
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of bicyclic polycyclic aromatic hydrocarbons (PAHs) from the reaction of a phenyl radical with cis-3-penten-1-yne†
Mingrui Weia,
Tingting Zhang a,
Xianfeng Chenb,
Fuwu Yana,
Guanlun Guo*a and
Dongju Zhang*c
a,
Xianfeng Chenb,
Fuwu Yana,
Guanlun Guo*a and
Dongju Zhang*c
aHubei Key Laboratory of Advanced Technology for Automotive Components, Hubei Collaborative Innovation Center for Automotive Components Technology, Wuhan University of Technology, Wuhan 430070, PR China. E-mail: glguo@whut.edu.cn
bSchool of Resources and Environmental Engineering, Wuhan University of Technology, Wuhan 430070, China
cKey Lab of Colloid and Interface Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250000, PR China
First published on 10th April 2018
Abstract
The formation of polycyclic aromatic hydrocarbons (PAHs) on the C11H11 potential energy surface involved in the reactions of a phenyl radical (C6H5) with cis-3-penten-1-yne (cis-C1H![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C2–C3H
C2–C3H![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C4H–C5H3, referred to as C5H6) and its three radicals (CHC–ĊCH–CH3, CHC–CHĊ–CH3, and cis-CHC–CHCH–ĊH2, referred to as the C3-, C4-, and C5-radicals with the same chemical components, C5H5) assisted by H atoms is investigated by performing combined density functional theory (DFT) and ab initio calculations. Five potential pathways for the formation of PAHs have been explored in detail: Pathways I–II correspond to the reaction of C6H5 with C5H6 at the C1 and C2 position, and Pathways III–V involve the reaction of C6H5 with the C3-, C4-, and C5-radicals with the assistance of H atoms. The initial association of C6H5 with C5H6 or C5H5 is found to be highly exothermic with only minor barriers (1.4–7.1 kcal mol−1), which provides a large driving force for the formation of PAHs. The hydrogen atom is beneficial for the ring enlargement and ring formation processes. The present calculations predict 9 potential PAHs, six (CS6, CS10, CS13, CS26, CS28 and CS29) of which are indicated to be energetically more favorable along Pathways I, III, IV and V at low temperature. The calculated barriers for the formation of these PAHs are around 19.2–38.0 kcal mol−1. All PAHs products could be formed at flame temperature, for the medium barriers are easily overcome in various flame conditions. The theoretical results supplement the PAH formation pathway and provide help to understand PAH growth mechanism.
C4H–C5H3, referred to as C5H6) and its three radicals (CHC–ĊCH–CH3, CHC–CHĊ–CH3, and cis-CHC–CHCH–ĊH2, referred to as the C3-, C4-, and C5-radicals with the same chemical components, C5H5) assisted by H atoms is investigated by performing combined density functional theory (DFT) and ab initio calculations. Five potential pathways for the formation of PAHs have been explored in detail: Pathways I–II correspond to the reaction of C6H5 with C5H6 at the C1 and C2 position, and Pathways III–V involve the reaction of C6H5 with the C3-, C4-, and C5-radicals with the assistance of H atoms. The initial association of C6H5 with C5H6 or C5H5 is found to be highly exothermic with only minor barriers (1.4–7.1 kcal mol−1), which provides a large driving force for the formation of PAHs. The hydrogen atom is beneficial for the ring enlargement and ring formation processes. The present calculations predict 9 potential PAHs, six (CS6, CS10, CS13, CS26, CS28 and CS29) of which are indicated to be energetically more favorable along Pathways I, III, IV and V at low temperature. The calculated barriers for the formation of these PAHs are around 19.2–38.0 kcal mol−1. All PAHs products could be formed at flame temperature, for the medium barriers are easily overcome in various flame conditions. The theoretical results supplement the PAH formation pathway and provide help to understand PAH growth mechanism.
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs) are commonly believed to mainly come from the incomplete combustion of fuels. Due to their toxicity and carcinogenic properties,1,2 the study of the formation mechanism of PAHs has become a major subject in the fields of combustion and environmental science during the past decades.3 The most widely accepted theory for PAH formation and growth involves a hydrogen-abstraction-acetylene-addition (HACA) mechanism,4–6 which has been extensively studied by both experimentalists and theorists and was popularly used in combustion models.7–12 However, the HACA mechanism was found to underestimate the concentrations of PAHs and soot when compared with the experimental results.12,13 Further experimentalists found that many non-acetylene type organic compounds, such as CH3, C3H3, C3H4, C3H6, C4H2, C4H4, C4H6 and C5H8 (ref. 14 and15) also contributed to PAH growth in various flame configurations. Therefore, the PAH formation from non-acetylene compounds has attracted remarkable attention.16–26 Furthermore, the introduction of reaction mechanism of odd-carbon chemistries as additions, especially C3 and C5 species, into HACA mechanism were found to reproduce the synergistic effect of some mixing fuels on PAH and soot formations.27–35 Hence, the roles of C3 and cyclo-C5 species as additions on the PAH growth have be studied experimentally and theoretically.17–19,36–40 Different from cyclo-C5 species, the linear C5 species draw little attentions on the PAHs formation mechanism studies, except that reactions of C5H3 and C5H5 with CH3 have been postulated as possible routes of benzene formation.11–13 And, isoprene (C5H8), the methyl-substituted 1,3-butadiene, was found to react with phenyl radical barrier-lessly.26,41 However, Hansen et al.42 identified the isomers of linear C5 chemicals, and found that cis-3-penten-1-yne (CHC–CHCH–CH3) was the most stable linear C5H6, and CHC–CHCH–CH2 radical the most stable linear C5H5 radical. Structurally, CHC–CHCH–CH3 is the methyl-substituted product of vinylacetylene (CHCCHCH2), which can form naphthalene via the bimolecular reactions with para-tolyl and phenyl radicals without entrance barriers under single collision conditions.21–23 Thus, it is meaningful to investigate the reaction of phenyl radical with CHCCHCHCH3.
The main objective of this work is to explore the formation of potential PAHs within 4-, 5-, 6- and 7-membered rings on C11H11 potential energy surface (PES) from phenyl radical (C6H5) with cis-3-penten-1-yne and its radicals. These 4-, 5- and 7-membered PAHs were all detected from vehicle emissions43,44 and confirmed as major contributors to soot formation in the post-flame region.45–47 By performing combined density functional theory (DFT) and ab initio calculations, we discuss the influences of reactive sites and methyl group of CHCCHCHCH3 on the formation of bicyclic PAHs, and the roles of hydrogen atoms in ring formation and ring enlargement steps.
