
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free C–H arylation of imidazoheterocycles with aryl hydrazines†
Sourav Jana a,
Sadhanendu Samantaa,
Avik K. Bagdib,
Valerii Z. Shirinianc and
Alakananda Hajra*a
a,
Sadhanendu Samantaa,
Avik K. Bagdib,
Valerii Z. Shirinianc and
Alakananda Hajra*a
aDepartment of Chemistry, Visva-Bharati (A Central University) Santiniketan, 731235, West Bengal, India. E-mail: alakananda.hajra@visva-bharati.ac.in; Web: http://www.visvabharati.ac.in/AlakanandaHajra.html
bDepartment of Chemistry, TDB College, Raniganj, Burdwan 713347, India
cZelinsky Institute of Organic Chemistry RAS, 47 Leninsky Prosp., 119991, Moscow, Russian Federation
First published on 3rd April 2018
Abstract
A simple and efficient metal-free arylation of imidazo[1,2-a]pyridines at the C-3 position with arylhydrazine has been achieved at room temperature under ambient air conditions. Various 2,3-disubstituted imidazopyridines and imidazothiazoles were synthesized with high yields. The present methodology demonstrates the usefulness of commercially available aryl hydrazine as an arylating agent.
Introduction
The development of efficient methodologies for the arylation of biological active heterocycles has been of great importance over the years.1 Traditionally, arylation is achieved via transition-metal-catalyzed cross-coupling reactions.2 In the last decade, transition-metal-catalyzed direct C–H arylation has emerged as an alternative to the conventional cross-coupling reaction.3 However, use of a metal catalyst, ligand, and additives limits the application of these methodologies. As such, it is desirable to develop transition-metal-free methods for the arylation of bioactive heterocycles.4 Arylhydrazine has recently been used as an arylating agent due to its ready availability.5 Few methodologies have been developed using arylhydrazine for the arylation of various heterocycles.5b,d,f,i,jImidazo[1,2-a]pyridine has attracted much interest due to its wide range of applications in pharmaceuticals and material science.6 The pharmacological activity of this moiety is dependent on its substituents. Several bioactive compounds such as γ-secretase modulators (GSMs) (1), liver X receptor (LXR) agonists (2), positive allosteric modulators (PAMs) of metabotropic glutamate 2 receptor (3), GABAAα2/α3 agonists (4), antileishmanial agents (5 and 6), and kinase inhibitors (7) contain the arylimidazo[1,2-a]pyridine moiety as the core structure (Fig. 1).7 As a consequence, a number of methodologies have been developed for the synthesis and functionalization of this moiety.8 Conventionally, arylation of imidazo[1,2-a]pyridine is carried out by transition-metal-catalyzed cross-coupling reactions using aryl halide/tosylate/mesylate as the aryl source.9 Despite these advances in the functionalization of this moiety, to the best of our knowledge, there is no metal-free protocol for the arylation of imidazo[1,2-a]pyridines. This prompted us to develop a transition-metal-free method for the arylation of imidazo[1,2-a]pyridines. Herein, we report a direct C–H arylation of imidazo[1,2-a]pyridines using easily accessible arylhydrazine hydrochloride in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) at room temperature under ambient air (Scheme 1).
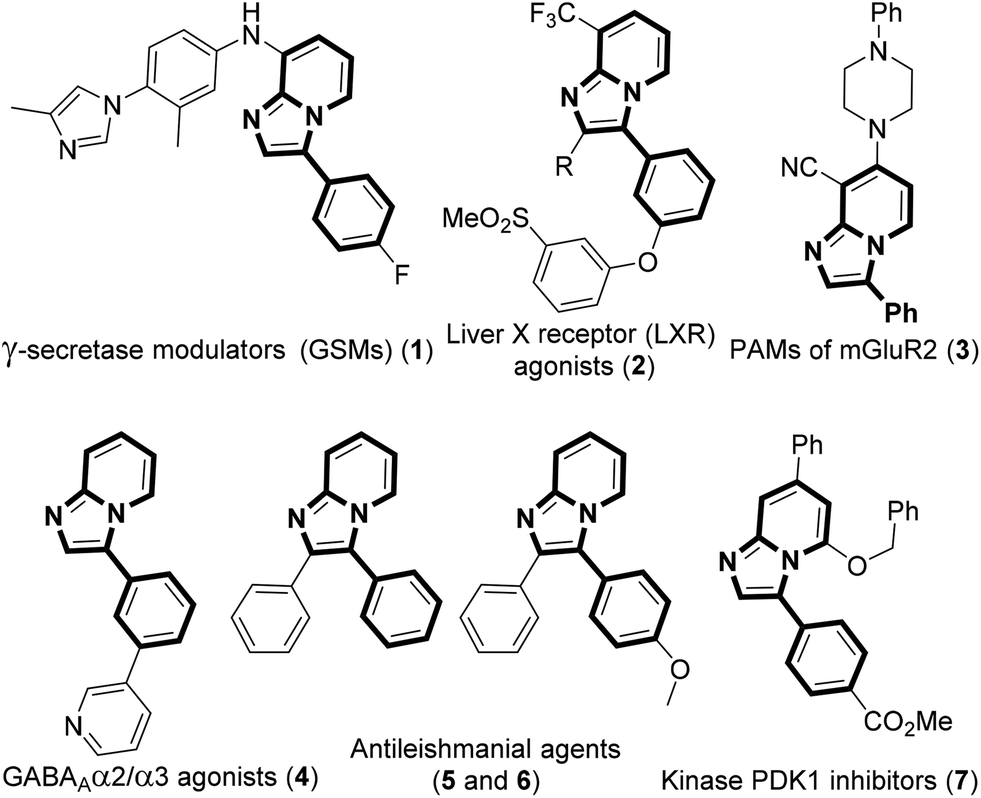 | ||
| Fig. 1 Arylimidazo[1,2-a]pyridine containing bioactive molecules. | ||
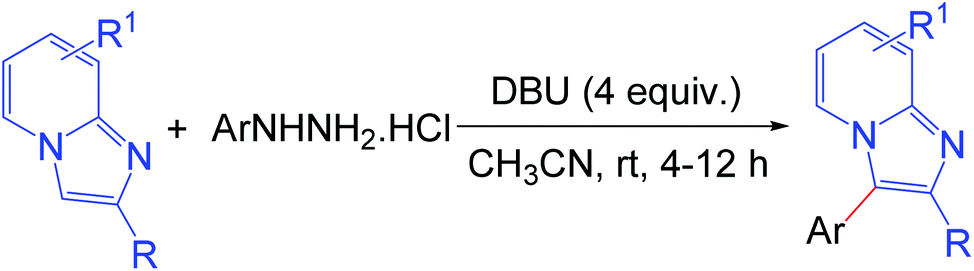 | ||
| Scheme 1 Arylation of imidazopyridines. | ||
Results and discussion
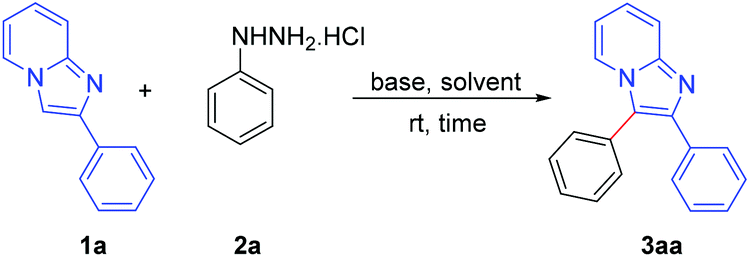
We commenced our study by taking 2-phenylimidazo[1,2-a]pyridine (1a) and phenyl hydrazine hydrochloride (2a) as the model substrates for the arylation of imidazopyridines. Initially we carried out the reaction using Et3N (4 equiv.) as a base in MeCN. Gratifyingly, the expected 2,3-diphenyl imidazo[1,2-a]pyridine was obtained in 56% yield within 4 h (Table 1, entry 1). On screening with other organic bases such as Et2NH, iPr2NH, DBU and 1,4-diazabicyclo[2.2.2]octane (DABCO), it was found that DBU was the optimal base, affording the desired product in 83% yield (Table 1, entries 2–5). Inorganic bases (K2CO3, Cs2CO3, K3PO4 and KOtBu) were also tested, but none were as effective (Table 1, entries 6–9). Other solvents including dimethyl sulfoxide (DMSO), dimethyl formamide (DMF), dimethyl acetamide (DMA), MeNO2, dichloroethane (DCE), H2O, MeOH and EtOH were not as effective as MeCN (Table 1, entries 10–17). Increment of base loading did not improve the yield, whereas decrement of base loading diminished the yield significantly (Table 1, entries 18 and 19). The reaction did not occur in the absence of base, which suggests a significant role for the base in this arylation reaction (Table 1, entry 20). When the reaction was carried in an oxygen atmosphere, no further improvement in yield was observed (Table 1, entry 21); however, in an inert atmosphere, only a trace amount of the product was obtained (Table 1, entry 22). Thus, the optimum yield was obtained by carrying out the reaction using 4 equiv. of DBU in MeCN in ambient air (Table 1, entry 4).
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | Time | Solvent | Yieldb |
| a Reaction conditions: 0.2 mmol of 1a, 1.3 equiv. of 2a and base in 3 mL solvent at rt.b Isolated yield.c NR = no reaction.d O2 atmosphere.e Ar atmosphere. | ||||
| 1 | Et3N (4) | 4 h | MeCN | 56% |
| 2 | Et2NH (4) | 4 h | MeCN | 45% |
| 3 | iPr2NH (4) | 4 h | MeCN | 35% |
| 4 | DBU (4) | 4 h | MeCN | 83% |
| 5 | DABCO (4) | 4 h | MeCN | 48% |
| 6 | K2CO3 (4) | 4 h | MeCN | 45% |
| 7 | Cs2CO3 (4) | 4 h | MeCN | 48% |
| 8 | K3PO4 (4) | 4 h | MeCN | 39% |
| 9 | KOtBu (4) | 4 h | MeCN | 51% |
| 10 | DBU (4) | 4 h | DMSO | 60% |
| 11 | DBU (4) | 4 h | DMF | 40% |
| 12 | DBU (4) | 4 h | DMA | 30% |
| 13 | DBU (4) | 4 h | MeNO2 | Trace |
| 14 | DBU (4) | 4 h | DCE | 22% |
| 15 | DBU (4) | 4 h | H2O | NR |
| 16 | DBU (4) | 4 h | MeOH | 54% |
| 17 | DBU (4) | 4 h | EtOH | 41% |
| 18 | DBU (5) | 4 h | MeCN | 83% |
| 19 | DBU (3) | 4 h | MeCN | 55% |
| 20 | — | 4 h | MeCN | NRc |
| 21 | DBU (4) | 4 h | MeCN | 82%d |
| 22 | DBU (4) | 4 h | MeCN | Tracee |
After establishing the optimized reaction conditions, we investigated the substrate scope of this arylation reaction. For this purpose, the effect of the substituent present at the 2-position on the imidazo[1,2-a]pyridine moiety was first tested (Scheme 2). Imidazo[1,2-a]pyridines bearing a phenyl ring with electron-donating as well as electron-withdrawing functionality afforded the desired products with excellent yields (3aa–3ia). The commercially available drug, zolimidine, was also arylated under the present reaction conditions with 87% yield (3ja). 2-Naphthyl imidazo[1,2-a]pyridine and 2-thiophenyl imidazo[1,2-a]pyridine also yielded the corresponding products (3ka and 3la). Imidazo[1,2-a]pyridine containing an aliphatic substituent at the 2-position effectively reacted with phenyl hydrazine to generate the desired product (3ma). Moreover, unsubstituted imidazo[1,2-a]pyridine afforded the regioselectively 3-arylated product with good yield (3na). However, 3-phenylimidazo[1,2-a]pyridine did not react under the present reaction conditions. It should also be mentioned that 2,3-diphenylimidazo[1,2-a]pyridine (3aa) and 2-(4-methoxyphenyl)-3-phenylimidazo[1,2-a]pyridine (3ca) are known as potent antileishmanial agents.7d
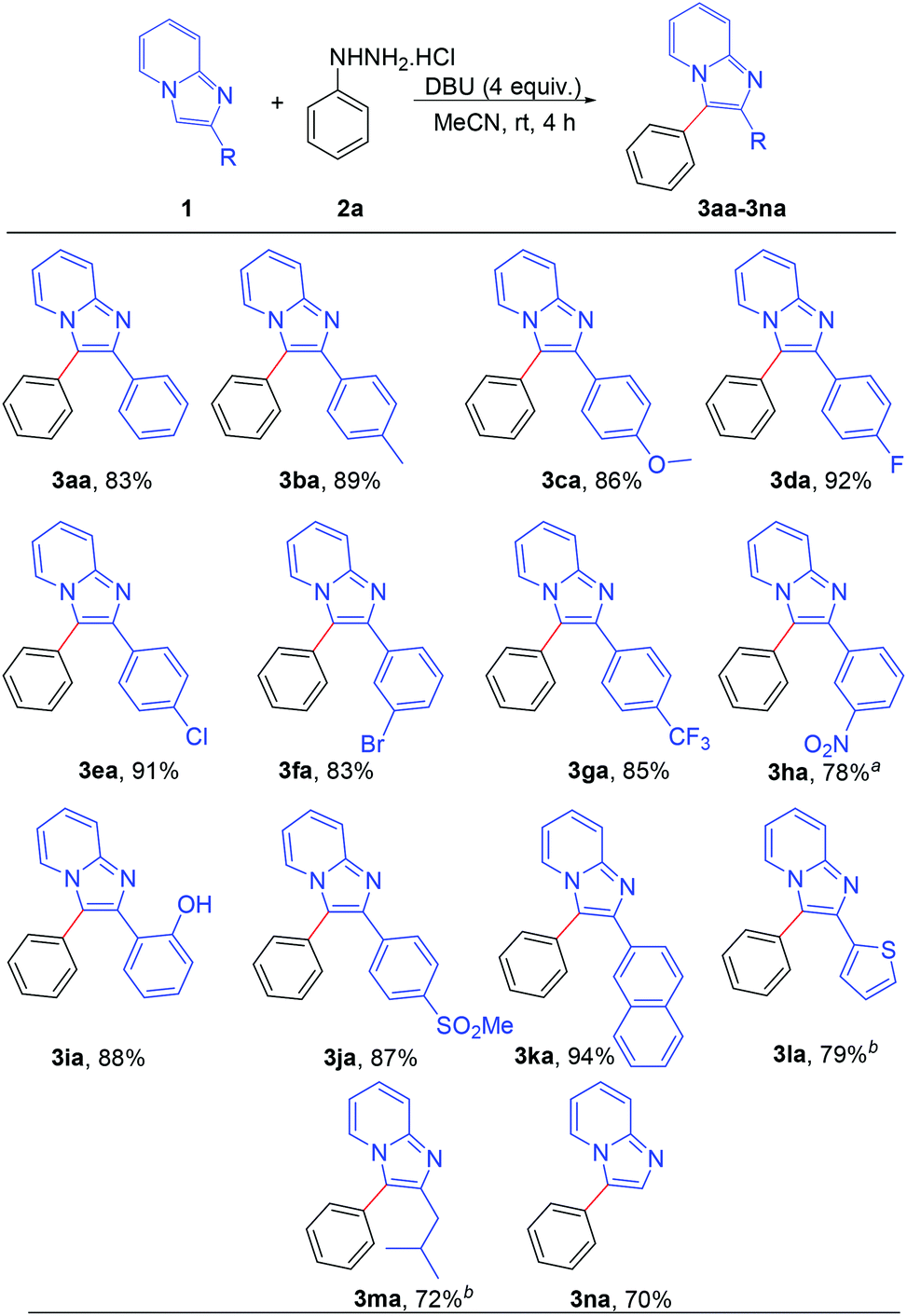 | ||
| Scheme 2 Scope of substrates: variation of C-2 substituents on the imidazo[1,2-a]pyridine. Reaction conditions: 0.2 mmol of 1, 1.3 equiv. of 2a and 4 equiv. of DBU in 3 mL MeCN at rt (27 °C) for 4 h. aReaction completed in 12 h. bReaction completed in 9 h. | ||
Next, the effect of the substituent on the pyridine ring of the imidazo[1,2-a]pyridine moiety was tested (Scheme 3). Imidazo[1,2-a]pyridines bearing different substituents such as –Me, –Cl, –Br and –CN on the pyridine rings successfully afforded the corresponding arylated products with high to excellent yields (3oa–3ta). The structure of 8-methyl-2,3-diphenylimidazo[1,2-a]pyridine (3oa) was confirmed by single crystal X-ray crystallography analysis.10 Applicability of the present methodology was also demonstrated on a gram scale. 6-Bromo-2,3-diphenylimidazo[1,2-a]pyridine reacted efficiently with phenyl hydrazine to afford the product 3ra without significant decrement of the yield (77%).
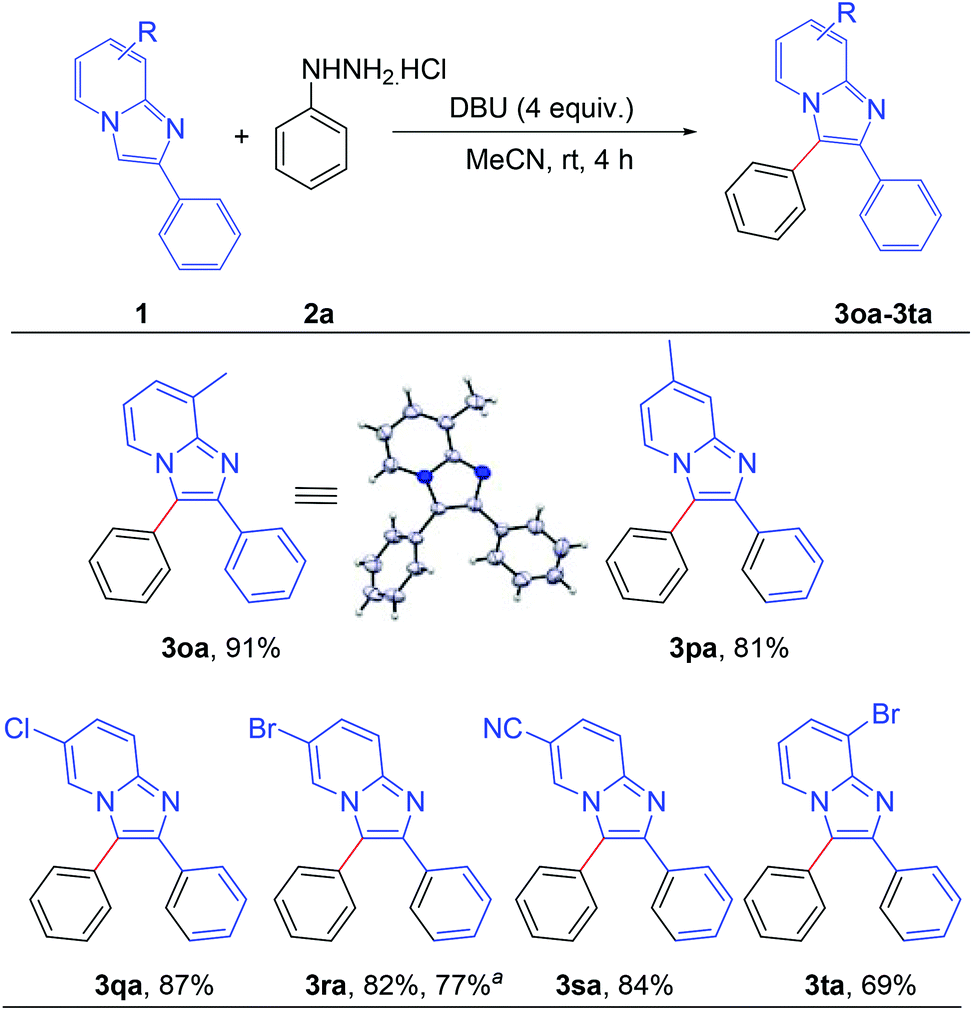 | ||
| Scheme 3 Scope of substrates: variation of substituents on imidazo[1,2-a]pyridine at pyridine rings: reaction conditions: 0.2 mmol of 1, 1.3 equiv. of 2a and 4 equiv. of DBU in 3.0 mL MeCN at rt (27 °C) for 4 h. a Reaction at 5.0 mmol scale. | ||
To assess the general applicability of the protocol, different arylhydrazine hydrochlorides were also reacted with 2-phenylimidazo[1,2-a]pyridine, as shown in Scheme 4. Different arylhydrazines bearing substituents such as –Me, –Cl, –Br afforded the corresponding arylated products (3ab–3ae) with good to excellent yields. However, arylhydrazines with strong electron-withdrawing groups such as –NO2 and –CN were unable to produce the desired products under the present reaction conditions.
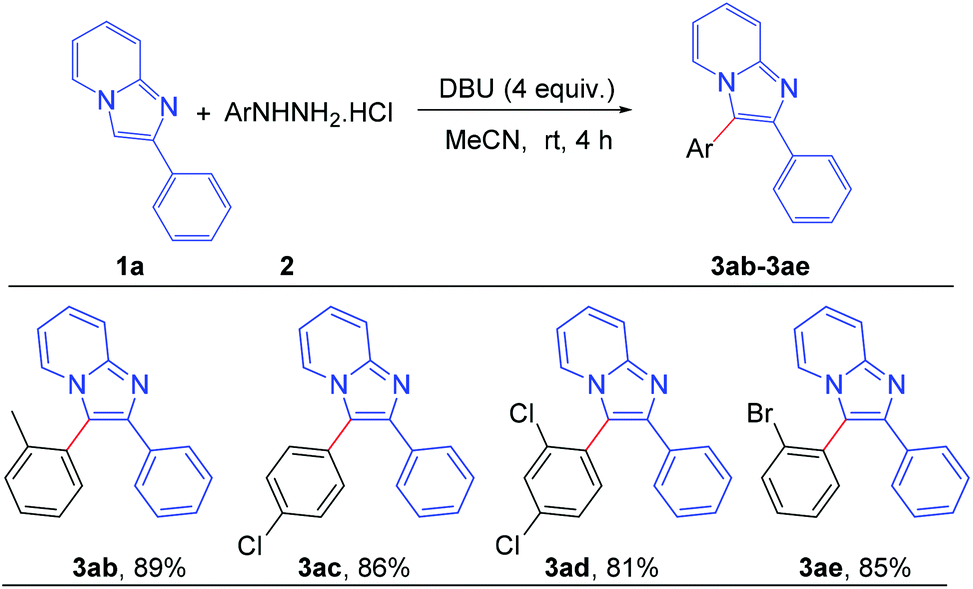 | ||
| Scheme 4 Scope of substrates: variation of substituents on arylhydrazine: reaction conditions: 0.2 mmol of 1a, 1.3 equiv. of 2 and 4 equiv. of DBU in 3.0 mL MeCN at rt (27 °C) for 4 h. | ||
Next, the applicability of this arylation method was extended to other imidazoheterocycles such as imidazo[1,2-a]thiazole and benzo[d]imidazo[2,1-b]thiazole (Scheme 5). Gratifyingly, this methodology successfully afforded the corresponding arylated products (4a and 4b) without any difficulties.
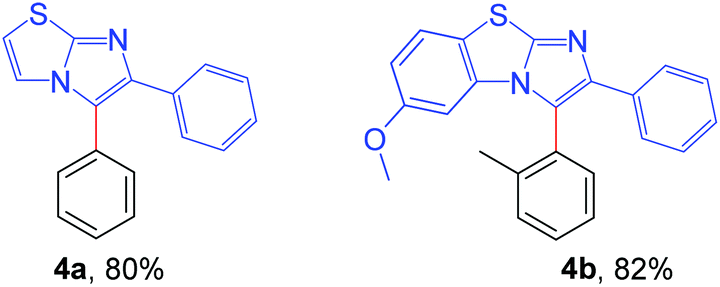 | ||
| Scheme 5 Substrate scope of imidazo[2,1-b]thiazole: reaction conditions: 0.2 mmol of imidazo[1,2-a]thiazole, 1.3 equiv. of 2 and 4 equiv. of DBU in 3.0 mL MeCN at rt (27 °C) for 4 h. | ||
The synthesized product was further functionalized employing Sonogashira and Suzuki coupling reactions (Scheme 6).8c 6-Bromo-2,3-diphenylimidazo[1,2-a]pyridine (3ra) was reacted with ethynylbenzene (Scheme 6, eqn A) to afford 2,3-diphenyl-6-(phenylethynyl)imidazo[1,2-a]pyridine (5) in excellent yield (95%). 2,3,6-Triphenylimidazo[1,2-a]pyridine (6) was synthesized in good yield (75%) via a Suzuki reaction between 6-bromo-2,3-diphenylimidazo[1,2-a]pyridine (3ra) and phenyl boronic acid (Scheme 6, eqn B).
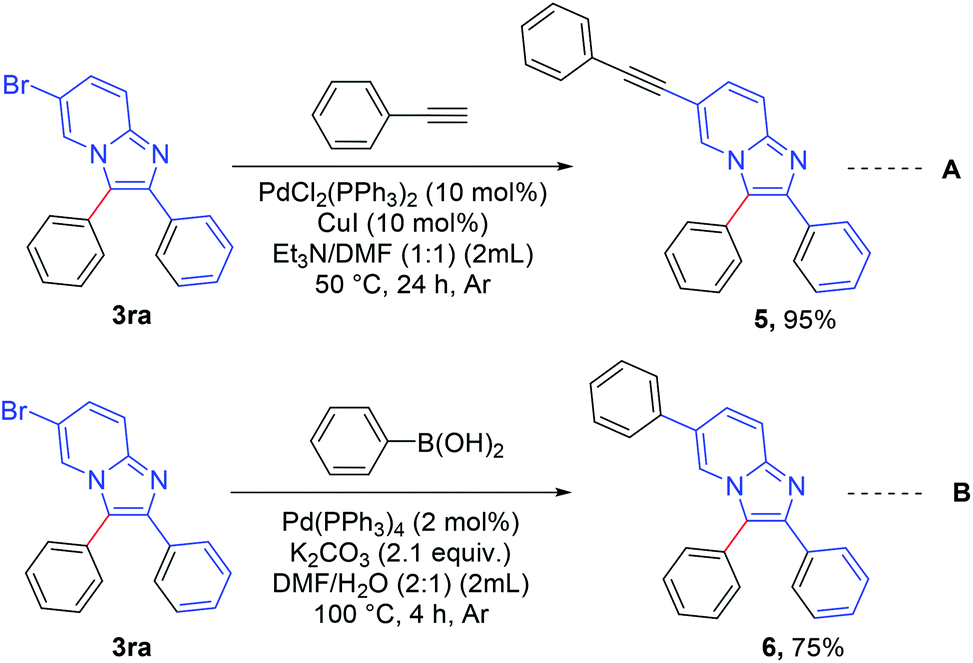 | ||
Scheme 6 Synthetic application: reaction conditions (A): 0.2 mmol of 3ra and 0.2 mmol of phenyl acetylene with 10 mol% PdCl2(PPh3)2 and 10 mol% Cu I dissolved in 2 mL Et3N/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under Ar atmosphere at 50 °C for 24 h. Reaction conditions (B): 0.2 mmol of 3ra and 1 equiv. of phenyl boronic acid with 2 mol% Pd(PPh3)4 and 2.1 equiv. of K2CO3 in 2 mL DMF/H2O (2:1) under Ar atmosphere at 100 °C for 4 h. :1) under Ar atmosphere at 50 °C for 24 h. Reaction conditions (B): 0.2 mmol of 3ra and 1 equiv. of phenyl boronic acid with 2 mol% Pd(PPh3)4 and 2.1 equiv. of K2CO3 in 2 mL DMF/H2O (2:1) under Ar atmosphere at 100 °C for 4 h. | ||
A number of control experiments were performed to investigate the reaction pathway. The reactions were carried out in the presence of radical scavengers such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and benzo-1,4-quinone (BQ). The formation of a trace amount only of the products indicates that the reaction probably proceeds through a radical pathway (Scheme 7, eqn (A)). Furthermore, the formation of a trace amount of the product in an argon atmosphere suggests that aerial oxygen plays a vital role in this reaction (Scheme 7, eqn (B)).
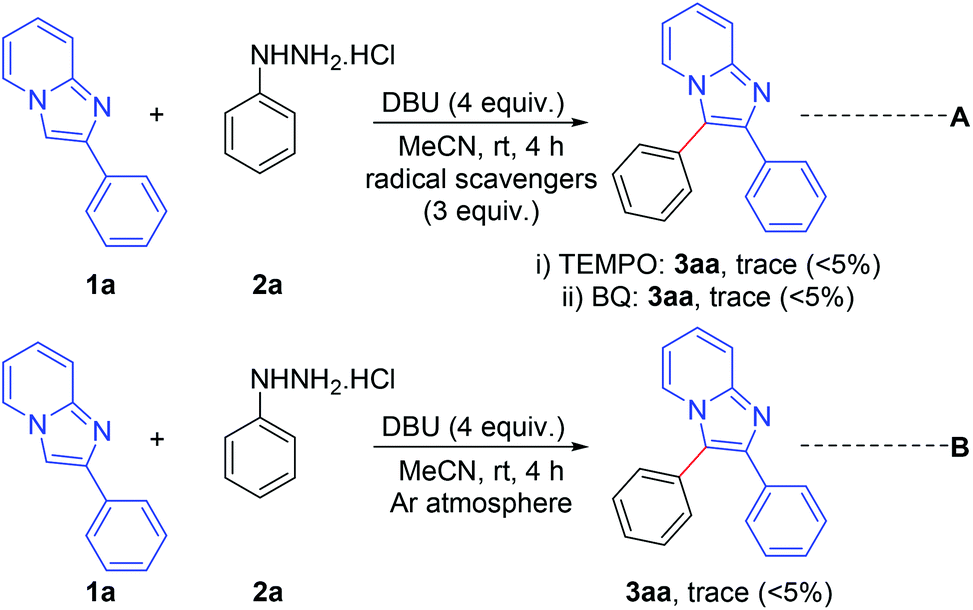 | ||
| Scheme 7 Control experiments. | ||
On the basis of the control experiments and literature reports,5b,f,i the probable mechanism of the reaction is outlined in Scheme 8. Initially the phenyl radical is formed from the phenyl hydrazine in the presence of base under aerobic conditions. The generated phenyl radical reacts with the imidazo[1,2-a]pyridine moiety to afford the radical intermediate A. Intermediate A is oxidized into the intermediate B under aerobic conditions. Finally, the product is obtained from the intermediate B via elimination of a proton.
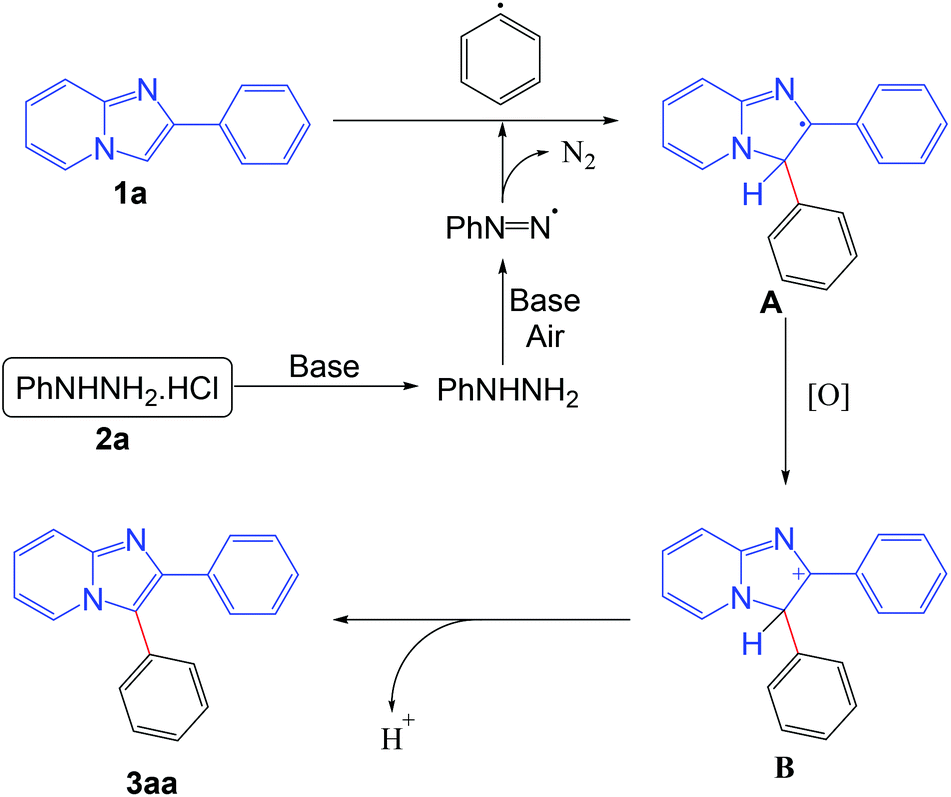 | ||
| Scheme 8 Plausible reaction pathway. | ||
Conclusions
In summary, we have developed a metal-free convenient methodology for the arylation of imidazo[1,2-a]pyridines employing arylhydrazine as an arylating agent at room temperature. The present methodology offers a practical route for the synthesis of various 2,3-disubstituted imidazo[1,2-a]pyridines with a wide range of functional groups. Imidazo[2,1-b]thiazole and benzo[d]imidazo[2,1-b]thiazole were also arylated under the present reaction conditions in good yields. We believe our present protocol for arylation will find useful applications in organic synthesis, the pharmaceutical industry, and material science.Experimental section
General information
Reagents were purchased from commercial sources and used without further purification. 1H and 13C{1H} nuclear magnetic resonance (NMR) spectra were determined on a 400 MHz spectrometer. 1H NMR spectra were determined on a 400 MHz spectrometer as solutions in CDCl3. Chemical shifts are expressed in parts per million (δ) and the signals are reported as s (singlet), d (doublet), t (triplet), m (multiplet), dd (double doublet), and coupling constants (J) are given in Hz. 13C{1H} NMR spectra were recorded at 100 MHz in CDCl3 solution. Chemical shifts as internal standard are referenced to CDCl3 (δ = 7.26 for 1H and δ = 77.16 for 13C{1H} NMR) as the internal standard. Thin layer chromatography (TLC) was performed on a silica gel-coated glass slide. Commercially available solvents were freshly distilled before the reaction. All reactions involving moisture-sensitive reactants were executed using oven-dried glassware. X-ray single crystal data were collected using MoKα (λ = 0.71073 Å) radiation with Charged Coupled Device (CCD) area. All the imidazoheterocycles were prepared by our reported methods.8e,fGeneral experimental procedure for the arylation of imidazo[1,2-a]pyridines (3)
A mixture of imidazo[1,2-a]pyridine (1, 0.20 mmol, 1 equiv.) and arylhydrazine hydrochloride (2, 0.26 mmol, 1.3 equiv.) was dissolved in 3 mL MeCN at room temperature (rt) in a reaction tube. Then, 4 equiv. of DBU (120 μL) was added to the reaction mixture and stirred in air for 4 h unless otherwise mentioned. After completion of the reaction, the reaction mixture was extracted with ethyl acetate and washed with water. The organic phase was dried over anhydrous Na2SO4. The crude residue was obtained after evaporating the solvent under reduced pressure and was finally purified by column chromatography on silica gel (60–120 mesh) using petroleum ether and ethylacetate as an eluent to afford the pure product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. H. and V. Z. S. acknowledge the financial support from DST, New Delhi (Indo-Russian joint project # INT/RUS/RFBR/P-295) and Russian Foundation for Basic Research, Russia (Ref. # 17-53-45127).Notes and references
- (a) C. Fischer and B. Koenig, Beilstein J. Org. Chem., 2011, 7, 59 CrossRef CAS PubMed; (b) R. Rossi, M. Lessi, C. Manzini, G. Marianetti and F. Bellina, Synthesis, 2016, 48, 3821 CrossRef CAS; (c) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed.
- (a) A. d. Meijere and F. Diederich, Metal-catalyzed cross-coupling reactions, completely revised and enlarged, Wiley-VCH: Weinheim, 2nd ed, 2004 Search PubMed; (b) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed.
- (a) G. Dyker, Handbook of C–H Transformations; Wiley-VCH: Weinheim, 2005 Search PubMed; (b) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC; (c) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed; (d) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2013, 46, 412 CrossRef CAS PubMed; (e) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed.
- (a) R. Rossi, M. Lessi, C. Manzini, G. Marianetti and F. Bellina, Adv. Synth. Catal., 2015, 357, 3777 CrossRef CAS; (b) A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140 RSC; (c) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed.
- (a) A. S. Demir, Ö. Reis and E. Özgül-Karaaslan, J. Chem. Soc., Perkin Trans. 1, 2001, 3042 RSC; (b) M. Ravi, P. Chauhan, R. Kant, S. K. Shukla and P. P. Yadav, J. Org. Chem., 2015, 80, 5369 CrossRef CAS PubMed; (c) M. Li and Y. Ye, ChemCatChem, 2015, 7, 4137 CrossRef CAS; (d) P. Patil, A. Nimonkar and K. G. Akamanchi, J. Org. Chem., 2014, 79, 2331 CrossRef CAS PubMed; (e) H. Zhang, C. Wang, Z. Li and Z. Wang, Tetrahedron Lett., 2015, 56, 5371 CrossRef CAS; (f) T. Taniguchi, M. Imoto, M. Takeda, F. Matsumoto, T. Nakai, M. Mihara, T. Mizuno, A. Nomoto and A. Ogawa, Tetrahedron, 2016, 72, 4132 CrossRef CAS; (g) T. Taniguchi, A. Murata, M. Takeda, T. Mizuno, A. Nomoto and A. Ogawa, Eur. J. Org. Chem., 2017, 2017, 4928 CrossRef CAS; (h) Y. Wang, B. Du, W. Sha, H. Mei, J. Han and Y. Pan, Org. Chem. Front., 2017, 4, 1313 RSC; (i) P. Chauhan, M. Ravi, S. Singh, P. Prajapati and P. P. Yadav, RSC Adv., 2016, 6, 109 RSC; (j) E. Kocaoğlu, M. A. Karaman, H. Tokgöz and O. Talaz, ACS Omega, 2017, 2, 5000 CrossRef; (k) H. Jasch, J. Scheumann and M. R. Heinrich, J. Org. Chem., 2012, 77, 10699 CrossRef CAS PubMed; (l) J.-W. Yuan, W.-J. Li and Y.-M. Xiao, Tetrahedron, 2017, 73, 179 CrossRef CAS; (m) Z.-Y. He, C.-F. Huang and S.-K. Tian, Org. Lett., 2017, 19, 4850 CrossRef CAS PubMed.
- (a) C. Enguehard-Gueiffier and A. Gueiffier, Mini-Rev. Med. Chem., 2007, 7, 888 CrossRef CAS PubMed; (b) A. T. Baviskar, S. M. Amrutkar, N. Trivedi, V. Chaudhary, A. Nayak, S. K. Guchhait, U. C. Banerjee, P. V. Bharatam and C. N. Kundu, ACS Med. Chem. Lett., 2015, 6, 481 CrossRef CAS PubMed; (c) A. J. Stasyuk, M. Banasiewicz, M. K. Cyrański and D. T. Gryko, J. Org. Chem., 2012, 77, 5552 CrossRef CAS PubMed.
- (a) F. Bischoff, D. Berthelot, M. De Cleyn, G. Macdonald, G. Minne, D. Oehlrich, S. Pieters, M. Surkyn, A. A. Trabanco, G. Tresadern, S. V. Brandt, I. Velter, M. Zaja, H. Borghys, C. Masungi, M. Mercken and H. J. M. Gijsen, J. Med. Chem., 2012, 55, 9089 CrossRef CAS PubMed; (b) S. C. Goodacre, L. J. Street, D. J. Hallett, J. M. Crawforth, S. Kelly, A. P. Owens, W. P. Blackaby, R. T. Lewis, J. Stanley, A. J. Smith, P. Ferris, B. Sohal, S. M. Cook, A. Pike, N. Brown, K. A. Wafford, G. Marshall, J. L. Castro and J. R. Atack, J. Med. Chem., 2006, 49, 35 CrossRef CAS PubMed; (c) S. Follot, J.-C. Debouzy, D. Crouzier, C. Enguehard-Gueiffier, A. Gueiffier, F. Nachon, B. Lefebvre and F. Fauvelle, Eur. J. Med. Chem., 2009, 44, 3509 CrossRef CAS PubMed; (d) S. Marhadour, P. Marchand, F. Pagniez, M.-A. Bazin, C. Picot, O. Lozach, S. Ruchaud, M. Antoine, L. Meijer, N. Rachidi and P. L. Pape, Eur. J. Med. Chem., 2012, 58, 543 CrossRef CAS PubMed; (e) S. Kona, R. S. Ravi, M. Chakravarty and V. N. R. Chava, J. Chem., 2013, 2013, 305934 Search PubMed.
- (a) A. K. Bagdi, S. Santra, K. Monir and A. Hajra, Chem. Commun., 2015, 51, 1555 RSC; (b) K. Pericherla, P. Kaswan, K. Pandey and A. Kumar, Synthesis, 2015, 47, 887 CrossRef CAS; (c) J. Koubachi, S. E. Kazzouli, M. Bousmina and G. Guillaumet, Eur. J. Org. Chem., 2014, 2014, 5119 CrossRef CAS; (d) S. Samanta, S. Jana, S. Mondal, K. Monir, S. K. Chandra and A. Hajra, Org. Biomol. Chem., 2016, 14, 5073 RSC; (e) A. K. Bagdi, M. Rahman, S. Santra, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1741 CrossRef CAS; (f) S. Mishra, K. Monir, S. Mitra and A. Hajra, Org. Lett., 2014, 16, 6084 CrossRef CAS PubMed; (g) O. Barun, H. Ila and H. Junjappa, J. Org. Chem., 2000, 65, 1583 CrossRef CAS PubMed; (h) P. Sun, M. Jiang, W. Wei, Y. Min, W. Zhang, W. Li, D. Yang and H. Wang, J. Org. Chem., 2017, 82, 2906 CrossRef CAS PubMed; (i) R. R. Donthiri, V. Pappula, N. N. K. Reddy, D. Bairagi and S. Adimurthy, J. Org. Chem., 2014, 79, 11277 CrossRef CAS PubMed; (j) Z. Chen, Y. Wen, G. Luo, M. Yea and Q. Wang, RSC Adv., 2016, 6, 86464 RSC; (k) I. I. Roslan, K.-H. Ng, J.-E. Wu, G.-K. Chuah and S. Jaenicke, J. Org. Chem., 2016, 81, 9167 CrossRef CAS PubMed.
- (a) Y. Wang, B. Frett and H.-Y. Li, Org. Lett., 2014, 16, 3016 CrossRef CAS PubMed; (b) P. Y. Choy, K. C. Luk, Y. Wu, C. M. So, L.-l. Wang and F. Y. Kwong, J. Org. Chem., 2015, 80, 1457 CrossRef CAS PubMed; (c) D. Nandi, Y.-M. Jhou, J.-Y. Lee, B.-C. Kuo, C.-Y. Liu, P.-W. Huang and H. M. Lee, J. Org. Chem., 2012, 77, 9384 CrossRef CAS PubMed; (d) C. Enguehard, J.-L. Renou, V. Collot, M. Hervet, S. Rault and A. Gueiffier, J. Org. Chem., 2000, 65, 6572 CrossRef CAS PubMed; (e) H. Cao, H. Zhan, Y. Lin, X. Lin, Z. Du and H. Jiang, Org. Lett., 2012, 14, 1688 CrossRef CAS PubMed; (f) B.-T. Luo, H. Liu, Z.-J. Lin, J. Jiang, D.-S. Shen, R.-Z. Liu, Z. Ke and F.-S. Liu, Organomettalics, 2015, 34, 4881 CrossRef CAS.
- Further information can be found in the CIF file. This crystal was deposited in the Cambridge Crystallographic Data Centre and assigned as CCDC 1589816.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1589816. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra01474d |
| This journal is © The Royal Society of Chemistry 2018 |