DOI:
10.1039/C8RA01740A
(Paper)
RSC Adv., 2018,
8, 17606-17615
Simultaneous reinforcement and toughness improvement of an epoxy–phenolic network with a hyperbranched polysiloxane modifier†
Received
27th February 2018
, Accepted 6th May 2018
First published on 15th May 2018
Abstract
An epoxy–phenolic network is modified with hyperbranched polysiloxane (HBPSi). The addition of HBPSi-2, which has medium molecular weight, can significantly decrease the viscosity of the uncured epoxy–phenolic system and increase the crosslinking density and homogeneity of the cured crosslinking network. With 10% HBPSi-2, the mechanical properties of the samples are improved comprehensively: tensile modulus and maximum strength increase by 11.4% and 36.2%, respectively, while elongation at break and impact strength increase by 153.8% and 186.7%, respectively. The comprehensive improvements in the mechanical properties are attributed to combined effects of crosslinking density, network rigidity, cohesive density and the matrix-modifier compatibility. What is more, HBPSi-2 also significantly increases the char yield of the material and decreases the thermal weight loss rate, indicating an improved thermal stability. All these results may provide a new strategy for toughness and strength improvement of the epoxy–phenolic network.
Introduction
Epoxy resin is an important and versatile subset of thermoset materials. According to different applications, epoxy resins with required properties can be prepared by adjusting the type and dosage of each ingredient in the epoxy formula. Phenolic curing agent has been widely used in the preparation of epoxy resins for the encapsulation of microelectronic devices because of its well-balanced properties including excellent chemical resistance, superior mechanical properties, ideal electrical properties and good adherence to many substrates.1 However, the rigid structure and high phenolic hydroxyl density of the phenolic curing agent often result in high brittleness of the cured resin, making the material prone to cracking under external force.2–4
Toughening of epoxy resin has been a subject of intense investigation. Over the past few decades, tremendous efforts have been made and different kinds of modifiers have been developed to improve the toughness of epoxy resins, such as liquid rubber or rubber particles,5,6 thermoplastics,7 and nanoparticles.8–10 In most cases, blending modifiers into the epoxy matrix brings about the improvement of toughness but often significantly compromises the thermal properties,11,12 modulus and strength of the resins.13,14 Moreover, phase separation before or during the curing process after incorporation of these modifiers may result in great difficulty in the design of interfacial chemistry and cure schemes to guarantee a proper morphology of the cured networks.15 Compared with these traditional modifiers, hyperbranched polymers have shown great potential.16,17 With a highly branched structure, ample terminal groups, and inherent cavities, hyperbranched molecules possess advantages over conventional modifiers, for example, better miscibility, lower viscosity, and higher reactivity. Generally, aliphatic polyesters backbones are most popular in the design of hyperbranched molecules because of the mature craft of esterification reaction. However, epoxies modified with those hyperbranched molecules tend to show decreased modulus or Tg. Boltorn™, the commercially available hydroxyl-terminated hyperbranched polymers with aliphatic polyesters backbones have been extensively studied.18–21 Results show that Boltorn™ can effectively improve toughness or the epoxy matrix. However, the strength and Tg were significantly compromised. Moreover, phase separation also appeared because of the poor miscibility between the epoxy matrix and the modifier. Sarah E. Morgan synthesized an aliphatic, epoxide-functional hyperbranched polymer and cured it into an aromatic epoxy–amine network without cure-induced phase separation.22 It is found that the hyperbranched molecules allowed for simultaneous enhancement in both fracture toughness and Young's modulus. But the flexible hyperbranched structure drastically decreased Tg of the network. Hyperbranched molecules with other backbones have also been studied. Jin and Park23 reported a hyperbranched polyimide which could increase the toughness of diglycidyl ether of bisphenol A/diaminodiphenylmethane (DGEBA/DDM) system without forming phase separation and lowering Tg. X. Miao et al. reported an epoxide-terminated hyperbranched polyether sulphone which simultaneously improved the toughness, strength and thermal properties.24 However, the synthetic processes of these hyperbranched modifiers tend to be complicated, which make them less promising for industrial mass production.
Hyperbranched polysiloxane (HBPSi) is a class of hyperbranched molecules with siloxane backbone and reactive end groups. Because of its simple preparation procedure and high designability, HBPSi has been used in the improvement of thermal and mechanical property for epoxy,25 cyanate ester resins,26,27 bismaleimide28 and polyamine.29 However, in the synthesis of HBPSi, crosslinking often occurs among tri-functional or tetra-functional precursors, resulting in a highly crosslinked structure. Moreover, the high polarity of the HBPSi backbone often causes poor miscibility with epoxy matrix. Thus, report involving modification of epoxy resins with HBPSi is rare. Songqi Ma et al. reported a novel dendritic polysiloxane (DPSO) bearing epoxide groups modifying the diglycidyl ether of DGEBA-aromatic amine system.30 Toughness and thermal stability of epoxy resin were improved after the addition of the DPSO, while the effects of the modifier on strength and modulus were not reported. Moreover, the synthesis procedure of the dendritic molecules is complicated.
In this paper, a series of novel HBPSi was synthesized and then cured into an epoxy–phenolic network. The HBPSi and the network of the modified resin were fully characterized. Moreover, the mechanical and thermal properties of the modified epoxy resins were systematically investigated. Involving mechanisms of the improved properties were fully discussed.
Experimental
Materials
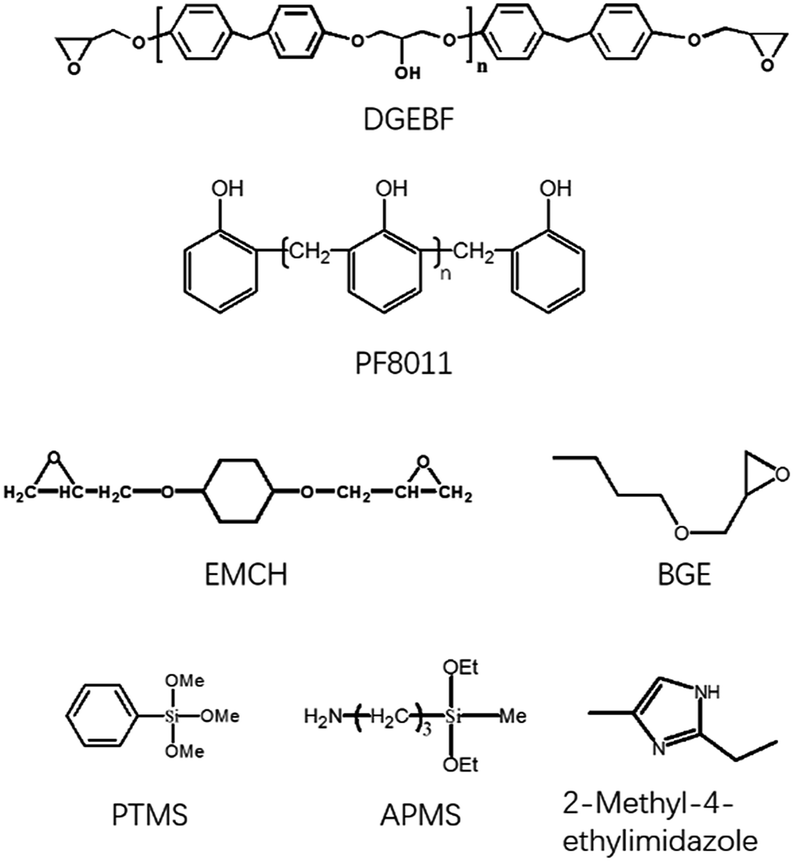
DGEBF, with an epoxy equivalent weight of 170 g (equiv.−1), butyl glycidyl ether (BGE) and 1,4-bis((2,3-epoxypropoxy) methyl) cyclohexane (EMCH) were purchased from Luohe Chemistry Co. Ltd., China. Curing agent, PF8011 (PF) with a hydroxyl equivalent weight of 105 g (equiv.−1), was purchased from Shandong Shengquan Co. Ltd., China. γ-Aminopropylmethyldiethoxysilane (APMS) and phenyltrimethoxysilane (PTMS) were purchased from Nanjing Kuncheng Co., Ltd. Tetrahydrofuran (THF) and 2-methyl-4-ethylimidazole (EMI) were purchased from Tansoole Co. Ltd. The chemical structures of these reactants are shown in Scheme 1. All materials were used as received without further purification.
