
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem blocking of PCR extension to form a single-stranded overhang for facile, visual, and ultrasensitive gene detection†
Zhe Suia,
Tong Lia,
Ran Anab,
Wei Wu a,
Makoto Komiyama*ac and
Xingguo Liang*ab
a,
Makoto Komiyama*ac and
Xingguo Liang*ab
aCollege of Food Science and Engineering, Ocean University of China, Qingdao 266003, China. E-mail: liangxg@ouc.edu.cn
bLaboratory for Marine Drugs and Bioproducts of Qingdao National Laboratory for Marine Science and Technology, Qingdao 266235, China
cNational Institute for Materials Science (NIMS), Namiki, Tsukuba 305-0044, Japan. E-mail: makotokomiyama@yahoo.com
First published on 25th April 2018
Abstract
In order to detect a predetermined gene in a field test, a facile, visual, and ultrasensitive approach without the need of special and expensive machines is required. In this study, a gene in the Ebola virus was targeted as an example for diagnosis. The key strategy is to incorporate molecular blockers (azobenzene-bearing moieties or thymine dimers) in tandem in one of the PCR primers and stop the polymerase extension there to form a single-stranded overhang. The PCR product was added to the dispersion of gold nanoparticles which were labelled with a probe oligonucleotide. When the Ebola virus-specific gene existed in the specimen, the oligonucleotide on the gold particles formed a double-helix with the single-stranded overhang, and thus the dispersion remained red in color. In the absence of the gene, however, the dispersion rapidly turned to blue because of nanoparticle aggregation. The difference was explicit even when the initial specimen involved only 1 copy of the gene. Accordingly, “whether the patient is infected by the virus or not” can be easily and visually judged by the naked eye.
Introduction
Facile and economical testing of viral infectious diseases at the bedside and their immediate treatment (Point-of-Care) have been some of the primary targets of today's science.1–3 There, the presence of a specific pathogen or its absence must be unanimously concluded outside the laboratory. Thus, the methods to detect a target gene easily and precisely without using complicated machines are crucially important. Visual detection with the naked eye, if possible, is still more desirable. Conventional PCR is undoubtedly the most important pioneer in this field. For practical applications, however, the analysis of the PCR products is often both time consuming and effortful. Real-time PCR is a more convenient alternative, and is currently the major diagnosis method.4,5 Nevertheless, this method requires special equipment and skills, and is not sufficiently user-friendly. Developing countries, where the worst outbreak of diseases like Ebola fever prevails, cannot afford the required laboratory capacity and logistical infrastructure. To overcome these problems, still more economical and easier methods are necessary. PCR-ELISA, the combination of enzyme-linked immunosorbent assay and PCR, is one of the promising methods.6–8 Alternatively, single-stranded amplification products are directly synthesized by unsymmetrical PCR (or heat treatments), and detected through hybridization with a probe.9 It is simple and attractive, but, in most cases, the amplification is not very effective. In spite of many other elegant attempts, complete answer to visual, sensitive, and inexpensive detection of designated gene for Point-of-Care (POC) has not yet been envisioned.10–14 The difficulty in advancing the PCR technology for POC applications lies in the difficulty of field testing of amplification products.Gold nanoparticles (AuNps) have been widely used for biosensors due to the unique optical properties which make them easily observable by naked eyes or detectable by costless equipment like UV-vis spectrometer.15 As compared with AuNps coated with proteins (e.g. streptavidin), the DNA-labelled AuNps are much more efficient for POC, because large amount of DNA strands could be attached onto the surface of AuNps and enable sequence specific detection with high sensitivity to nM. In addition, the DNA-labelled AuNPs are more easily prepared than protein-coated ones.15–18 However, AuNps are difficult to be used for direct detection of normal PCR products because they are dsDNA without a single-stranded overhang. Although single-stranded DNA products have been detected by using DNA-labelled AuNps after rolling circle amplification (RCA), hybridization chain reaction (HCR), and strand displacement amplification (SDA), these amplification approaches are not comparable with PCR.19–23
Here we propose a more straightforward methodology in which target gene is visually detected in extremely high sensitivity without using complicated machines. The point is to attach single stranded overhangs onto the end of PCR products by using modified primers. In this way we could detect PCR products directly with DNA-labelled AuNps, thus our new approach combines the advantages of both PCR technique and colloid Au science.
As presented in Scheme 1, the procedure is composed of two steps. In the first step (a), DNA specimen, obtained from potential patients by conventional methods, is amplified by “indenting PCR” to the products which bear a single-stranded overhang. The key feature of this newly developed PCR is to incorporate to pathogen-specific primers either azobenzene-bearing moieties or thymine dimers which function as “molecular blockers” of polymerase extension (the blue circles in Scheme 1a). During the amplification, the polymerase stalls at the position of these blockers to form required overhang structure. In order to suppress the run-through extension completely and confirm the reproducibility of diagnosis, multiple blockers are introduced in tandem to the primer (vide infra). Accordingly, the products bearing a single-stranded overhang at the end are obtained, only when a gene from the pathogen exists in the primary specimen. Our approach is better than using asymmetric PCR which gives single-stranded DNA products.9 In some cases it is hard to carry out asymmetric PCR because the concentration of one primer is usually much lower. In addition, the long ssDNA products may cause aggregation of AuNPs.23 For indenting PCR, the products are dsDNA with high rigidity, which is much more efficient for avoiding AuNP aggregation.
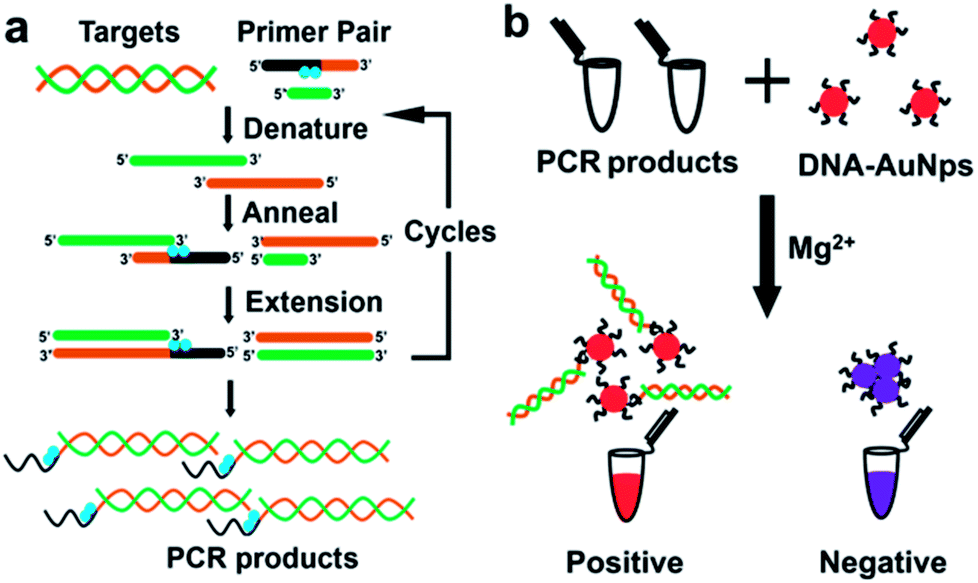 | ||
| Scheme 1 Outline of the visual detection of designated gene proposed in this paper. In the “indenting PCR” (a), PCR products having an overhang are prepared by placing multiple “molecular blockers” (blue circles) in tandem in one of the primers. In (b), these PCR products are treated with AuNps, labelled with the probe oligonucleotide, in the presence of appropriate concentration of Mg2+. The overhang having complementary sequence, if any, should bind to the probe DNA on the AuNps and prevent the particles from aggregation (the left in (b)). Otherwise, the salt-induced aggregation promptly occurs (the right). Thus, the presence or the absence of the designated gene in the original specimen is differentiated simply in terms of the color of the solution (either red or blue). | ||
In the second step (b), the PCR products are added to the dispersion of gold nanoparticles (AuNps) labelled with a single-stranded oligonucleotide as probe. If PCR products are produced in the step (a), their single-stranded overhangs are complementary with the probe on the AuNps, and thus double-stranded helixes are formed between them. Mg2+ was then added into the mixed solution of AuNps and PCR products. It neutralized negative charge of labelled DNA on Au nanoparticles.17,20,24,25 If the PCR products with single overhangs hybridized to the surface of AuNps, it provides additional negative charges and steric hindrance to protect AuNps from aggregation. Otherwise, if there exist no PCR products, the AuNps aggregated rapidly since the electrostatic stabilization effect is lost. In order to enhance the difference between these two situations, appropriate concentration of Mg2+ is added to the system. Thus, the presence of the designated gene in the original specimen or its absence can be differentiated simply by observing the color change of the solution. The present strategy employs the advantages of both PCR and Au colloidal science, and thus satisfactorily fulfils all the factors required for Point-of-Care detection (easiness, visibility, low cost, promptness, and no need of expensive machines).3
Experimental
Materials
The azobenzene tethered XF39 was synthesized and purified by Nihon Techno Service Co. LTD (Ibaraki, Japan). All the other oligonucleotides (including thiol modified ones) were synthesized by Genewiz (Suzhou, China). Taq DNA polymerase and deoxy ribonucleotides (dNTPs) were purchased from Thermo Fisher Scientific (Pittsburgh, USA). All other reagents were of analytical reagent grade. The aqueous solutions were prepared using Milli-Q purified water (resistance of 18.2 MΩ cm−1).Primer extension by DNA polymerase on a modified DNA template
A solution (10 μL) of DNA template (0.5 μM), FAM-labelled DNA primer (2 μM), and Taq DNA polymerase (0.02 U μL−1) was prepared in 1 × buffer of Taq DNA polymerase. The solution was then transferred to a PCR apparatus, and the reaction was allowed to proceed at 95 °C for 5 min, 50 °C for 20 min, and 72 °C for 20 min. After the reaction, a sample (5 μL) was added to loading buffer (2.5 μL) containing formamide (80%), EDTA (50 mM), and bromophenol blue (0.025%). The mixture was subjected to electrophoresis on a denaturing polyacrylamide gel (14%) containing urea (7 M) and methanamide (24%). The gel was photographed under ultraviolet illumination at 302 nm by a Bio-rad molecular imager without stain.PCR amplification
The first-round primer was to recognize and amplify a 163-bp cloned DNA fragment chosen from the NP encapsidation of genomic RNA of the Sudan Ebola virus. The second-round primers targeted the 103 bp gene inside of the amplified fragments obtained from the first round PCR. The solution was prepared as follows: cloned gene of Ebola virus (163 bp, different copies), the unmodified primer LongF23 (0.2 μM), XF39 or T10F50 (0.4 μM), R24 (0.6 μM), each of dNTPs (0.3 mM), Taq DNA polymerase (0.03 U μL−1), 1 × buffer (MgCl2, 2.5 mM). The sequence of ELongF23 is 5′-ATGTCCGGGCCGCCCTCTCCTTG-3′ PCR conditions: 95 °C for 10 s, 55 °C for 30 s, and 72 °C for 45 s, ended with a final extension at 72 °C for 10 min.Manufacture of AuNps and their labelling with thiol modified oligonucleotides
