
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg–K/MnO2 nanorods as highly efficient catalysts for formaldehyde oxidation at low temperature
Suhong
Lu
 *a,
Xue
Wang
a,
Qinyu
Zhu
a,
Canchang
Chen
a,
Xuefeng
Zhou
a,
Fenglin
Huang
a,
Kelun
Li
b,
Lulu
He
a,
Yanxiong
Liu
a and
Fanjue
Pang
a
*a,
Xue
Wang
a,
Qinyu
Zhu
a,
Canchang
Chen
a,
Xuefeng
Zhou
a,
Fenglin
Huang
a,
Kelun
Li
b,
Lulu
He
a,
Yanxiong
Liu
a and
Fanjue
Pang
a
aCollege of Chemistry and Chemical Engineering, Xi'an Shiyou University, Xi'an 710065, China. E-mail: lusuhong@xsyu.edu.cn
bShaanxi Coal and Chemical Technology Institute Co., Ltd, Xi'an 710070, China
First published on 17th April 2018
Abstract
A series of Ag–K/MnO2 nanorods with various molar ratios of K/Ag were synthesized by a conventional wetness incipient impregnation method. The as-prepared catalysts were used for the catalytic oxidation of HCHO. The Ag–K/MnO2 nanorods with an optimal K/Ag molar ratio of 0.9 demonstrated excellent HCHO conversion efficiency of 100% at a low temperature of 60 °C. The structures of the samples were investigated by BET, TEM, SEM, XRD, H2-TPR, O2-TPD and XPS. The results showed that Ag–0.9K/MnO2-r exhibited more facile reducibility and greatly abundant surface active oxygen species, endowing it with the best catalytic activity of the studied catalysts. This work provides new insights into the development of low-cost and highly efficient catalysts for the removal of HCHO.
1 Introduction
Formaldehyde (HCHO) is commonly released from building/decorative materials and unregulated indoor smoking; it is regarded as a major pollutant.1,2 It has been reported that long-term exposure to indoor air containing even a few ppm of HCHO threatens human health and causes health problems, such as nasal tumors, skin irritation, allergies, and decreased concentration.3,4 Thus, numerous technologies have been investigated to eliminate indoor emission of HCHO to satisfy stringent environmental regulations; these techniques include adsorption,5 photo-catalysis,6 plasma technology,7 and thermal catalytic oxidation.8In recent years, complete oxidation of HCHO over heterogeneous catalysts has been considered to be the most promising strategy for indoor HCHO removal due to its properties of high efficiency and lack of pollution. Highly effective catalysts have been prepared to reduce HCHO, such as transition metal oxides9–11 and supported noble metals (Pt,12–15 Au,16 and Pd17). Generally, transition metal oxides can achieve complete oxidation of HCHO at very high temperatures (>100 °C). In contrast, supported noble metal catalysts such as Pt/TiO2,18 Na-Pd/TiO2 (ref. 19) and Au/Co3O4–CeO2 (ref. 20) have been proved to have excellent catalytic activity at relatively low temperatures, even room temperature. However, the high cost of the supported noble metals limits their widespread application. Therefore, it is still a challenge to develop efficient and low cost catalysts.
On the basis of current research, Ag-supported catalysts, which are much less expensive than the abovementioned noble metals, have been demonstrated to possess efficient low-temperature catalytic activities for HCHO oxidation. For example, the price of Pt per gram for H2PtCl6·6H2O is about 20 times that of Ag per gram for AgNO3 on the Sigma-Aldrich website. It was reported that complete oxidation of HCHO was obtained over Ag/MnOx–CeO2 catalysts at a temperature as low as 100 °C; the catalysts were synthesized by a deposition precipitation method.21 Ma et al.22 successfully prepared Ag/CeO2 nanospheres by a one-step hydrothermal method which could achieve complete HCHO oxidation at temperatures above 110 °C under relatively high space velocity. Huang et al.23 demonstrated that Ag nanoparticles-supported hollandite-type manganese oxide nanorods exhibited good activity for HCHO oxidation; complete conversion of HCHO was obtained at 110 °C. In order to improve the activity of Ag-supported catalysts, Li et al.24 indicated that Ag/Fe–MnOx possesses higher catalytic activity than Ag/MnOx due to the enhanced interaction between Ag and the support. The conversion of HCHO was 100% at 90 °C. Qu et al.25 reported a bimetallic AgCo/APTES@MCM-41 catalyst that could achieve complete oxidation of HCHO at temperatures as low as 90 °C because of the formation of strong metal–metal interactions between Ag and Co. Bai et al.26 demonstrated that K–Ag/Co3O4 displayed complete conversion of HCHO at 70 °C due to additional surface OH− provided by K+ ions. However, the performance of Ag-supported catalysts at low temperature still requires improvement in comparison with other noble metal catalysts.
Due to its remarkable oxygen storage ability, manganese oxide has attracted much attention and is regarded as an effective catalyst for the catalytic oxidation of HCHO. For example, Torres et al.27 fabricated mesoporous MnOx catalysts and observed their good catalytic activities for the complete oxidation of HCHO; using these catalysts, complete conversion of HCHO was achieved at 145 °C. Zhou et al.28 prepared porous cellulose-supported MnO2 nanosheet catalysts and observed their excellent performance in the catalytic oxidation of HCHO at a temperature of 140 °C. Previous literature studies have reported that manganese oxides with various morphologies showed obvious differences in catalytic performance.29 For example, flower-like manganese oxide nanospheres (α-MnO2) outperformed other crystalline manganese oxides below 120 °C for the oxidation of HCHO.30 Studying the preparation and catalysis of materials for rod-like tetragonal α-MnO2, flower-like hexagonal ε-MnO2, and dumbbell-like tetragonal β-MnO2, Shi et al.31 discovered that the catalytic activity decreased in the order of α-MnO2 > ε-MnO2 > β-MnO2. To the best of our knowledge, the catalytic application of Ag/MnO2 nanorods for the catalytic oxidation of HCHO has not been reported. Moreover, it has been well acknowledged that alkali metals can serve as electronic or textural promoters for catalysis.19,32 The addition of alkali metals (Li, Na and K) to Pt/TiO2 catalysts resulted in remarkable enhancements of their performance in HCHO oxidation.33 It was further found that addition of K has an obvious enhancing effect on Ag/Co3O4 catalysts for HCHO oxidation.26
Therefore, in this work, MnO2 nanorods were synthesized by a hydrothermal preparation method and Ag–K/MnO2 nanorods were prepared by a conventional wetness incipient impregnation method. These catalysts were applied for the catalytic oxidation of HCHO. The catalysts were characterized by BET, TEM, SEM, XRD, H2-TPR and O2-TPD. It is proposed that the abundant surface oxygen species and low-temperature reducibility enhanced the catalytic oxidation activity of the Ag–K/MnO2 nanorods.
2 Experimental
Materials
KMnO4, MnSO4, AgNO3 and KNO3 (analytically pure reagents) were obtained from Sinopharm Chemical Reagent Co., Ltd. The reagents were used as received without further purification.Preparation of catalysts
The MnO2 nanorods were synthesized by a hydrothermal method similar to previous studies.31,34 16 mmol of KMnO4 and 6 mmol of MnSO4 precursor were dissolved in 160 mL of deionized water and then stirred for 1 h to form a homogeneous solution. Subsequently, the mixture was transferred into a stainless steel autoclave for thermal treatment at 160 °C for 12 h. After cooling, the resulting precipitate was collected by filtration, washed with deionized water to remove any possible residual reactants and dried at 60 °C for 24 h. The acquired powder was calcined in a muffle oven at 300 °C for 4 h with a heating rate of 1 °C min−1, thus affording the final MnO2 nanorods (designed as MnO2-r).Ag–K/MnO2 nanorods with different molar ratios (K/Ag = 0.5, 0.9, 1.3) were prepared by a conventional wetness incipient impregnation method, and the general steps were as follows: 1 g MnO2-r was dispersed in 40 mL deionized water under vigorous stirring for 30 min, and then the appropriate amounts of AgNO3 and KNO3 solution were added dropwise. After that, the resulting suspension was stirred at room temperature for 24 h and then evaporated at 80 °C with continuous stirring. The obtained powder was dried at 80 °C for 12 h. The amount of Ag in the final catalyst was 0.1 wt%, measured via ICP-AES. The as-prepared samples were thus represented as Ag–xK/MnO2-r (x indicates the molar ratio of K/Ag). Ag/MnO2-r was also synthesized by a similar process to that described above.
Characterization
The Ag contents of the prepared catalysts were determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES, Vista-MPX, Varian).Nitrogen adsorption and desorption isotherms were measured at −196 °C using a Micromeritics apparatus (ASAP2020HD88). The specific surface areas were computed by applying the Brunauer–Emmett–Teller (BET) method. The pore size distribution was estimated from the desorption branch of the isotherms using the BJH model. All samples were outgassed under vacuum at 250 °C for 4 h prior to analysis.
The morphologies of the products were determined by scanning electron microscopy (SEM, America FET Quanta 600 FEG) and transmission electron microscopy (TEM, Tecnai G2 F20).
Powder X-ray diffraction (XRD) measurements of the catalysts were carried out using a Panalytical Empyrean X-ray diffractometer with Cu-Kα radiation (λ = 0.154![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 056 nm) at 40 kV and 30 mA with a scanning speed of 2° min−1 from 10.0° to 80.0°.
056 nm) at 40 kV and 30 mA with a scanning speed of 2° min−1 from 10.0° to 80.0°.
The temperature programmed reduction (H2-TPR) equipment consisted of a thermal conductivity detector (TCD) connected to a flow-control system and a programmed heating unit. About 50 mg of catalyst was used in each measurement. The reduction agent of 10% H2/N2 was introduced with a flow rate of 60 mL min−1; each sample was heated to 650 °C from 40 °C at a rate of 10 °C min−1.
Oxygen temperature-programmed desorption (O2-TPD) tests were conducted using the same equipment as the H2-TPR tests. Prior to each run, 60 mg of sample was loaded and pretreated with 21 vol% O2/N2 (30 mL min−1) at 300 °C for 1 h, then prereduced in a stream of 10 vol% H2/N2 flow at 200 °C for 1 h with a heating rate of 10 °C min−1. After cooling to 65 °C in the same flow, the adsorption of O2 was operated in a flow of 21% O2/N2 at a rate of 20 mL min−1 for 1 h at 65 °C. Finally, the catalyst was heated from 65 °C to 500 °C at a constant heating rate of 10 °C min−1 in a flow of He (60 mL min−1). The desorption of oxygen was calculated from the signal of a TCD detector.
X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific K-Alpha instrument using MgKα radiation (1653.6 eV) at a beam power of 250 W. The binding energy of C 1s (284.6 eV) was used as the internal standard.
Catalytic activity tests
The catalytic activity tests of the as-prepared catalysts for HCHO oxidation were performed in a fixed-bed reactor under atmospheric pressure. Before the catalytic activity tests, the Ag–K/MnO2 nanorods (50 mg) were pretreated with 21 vol% O2/N2 at 300 °C for 1 h and then cooled to 200 °C. The gas was switched to 10 vol% H2/N2 for 1 h at a temperature of 200 °C. After that, the temperature was cooled to 25 °C. Gaseous HCHO was generated by flowing 21% O2/N2 over paraformaldehyde in a thermostatic water bath. The standard feed gas containing 300 ppm of HCHO, 21% O2 and N2 comprising the balance was introduced into the reactor. The total flow rate was 30 mL min−1 in a mass space velocity of 36000 mL gcat−1 h−1. The analyses of the products, such as CO and CO2, were executed using a gas chromatograph equipped with a hydrogen flame ionization detector (FID) and Ni catalyst convertor. No carbon-containing compounds other than CO2 in the products were detected for the tested catalysts. Thus, HCHO conversion was calculated as follows:where [CO2]out and [HCHO]in in the formula correspond to the CO2 concentration in the products and the concentration of HCHO in the gas flow, respectively.