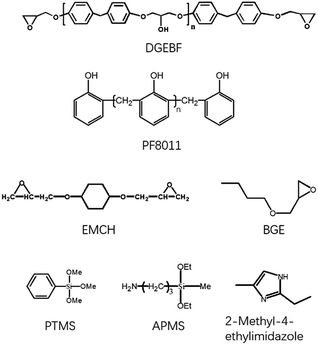 |
| | Scheme 1 The chemical structures of reactants. | |
Synthesis of the HBPSi
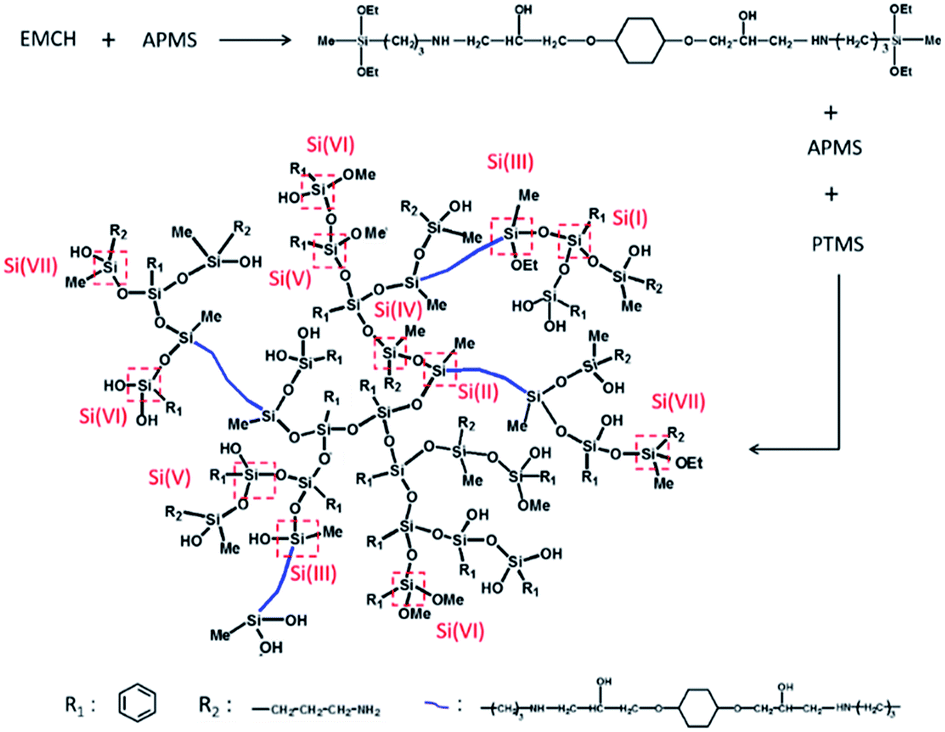
48.2 mmol of APMS, 15.6 mmol of EMCH and 68.5 mmol of PTMS were mixed in a flask containing 0.97 mol of THF. Then the mixture was stirred at 60 °C for 4 h. The control of hydrolysis and condensation of the precursors was based on previous literature.31,32 Typically, the mixture was firstly cooled in ice water. Then 0.32 mol of deionized water was injected during a period of 30 min into the mixture. The mixture was stirred in an ice-water bath and allowed to hydrolyze for 1 h. Subsequently, the system was heated to 50 °C and stirred for 1–4 h. The mixture was then evaporated under a reduced pressure to remove THF and small molecules. Finally, a transparent and viscous liquid mixture was obtained. By controlling the reaction time in the last synthesis step and changing the aging time of the product solution, HBPSi with different molecular weights can be prepared. In this paper, HBPSi with Mn of 1516 g mol−1, 3219 g mol−1 and 7635 g mol−1 are denoted as HBPSi-1, HBPSi-2 and HBPSi-3, respectively. To study the effect of EMCH on the property of the hyperbranched molecule, HBPSi-0 was synthesized as control sample with only APMS and PTMS. The synthesize scheme of HBPSi is shown in Scheme 2 and the characterization results are shown in Table 1. Detailed characterization of the HBPSi can be seen in the ESI.†
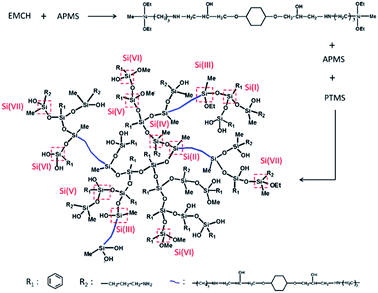 |
| | Scheme 2 Synthetic scheme and representative structure of HBPSi. | |
Table 1 The characterization results of HBPSi
| Sample |
Reaction/aging time |
Mn |
Mw |
PDI |
| HBPSi-0 |
50 °C for 4 h |
3120 |
3740 |
1.20 |
| HBPSi-1 |
50 °C for 1 h |
1516 |
2016 |
1.33 |
| HBPSi-2 |
50 °C for 3 h |
3219 |
4120 |
1.28 |
| HBPSi-3 |
50 °C for 4 h, aging for 12 h at 25 °C |
7635 |
11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 412 412 |
1.49 |
Preparation of curing system
The HBPSi solution was directly used for the preparation of hybrid curing system. A certain amount of HBPSi solution was mixed into the mixture of DGEBF, BGE and PF. The mass ratio of DGEBF and BGE was 3:1. The mixture was mechanically stirred for 20 min before it was evaporated in a vacuum to remove THF and air bubbles. Then a certain amount of accelerant EMI was added into the mixture. Considering that during the temperature programming process, the high post curing temperature may be unfavorable for the stability of the matrix for applications like bonding and packaging, isothermal curing procedure was conducted for the epoxy system. Specifically, the degassed mixture was poured into a preheated PTFE mold and cured for 48 h at 150 °C. In this article, the hybrid resins modified with HBPSi-0, HBPSi-1, HBPSi-2 and HBPSi-3 are denoted as E-HBPSi-0, E-HBPSi-1, E-HBPSi-2 and E-HBPSi-3, respectively. For all samples, the HBPSi content in the hybrid is expressed as weight percent.
Characterization
Fourier transform infrared (FTIR) spectroscopy was performed on a Thermo Scientific Nicolet 6700 spectrometer in the range from 400 to 4000 cm−1 using KBr pellets. 29Si NMR spectra were collected using a Bruker AV-600 spectrometer (600 MHz) with CDCl3 as the solvent. HPLC grade tetrahydrofuran (THF) was used as the eluent with a flow rate of 1 mL min−1 at 25 °C. Rheology study of HBPSi and uncured epoxy–phenolic mixtures were conducted on a AR2000 rheometer. The measurements were performed at a steady flow mode with shear rate between 0.1 and 1000 s−1 at room temperature. Differential scanning calorimetry (DSC) measurements were conducted under nitrogen using a DSC Q1000 (TA instruments). The samples were heated from 30 °C to 280 °C at 10 °C min−1. Dynamic mechanical analysis (DMA) experiments were conducted in a three-point bending mode using a DMA Q800 (TA instruments). All non-isothermal runs (temperature scans) were performed in the “multifrequency, strain” mode at 1 Hz, from 30 °C to 250 °C at a heating rate of 3 °C min−1. The temperature where the maximum of the loss factor tanδ showed was evaluated as the glass transition temperature (Tg). The specimen was made into dumbbell shaped strip (150 × 10 × 4 mm3) and the tensile measurement of mechanical property was conducted on a universal testing machine (Instron 5567, US) at room temperature with a cross-head speed of 1 mm min−1. The tensile fractured surfaces of samples were examined by a scanning electron microscopy (SEM) instrument with an acceleration voltage of 15 kV. All the surfaces were sputtered with a thin gold film before observation. The thermal stability was measured by thermalgravimetric analysis (TG209F1, Netzsch, Germany) under nitrogen atmosphere. The heating rate was 10 °C min−1.