Gold nanoparticles with an average diameter of 15 ± 2 nm were synthesized using the citrate reduction protocol. The concentration of AuNPs was calculated to be ∼2.4 nM by Beer-Lambert Law (K = 2.4 × 108 M−1 cm−1) at 520 nm. These AuNps were labelled with 3′ thiol modified oligonucleotides by gradually increasing the concentration of NaCl up to 0.3 M. The functionalized AuNPs were purified by centrifugation and removal of supernatant. Then the final particles were resuspended in sodium phosphate buffer (10 mM, 0.3 M NaCl, pH 7.5). The number of DNA strands on each Au particle was 50–55.26Colorimetric detection of PCR products with labelled AuNps
After the PCR reaction, the product (10 μL) was mixed with 90 μL of labelled AuNps solution and incubated at room temperature for 10 min. Then the mixture was ultra-sonicated for 10 s, and 2 μL of 1 M MgCl2 was added. The color changes of solutions were observed by the naked eyes and photographed by a digital camera. Finally, the absorption spectra were recorded with a Thermo Varioskan Flash Microplate Reader in 20 min.Results and discussion
Regulation of polymerase extension by chemical and photochemical modifications of template strand has been already reported.27–31 In most of the cases, however, only some suppression of extension was evidenced, and complete absence of run-through products was not confirmed. In order to diagnose the presence or the absence of a designated gene precisely and reproducibly by the strategy in Scheme 1, complete blocking of extension must be guaranteed under variety of operation conditions (e.g., changes of temperatures, minor handling mistakes, and others). This factor is critically important especially when only small amount of gene is available, since indenting products are otherwise formed in insufficient amount and thus real virus carriers could be wrongly diagnosed as uninfected. Accordingly, we have incorporated two (or more) blocking molecules in tandem to the primer and take advantage of their cooperative blocking of extension. As candidate blockers of extension by Taq DNA polymerase, either (i) azobenzene-bearing moieties or (ii) thymine dimers were employed. First, two azobenzene-bearing moieties X's were introduced to the template strand (XF39 in Table 1), and the blocking activity of these tandem blockers was evaluated by primer extension experiments (Fig. 1a). In the X moiety, an azobenzene is tethered to D-threoninol on the phosphate backbone (Fig. S1a†).28 Because both the backbone and the side chain were unnatural, notable blocking of polymerase extension was expected. Two X moieties were introduced in tandem to the template. As a control, template NF39, which has no X residues, was used. Three primers Pr19, Pr20, and Pr21 (19, 20, and 21 nt) containing no X groups were 5′-FAM labelled (Table 1). As shown in lanes 2 and 5 in Fig. 1b, the extension using either Pr19 or Pr20 primer provided only one product of 21 nt length, which was two-bases and one-base longer than the corresponding primers, respectively. Apparently, the extension of both primers was stopped exactly at the position of the X moiety which the DNA polymerase first encountered (the 3′-side one). Consistently, with the use of Pr21 primer, which is completely complementary with XF39 down to this X moiety, no extension occurred (lane 8). As expected, the primer extension on natural template NF39 without the X modification proceeded thoroughly down to the end of the template, giving the 39 nt product (lanes 3, 6, and 9).| ODNs | Sequence (5′ → 3′) | Length/nt |
|---|---|---|
