
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene nanoplatelets-sericin surface-modified Gum alloy for improved biological response†
Valentina
Mitran
 a,
Valentina
Dinca
b,
Raluca
Ion
*a,
Vasile D.
Cojocaru
c,
Patricia
Neacsu
a,
Cerasela Zoica
Dinu
d,
Laurentiu
Rusen
b,
Simona
Brajnicov
be,
Anca
Bonciu
bf,
Maria
Dinescu
b,
Doina
Raducanu
c,
Ioan
Dan
g and
Anisoara
Cimpean
*a
a,
Valentina
Dinca
b,
Raluca
Ion
*a,
Vasile D.
Cojocaru
c,
Patricia
Neacsu
a,
Cerasela Zoica
Dinu
d,
Laurentiu
Rusen
b,
Simona
Brajnicov
be,
Anca
Bonciu
bf,
Maria
Dinescu
b,
Doina
Raducanu
c,
Ioan
Dan
g and
Anisoara
Cimpean
*a
aUniversity of Bucharest, Department of Biochemistry and Molecular Biology, 91-95 Spl. Independentei, 050095, Bucharest, Romania. E-mail: anisoara.cimpean@bio.unibuc.ro; rciubar@yahoo.com; Fax: +40 21 3181575/102; Tel: +40 21 3181575/106
bNational Institute for Lasers, Plasma and Radiation Physics, 409 Atomistilor Street, Magurele, 077125, Romania
cUniversity POLITEHNICA of Bucharest, 313 Spl. Independentei, 060042 Bucharest, Romania
dWest Virginia University, Department of Chemical and Biomedical Engineering, Morgantown, West Virginia 26506, USA
eUniversity of Craiova, Faculty of Mathematics and Natural Science, 13 Alexandru Ioan Cuza, 200585 Craiova, Romania
fUniversity of Bucharest, Faculty of Physics, 405 Atomistilor, Magurele, 077125, Romania
gSC R&D ConsultantasiServicii SRL, 45 Maria Ghiculeasa, 020943 Bucharest, Romania
First published on 21st May 2018
Abstract
In this study a “Gum Metal” titanium-based alloy, Ti-31.7Nb-6.21Zr-1.4Fe-0.16O, was synthesized by melting and characterized in order to evaluate its potential for biomedical applications. The results showed that the newly developed alloy presents a very high strength, high plasticity and a low Young's modulus relative to titanium alloys currently used in medicine. For further bone implant applications, the newly synthesized alloy was surface modified with graphene nanoplatelets (GNP), sericin (SS) and graphene nanoplatelets/sericine (GNP–SS) composite films via Matrix Assisted Pulsed Laser Evaporation (MAPLE) technique. The characterization of each specimen was monitored by scanning electron microscopy (SEM), atomic force microscopy (AFM), contact angle (CA) measurements, and Fourier Transform Infrared Spectroscopy (FTIR). The materials' surface analyses suggested the successful coating of GNP, SS and GNP–SS onto the alloy surface. Additionally, the activities of pre-osteoblasts such as cell adhesion, cytoskeleton organization, cell proliferation and differentiation potentials exhibited on these substrates were investigated. Results showed that the GNP–SS-coated substrate significantly enhanced the growth and osteogenic differentiation of MC3T3-E1 cells when compared to bare and GNP-coated alloy. Collectively, the results show that GNP–SS surface-modified Gum alloy can modulate the bioactivity of the pre-osteoblasts holding promise for improved biological response in vivo.
1 Introduction
During the last decade, a special class of titanium-based alloys called Gum-type alloys was investigated1–4 in order to develop new materials targeted for use in biomedical domain as dental and orthopaedic materials, due to their superior mechanical properties, corrosion resistance and biocompatibility. The uniqueness of Gum-type alloys lies in their combination of mechanical properties. Their low elastic modulus, high strength and large elastic strain characteristics are due to oxygen addition in Ti-β phase (bcc – body centred cubic) crystalline structure, with oxygen acting like an interstitial hardening element, which is capable of increasing the strength of the Ti-β phase and partially suppressing the Ti-β → Ti-α′′ phase transformation.5–8 Low modulus of elasticity allows proper loading of the bone, inhibiting bone atrophy and enhancing bone formation.9–11 Furthermore, the high corrosion resistance and biocompatibility limit failures such as fatigue fractures, implant loosening, and adverse reactions due to ion release. To further improve the quality of the biomaterial integration in host tissue and of the biocompatibility in general, most of metallic implants are subjected to surface treatments.Over the past few years, graphene (G) has received increased attention due to its unique physical and chemical properties, and its resulting biocompatibility. G, the elementary structure of graphite, is an atomically-thick sheet composed of sp2 carbon atoms arranged in a flat honeycomb structure. In addition to (monolayer) G, various G-based materials (GBMs) with different numbers of layers, lateral dimensions, and chemical modifications have also been developed. These include ultrathin multilayer materials made by graphite exfoliation, few-layer materials, graphene oxide (GO) and other chemically modified G, as well as carbon materials made from both G or GO as atomically-thin precursors to be used for the design of various three-dimensional (3D) architectures.12 Beyond the broad range of technical applications, the biomedical application of G and GBMs is an area with very rapid expansion. So far, investigations have been carried out to explore the use of G and GBMs for drug delivery,13–16 biological sensing and imaging,17–21 and as antibacterial materials,22–25 and biocompatible scaffolds for cell culture.26–28 Some examples of the reported advantages of G and GBMs are: (a) improvement of biomaterials' mechanical/electrical properties; (b) enhancement of cellular attachment and growth at biomaterials surface; (c) production of efficient biosensors, and (d) better imaging, tracking and monitoring of drug delivery.29,30 Recently, another promising area of interest for G was as coating for metallic substrates. Several reports confirmed the capability of G to interfere positively in several processes related to osteogenic differentiation and cellular maturation toward osteoblastic phenotype.31–35 Moreover, G could be used as platform for the release of therapeutic molecules at the surgical sites to improve bone formation and integration of implants. G, GO and reduced GO (rGO) are for instance known to bind to dexamethasone, serum, insulin, albumin, bone morphogenetic protein (BMP)-2, and others.36–38 As such, the application of rGO-based coating loaded dexamethasone onto Ti13Nb13Zr alloy was shown to enhance the differentiation of MC3T3-E1 cells into osteoblasts. Notably, cells seeded on the rGO/dexamethasone-coated alloy presented higher expression of collagen I, osteocalcin (OCN), Runt-related transcription factor 2 (RUNX2) and osteopontin (OPN) genes after seven days in culture. Moreover, it was observed higher alkaline phosphatase (ALP) activity and increased calcium nodule formation when compared to the uncoated alloy or unloaded rGO coating.39
In this context, the present study explored the potential of graphene nanoplatelets (GNP), sericin (SS) and graphene nanoplatelets/sericin (GNP–SS) composite coatings deposited onto the Ti-31.7Nb-6.21Zr-1.4Fe-0.16O Gum alloy to enhance osteogenesis in vitro. SS, a hydrophilic, water soluble macromolecular glycoprotein from silk worm cocoon, was proposed to be used in the composite coatings due to its demonstrated bioactivities, including cell adhesion, biodegradability, biocompatibility, and low-immunogenicity.40–42 An increasing number of studies are directed towards the applications of SS in the field of tissue engineering and regenerative medicine. Thus, it has been explored in the neuronal regeneration,43,44 skin regeneration,45 myocardial repair,46 SS capped particles47,48 and even fabrication of self-assembled nanoparticles for cancer treatment.49 Nayak et al.50 demonstrated for instance that surface modification with SS and arginine–glycine–aspartic acid (RGD) peptide enhanced the osteointegration and osteoconductivity of the orthopaedic titanium implants. However, SS has never been investigated in composite coatings with GNP for applications in orthopedics. Here we demonstrate that GNP–SS composite coatings support cell viability/proliferation and promote MC3T3-E1 pre-osteoblast differentiation to a significantly higher extent than GNP alone.
2 Experimental
2.1. Alloy synthesis and characterization
The Ti-31.7Nb-6.21Zr-1.4Fe-0.16O (% wt) Gum-type alloy was produced using the high-purity individual elemental components, in a cold crucible levitation melting furnace (FIVES CELES MP25) under argon protective atmosphere. Given the high difference in specific weight and melting temperature of the incorporated alloying elements, three re-melts were performed to increase resulting alloy's homogeneity. The melt was cast in 20 × 45 mm diameter ingots inside melting furnace chamber.The samples were cut in the shape of disks using an automatic cutting machine (Metkon Micra Cut 200) and diamond disks. Subsequently, the samples surfaces were grinded down to 600-grit SiC paper (Buehler CarbiMet) in order to remove any surface defects induced by cutting. The Gum alloy's microstructure and chemical homogeneity was metallographically investigated using Scanning Electron Microscopy – SEM (TESCAN Vega II-XMU SEM microscope fitted with a Bruker Quantax 6/30 XFlash EDS detector). The samples for metallographic analysis were hot mounted (Buehler SimpliMet) in conductive phenolic resin (Buehler KonductoMet) and grinded down (Metkon DigiPrep ACCURA) to 1200-grit SiC paper. The samples were polished with 6 μm and 1 μm polycrystalline diamond suspensions (Buehler MetaDi) followed by super-polishing with 0.02 μm colloidal silica (Buehler MasterMet2) and mixed with 10% H2O2 as mild oxidizing agent. For SEM analysis, the metallographic prepared samples were etched by swabbing for 10–15 s with a mixture of HNO3, HF and H2O (6–3–91%).
The X-ray diffraction (XRD) investigations were performed using a PANalytical X'Pert PRO MRD diffractometer with a wavelength of Cu k-alpha (λ = 1.5418 Å). The recorded XRD spectra were fitted to determine the phase structure and to calculate the lattice parameters of the observed phases. The fitting procedure was performed using PeakFit v4.11 software package, which allowed to determine for each of the observed diffracted peak its position, intensity and broadening respectively. A pseudo-Voigt diffraction line profile was used in the fitting procedure.
The mechanical characterization of the alloy was performed using a DEBEN MicroTest 2000N universal micromechanical testing module, at a 0.4 mm min−1 cross-head speed. The tests were repeated 10 times, all computed data being statistically analysed in order to determine for each parameter its standard deviation.
The obtained samples were further cleaned by sonication with ethanol and double distilled water (30 min each step) before being functionalized by Matrix Assisted Pulsed Laser Evaporation (MAPLE) with SS-, GNP- and GNP–SS and prepared to assess the in vitro osteogenic response.
2.2. Target preparation
SS (Sericin Bombyx mori (silkworm), code S5201) and GNPs (code 799084) were obtained from Sigma-Aldrich and were suspended, respectively dissolved in double distilled water to lead to 0.01 (for GNP) and 2 (for SS) weight% solutions. The GNPs (as micron sized aggregates of stacks of 2–5 microns large platelets – see Fig. S1a in ESI†) solutions were subsequently sonicated for several hours (10 h) (Sharpertek Digital Ultrasonic cleaner XP PRO) and kept overnight at room temperature. As shown in Fig. S1b,† no significant changes were observed for GNPs after the sonication process, all relative to both powder and suspended GNPs.To form the single element target, the solutions were again sonicated for 15 min and rapidly frozen drop-by-drop in a liquid nitrogen cooled copper container. In the case of the composite target, equal volumes of SS and GNP solutions were mixed together and frozen. The container was then mounted on a cryogenic holder inside the deposition chamber (Neocera spherical vacuum chamber). The target was maintained frozen during deposition by a circulating liquid nitrogen system.
2.3. Substrates preparation
Two types of substrates, namely double polished silicon (Si) (100) transparent in the infrared (Neyco) and Gum alloy, were used in this study. Si was used for FTIR measurements as well as for thickness measurements. The substrates were carefully cleaned in an ultrasonic bath in acetone (99.5% purity, Chimreactiv SRL) ethyl alcohol (99.5% purity, Chimreactiv SRL) and deionized water and subsequently blow-dried under N2 gas before use. All substrates were placed at a distance of 3.5 cm from the frozen target and kept at ambient temperature during the deposition; no post deposition annealing was carried out.2.4. Matrix assisted pulsed laser evaporation (MAPLE) system
A “Surelite II” pulsed Nd: YAG laser system (Continuum Company) (5–7 ns pulse duration) at 266 nm and 10 Hz repetition rate was used to irradiate the frozen targets. The laser fluence was 0.6 J cm−2 and the number of pulses used was 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for GNP and 72000 for SS and GNP–SS coatings, leading to 91, 424 and 458 nm thick coatings, respectively. In order to avoid the damage of the target by local overheating and drilling following multiple pulses during the laser irradiation, the target was rotated with 20 rpm using a motion feed driven motor contained within the system. The background pressure in the deposition chamber was adjusted at 1–2 × 10−3 Pa by TPU 170 turbomolecular pump (Pfeiffer-Balzers). Laser beam interacted with the target surfaces, leading to the evaporation process, during which GNP and SS materials were transported by the formed vapours and deposited onto the substrate placed parallel to the target at a distance of 3 cm.
000 for GNP and 72000 for SS and GNP–SS coatings, leading to 91, 424 and 458 nm thick coatings, respectively. In order to avoid the damage of the target by local overheating and drilling following multiple pulses during the laser irradiation, the target was rotated with 20 rpm using a motion feed driven motor contained within the system. The background pressure in the deposition chamber was adjusted at 1–2 × 10−3 Pa by TPU 170 turbomolecular pump (Pfeiffer-Balzers). Laser beam interacted with the target surfaces, leading to the evaporation process, during which GNP and SS materials were transported by the formed vapours and deposited onto the substrate placed parallel to the target at a distance of 3 cm.
2.5. Structural and morphological characterization of the deposited graphene, graphene–sericin thin films
FTIR was performed using a Jasco FT/IR-6300 type spectrometer in the 400–7800 cm−1 range, with a resolution of 4 cm−1, and was used to analyse the characteristic vibrations of the functional groups of the SS deposited thin films. The materials deposited by drop cast were used as controls for the FTIR measurements.Atomic Force Microscopy (AFM) and SEM were used for the morphological evaluation of the samples. An AFM-E 100, Park system was used for performing measurements in non-contact mode and surface roughness analyses. Qualitative analysis of surface morphological characteristics was carried out by using a scanning electron microscope (JSM-531 microscope (FEI Inspect S)) equipped with energy dispersive X-ray spectrometer EDAX (Element 2CB detector), operating at 5 kV. Samples were coated with 10 nm Au prior to imaging to obtain electrical conductivity.
Contact Angle (CA) measurements were performed with KSV CAM101 microscope equipped with FireWire interface video camera. The sessile drop method was applied at constant room temperature (24 °C) using a syringe with double distilled water, which ensured droplets with a volume of 2 μl. The reported values for the CAs were obtained by averaging five measurements performed on different areas of the samples.
2.6. Cell culture conditions
Cell culture experiments were performed with mouse pre-osteoblast MC3T3-E1 cell line, subclone 4 (CRL-2593™, American Type Culture Collection). The cells were seeded onto sterilized metallic specimens placed in 12-well cell culture plates at a density of 104 cells per cm2 and maintained in a humidified atmosphere of 5% CO2 at 37 °C in Dulbecco's Minimal Essential Medium supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin (10000 units per mL penicillin and 10 mg mL−1 streptomycin).
2.7. Cell viability and proliferation
The cytotoxic potential of the analysed specimens was evaluated by cell staining with LIVE/DEAD Cell Viability/Cytotoxicity Assay Kit (Molecular Probes), in accordance with the manufacturer's instructions. Briefly, after 1 and 5 days of culture, the analysed samples were incubated in a solution consisting of calcein-AM (2 μM) and ethidium homodimer-1 (4 μM) for 15 min at room temperature, protected from the light.51 The samples were then washed with phosphate buffered saline (PBS) and examined under an inverted microscope Olympus IX71 to detect the bright green fluorescent living cells and red fluorescent dead cells. The fluorescent images were captured using Cell F Image acquiring system.The capacity of cells to proliferate onto these substrates was investigated at 1 and 5 days post-seeding by cell incubation with 1 mg mL−1 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich Co.) solution, as previously described.52 The amount of formazan produced by metabolically active viable cells was solubilized with dimethyl sulfoxide and absorbance of dye was recorded at 550 nm using a microplate reader (Thermo Scientific Appliskan).
2.8. Microscopic evaluation of MC3T3-E1 cell morphology
The morphological features of MC3T3-E1 cells grown on each sample were examined using immunofluorescent labeling of actin and vinculin at 1 day and 5 days post seeding. For this, the pre-osteoblast cells grown on the above substrates were fixed, permeabilized, blocked and incubated with specific antibodies, as previously reported.53 Subsequently, phalloidin coupled with Alexa Fluor 488 was added in order to label actin filaments and, then, 4′,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei. Labelled samples were washed three times with PBS and examined under an inverted microscope equipped with epifluorescence (Olympus IX71). The microscopic images were captured using the Cell F software.2.9. Evaluation of the pre-osteoblast differentiation potential
For differentiation assay, the pre-osteoblastic cells were seeded onto samples at a density of 5 × 104 cells per cm2. At 24 h post-seeding, differentiation was induced by maintaining the cells in culture media supplemented with 50 μg mL−1 ascorbic acid (Sigma-Aldrich) and 5 mM beta-glycerophosphate (Sigma-Aldrich). The medium was changed every two days during the culture period.Cell differentiation was evaluated by analysing alkaline phosphatase (ALP) activity at 7 and 14 days post-seeding and by Alizarin Red S (ARS) staining for matrix mineralization after 3 weeks of incubation. The intracellular ALP activity was determined with an Alkaline Phosphatase Activity Colorimetric Assay Kit (BioVision, Milpitas, CA, USA), as previously described.54 For ARS staining, cell layers were washed three times with PBS and fixed in 10% paraformaldehyde. Following three rinses in deionized water, cells were placed in a 1 mg mL−1 ARS (Sigma-Aldrich) solution for 30 min. After washing the samples with deionized water, cells were air-dried for 24 h. Subsequently, ARS stain on specimens was dissolved in 5% v/v perchloric acid in order to measure the optical density at 405 nm.
Immunofluorescence staining was also performed to detect the expression of osteocalcin (OCN). Briefly, the samples were rinsed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 in PBS, and blocked in PBS containing 2% Bovine Serum Albumin (BSA). The samples were then incubated with mouse anti-osteocalcin monoclonal antibody (Santa Cruz Biotechnology) in PBS containing 1.2% BSA. After washes with PBS, they were further incubated with Alexa Fluor 546-conjugated goat anti-mouse IgG antibody (Invitrogen)in PBS containing 1.2% BSA, followed by washing with PBS. A 2 μg mL−1 DAPI solution was used to stain cell nuclei. Fluorescent images were taken with an Olympus IX71 inverted microscope.
2.10. Statistical analysis
Triplicate samples were used in the experiments to ensure the reproducibility of the results. Statistical analysis of the data was performed with GraphPrism software using one-way ANOVA with Bonferroni's multiple comparison tests. All results are expressed as means ± SD (standard deviation) and p values of less than 0.05 were considered statistically significant.3. Results and discussion
3.1. Alloy's characterization
As observed in Fig. 1a, the Ti-31.7Nb-6.21Zr-1.4Fe-0.16O (% wt) Gum-type alloy microstructure consists of polyhedral equiaxed grains, with an average grain-size of about 120 μm. Grains seem to be formed from a mixture of zones, some enriched in Ti, Zr and lean in Nb, and others enriched in Nb and lean in Ti and Zr respectively (Fig. 1b–d). The Fe distribution was non-uniform (Fig. 1e), whereas the O one was more uniform (Fig. 1f) all relative to the other elements, namely Ti, Zr, Fe and Nb.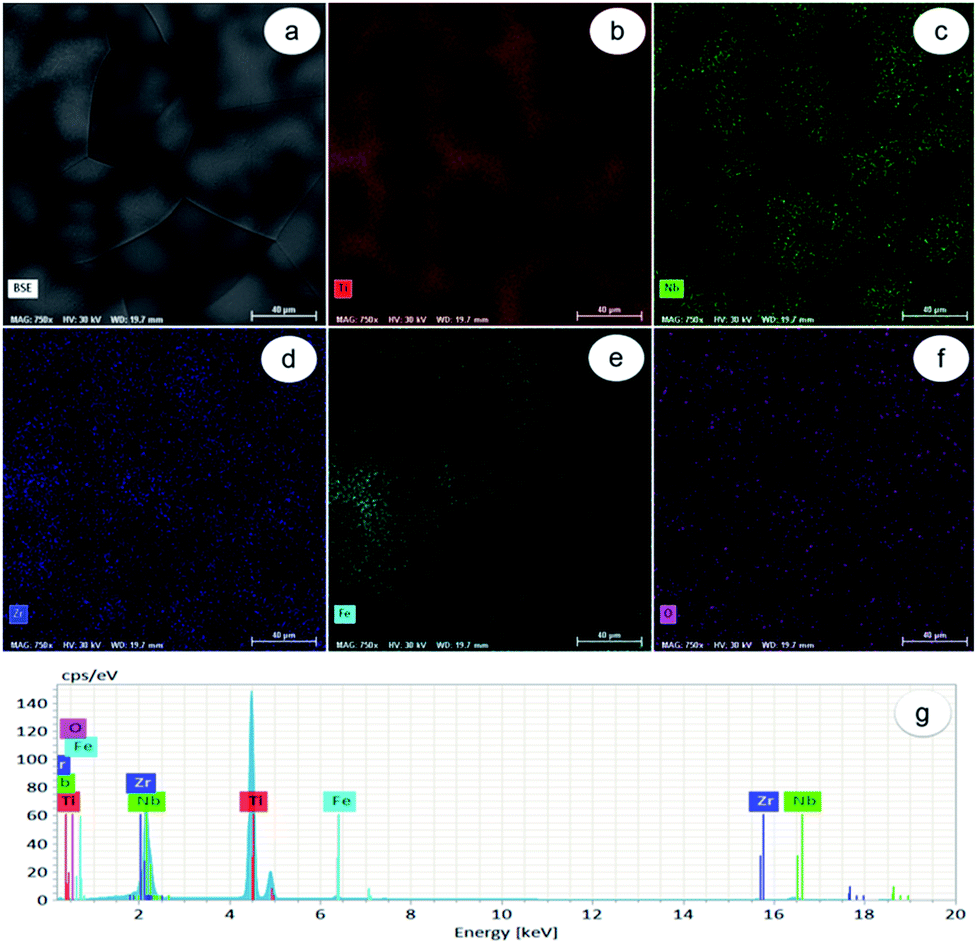 | ||
| Fig. 1 Dispersion of alloying elements in Ti-31.7Nb-6.21Zr-1.4Fe-0.16O (% wt) Gum-type alloy microstructure: (a) SEM-BSE image of investigated field; (b) dispersion map of Ti; (c) dispersion map of Nb; (d) dispersion map of Zr; (e) dispersion map of Fe; (f) dispersion map of O; (g) EDS spectrum of investigated field. Scale bar: 40 μm. | ||
Energy-dispersive X-ray (EDS) spectrum (Fig. 1g) identified separate peaks for Ti, Nb, Zr, Fe and O. The chemical composition of Gum-type alloy computed based on this spectrum is shown in Table 1. Overall, the Gum-type alloy microstructure showed the presence of chemically inhomogeneous zones, which can be diminished/removed either by increasing the number of re-melts during synthesis or by further thermal processing the ingots and homogenization heat treatment.
| Element | Composition [% wt] | Composition [% at] | Absolute error [%] | Relative error [%] |
|---|---|---|---|---|
| Titanium (Ti) | 60.1603 | 73.6959 | 1.6146 | 2.1909 |
| Niobium (Nb) | 31.7612 | 20.0467 | 0.6400 | 3.1925 |
| Zirconium (Zr) | 6.5123 | 4.1859 | 0.1270 | 3.0340 |
| Iron (Fe) | 1.4028 | 1.4728 | 0.0557 | 3.7819 |
| Oxygen (O) | 0.1634 | 0.5987 | 0.0861 | 14.3812 |
The XRD spectra corresponding to the as-obtained alloy are shown in Fig. 2. It was found that β-Ti phase shows the presence of (110), (200) and (211) diffraction lines. The β-Ti phase was indexed in Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m body centred cubic system, with a calculated lattice parameter of about a = 3.31 Å.
m body centred cubic system, with a calculated lattice parameter of about a = 3.31 Å.
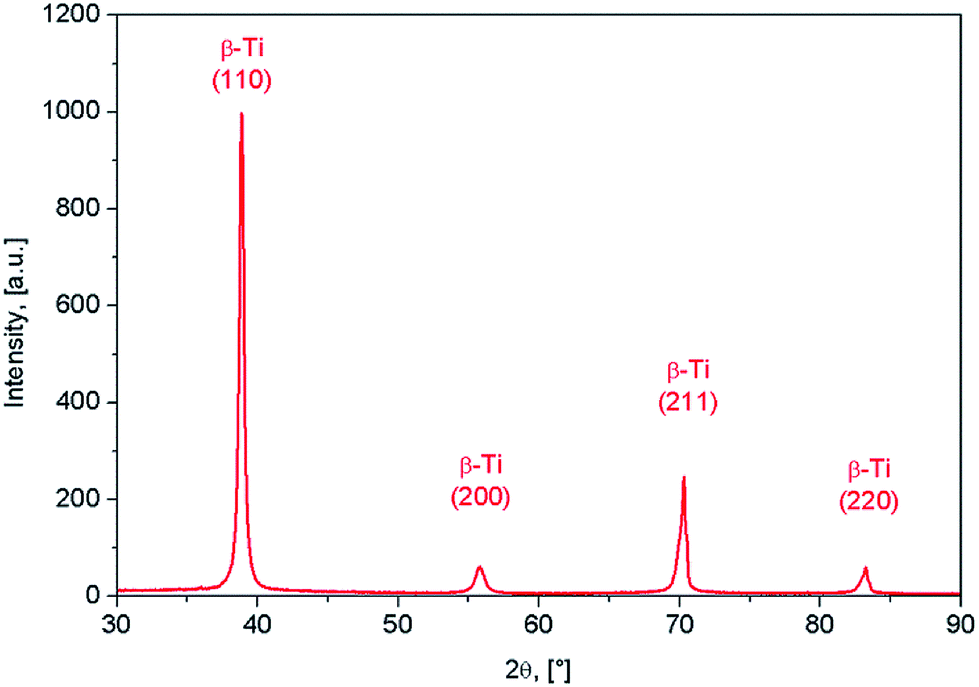 | ||
| Fig. 2 XRD spectra of Ti-31.7Nb-6.21Zr-1.4Fe-0.16O (% wt) Gum-type alloy. | ||
Mechanical characterization aimed to determine the mechanical properties of the Gum-type alloy, which were expressed by 0.2 yield strength (σ#0.2), ultimate tensile strength (σUTS), elongation to fracture (εf), elastic modulus (E) and microhardness (HV0.1). The strain–stress diagram of Gum-type alloy in as-obtained condition is shown in Fig. 3 and indicates high plasticity of the alloy. Also, a high necking process was observed prior to fracture, most likely indicating the presence of a ductile fracture mechanism.
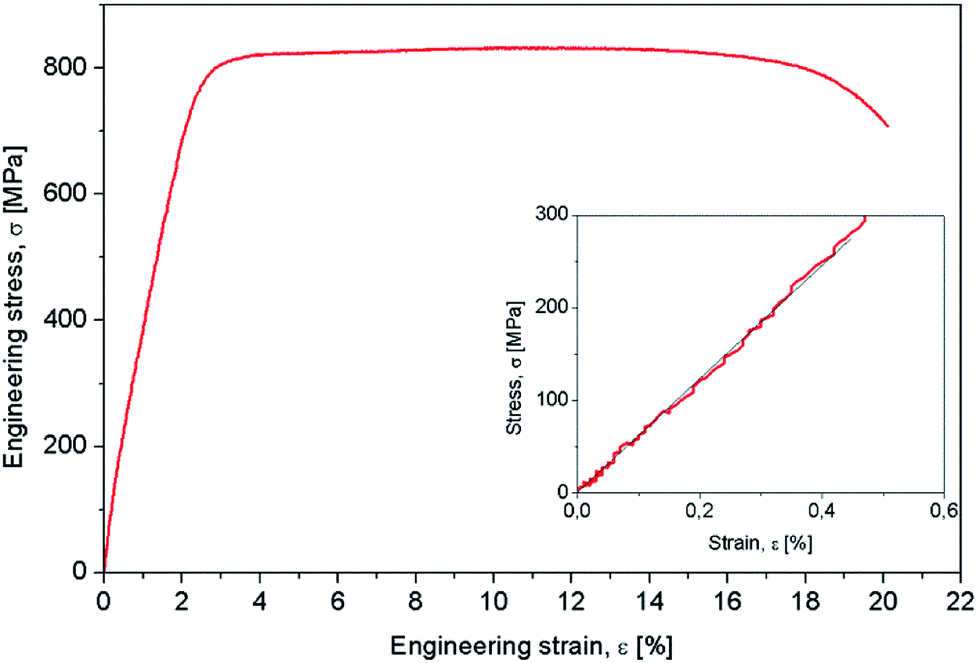 | ||
| Fig. 3 Strain–stress curve of Ti-31.7Nb-6.21Zr-1.4Fe-0.16O (% wt) Gum-type alloy. | ||
Table 2 shows that the Gum-type alloy exhibits a low elastic modulus, close to 58 ± 3,4 GPa, much lower than that of commercial pure titanium (cpTi) (102–104 GPa) which is a conventionally used metallic biomaterial.
| Ultimate tensile strength, σUTS [MPa] | Yield strength, σ0.2 [MPa] | Elongation to fracture, εf [%] | Elastic modulus, E [GPa] |
|---|---|---|---|
| 832 ± 13.8 | 331 ± 6.2 | 19,3 ± 3.1 | 58 ± 3,4 |
3.2. Coatings deposition and characterization
MAPLE was chosen as a suitable method for the coating deposition; previous results showed that a good control on coating composition, morphology, higher adhesion onto different types of substrates,55,56 and, most importantly, the assembling of an unlimited class of compounds, even immiscible ones within the same coating, can be achieved using such technique.57,58 Since its first implementation in the late 1990s, a variety of materials from smart polymers to bioactive compounds, ceramics and proteins were used to enhance cellular responses toward a surface.59–62FTIR analysis of the SS and GNP–SS samples deposited by MAPLE showed mainly typical protein absorption bands corresponding to amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), amide II and amide III (C–N stretching, N–H in-plane bending) bands in the range of 1710–1590 cm−1, 1570–1480 cm−1 and 1270–1200 cm−1, respectively. Despite the observed difference in peaks intensity associated with different thickness of the measured samples which were observed as well in our previous works,57,60 the typical signatures for SS were observed for both control and MAPLE coatings (Fig. 4). Specifically, bands were observed at 1646 cm−1 (νCO for amide I); 1535 cm−1 (νN–H for amide II); and 1230–1400 cm−1 (N–H in plane bend and νC–N for amide III) respectively. The characteristic amide I peak (1646 cm−1) shows the presence of α-helix structures in the protein. The band at 2700–3200 cm−1 corresponds to N–H bend vibration from SS material and was merged with the 3450 cm−1 characteristic to O–H vibration band.63
O stretching), amide II and amide III (C–N stretching, N–H in-plane bending) bands in the range of 1710–1590 cm−1, 1570–1480 cm−1 and 1270–1200 cm−1, respectively. Despite the observed difference in peaks intensity associated with different thickness of the measured samples which were observed as well in our previous works,57,60 the typical signatures for SS were observed for both control and MAPLE coatings (Fig. 4). Specifically, bands were observed at 1646 cm−1 (νCO for amide I); 1535 cm−1 (νN–H for amide II); and 1230–1400 cm−1 (N–H in plane bend and νC–N for amide III) respectively. The characteristic amide I peak (1646 cm−1) shows the presence of α-helix structures in the protein. The band at 2700–3200 cm−1 corresponds to N–H bend vibration from SS material and was merged with the 3450 cm−1 characteristic to O–H vibration band.63
 | ||
| Fig. 4 FTIR spectra of SS protein (a), GNP (b) and GNP–SS (c) deposited by MAPLE (red line) as compared with the drop cast (black line). | ||
The important vibration peaks of GNP material at 1585 cm−1νCC and in the region of 1050–1400 cm−1νC–C, skeleton vibration and also νC–O (related to the presence of water in the control samples) are also shown in the IR spectra of GNP coatings (Fig. 4b). The large absorption band observed in the 2500–3500 cm−1 region is most likely related to the O–H vibration. In the case of composite coatings, due to GNP being highly absorbent of IR radiation, the fingerprint of the SS was mainly observed, with weak vibrations in the range of 1050–1400 cm−1 from GNP coatings.
The typical SEM and AFM micrographs of bare alloy and alloy functionalized with GNP and GNP–SS respectively, are shown in Fig. 5. The bare sandblasted alloy revealed the presence of irregular shapes on the surface (Fig. 5a–d). Due to the substrate morphology, and relatively small thickness of the coatings (from 91 to 442 nm), similar morphologies to that of bare alloy surface were observed overall for GNP- (Fig. 5b–e) and GNP–SS- (Fig. 5c–f) functionalized alloy. While the Ra value for the alloy samples functionalized with GNP films was similar to the initial roughness (∼640 nm), the presence of SS led to a decrease in roughness of up to ∼495 nm. As a general observation, the presence of SS in the films produced by MAPLE also led to the improvement of the film homogeneity and more than 20% reduction in surface roughness.
 | ||
| Fig. 5 SEM (a–c) and AFM (d–f) micrographs of the Gum alloy (a and d) functionalized with GNP (b and e), and GNP–SS (c and f) by MAPLE (266 nm, 600 mJ cm−2). The scanned area by AFM was 40 × 40 μm2. Scale bar: 20 μm. | ||
In order to confirm these observations and to have a better understanding on the nanoscale topography characteristic of each coating, higher magnification SEM micrographs of SS immobilized on alloy by MAPLE, with and without GNP, were taken (Fig. 6). A uniform non-porous hydrogel like appearance was noticed for SS, which might explain the observed decrease of roughness values.
 | ||
| Fig. 6 SEM micrographs of alloy functionalized with SS (a), GNP (b) and GNP–SS (c) by MAPLE (266 nm, 600 mJ cm−2). Scale bar: 4 μm. | ||
Contact angle was also evaluated on the analysed specimens. No significant differences in CA values were noticed among the bare and GNP-coated material (98° and 92°, respectively). However, SS and GNP–SS exhibited a considerable decrease in the CA to 47° and 65°, respectively.
3.3. In vitro pre-osteoblast response
Biological studies used the MC3T3-E1 pre-osteoblasts to evaluate the cellular response to the bare, GNP-, SS- and GNP–SS-coated alloy surfaces. The cell–material interactions were studied in terms of cell adhesion and morphological features, viability, proliferation and differentiation.The morphology and adhesion of MC3T3-E1 pre-osteoblasts on the synthesized substrates were visualized by fluorescence microscopy after culture for 1 day and 5 days respectively. As shown in Fig. 7a as well as in the ESI (Fig. S2a†), a significant number of cells colonized the substrate's surface at 1 day post-seeding and adopted different morphologies. Analysis showed that the cells attached to the control surfaces presented a cuboidal morphology and underwent less stretching when compared to the cells adhered onto the coated surfaces, which exhibited more stretched and elongated shapes. The cells also seemed to be settled faster on GNP-, SS- and GNP–SS-coated substrates, and that they have quickly interacted and became adapted to these surfaces earlier than on bare alloy. In addition, well-defined arrangements of actin stress fibers that appear to be oriented in a parallel direction along to the long axis of the cells were more visible on GNP-, SS- and GNP–SS-coated alloy. The formation and contractility of stress fibers has been shown to be associated with stem cell differentiation.64 Specifically, the cells grown on substrates that promote increased stress fiber and focal adhesion formation also exhibited increased osteoblastic differentiation. Our finding indirectly indicates that GNP-, SS- and GNP–SS-coated substrates might be helpful for stimulating cell differentiation. Furthermore, immunoreactive sites for vinculin, displaying punctiform pattern, localized at the peripheral regions of the cells, predominantly at the termini of actin microfilament bundles, were more numerous on the modified surfaces relative to control substrate (uncoated alloy). It is well known that vinculin is a major protein of focal adhesions playing a critical role in initiating and establishing cell adhesion, cell shape, and cytoskeletal development.65,66 Therefore, it is suggested that GNP-, SS- and GNP–SS-coated substrates are more effective in promoting cell adhesion and establishing tight interactions with MC3T3-E1 cells than the control surface. Fig. 7b and Fig. S2b (ESI†) show the fluorescence micrographs representing the expression and dense distribution of vinculin-associated actin stress fibers in MC3T3-E1 pre-osteoblasts after 5 days of culture. It can be observed that the coated surfaces enabled actin filaments to form a strong network with well-defined and elongated fibers, better represented than on bare alloy.
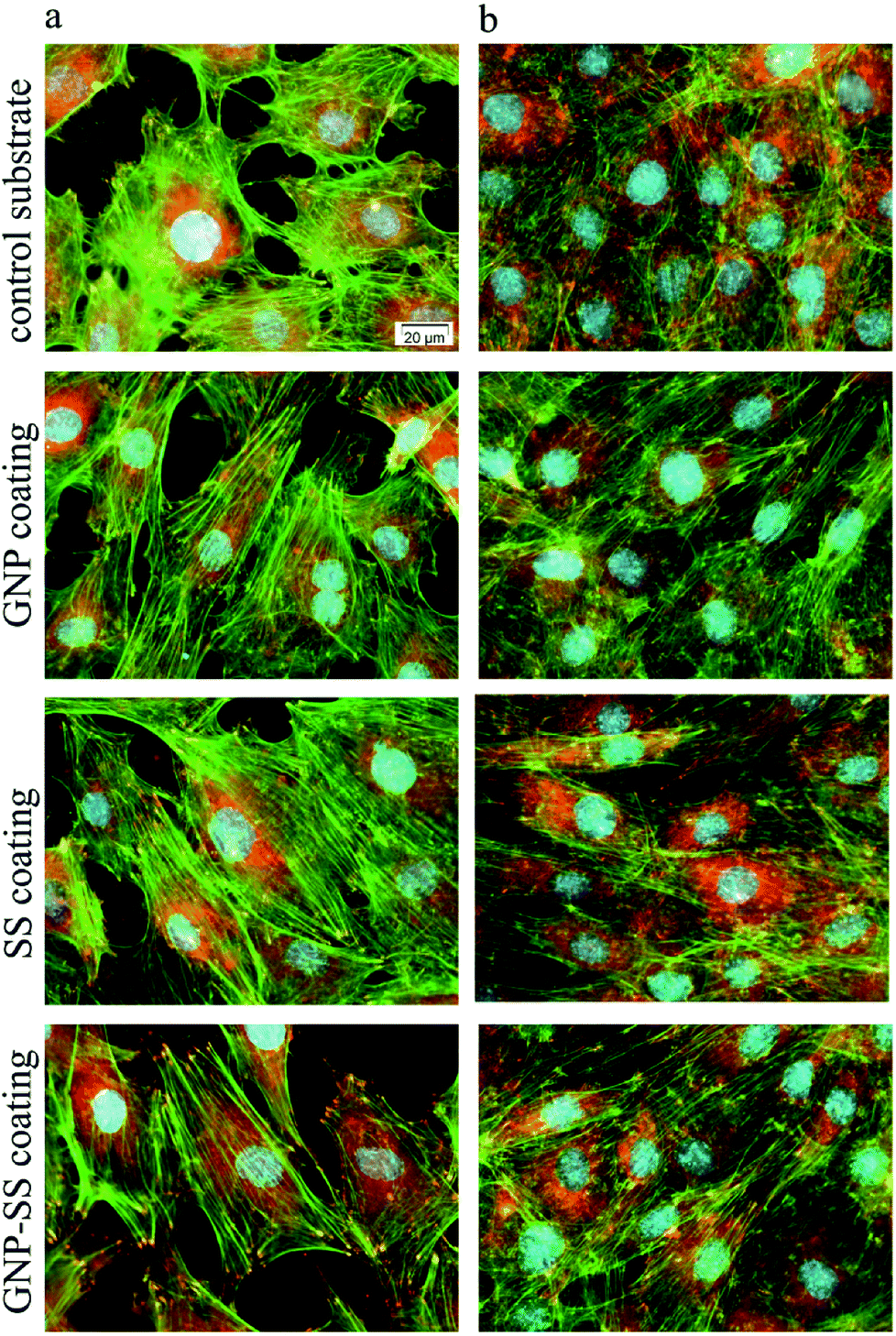 | ||
| Fig. 7 Fluorescent images of MC3T3-E1 pre-osteoblasts grown on control substrate and GNP-, SS- and GNP–SS-coated alloy for 1 day (a) and 5 days (b) respectively. Green fluorescence: actin cytoskeleton; red fluorescence: vinculin signals. Scale bar: 20 μm. | ||
MC3T3-E1 cell proliferation on the substrates was investigated by MTT assay. As shown in Fig. 8a, the metabolic activity (OD values) of the MC3T3-E1 cells of all four experimental groups increased with the prolonged culture time. On the other hand, the proliferation of cells grown for 5 days on the GNP-, SS- and GNP–SS-coated substrates was significantly higher than that expressed on the control surfaces (p < 0.05 for GNP coated alloy and p < 0.01 for both SS- and GNP–SS-coated alloy). This finding indicates that all modified alloy surfaces stimulate cell proliferation, although to a different extent, namely with the SS- and GNP–SS-coated substrates having a much more prominent impact on cell proliferation than the GNP-coated surface.
 | ||
| Fig. 8 Viability/proliferation of MC3T3-E1 cells grown on test substrates for 1 day and 5 days respectively. (a) MTT assay (n = 3, mean ± SD), *p < 0.05 for GNP-coated alloy versus control, **p < 0.01 for SS and GNP–SS-coated alloy versus control; (b) fluorescent cell staining with LIVE/DEAD Cell Viability/Cytotoxicity Assay Kit (live cells fluorescence green and dead cells fluorescence red). Scale bar: 100 μm. | ||
The presence of SS in the coatings is assumed to be the major reason for the enhancement of cell proliferation because of its attractive bioactive properties in tissue engineering field.41 The compound has been previously shown to increase cell attachment and proliferation.41,43,45,46,51,67 In our experiments, the highest OD values corresponded to MC3T3-E1 cells grown for 5 days on the GNP–SS-coated alloy. This behaviour could be partially ascribed to the moderately hydrophilic character exhibited by this coating (CA of ∼65°). It is well known that moderate hydrophilic surfaces are more adequate for cell growth relative to hydrophobic surfaces.68,69
In addition, calcein AM/EthD-1 staining revealed a high percentage of viable cells converting non-fluorescent calcein AM to green-fluorescent calcein as well as a low number of red fluorescent dead cells (Fig. 8b; see also separate channel images in Fig. S3 – ESI†). It can be also observed, on all analysed substrates, an increasing pre-osteoblast density with the increase in the culture period. Specifically, at 5 days post-seeding cell density increased in the following order: bare alloy < GNP < SS < GNP–SS indicating a good agreement with the cell proliferation experimental data. Taken together, the data clearly indicate that the GNP-, SS- and, especially, GNP–SS-coated samples favour the growth of MC3T3-E1 pre-osteoblasts.
We further assessed the potential of both uncoated and GNP-, SS-, and GNP–SS-coated alloy to promote osteoblast differentiation in the presence of soluble factors (i.e., osteogenic medium). ALP expression was measured on incubation-days 7 and 14 (Fig. 9a), and used as an early marker of osteogenic differentiation. A significantly higher ALP activity was detected in cells cultured on the GNP–SS substrate than in those in contact with GNP and control samples at both time points. SS-coated substrate was also found to have a significant impact on ALP activity after 14 days of culture suggesting that the presence of SS stimulated the early osteoblastic differentiation. Moreover, the OD values corresponding to ALP activity exhibited by MC3T3-cells grown on the GNP and bare alloy substrates did not show any significant difference, suggesting that GNP coating had no effect on the potential of the Gum alloy to induce the early stages of osteoblast differentiation. Deposition of calcium phosphate by the pre-osteoblasts maintained in contact with the developed biomaterials was demonstrated by ARS staining (Fig. 9b). The OD values recorded indicated a significantly higher mineral content on SS- and GNP–SS-coated Gum alloy than on control and GNP surfaces after three weeks.
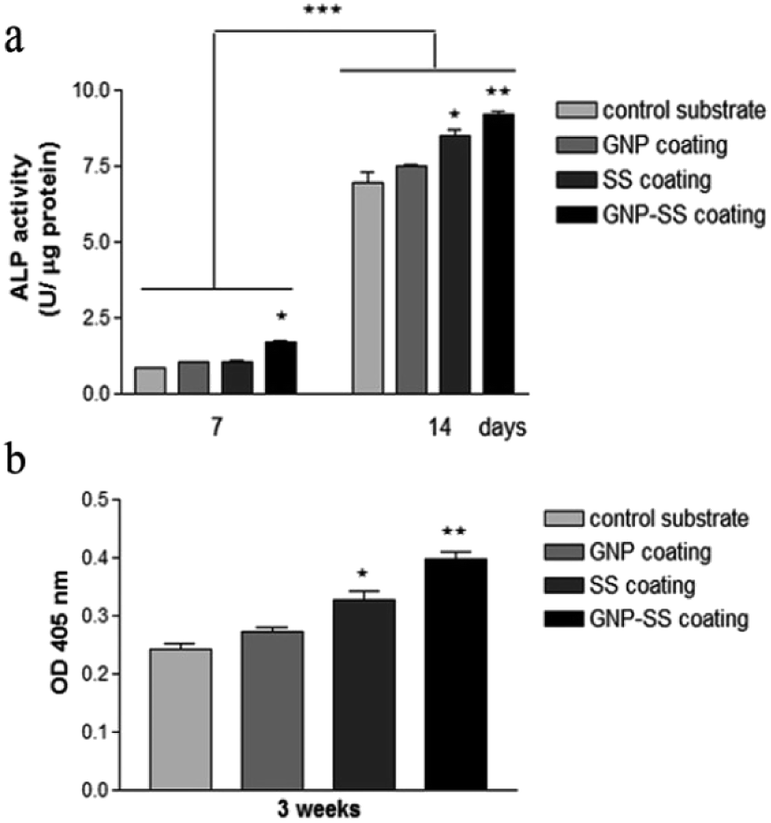 | ||
| Fig. 9 Differentiation of MC3T3-E1 pre-osteoblasts grown on bare and coated Gum alloy. (a) The levels of intracellular ALP activity at 7 and 14 days post-seeding (*p < 0.05 for GNP–SS-coated alloy vs. control substrate at 7 days post-seeding; *p < 0.05 for SS-coated alloy and **p < 0.01 for GNP–SS-coated alloy vs. control substrate at 14 days post-seeding; ***p < 0.001 ALP activity at 14 days vs. ALP activity at 7 days post-seeding). (b) Quantitative colorimetric analysis of extracellular matrix mineralization after 3 weeks of culture; *p < 0.05 for SS-coated alloy, **p < 0.01 for GNP–SS-coated alloy vs. control sample. Results are presented as means ± SD (n = 3). | ||
Furthermore, no statistically significant differences were detected between SS- and GNP–SS-coated substrates, suggesting once again that GNP played little role in the osteogenic differentiation of MC3T3-E1 cells.
To further confirm osteogenic commitment, the protein expression of late osteoblast differentiation marker OCN was studied. It is known that OCN is essential for hydroxyapatite binding and deposition in the extracellular matrix of bone.70 The OCN expression was evaluated by using fluorescent immunocytochemistry. As shown in Fig. 10 and S4 – ESI,† GNP–SS-coated surface displayed the strongest OCN immunoreactivity. These results strengthen the findings of ALP assay and mineral quantification.
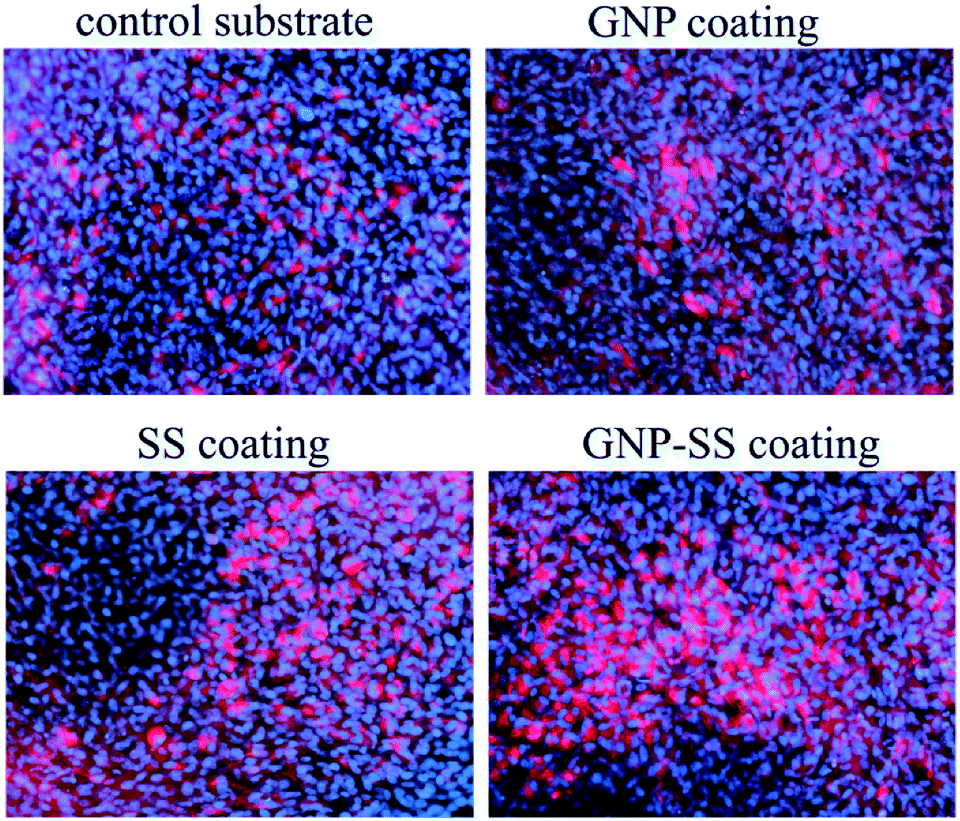 | ||
| Fig. 10 Immunofluorescence detection of osteocalcin expression by MC3T3-E1 cells grown for 3 weeks on Gum alloy and GNP-, SS- and GNP–SS-coatings. Scale bar: 50 μm. | ||
Overall, our data are in line with the results obtained by Nayak et al.,50 which demonstrated that the immobilization of SS on Ti surface generates a bioactive nano-topography that induces effects on early events of osteogenesis and could possibly augment bone formation. However, it was surprising to achieve a similar outcome of cell differentiation experiments for bare and GNP-functionalized specimens. Several previous studies have shown that G, mostly grown by chemical vapor deposition (CVD), can enhance osteogenic cell differentiation when used either as two-dimensional or three-dimensional substrates.31–34 For example, Nayak et al.34 demonstrated that Si/SiO2 substrates treated with G were able to accelerate human mesenchymal stem cells (hMSCs) differentiation into bone cells. Moreover, Xie et al.32 showed that two- and three-dimensional G substrates upregulated bone-related genes and proteins regardless the use of osteogenic medium, all relative to control glass slides and polystyrene scaffolds. In another study,33 polydimethylsiloxane was used as a reference material and support for G and GO. The authors outlined that the strong noncovalent binding abilities of G allow it to act as a preconcentration platform for osteogenic inducers, which accelerate MSCs commitment toward the osteogenic lineage.
Unlike previously mentioned studies, in the present work the control sample was represented by a Ti-based alloy which was subjected to sandblasting, an effective method to greatly improve the surface bio-performances. Moreover, GNP coatings were used, which are different as morphology from those based on G used in other studies. Therefore, the differences between our results and those reported by other groups regarding mineral deposition on GNP-coated substrates could be attributed to the comparison of the outcomes obtained using GNP coatings on other substrates that differ physically and chemically from Ti based materials. Moreover, the use of osteogenic medium could have been masking the effects promoted by GNP alone.
4 Conclusions
In this work, a new Ti-31.7Nb-6.21Zr-1.4Fe-0.16O Gum alloy through a melting process for bone implant applications, was successfully synthesized. This alloy exhibited a lower elastic modulus than that of conventionally used metallic biomaterials however, a higher plasticity. The surface of Ti-31.7Nb-6.21Zr-1.4Fe-0.16O alloy was easily modified with GNP, SS and GNP–SS by MAPLE technique. Analysis showed that GNP–SS coated substrate promoted cell differentiation to a higher extent than bare, SS- and GNP-modified substrates. Collectively, the results presented herein reveal the potential of GNP–SS surface-modified Ti-31.7Nb-6.21Zr-1.4Fe-0.16O alloy to enhance the osseointegration in orthopaedic applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was financially supported by research grant from the Romanian Ministry of National Education, CNCS-UEFISCDI (projects PCCA 213/2014, 63PCCDI/2018) and National Science Foundation (grant 1454230).References
- M. Besse, P. Castany and T. Gloriant, Acta Mater., 2011, 59, 5982–5988 CrossRef CAS.
- H. P. Duan, H. X. Xu, W. H. Su, Y. B. Ke, Z. Q. Liu and H. H. Song, Int. J. Miner., Metall. Mater., 2012, 19, 1128–1133 CrossRef CAS.
- M. Nakai, M. Niinomi, T. Akahori, H. Tsutsumi and M. Ogawa, Mater. Trans., 2009, 50, 2716–2720 CrossRef CAS.
- D. M. Gordin, R. Ion, C. Vasilescu, S. I. Drob, A. Cimpean and T. Gloriant, Mater. Sci. Eng., C, 2014, 44, 362–370 CrossRef CAS.
- L. S. Wei, H. Y. Kim and S. Miyazaki, Acta Mater., 2015, 100, 313–322 CrossRef CAS.
- F. B. Vicente, D. R. N. Correa, T. A. G. Donato, V. E. Arana-Chavez, M. A. R. Buzalaf and C. R. Grandini, Materials, 2014, 7, 542–553 CrossRef PubMed.
- K. Y. Xie, Y. Wang, Y. Zhao, L. Chang, G. Wang, Z. Chen, Y. Cao, X. Liao, E. J. Lavernia, R. Valiev, B. Sarrafpour, H. Zoellner and S. Ringer, Mater. Sci. Eng., C, 2013, 33, 3530–3536 CrossRef CAS.
- Y. Li, C. Yang, H. Zhao, S. Qu, X. Li and Y. Li, Materials, 2014, 7, 1709–1800 CrossRef.
- N. Sumitomo, K. Noritake, T. Hattori, K. Morikawa, S. Niwa, K. Sato and M. Niinomi, J. Mater. Sci.: Mater. Med., 2008, 19, 1581–1586 CrossRef CAS.
- M. Niinomi, Bull. Iron Steel Inst. Jpn., 2010, 15, 661–670 CAS.
- M. Niinomi and T. Hattori, in Interface Oral Health Science, ed. T. Sasano and O. Suzuki, Springer, 2010, pp. 90–99 Search PubMed.
- A. Bianco, H.-M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1–6 CrossRef CAS.
- J. Liu, L. Cui and D. Losic, Acta Biomater., 2013, 9, 9243–9257 CrossRef CAS.
- Q. Zhang, Z. Wu, N. Li, Y. Pu, B. Wang, T. Zhang and J. Tao, Mater. Sci. Eng., C, 2017, 77, 1363–1375 CrossRef CAS.
- C. McCallion, J. Burthem, K. Rees-Unwin, A. Golovanov and A. Pluen, Eur. J. Pharm. Biopharm., 2016, 104, 235–250 CrossRef CAS.
- Z. Wang, L. C. Ciacchi and G. Wei, Appl. Sci., 2017, 7, 1175 CrossRef.
- C. I. L. Justino, A. R. Gomes, A. C. Freitas, A. C. Duarte and T. A. P. Rocha-Santos, TrAC, Trends Anal. Chem., 2017, 91, 53–66 CrossRef CAS.
- D. Rodrigo, O. Limaj, D. Janner, D. Etezadi, F. J. García de Abajo, V. Pruneri and H. Altug, Science, 2015, 349, 165–168 CrossRef CAS.
- P. Suvarnaphaet and S. Pechprasarn, Sensors, 2017, 17, 2161 CrossRef.
- J. Lin, X. Chen and P. Huang, Adv. Drug Delivery Rev., 2016, 105, 242–254 CrossRef CAS.
- K. Li, X. Zhao, G. Wei and Z. Su, Curr. Med. Chem., 2017 DOI:10.2174/0929867324666170223154145.
- S. Szunerits and R. Boukherroub, J. Mater. Chem. B, 2016, 4, 6892–6912 RSC.
- P. C. Henriques, I. Borges, A. M. Pinto, F. D. Magalhães and I. C. Gonçalves, Carbon, 2018, 132, 709–732 CrossRef CAS.
- L. Shi, J. Chen, L. Teng, L. Wang, G. Zhu, S. Liu, Z. Luo, X. Shi, Y. Wang and L. Ren, Small, 2016, 12, 4165–4184 CrossRef CAS.
- V. Palmieri, M. Papi, C. Conti, G. Ciasca, G. Maulucci and M. De Spirito, Expert Rev. Med. Devices, 2016, 13, 1013–1019 CrossRef CAS PubMed.
- N. Shadjou, M. Hasanzadeh and B. Khalilzadeh, Bioengineered, 2018, 9, 38–47 CrossRef CAS PubMed.
- G. Lalwani, M. D'agati, A. Gopalan, M. Rao, J. Schneller and B. Sitharaman, J. Biomed. Mater. Res., Part A, 2017, 105, 73–83 CrossRef CAS PubMed.
- O. Akhavan, J. Mater. Chem. B, 2016, 4, 3169–3190 RSC.
- A. M. Pinto, I. C. Gonçalves and F. D. Magalhães, Colloids Surf., B, 2013, 111, 188–202 CrossRef CAS.
- S. Syama and P. V. Mohanan, Int. J. Biol. Macromol., 2016, 86, 546–555 CrossRef CAS.
- N. Dubey, R. Bentini, I. Islam, T. Cao, A. H. Castro Neto and V. Rosa, Stem Cells Int., 2015, 2015(804213), 1–13 Search PubMed.
- H. Xie, T. Cao, J. V. Gomes, A. H. Castro Neto and V. Rosa, Carbon, 2015, 93, 266–275 CrossRef CAS.
- W. C. Lee, C. H. Y. Lim, H. Shi, L. A. Tang, Y. Wang, C. T. Lim, H. Shi, L. A. L. Tang, Y. Wang, C. T. Lim and K. P. Loh, ACS Nano, 2011, 5, 7334–7341 CrossRef CAS.
- T. R. Nayak, H. Andersen, V. S. Makam, C. Khaw, S. Bae, X. Xu, P. L. Ee, J. H. Ahn, B. H. Hong, G. Pastorin and B. Özyilmaz, ACS Nano, 2011, 5, 4670–4678 CrossRef CAS.
- V. Rosa, N. Dubey, S. S. Rajan and H. Xie, in Smart materials for tissue engineering: fundamental principles, ed. Q. Wang, 2016, ch. 12 Search PubMed.
- O. Akhavan, E. Ghaderi and M. Shahsavar, Carbon, 2013, 59, 200–211 CrossRef CAS.
- W. G. La, M. Jin, S. Park, H. H. Yoon, G. J. Jeong, S. H. Bhang, H. Park, K. Char and B. S. Kim, Int. J. Nanomed., 2014, 9, 107–116 Search PubMed.
- W. G. La, S. Park, H. H. Yoon, G. J. Jeong, T. J. Lee, S. H. Bhang, J. Y. Han, K. Char and B. S. Kim, Small, 2013, 9, 4051–4060 CrossRef CAS.
- H. S. Jung, T. Lee, I. K. Kwon, H. S. Kim, S. K. Hahn and C. S. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 9598–9607 CrossRef CAS.
- M. Li and J. Li in Silk Biomaterials for Tissue Engineering and Regenerative Medicine, ed. S. C. Kundu, Woodhead Publishing, UK, 2014, pp. 330–348 Search PubMed.
- L. Lamboni, M. Gauthier, G. Yang and Q. Wang, Biotechnol. Adv., 2015, 33, 1855–1967 CrossRef CAS.
- Z. Wang, Y. Zhang, J. Zhang, L. Huang, J. Liu, Y. Li, G. Zhang, S. C. Kundu and L. Wang, Sci. Rep., 2014, 4, 7064 CrossRef PubMed.
- H. Xie, W. Yang, J. Chen, J. Zhang, X. Lu, X. Zhao, K. Huang, H. Li, P. Chang, Z. Wang and L. Wang, Adv. Healthcare Mater., 2015, 4, 2195–2205 CrossRef CAS.
- Z. Wang, J. Wang, Y. Jin, Z. Luo, W. Yang, H. Xie, K. Huang and L. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 24629–24640 CrossRef CAS PubMed.
- T. Siritientong, A. Angspatt, J. Ratanavaraporn and P. Aramwit, Pharm. Res., 2014, 31, 104–116 CrossRef CAS PubMed.
- Y. Song, C. Zhang, J. Zhang, N. Sun, K. Huang, H. Li, Z. Wang, K. Huang and L. Wang, Acta Biomater., 2016, 41, 210–223 CrossRef CAS.
- P. Aramwit, N. Bang, J. Ratanavaraporn and S. Ekgasit, Nanoscale Res. Lett., 2014, 9, 79 CrossRef.
- D. J. Belton, A. J. Mieszawska, H. A. Currie, D. L. Kaplan and C. C. Perry, Langmuir, 2012, 28, 4373–4381 CrossRef CAS.
- L. Huang, K. Tao, J. Liu, C. Qi, L. Xu, P. Chang, J. Gao, X. Shuai, G. Wang, Z. Wang and L. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 6577–6585 CrossRef CAS.
- S. Nayak, T. Dey, D. Naskar and S. C. Kundu, Biomaterials, 2013, 34, 2855–2864 CrossRef CAS.
- S. I. Voicu, R. M. Cotruz, V. Mitran, A. Cimpean, F. Miculescu, C. Andronescu and M. Miculescu, ACS Sustainable Chem. Eng., 2016, 4, 1765–1774 CrossRef CAS.
- R. Ion, S. Vizireanu, C. E. Stancu, C. Luculescu, A. Cimpean and G. Dinescu, Mater. Sci. Eng., C, 2015, 48, 118–125 CrossRef CAS.
- V. Mitran, C. Vasilescu, S. I. Drob, P. Osiceanu, J. M. Calderon-Moreno, M. C. Tabirca, D. M. Gordin, T. Gloriant and A. Cimpean, BioMed Res. Int., 2015, 2015, 261802 Search PubMed.
- P. Neacsu, A. I. Staras, S. I. Voicu, I. Ionascu, T. Soare, S. Uzun, V. D. Cojocaru, A. M. Pandele, S. M. Croitoru, F. Miculescu, C. M. Cotrut, I. Dan and A. Cimpean, Materials, 2017, 10, 686 CrossRef.
- E. Axente, F. Sima, L. E. Sima, M. Erginer, M. S. Eroglu, N. Serban, C. Ristoscu, S. M. Petrescu, E. ToksoyOner and I. N. Mihailescu, Biofabrication, 2014, 3, 035010 CrossRef.
- L. Rusen, V. Dinca, B. Mitu, C. Mustaciosu and M. Dinescu, Appl. Surf. Sci., 2014, 302, 134–140 CrossRef CAS.
- V. Dinca, P. E. Florian, L. E. Sima, L. Rusen, C. Constantinescu, R. W. Evans, M. Dinescu and A. Roseanu, Biomed. Microdevices, 2014, 16, 11–21 CrossRef CAS.
- R. A. McGill, D. B. Chrisey, Method of producing a film coating by matrix assisted pulsed laser deposition, US Pat., US6025036 A, 2000.
- F. Sima, P. M. Davidson, J. Dentzer, R. Gadiou, E. Pauthe, O. Gallet, I. N. Mihailescu and K. Anselme, ACS Appl. Mater. Interfaces, 2014, 7, 911–920 CrossRef.
- L. Rusen, P. Neacsu, A. Cimpean, I. Valentin, S. Brajnicov, L. N. Dumitrescu, J. Banita, V. Dinca and M. Dinescu, Appl. Surf. Sci., 2016, 374, 23–30 CrossRef CAS.
- L. E. Sima, G. E. Stan, C. O. Morosanu, A. Melinescu, A. Ianculescu, R. Melinte, J. Neamtu and S. M. Petrescu, J. Biomed. Mater. Res., Part A, 2010, 95, 1203–1214 CrossRef.
- L. Rusen, S. Brajnicov, P. Neacsu, V. Marascu, A. Bonciu, M. Dinescu, V. Dinca and A. Cimpean, Surf. Coat. Technol., 2017, 325, 397–409 CrossRef CAS.
- X. Zhang, M. Tsukada, H. Morikawa, K. Aojima, G. Zhang and M. Miura, Nanoscale Res. Lett., 2011, 6, 510 CrossRef.
- K. A. Kilian, B. Bugarija, B. T. Lahn and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4872–4877 CrossRef CAS.
- M. Bailly, Trends Cell Biol., 2003, 13, 163–165 CrossRef CAS.
- J. D. Humphries, P. Wang, C. Streuli, B. Geiger, M. J. Humphries and C. Ballestrem, J. Cell Biol., 2007, 179, 1043–1057 CrossRef CAS.
- V. Mitran, M. G. Albu, E. Vasile, A. Cimpean and M. Costache, Prog. Nat. Sci.: Mater. Int., 2015, 25, 122–130 CrossRef CAS.
- Y. Arima and H. Iwata, Biomaterials, 2017, 28, 3074–3082 CrossRef.
- E. Torres, A. Vallés-Lluch, V. Fombuena, B. Napiwocki and T. Lih-Sheng, Macromol. Mater. Eng., 2017, 302, 1700259 CrossRef.
- Y. T. Tsao, Y. J. Huang, H. H. Wu, Y. A. Liu, Y. S. Liu and O. K. Lee, Int. J. Mol. Sci., 2017, 18, 159 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01784k |
| This journal is © The Royal Society of Chemistry 2018 |