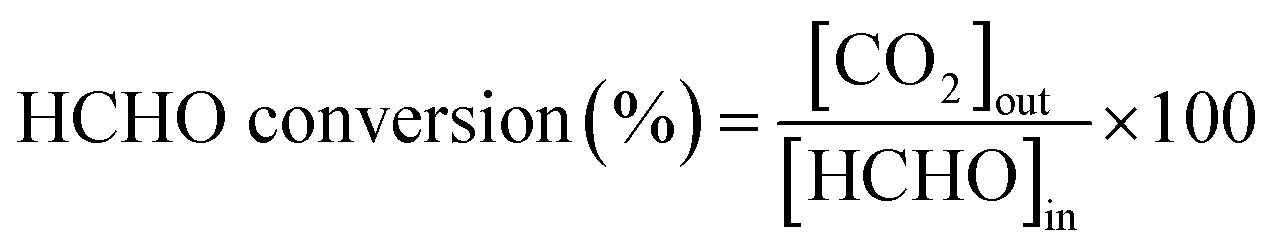
3 Results and discussion
N2 adsorption and desorption
The specific surface areas, pore diameters and pore volumes of the Ag–xK/MnO2-r samples, along with those of Ag/MnO2-r and MnO2-r, are listed in Table 1. Among all the samples, pure MnO2-r possessed the highest surface area (102.1 m2 g−1) and pore volume (0.37 cm3 g−1). However, the surface area and pore volume showed remarkable decreases after the addition of Ag and K; especially, when the molar ratio of K/Ag was 1.3, these values were 45.9 m2 g−1 and 0.25 cm3 g−1, respectively, for Ag–1.3K/MnO2-r. Meanwhile, the average pore diameters of these samples increased from 12.2 to 19.2 nm, which indicates that Ag and K nanoparticles formed on the surface and blocked the small pores.22,24SEM and TEM images
The morphologies of the MnO2-r and Ag–0.9K/MnO2-r samples were investigated using SEM (Fig. 1). It was observed that the MnO2-r sample derived hydrothermally at 160 °C displayed a rod-like morphology, in good agreement with previous reports for MnO2 in the literature.31,34 The MnO2-r samples had diameters of about 80 nm and lengths ranging from 2 to 5 µm. Moreover, the Ag–0.9K/MnO2-r catalyst also demonstrated a rod-like morphology similar to that of MnO2-r. | ||
| Fig. 1 SEM images of (a and b) MnO2-r and (c and d) Ag–0.9K/MnO2-r. TEM image of (e) MnO2-r. | ||
XRD patterns
Fig. 2 displays the XRD patterns of the as-prepared Ag–xK/MnO2-r catalysts, Ag/MnO2-r and MnO2-r. It can be seen that all the samples consisted of single-phase α-MnO2 (JCPDS PDF 44-0141). However, no feature peaks corresponding to the Ag and K species could be found in the XRD patterns; the absence of peaks related to Ag and K species probably resulted from the low loading content and high dispersion of Ag and K with small diameters.22,35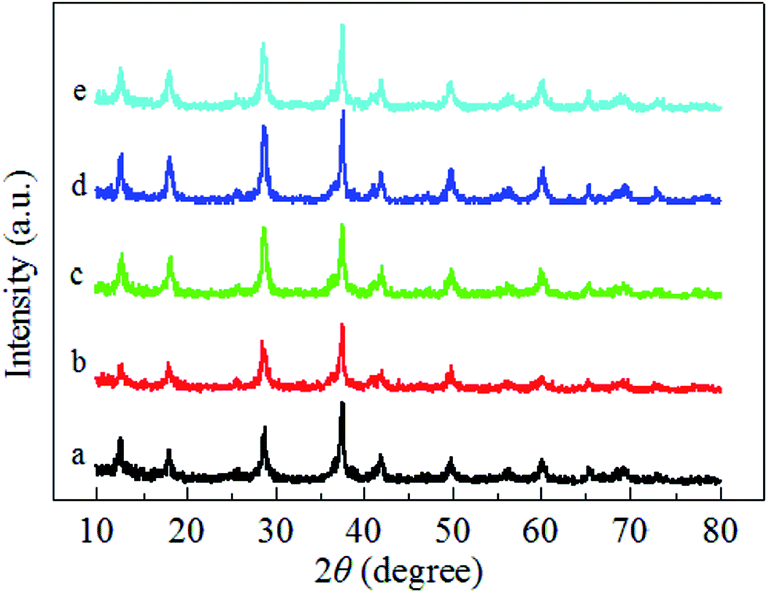 | ||
| Fig. 2 XRD patterns of (a) MnO2-r, (b) Ag/MnO2-r, (c) Ag–0.5K/MnO2-r, (d) Ag–0.9K/MnO2-r and (e) Ag–1.3K/MnO2-r. | ||
H2-TPR
Fig. 3 illustrates the H2-TPR profiles of the pure MnO2-r, Ag/MnO2-r and Ag–xK/MnO2-r samples. For the sample of pure MnO2-r, a strong reduction peak was observed in the temperature range from 149–450 °C with an overlap peak centered at 327 °C; this can be assigned to the combined reduction of MnO2/Mn2O3 to Mn3O4 and of Mn3O4 to MnO. This is similar to the observation of Li et al.35 For Ag/MnO2-r, a shoulder peak was centered at 306 °C and a strong broad reduction peak appeared between 324 °C and 432 °C. Obviously, the addition of Ag to MnO2-r dramatically shifted the reduction temperatures to lower regions, indicating the occurrence of metal-support interactions between Ag and MnO2-r. It has been reported that the reducibility of a material can be significantly improved by noble metal loading, which is often interpreted in terms of the activation and spillover of hydrogen from Ag atoms to MnO2-r.24 The H2 spillover phenomenon showed an ability to adsorb, activate, and migrate hydrogen. Activated hydrogen on the Ag surface can readily migrate to the surface of the MnO2-r support and participate in the reduction reaction.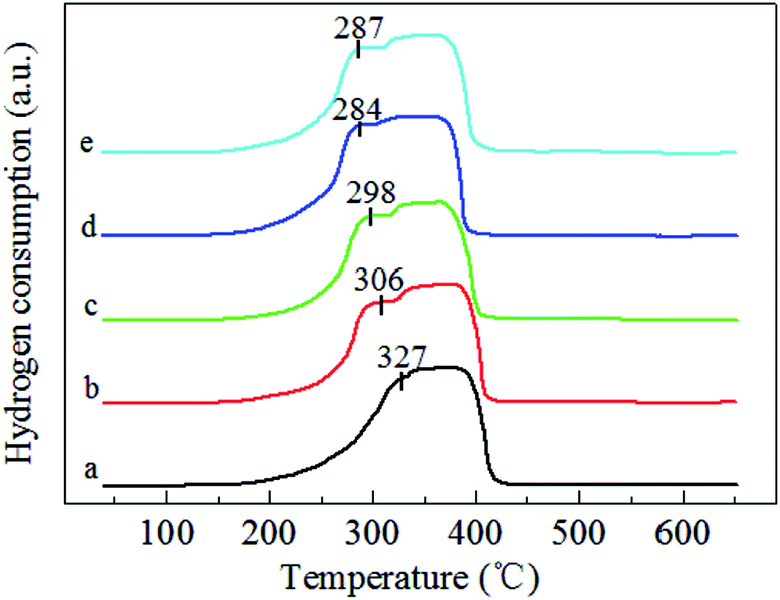 | ||
| Fig. 3 H2-TPR profiles of (a) MnO2-r, (b) Ag/MnO2-r, (c) Ag–0.5K/MnO2-r, (d) Ag–0.9K/MnO2-r and (e) Ag–1.3K/MnO2-r. | ||