| a The bold characters refer to the portion which is not covered by the primer and potentially subjected to elongation by the polymerase. | ||
| Pr19 | CAATTTGGACGACGACGAT | 19 |
| Pr20 | CAATTTGGACGACGACGATG | 20 |
| Pr21 | CAATTTGGACGACGACGATGT | 21 |
| XF39 | CCATGTCTAATGCTGATXGXACAT-CGTCGTCGTCCAAATTG | 39 |
| T10F50 | CCATGTCTAATGCTGATGAT(10)ACAT-CGTCGTCGTCCAAATTG | 50 |
| NF39 | CCATGTCTAATGCTGATGACAT-CGTCGTCGTCCAAATTG | 39 |
| R24 | GTTGACCCGTATGATGATGAGAGT | 24 |
| F21 | ACATCGTCGTCGTCCAAATTG | 21 |
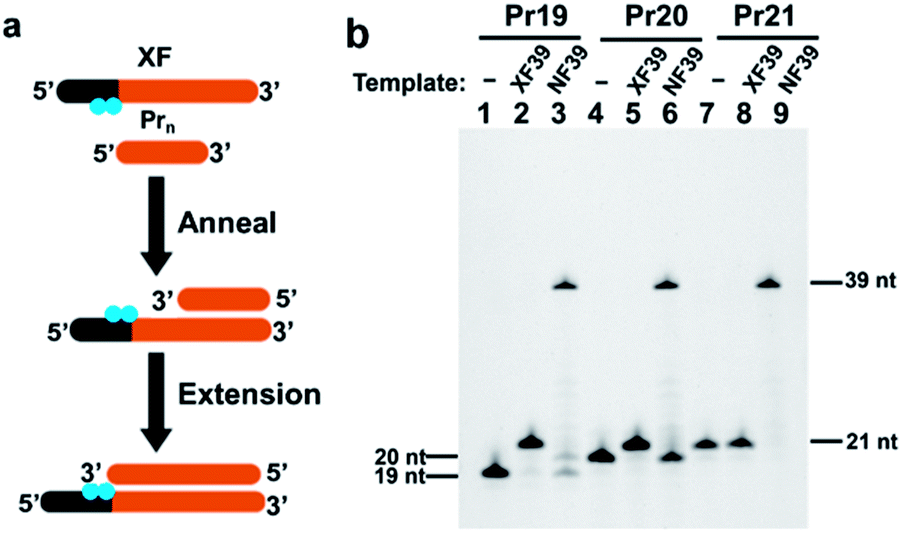 | ||
| Fig. 1 Primer extension experiments to evaluate the blocking activity of two tandem azobenzene-bearing moieties X's (a) and the results of electrophoresis analysis (b). Lane 1, Pr19 primer (19 nt) alone; lane 2, Pr19 on template XF39 (involving two X moieties); lane 3, Pr19 on natural template NF39; lane 4, Pr20 primer (20 nt) alone; lane 5, Pr20 on template XF39, lane 6, Pr20 on template NF39; lane 7, Pr21 primer (21 nt) alone; lane 8, Pr21 on template XF39; lane 9, Pr21 on template NF39. | ||
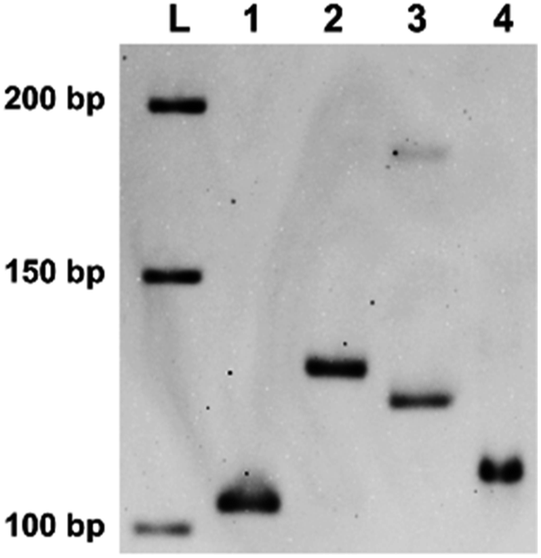 | ||
| Fig. 2 The product of “indenting PCR” of a gene in Ebola virus using XF39 primer involving two X residues. Lane 1, PCR product (103 bp) using non-modified primers F39 (39 nt without X) and R24 (24 nt without X); lane 2, the product of indenting PCR (103 bp duplex bearing 18 nt overhang) using XF39 (39 nt + X2) and R24 primers; lane 3, PCR product (121 bp) using F21 (21 nt without X) and R24. In lane 4, the product in lane 2 was treated with nuclease S1 to remove the overhang. Note that the mobility was close to that in lane 1. Lane L is a sequencing ladder. | ||
Indenting PCR
On the basis of these findings, “indenting PCR” was conducted to construct the products having desired single-stranded overhang. As the template, a gene in Ebola virus was chosen (Ebola S-EBOV NC_006432). In the upstream primer, two X residues were incorporated in the “tag” sequence (XF39, which was used for the primer extension experiments in Fig. 1). The downstream primer R24 contained no X groups. Importantly, only one product was formed by the “indenting PCR” (lane 2). Formation of the fragment of targeted structure (103 bp double-strand bearing 18 nt overhand) was confirmed by the treatment of this product with nuclease S1. Upon selective removal of its single-stranded overhang by this enzyme, the band was converted to new one (in lane 4), which showed the similar mobility as the 103 bp double-stranded DNA (lane 1). Interestingly, the indenting product in lane 2 migrated more slowly than the 121 bp double-stranded DNA which was produced by PCR using NF39/R24 combination (lane 3). This smaller mobility of the indenting fragment is probably due to higher flexibility of the single-stranded overhang than the corresponding double-helix.In addition to remarkable blocking activity of tandem X residues for “indenting PCR”, we have found that thymine–thymine dimers (Fig. S1b†), placed in tandem in the primer, also satisfactorily work as “molecular blockers” of the extension of Taq DNA polymerase. Although these dimers (both the cyclobutane dimers and (6–4) adducts) were previously reported to suppress primer extension,29–31 complete blocking of polymerase extension has been for the first time envisioned here. From the viewpoints of practical applications, these thymine dimer-based molecular blockers are advantageous in that they can be easily prepared simply by photo-irradiating oligonucleotides obtained by DNA synthesizers. In order to construct thymine dimers in tandem, consecutive thymine sequences were incorporated to the precursors of primers, and these oligonucleotides were photo-irradiated under air by using a simple handheld UV lamp with the intensity of 0.4 W m−2 at 254 nm (measured by a UV-c light meter). This light was much weaker than the ones (>10 W m−2) employed previously.
After careful optimization of conditions (Fig. S2 and S3†), the primer for the present indenting PCR was prepared by irradiating T10F50 (involving 10 consecutive thymine residues) for 28 h. Complete blocking of polymerase extension by this primer was confirmed by primer extension experiments (Fig. S3†). It is noteworthy that the blocking effect was minimal when T2F42 and T4F44 (involving 2 and 4 consecutive thymines) were irradiated for 28 h. The blocking effect gradually increased with the increase of the length of consecutive thymine sequence, and was notable for T6F46 and T8F48. Finally, almost complete inhibition of the polymerase extension was accomplished by T10F50 which was photo-irradiated for 28 h. These results substantiate that two or more thymine dimers in the primer are required to block the polymerase extension effectively.
In indenting PCR using the combination of the irradiated T10F50 and R24 as the primers, several amplification products were produced (lane 2 in Fig. 3). All of them migrated more slowly than the 103 bp double-stranded DNA in lane 1. In the T10F50-derived primer, thymine dimers are formed by photoirradiation at various positions in the T10 sequence, and block the polymerase extension there. Thus, the lengths of both double-helix portions and overhanging tails of the PCR products should be different from each other, providing multiple bands of different gel-mobility. Consistently, when the products in lane 2 were treated with nuclease S1, all of them were converted to new bands which show similar mobility as the 103 bp duplex (compare lane 4 with lane 1). The smearing character of the bands for these enzymatic digests is probably reflecting the different lengths of the double-helix portions of the indenting PCR products. It is noteworthy that the band for the run-through product in lane 3 was never perceived in this nuclease S1 digest. Apparently, tandem thymine dimers in one of the primers can almost completely block the polymerase extension with Taq polymerase, and is successfully applicable to indenting PCR.
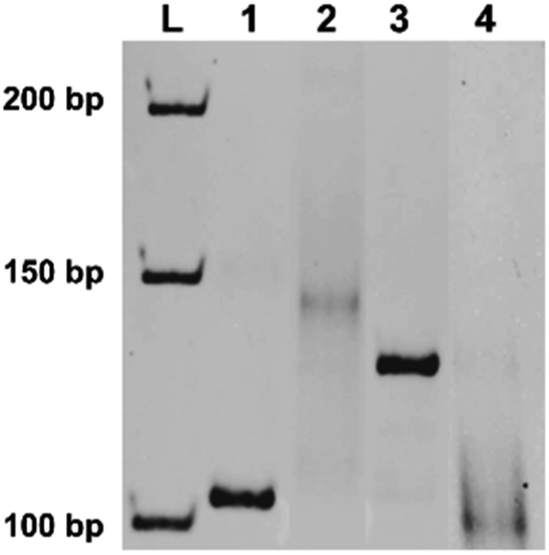 | ||
| Fig. 3 Indenting PCR using the primer bearing tandem thymine dimers. The gene in Ebola virus, used in Fig. 2, was also the template. In lane 2, T10F50 was first irradiated by a handheld UV lamp for 28 h, and used as the primer, together with R24 primer. In lane 4, the product in lane 2 was digested by nuclease S1. Lane 1, the amplification product (103 bp) obtained by using unmodified primers (F21 and R24); lane 3, the product using primer precursor T10F50 (without irradiation) and R24 as the primers. | ||
Colorimetric detection of indenting PCR products
These two kinds of indenting PCR using tandem blockers were employed for facile and visual detection of the designated gene in Ebola virus ((b) in Scheme 1). In Fig. 4, tandem X residues were used as the blockers. Au nanoparticles (average diameter = 15 ± 2 nm) were labelled with the oligonucleotide (18 nt) which was complementary with the single-stranded overhang constructed by the indenting PCR, and mixed with the amplification products. Then, MgCl2 was added to the mixture to increase the total Mg2+ concentration to 20 mM (this concentration was chosen as described in Fig. S4†).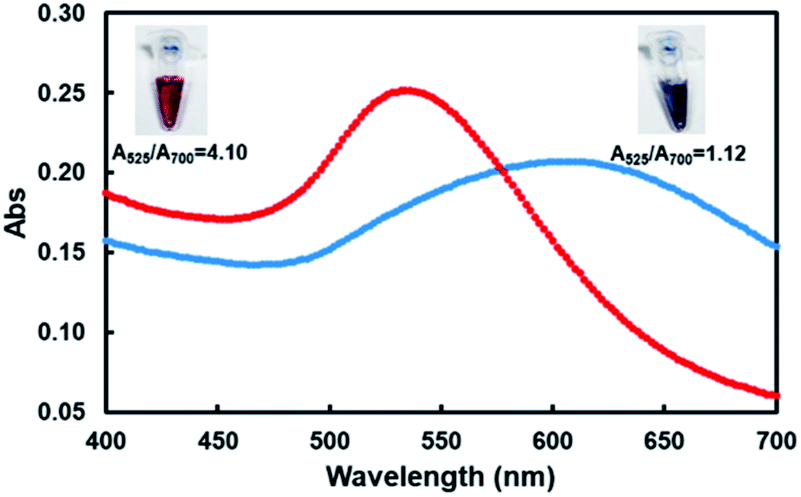 | ||
| Fig. 4 Visual and spectral detection of the product of indenting PCR using XF39 involving two X groups and R24 as the primers (red line). The product of this indenting PCR (lane 2 of Fig. 2) was treated with AuNps dispersion. For the purpose of comparison, the results with the product of conventional PCR using unmodified primers (bearing no single-stranded overhang) are shown by blue line. The inserts are the corresponding pictures. Almost the same results were obtained for indenting PCR using thymine dimers as molecular blockers (see Fig. S5†). | ||
The color of the solution remained red (the picture inserted in top-left). As shown by the UV-vis spectra (red line), the absorption maximum was located at around 535 nm. The ratio of absorbance at the plasmon peak (525 nm) to that at 700 nm (A525/A700) was 4.10.9 On the other hand, when the PCR product using non-modified primers which had no single-stranded overhang was mixed with AuNps, the red solution rapidly turned to blue upon the addition of MgCl2 (the right-top picture). The absorbance maximum was at 610 nm (blue line), and the A525/A700 ratio was 1.12. This color change of the solution occurred within minutes. Almost the same results were obtained when thymine dimers were used as molecular blockers of polymerase extension (Fig. S5†). The principle of the gene detection in Scheme 1 has been confirmed. It is well known that highly concentrated salts screen the electrostatic repulsion between AuNps and lead to their aggregation. Without any protecting factors, the AuNps dispersions thus immediately turn from red to blue.9,20,24 When the product of indenting PCR is added to the solution, however, its complementary overhangs form duplexes with the probes on the AuNps, and stabilize the dispersion through increased negative charges and steric repulsion. Furthermore, the PCR products cover the particles by thick polymeric barriers, inhibiting their mutual access.24 Notably, the selectivity of this assay detection is not only limited to the primers, DNA-labelled AuNps ensures specific detection due to the specificity of base pairing.32 We confirmed that the detection of PCR products is positive only when the labelled probe is fully complementary to the overhangs attached on the PCR products, which may be used for multiplex PCR by using several kinds of AuNPs (Fig. S6†). As comparing to other assays combining PCR technique and colloid Au technique, we were the first to detect PCR products directly and specifically with labelled AuNPs.9,33 Besides, the effect of pH was also checked, and found that our method works well at a pH range of 5–9 (Fig. S7†).
Applicability and sensitivity of this assay
In order to assess real applicability of the present method to on-site detection of Ebola virus, its detection sensitivity was evaluated by varying the copy number of the virus gene down to extremely small values (Fig. 5). The specimen was amplified by conventional PCR using non-modified primers and the indenting PCR using XF39 or T10F50. | ||
| Fig. 5 Sensitivity of the present visual detection of the gene from Ebola virus. The copy number of Ebola virus gene in the starting specimens is shown in the top. In (a), two azobenzene-bearing groups were introduced to one of the primer (XF39 in Table 1), which was combined with R24. In (b), T10F50 was irradiated for 28 h and used as the primer together with R24. In both cases, the specimen containing the designated copy number of Ebola gene was amplified by nested PCR (38 cycles) where the conventional PCR and the “indenting PCR” were simultaneously carried out in the specimen. The final products were added to AuNps dispersions without purification. The A525/A700 ratios as the measure of suppression of AuNps aggregation are presented above each sample tube. | ||
According to the strategy of nested PCR, the upstream primer (ElongF23) of the conventional PCR was bound to 60 nt upstream from the part amplified by the indenting PCR. The upstream primers ElongF23, XF39 and downstream R24 (or the combination of T10F50, irradiated for 28 h, with ElongF23 and R24) were simultaneously added to one vessel, and PCR (38 cycles) was carried out. Then, a portion of the final product of the PCR amplification was added (without purification) to the AuNps dispersion, and the color change was checked by our naked eyes. In a wide range of the number of Ebola virus gene (from 5 × 101 to 5 × 106), the presence of this gene was clearly detected in terms of the appearance of red color (Fig. 5a and b). In addition to this facile diagnosis by our naked eyes, further reconfirmation of the diagnostic result was possible, if necessary, with the use of UV-Vis spectroscopy. For the specimens containing the gene, the A525/A700 ratios were 2.0–4.0 depending on the copy number, and always much larger than the values for the non-gene samples (about 1.0), as shown in Fig. 5. For example, the two samples starting from 5 copies (the 2nd vessels from the left) showed the A525/A700 ratios around 1.4.
On the basis of these results, the diagnostic procedures were further optimized to allow the detection of minimum amount of designated gene. In Fig. 6, the initial specimen contained only 1 copy of Ebola gene (see Fig. S8† for details). It was amplified by the nested PCR (45 cycles) involving the conventional PCR and the indenting PCR. Except for the increased number of PCR cycles, the primers and the other conditions were exactly the same as descried for Fig. 5. In both off the molecular blockers, the difference in color from the non-virus sample was still more clear-cut, and a concrete conclusion could be easily obtained on the crucially important question: “whether this patient is infected by Ebola virus or not”. The sensitivity is similar to the previous reports, e.g. 1.9 × 104 PFU mL−1 of Ebola virus by one step qRT-PCR, 10 copies per cDNA reaction by nested RT-PCR.2,34 We also confirmed the applicability and selectivity of this method in detection of pathogenic bacteria (Fig. S9†). High sensitivity was obtained and even 1 CFU (a multicopy gene target was selected) could be detected without non-specific signals.
 | ||
| Fig. 6 Visual detection of 1 copy of Ebola virus gene by the present method. The primers used for indenting PCR are (a) XF39 involving two X residues and (b) T10F50, which was irradiated for 28 h and contained thymine dimers. The specimen was amplified by nested PCR (45 cycles) involving both the conventional PCR and the indenting PCR, and visualized by Au nanoparticles (see text for more details). For both of the molecular blockers ((a) and (b)), the diagnostic results starting from the sample containing about 1 copy of Ebola gene are shown in the right (see Fig. S8† for experimental details). The results of control experiment starting from non-gene specimen are in the left. Note that these two diagnostic results can be unanimously differentiated with the use of our naked eyes, although reconfirmation of the result using A525/A700 ratio is also possible as described above. | ||
Conclusions
In conclusion, a simple, visual, highly sensitive, and selective method to detect predetermined gene has been developed. The key is to incorporate azobenzene-bearing moieties or thymine dimers in tandem in one of the primers, and stop the polymerase extension at these sites. The resultant formation of single-stranded overhang at the end of PCR product is easily and straightforwardly visualized by using gold nanoparticle dispersions bearing an oligonucleotide as probe. Although the present analysis involves two PCR processes (conventional PCR and indenting PCR), no purification procedure is necessary throughout the analysis, and two sets of primer pair are simply added to the reaction vessels. Even 1 copy of a gene from Ebola virus can be successfully detected. The present method should be promising for the applications to on-site detection of versatile genes for Point of Care treatments.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China [31571937 to X. L.], and the Fundamental Research Funds for the Central Universities [201713050 to R. A.].Notes and references
- H. Cai, J. W. Parks, T. A. Wall, M. A. Stott, A. Stambaugh, K. Alfson, A. Griffiths, R. A. Mathies, R. Carrion, J. L. Patterson, A. R. Hawkins and H. Schmidt, Sci. Rep., 2015, 5, 14494 CrossRef CAS PubMed.
- J. T. Baca, V. Severns, D. Lovato, D. W. Branch and R. S. Larson, Sensors, 2015, 15, 8605–8614 CrossRef CAS PubMed.
- S. Petralia and S. Conoci, ACS Sens., 2017, 2, 876–891 CrossRef CAS PubMed.
- S. A. Deepak, K. R. Kottapalli, R. Rakwal, G. Oros, K. S. Rangappa, H. Iwahashi, Y. Masuo and G. K. Agrawal, Curr. Genomics, 2007, 8, 234–251 CrossRef CAS PubMed.
- E. M. Leroy, S. Baize, C. Y. Lu, J. B. McCormick, A. J. Georges, M. -C. Georges-Courbot, J. Lansoud-Soukate and S. P. Fisher-Hoch, J. Med. Virol., 2000, 60, 463–467 CrossRef CAS PubMed.
- D. Y. Ryazantsev, D. V. Voronina and S. K. Zavriev, Biochemistry, 2016, 81, 1754–1770 CAS.
- C. M. Niemeyer, M. Adler and R. Wacker, Trends Biotechnol., 2005, 23, 208–216 CrossRef CAS PubMed.
- C. Parolo and A. Merkoci, Chem. Soc. Rev., 2013, 42, 450–457 RSC.
- H. Deng, Y. Xu, Y. Liu, Z. Che, H. Guo, S. Shan, Y. Sun, X. Liu, K. Huang, X. Ma, Y. Wu and X. J. Liang, Anal. Chem., 2012, 84, 1253–1258 CrossRef CAS PubMed.
