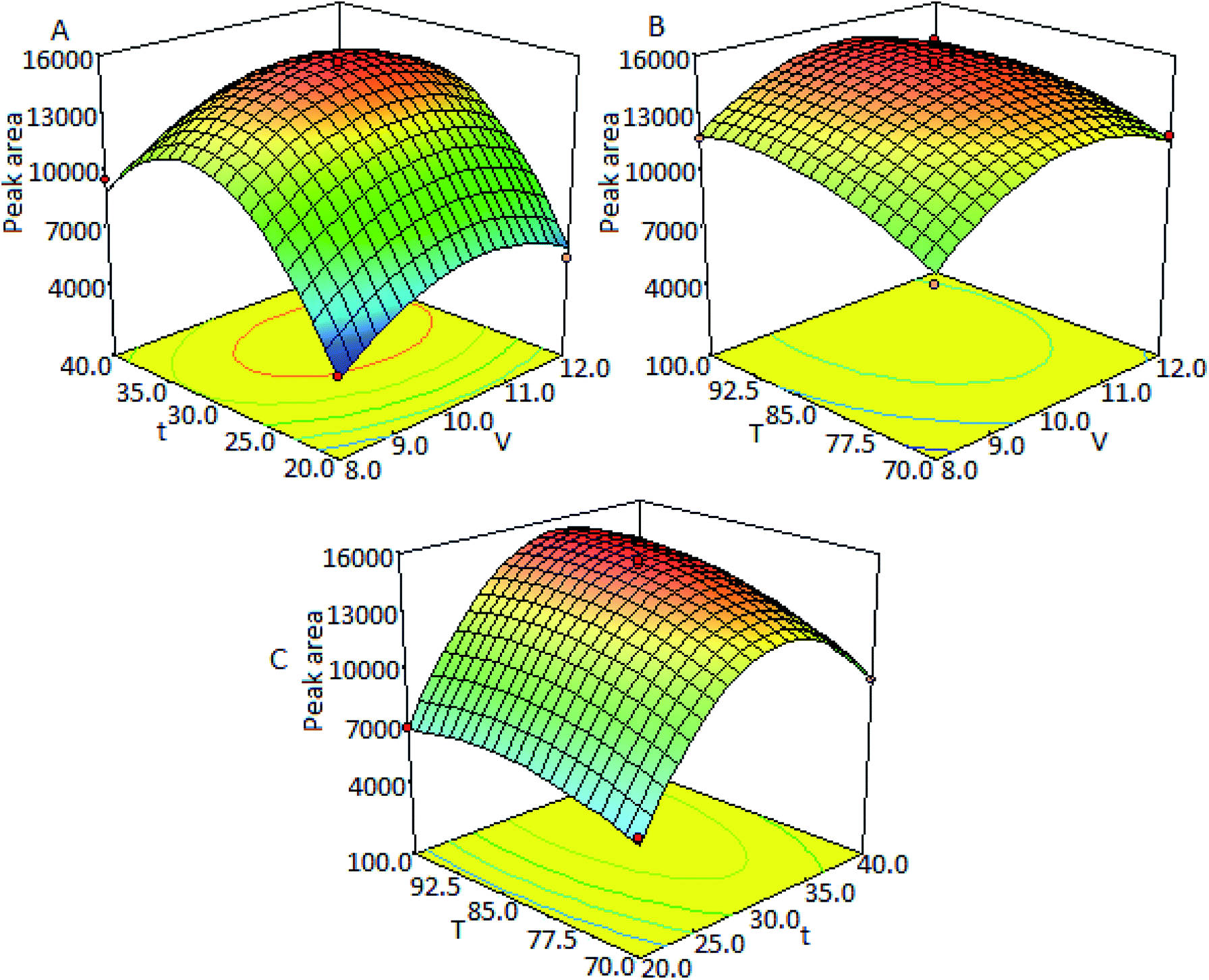 | ||
| Fig. 3 The 3D response surface plots showing effects of the examined factors on the response. (A) The effect of reaction time and TSTPE amounts on the peak area; (B) the effect of derivatization temperature and TSTPE amounts on the peak area; (C) the effect of derivatization temperature and reaction time on the peak area. | ||
The optimal values of the selected variables were obtained by solving the regression equation using Design-Expert software. The optimal derivatization conditions estimated by the model equation were molar ratio of TSTPE to fatty acid = 10.4, derivatization temperature = 94 °C, and derivatization time = 32.6 min. According to the results of response surface methodology, the peak area changed little in the range of 90–100 °C with RSD = 0.37% which means that there is little change for derivatization yield in this temperature range. The peak area also changed little in the range of 30–35 min with RSD = 1.4%. When reaction time was more than 35 min, the peak area dramatically decreased which maybe caused by the decomposition of derivatives. Therefore the optimal derivatization procedure is as follows: 20 mg of K2CO3 and 400 μL of DMF are added into a 2.0 mL vial, and then 100 μL of fatty acid solution and 270 μL of TSTPE are added successively. The vial is sealed and shaken for 1 min and then heated at 94 °C for 32.6 min. After derivatization, 230 μL acetonitrile is added into the mixture. The final solution is directly injected into the HPLC system for analysis.