Compared with the reduction features of Ag/MnO2-r, the reduction peaks of the Ag–xK/MnO2-r samples shifted towards lower temperatures; this shift was most noticeable for the Ag–0.9K/MnO2-r catalyst. Moreover, without changes in its peak pattern, the reduction temperature of Ag–0.9K/MnO2-r showed the best low-temperature reducibility, which was 14 °C and 3 °C lower than those of Ag–0.5K/MnO2-r and Ag–1.3K/MnO2-r, respectively. A similar enhancing effect of K addition on catalytic reducibility was studied previously. Bai et al.26 reported that the presence of K+ enhanced the reducibility of K–Ag/Co3O4 due to the stronger metal-support interaction. They also found that the presence of K+ ions increased the number of Co3+ cations. Pan et al.36 indicated that acid-treated MnOx samples showed the best redox properties due to the higher oxidation states of the manganese ions, which appeared to be responsible for their enhanced catalytic properties in HCHO oxidation. Hence, it is reasonable to speculate that the addition of K+ improved the low-temperature reducibility and higher oxidation states of manganese ions of the Ag–0.9K/MnO2-r sample in this study. The sufficient manganese cations implied that the oxygen species in the Ag–0.9K/MnO2-r sample could be more readily activated and that they migrated and improved the catalytic performance of HCHO oxidation (see Fig. 5).37
In addition, previous studies reported that catalyst reducibility is related to oxygen vacancies. Catalysts with better reducibility can yield more oxygen vacancies.24,38 Thus, Ag–0.9K/MnO2-r could produce more oxygen vacancies with the assistance of K than Ag/MnO2-r. Gas-phase oxygen molecules could be readily adsorbed in the oxygen vacancies, forming active oxygen species.20,39 This result is in agreement with the O2-TPD results (see Fig. 4).
 | ||
| Fig. 4 O2-TPD profiles of (a) MnO2-r, (b) Ag/MnO2-r, (c) Ag–0.5K/MnO2-r, (d) Ag–0.9K/MnO2-r and (e) Ag–1.3K/MnO2-r. | ||
O2-TPD
O2-TPD experiments over the pure MnO2-r, Ag/MnO2-r, Ag–0.5K/MnO2-r, Ag–0.9K/MnO2-r and Ag–1.3K/MnO2-r catalysts were carried out to determine the mobility of oxygen species, as shown in Fig. 4. According to the literature,40,41 the desorption oxygen species of the oxides can be generally ranked in the sequence of oxygen molecule (O2) > oxygen molecule anion (O2−) > oxygen anion (O−) > lattice oxygen (O2−). The desorption of O2 can occur at a very low temperature (<200 °C), while surface active oxygen species such as O2− and O− can desorb between 200 °C and 400 °C and the lattice oxygen from MnO2 can desorb above 400 °C.20,42 As shown in Fig. 4, for pure MnO2-r, the large peak between 150 °C and 350 °C can be assigned to O2, O2− and O− desorption. After the introduction of Ag, significant increases of the desorption peaks in the range of 175–400 °C were noted for Ag/MnO2-r. Moreover, the desorption peak positions shifted to a lower temperature range. These data suggest that the presence of Ag is beneficial to produce surface active oxygen in the Ag/MnO2-r catalyst. Similar phenomena were observed by Huang et al.40 and Ma et al.22 Huang et al.40 observed that the formation of Ru–O–Ce in Ru/CeO2 can remarkably increase the amount of oxygen vacancies (a reflection of the surface active oxygen). Ma et al.22 observed that the presence of Ag in Ag/CeO2 nanospheres facilitates the generation of much more surface active oxygen desorption during the O2-TPD process.With the addition of K, the original desorption temperature of the surface active oxygen species shifted to lower temperatures for Ag–xK/MnO2-r in comparison with those of Ag/MnO2-r. For example, the desorption of oxygen from the surface active oxygen species appeared at 218 °C on Ag–0.5K/MnO2-r, 197 °C on Ag–0.9K/MnO2-r and 207 °C on Ag–1.3K/MnO2-r. This indicates that the Ag–0.9K/MnO2-r catalyst most readily desorbed the surface active oxygen species. This is in good accordance with the TPR measurements (see Fig. 3). As reported,43,44 the oxygen desorption temperature of catalytic materials is related to their catalytic oxidation activity. Lower starting oxygen desorption temperatures may be significantly favorable for high activity in HCHO oxidation due to the abundance of active surface oxygen species that can readily participate in the catalytic oxidation reaction.45 Here, it can be expected that Ag–0.9K/MnO2-r will possess excellent catalytic performance for HCHO oxidation at low temperature.
XPS spectra
The XPS spectra of the MnO2-r, Ag/MnO2-r and Ag–0.9K/MnO2-r samples are displayed in Fig. 5. As shown in Fig. 5A, the two peaks of binding energy around 368.0 eV and 375.0 eV can be assigned to the escape of electrons from the 3d5/2 and 3d3/2 core levels. The Ag 3d5/2 binding energies of Ag/MnO2-r and Ag–0.9K/MnO2-r are 368.3 eV, reflecting the metallic state. The Ag atoms of Ag–0.9K/MnO2-r have a relatively higher electronic density of states than those of Ag/MnO2-r, originating from the different density of states of the Ag d orbitals.46 In the Mn 2p spectra (Fig. 5B), two obvious peaks were found. The splitting energy of the Mn 2p peak (11.8 eV) approached that of Mn4+, in accord with the energy separation between Mn 2p3/2 and Mn 2p1/2 reported previously.37 In comparison with MnO2-r, apparent upshifts of the Mn 2p3/2 and Mn 2p1/2 peaks occurred for Ag/MnO2-r and Ag–0.9K/MnO2-r, which indicates that the addition of K increased the number of Mn4+ ions. Fig. 5C displays that K 2p has two components at BE = 291.8 and 295.0 eV, which demonstrates the presence of K+ ions.26 The BE of O 1s in Fig. 5D shows two surface oxygen species by deconvolution of the O 1s spectra. A shoulder on a higher BE (532.3 eV) can be assigned to adsorbed oxygen or surface hydroxyl species, while the main peak at about 529.9 eV can be attributed to surface lattice oxygen atoms. Moreover, discernible shifts to a lower binding energy for Ag/MnO2-r and Ag–0.9K/MnO2-r were observed in comparison to MnO2-r, accounting for an increase in their negative charge.46 It has been reported that increased surface adsorbed oxygen and hydroxyl species play crucial roles to promote catalytic activity for HCHO oxidation.26,46 From Fig. 5D, it can be observed that the intensity of the higher BE (531.8 eV) of the Ag–0.9K/MnO2-r catalyst is the strongest, implying that it contains more surface active oxygen. The result is in good agreement with the O2-TPD analysis (Fig. 4).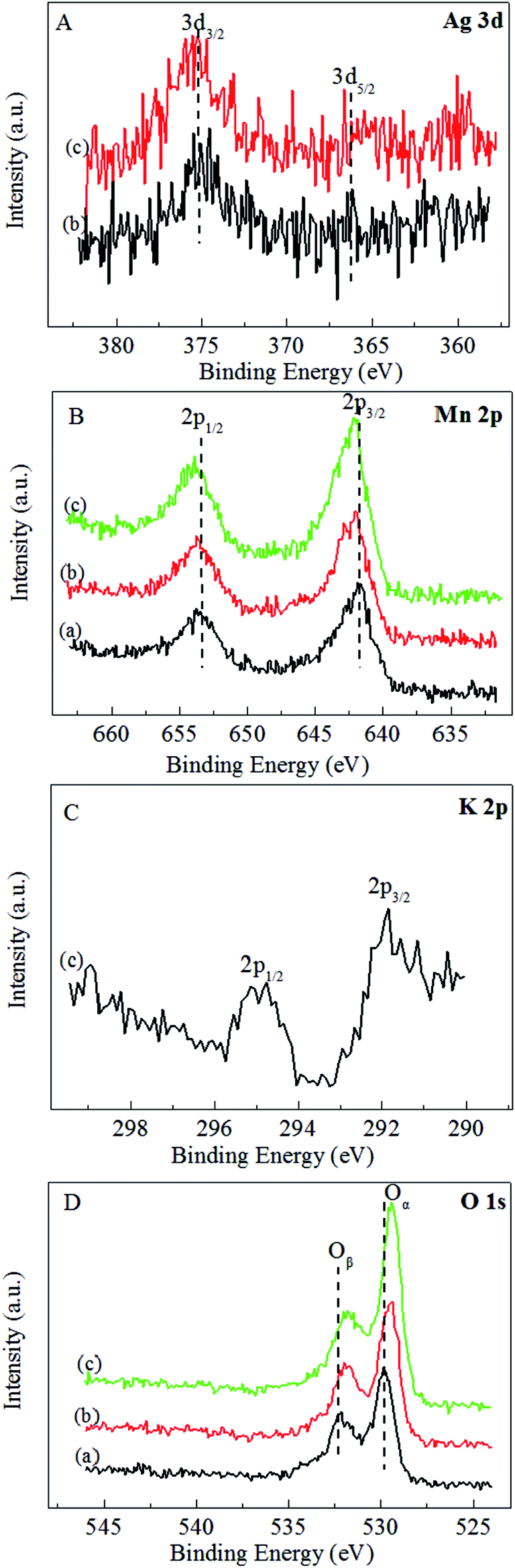 | ||
| Fig. 5 XPS spectra of (a) MnO2-r, (b) Ag/MnO2-r and (c) Ag–0.9K/MnO2-r. (A) Ag 3d, (B) Mn 2p, (C) K 2p and (D) O 1s. | ||
Catalytic activity
Formaldehyde conversion was measured as a function of reaction temperature over the Ag–xK/MnO2-r catalysts together with Ag/MnO2-r and pure MnO2-r. As shown in Fig. 6, pure MnO2-r afforded HCHO conversion of less than 89% in the temperature range investigated. With 0.1% Ag addition, 85.4% HCHO conversion was tested at 60 °C over Ag/MnO2-r. Obviously, the catalytic performance of Ag/MnO2-r was promoted by the presence of K+ ions. Ag–0.5K/MnO2-r achieved 92.7% HCHO conversion at 60 °C. With increasing K+ ion content, Ag–0.9K/MnO2-r showed the highest performance, with 100% HCHO conversion at 60 °C. The conversion of HCHO slightly decreased with further increase of the K+ ion loading amount. 99.6% conversion of HCHO was obtained at 60 °C over Ag–1.3K/MnO2-r. The results revealed that the catalytic activity followed the decreasing order of Ag–0.9K/MnO2-r > Ag–1.3K/MnO2-r > Ag–0.5K/MnO2-r > Ag/MnO2-r > MnO2-r. The optimal molar ratio of K/Ag was 0.9. Thus, the addition of K enhanced the catalytic activity for HCHO oxidation, which is similar to Na or K-promoted catalysts.19,26 It is well known that the presence of K+ ions provides surface OH− species, which may play an important role in the catalytic oxidation of HCHO. Nie et al.47 investigated the reactivity of Pt/TiO2 catalysts with the assistance of NaOH toward HCHO oxidation; they found that the catalyst showed higher activity than that without NaOH due to the introduction of additional surface hydroxyl groups (OH−), which can favor HCHO adsorption via strong hydrogen-bond interactions between HCHO and OH− present on TiO2. Bai et al.26 prepared 3D K-Ag/Co3O4 catalyst and discovered that the addition of K+ ions obviously promoted the catalytic performance for HCHO oxidation because of the surface OH− species provided by K+ ions. Yang et al.48 reported that the superior performance of PtNi(OH)x/γ-Al2O3 could be attributed to the preferred hydroxyl-facilitated HCHO oxidation pathway through formate oxidation by the abundant associated hydroxyl groups near the Pt active sites.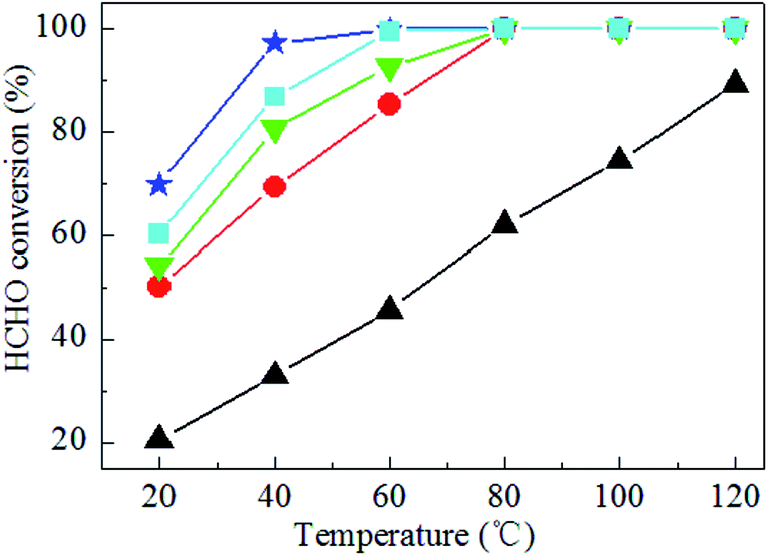 | ||
| Fig. 6 HCHO conversion over (▲) MnO2-r, (●) Ag/MnO2-r, (▼) Ag–0.5K/MnO2-r, (★) Ag–0.9K/MnO2-r and (■) Ag–1.3K/MnO2-r catalysts. Reaction conditions: 300 ppm HCHO, 21 vol% O2 and N2 (balance). The total flow rate and WHSV are 30 mL min−1 and 36000 mL gcat−1 h−1, respectively. | ||
The Ag-based catalysts currently being applied for HCHO oxidation are summarized in Table 2. It can be clearly seen that the Ag–0.9K/MnO2-r catalysts show the best catalytic activities at the lowest temperature.
| Catalyst | Reaction conditions | T 100 (°C) | Ref. |
|---|---|---|---|
| Ag/MnOx–CeO2 | 580 ppm HCHO, 18% O2, 30000 mL gcat−1 h−1 |
100 | 21 |
| Ag/CeO2 nanosphere | 810 ppm HCHO, 20% O2, 84000 h−1 |
110 | 22 |
| Ag/Fe0.1–MnOx | 230 ppm HCHO, 21% O2, 30000 mL gcat−1 h−1 |
90 | 24 |
| AgCo/APTES@MCM-41 | 500 ppm HCHO, 20% O2, 9000 mL gcat−1 h−1 | 90 | 25 |
| 3D K–Ag/Co3O4 | 100 ppm HCHO, 20% O2, 30000 h−1 |
70 | 26 |
| Ag–0.9K/MnO2-r | 300 ppm HCHO, 21% O2, 36000 mL gcat−1 h−1 |
60 | Herein |
Catalytic stability of the 0.9K/MnO2-r samples is very important for HCHO decomposition; therefore, cycle and stability tests were carried out at 60 °C for 60 h with a WHSV of 36000 mL gcat−1 h−1, as displayed in Fig. 7 and 8. No noticeable decrease in the catalytic activity was found during five on/off cycles. As shown in Fig. 7, the catalytic activity of Ag–0.9K/MnO2-r showed no obvious change up to the end of the experiments, indicating the outstanding stability and efficiency of this catalyst for HCHO removal.
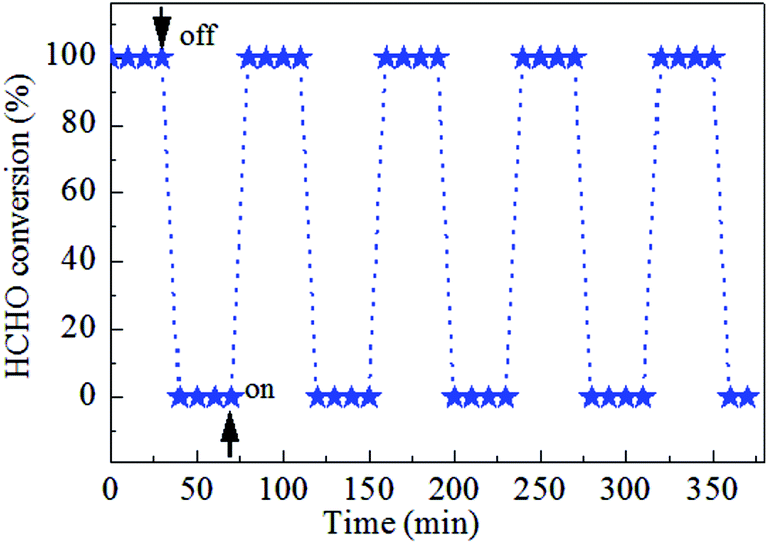 | ||
| Fig. 7 Cycle test of Ag–0.9K/MnO2-r for HCHO oxidation at 60 °C under the reaction conditions of 300 ppm HCHO, 21 vol% O2 and N2 (balance). | ||
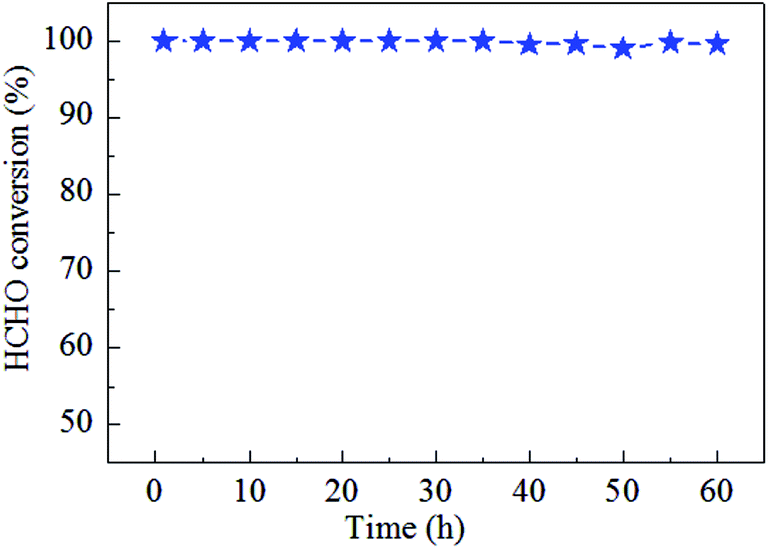 | ||
| Fig. 8 Stability test of Ag–0.9K/MnO2-r for HCHO oxidation at 60 °C with the reaction conditions of 300 ppm HCHO, 21 vol% O2 and N2 (balance). | ||
Generally, it is well acknowledged that the surface area, surface active oxygen species, and reducibility are crucial parameters influencing the catalytic performance of a catalyst.49 Notably, the Ag–xK/MnO2-r samples, which have slightly smaller specific surface areas, exhibited higher catalytic activity than Ag/MnO2-r; this was also mentioned in previous reports.50 This observation implied that the specific surface area was not the decisive factor for HCHO oxidation. In addition to the specific surface area, better low-temperature reducibility and abundant surface active oxygen species are significantly beneficial for enhanced catalytic activity. In our case, by comparing the activity data and characterization results, it can be seen that there are obvious relationships of the reducibility (Fig. 3) and surface active oxygen species (Fig. 4) with the catalytic activity (Fig. 5). The decreasing order of reducibility and oxygen desorption is Ag–0.9K/MnO2-r > Ag–1.3K/MnO2-r > Ag–0.5K/MnO2-r > Ag/MnO2-r > MnO2-r, which is the same as the sequence of the HCHO catalytic oxidation tests. Research found that the oxidation of HCHO followed a redox reaction mechanism;46 an efficient catalyst requires good reducibility. Simultaneously, the enriched surface active oxygen species generated on the catalyst interface can take part in the reaction of HCHO at low temperature.20,51 It is reasonable to deduce that the good performance of the catalyst is associated with its better low-temperature reducibility and higher number of surface active oxygen species.
4 Conclusions
In summary, Ag–K/MnO2 nanorods have been demonstrated as a new and high-performance catalyst for efficient catalytic oxidation of HCHO. The molar ratio of K/Ag greatly influenced the catalytic activity of the Ag–K/MnO2 nanorods, and the optimal K/Ag ratio was found to be 0.9. It was observed that Ag–0.9K/MnO2-r presented outstanding catalytic activity; this catalyst achieved 100% HCHO conversion at a low temperature of 60 °C. The excellent performance of Ag–0.9K/MnO2-r resulted from its facile reducibility and highly abundant surface active oxygen species. This work provides new insight into the development of low-cost and high-efficiency catalysts for low-temperature catalytic oxidation of HCHO.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored financially by the Science & Technology Plan Project of Xi'an City (No. 2017081CG/RC044 (XASY006)), the Special Scientific Research of Shaanxi Educational Committee (No. 17JK0608), the College Students' Innovative Entrepreneurial Training Program of Xi'an Shiyou University (No. 201610705027 and YCS17221013) and Shaanxi Province (No. 2017107051486), and the National Nature Science Foundation of China (No. 21606177).References
- B. Y. Bai, Q. Qiao, J. H. Li and J. M. Hao, Chin. J. Catal., 2016, 37, 102–122 CrossRef.
- L. H. Nie, J. G. Yu, M. Jaroniec and F. F. Tao, Catal. Sci. Technol., 2016, 6, 3649–3669 Search PubMed.
- J. Q. Torres, S. Royer, J. P. Bellat, J. M. Giraudon and J. F. Lamonier, ChemSusChem, 2013, 6, 578–592 CrossRef PubMed.
- H. M. Guo, M. Li, X. Liu, C. G. Meng, R. Linguerri, Y. Han and G. Chambaud, Catal. Sci. Technol., 2017, 7, 2012–2021 Search PubMed.
- L. R. Wu, Z. Y. Qin, L. X. Zhang, T. Meng, F. Yu and J. Ma, New J. Chem., 2017, 41, 2527–2533 RSC.
- L. K. Gao, W. T. Gan, S. L. Xiao, X. X. Zhan and J. Li, RSC Adv., 2015, 5, 52985–52992 RSC.
- X. X. Feng, H. X. Liu, C. He, Z. X. Shen and T. B. Wang, Catal. Sci. Technol., 2018, 8, 936–954 Search PubMed.
- H. B. Huang, Y. Xu, Q. Y. Feng and D. Y. C. Leung, Catal. Sci. Technol., 2015, 5, 2649–2669 Search PubMed.
- Z. Wang, W. Z. Wang, L. Zhang and D. Jiang, Catal. Sci. Technol., 2016, 6, 3845–3853 Search PubMed.
- Z. J. Dai, X. W. Yu, C. Huang, M. Li, J. F. Su, Y. P. Guo, H. Xu and Q. F. Ke, RSC Adv., 2016, 6, 97022–97029 RSC.
- Y. X. Wu, M. Ma, B. Zhang, Y. H. Gao, W. P. Lu and Y. C. Guo, RSC Adv., 2016, 6, 102127–102133 RSC.
- W. Y. Cui, X. L. Yuan, P. Wu, B. Zheng, W. X. Zhang and M. J. Jia, RSC Adv., 2015, 5, 104330–104336 RSC.
- J. W. Ye, B. Cheng, S. Wageh, A. A. Al-Ghamdi and J. G. Yu, RSC Adv., 2016, 6, 34280–34287 RSC.
- L. H. Nie, J. Wang and J. G. Yu, RSC Adv., 2017, 7, 21389–21397 RSC.
- G. J. Kim, S. M. Lee, S. C. Hong and S. S. Kim, RSC Adv., 2018, 8, 3626–3636 RSC.
- G. N. Li and L. Li, RSC Adv., 2015, 5, 36428–36433 RSC.
- Y. B. Li, C. B. Zhang, H. He, J. H. Zhang and M. Chen, Catal. Sci. Technol., 2016, 6, 2289–2295 Search PubMed.
- C. B. Zhang, H. He and K. Tanaka, Appl. Catal., B, 2006, 65, 37–43 CrossRef.
- C. B. Zhang, Y. B. Li, Y. F. Wang and H. He, Environ. Sci. Technol., 2014, 48, 5816–5822 CrossRef PubMed.
- C. Y. Ma, D. H. Wang, W. J. Xue, B. J. Dou, H. L. Wang and Z. P. Hao, Environ. Sci. Technol., 2011, 45, 3628–3634 CrossRef PubMed.
- X. F. Tang, J. L. Chen, Y. G. Li, Y. Li, Y. D. Xu and W. J. Shen, Chem. Eng. J., 2006, 118, 119–125 CrossRef.
- L. Ma, D. S. Wang, J. H. Li, B. Y. Bai, L. X. Fu and Y. D. Li, Appl. Catal., B, 2014, 148–149, 36–43 Search PubMed.
- Z. W. Huang, X. Gu, Q. Q. Cao, P. P. Hu, J. M. Hao, J. H. Li and X. F. Tang, Angew. Chem., 2012, 124, 4274–4279 CrossRef.
- D. D. Li, G. L. Yang, P. L. Li, J. L. Wang and P. Y. Zhang, Catal. Today, 2016, 277, 257–265 CrossRef.
- Z. P. Qu, D. Chen, Y. H. Sun and Y. Wang, Appl. Catal., A, 2014, 487, 100–109 CrossRef.
- B. Y. Bai and J. H. Li, ACS Catal., 2014, 4, 2753–2762 CrossRef.
- J. Q. Torres, J. M. Giraudon and J. F. Lamonier, Catal. Today, 2011, 176, 277–280 CrossRef.
- L. Zhou, J. H. He, J. Zhang, Z. C. He, Y. C. Hu, C. B. Zhang and H. He, J. Phys. Chem. C, 2011, 115, 16873–16878 Search PubMed.
- S. C. Kim and W. G. Shim, Appl. Catal., B, 2010, 98, 180–185 CrossRef.
- L. Zhou, J. Zhang, J. H. He, Y. C. Hu and H. Tian, Mater. Res. Bull., 2011, 46, 1714–1722 CrossRef.
- F. J. Shi, F. Wang, H. X. Dai, J. X. Dai, J. G. Deng, Y. X. Liu, G. M. Bai, K. M. Ji and C. T. Au, Appl. Catal., A, 2012, 433–434, 206–213 CrossRef.
- X. L. Zhu, M. Shen, L. L. Lobban and R. G. Mallinson, J. Catal., 2011, 278, 123–132 CrossRef.
- C. B. Zhang, F. D. Liu, Y. P. Zhai, H. Ariga, N. Yi, Y. C. Liu, K. Asakura, M. Flytzani-Stephanopoulos and H. He, Angew. Chem., Int. Ed., 2012, 51, 9628–9632 CrossRef PubMed.
- J. Zhou, L. F. Qin, W. Xiao, C. Zeng, N. Li, T. Lv and H. Zhu, Appl. Catal., B, 2017, 207, 233–243 CrossRef.
- J. M. Li, Z. P. Qu, Y. Qin and H. Wang, Appl. Surf. Sci., 2016, 385, 234–240 CrossRef.
- Y. M. Pan, Z. S. Mei, Z. H. Yang, W. X. Zhang, B. Pei and H. X. Yao, Chem. Eng. J., 2014, 242, 397–403 CrossRef.
- X. F. Tang, Y. G. Li, X. M. Huang, Y. D. Xu, H. Q. Zhu, J. G. Wang and W. J. Shen, Appl. Catal., B, 2006, 62, 265–273 CrossRef.
- Q. Y Liu, Y. W. Bie, S. B. Qiu, Q. Zhang, J. Sainio, T. J. Wang, L. L. Ma and J. Lehtonen, Appl. Catal., B, 2014, 147, 236–245 CrossRef.
- B. C. Liu, Y. Liu, C. Y. Li, W. T. Hu, P. Jing, Q. Wang and J. Zhang, Appl. Catal., B, 2012, 127, 47–58 CrossRef.
- H. Huang, Q. G. Dai and X. Y. Wang, Appl. Catal., B, 2014, 158–159, 96–105 CrossRef.
- T. Cai, H. Huang, W. Deng, Q. G. Dai, W. Liu and X. Y. Wang, Appl. Catal., B, 2015, 166–167, 393–405 CrossRef.
- L. Xue, C. B. Zhang, H. He and Y. Teraoka, Appl. Catal., B, 2007, 75, 167–174 CrossRef.
- C. Y. Ma, Z. Mu, J. J. Li, Y. G. Jin, J. Cheng, G. Q. Lu, Z. P. Hao and S. Z. Qiao, J. Am. Chem. Soc., 2010, 132, 2608–2613 CrossRef PubMed.
- Z. P. Hao, D. Y. Cheng, Y. Guo and Y. H. Liang, Appl. Catal., B, 2011, 33, 217–222 CrossRef.
- S. S. Sun, D. S. Mao and J. Yu, J. Rare Earths, 2015, 33, 1268–1274 CrossRef.
- P. P. Hu, Z. Amghouz, Z. W. Huang, F. Xu, Y. X. Chen and X. F. Tang, Environ. Sci. Technol., 2015, 49, 2384–2390 CrossRef PubMed.
- L. H. Nie, J. G. Yu, X. Y. Li, B. Cheng, G. Liu and M. Jaroniec, Environ. Sci. Technol., 2013, 47, 2777–2783 CrossRef PubMed.
- T. F. Yang, Y. Huo, Y. Liu, Z. B. Rui and H. B. Ji, Appl. Catal., B, 2017, 200, 543–551 CrossRef.
- Y. S. Xia, H. X. Dai, H. Y. Jiang, L. Zhang, J. G. Deng and Y. X. Liu, J. Hazard. Mater., 2011, 186, 84–91 CrossRef PubMed.
- L. Qi, W. Ho, J. Wang, P. Zhang and J. Yu, Catal. Sci. Technol., 2015, 5, 2366–2377 Search PubMed.
- F. Yu, Z. Qu, X. Zhang, Q. Fu and Y. Wang, J. Energy Chem., 2013, 22, 845–852 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |