DOI:
10.1039/C8RA01506F
(Paper)
RSC Adv., 2018,
8, 12791-12798
Ascorbic acid/Fe0 composites as an effective persulfate activator for improving the degradation of rhodamine B
Received
19th February 2018
, Accepted 26th March 2018
First published on 3rd April 2018
Abstract
An ascorbic acid/Fe0 composite (H2A/Fe0)-activated persulfate (PS) process was provided for rhodamine B (RhB) removal. Experimental results demonstrated that the H2A/Fe0–PS system exhibits a rapid and continuous oxidation of organic contaminants, and shows great advantages over the conventional Fe0–PS system by significantly improving removal efficiency. This H2A induced dramatic enhancement for RhB degradation could be attributed to both the reduction and chelating ability of H2A. The H2A/Fe0 composites were characterized using TEM, FE-SEM, FTIR and XPS, indicating that the obtained H2A/Fe0 composites were prepared successfully. Key factors affecting the treatment were determined for the H2A/Fe0–PS system, including H2A/Fe0 dosage, PS dosage, initial solution pH and temperature. Moreover, radical quenching tests revealed that sulfate radicals (SO4˙−), hydroxyl radicals (˙OH) and superoxide radicals (O2˙−) were generated in the H2A/Fe0–PS system, and SO4˙− was the main radical species responsible for RhB degradation. Finally, possible degradation pathways are proposed. These findings prove that the H2A/Fe0–PS system may provide a simple and effective technology for improving the degradation of refractory organic pollutants.
1. Introduction
Nowadays, advanced oxidation processes (AOPs) are regarded as good alternatives for the in situ oxidation of various organic pollutants.1 In particular, sulfate radical (SO4˙−, E0 = 2.5–3.1 V) based AOPs have become hot topics of research due to their high oxidizing ability, wide pH range and long lifetime.2 The highly reactive SO4˙− can be generated by activation of persulfate (PS) with heat,3 UV,4 metal oxides (Fe3O4, CuFe2O4),5,6 or transition metals (e.g., Fe2+, Co2+, Cu2+).7,8 Among these methods, Fe2+ has been commonly selected as the activator for PS to generate SO4˙− via eqn (1) due to its non-toxicity, low cost and abundance.9 However, in the Fe2+/PS system, Fe2+ is hard to regenerate after conversion to Fe3+. In addition, excess Fe2+ will scavenge the generated SO4˙− via eqn (2), resulting in reduced degradation efficiency for target contaminants.| | |
S2O82− + Fe2+ → SO4˙− + Fe3+ + SO42−
| (1) |
| | |
SO4˙− + Fe2+ → Fe3+ + SO42−
| (2) |
Zero-valent iron (Fe0) can be an alternative source of Fe2+ by gradually releasing Fe2+ into an aqueous solution so that Fe2+ induced scavenging is minimal.10 Moreover, in the Fe0/PS system, Fe0 can not only lessen the generation of ferric hydroxides by recycling Fe3+ at the surface of ZVI via eqn (3), but also directly transfer electrons to PS, resulting in the generation of SO4˙− via eqn (4). Peluffo et al. reported that the Fe0/PS process yielded a higher removal rate of PAHs than the Fe2+–PS process under optimal conditions.11
| | |
S2O82− + Fe0 → 2SO4˙− + Fe2+ + 2SO42−
| (4) |
Recently, an Fe0–PS system has been employed to remove various organic pollutants, such as trichloroethylene (TCE),12 orange G (OG),13 and bentazon (BTZ).10 However, the biggest drawback of this technology is that it requires high concentrations of PS and Fe0 dosages to achieve favorable activation performance, resulting in increased treatment cost and extra disposal,14 which may be due to the slow conversion from Fe3+ to Fe2+.15 Therefore, accelerating the redox cycle of Fe3+/Fe2+is crucial to improving the oxygen activation performance of the Fe0–PS system for practical application.
To solve the above-mentioned problems, we conducted a new attempt to provide a novel ascorbic acid/Fe0–PS system to degrade organic pollutants in an aqueous system. In this research work, Fe3+/Fe2+ cycles were successfully promoted by employing a reducing agent, ascorbic acid (C6H8O6, H2A), in the Fe0–PS system. Ascorbic acid (H2A), which is also known as vitamin C, is an eco-friendly reducing agent and a natural antioxidant. H2A has shown great promise in water and wastewater treatment.16 Many high oxidation states like H2O2, Fe3+, and Cr6+ could be reduced using H2A as an electron donor.17,18 Fukuchi et al. reported that the dosages of H2O2 and Fe2+ could be largely reduced with the addition of H2A into traditional Fenton-like processes, leading to a relatively steady Fe2+ concentration and higher production of free radicals.19 In addition, H2A is a bidentate ligand with a bifunctional enediol group that has the ability to solubilize metal ions by chelate formation.20 Therefore, it might be reasonable to introduce H2A into the Fe0–PS system to improve the degradation efficiency for pollutants. Although it has also been reported that Fe0 nanoparticles can be stabilized with ascorbic acid to increase the durability of synthesized Fe0.21 To the best of our knowledge, the use of H2A coated Fe0 nanoparticles as an activator to enhance the activation performance of PS has not been reported so far.
To investigate the degradation efficiency of the H2A/Fe0–PS system, rhodamine B (RhB) was selected as a model pollutant because it is an extremely stable and resistant aromatic compound, and hard to degrade using conventional wastewater treatment methods. The additional purposes of this study were to (1) investigate the degradation performance of RhB in the H2A/Fe0–PS system; (2) explore the mechanism of enhanced RhB degradation in the H2A/Fe0–PS system; and (3) reveal the important parameters (H2A/Fe0 dosage, PS dosage, pH and temperature) affecting the H2A/Fe0–PS system.
2. Materials and methods
2.1. Chemicals and materials
Potassium borohydride (KBH4, 99%), ferrous sulfate heptahydrate (FeSO4·7H2O), sodium persulfate (Na2S2O8), sodium hydroxide (NaOH, 99%), rhodamine B (RhB), sulfuric acid (H2SO4, 95% to 98%), sodium acetate anhydrous (CH3COONa), ethanol, 1,10-phenanthroline (C12H8N2), tert-butyl alcohol (TBA), methanol (MeOH), 1,4-benzoquinone (BQ), acetone and ascorbic acid (H2A) were of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd. All the solutions were prepared with deionized water before each run.