There are several advantages of RSM over traditional methods for derivatization optimization based on single-factor experiments. It is an efficient tool that can decrease labor time and reagent consumption and includes interaction effects of derivatization conditions in addition to single-factor effects.
3.4 Stability of TSTPE and derivatives
There was no obvious difference for the derivatization yields when solution of TSTPE in DMF was stored at 4 °C for at least 3 days. The corresponding neutral derivatives solution was stored at 4 °C for a period of 24 hours, during which time the solutions were analyzed six times. Results showed that derivatives were stable enough for accurate HPLC with normalized peak areas varying <4.5%.3.5 Solubility of TSTPE
In order to evaluate the solubility of TSTPE in DMF, 10 mg of TSTPE was added into a 2.0 mL vial, then, a definite proportion of DMF was added. The vial was treated by ultrasonic for 5 min, and no solid particle was observed when 200 μL DMF was added. Therefore, we inferred that TSTPE dissolve in DMF easily, and the solubility of TSTPE in DMF is >0.1 mol L−1 which could well meet the requirement of derivatization.3.6 Chromatographic separation
The polarity of fatty acid derivatives decreases when the length of carbon chain increases, which would increase the retention time of analytes. In order to shorten analysis time, the separation of fatty acid derivatives was carried out on a hypersil BDS-C8 column, rather than C18 column. In order to obtain complete baseline resolution and good peak shape within shorter time, gradient elution was utilized with water–acetonitrile as mobile phase. The chromatogram of twenty-six fatty acid derivatives was obtained within 50 min (Fig. 4).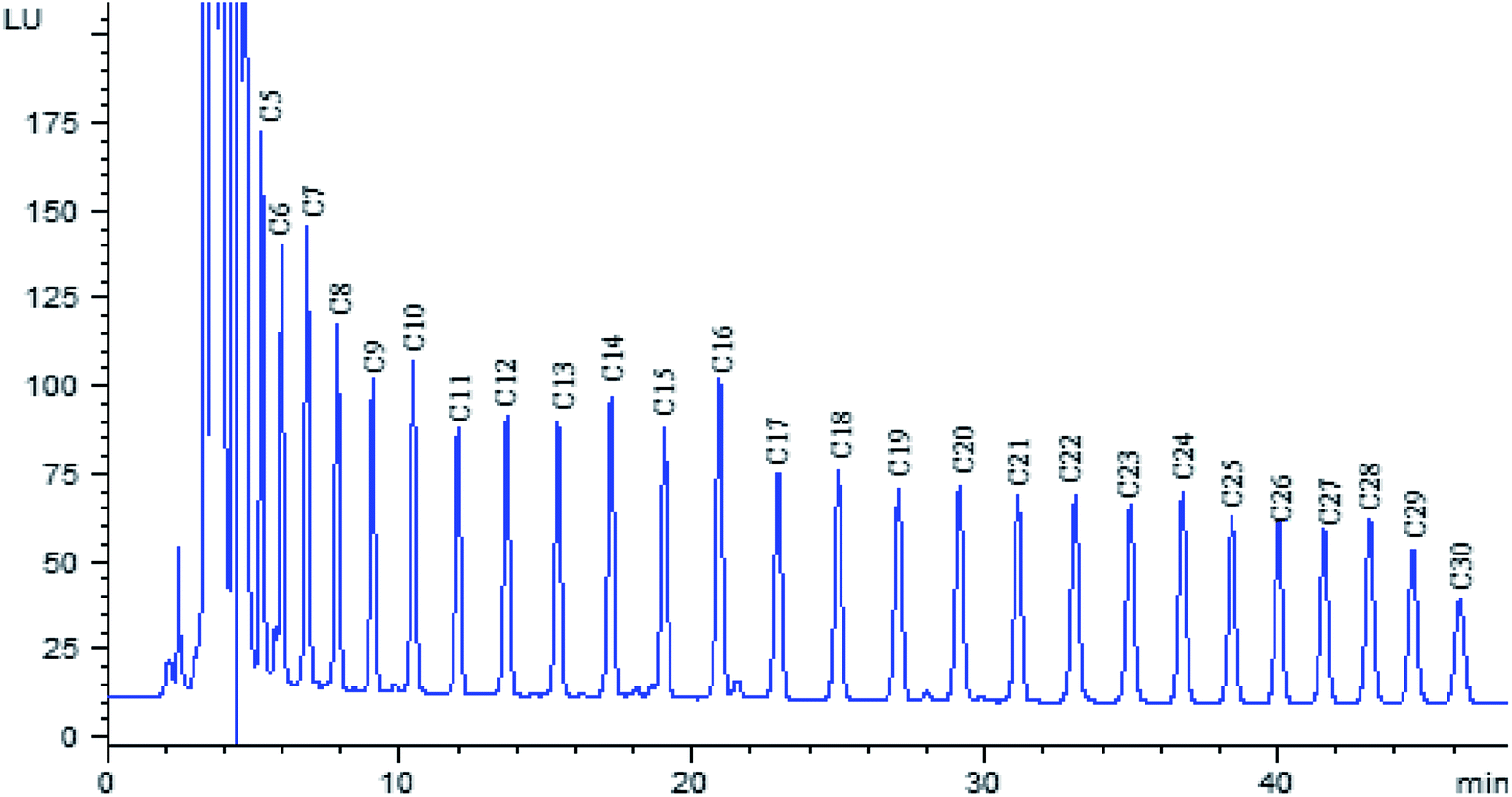 | ||
| Fig. 4 Chromatogram of standard fatty acids derivatives. Chromatographic conditions: Hypersil BDS-C8 column (4.6 × 150 mm, 5 μm), column temperature 30 °C; flow rate 1.0 mL min−1; excitation and emission: λex/λem = 314/398 nm. Peaks are labeled with abbreviations for all fatty acids: C5 (pentanoic acid); C6 (hexanoic acid); C7 (heptanoic acid); C8 (caprylic acid); C9 (pelargonic acid); C10 (decanoic acid); C11 (undecanoic acid); C12 (dodecanoic acid); C13 (tridecanoic acid); C14 (myristic acid); C15 (pentadecanoic acid); C16 (hexadecanoic acid); C17 (heptadecanoic acid); C18 (octadecanoic acid); C19 (nonadecanoic acid); C20 (arachidic acid); C21 (heneicosoic acid); C22 (docosanoic acid); C23 (tricosanoic acid); C24 (tetracosanoic acid); C25 (pentacosanoic acid); C26 (hexacosanoic acid); C27 (carboceric acid); C28 (montanic acid); C29 (motanic acid); C30 (triacontanoic acid). | ||
3.7 Validation parameters
The developed method was validated in terms of linearity, limit of detection (LOD), limit of quantitation (LOQ) and precision calculated as relative standard deviation (RSD).The calibration graph was established with the peak area (Y) versus fatty acid concentration (X, pmol, injected amount), and injection amount varied from 100 pmol to 40 fmol. The linear regression equations were shown in Table 2. All fatty acids were found to give excellent linear responses over this range, with correlation coefficients of >0.993. LODs (at signal-to-noise ratio of 3:1) were 8.8–45.5 fmol for the labeled fatty acids and LOQs (at signal-to-noise ratio of 10:1) were 29.5–152 fmol. A standard solution consisting of 25 pmol fatty acid derivatives was prepared and analyzed to examine the method repeatability. The relative standard deviations (RSDs; n = 6) for the peak areas was <3.1%. A high repeatability in the retention time was obtained with RSD values lower than 0.18%. The recoveries of fatty acids were evaluated by the addition of a known amount of standard into the sample. The sample was treated according to the method as described above and derivatized with TSTPE. The analysis was carried out in triplicate. The experimental recoveries were in the range of 87.29–96.27% (see Table 2) and the method was sufficiently accurate for the determination of free fatty acids.
| Fatty acids | Y = AX + B | Correlation coefficient | LOD (fmol) | LOQ (fmol) | Recovery (%) | RSD (%) |
|---|---|---|---|---|---|---|
| C5 | Y = 32.46X − 17.60 | 0.9957 | 8.82 | 29.41 | 92.24 | 4.5 |
| C6 | Y = 31.25X − 28.20 | 0.9932 | 11.06 | 36.87 | 91.54 | 3.9 |
| C7 | Y = 32.21X − 28.34 | 0.9971 | 10.82 | 36.08 | 89.42 | 5.1 |
| C8 | Y = 26.27X − 22.43 | 0.9983 | 13.44 | 44.80 | 94.49 | 4.2 |
| C9 | Y = 23.71X − 24.20 | 0.9983 | 15.76 | 52.52 | 92.87 | 3.1 |
| C10 | Y = 26.82X − 27.24 | 0.9985 | 14.79 | 49.31 | 87.29 | 4.7 |
| C11 | Y = 22.02X − 22.25 | 0.9985 | 18.52 | 61.73 | 94.60 | 2.9 |
| C12 | Y = 24.41X − 21.55 | 0.9991 | 17.28 | 57.60 | 96.27 | 6.5 |
| C13 | Y = 24.52X − 20.60 | 0.9992 | 17.61 | 58.69 | 92.28 | 4.6 |
| C14 | Y = 27.39X − 21.33 | 0.9993 | 16.20 | 54.00 | 95.40 | 5.2 |
| C15 | Y = 25.31X − 20.35 | 0.9992 | 18.52 | 61.73 | 89.74 | 4.9 |
| C16 | Y = 31.60X − 24.53 | 0.9993 | 15.69 | 52.30 | 92.78 | 3.8 |
| C17 | Y = 24.04X − 14.70 | 0.9998 | 21.74 | 72.46 | 94.00 | 3.7 |
| C18 | Y = 25.09X − 16.94 | 0.9994 | 20.78 | 69.25 | 91.25 | 4.1 |
| C19 | Y = 23.55X − 15.91 | 0.9994 | 22.94 | 76.45 | 90.75 | 3.1 |
| C20 | Y = 24.96X − 17.23 | 0.9994 | 21.93 | 73.10 | 92.54 | 3.9 |
| C21 | Y = 24.05X − 17.01 | 0.9994 | 22.59 | 75.30 | 90.22 | 5.3 |
| C22 | Y = 24.47X − 17.88 | 0.9994 | 22.52 | 75.08 | 87.98 | 3.5 |
| C23 | Y = 23.12X − 17.81 | 0.9993 | 23.29 | 77.64 | 90.42 | 6.2 |
| C24 | Y = 24.63X − 18.82 | 0.9993 | 21.80 | 72.67 | 92.22 | 3.6 |
| C25 | Y = 21.95X − 18.38 | 0.9993 | 24.43 | 81.43 | 91.29 | 5.9 |
| C26 | Y = 21.40X − 19.05 | 0.9992 | 24.83 | 82.78 | 96.42 | 6.1 |
| C27 | Y = 20.83X − 21.39 | 0.9990 | 26.79 | 89.29 | 92.87 | 6.3 |
| C28 | Y = 23.42X − 22.61 | 0.9991 | 23.89 | 79.62 | 92.04 | 5.8 |
| C29 | Y = 20.56X − 20.36 | 0.9990 | 30.61 | 102.04 | 87.49 | 6.7 |
| C30 | Y = 16.38X − 18.60 | 0.9990 | 45.45 | 151.52 | 90.98 | 6.2 |
3.8 Analysis of free fatty acids
The proposed method was applied to determine free fatty acids extracted from ginkgo nut and ginkgo leaf samples. The obtained chromatogram is shown in Fig. 5, and the analytical results are listed in Table 3. It was found that the content of fatty acids with even number carbon atom was much higher than that of fatty acids with odd number carbon atom, and C16 and C18 are the main saturated fatty acids in both samples. These two kinds of fatty acids are widely used in chemical enterprises. It is clear that the established method is suitable for the determination of these components from ginkgo nut and ginkgo leaf with satisfactory results. It can also be used to determinate free fatty acids in other plant samples.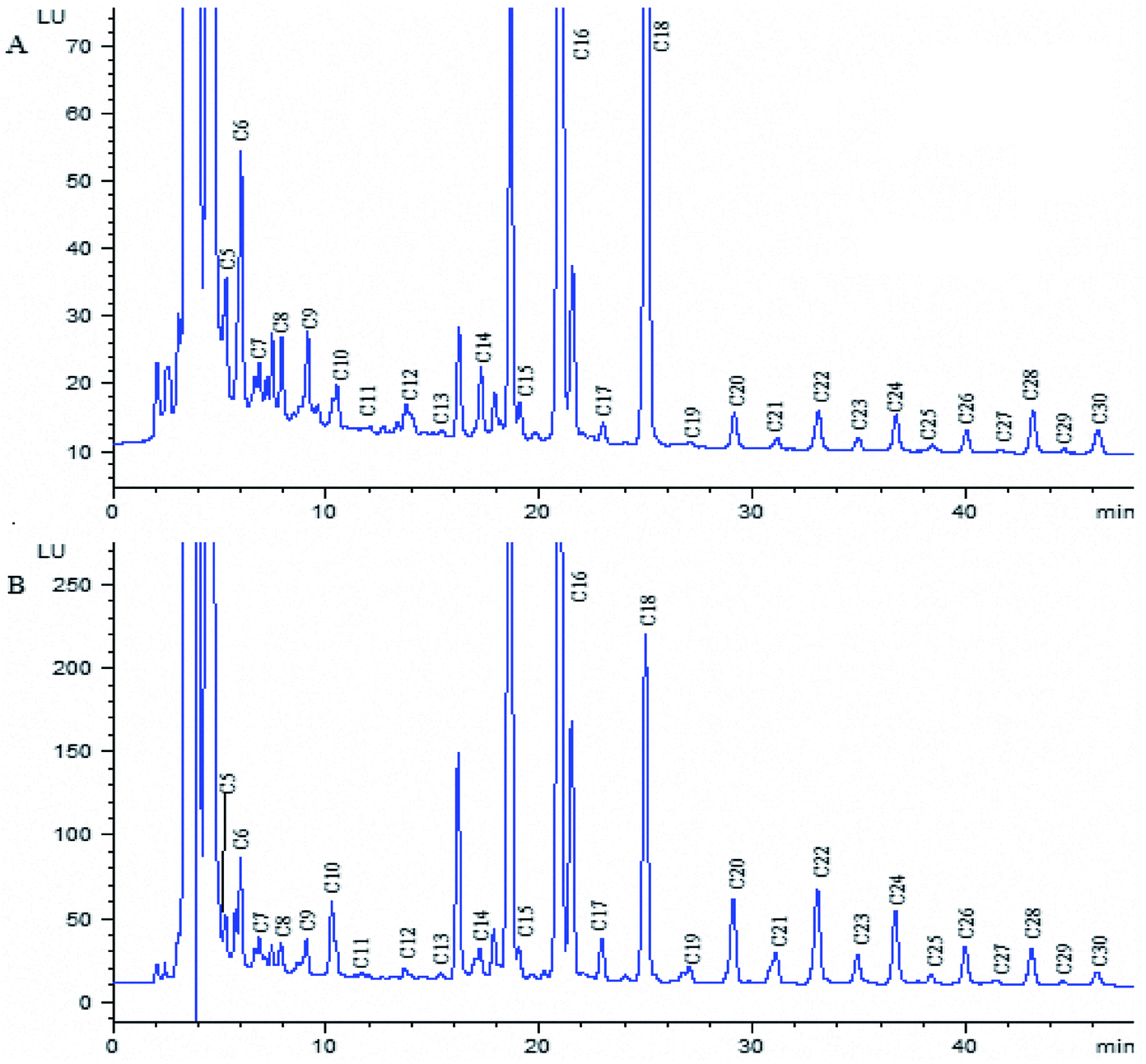 | ||
| Fig. 5 Chromatogram of fatty acid derivatives from the extracted ginkgo leaf sample (A) and ginkgo nut sample (B). Chromatographic conditions and peaks labels are the same with Fig. 4. | ||
| Fatty acids | Shandong (μg g−1) | Jiangsu (μg g−1) | Henan (μg g−1) | |||
|---|---|---|---|---|---|---|
| Gingko nut | Gingko leaf | Gingko nut | Gingko leaf | Gingko nut | Gingko leaf | |
| C5 | 7.88 | 6.07 | 9.28 | 4.46 | 12.43 | 5.14 |
| C6 | 12.97 | 11.02 | 17.88 | 9.12 | 18.18 | 9.05 |
| C7 | 6.17 | 2.55 | 3.78 | 1.41 | 4.48 | 1.82 |
| C8 | 7.52 | 5.17 | 10.99 | 4.14 | 12.64 | 5.40 |
| C9 | 11.15 | 6.21 | 14.14 | 7.96 | 12.05 | 6.88 |
| C10 | 6.47 | 2.87 | 37.63 | 3.20 | 5.40 | 3.61 |
| C11 | 2.53 | 1.89 | 3.48 | 1.37 | 3.45 | 1.28 |
| C12 | 5.12 | 2.91 | 4.80 | 3.05 | 5.78 | 3.01 |
| C13 | 2.57 | 1.93 | 3.65 | 1.75 | 4.06 | 1.61 |
| C14 | 8.58 | 5.04 | 12.22 | 5.05 | 16.68 | 3.90 |
| C15 | 8.97 | 2.82 | 10.91 | 3.9 | 14.45 | 3.91 |
| C16 | 176.14 | 70.86 | 228.92 | 82.96 | 181.59 | 102.8 |
| C17 | 12.15 | 2.11 | 16.12 | 6.21 | 12.69 | 4.06 |
| C18 | 90.05 | 48.68 | 112.38 | 43.37 | 108.14 | 50.21 |
| C19 | 5.66 | 0.73 | 10.94 | 4.09 | 4.59 | 1.86 |
| C20 | 26.11 | 2.90 | 34.00 | 2.75 | 44.14 | 4.12 |
| C21 | 8.51 | 1.26 | 15.14 | 5.93 | 14.89 | 2.63 |
| C22 | 34.25 | 3.65 | 36.56 | 1.91 | 46.67 | 1.76 |
| C23 | 11.72 | 1.61 | 13.35 | 2.78 | 14.76 | 2.45 |
| C24 | 25.59 | 3.24 | 24.86 | 2.91 | 50.42 | 2.47 |
| C25 | 5.72 | 1.17 | 6.61 | 2.92 | 8.03 | 2.65 |
| C26 | 16.23 | 2.53 | 12.67 | 6.74 | 49.35 | 4.15 |
| C27 | 4.17 | 0.97 | 6.39 | 2.11 | 5.62 | 1.74 |
| C28 | 16.04 | 4.62 | 15.96 | 4.09 | 17.53 | 2.67 |
| C29 | 3.09 | 1.20 | 3.03 | 1.82 | 2.67 | 1.58 |
| C30 | 10.50 | 4.59 | 13.15 | 6.98 | 5.39 | 4.19 |
3.9 Comparisons of the proposed method with the reported methods
As mentioned above, there were several GC and HPLC methods reported in other papers for the determination of free fatty acids. The comparison of those methods with this work is listed in Table 4. The main advantage of this study is that the LOD of the proposed method was lower than those of GC,36 GC-MS6,37 and HPLC,12,13,38 which means a higher sensitivity. The commonly-used detector for HPLC is UV-vis detector. However, fatty acids show weak UV absorption at the wavelength of 210 nm nearby, so the LOD of the method for direct determination of fatty acids without derivatization was only 6.4–9.4 nmol.19 To increase the sensitivity, fatty acids were usually derivatived by UV39 or fluorescence reagent.21,31,40–47 TSTPE was evaluated by comparing with those labeling reagents used for the derivatization of free fatty acids in other reports in term of derivatization condition and detection limit (see Table 4). The LOD for free fatty acids in this study is 8.8–45.5 fmol which is lower than most other methods. Therefore, TSTPE can act as a new derivatization reagent with a high sensitivity for free fatty acids.| Analytes | Method | Labeling reagents | Derivatization conditions | Injection volume | LOD | Ref |
|---|---|---|---|---|---|---|
| C6; C8; C10 | GC-FID | No | No | 0.8 μL | 4.2–5.9 pmol | 36 |
| C12–C26 | GC-MS | PFBBr | Room temperature, 20 min | 1 μL | 0.25–17.6 fmol | 37 |
| C16; C18 | GC-MS | Dimethylamine | 0 °C, 30 min | 1 μL | 1.95; 1.69 pmol | 6 |
| C12–C22 (even) | HPLC-ELSD | No | No | 10 μL | 11–150 pmol | 12 |
| C14; C16; C18 | HPLC-C4D | No | No | 20 μL | 7.0–53 pmol | 13 |
| C14; C16; C18 | HPLC-CL | PROB | Room temperature, 40 min | 20 μL | 11.8–64.4 fmol | 38 |
| C2; C3; C4 | HPLC-UV (210 nm) | No | No | 20 μL | 6.4–9.4 nmol | 19 |
| C10–C18 | HPLC-UV (400 nm) | 2-NPH | 60 °C, 35 min | — | 100–200 fmol | 39 |
| C14–C20 (even) | HPLC-FL (λex/λem = 315/435 nm) | APIETS | 85 °C, 35 min | 10 μL | 13–27 fmol | 21 |
| C12–C19 | HPLC-FL (λex/λem = 353/451 nm) | ADC | 25 °C, 5 min | 10 μL | 20–50 fmol | 27 |
| C1–C19 | HPLC-FL (λex/λem = 333/390 nm) | BCE | 60 °C, 60 min | 10 μL | 11–33 fmol | 31 |
| C20–C30 (even) | HPLC-FL (λex/λem = 260/380 nm) | TSPP | 90 °C, 30 min | 10 μL | 26–77 fmol | 40 |
| C4–C20 (even) | HPLC-FL (λex/λem = 430/570 nm) | APZBD-NHMe | 60 °C, 30 min | 10 μL | 160–880 fmol | 41 |
| C12–C18 | HPLC-FL (λex/λem = 467/512 nm) | APF | 60 °C, 60 min | 10 μL | 1–64 fmol | 42 |
| C12–C24 | HPLC-FL (λex/λem = 330/380 nm) | BCEHC | 100 °C, 60 min | 10 μL | 31.6–32.8 fmol | 43 |
| C7–C24 | HPLC-FL (λex/λem = 330/380 nm) | 6AQ | Room temperature, 750 min | 10 μL | 50 fmol | 44 |
| C6–C24 | HPLC-FL (λex/λem = 247/401 nm) | BQETS | 90 °C, 20 min | 10 μL | 8–28 fmol | 45 |
| C10–C22 | HPLC-FL (λex/λem = 265/315 nm) | FMOC-Cl | 60 °C, 10 min | 10 μL | 0.7–3.0 pmol | 46 |
| ALA, PLA, AA, LA, OA | HPLC-FL (λex/λem = 450/560 nm) | DBD-ED | Room temperature, 120 min | 10 μL | 2.29–4.75 fmol | 47 |
| C5–C30 | HPLC-FL (λex/λem = 314/398 nm) | TSTPE | 94 °C, 32.6 min | 10 μL | 8.8–45.5 fmol | This work |
4. Conclusion
In this paper, a new fluorescence labeling reagent, toluene-4-sulfonic acid 2-(2-thiophen-2-yl-phenanthro[9,10-d]imidazol-1-yl)-ethyl ester (TSTPE), was designed and synthesized. A simple and highly sensitive method to determine twenty-six fatty acids was developed utilizing TSTPE as the novel pre-column derivatization reagent followed by high-performance liquid chromatography with fluorescence detection. TSTPE was stable enough for the derivatization procedure. The derivatization reaction was carried out under mild conditions and the derivative was stable enough for HPLC analysis. The described method shows good correlation, precision and accuracy which were useful in the analysis of free fatty acids in ginkgo nut and ginkgo leaf. In contrast to the existing methods using other derivatization reagents, the method established in this paper has advantage such as shorter derivatization reaction times, simplified sample preparation and higher sensitivity.Conflicts of interest
There are no conflicts to declare.References
- A. A. Nanji, K. Jokelainen, G. L. Tipoe, A. Rahemtulla and A. J. Dannenberg, J. Pharmacol. Exp. Ther., 2001, 299, 638–644 Search PubMed.
- M. J. J. Ronis, S. Korourian, M. Zipperman, R. Hakkak and T. M. Badger, J. Nutr., 2004, 134, 904–912 CrossRef PubMed.
- H. Iso, M. J. Stampfer, J. E. Manson, K. Rexrode, F. B. Hu, C. H. Hennekens, G. A. Colditz, F. E. Speizer and W. C. Willett, Circulation, 2001, 103, 856–863 CrossRef PubMed.
- L. I. Mennen, L. Lafay, E. J. M. Feskens, M. Novak, P. Lépinay and B. Balkau, Nutr. Res., 2000, 20, 335–347 CrossRef.
- M. A. Pereira, D. R. Jacobs, L. Van Horn, M. L. Slattery, A. I. Kartashov and D. S. Ludwig, JAMA, J. Am. Med. Assoc., 2002, 287, 2081–2089 CrossRef PubMed.
- C. O. Kangani, D. E. Kelley and J. P. Delany, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 873, 95–101 CrossRef PubMed.
- S. G. Woo, K. Yoo, J. Lee, S. Bang, M. Lee, K. On and J. Park, Talanta, 2012, 97, 103–110 CrossRef PubMed.
- J. Ecker, M. Scherer, G. Schmitz and G. Liebisch, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 897, 98–104 CrossRef PubMed.
- M. Vosough and A. Salemi, Talanta, 2007, 73, 30–36 CrossRef PubMed.
- A. G. Casado, E. J. A. Hernández and J. L. Vílchez, Water Res., 1998, 32, 3168–3172 CrossRef.
- T. Toyo'oka, Anal. Chim. Acta, 2002, 465, 111–130 CrossRef.
- J. Zhao, S. P. Li, F. Q. Yang, P. Li and Y. T. Wang, J. Chromatogr. A, 2006, 1108, 188–194 CrossRef PubMed.
- A. Makahleh, B. Saad, G. H. Siang, M. I. Saleh, H. Osman and B. Salleh, Talanta, 2010, 81, 20–24 CrossRef PubMed.
- C. Orellana-Coca, D. Adlercreutz, M. M. Andersson, B. Mattiasson and R. Hatti-Kaul, Chem. Phys. Lipids, 2005, 135, 189–199 CrossRef PubMed.
- M. Špitsmeister, K. Adamberg and R. Vilu, J. Microbiol. Methods, 2010, 82, 288–295 CrossRef PubMed.
- J. P. C. L. Lacaze, L. A. Stobo, E. A. Turrell and M. A. Quilliam, J. Chromatogr. A, 2007, 1145, 51–57 CrossRef PubMed.
- Y. Ohba, N. Kuroda and K. Nakashima, Anal. Chim. Acta, 2002, 465, 101–109 CrossRef.
- E. Bravi, G. Perretti and L. Montanari, J. Chromatogr. A, 2006, 1134, 210–214 CrossRef PubMed.
- L. R. V. de Sá, M. A. L. de Oliveira, M. C. Cammarota, A. Matos and V. S. Ferreira-Leitão, Int. J. Hydrogen Energy, 2011, 36, 15177–15186 CrossRef.
- B. Bravo, G. Chavez, N. Pina, F. Ysambertt, N. Marquez and A. Caceres, Talanta, 2004, 64, 1329–1334 CrossRef PubMed.
- Z. Sun, J. You, C. Song and L. Xia, Talanta, 2011, 85, 1088–1099 CrossRef PubMed.
- G. Li, J. You, Y. Suo, C. Song, Z. Sun, L. Xia, X. Zhao and J. Shi, Food Chem., 2011, 125, 1365–1372 CrossRef.
- N. Jing, J. Shi, G. Li, Z. Sun and J. You, Food Res. Int., 2012, 48, 155–163 CrossRef.
- T. Yoshida, A. Uetake, H. Yamaguchi, N. Nimura and T. Kinoshita, Anal. Biochem., 1988, 173, 70–74 CrossRef PubMed.
- M. A. Quiliam, A. Gago-martínez and J. A. Rodríguez-Vázquez, J. Chromatogr. A, 1998, 807, 229–239 CrossRef.
- Y. Amet, F. Berthou and J. F. Menez, J. Chromatogr. B: Biomed. Sci. Appl., 1996, 681, 233–239 CrossRef PubMed.
- K. Sasamoto, T. Ushijima, M. Saito and Y. Ohkura, Anal. Sci., 1996, 12, 189–193 CrossRef.
- P. Pradosa, T. Fukushima, T. Santa, H. Homma, M. Tsunoda, S. Al-Kindy, S. Mori, H. Yokosu and K. Imai, Anal. Chim. Acta, 1997, 344, 227–232 CrossRef.
- S. Uchiyama, T. Santa and K. Imai, Anal. Chem., 2001, 73, 2165–2170 CrossRef PubMed.
- J. You, W. Zhang and Y. Zhang, Anal. Chim. Acta, 2001, 436, 163–172 CrossRef.
- X. Chen, X. Dong and J. You, Chromatographia, 2010, 72, 171–175 Search PubMed.
- Y. Cao and Y. Suo, J. Food Compos. Anal., 2010, 23, 100–106 CrossRef.
- J. H. Wolf and J. Korf, J. Pharm. Biomed. Anal., 1992, 10, 99–107 CrossRef PubMed.
- T. Rezanka and J. Votruba, Anal. Chim. Acta, 2002, 465, 273–297 CrossRef.
- T. A. van Beek and P. Montoro, J. Chromatogr. A, 2009, 1216, 2002–2032 CrossRef PubMed.
- K. Takahashi and N. Goto-Yamamoto, J. Chromatogr. A, 2011, 1218, 7850–7856 CrossRef PubMed.
- O. Quehenberger, A. M. Armando and E. A. Dennis, BBA, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2011, 1811, 648–656 Search PubMed.
- H. Yoshida, K. Ureshino, J. Ishida, H. Nohta and M. Yamaguchi, Anal. Sci., 1999, 15, 937–941 CrossRef.
- H. Miwa and M. Yamamoto, J. Chromatogr., 1987, 416, 237–245 CrossRef PubMed.
- J. You, X. Zhao, Y. Suo, Y. Li, H. Wang and G. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 848, 283–291 CrossRef PubMed.
- Y. Tsukamoto, T. Santa, H. Saimaru, K. Imai and T. Funatsu, Biomed. Chromatogr., 2005, 19, 802–808 CrossRef PubMed.
- X. Du, H. Zhang, X. Guo, Y. Deng and H. Wang, J. Chromatogr. A, 2007, 1169, 77–85 CrossRef PubMed.
- Z. Zeng, Z. Ji, N. Hu, B. Bai, H. Wang and Y. Suo, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1064, 151–159 CrossRef PubMed.
- M. Jonnada, G. D. E. Rassi and Z. E. Rassi, Electrophoresis, 2017, 38, 1592–1601 CrossRef PubMed.
- Y. Sun, X. Zhang, Z. Ji, C. Song, Z. Sun and J. You, Chromatographia, 2016, 79, 547–559 Search PubMed.
- M. B. Majnooni, B. Mohammadi, R. Jalili, A. Babaei and G. Bahrami, J. Liq. Chromatogr. Relat. Technol., 2016, 39, 877–881 CrossRef.
- M. Nishikiori, H. Iizuka, H. Ichiba, K. Sadamoto and T. Fukushima, J. Chromatogr. Sci., 2015, 53, 537–541 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |