
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determination of the Lewis acidity of amide–AlCl3 based ionic liquid analogues by combined in situ IR titration and NMR methods
Pengcheng Hu *,
Wei Jiang,
Lijuan Zhong and
Shu-Feng Zhou
*,
Wei Jiang,
Lijuan Zhong and
Shu-Feng Zhou
College of Chemical Engineering, Huaqiao University, Xiamen, 361021, Fujian, China. E-mail: hupc1987@hqu.edu.cn; szhou@hqu.edu.cn
First published on 10th April 2018
Abstract
A combinatorial method to determine both acidic strength and acidic amount of each Lewis acid site in amide–AlCl3 based ionic liquid (IL) analogues was developed by the combination of in situ IR titration and NMR analysis. 31P NMR was used to distinguish effectively the acidic strength of each Lewis acid site in the amide–AlCl3 based IL analogues. Nitrobenzene was used as a molecular probe to measure the total Lewis acidic amount of the amide–AlCl3 based IL analogues by in situ IR titration. The acidic amount of each Lewis acid site in the amide–AlCl3 based IL analogues was calculated with the assistance of 27Al NMR analysis.
Acidic amide–AlCl3 based ionic liquid (IL) analogues have attracted significant attention as good alternatives to traditional imidazolium and pyridinium based halometallate ILs due to their broad acidity-adjusting range, high catalytic activity, low toxicity and cost, and easy preparation.1–3 Amide–AlCl3 based IL analogues exhibit a mixture of neutral molecular Al species, and cationic and anionic Al species in equilibrium, which contribute to the incomplete asymmetric splitting of Al2Cl6 under the induction of amide.4,5 Therefore, multiple Lewis acidic species with catalytic activity exist in these ILs analogues.6 The Lewis acidity of the amide–AlCl3 based IL analogues, including acidic strength and amount, is correlated with their catalytic activity and selectivity.7–9 Hence, it is necessary to establish a suitable method to determine the acidic strength and amount of each Lewis acid in these IL analogues, which can guide corresponding acid-catalyzed reactions.
For traditional ILs, the spectral measurement methods of the acidity are mainly UV-vis, NMR and IR spectroscopies. The UV-vis spectroscopy method determines semi-quantitatively the acidic strength of total Brønsted acid in ILs according to the Hammett function,10–12 but it could not be applied in the analysis of Lewis acid in ILs, such as [Al2Cl7]− in chloroaluminate ILs. The Lewis acidic strength can be quantified by the Gutmann acceptor number, which is directly proportional to the 31P NMR chemical shift of triethylphosphineoxide (TEPO) dissolved in ILs.13,14 The 31P NMR method can distinguish effectively the acidic strength of each Lewis acid in ILs with multiple Lewis acids, but it could not measure the acidic amount of each Lewis acid.15–17 The traditional KBr tabletting IR uses nitrogen-containing compounds as molecular probes, such as pyridine and ethanenitrile. The change in the IR frequencies of the molecular probes is correlated to the acidic strength of the acid species in ILs. The tabletting IR method can distinguish evidently the Brønsted and Lewis acid according to the wavenumber of the characteristic peaks.18,19 For example, two peak at 1450 cm−1 and 1540 cm−1 were the indication of pyridine coordinated to Lewis and Brønsted acid, respectively.20 But this method neither distinguishes easily the acidic strength of each Lewis acid in ILs with multiple Lewis acids because of the overlap of characteristic peaks, nor can it measure the acidic amount of each Lewis acid. In addition, infrared studies of ammonia adsorption and microcalorimetry were also used by Dupont Company to investigate the acidity of zeolite.21
In this communication, we first establish a combinatorial method to determine the acidity of amide–AlCl3 based IL analogues with multiple Lewis acids by combining in situ IR titration with NMR analysis. This method not only distinguished effectively the acidic strength of each Lewis acid in amide–AlCl3 based IL analogues, but also measured the acidic amount of each Lewis acid.
Firstly, 31P NMR was used to identify the acidic strength of each Lewis acid in amide–AlCl3 based IL analogues, as shown in Fig. 1. A single peak at 83.48 ppm was observed in the 31P NMR spectra of molecular probe (TEPO) dissolved in neat Et3NHCl–AlCl3 IL (molar ratio of Et3NHCl to AlCl3 was 0.65), which was assigned to the coordination of TEPO to Lewis acid. This result indicated that neat Et3NHCl–AlCl3 IL only contained single Lewis acid, namely [Al2Cl7]−. However, two peaks at 83.48 and 84.92 ppm were observed in neat NMA–AlCl3 IL analogue (molar ratio of N-methylacetamide to AlCl3 was 0.65, marked as 0.65NMA–1.0AlCl3) with the addition of TEPO. This phenomenon indicated that another Lewis acid in addition to [Al2Cl7]− existed in 0.65NMA–1.0AlCl3. The peak at 84.92 ppm was assigned to the cationic Al species because the molecule Al species was neutral.5 Meanwhile, the acidic strength of cationic Al species located in low field was stronger than that of [Al2Cl7]−.
 | ||
| Fig. 1 31P NMR spectra of three amide–AlCl3 based IL analogues and Et3NHCl–AlCl3 IL with 1 mol% TEPO (ligand/AlCl3 molar ratio was 0.65). | ||
Subsequently, in situ IR titration method was used to measure the acidic mount of two Lewis acids in neat 0.65NMA–1.0AlCl3. The principle of this method is based on the online monitoring of the variation in the characteristic peaks formed by the coordination of indicator (nitrobenzene) with 0.65NMA–1.0AlCl3.22,23 A quantitative measurement of the acidic amount of 0.65NMA–1.0AlCl3 was made based on the typical procedure. 0.65NMA–1.0AlCl3 (10 g) was placed into a 25 mL two-necked flask equipped with a stirrer. The silicon probe of the in situ IR apparatus was inserted into the 0.65NMA–1.0AlCl3, and then the data on the IR spectra were collected. Next, nitrobenzene (0.25 g) was added dropwise to the flask and IR spectra were collected continuously until the absorbance of the characteristic peaks remained constant, meanwhile, the peaks of nitrobenzene itself were observed. The aforementioned steps were repeated until the absorbance of the characteristic peaks did not change with the addition of nitrobenzene. This point was marked as the terminal point of titration, the total mass of nitrobenzene added into 0.65NMA–1.0AlCl3 was collected.
As a premise of the in situ IR titration method, the characteristic peak formed by the coordination of nitrobenzene with 0.65NMA–1.0AlCl3 and the peak of nitrobenzene itself needed to be marked. Fig. 2 shows the IR spectra of neat nitrobenzene, neat 0.65NMA–1.0AlCl3, and the mixture of 0.65NMA–1.0AlCl3 with nitrobenzene. Two peaks at 1520 and 1346 cm−1 were observed in neat nitrobenzene, which were assigned to the υas(O–N–O) and υs(O–N–O) stretching vibration of –NO2 group, respectively.24,25 A new peak at 1260 cm−1 was observed in the mixture of 0.65NMA–1.0AlCl3 with nitrobenzene, which should be assigned to the coordination of nitrobenzene with Lewis acids. Meanwhile, the υas(O–N–O) stretching vibration at 1520 cm−1 shifted to higher wavenumber 1537 cm−1. The υs(O–N–O) stretching vibration at 1346 cm−1 appeared only in the case that excess nitrobenzene were added into 0.65NMA–1.0AlCl3. Therefore, the peaks at 1260 and 1346 cm−1 were chosen as the characteristic peaks to observe in the following in situ IR titration method.
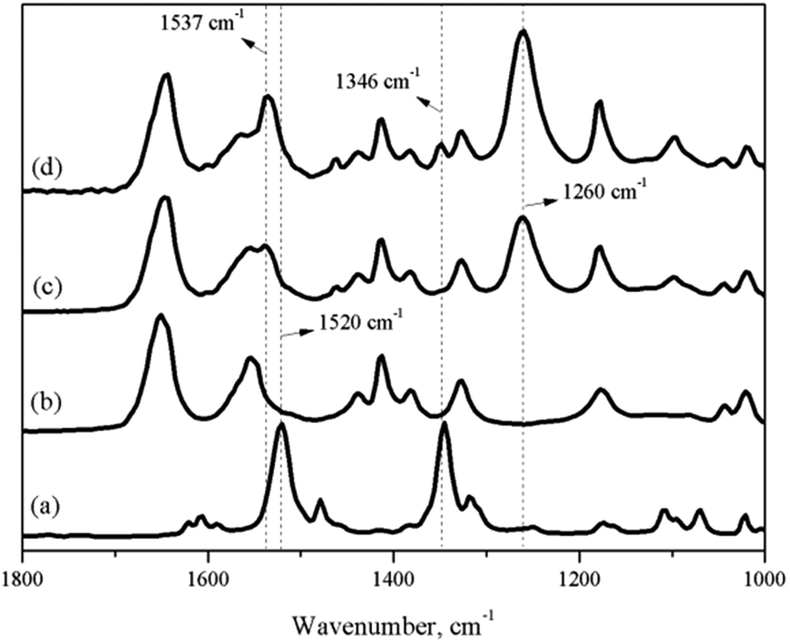 | ||
Fig. 2 IR spectra of (a) pure nitrobenzene; (b) neat 0.65NMA–1.0AlCl3 based IL analogue; (c) nitrobenzene + 0.65NMA–1.0AlCl3 based IL analogue (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 by mass ratio); (d) nitrobenzene + 0.65NMA–1.0AlCl3 based IL analogue (1:4 by mass ratio). :10 by mass ratio); (d) nitrobenzene + 0.65NMA–1.0AlCl3 based IL analogue (1:4 by mass ratio). | ||
Fig. 3 shows the variation of the characteristic peaks at 1260 cm−1 and 1346 cm−1 from the coordination of nitrobenzene with 0.65NMA–1.0AlCl3 and υs(O–N–O) stretching vibration of nitrobenzene, respectively. A surface plot was generated during the continuous addition of nitrobenzene into 0.65NMA–1.0AlCl3 (Fig. 4). The absorbance of the peak at 1260 cm−1 increased with the increasing addition of nitrobenzene, while the absorbance of the peak at 1346 cm−1 remained almost constant before the terminal point, which attributed that the Lewis acidic amount of 0.65NMA–1.0AlCl3 was continuously consumed by nitrobenzene. When the Lewis acidic amount of 0.65NMA–1.0AlCl3 was used up, the absorbance of the peak at 1346 cm−1 had a significantly increase with the addition of nitrobenzene. The total mass of nitrobenzene from start to terminal point was recorded, and “the molar consumption of nitrobenzene per 1000 g IL analogue” was defined as “activity index” to evaluate the acidic amount of 0.65NMA–1.0AlCl3.26,27
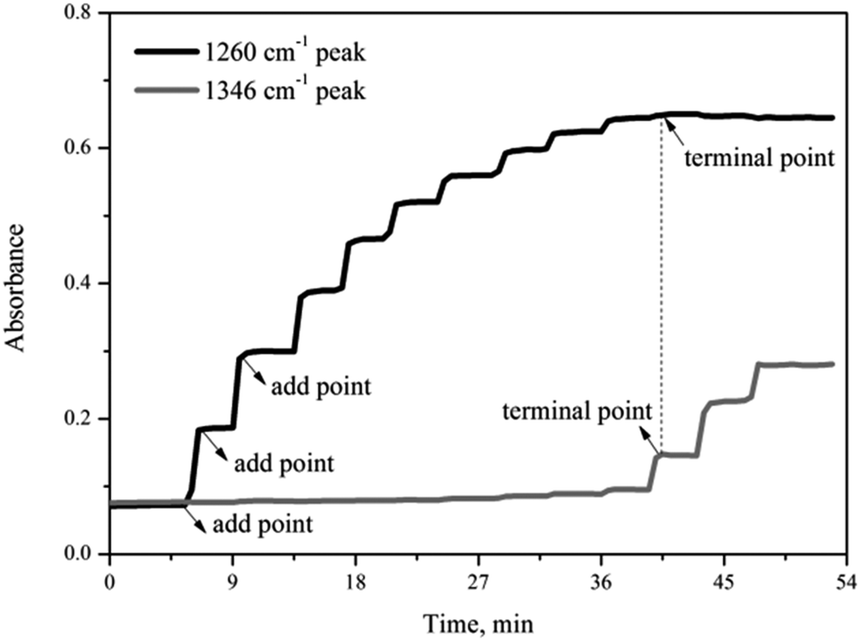 | ||
| Fig. 3 Trend of the characteristic peaks at 1260 cm−1 and 1346 cm−1 for the addition of nitrobenzene into 0.65NMA–1.0AlCl3 based IL analogue. | ||
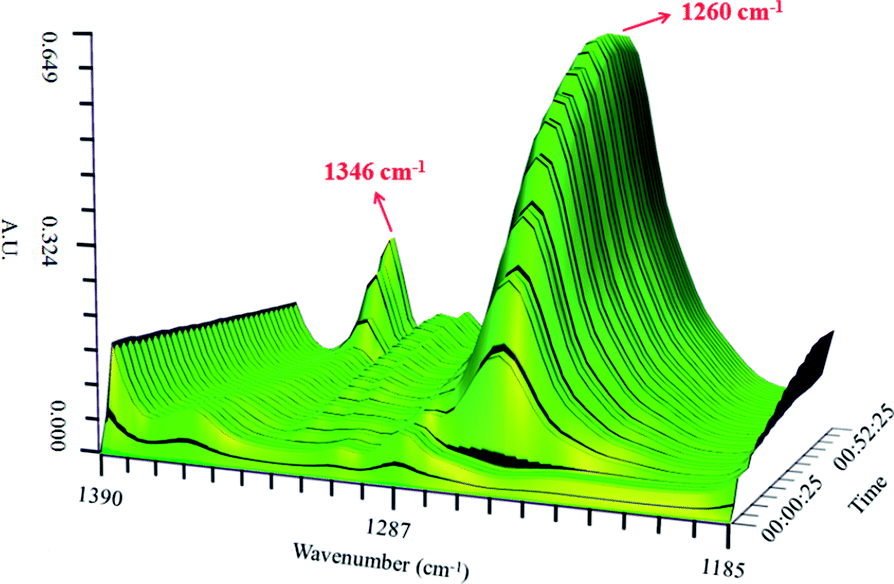 | ||
| Fig. 4 Surface plot in the 1390–1185 cm−1 range for the 0.65NMA–1.0AlCl3 based IL analogue with the addition of nitrobenzene. | ||
The Lewis acidic amount of several amide–AlCl3 based IL analogues with different amide structures and amide/AlCl3 molar ratios were measured by in situ IR titration method, as shown in Fig. 5. The amide structure affected the Lewis acidic amount of amide–AlCl3 based IL analogues,28 for example, the Lewis acidic amount of amide–AlCl3 based IL analogues (NMA–AlCl3 and DMA–AlCl3) with bidentate coordination was higher than that of amide–AlCl3 based IL analogues (AA–AlCl3 and Ur–AlCl3) with monodentate coordination under the same amide/AlCl3 molar ratio.29 This phenomenon was attributed to the fact that the bidentate coordination was more favorable to the asymmetric splitting of AlCl3 than the monodentate coordination with the same amide/AlCl3 molar ratio, resulting in the more active Lewis species (anionic Al species and cationic Al species). On the other hand, the amide/AlCl3 molar ratio also affected the Lewis acidic amount of amide–AlCl3 based IL analogues. The Lewis acidic amount of amide–AlCl3 based IL analogues increased with the decreasing amide/AlCl3 molar ratio. For amide–AlCl3 based IL analogues, the balance between neutral molecular Al species and ionic Al species was readily broken with the change of amide/AlCl3 molar ratio. The asymmetric splitting degree of Al2Cl6 increased and the molecular species transformed into ionic species as the amide/AlCl3 molar ratio decreased, so the Lewis acidic amount of amide–AlCl3 based IL analogue also increased.
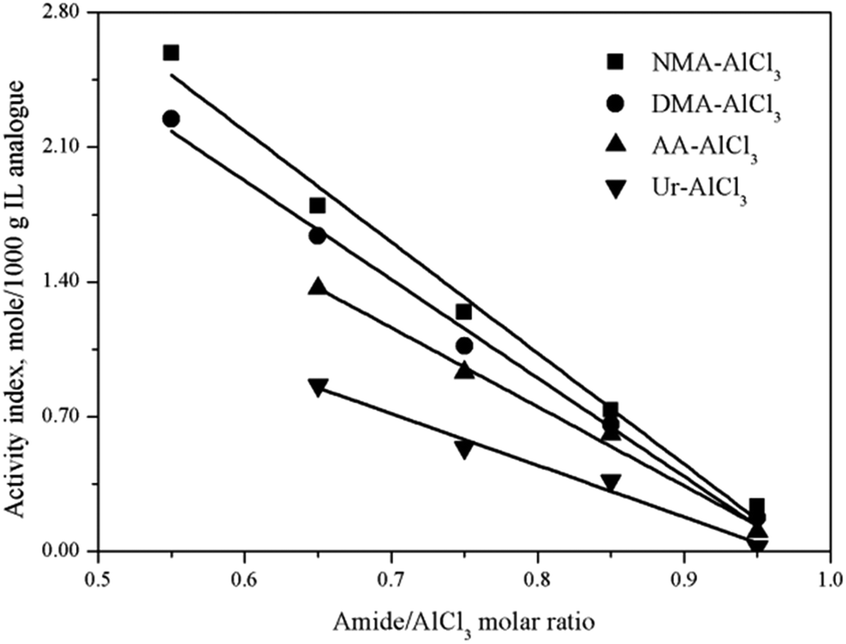 | ||
| Fig. 5 Activity index of four amide–AlCl3 based IL analogues at different amide/AlCl3 molar ratios. | ||
The total Lewis acidic amount of amide–AlCl3 based IL analogues could be measured by in situ IR titration method, but the acidic amount of anionic Al species and cationic Al species needed to be further determined. 27Al NMR is a good tool to distinguish these Al species, and the peaks at 102.75, 89.30 and 77.05 ppm should be assigned to the anionic Al species ([Al2Cl7]− and [AlCl4]−), molecular Al species [AlCl3Ln], and cationic Al species [AlCl2Ln]+, respectively.29 The integral area ratio of anionic Al species ([Al2Cl7]−, [AlCl4]−) to cationic Al species ([AlCl2Ln]+) was obtained by the normalization method of the peak areas, as shown in Fig. 6. The integral area represented the number of Al nucleus (note: [Al2Cl7]− had two Al nuclei). Therefore, the integral area ratio of anionic Al species to cationic Al species represented the molar ratio of 2 × [Al2Cl7]− + [AlCl4]− to [AlCl2Ln]+. The mole of [Al2Cl7]− + [AlCl4]− was equal to that of [AlCl2Ln]+ according to the conservation law of charge, so the molar ratio of [Al2Cl7]− to [AlCl2Ln]+ could be calculated. Taking NMA–AlCl3 based IL analogue with different NMA/AlCl3 molar ratios as an example, the acidic amount of two Lewis acids ([Al2Cl7]− and [AlCl2Ln]+) in NMA–AlCl3 based IL analogue was calculated from the results of both in situ IR titration and 27Al NMR analysis, as listed in Table 1.
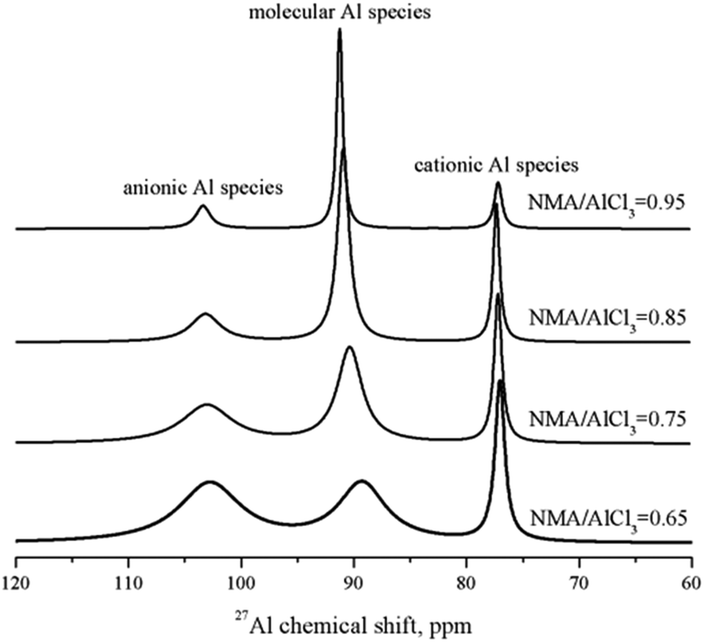 | ||
| Fig. 6 27Al NMR spectra of the NMA–AlCl3 based IL analogue with different NMA/AlCl3 molar ratios. | ||
| NMA/AlCl3 | Molar ratio, mol/mol | Acidic amount, mol nitrobenzene/1000 g IL analogue | |||
|---|---|---|---|---|---|
| (2 × [Al2Cl7]− + [AlCl4]−)/[AlCl2Ln]+ | [Al2Cl7]−/[AlCl2Ln]+ | [AlCl4]−/[AlCl2Ln]+ | [Al2Cl7]− | [AlCl2Ln]+ | |
| 0.65 | 1.77 | 0.77 | 0.23 | 0.7808 | 1.0140 |
| 0.75 | 1.58 | 0.58 | 0.42 | 0.4562 | 0.7866 |
| 0.85 | 1.41 | 0.41 | 0.59 | 0.2134 | 0.5205 |
| 0.95 | 1.19 | 0.19 | 0.81 | 0.0372 | 0.1958 |
Conclusions
An efficient method to determine the acidic strength and acidic amount of each Lewis acid in amide–AlCl3 based IL analogue was proposed in this study. The 31P NMR using triethylphosphineoxide as a molecular probe showed that two active Lewis acids ([AlCl2Ln]+, [Al2Cl7]−) existed in amide–AlCl3 based IL analogues, and the acidic strength of [AlCl2Ln]+ was stronger than that of [Al2Cl7]−. The principle of the in situ IR titration method was described in detail, and the total Lewis acidic amount of these IL analogues was measured with nitrobenzene as indicator. The results indicated that the total Lewis acidic amount of amide–AlCl3 based IL analogues was related with both the amide structure and the amide/AlCl3 molar ratio. The amide–AlCl3 based IL analogues with bidentate coordination structure and low amide/AlCl3 molar ratio had a high Lewis acidic amount. The Lewis acidic amount of each Lewis acid was calculated further by combining 27Al NMR analysis with in situ IR titration.Experiments
The general route for the synthesis of 0.65amide–1.0AlCl3 IL analogue was as follows: anhydrous AlCl3 (0.2 mol) was placed in 250 mL two-necked flask; then, amide (0.13 mol; N-methylacetamide, NMA; N,N-dimethylacetamide, DMA; acetamide, AA; urea, Ur) was added slowly while stirring for 30 min. The mixture was then heated to 80 °C and maintained at that temperature until all solids “dissolved” (approximately 4 h).30IR spectra over the 4000 cm−1 to 650 cm−1 frequency range were obtained at room temperature and at 8 cm−1 resolution using an in situ IR spectrometer (Mettler-Toledo) equipped with an attenuated total reflectance based silicon probe and a liquid nitrogen-cooled mercury-cadmium-tellurium (MCT) detector. IL analogue (10 g) was placed into a 25 mL two-necked flask equipped with a stirring bar at room temperature. The silicon probe was then inserted into the IL analogue, after which date on the IR spectra were collected. Next, the indicator (0.05 g) was added dropwise to the flask and IR spectra were collected continuously until the characteristic peaks remained constant. The aforementioned steps were repeated. During the measurement, the optical path of the spectrometer was continuously purged with dry N2 at a flow rate of 2 mL min−1 to eliminate moisture and CO2. 27Al and 31P NMR spectra were obtained using a Bruker Avance spectrometer.
The samples were placed into a 10 mm standard tube by inserting a well-centered capillary. Thereafter, the NMR tube was capped and sealed with parafilm. The aqueous solutions of Al(NO3)3 (1.0 mol L−1) and H3PO5 (85 wt%) in the capillary was used as an external reference for the 27Al NMR and 31P NMR chemical shift, respectively. Peak intensities and areas were carefully measured using the Brucker-NMR software package.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support of the Scientific Research Funds of Huaqiao University (No. 600005-Z17Y0073), Xiamen, China.References
- A. P. Abbott, J. C. Barron, K. S. Ryder and D. Wilson, Chem.–Eur. J., 2007, 13, 6495–6501 CrossRef CAS PubMed.
- A. P. Abbott, R. C. Harris, Y. T. Hsieh, K. S. Ryder and I. W. Sun, PCCP Phys. Chem. Chem. Phys., 2014, 16, 14675–14681 RSC.
- M. Li, B. L. Gao, C. Y. Liu, W. T. Chen, Z. N. Shi, X. W. Hu and Z. W. Wang, Electrochim. Acta, 2015, 180, 811–814 CrossRef CAS.
- H. M. A. Abood, A. P. Abbott, A. D. Ballantyne and K. S. Ryder, Chem. Commun., 2011, 47, 3523–3525 RSC.
- F. Coleman, G. Srinivasan and M. Swadźba-Kwaśny, Angew. Chem., Int. Ed., 2013, 52, 12582–12586 CrossRef CAS PubMed.
- Y. X. Fang, K. Yoshii, X. G. Jiang, X. G. Sun, T. Tsuda, N. Mehio and S. Dai, Electrochim. Acta, 2015, 160, 82–88 CrossRef CAS.
- J. M. Hogg, F. Coleman, A. Ferrer-Ugalde, M. P. Atkins and M. Swadźba-Kwaśny, Green Chem., 2015, 17, 1831–1841 RSC.
- K. Matuszek, A. Chrobok, J. Hogg, F. Coleman and M. Swadźba-Kwaśny, Green Chem., 2015, 17, 4225–4262 RSC.
- P. C. Hu, Y. D. Wang, X. H. Meng, R. Zhang, H. Y. Liu, C. M. Xu and Z. C. Liu, Fuel, 2017, 189, 203–209 CrossRef CAS.
- G. P. Smith, A. S. Dworkin, R. M. Pagni and S. P. Zingg, J. Am. Chem. Soc., 1989, 111, 525–530 CrossRef CAS.
- G. P. Smith, A. S. Dworkin, R. M. Pagni and S. P. Zingg, J. Am. Chem. Soc., 1989, 111, 5075–5077 CrossRef CAS.
- C. Thomazeau, H. Olivier-Bourbigou, L. Magna, S. Luts and B. Gilbert, J. Am. Chem. Soc., 2003, 125, 5264–5265 CrossRef CAS PubMed.
- R. A. Osteryoung and T. A. Zawodzinski Jr, Inorg. Chem., 1989, 28, 1710–1715 CrossRef.
- M. Schmeisser, P. Illner, R. Puchta, A. Zahl and R. van Eldik, Chem.–Eur. J., 2012, 18, 10969–10982 CrossRef CAS PubMed.
- J. Estager, A. A. Oliferenko, K. R. Seddon and M. Swadźba-Kwaśny, Dalton Trans., 2010, 39, 11375–11382 RSC.
- R. A. Mantz, P. C. Trulove, R. T. Carlin, T. L. Theim and R. A. Osteryoung, Inorg. Chem., 1997, 36, 1227–1232 CrossRef CAS PubMed.
- M. Currie, J. Estager, P. Licence, S. Men, P. Nockemann, K. R. Seddon, M. Swadźba-Kwaśny and C. Terrade, Inorg. Chem., 2013, 52, 1710–1721 CrossRef CAS PubMed.
- S. Tait and R. A. Osteryoung, Inorg. Chem., 1984, 23, 4352–4360 CrossRef CAS.
- F. H. Cao, L. Tian, N. Luo, D. Y. Fang, W. Y. Ying and J. A. Wang, Catal. Commun., 2009, 10, 1310–1312 CrossRef CAS.
- Y. L. Yang and Y. Kou, Chem. Commun., 2004, 226–227 RSC.
- R. D. Shannon, R. H. Staley and A. Auroux, Zeolites, 1987, 7, 301–306 CrossRef CAS.
- P. C. Hu, R. Zhang, Z. C. Liu, H. Y. Liu, C. M. Xu, X. H. Meng, M. Liang and S. S. Liang, Energy Fuels, 2015, 29, 6019–6024 CrossRef CAS.
- D. D. Dunuwila, L. B. Carroll Il and K. A. Berglund, J. Cryst. Growth, 1994, 137, 561–568 CrossRef CAS.
- J. Clarkson and W. Ewen Smith, J. Mol. Struct., 2003, 655, 413–422 CrossRef CAS.
- J. D. Laposa, Spectrochim. Acta. A, 1979, 35, 65–71 CrossRef.
- X. Zhang, R. Zhang, H. Y. Liu, X. H. Meng, C. M. Xu, Z. C. Liu and P. A. A. Klusener, Ind. Eng. Chem. Res., 2016, 55, 11878–11886 CrossRef CAS.
- R. Zhang, Z. C. Liu, X. Zhang, X. H. Meng, H. Y. Liu, C. M. Xu and P. A. A. Klusener, WO Patent 202905 A1, 2016.
- M. P. Atkins, K. R. Seddon, M. Swadźba-Kwaśny and F. Coleman, US Pat. 0052838 A1, 2016.
- P. C. Hu, R. Zhang, X. H. Meng, H. Y. Liu, C. M. Xu and Z. C. Liu, Inorg. Chem., 2016, 55, 2374–2380 CrossRef CAS PubMed.
- A. P. Abbott, WO Patent 003956 A2, 2007.
| This journal is © The Royal Society of Chemistry 2018 |