2.2. Synthesis of H2A/Fe0
The H2A/Fe0 was synthesized in an anaerobic glove box under the protection of purified N2. First, a 0.5 M solution of Fe2+ was prepared by dissolving FeSO4·7H2O in deionized water (100 mL). Then, a certain volume stabilizer (H2A, 10 g L−1) solution was added into the FeSO4 solution with electromagnetic stirring for 15 min. After intensive mixing, 100 mL of 1.5 M of KBH4 solution was added dropwise into the mixture solution. After delivering all of the KBH4 solution, the solution was stirred continuously for 30 min. Subsequently, the solid precipitation was filtered with a 0.45 μm membrane filter under vacuum, followed by rinsing with deionized water, absolute ethanol, and acetone alternately 3 times.
2.3. Experimental procedures
Degradation experiments were carried out in 250 mL serum bottles, which were filled with 50 mL of RhB solution. A predetermined amount of PS was quickly added into the pH-adjusted RhB solution. Subsequently, a certain amount of H2A/Fe0 was quickly added into the above solution to trigger the reaction. The initial pH value of the solution was adjusted with NaOH (0.1 M) or H2SO4 (0.1 M). The bottles were then placed into a temperature-controlled orbital shaker with a constant shaking speed (170 rpm) to ensure a complete mixing state. At predetermined time intervals, solution samples were taken using disposable syringes, and then filtered with a 0.45 μm filter membrane. Subsequently, the filtered sample was mixed with excess methanol (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to quench the reaction before analysis.
:1) to quench the reaction before analysis.
The concentration of RhB was determined with a UV-2450 visible spectrophotometer (Shimadzu, Japan), and its maximum characteristic wavelength was adopted at 554 nm. The concentrations of total iron and ferrous ions were determined according to the 1,10-phenanthroline colorimetric method.22 Radical scavenging tests were conducted and three radical scavengers, MeOH, TBA and BQ, were used to determine the type of the predominant reactive oxygen species for RhB degradation.
2.4. Characterizations
The morphology and particle structure of H2A/Fe0 and Fe0 were studied with a high-resolution field emission scanning electron microscope (FE-SEM, JEOL Ltd., Japan) at an accelerating voltage of 5.0 kV with an X-ray energy-dispersive spectroscopy (EDS) detector (IE300X, Oxford, United Kingdom) and transmission electron microscopy (TEM, JEOL Ltd., Japan). The surface chemical structure and composition of the H2A/Fe0 and Fe0 particles were studied by Fourier transform infrared spectroscopy (FTIR, Perkin-Elmer Instrument Co. Ltd., USA). The element valence and chemical groups of H2A/Fe0 and Fe0 were determined by X-ray photoelectron spectroscopy using monochromatic Mg Kα radiation (XPS, Model PHI 5700, Physical Electronics, USA).
3. Results and discussion
3.1. Comparison of RhB degradation in different systems
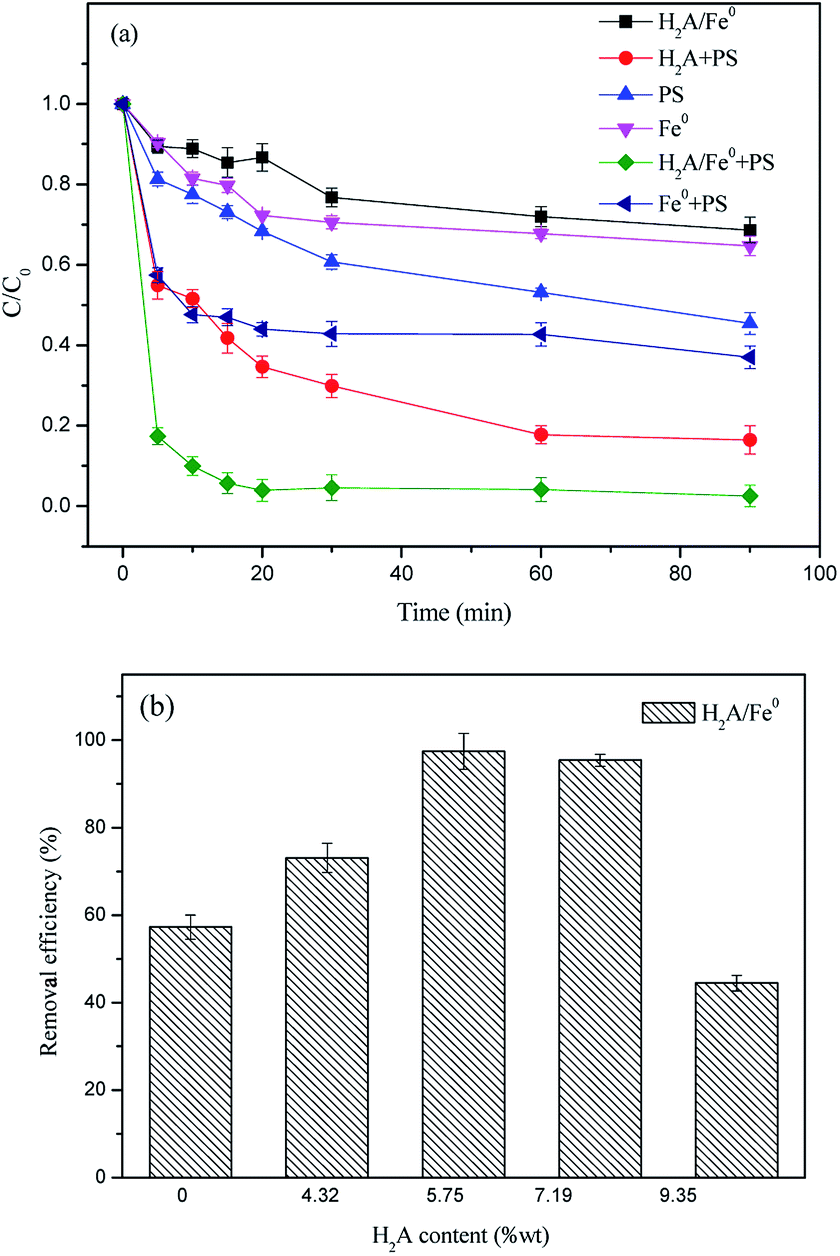
Comparative experiments were carried out in different systems to determine the effects of the synthesized H2A/Fe0 particles on PS activation for RhB degradation under the same operating conditions. As shown in Fig. 1a, about 35.28% RhB was removed by Fe0 alone, and only 31.33% RhB was removed by H2A/Fe0 within 90 min, indicating that some of the reactive sites might be blocked by the excessive dispersant coated on the Fe0 surfaces. The removal efficiency for RhB was apparently improved when the reaction time was prolonged, mainly because of desorption of H2A into the aqueous solution. In contrast, PS alone could degrade 54.59% of RhB within 90 min. The carboxyl and phenyl groups of RhB might be capable of PS activation.23 In addition, activation of PS by Fe0 was also investigated, and the results showed that approximately 18% of the RhB was removed over 60 min in the Fe0/PS system. A slightly improved removal efficiency of RhB was achieved in the H2A/PS system because H2A can react with PS to generate SO4˙−, according to eqn (5) and (6).24 However, more than 90% of the RhB was removed with H2A/Fe0 activated PS. Therefore, the high activity of the H2A/Fe0 composites for PS activation was assumed to be the result of the existence of a synergistic effect in this system.| | |
H2A + S2O82− → 2H+ + SO42− + SO4˙− + A˙−
| (5) |
| | |
HA− + S2O82− → H+ + SO42− + SO4˙− + A˙−
| (6) |
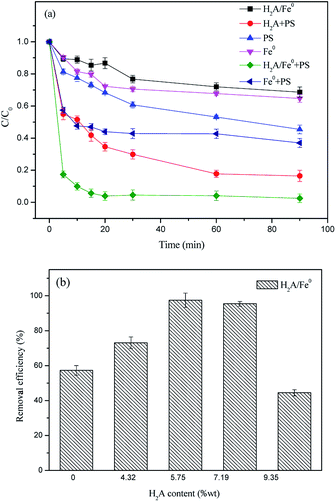 |
| | Fig. 1 (a) Comparison of removal efficiency of RhB in different systems (C0 = 50 mg L−1, PS dosage = 1.4 g L−1, Fe0 dosage = 1 g L−1, H2A/Fe0 dosage = 1 g L−1, H2A dosage = 1.6 g L−1 and T = 298 K); (b) effect of H2A concentration on removal efficiency of RhB in the H2A/Fe0–PS system (C0 = 50 mg L−1, Fe0 dosage = 0.8 g L−1, T = 298 K and the solution volume is 50 mL). | |
The reactivity of H2A/Fe0 particles with different H2A content was further investigated. As shown in Fig. 1b, the highest removal efficiency was achieved with an H2A solution addition amount of 3.60‰ wt (to Fe2+) during the synthesis process of Fe0. For an H2A addition content higher than 3.60‰, the excess H2A might occupy the available reactive sites of Fe0 particles and then block the target pollutants diffusing onto the surface sites. In contrast, insufficient loading of the coating onto the surface of Fe0 might result in ineffective dispersion of the Fe0, indicating that the optimal H2A concentration would be 3.60‰. Therefore, 3.60‰ was selected in the following characterization and degradation experiments.
3.2. Characterization of H2A/Fe0
The morphology of the Fe0 and H2A/Fe0 was observed by TEM and FE-SEM. The TEM observation (Fig. 2a) on the Fe0 shows that bare Fe0 particles were aggregated and featured as a necklace-like chain of length 80–100 nm. When H2A was added, the dispersion of Fe0 was significantly increased (Fig. 2b), indicating that H2A could act as a dispersant for Fe0 nanoparticles. The particle size of H2A/Fe0 ranges from 20 to 80 nm. The reason might be that the added H2A could coordinate with ferrous ions to indirectly control the growth of iron particles, resulting in the smaller size of the H2A/Fe composite.
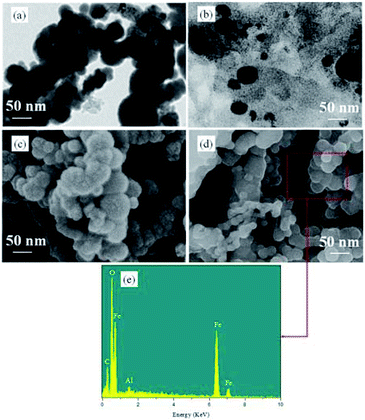 |
| | Fig. 2 TEM images of (a) Fe0 and (b) H2A/Fe0, FE-SEM images of (c) Fe0 and (d) H2A/Fe0, and (e) electron diffusion spectroscopy elemental analysis. | |
Fig. 2c shows the representative FE-SEM image of the as-synthesized bare Fe0, which exhibits a spherical morphology with a size in the range 80–100 nm, and which tends to aggregate together due to the magnetic interactions between iron particles.21 Comparatively, with the coating of H2A, it can be observed that the H2A/Fe0 had a smaller size than bare Fe0 (Fig. 2d). Simultaneously, the EDS spectrum further revealed the successful incorporation of H2A and iron nanoparticles (Fig. 3e).
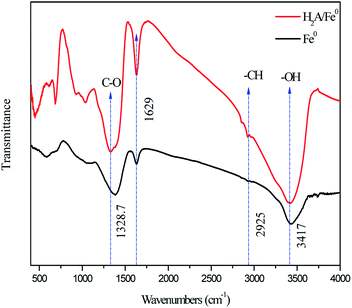 |
| | Fig. 3 FTIR spectra of Fe0 and H2A/Fe0. | |
In order to prove the existence of functional groups of H2A on the surface of the Fe nanoparticles, Fe0 and H2A/Fe0 were analyzed by FTIR spectroscopy. As shown in Fig. 3, the absorption peak on H2A/Fe0 at about 3417 cm−1 was significantly larger than that on Fe0, which was attributed mainly to stretching vibrations of –OH.25 In addition, compared with the spectrum of Fe0, there are several inconspicuous infrared characteristic peaks for H2A/Fe0, among which the absorption peak appearing at approximately 2925 cm−1 was assigned to –CH. The bands at 1328.7 cm−1 are attributed to C–O vibrations. Meanwhile, an additional adsorption peak at 1629 cm−1 corresponded to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups, indicating the presence of the H2A constituent.15
C groups, indicating the presence of the H2A constituent.15
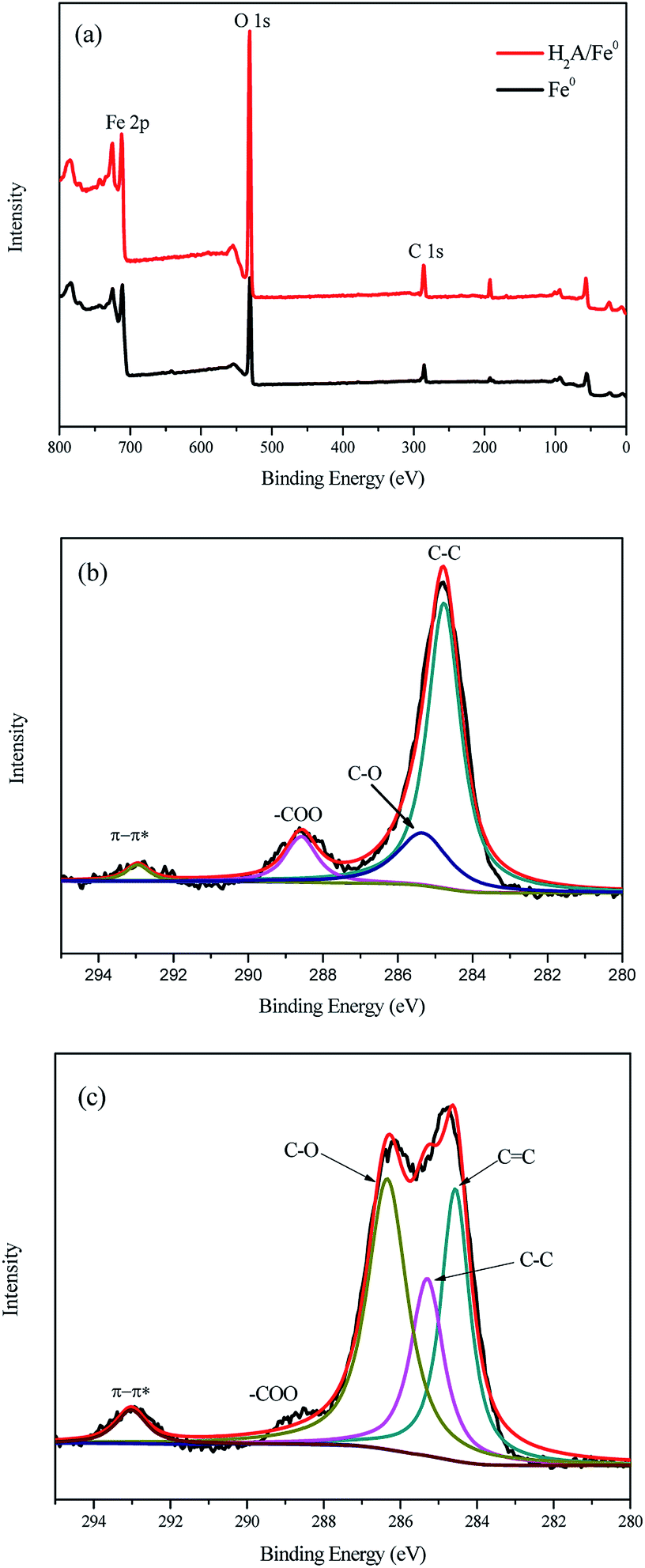
XPS was used to investigate the composition and chemical state of Fe0 before and after coating by H2A. The full-range survey spectra of Fe0 and H2A/Fe0 particles are illustrated in Fig. 4a, showing that the nanoparticle surface consists mainly of iron (Fe), oxygen (O), and carbon (C). The small amount of carbon appearing in the Fe0 spectrum is likely to be attributed to carbon dioxide, water, and/or organic compounds (such as ethanol and acetone) during the fabrication, drying and preserving processes.26 Detailed XPS patterns for the C 1s regions of the Fe0 and H2A/Fe0 particles are presented in Fig. 4b and c. Compared with Fe0, a conspicuous peak at 284.7 eV appeared in the spectrum of H2A/Fe0, corresponding to CC species, verifying the presence of H2A. All the above characterization results of FTIR and XPS confirmed that H2A had been successfully coated onto Fe0.
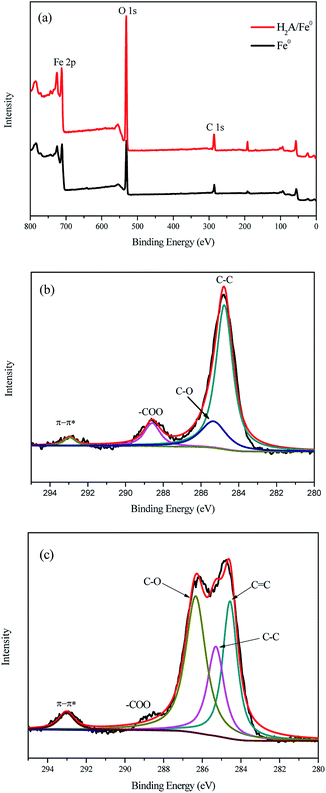 |
| | Fig. 4 XPS spectra of Fe0 and H2A/Fe0: (a) full-range XPS spectra, (b) C 1s regions of Fe0, and (c) C 1s regions of H2A/Fe0. | |
3.3. Factors affecting RhB degradation in the H2A/Fe0–PS system
The effects of PS dosage, H2A/Fe0 dosage, pH value, and temperature on RhB degradation were investigated and the results are shown in Fig. 5. Fig. 5a shows the effect of PS dosage on RhB degradation. The results clearly show that the degradation of RhB accelerated as the PS dosage increased from 0.1 to 0.3 g L−1. Since PS was the SO4˙− donor, a higher PS dosage would improve the formation of SO4˙−. However, when the PS dosage was further increased to 0.5 g L−1, it led to a decline in RhB degradation efficiency. Similar results were also reported in other studies.27 A reasonable explanation for this phenomenon is that the excess S2O82− may lead to SO4˙− scavenging (eqn (7)) and the reaction between two SO4˙− (eqn (8)).28 Hence, 0.3 g L−1 PS was selected as the optimal dosage in the following experiments.| | |
SO4˙− + S2O82− → SO42− + S2O8˙−
| (7) |
| | |
SO4˙− + SO4˙− → S2O8˙−
| (8) |
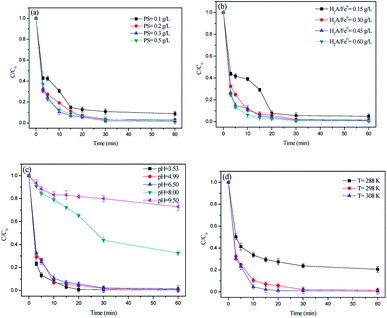 |
| | Fig. 5 Parameters affecting the degradation of RhB: (a) PS dosage, (b) H2A/Fe0 dosage, (c) pH value, and (d) temperature. Except for the investigated parameter, other parameters were fixed at C0 = 50 mg L−1, PS = 0.3 g L−1, H2A/Fe0 = 0.3 g L−1, pH = 6.50 and T = 298 K. | |
The effect of H2A/Fe0 dosage on RhB degradation was evaluated with the H2A/Fe0 dosage ranging from 0.15 to 0.60 g L−1. As shown in Fig. 5b, the increase in the activator dosage significantly promoted the degradation of RhB, which may be because the increase in H2A/Fe0 dosage could provide more active sites for the activation of PS, thus producing more reactive free radicals for more efficient RhB degradation. At a lower dosage of H2A/Fe0 (0.15 g L−1), 94.44% removal could be obtained within 30 min, indicating the excellent activity of H2A/Fe0 for PS activation with 50 mg L−1 RhB.
Fig. 5c shows the RhB degradation in the H2A/Fe0–PS system with different initial pH values ranging from 3.53 to 9.50. The pH value played an important parameter in the degradation of contaminants.29 It could be observed that the highest RhB removal was achieved at an initial pH value of 3.53, and the removal efficiency obviously increased as the pH value decreased. This result is in accordance with Leng et al.’s report, which suggested that the degradation rate was lowered under neutral and alkaline conditions.30 The better performance under acidic conditions in the present study may be because: (1) At lower pH values, the Fe0 corrosion rate could be accelerated, leading to much faster release of Fe2+, which favors the induction of eqn (1) to produce more SO4˙−. (2) At a pH higher than 7, the formation of Fe2+ complexes could inhibit PS activation by eqn (9). Also, the reactions between SO4˙− and H2O/OH− could result in the rapid decay of SO4˙− (eqn (10) and (11)).31 Overall, acidic and weakly alkaline conditions were beneficial for RhB destruction. Compared with the Fenton process that must be carried out over a narrow pH range, the results suggested that the novel method we provide in this research possesses tremendous advantages for applications in practice.
| | |
Fe2+ + H2O → FeOH+ + H+
| (9) |
| | |
All pH: SO4˙− + H2O → ˙OH + SO42− + H+
| (10) |
| | |
Alkaline pH: SO4˙− + OH− → ˙OH + SO42−
| (11) |
The degradation reaction of RhB was also found to be influenced by solution temperature. As shown in Fig. 5d, higher temperatures favored the degradation of RhB by PS activated with H2A/Fe0, which could be associated with the thermal activation of PS to generate free radicals (eqn (12)),32 as well as the acceleration of the reaction between H2A/Fe0 and PS, probably because the diffusion and mobility of RhB molecules increased.
| | |
S2O82− + heat → SO4˙−
| (12) |
3.4. Degradation mechanism
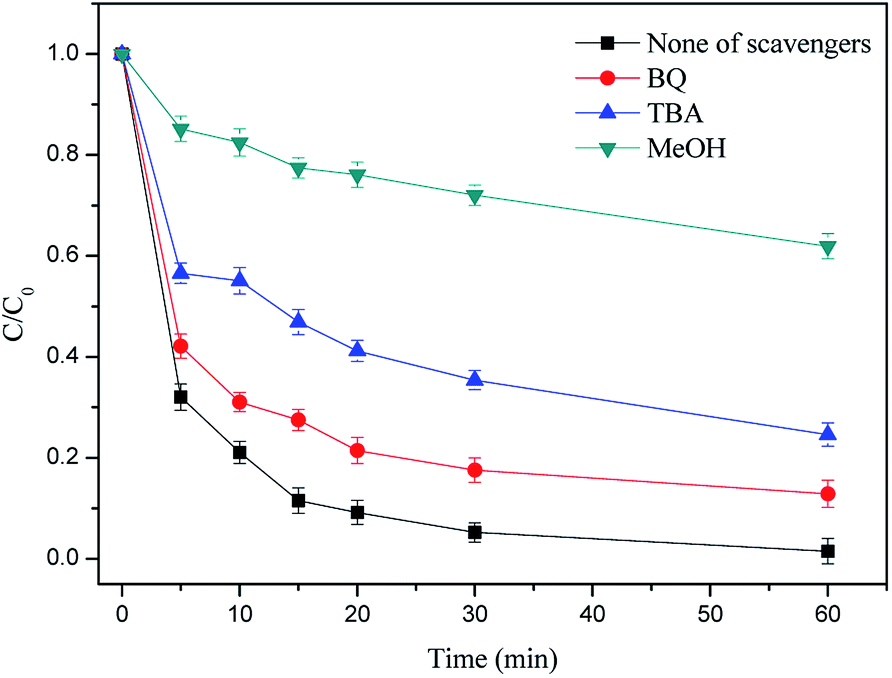
3.4.1 Identification of free radicals in the H2A/Fe0–PS system. It has been reported that a series of reactive oxygen species, e.g., SO4˙−, ˙OH and superoxide anion radicals (O2˙−), were generated in a PS/Fe(II)/hydroxylamine (HA) system and may also be generated in the H2A/Fe0–PS system.33 In order to evaluate the major radical species for the RhB oxidation reaction, scavenging experiments were performed in the H2A/Fe0–PS system by adding MeOH, TBA and BQ into the reaction solution. MeOH (with α-hydrogen) is an effective quencher for both ˙OH (1.2 × 109 to 2.8 × 109 M−1 s−1) and SO4˙− (1.6 × 107 to 7.7 × 107 M−1 s−1). In contrast, TBA (without α-hydrogen) has an approximately 1000 times greater rate constant with ˙OH (3.8 × 108 to 7.6 × 108 M−1 s−1) than with SO4˙− (4.0 × 105 to 9.1 × 105 M−1 s−1).6,34 Therefore, MeOH was used to scavenge both radicals, and TBA was used to selectively scavenge ˙OH. Moreover, we used BQ as an effective scavenger of O2˙− by electron transfer to generate benzoquinone radicals.35As shown in Fig. 6, 94.78% of RhB was degraded in 30 min when no quenching agent was added. However, with the addition of BQ and TBA, the removal efficiency of RhB decreased to 82.44% and 64.63% in 30 min, respectively. Meanwhile, the degradation of RhB was almost completely inhibited (27.98%) by the addition of MeOH. These data indicate that O2˙−, SO4˙− and ˙OH are generated in the H2A/Fe0–PS system. Similar findings were reported by a previous study.36 It can be preliminarily inferred that O2˙− may be derived from catalytic hydrolysis of PS in accordance with eqn (13) and (14). Hence, the scavenging studies clearly prove the simultaneous participation of O2˙−, SO4˙− and ˙OH in the H2A/Fe0–PS oxidation process, and SO4˙− played a major role in achieving a good oxidation reaction.
| | |
S2O82− + 2H2O → 2SO42− + HO2− + 3H+
| (13) |
| | |
S2O82− + HO2− → SO42− + SO4˙− + O2˙− + H+
| (14) |
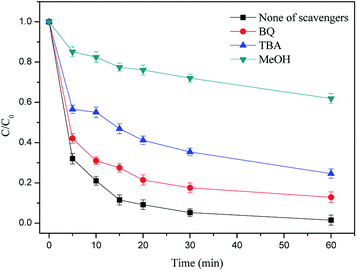 |
| | Fig. 6 Effect of different radical quenching agents on RhB removal. Reaction conditions: C0 = 50 mg L−1, PS = 0.3 g L−1, H2A/Fe0 = 0.3 g L−1, pH = 6.50 and T = 298 K. | |
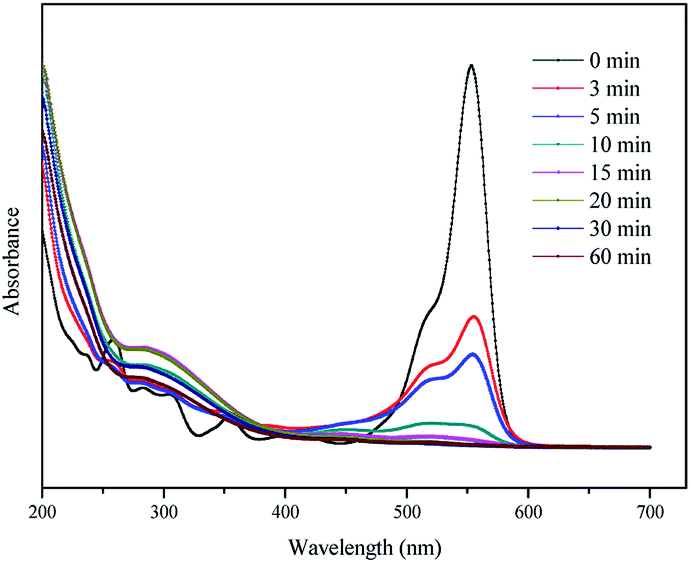
3.4.2 UV-vis spectral changes. The UV-vis spectra were used to observe changes during the degradation of RhB by the H2A/Fe0–PS system at different reaction times. As shown in Fig. 7, there were two main characteristic absorption bands of RhB in the UV region (259 nm) and visible light region (554 nm), respectively. The UV absorbance at 259 nm corresponds to the existence of aromatic rings in RhB. The visible absorbance at 554 nm is responsible for the RhB coloration due to the conjugated structure of the CN and CO groups. COD removal of the RhB aqueous solution was measured to evaluate the mineralization of RhB. The COD removal efficiency of RhB reached 87.6% in 60 min, indicating that the majority of RhB was mineralized and the H2A/Fe0–PS system has a high capacity for degradation of RhB. This result is further confirmed by the UV-vis adsorption spectra of RhB, as shown in Fig. 7. Usually for RhB degradation, two possible competitive mechanisms occur, i.e. N-demethylation and cleavage of the chromophore structure.37 First, the decrease in the main chromophore band of RhB at 554 nm with an increase in the reaction time indicates the destruction of the xanthene structure of the chromophore. Second, the decrease in absorbance at 259 nm indicates the destruction of the aromatic part. Third, some new peaks appeared at around 210 nm and 328 nm, possibly demonstrating the yield of intermediate or final products.38,39
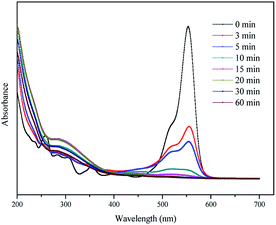 |
| | Fig. 7 UV-vis adsorption spectra of RhB at various times by the H2A/Fe0–PS system. | |
3.4.3 The role of H2A in enhancing RhB degradation. The role of H2A on enhancing RhB degradation in the H2A/Fe0–PS system was investigated. First of all, it is generally recognized that H2A is a potent antioxidant agent with the ability to prevent the aggregation of Fe0 by the effect of coating (Fig. 2b and d).40 In addition, H2A is a weak acid with the ability to solubilize metal ions by Fe2+–H2A (Fe2+/H2Ax) complex (eqn (15)) formation either in solution or on the surface of iron based materials, which can not only further activate PS (eqn (16)) and account for the generation of SO4˙−, but also react with O2 to produce O2˙− in accordance with eqn (17).40 Besides its acidity, H2A can also serve as a reducing agent, which is capable of effectively reducing Fe3+ and realizing Fe3+/Fe2+ redox cycles in the system (eqn (18)).| | |
Fe2+ + H2A → Fe2+H2Ax
| (15) |
| | |
Fe2+H2Ax + S2O82− → Fe3+H2Ay + SO42− + SO4˙−
| (16) |
| | |
Fe2+H2Ax + O2 → Fe3+H2Ay + O2˙−
| (17) |
| | |
H2A + 2Fe3+ → Fe2+ + A + 2H+
| (18) |
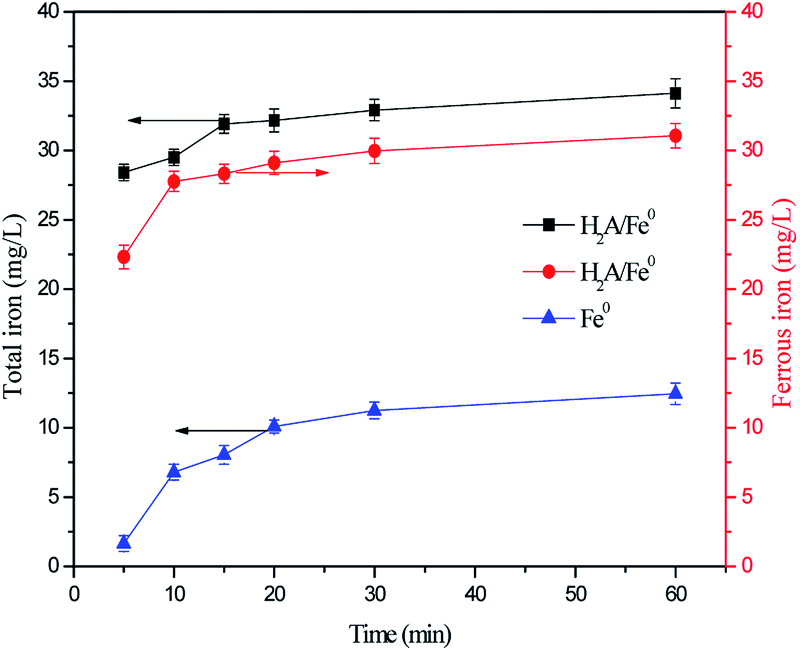
To confirm this opinion, the changes in concentration of total iron and ferrous iron in the solution were monitored during the oxidation of RhB. Fig. 8 shows that the total iron concentration increased as the reaction time increased, with no period of decline. This phenomenon could be attributed to the continuous leaching of iron from H2A/Fe0. By contrast, the leaching of iron was faster during the oxidation of RhB by the H2A/Fe0 composite than by pristine Fe0, and this result further confirms the pronounced weak acid effect of H2A with the ability to solubilize metal ions. Moreover, the ferrous iron concentration was found to increase from 22.32 to 29.11 mg L−1 in the first 20 min, and then remained relatively stable over the later reaction time without decreasing. This was due to the electron transfer from H2A to both the surface and dissolved Fe3+ via eqn (18), resulting in an effective Fe3+/Fe2+ cycle. As expected, the reductive effect of H2A was also promoted to form more and more Fe2+ and SO4˙− in the H2A/Fe0–PS system, which results in an acceleration in RhB degradation.
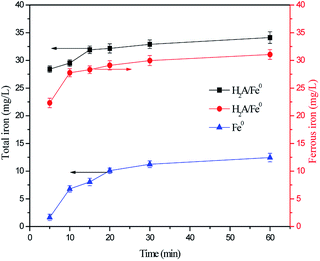 |
| | Fig. 8 Variation in total iron and ferrous iron concentrations in solution. Reaction conditions: C0 = 50 mg L−1, PS = 0.3 g L−1, H2A/Fe0 = 0.3 g L−1, pH = 6.50 and T = 298 K. | |
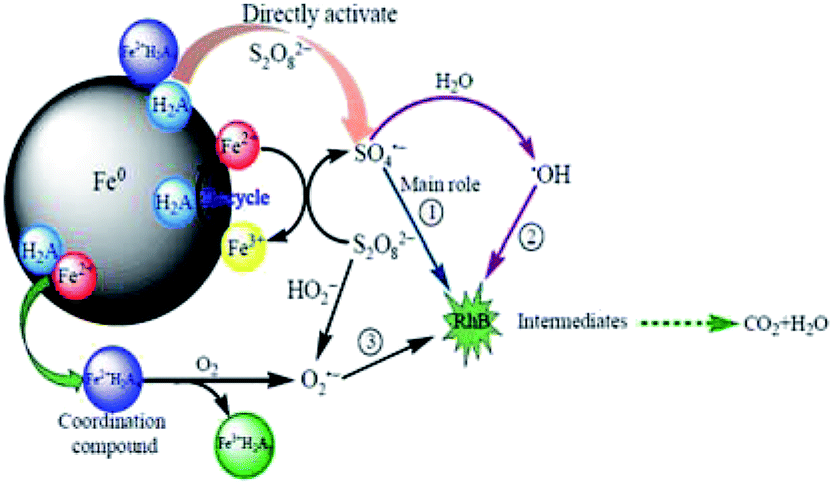
The H2A/Fe0–PS system exhibits significant superiority for the removal of RhB. Fig. 9 illustrates the proposed conceptual model of the RhB removal process by the H2A/Fe0–PS system. The superiority of the H2A/Fe0–PS system is mainly attributed to: (1) H2A can reduce the structure size of Fe0 particles and increase their dispersibility: however, considering that the aggregation of H2A/Fe0 was still remarkable (Fig. 2b and d), we believe that the high activation performance of H2A/Fe0 was not mainly due to the increased dispersibility in solution. (2) H2A is a weak acid that has the ability to solubilize metal ions by Fe2+/H2Ax complex formation, resulting in another pathway for the production of O2˙− by eqn (17), which is helpful for improving the degradation. (3) H2A can also serve as a reducing agent, and the presence of H2A accelerated the redox cycle of Fe3+/Fe2+ and robust Fe2+ regeneration to promote RhB degradation. Therefore, this H2A/Fe0–PS system can produce more SO4˙− and O2˙− than the Fe0–PS system alone. In a word, the H2A/Fe0–PS system involved the generation of ˙OH, SO4˙− and O2˙− species, among which SO4˙− played a key role in the degradation of RhB.
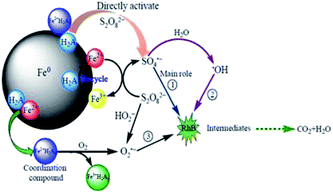 |
| | Fig. 9 Conceptual model of the RhB removal process by the H2A/Fe0–PS system. | |
4. Conclusions
In this study, we present H2A/Fe0 nanoparticles as a heterogeneous PS activator for the effective degradation of RhB. The physical–chemical properties of H2A/Fe0 nanoparticles were characterized by TEM, FE-SEM, FTIR and XPS. Compared with bare Fe0, the H2A/Fe0 composites showed much higher performance for PS activation for RhB degradation. We explored the role of H2A and concluded that both its reduction and chelating ability contribute to the good performance of the H2A/Fe0–PS system. In addition, the radical scavenging tests clearly confirmed the generation of SO4˙−, ˙OH and O2˙− and that the primary reactive oxygen species is SO4˙− in the H2A/Fe0–PS system. Batch experiments indicated that the optimal PS dosage is 0.3 g L−1. RhB removal efficiency increased with increasing H2A/Fe0 dosage and solution temperature, and it increased with a decrease in pH values. The findings in this study strongly suggested that the H2A/Fe0 activated PS process is a highly promising technique for the degradation of dye wastewater and other refractory organic toxic pollutants.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support by National Natural Science Foundation of China (NSFC, No. 51368025 and No. 51068011).
References
- Y. Deng and R. Zhao, Curr. Pollut. Rep., 2015, 1, 167–176 CrossRef CAS.
- J. Yan, M. Lei, L. Zhu, M. N. Anjum, J. Zou and H. Tang, J. Hazard. Mater., 2011, 186, 1398–1404 CrossRef CAS PubMed.
- Y. Fan, Y. Ji, D. Kong, J. Lu and Q. Zhou, J. Hazard. Mater., 2015, 300, 39–47 CrossRef CAS PubMed.
- J. Y. Fang and C. Shang, Environ. Sci. Technol., 2012, 46, 8976–8983 CrossRef CAS PubMed.
- G.-D. Fang, D. D. Dionysiou, S. R. Al-Abed and D.-M. Zhou, Appl. Catal., B, 2013, 129, 325–332 CrossRef CAS.
- X. Zhang, M. Feng, R. Qu, H. Liu, L. Wang and Z. Wang, Chem. Eng. J., 2016, 301, 1–11 CrossRef CAS.
- X. Jiang, Y. Wu, P. Wang, H. Li and W. Dong, Environ. Sci. Pollut. Res. Int., 2013, 20, 4947–4953 CrossRef CAS PubMed.
- C. Wang, J. Wan, Y. Ma and Y. Wang, Res. Chem. Intermed., 2015, 42, 481–497 CrossRef.
- F. Gong, L. Wang, D. Li, F. Zhou, Y. Yao, W. Lu, S. Huang and W. Chen, Chem. Eng. J., 2015, 267, 102–110 CrossRef CAS.
- X. Wei, N. Gao, C. Li, Y. Deng, S. Zhou and L. Li, Chem. Eng. J., 2016, 285, 660–670 CrossRef CAS.
- M. Peluffo, F. Pardo, A. Santos and A. Romero, Sci. Total Environ., 2016, 563–564, 649–656 CrossRef CAS PubMed.
- J. Yan, L. Han, W. Gao, S. Xue and M. Chen, Bioresour. Technol., 2015, 175, 269–274 CrossRef CAS PubMed.
- X. Xiong, B. Sun, J. Zhang, N. Gao, J. Shen, J. Li and X. Guan, Water Res., 2014, 62, 53–62 CrossRef CAS PubMed.
- B. M. Voelker and B. Sulzberger, Environ. Sci. Technol., 1996, 30, 1106–1114 CrossRef CAS.
- C. Sun, R. Zhou, J. E, J. Sun, Y. Su and H. Ren, RSC Adv., 2016, 6, 10633–10640 RSC.
- Y. T. Lin and C. Liang, Environ. Sci. Technol., 2013, 47, 3299–3307 CrossRef CAS PubMed.
- X. Hou, X. Huang, Z. Ai, J. Zhao and L. Zhang, J. Hazard. Mater., 2016, 310, 170–178 CrossRef CAS PubMed.
- X.-R. Xu, H.-B. Li, X.-Y. Li and J.-D. Gu, Chemosphere, 2004, 57, 609–613 CrossRef CAS PubMed.
- S. Fukuchi, R. Nishimoto, M. Fukushima and Q. Zhu, Appl. Catal., B, 2014, 147, 411–419 CrossRef CAS.
- Y.-T. Lin, C. Liang and C.-W. Yu, Ind. Eng. Chem. Res., 2016, 55, 2302–2308 CrossRef CAS.
- M. Savasari, M. Emadi, M. A. Bahmanyar and P. Biparva, J. Ind. Eng. Chem., 2015, 21, 1403–1409 CrossRef CAS.
- Z.-H. Diao, X.-R. Xu, D. Jiang, L.-J. Kong, Y.-X. Sun, Y.-X. Hu, Q.-W. Hao and H. Chen, Chem. Eng. J., 2016, 302, 213–222 CrossRef CAS.
- J. Du, J. Bao, X. Fu, C. Lu and S. H. Kim, Sep. Purif. Technol., 2016, 163, 145–152 CrossRef CAS.
- Y. Lei, H. Zhang, J. Wang and J. Ai, Chem. Eng. J., 2015, 270, 73–79 CrossRef CAS.
- X. Wang, Y. Du and J. Ma, Appl. Surf. Sci., 2016, 390, 50–59 CrossRef CAS.
- X. Wang, P. Wang, J. Ma, H. Liu and P. Ning, Appl. Surf. Sci., 2015, 345, 57–66 CrossRef CAS.
- H. Chen, Z. Zhang, M. Feng, W. Liu, W. Wang, Q. Yang and Y. Hu, Chem. Eng. J., 2017, 313, 498–507 CrossRef CAS.
- L. Hou, H. Zhang and X. Xue, Sep. Purif. Technol., 2012, 84, 147–152 CrossRef CAS.
- C. Tan, N. Gao, W. Chu, C. Li and M. R. Templeton, Sep. Purif. Technol., 2012, 95, 44–48 CrossRef CAS.
- Y. Leng, W. Guo, X. Shi, Y. Li and L. Xing, Ind. Eng. Chem. Res., 2013, 52, 13607–13612 CrossRef CAS.
- J. Li, Q. Liu, Q. Q. Ji and B. Lai, Appl. Catal., B, 2017, 200, 633–646 CrossRef CAS.
- C. Tan, N. Gao, Y. Deng, N. An and J. Deng, Chem. Eng. J., 2012, 203, 294–300 CrossRef CAS.
- X. Wu, X. Gu, S. Lu, Z. Qiu, Q. Sui, X. Zang, Z. Miao and M. Xu, Sep. Purif. Technol., 2015, 147, 186–193 CrossRef CAS.
- A. Farhat, J. Keller, S. Tait and J. Radjenovic, Environ. Sci. Technol., 2015, 49, 14326–14333 CrossRef CAS PubMed.
- R. Kumar, E. H. Gleissner, E. G. Tiu and Y. Yamakoshi, Org. Lett., 2016, 18, 184–187 CrossRef CAS PubMed.
- Y. Xu, J. Ai and H. Zhang, J. Hazard. Mater., 2016, 309, 87–96 CrossRef CAS PubMed.
- J. Hong, S. Lu, C. Zhang, S. Qi and Y. Wang, Chemosphere, 2011, 84, 1542–1547 CrossRef CAS PubMed.
- M. F. Hou, L. Liao, W. D. Zhang, X. Y. Tang, H. F. Wan and G. C. Yin, Chemosphere, 2011, 83, 1279–1283 CrossRef CAS PubMed.
- K. P. Mishra and P. R. Gogate, Sep. Purif. Technol., 2010, 75, 385–391 CrossRef CAS.
- X. Hou, W. Shen, X. Huang, Z. Ai and L. Zhang, J. Hazard. Mater., 2016, 308, 67–74 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yi Dua,
Huiling Liub and
Jun Mab
*a,
Yi Dua,
Huiling Liub and
Jun Mab