Results and discussion
Effect of PF content on neat epoxy system
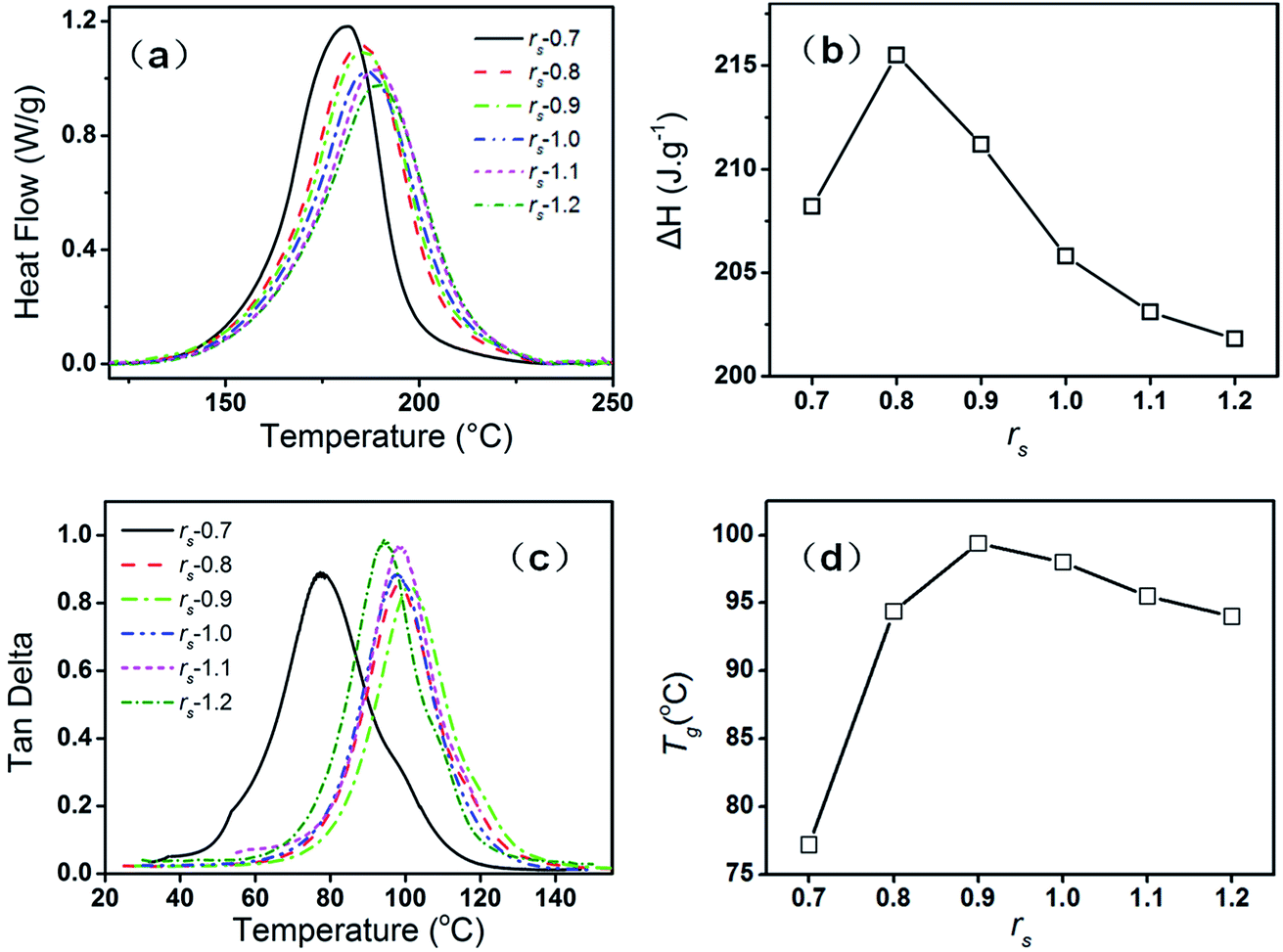
The curing mechanism and property of the epoxy–phenolic–EMI system are dependent on the ratio between the epoxy and phenolic. In epoxy–phenolic–EMI system, when the amount of phenolic is insufficient, reaction between epoxy groups and aliphatic hydroxyl groups, or reaction among epoxy groups themselves, can still happen under the acceleration of EMI. In other words, epoxy–phenolic-EMI system may also reach complete cure even with insufficient phenolic. DCS thermographs and ΔH of curing systems with various stoichiometric ratios (rs) from 0.7 to 1.2 are shown in Fig. 1(a) and (b). As the rs increases from 0.7 to 1.2, the initial reaction temperature rises stepwise and the exothermal peak becomes wider (Fig. 1(a)), which are derived from the high viscosity and steric effect of the phenolic, hindering the curing process of the system. The heat enthalpy first increases and then decreases as the rs increases from 0.7 to 1.2 (Fig. 1(b)). When rs reaches 0.8, the heat enthalpy reaches maximum, indicating the addition reaction between epoxy and phenolic and the side reactions as mentioned contribute to a complete reaction of the curing system. When rs increases to 1.0, however, the high viscosity results in incomplete cure, leading to a decreased enthalpy. Loss tangent and the dependence of Tg on rs are shown in Fig. 1(c) and (d). The temperature where the maximum of the loss factor tanδ showed is evaluated as Tg. As shown, Tg first increases and then decreases as the rs increases from 0.7 to 1.2. When rs is 0.9, Tg reaches maximum. This result is not completely consistent with that from the DSC test because the polymer network formed by the reactions between epoxy and phenolic is more rigid and compact than that formed by the self-condensing ring-opening polymerization. When rs is 0.9, though the high viscosity may lead to incomplete reaction, the sample still shows a high Tg due to the rigid and compact structure. In this paper, rs is fixed at 0.9 for whether neat DGEBF or hybrid samples.
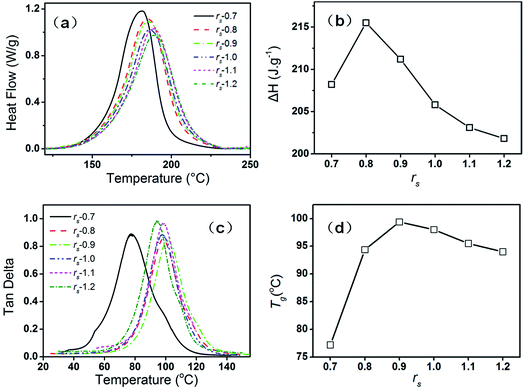 |
| | Fig. 1 DCS thermographs (a) and ΔH (b) of curing systems with varying rs; loss tangent (c) and the dependence of Tg (d) on rs. | |
Rheology of modified system
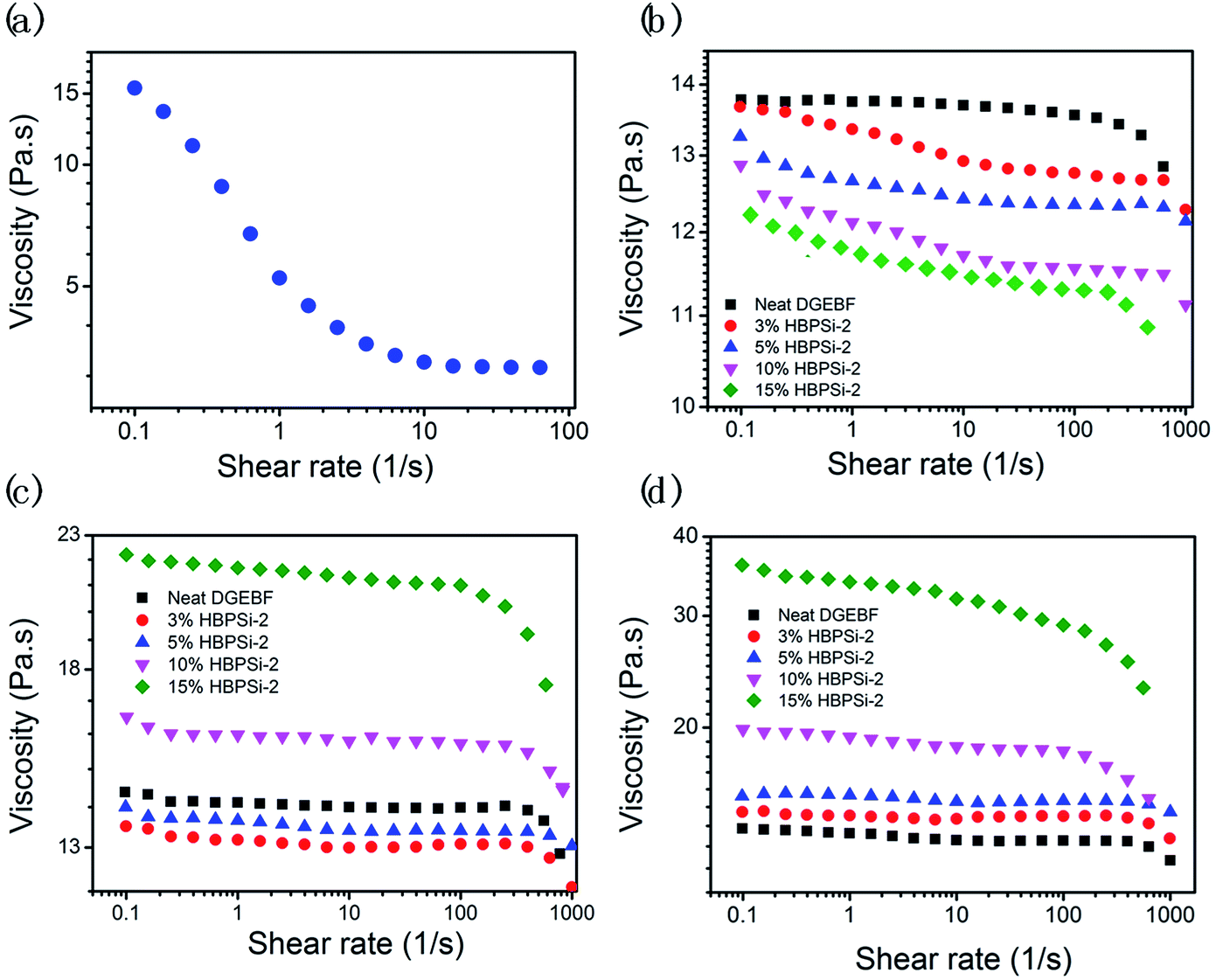
The viscosity, shear response, and viscosity stability are important key parameters reflecting the processibility, flowability and pot life of the epoxy mixture. The study of the shear response behaviors of HBPSi and epoxy–phenolic mixtures was conducted at 0 h, 24 h, and 48 h under room temperature. Because the modified epoxies containing HBPSi-2 show the best overall performance, which will be shown next, the rheology results are only demonstrated for the case of HBPSi-2 and the modified mixtures with HBPSi-2. As shown in Fig. 2(a), HBPSi-2 shows significant shear thinning behavior: as the shear rate increases from 0.1 to 100 s−1, the viscosity decreases from 15.5 Pa s to 3.2 Pa s. The shear thinning behavior of HBPSi-2 is believed to be derived from the hydrogen bonds between HBPSi-2 molecules, which are easy to be destroyed under the external shear forces.33,34 For epoxy mixtures with varying HBPSi-2 loadings, the viscosity of the mixture decreases as the HBPSi-2 loading increases, indicating an improved flowability after the addition of HBPSi-2 (Fig. 2(b)). In the mixture, because of the dilution effect, the hydrogen bonds between HBPSi-2 molecules are weakened. Thus, the viscosity of epoxy mixtures decreases evidently with the increasing HBPSi-2 loading because of the intrinsic low viscosity of HBPSi-2. After addition of HBPSi-2, epoxy mixture also exhibits different shear response. As shown in Fig. 2(b), neat DGEBF exhibits Newtonian behavior with a slight shear thinning behavior at high shear rate, which is due to the alignment of the epoxy and phenolic molecules under the shear force.35 After addition of HBPSi-2, the shear thinning behaviors of the mixtures begin at lower shear rate, which is attributed to the destruction of the hydrogen bond within the system.35 As the HBPSi-2 content increases to 15%, the high HBPSi-2 content leads to strong non-bond interaction. Thus, the shear thinning behavior is much more pronounced.
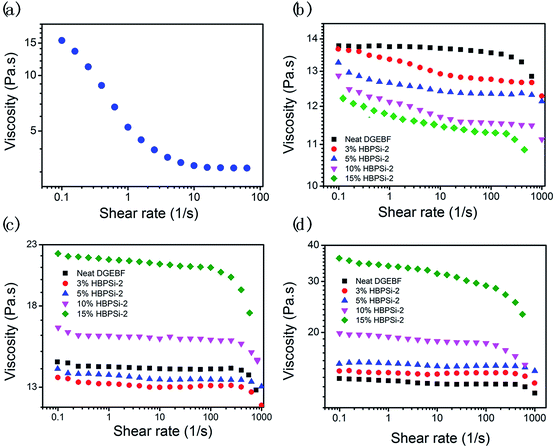 |
| | Fig. 2 The shear response behaviors of HBPSi-2 (a) and epoxy–phenolic mixtures with varying HBPSi-2 contents under room temperature at 0 h (b), 24 h (c), and 48 h (d). | |
The rheology property of neat DGEBF and mixtures with varying HBPSi-2 loadings after 24 h and 48 h are shown in Fig. 2(c) and (d), respectively. As shown, the shear response of the neat EP still maintains a Newtonian characteristic and the viscosity of the neat EP barely changes, indicating a long pot life of the epoxy–phenolic mixture. However, after addition of HBPSi-2, the viscosity of the mixture increases apparently as the storage time increases. After 48 h, all mixtures with HBPSi-2 exhibit higher viscosity than that of the neat DGEBF. Especially, after 24 h and 48 h, drastic increases of viscosities can be seen for mixtures with 10% and 15% HBPSi-2 loading. The increase of viscosity is believed to be caused by the formation of oligomer. Due to the higher reactivity of the amine groups on HBPSi-2, epoxy groups react with HBPSi-2 slowly even at room temperature (ESI, Fig. S3†). Meanwhile, the generated tertiary amine can act as accelerator for the reaction between epoxy and phenolic. When the HBPSi-2 loading is high, entanglements among molecular chains and slight crosslinking caused by the reaction between epoxy and HBPSi-2 both drastically increase the viscosity of the mixture. Also, with high HBPSi-2 content (10% and 15%), the fluid is more sensitive to the shear rate after long period of storage because of the large amounts of physical association, leading to more significant shear thinning behavior (Fig. 2(d)).
DMA characterization
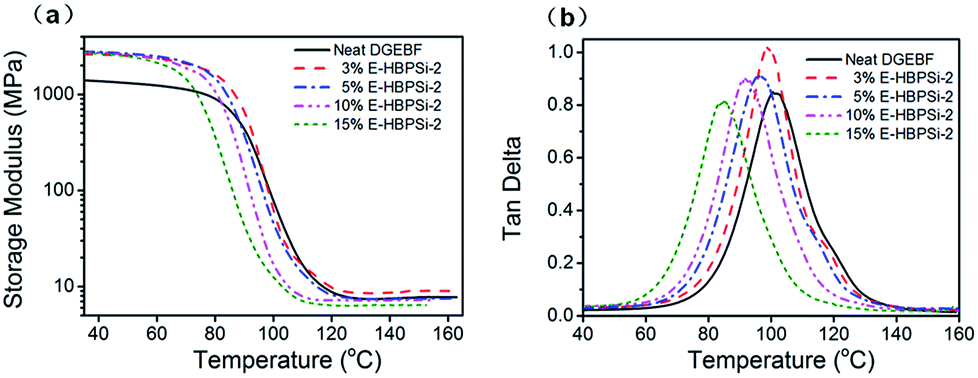
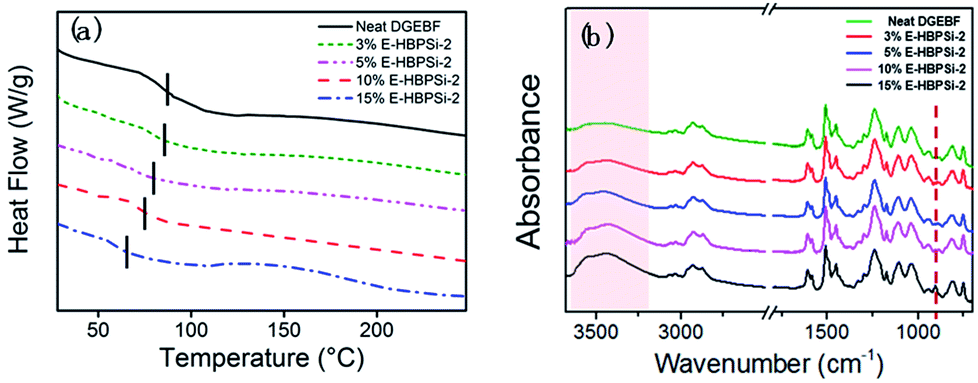
Effects of HBPSi-2 loading on storage moduli (E′) and loss tangent (tanδ) as a function of temperature are shown in Fig. 3. Obviously, for all samples, only one step change in E′ and one peak in tanδ can be observed. This result suggests no signs of phase separation, indicating a good compatibility between HBPSi-2 and the epoxy matrix. As shown in Fig. 3(a), the glassy storage modulus (Eg) of each hybrid is much higher than that of the neat DGEBF, which is attributed to the large amounts of benzene rings in the HBPSi-2, contributing to the rigidity of the network and the restriction of short-range molecular motions. The rubbery storage modulus (Er) increases as HBPSi-2 loading increases from 0% to 3%. According to the classical theory of rubber elasticity,36 the average crosslinking density increases with 3% HBPSi-2 loading, which is due to the higher functionality (terminal amine groups) of HBPSi-2. However, as the HBPSi-2 loading increases from 3% to 15%, the average crosslinking density decreases stepwise. This can be attributed to the shifts of the resin/hardener ratio after introducing HBPSi-2 into the system. At 10% and 15% HBPSi-2 loading, the hybrid's average crosslinking density decreases below that of the neat epoxy, suggesting the effect of the excessive hardener overwhelming that of the high functionality of the HBPSi-2. As shown in Fig. 3(b), as the HBPSi-2 loading increases from 0% to 15%, Tg of E-HBPSi-2 decreases stepwise. Various research groups37–42 have reported a decrease in Tg when silica or organosilicone are added to epoxy or some other matrix. Generally, this behavior has been attributed to plasticizing effect or to a limited amount of unreacted reactant.43 For the present curing system, when HBPSi-2 loading