2. Methods
The molecular structures involved in the reactions under study were optimized on C11H11 PES by using the hybrid B3LYP functional with the 6-311++G(d,p) basis set.48–50 The vibrational frequency calculations were performed at the same level to identify the optimized structures as local minimum or first-point saddle points and to provide their zero-point vibrational energies (ZPE). The intrinsic reaction coordinate calculations were implemented to ensure that the transition states connect to the relevant reactants and products correctly. The final single-point energies of all species were gained using the G3(MP2,CC) method,23,25 which is expected to generate relative energies of various species within the accuracy of 1–2 kcal mol−1,51 and extensively used to study PAH growth mechanism.8,9,18,21–23 The majority of the intermediate species and transition structures found in this study were either closed shell singlets or open shell doubles. The diradical species were found by using triplet spin multiplicity, and the calculations of diradical species contain negligible spin contamination. A high spin contamination can affect the geometry and energy of the molecule.52 As a check for the presence of spin contamination, ab initio calculations give the expectation value of the total spin 〈S2〉. If there is no spin contamination, 〈S2〉 should equal s(s + 1), where s equals 1/2 times the number of unpaired electrons. One rule of thumb, which was derived from experience with organic molecule calculations, is that the spin contamination is negligible if the value of 〈S2〉 differs from s(s + 1) by less than 10%. In addition, spin contamination has little effect on DFT, CC and CI calculations.52,53 All 〈S2〉 values of chemical species or transition states with 2 unpaired electrons in this study were shown in ESI.† Asterisks were added to energies corresponding to diradical species with spin contamination. The G3(MP2,CC) energies are calculated according to the following equation:| E[G3(MP2,CC)] = E[CCSD/6-311++(d,p)] + ΔE(MP2) + ΔE(SO) + ΔE(HLC) + E(ZPE) | (1) |
3. Results and discussion
cis-3-Penten-1-yne with one C–C triple bond, one C–C double bond, and two C–C single bonds, contains five chemically unequivalent carbon atoms, marked as C1, C2, C3, C4 and C5 (Fig. 1), and its reaction with phenyl radical may initially occur at any one of these carbon atoms, corresponding to five possible pathways shown in Fig. 2–6. It is known that a vinyl-type group can conjugate to the –CCH group to form a resonantly stabilized free radical (RSFR) intermediate, which was confirmed to play important role in PAHs formation.26,55 Thus, phenyl radical (C6H5), a typical vinyl-type group, can directly conjugate to C1 or C2 position of cis-3-penten-1-yne via Pathway I or Pathway II. Alternatively, it can also react with three C5H5 radicals of cis-3-penten-1-yne with the single electron respectively located at C3, C4 and C5 positions, via Pathways III–V. All pathways were on C11H11 PES, and only reactions lead to the PAHs products were considered in this study. Fig. 1 shows the formation of these three radicals, denoted as CSa, CSb, and CSc from cis-3-penten-1-yne via the H-abstraction reaction by an external H atom. It is found that the barriers involved in the H-abstraction reactions are 12.9, 14.6 and 7.6 kcal mol−1, respectively, implying that CSc is easier to be produced than CSa and CSb.
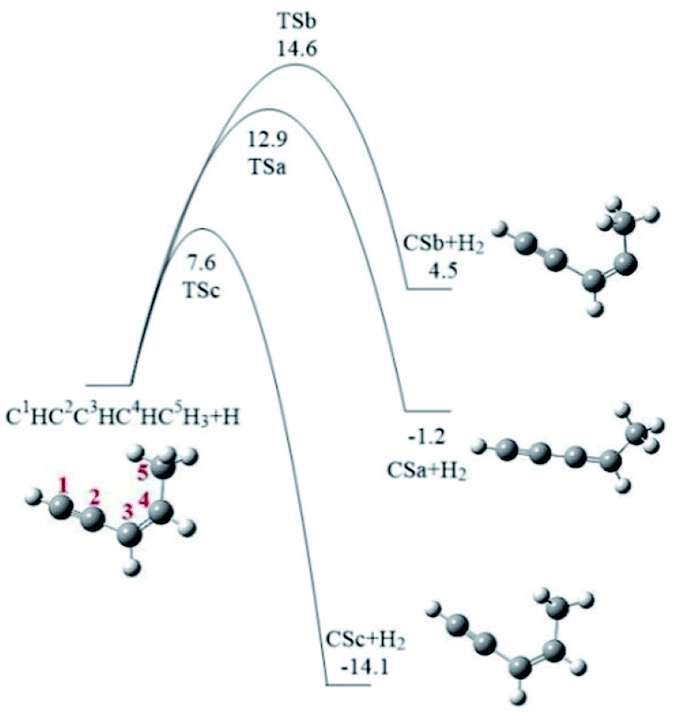 | ||
| Fig. 1 Relative energy surfaces for the formation of the three C5H5 radicals of cis-3-penten-1-yne via the H-abstraction reactions at the C3, C4 and C5 positions by an external H atom. Relative energies are in kcal mol−1. | ||
 | ||
| Fig. 2 Pathway I: the reaction of phenyl radical with cis-3-penten-1-yne at the C1 position. | ||
 | ||
| Fig. 3 Pathway II: the reaction of phenyl radical with cis-3-penten-1-yne at the C2 position. | ||
 | ||
| Fig. 4 Pathway III: the reaction of phenyl radical with the C3-radical of cis-3-penten-1-yne. | ||
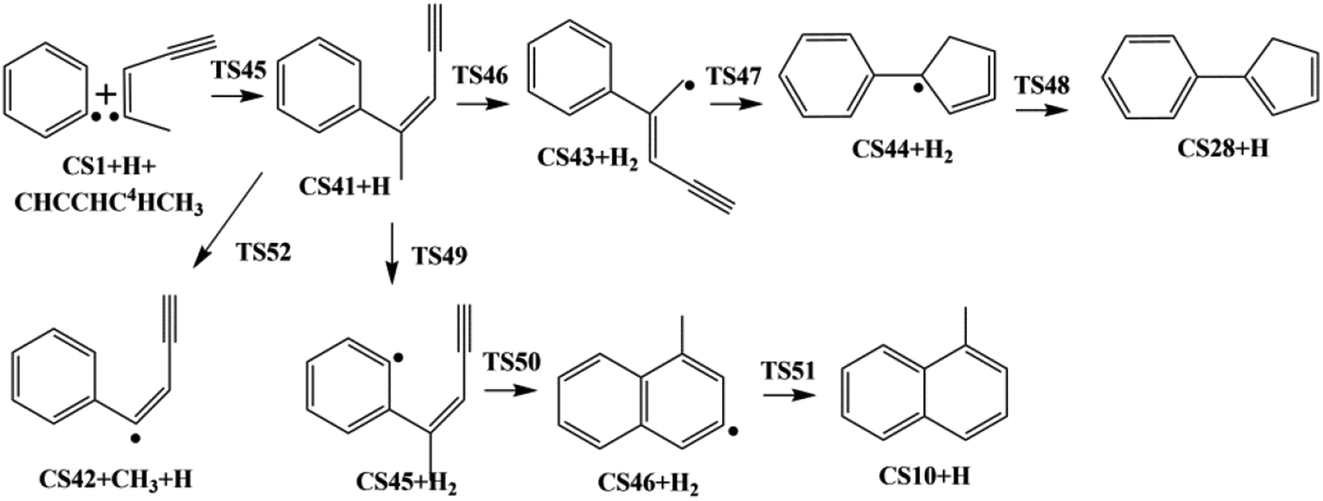 | ||
| Fig. 5 Pathway IV: the reaction of phenyl radical with the C4-radical of cis-3-penten-1-yne. | ||
 | ||
| Fig. 6 Pathway V: the reaction of phenyl radical with the C5-radical of cis-3-penten-1-yne. | ||
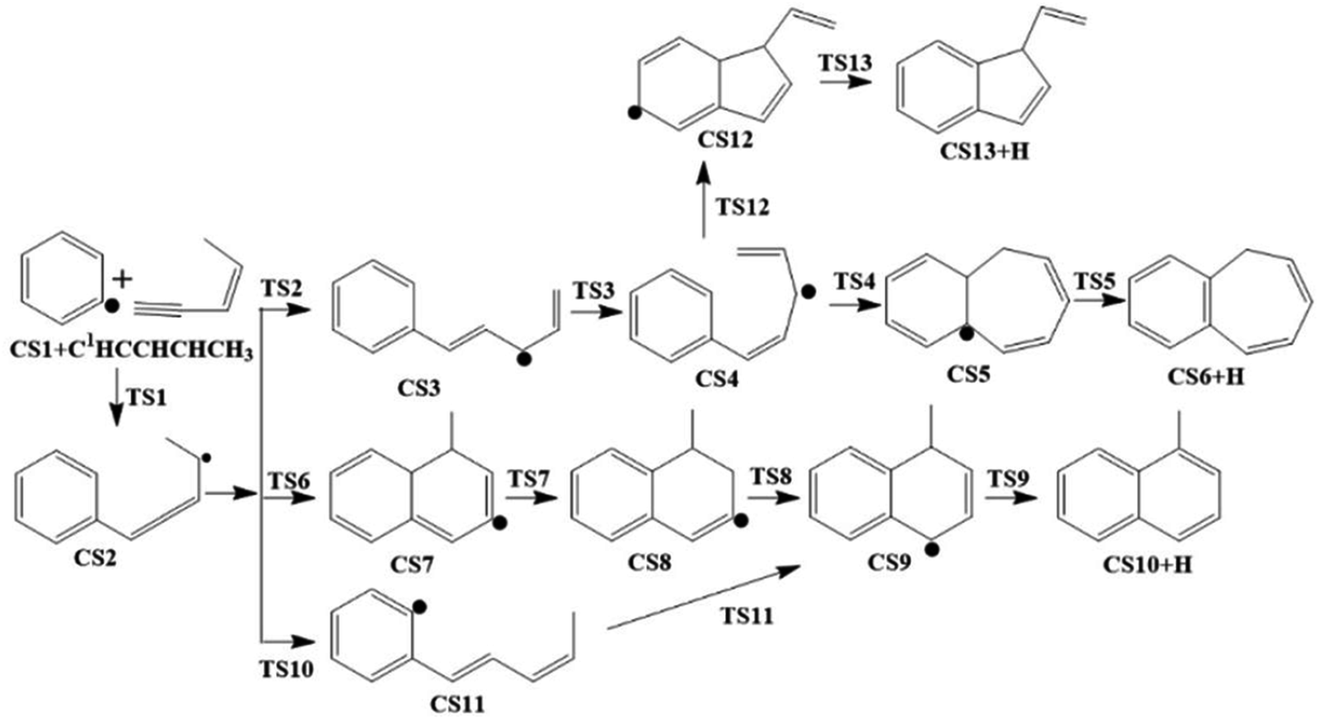
The present work studied five possible pathways, as schematically shown in Fig. 2–6. The resulting PAHs include benzocycloheptatriene (CS6), 1-methylnaphthalene (CS10), 1-vinyl-1H-indene (CS13), 3a,7b-dihydro-1H-cyclopenta[3,4]cyclobuta[1,2]benzene (CS19), 1-methylene-1,4-dihydronaphthalene (CS26), cyclopenta-1,3-dien-1-ylbenzene (CS28), cyclopenta-1,4-dien-1-ylbenzene (CS29), (Z)-7-ethylidene-8-methylenebicyclo[4.2.0]octa-1(6),2,4-triene (CS34), and 7H-Benzo[7]annulene (CS52). The following discussion shows the mechanism details for the formation of these PAHs.
3.1. Pathway I
As shown in Fig. 2 and 7, there are four potential channels for the reaction of C6H5 with C1HCCHCHCH3 to the C1 position, resulting in three PAHs, CS6, CS10, and CS13. The initial association of C6H5 to the C1 position of C1HCCHCHCH3 results in the formation of CS2. This process is found to be a thermodynamically and kinetically favorable process, highly exothermic (51.7 kcal mol−1) with a very small barrier (3.0 kcal mol−1), providing a large driving force for the subsequent formation of PAHs. The result is comparable with that for the reaction of C6H5 with C2H2 with a barrier of ∼6 kcal mol−1 and a reaction energy of 39 kcal mol−1.56 Once formed, CS2 can evolves into PAHs via different channels. The blue one, CS1 + C5H6 → CS2 → CS3 → CS4 → CS5 → CS6, involves H-migration, cis–trans conversion, ring-closure, and H-dissociation steps, leading to the formation of benzocycloheptatriene (CS6). The barriers of these four elementary steps are 34.3, 11.9, 22.5 and 34.6 kcal mol−1, respectively. Note that CS4, instead of evolving into CS6 along the bule channel, can evolve into a relative more stable PAH, CS13 alone the green sub-pathway in Fig. 7, CS4 → CS12 → CS13, via ring-closure and H-dissociation steps. The ring-closure reaction occurs at C3 position with a barrier of 34.9 kcal mol−1, leading to CS12, a 5-membered bicyclo-PAH, which then dissociates into CS13 + H with a barrier of 31.0 kcal mol−1.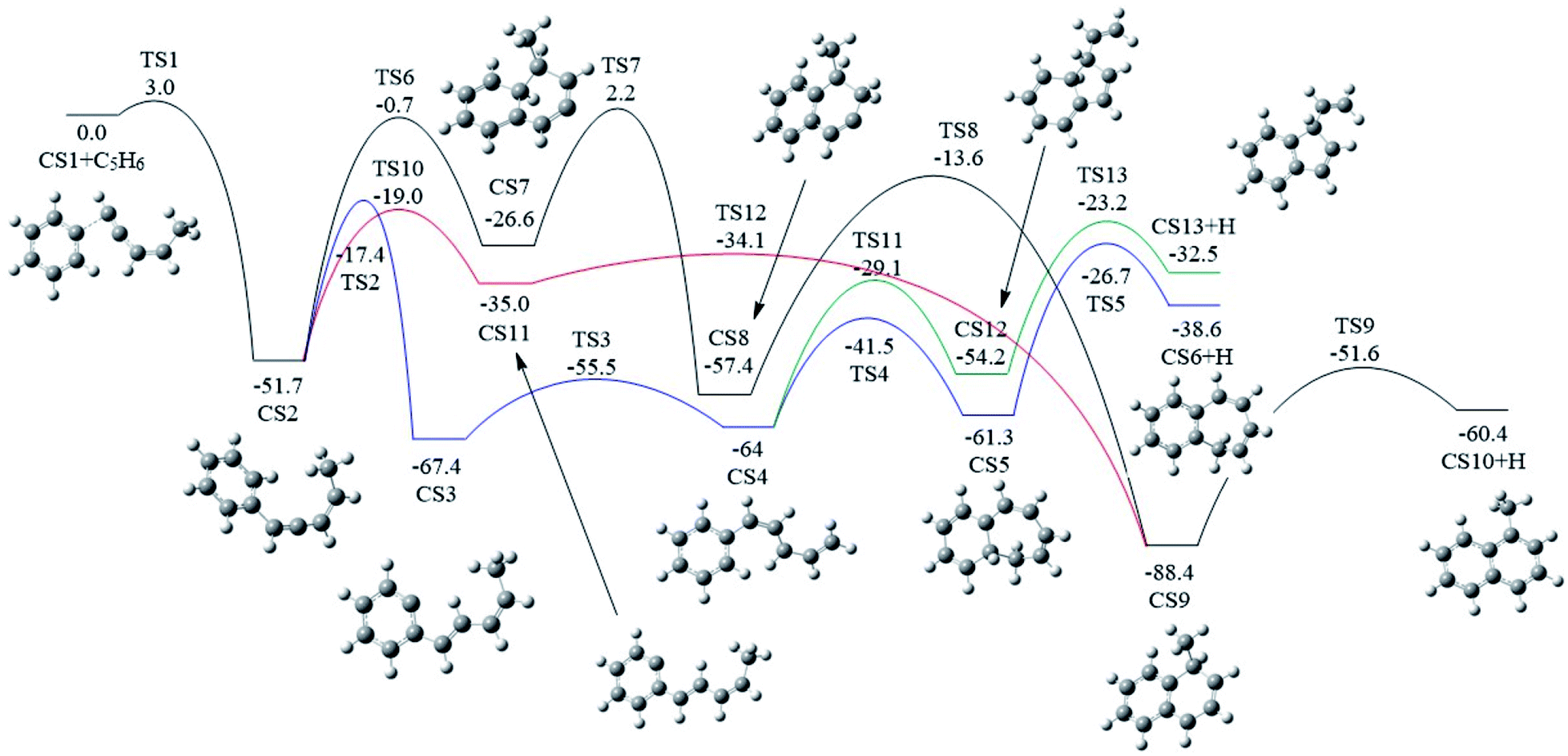 | ||
| Fig. 7 Calculated potential energy surface of Pathway I started from the association of C6H5 to the C1 position of C1HC2C3HC4HC5H3. Relative energies are in kcal mol−1. | ||
The black channel in Fig. 7, CS2 → CS7 → CS8 → CS9 → CS10, shows the formation of 1-methylnaphthalene (CS10), involving ring closure, twice H-migration, and H-dissociation steps via TS6-TS9. The barriers of these four steps are calculated to be 51.0, 28.8, 43.8 and 36.8 kcal mol−1, respectively. Clearly, such a channel is energetically very unfavorable for the formation of CS10. Alternatively, a relatively more viable channel has been identified, as shown by the red line in Fig. 7, i.e. CS2 → CS11 → CS9 → CS10. Along this channel, CS2 is converted to CS10, undergoing H-migration, ring closure, and H-dissociation steps. The barriers involved in these three steps are 32.7, 0.9 and 36.8 kcal mol−1, respectively.