- C. Aslanidis and P. J. de Jong, Nucleic Acids Res., 1990, 18, 6069–6074 CrossRef CAS PubMed.
- J. Bitinaite, M. Rubino, K. H. Varma, I. Schildkraut, R. Vaisvila and R. Vaiskunaite, Nucleic Acids Res., 2007, 35, 1992–2002 CrossRef CAS PubMed.
- B. Zhu, G. Cai, E. Hall and G. J. Freeman, BioTechniques, 2007, 43, 354–359 CrossRef CAS PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, E63 CrossRef CAS PubMed.
- C. C. Chiou, J. D. Luo and T. L. Chen, Nat. Protoc., 2006, 1, 2604–2612 CrossRef CAS PubMed.
- H. Jans and Q. Huo, Chem. Soc. Rev., 2012, 41, 2849–2866 RSC.
- K. M. Abu-Salah, M. M. Zourob, F. Mouffouk, S. A. Alrokayan, M. A. Alaamery and A. A. Ansari, Sensors, 2015, 15, 14539–14568 CrossRef CAS PubMed.
- K. Sato, K. Hosokawa and M. Maeda, J. Am. Chem. Soc., 2003, 125, 8102–8103 CrossRef CAS PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS PubMed.
- X. M. Li, J. Luo, N. B. Zhang and Q. L. Wei, Anal. Chim. Acta, 2015, 881, 117–123 CrossRef CAS PubMed.
- C. Ma, W. Wang, A. Mulchandani and C. Shi, Anal. Biochem., 2014, 457, 19–23 CrossRef CAS PubMed.
- Z. Fang, W. Wu, X. Lu and L. Zeng, Biosens. Bioelectron., 2014, 56, 192–197 CrossRef CAS PubMed.
- Y. Chen, Z. Wang, Y. Liu, X. Wang, Y. Li, P. Ma, B. Gu and H. Li, Eur. J. Clin. Microbiol. Infect. Dis., 2018 DOI:10.1007/s10096-018-3230-x.
- B. Hu, J. Guo, Y. Xu, H. Wei, G. Zhao and Y. Guan, Anal. Bioanal. Chem., 2017, 409, 4819–4825 CrossRef CAS PubMed.
- W. Zhao, M. A. Brook and Y. Li, ChemBioChem, 2008, 9, 2363–2371 CrossRef CAS PubMed.
- W. Zhao, W. Chiuman, M. A. Brook and Y. Li, ChemBioChem, 2007, 8, 727–731 CrossRef CAS PubMed.
- S. J. Hurst, A. K. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2006, 78, 8313–8318 CrossRef CAS PubMed.
- K. Tanaka, H. Katada, N. Shigi, A. Kuzuya and M. Komiyama, ChemBioChem, 2008, 9, 2120–2126 CrossRef CAS PubMed.
- X. Liang, K. Fujioka and H. Asanuma, Chemistry, 2011, 17, 10388–10396 CrossRef CAS PubMed.
- L. B. Rubin, T. N. Menshonkova, N. A. Simukova and E. I. Budowsky, Photochem. Photobiol., 1981, 34, 339–344 CrossRef CAS PubMed.
- R. B. Setlow, W. l. Carrier and F. J. Bollum, Proc. Natl. Acad. Sci. U. S. A., 1965, 53, 1111–1118 CrossRef CAS.
- G. L. Chan, P. W. Doetsch and W. A. Haseltine, Biochemistry, 1985, 24, 5723–5728 CrossRef CAS PubMed.
- M. S. Verma, J. L. Rogowski, L. Jones and F. X. Gu, Biotechnol. Adv., 2015, 33, 666–680 CrossRef CAS PubMed.
- Y. L. Jung, C. Jung, H. Parab, T. Li and H. G. Park, Biosens. Bioelectron., 2010, 25, 1941–1946 CrossRef CAS PubMed.
- J. S. Towner, P. E. Rollin, D. G. Bausch, A. Sanchez, S. M. Crary, M. Vincent, W. F. Lee, C. F. Spiropoulou, T. G. Ksiazek, M. Lukwiya, F. Kaducu, R. Downing and S. T. Nichol, J. Virol., 2004, 78, 4330–4341 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting information consists of: a figure of the structures of the molecular blockers, a figure on the influence of the irradiation time on the formation of thymine dimmers, a figure on the influence of the length of continuous thymines in primer precursors on the formation of thymine dimers, a table containing the sequence information of the oligonucleotides used for primer extension experiments to study the blocking effect of thymine dimers, an image showing the effect of the concentration of Mg2+ on AuNps dispersion-induced visual detection of the product of indenting PCR, a figure showing the visual and spectral detection of the products of indenting PCR when thymine dimers were used as molecular blockers and the figure on of the analysis of PCR products amplified from specimen containing low copies of target gene. See DOI: 10.1039/c8ra01471j |
| This journal is © The Royal Society of Chemistry 2018 |