is 3%, the crosslinking density increases. However, the addition of HBPSi may also bring some moistures and residual small molecules into the curing mixture,44 leading to a slight decrease of the hybrid's Tg. As HBPSi-2 loading furtherly increases, the excessive hardener results in the decrease of the crosslinking density. Together with the plasticizing effect, the further decrease of the Tg of the hybrid can be expected. The shape and width of tanδ peak are related to the homogeneity of the crosslinking network. As shown in Table 2, full width at half maximum (FWHM) for the glass transitions of hybrids with 3–10% HBPSi-2 loadings are narrower than that of the neat DGEBF, suggesting that hybrids have a more homogeneous network structure. Nevertheless, FWHM of the hybrid begins to increase at higher HBPSi-2 loading. The broadening of FWHM at high loadings may be related to the incomplete cure. As shown in Fig. 4(a), only one endothermic peak, i.e. glass transition, shows up for neat DGEBF and E-HBPSi-2 with HBPSi-2 loading between 3-10%. Clearly, Tg values obtained from DMA and DSC show the same trend: Tg decreases monotonically with increasing HBPSi-2 loading. However, for E-HBPSi-2 with 15% HBPSi-2 loading, an exothermic peak can be observed in the temperature range of 130 °C to 200 °C, indicating an incomplete cure of the isothermally cured sample. Similar results can be got from FTIR spectrum. As shown in Fig. 4(b), an absorption peak of epoxy group at 915 cm−1 can be clearly observed for 15% E-HBPSi-2, while no obvious peak at the same wavenumber is observed for any other sample. It can be concluded that when rs is 0.9, a complete consumption of epoxy can be got. However, because of the large steric hindrance that the high content of HBPSi-2 brings about, complete reaction between epoxy and cuing agent is hard to achieve without post cure. During the curing process, epoxy reacts with HBPSi-2 first, forming structures with higher crosslinking density. As the reaction goes on, the high steric hindrance leads to structures with lower crosslinking density.45 Thus, at high HBPSi-2 loadings, the heterogeneous structure is expected.
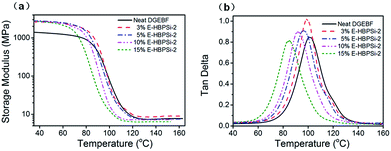 |
| | Fig. 3 Variation of storage modulus (a) and tanδ (b) with temperature in neat DGEBF and E-HBPSi-2 with varying HBPSi-2 loadings. | |
Table 2 Parameters of networks summarized and calculated from DMA
| Samples |
Tg (°C) |
Eg (MPa) |
Er (MPa) |
FWHM (°C) |
| Neat DGEBF |
101.5 |
1382.8 |
7.6 |
22.1 |
| 3% E-HBPSi-2 |
99.2 |
2610.2 |
9.5 |
18.1 |
| 5% E-HBPSi-2 |
96.3 |
2768.6 |
7.8 |
21.4 |
| 10% E-HBPSi-2 |
92.0 |
2722.1 |
7.2 |
22.0 |
| 15% E-HBPSi-2 |
84.5 |
2504.9 |
6.4 |
23.3 |
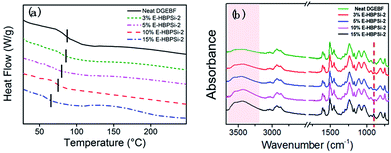 |
| | Fig. 4 DSC curves (a) and FTIR spectrums (b) of the neat DGEBA and hybrids at different E-HBPSi-2 loadings. | |
Mechanical properties
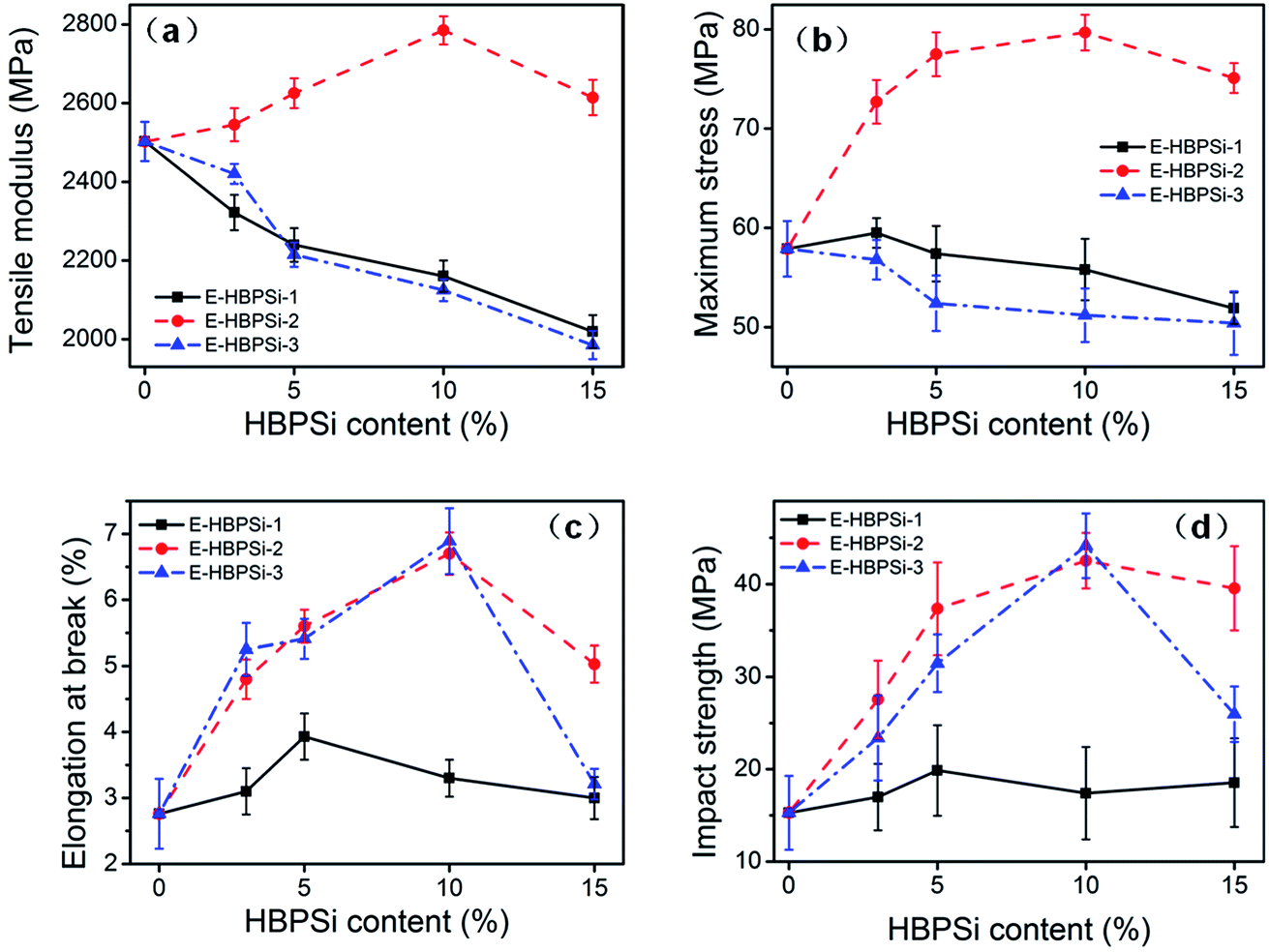
Effects of Mn and HBPSi loading on tensile modulus, strength, elongation at break and impact strength are shown in Fig. 5(a), (b), (c) and (d), respectively. Tensile modulus and maximum stress of samples modified by HBPSi-1 and HBPSi-3 both decrease as the HBPSi loading increase. However, incorporation of HBPSi-2 can significantly increase the modulus and strength: when HBPSi-2 content increases to 10%, tensile modulus and maximum strength increase by 11.4% and 36.2%, respectively. On the other hand, elongation at break and impact strength of samples modified by HBPSi-2 and HBPSi-3 are significantly increased: when HBPSi-2 content increases to 10%, elongation at break and impact strength increase by 153.8% and 186.7%, respectively. However, HBPSi-1 does not affect these two properties obviously. This result indicates that HBPSi with high Mn can improve sample's toughness but impact the modulus and strength, while HBPSi with medium Mn can simultaneously reinforce and toughen the epoxy matrix.