From the results above, it is found that the reaction of phenyl radical with cis-3-penten-1-yne at the C1 position can result in the formation of three PAHs, CS6, CS10, and CS13, and the highest barriers involved are 34.6, 36.8 and 34.9 kcal mol−1, respectively. These almost the same barriers indicate the formations of these three PAHs are competing each other.
3.2. Pathway II
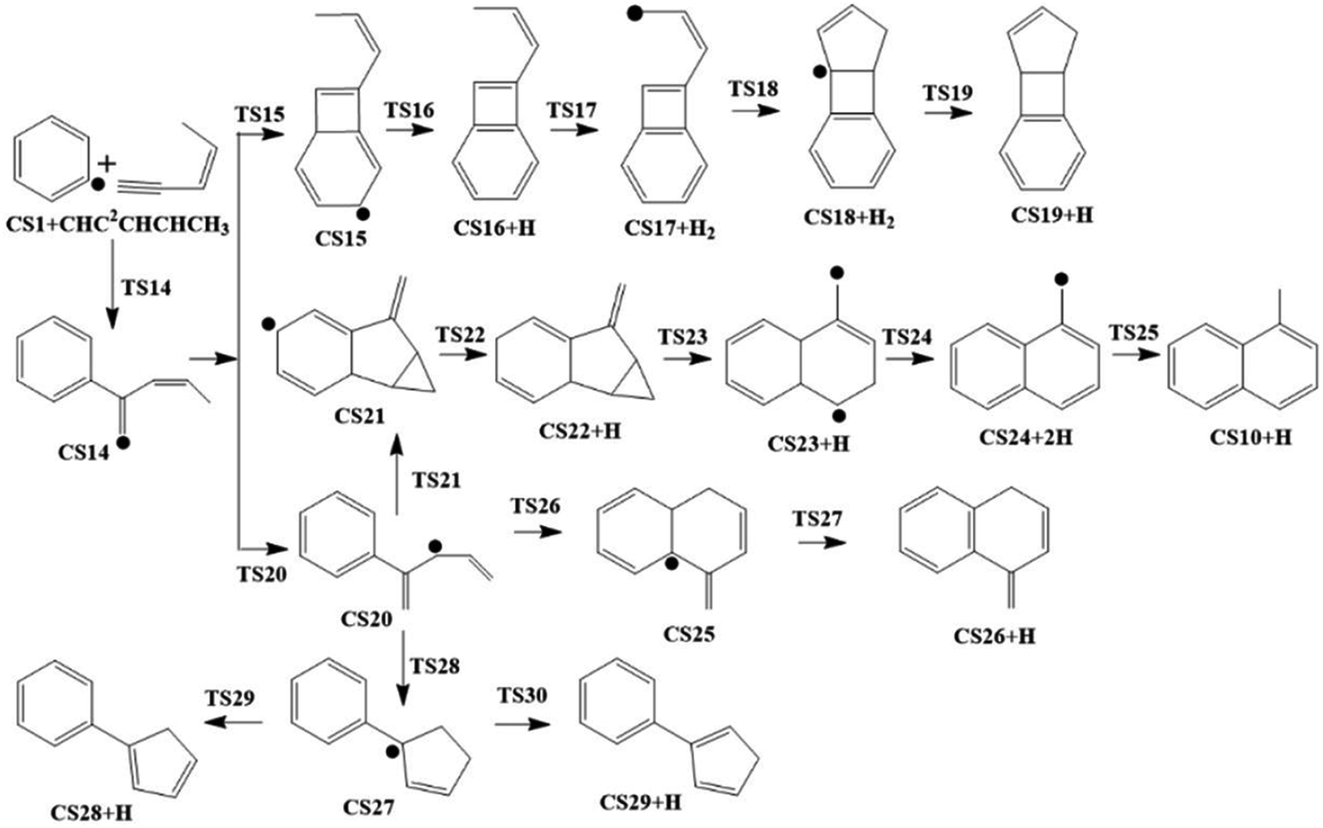
As shown in Fig. 2, Pathway II refers to the reaction of phenyl radical with cis-3-penten-1-yne at the C2 position. The calculated potential energy surface along this pathway is depicted in Fig. 8. Similar to the situation in Pathway I, the initial association of C5H6 with cis-3-penten-1-yne at the C2 position is also a thermodynamically and kinetically favorable process, exothermic by 33.3 kcal mol−1 with a barrier of 7.1 kcal mol−1, leading to the adduct CS14. This adduct with the radical site at the C1 position, can evolves into five different PAHs, CS10, CS19, CS26, CS28 and CS29, along five possible channels. The diradical species in this pathway were found by using triplet spin multiplicity, and hence the calculations of diradical species contain spin-contamination.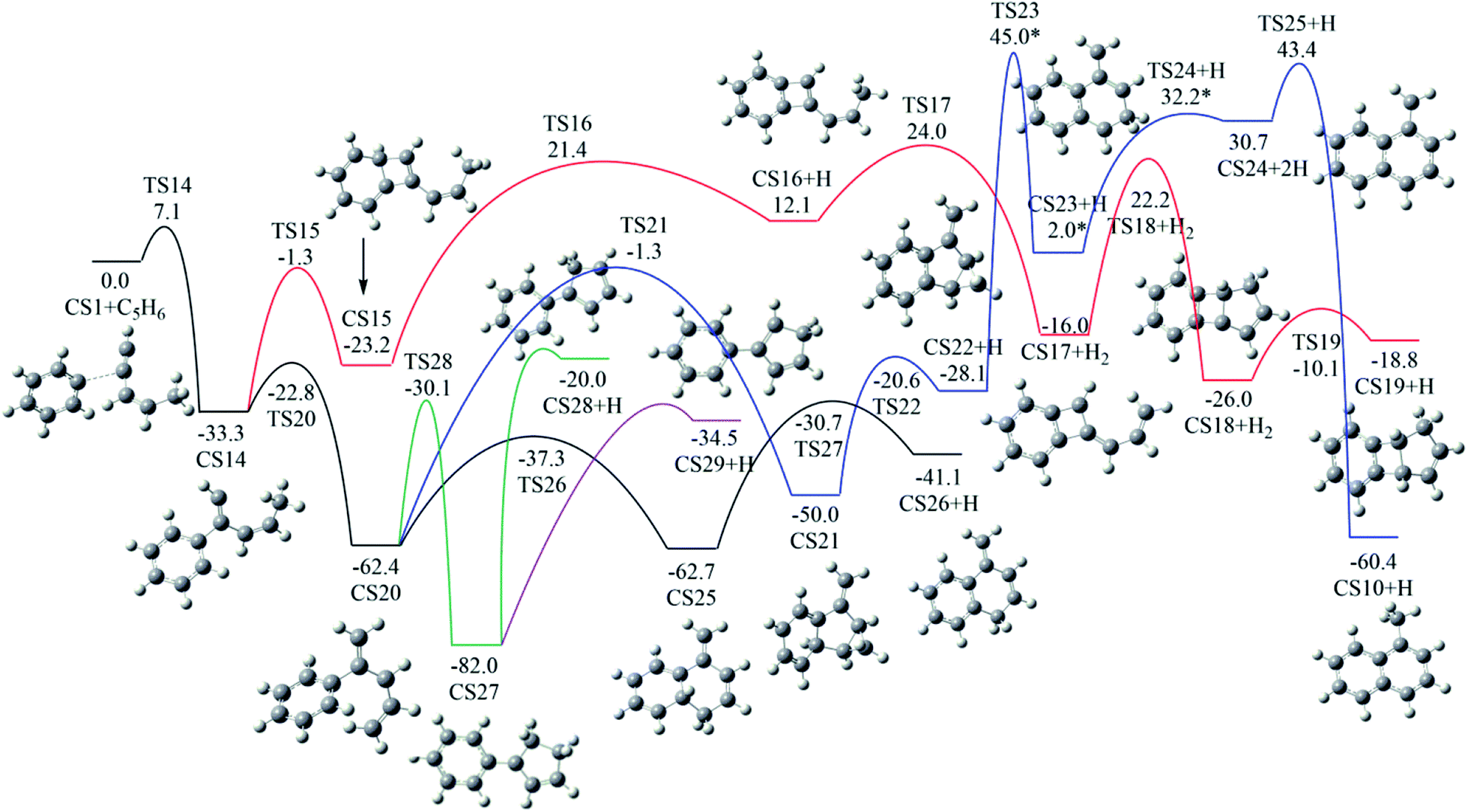 | ||
| Fig. 8 Calculated potential energy surface of Pathway II started from the association of C6H5 to the C2 position of C1HC2C3HC4HC5H3. Relative energies are in kcal mol−1. | ||
The red channel gives CS19 via five elementary steps, involving transition states TS15-TS19. The radical site at C1 in CS14 conjugates to the benzene ring via TS15, forming CS15 (a 4-membered PAH), with a barrier of 32.0 kcal mol−1. Subsequently, a H-dissociation process occurs via TS16 with a barrier of 44.6 kcal mol−1, followed by a H-abstract reaction via TS17 with a barrier of 11.9 kcal mol−1, leading to CS17 with the radical site at C5 position. Then the ring closure reaction occurs via TS18 with a barrier of 38.2 kcal mol−1, forming the 5-membered ring intermediate CS18. Finally, CS19 is formed via the reaction of CS18 with H2 with a barrier of 15.9 kcal mol−1. The overall energy barrier involved in this channel is 44.6 kcal mol−1.