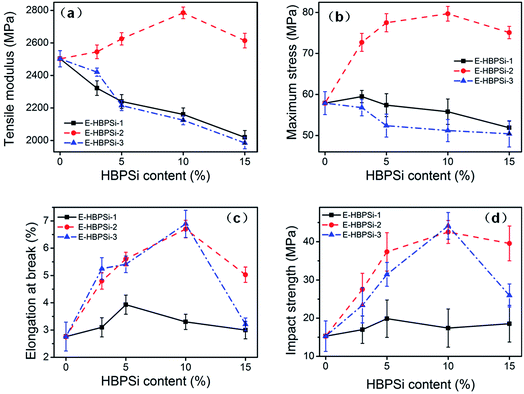 |
| | Fig. 5 Effects of HBPSi-2 loading on mechanical properties: tensile modulus (a); tensile strength (b); elongation at break (c) and impact strength (d). | |
Fracture surface morphology
The SEM images of tensile fracture surfaces of E-HBPSi-2 with varying HBPSi-2 contents are displayed in Fig. 6. The neat specimens exhibit characteristic brittle fracture surface morphology, with minimal deformation observed from the smooth surface pattern, indicative of poor fracture toughness. The fracture surfaces of E-HBPSi-2 show substantial increase in the surface roughness, indicating improved toughness. The rough fracture surface indicates deflection of crack path, i.e. the crack deviation from its original plane, increasing the area of the crack and the required energy for the propagation of the cracks on the fracture surfaces.46 Moreover, dimples appear in the fracture surface of E-HBPSi-2, indicating plastic deformation may be produced under tension and much fracture energy is likely dissipated.47,48 The neat DGEBF exhibits a smooth area with clear fracture boundaries, which means the crack propagation resistance is small and there is almost no plastic deformation, showing brittle fracture and poor toughness.
 |
| | Fig. 6 SEM images of fracture surfaces from fracture toughness specimens: neat DGEBF (a), 3% E-HBPSi-2 (b), 5% E-HBPSi-2 (c), 10% E-HBPSi-2 (d) and 15% E-HBPSi-2 (e). | |
Thermal stability
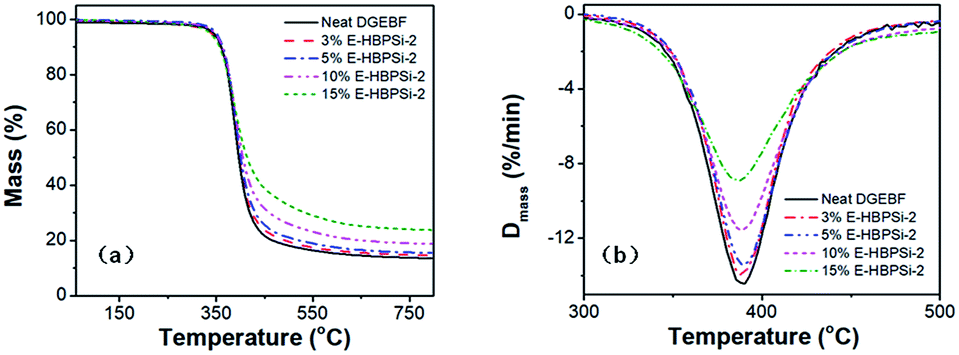
To investigate the thermal stability of the neat DGEBF and modified system, TGA was conducted and the TGA and DTG curves of samples with varying HBPSi-2 contents in a nitrogen atmosphere are shown in Fig. 7. The initial thermal decomposition temperatures [Ti (°C)], the temperatures of the maximum decomposition rate [Tmax (°C)], and the final char yields at 750 °C are also summarized in Table 3. Results show that all cured resins have one-stage decomposition process, demonstrating that the neat and modified resins have similar degradation processes. As the content of HBPSi increases, the maximum mass loss rate of the samples decreases stepwise, which is attributed to the thermal insulation effect of silicone-containing chains.49,50 As the HBPSi-2 content increases to 5%, Ti and Tmax increase due to the more stable structure derived from the increased crosslinking density and higher content of aromatic ring. However, as the HBPSi-2 content increases to 15%, Ti and Tmax decrease because of the incomplete curing. What is more, due to the high decomposition temperature of inorganic moieties, char residue at 750 °C remarkably increases from 14.2% to 23.8% with HPBSi-2 content increases from 0% to 15%, showing a similar trend to that reported in other literature.49,50
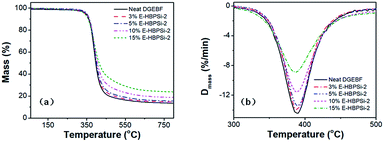 |
| | Fig. 7 TGA (a) and DTG (b) curves of samples with varying HBPSi-2 contents in a nitrogen atmosphere. | |
Table 3 Thermal stability properties of samples with varying HBPSi-2 contents calculated from TGA curves
| Sample |
Ti (°C) |
Tmax (°C) |
Char yield (%) |
| Neat DGEBF |
343.0 |
387.8 |
14.2 |
| 3% E-HBPSi-2 |
347.9 |
388.6 |
15.0 |
| 5% E-HBPSi-2 |
350.4 |
389.9 |
16.2 |
| 10% E-HBPSi-2 |
349.1 |
388.5 |
18.7 |
| 15% E-HBPSi-2 |
337.9 |
385.1 |
23.8 |
Discussion about the toughening and reinforcement mechanism
In most cases, the improvements in toughness of the resins modified with hyperbranched polymers are attributed to the increased fractional free volume.51–53 According to X. Miao et al.,24 the fractional free volume (fT) can be expressed as fT = fg + Δα(T − Tg), where Δα is the difference between the volumetric coefficients of thermal expansion (CTE) in the rubbery and glassy states, respectively, and fg is the fractional free volume “frozen” at Tg. Thus, Δα is directly proportional to fractional free volume. Results of TMA test are shown in Table 4. Δα increases as the HBPSi-2 content increases, indicating that samples with higher content of HBPSi-2 have higher fractional free volume, leading to improved toughness. When the HBPSi-2 loading increases to 15%, though the sample shows higher fractional free volume, the toughness begins to decrease due to the incomplete cure. In addition, Δα of the hybrid also depends on Mn of the HBPSi. When the HBPSi loading is 5%, the fractional free volume increases with Mn. This result is similar to those reported elsewhere, which could be explained by the bigger internal cavities and lower ultimate conversion in hybrids containing HBPSi of higher Mn.53–56 Thus, HBPSi-3 shows effective toughening effect just like HBPSi-2 does, while HBPSi-1 doesn't have obvious effect on the toughness improvement of the matrix because of its low contribution to the increase of the fractional free volume.
Table 4 Linear coefficients of thermal expansion of neat DGEBF and hybrids
| Sample |
αg (×106 K−1) |
αr (×106 K−1) |
Δα (×106 K−1) |
| 0% E-HBPSi-2 |
82.34 |
242.05 |
159.71 |
| 3% E-HBPSi-2 |
80.41 |
248.15 |
167.74 |
| 5% E-HBPSi-2 |
78.08 |
252.90 |
174.82 |
| 10% E-HBPSi-2 |
76.85 |
255.78 |
178.93 |
| 15% E-HBPSi-2 |
74.36 |
260.64 |
186.28 |
| 5% E-HBPSi-1 |
80.67 |
243.92 |
163.25 |
| 5% E-HBPSi-3 |
73.02 |
253.51 |
180.49 |
As shown in Fig. 5, with a certain loading of HBPSi-2, resin sample can be simultaneously reinforced and toughened. In HBPSi-2 molecule, the high density of aromatic ring leads to a structure of high rigidity. At 10% loading, the rigid network contributes to an increased modulus, while the high toughness of the sample also decreases the sensitivity to defect, leading to a high maximum stress. As the HBPSi-2 loading furtherly increases, incomplete cure results in the decrease of the strength, modulus and toughness. E-HBPSi-1 and E-HBPSi-3 don't show comprehensively improved mechanical property. Because of the higher reactivity, HBPSi can be more easily incorporated in the network during the early stage of cure, leading to acceleration of gelation.57 As the curing reaction goes on, the hyperbranched molecule can build up a percolation scaffold in the early stage of cure. According to Xiaoyu Li et al., each hyperbranched crosslink and its immediately connecting regions can be treated as an “effective sphere”.24 Once those effective spheres percolate through the epoxy network, toughness and other performance are greatly improved. However, for HBPSi-1, Mn is too low to create enough fractional free volume, resulting in poor toughening effect. Because of the low molecular weight, HBPSi-1 play a role of plasticizer rather than toughener, for which effective percolation is hard to form. Thus, neither the strength nor the toughness of E-HBPSi-1 are improved. For HBPSi-3, large fractional free volume derived from high molecular weight endows E-HBPSi-3 with significantly improved toughness. However, high molecular weight also results in increased viscosity and steric hindrance, hindering the reaction among reactants during the formation of the percolation network and the later stage of curing. Thus, for E-HBPSi-3, incomplete curing results in decrease of stress and modulus though toughness of the system is improved. The difference in the reinforcing effect of different HBPSi on the epoxy matrix can also be reflected by the DMA results (Fig. S6†). Er of 5% E-HBPSi-2 is higher than that of the neat DGEBF, indicating an increased crosslinking density. However, 5% E-HBPSi-1 and 5% E-HBPSi-3 show lower crosslinking densities than that of the neat DGEBF, resulting in a decreased modulus and stress. Also, Tg of 5% E-HBPSi-1 and 5% E-HBPSi-3 is much lower than that of E-HBPSi-2. The high density of aromatic ring and high functionality of HBPSi-2 contribute to the reinforcement of the modified epoxy, while the plasticizing effect and incomplete curing in the E-HBPSi-1 and E-HBPSi-3 lead to a decreased modulus and stress.
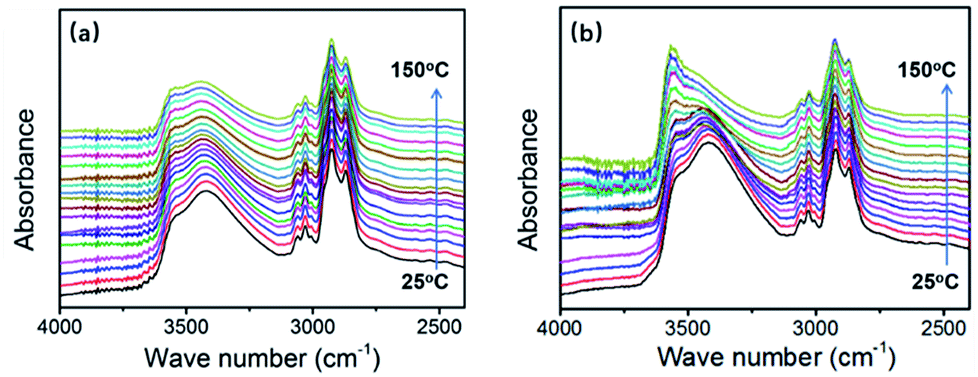
As mentioned in the “rheology of modified system” section, after the addition of HBPSi-2, strong hydrogen-bond interaction exists in the curing system between hydroxyl groups in DGEBF, phenolic and HBPSi. During the reaction, phenolic hydroxy is consumed due to the polymerization. However, at the same time new hydroxyl is generated because of the ring-opening of the epoxy group. Thus, the hydroxyl content still maintains at a high level, especially for systems with high HBPSi contents. As shown in Fig. 4(b), in the spectrum of E-HBPSi-2 with higher HBPSi-2 content, stronger absorption peak of hydroxyl group shows up, indicating a high possibility of the formation of hydrogen bonding network. For this reason, it is possible that this hydroxyl-rich system allows for an increase in secondary bonding and interaction.22,58 FTIR was conducted under ramping temperature for neat DGEBF and E-HBPSi-2. As shown in Fig. 8, for neat DGEBF, peak shape and peak height barely change as the temperature rises from 25 °C to 150 °C. However, for E-HBPSi-2, significant change of hydroxyl group absorption peak appears: peak ranging from 3350 cm−1 to 3450 cm−1 becomes weaker, while peak ranging from 3500 cm−1 to 3700 cm−1 becomes stronger. This result indicates that hydrogen bonding network, which is destroyed at high temperature, do exists in the modified system. Thus, it is believed that in addition to the aforementioned structural effect, the increased cohesive energy density derived from the secondary bonding and interaction also contributes to the increased modulus of the modified system.
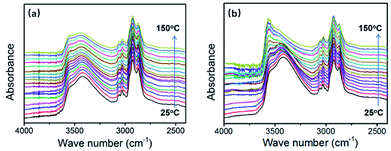 |
| | Fig. 8 FTIR spectrums of the neat DGEBA (a) and 10% E-HBPSi-2 (b) under ramping temperature. | |
Compatibility between the modifier and the matrix also has significant effect on the mechanical property of the hybrid. In the design of HBPSi molecular structure, EMCH was used to increase the miscibility between the HBPSi and epoxy matrix. Tensile curves of E-HBPSi-0 and E-HBPSi-2 with varying modifier contents are shown in Fig. S4.† The tensile modulus, strength and elongation at break are all increased after incorporation of HBPSi-0, which is similar to the effect of HBPSi-2. However, compared with E-HBPSi-2, E-HBPSi-0 is prone to break and no maximum in the stress is observed. By contrast, yielding is observed for E-HBPSi-2. Meanwhile, when the HBPSi-2 loading is 10%, the fracture strain exceeds 6.5%, showing a ductile behavior, which is drastically different from the commonly brittle behavior of neat DGEBF and E-HBPSi-0. For HBPSi-0, large amounts of hydroxyl groups result in strong polarity, making aggregation easy to happen. High magnification SEM images of morphology of E-HBPSi-0 and E-HBPSi-2 with 10% HBPSi content are shown in Fig. S5.† Small round spheres with diameter of about 100 nm, which are derived from the aggregation of HBPSi-0, can be observed in fracture surfaces of E-HBPSi-0. By contrast, no aggregations can be observed in fracture surfaces of E-HBPSi-2, showing no evidence of phase separation. Higher compatibility between resin matrix and modifier reduces the chance of aggregation and ensures the hyperbranched molecular chains spreading well, for which defects under tensile forces are decreased, contributing to the overall improvement of the mechanical property.
Conclusions
A series of hyperbranched polysiloxane (HBPSi) are synthesized and used for modification of the epoxy–phenolic network. The addition of HBPSi-2, which has medium Mn, can significantly decrease the viscosity and increase the shear thinning effect of the epoxy–phenolic mixture because of HBPSi's low intrinsic viscosity and the hydrogen bonding network in the modified mixture. After curing, for the hybrid with moderate content of HBPSi-2, the crosslinking density and the homogeneity of the crosslinking network show simultaneous increase. Mechanical tests show that HBPSi-2 can simultaneously improve the toughness and strength of the epoxy matrix: with 10% HBPSi-2, tensile modulus and maximum strength of the hybrid are increased by 11.4% and 36.2%, respectively, while elongation at break and impact strength are increased by 153.8% and 186.7%, respectively. However, for HBPSi-1 or HBPSi-3, which has relative low or high molecular weight, respectively, fails to comprehensively enhance resin's mechanical performance. Through dynamic mechanical analysis, thermomechanical analysis and FTIR, it is found that the higher free volume contributes to the improved toughness, while the higher strength and modulus are derived from the combined effects of the crosslinking density, network rigidity and cohesive density. The addition of HBPSi also significantly increases the char yield of the material and decreases the thermal weight loss rate, indicating an improved thermal stability. These findings may provide a new strategy for comprehensive improvement of epoxy–phenolic network.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the State Key Scientific Special Project (2016ZX05017-002) of China.
Notes and references
- H. Tsung-Han and C.-S. Wang, Eur. Polym. J., 2001, 37, 267–274 CrossRef.
- M. Rutnakornpituk, Eur. Polym. J., 2005, 41, 1043–1052 CrossRef.
- M. Ogata, N. Kinjo and T. Kawata, J. Appl. Polym. Sci., 1993, 48, 583–601 CrossRef.
- T. H. Ho and C. S. Wang, Eur. Polym. J., 2001, 37, 267–274 CrossRef.
- R. Thomas, D. Yumei, H. Yuelong, Y. Le, P. Moldenaers, Y. Weimin, T. Czigany and S. Thomas, Polymer, 2008, 49, 278–294 CrossRef.
- A. J. Kinloch, S. J. Shaw, D. A. Tod and D. L. Hunston, Polymer, 1983, 24, 1341–1354 CrossRef.
- R. A. Pearson and A. F. Yee, Polymer, 1993, 34, 3658–3670 CrossRef.
- W. Liu, R. Zhou, H. L. S. Goh, S. Huang and X. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 5810–5817 Search PubMed.
- X. Zhang, W. Xu, X. Xia, Z. Zhang and R. Yu, Mater. Lett., 2006, 60, 3319–3323 CrossRef.
- M. A. Rafiee, J. Rafiee, Z. Wang, H. Song, Z.-Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884–3890 CrossRef PubMed.
- L. Boogh, B. Pettersson and J. A. E. Månson, Polymer, 1999, 40, 2249–2261 CrossRef.
- W. Yu, J. Fu, X. Dong, L. Chen, H. Jia and L. Shi, ACS Appl. Mater. Interfaces, 2013, 5, 8897–8906 Search PubMed.
- D. Ratna, O. Becker, R. Krishnamurthy and R. J. Varley, Polymer, 2003, 44, 7449–7457 CrossRef.
- S. Wu, Polymer, 1985, 26, 1855–1863 CrossRef.
- T. Liu, Y. Nie, L. Zhang, R. Chen, Y. Meng and X. Li, RSC Adv., 2015, 5, 3408–3416 RSC.
- X. Zhang, Polym. Int., 2011, 60, 153–166 CrossRef.
- M. Morell, X. Ramis, F. Ferrando, Y. Yu and A. Serra, Polymer, 2009, 50, 5374–5383 CrossRef.
- T. Li, H. Qin, Y. Liu, X. Zhong, Y. Yu and A. Serra, Polymer, 2012, 53, 5864–5872 CrossRef.
- D. Ratna, R. Varley and G. P. Simon, J. Appl. Polym. Sci., 2003, 89, 2339–2345 CrossRef.
- D. Ratna, R. Varley, R. S. Raman and G. P. Simon, J. Mater. Sci., 2003, 38, 147–154 CrossRef.
- D. Foix, A. Serra, L. Amparore and M. Sangermano, Polymer, 2012, 15, 3084–3088 CrossRef.
- Q. Jin, J. M. Misasi, J. S. Wiggins and S. E. Morgan, Polymer, 2015, 73, 174–182 CrossRef.
- F. L. Jin and S. J. Park, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 3348–3356 CrossRef.
- X. Miao, Y. Meng and X. Li, Polymer, 2015, 60, 88–95 CrossRef.
- D. Ratna and G. P. Simon, Polym. Eng. Sci., 2001, 41, 1815–1822 Search PubMed.
- Z. Zhang, L. Yuan, G. Liang, A. Gu, Z. Qiang, C. Yang and X. Chen, J. Mater. Chem. A, 2014, 2, 4975–4988 Search PubMed.
- C. Zhou, A. Gu, G. Liang and L. Yuan, Polym. Adv. Technol., 2011, 22, 710–717 CrossRef.
- D. Zhuo, A. Gu, G. Liang, J. T. Hu, L. Yuan and X. Chen, J. Mater. Chem., 2011, 21, 6584–6594 RSC.
- Z. Qiang, G. Liang, A. Gu and L. Yuan, Mater. Lett., 2014, 115, 159–161 CrossRef.
- S. Ma, W. Liu, C. Hu, Z. Wang and C. Tang, Macromol. Res., 2010, 18, 392–398 CrossRef.
- X. F. Lei, Y. Chen, H. P. Zhang, X. J. Li, P. Yao and Q. Y. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 10207–10220 Search PubMed.
- X. Lei, Y. Chen, M. Qiao, L. Tian and Q. Zhang, J. Mater. Chem. C, 2016, 4, 2134–2146 RSC.
- L. Yang, S. L. Phua, J. K. Teo, C. L. Toh, S. K. Lau and J. Ma, ACS Appl. Mater. Interfaces, 2011, 3, 26–32 Search PubMed.
- P. Cassagnau, Polymer, 2008, 49, 2183–2196 CrossRef.
- D. Zhang, A. B. Karki, D. Rutman, D. P. Young, A. Wang, D. Cocke and Z. Guo, Polymer, 2009, 50, 4189–4198 CrossRef.
- K. L. Ngai and C. M. Roland, Macromolecules, 1993, 26, 6824–6830 CrossRef.
- Y. L. Liu, C. Y. Hsu, W. L. Wei and R. J. Jeng, Polymer, 2003, 44, 5159–5167 CrossRef.
- G. Ragosta, M. Abbate, P. Musto, G. Scarinzi and L. Mascia, Polymer, 2005, 46, 10506–10516 CrossRef.
- H. Zhang, Z. Zhang, K. Friedrich and C. Eger, Acta Mater., 2006, 54, 1833–1842 CrossRef.
- X. Lei, Y. Chen, M. Qiao, L. Tian and Q. Zhang, J. Mater. Chem. C, 2016, 4, 2134–2146 RSC.
- R. A. Pethrick, C. Miller and I. Rhoney, Polym. Int., 2010, 59, 236–241 Search PubMed.
- W. Yu, J. Fu, X. Dong, L. Chen, H. Jia and L. Shi, ACS Appl. Mater. Interfaces, 2013, 5, 8897–8906 Search PubMed.
- P. Rosso and L. Ye, Macromol. Rapid Commun., 2007, 28, 121–126 CrossRef.
- M. Preghenella, A. Pegoretti and C. Migliaresi, Polymer, 2005, 46, 12065–12072 CrossRef.
- R. Mezzenga, L. Boogh, J. A. E. Månson and B. Pettersson, Macromolecules, 2000, 33, 4373–4379 CrossRef.
- X. Chua, R. Huanga, H. Yanga, Z. Wua, J. Lua, Y. Zhoua, L. Li and X. Chu, Mater. Sci. Eng., A, 2011, 528, 3367–3374 CrossRef.
- A. J. Kinloch, D. Maxwell and R. J. Young, J. Mater. Sci. Lett., 1985, 4, 1276–1279 CrossRef.
- H. Zhang, Z. Zhang, K. Friedrich and C. Eger, Acta Mater., 2006, 54, 1833–1842 CrossRef.
- P. Jia, H. Liu, Y. Wang and X. Cai, RSC Adv., 2015, 5, 97413–97421 RSC.
- P. Jia, H. Liu, Q. Liu and X. Cai, Polym. Degrad. Stab., 2016, 134, 144–150 CrossRef.
- J. Lv, Y. Meng, L. He, T. Qiu, X. Li and H. Wang, J. Appl. Polym. Sci., 2013, 128, 907–914 CrossRef.
- L. Luo, Y. Meng, T. Qiu, Z. Li, J. Yang, X. Cao and X. Li, Polym. Compos., 2013, 34, 1051–1060 CrossRef.
- D. Ratna and G. P. Simon, J. Appl. Polym. Sci., 2010, 117, 557–564 Search PubMed.
- T. Liu, Y. Nie, R. Chen, L. Zhang, Y. Meng and X. Li, J. Mater. Chem. A, 2015, 3, 1188–1198 Search PubMed.
- L. Luo, Y. Meng, T. Qiu and X. Li, J. Appl. Polym. Sci., 2013, 130, 1064–1073 CrossRef.
- J. P. Yang, Z. K. Chen, G. Yang, S. Y. Fu and L. Ye, Polymer, 2008, 49, 3168–3175 CrossRef.
- Q. Li, X. Li and Y. Meng, Thermochim. Acta, 2012, 549, 69–80 CrossRef.
- E. Žagar and M. Žigon, Prog. Polym. Sci., 2011, 36, 53–88 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01740a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*