The black channel in Fig. 8 corresponds to the formation of CS26, undergoing H-migration, ring closure, and H-dissociation steps. The H-migration from C5 to C1 proceeds via TS20 with a barrier of 10.5 kcal mol−1, leading to intermediate CS20. The ring closure step produces the bicyclic PAH radical, CS25, and need to overcome a barrier of 25.1 kcal mol−1. The final H-dissociation reaction yields CS26, overcoming a barrier of 32.0 kcal mol−1, which is also the highest barrier along this channel.
Other three potential channels of the evolvement of CS20 was also explored, as shown by blue, green, and purple lines. The blue one corresponds the formation of CS10, which undergoes ring closure, H-abstraction, ring opening and H-addition steps. The overall barrier is calculated to be as high as 107.4 kcal mol−1 (the energy difference between TS23 and CS20), which is much higher than that involved in Pathway I. This result implies that CS10 is a less possible product from the reaction of phenyl radical with cis-3-penten-1-yne at the C2 position.
The green and purple channels show the formations of two PAHs (CS28 and CS29) that are structurally considered as “cyclopenta-benzene” (biphenyl with one benzene ring replaced by a cyclopenta ring). The involved overall barriers are calculated to be 62.4 and 57.5 kcal mol−1, respectively.
3.3. Pathway III
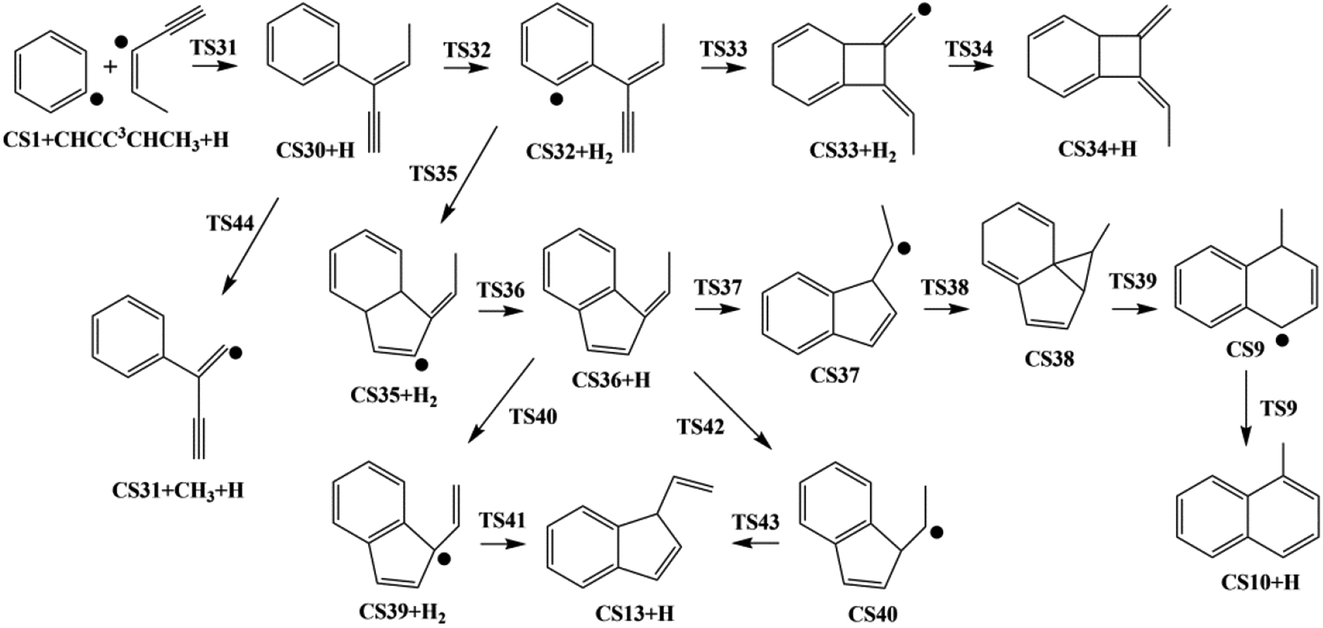
As shown in Fig. 4, Pathway III refers to the reaction of phenyl radical with the C3-radical of cis-3-penten-1-yne and H atom. The calculated potential energy surface is depicted in Fig. 9. | ||
| Fig. 9 Calculated potential energy surface of Pathway III started with the reaction of C6H5 to the C3 position of C1HC2C3C4HC5H3. Relative energies are in kcal mol−1. | ||
Initially, CS30 is formed via a low barrier of 2.4 kcal mol−1 by the direct association of phenyl radical and the C3-radical of cis-3-penten-1-yne, releasing an energy of 107.3 kcal mol−1. Subsequently, CS30 can evolves into different PAHs, CS10, CS13, and CS34. The blue channel in Fig. 9 shows the mechanism for the formation of CS34. The reaction occurs via H-abstraction, ring closure, and H-addition steps (CS30 → CS32 → CS33 → CS34). The H-abstraction of CS30 leading to intermediate CS32 need to overcome an energy barrier of 19.2 kcal mol−1, which is also the highest energy barrier along this channel.
As shown by green, red and purple lines in Fig. 9, the formations of CS10 and CS13 involve a common, stable intermediate, CS36, which can be formed via a ring closure and a H-addition steps (CS32 → CS35 → CS36) with an overall barrier of 15.5 kcal mol−1. Jasper and Hansen suggested that the ring enlargement from fulvene to benzene could be assisted by the presence of hydrogen atom.57 Fig. 9 shows the calculated results for the H-assisted transformation from CS36 to CS10 by H-addition, H-migration and H-loss steps (CS36 → CS37 → CS38 → CS9 → CS10). This process involves barrier of 36.9 kcal mol−1, which is also the overall barrier.
Instead of evolving into CS10, the intermediate CS36 can convert to CS13 undergoing intermediate CS39 with an overall barrier of 38.1 kcal mol−1 or undergoing intermediate CS40 with an overall of 38.1 kcal mol−1.
3.4. Pathway IV
The reaction of phenyl radical with the C4-radical of cis-3-penten-1-yne is referred as Pathway IV (Fig. 5), which can results in the formation of two PAHs, CS10 and CS28. As indicated by Fig. 10, potential channel with blue lines is open for the formation of CS10 from CS41, which is formed from the initial adduct of phenyl radical with the C4-radical of cis-3-penten-1-yne with very low barrier of 1.4 kcal mol−1, releasing an energy of 115.2 kcal mol−1. Along this channel, CS41 first evolves into CS45 via a H-abstraction step. This process with a barrier of 23.4 kcal mol−1 is indentified as the rate-determining step. Subsequently, the ring closure reaction leads to intermediate CS46, which undergoes a H-abstraction reaction via TS51, producing PAH, CS10.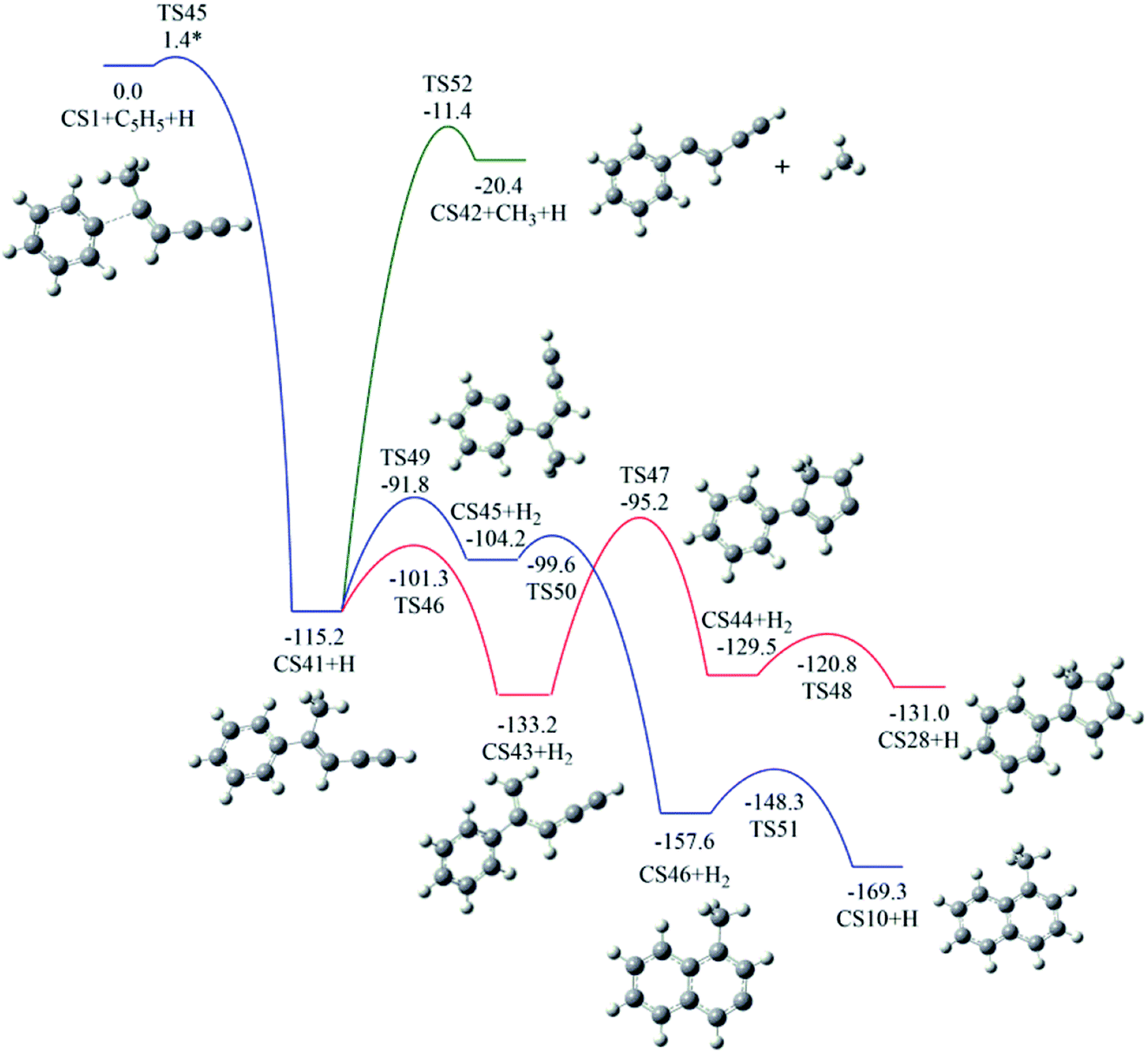 | ||
| Fig. 10 Calculated potential energy surface of Pathway IV started with the reaction of C6H5 to the C4 position of C1HC2C3HC4·C5H3. Relative energies are in kcal mol−1. | ||
Another possible PAH product from the reaction of phenyl radical with the C4-radical is CS28, a “cyclopenta-benzene”-like structure. Instead of evolving into CS10, CS41 may perform H-abstraction, ring closure and H-addition reactions, leading to CS28, with a barrier of 38.0 kcal mol−1.
3.5. Pathway V
As shown in Fig. 6, Pathway V refers to the reaction of phenyl radical with the C5-radical of cis-3-penten-1-yne. The present calculations locate six potential channels, leading to two seven-membered PAHs (CS6 and CS52) and two six-membered PAHs, (CS10 and CS26).As shown in Fig. 11, the initial association of phenyl radical with the C5-radical leads to the formation of CS47 with barrier of 5.4 kcal mol−1. Once formed, CS47 can evolve into CS52 along the blue channel via the H-addition, H-transfer, H-abstraction, ring closure and H-addition steps. The highest barrier involved is calculated to be 50.8 kcal mol−1.
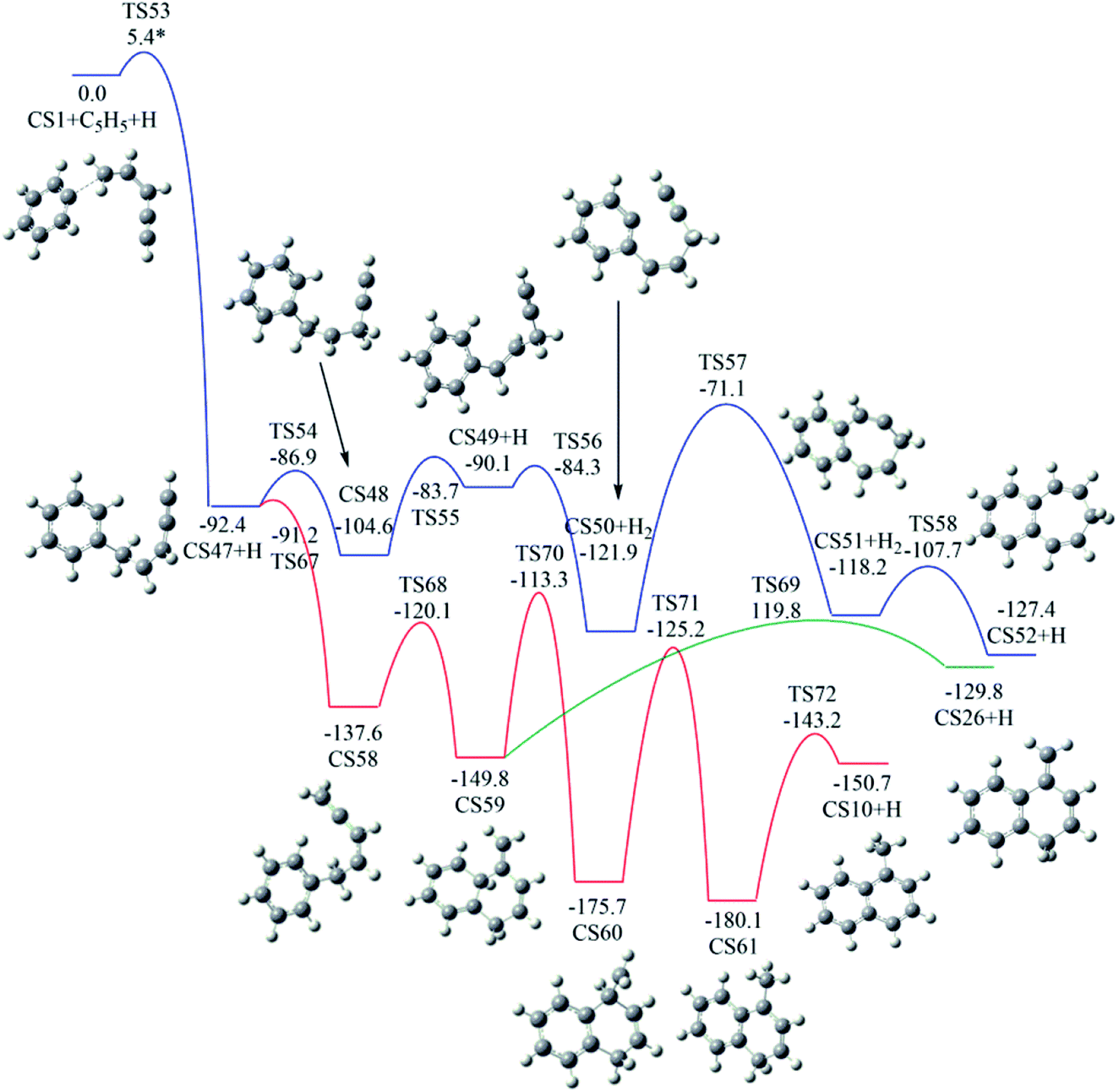 | ||
| Fig. 11 Calculated potential energy surface of Pathway V started with the reaction of C6H5 to the C5 position of C1HC2C3HC4HC5H2·. Relative energies are in kcal mol−1. | ||
In addition, CS47 can be converted to CS10 along the red channel. This channel consists of H-addition, ring closure, H-migrations and H-dissociation steps (CS47 + H → CS58 → CS59 → CS60 → CS61 → CS10 + H). From the calculated potential energy surface (Fig. 11), rate-determining step, CS60 → CS61, involves energy barrier of 50.5 kcal mol−1.
Fig. 12 shows calculated potential channels for the formations of CS6 and CS26 along Pathway V. The blue channel in Fig. 12 corresponds to the formation of CS26. This channel involves H-abstraction, ring closure and H-addition steps. The ring closure is identified as the rate-determining step with a barrier of 24.2 kcal mol−1. In Fig. 11, the intermediate CS59 can convert to CS26 with an overall barrier of 30.0 kcal mol−1. From the calculated potential energy surface (Fig. 11 and 12), it is clear that the channel in Fig. 12 is energetically more favorable.
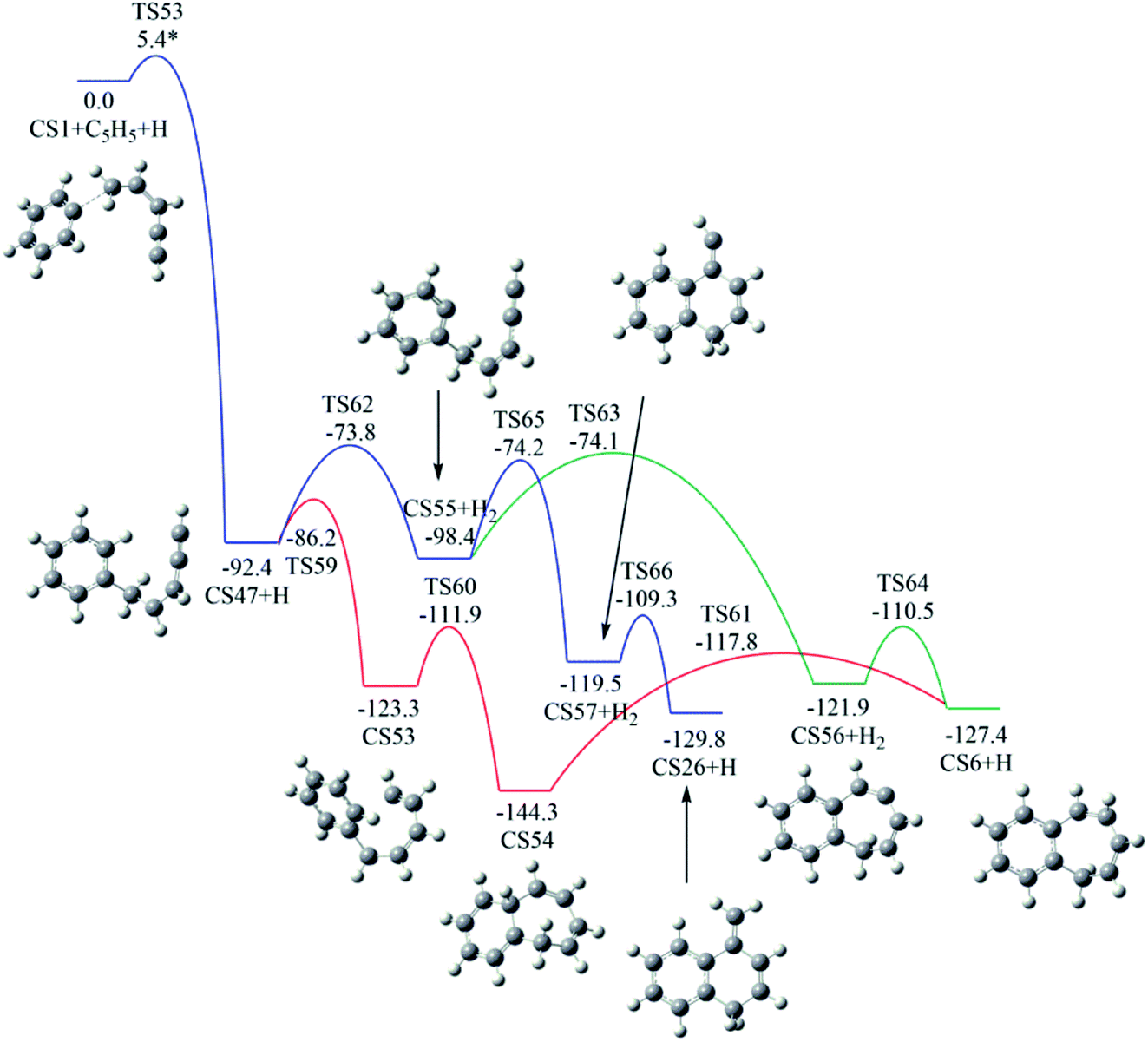 | ||
| Fig. 12 Calculated potential energy surface of Pathway V started with the reaction of C6H5 to the C5 position of C1HC2C3HC4HC5H2·. Relative energies are in kcal mol−1. | ||
As indicated by red and green lines in Fig. 12, CS47 is converted to CS6 undergoing intermediates CS53 and CS54 with an overall barrier of 26.5 kcal mol−1 or undergoing intermediate CS55 and CS56 with an overall of 24.3 kcal mol−1. The minima energy pathway forms a bicyclic PAH, CS6 which is formed via the H-abstraction, ring closure and H-addition steps. The ring closure step is indentified as the rate-determining step with a barrier of 24.3 kcal mol−1. The calculated overall barriers for all final products in Pathways I–V were listed in Table 1 in kcal mol−1.
| Products | Pathway I (kcal mol−1) | Pathway II (kcal mol−1) | Pathway III (kcal mol−1) | Pathway IV (kcal mol−1) | Pathway V (kcal mol−1) |
|---|---|---|---|---|---|
| CS6 | 34.6 | — | — | — | 24.3 |
| CS10 | 36.8 | 107.4 | 36.9 | 23.4 | 50.5 |
| CS13 | 34.9 | — | 38.1 | — | — |
| CS19 | — | 44.6 | — | — | — |
| CS26 | — | 32.0 | — | — | 24.2 |
| CS28 | — | 62.4 | — | 38.0 | — |
| CS29 | — | 57.5 | — | — | — |
| CS34 | — | — | 19.2 | — | — |
| CS52 | — | — | — | — | 50.8 |
4. Conclusions
In this work, the reaction mechanism of PAH growth to dual-ring structures on C11H11 surface initiated by C6H5 with linear C5 species (cis-3-penten-1-yne) were investigated using DFT and ab initio methods. Five bicyclic PAHs formation pathways are detailedly explored include reactions of phenyl radical (C6H5) with cis-3-penten-1-yne (cis-C1HC2–C3HC4H–C5H3, referred to C5H6) and its three radicals (CHC–ĊCH–CH3, CHC–CHĊ–CH3, and cis-CHC–CHCH–ĊH2, referred to the C3-, C4-, and C5-radicals with the same chemical components, C5H5). And 4-, 5-, 6- and 7- membered PAHs (referred to the species of CS6, CS10, CS13, CS19, CS26, CS28, CS29, CS34 and CS52) were designed as final products.
The results show that the initial combination of C6H5 with C5H6 or C5H5 is found to be highly exothermic with only minor barriers (1.4–7.1 kcal mol−1) in Pathways I–V, which provides a large driving force for PAH formation. The hydrogen atom is beneficial for the ring enlargement and ring formation processes.
The calculated Pathways I–V predicate 9 potential PAHs include 4-, 5- and 7-membered rings. Among them, CS6, CS10, CS13, CS26 and CS28 could be produced from two or more pathways, CS19, CS29, CS34 and CS52 from only one pathway. At low temperature, CS6 and CS26 are more energetically favorable along Pathway V, CS10 and CS28 along Pathway IV, CS13 along Pathway I and CS34 along Pathway III. The calculated barriers for the formation of these PAHs are around 19.2–38.0 kcal mol−1. Hence, at low temperature, Pathway I, Pathway III, Pathway IV and Pathway V are energetically favorable for bicyclic PAHs (CS6, CS10, CS13, CS26, CS28 and CS29) formation. Considering the high temperature in various combustion flames, all above medium and even higher barriers are easily overcome. Hence, all 9 potential PAHs are easy to be produced at flame temperature. The theoretical results supplement the PAH formation pathway and provide some new insights to understand PAH growth mechanism.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by the National Key R&D Program of China (No. 2017YFC0211201, and No. 2016YFC0802801), National Natural Science Foundation of China (No. 51276132, and No. 51774221) and the 111 Project (B17034). The Key Lab of Colloid and Interface Chemistry, at Shandong University would like to be acknowledged for providing computing resources that have contributed to the research results reported within this paper.References
- W. M. Baird, L. A. Hooven and B. Mahadevan, Environ. Mol. Mutagen., 2005, 45, 106 CrossRef CAS PubMed.
- F. P. Perera, Science, 1997, 278, 1068 CrossRef CAS PubMed.
- H. Wang, Proc. Combust. Inst., 2011, 33, 41 CrossRef CAS.
- M. Frenklach, D. W. Clary, W. C. Gardiner and S. E. Stein, Symp. (Int.) Combust., [Proc.], 1984, 20, 887 CrossRef.
- M. Frenklach and H. Wang, Proc. Combust. Inst., 1991, 23, 1559 CrossRef.
- H. Wang and M. Frenklach, J. Phys. Chem., 1994, 98, 11465 CrossRef CAS.
- C. W. Bauschlicher Jr and A. Ricca, Chem. Phys. Lett., 2000, 326, 283 CrossRef.
- C. W. Bauschlicher, A. Ricca and M. Rosi, Chem. Phys. Lett., 2002, 355, 159 CrossRef CAS.
- P. Ghesquière, D. Talbi and A. Karton, Chem. Phys. Lett., 2000, 595–596, 13 Search PubMed.
- D. S. N. Parker, R. I. Kaiser, T. P. Troy and M. Ahmed, Angew. Chem., Int. Ed., 2014, 53, 7740 CrossRef CAS PubMed.
- A. M. Mebel, Y. Georgievskii, A. W. Jasper and S. J. Kliooenstein, Proc. Combust. Inst., 2017, 36, 919 CrossRef CAS.
- V. V. Kislov, A. I. Sadovnikov and A. M. Mebel, J. Phys. Chem. A, 2013, 117, 4794 CrossRef CAS PubMed.
- A. Violi, A. D'Anna and A. D'Alessio, Chem. Eng. Sci., 1999, 54, 3433 CrossRef CAS.
- Y. Li, L. Wei, Z. Tian, B. Yang, J. Wang, T. Zhang and F. Qi, Combust. Flame, 2008, 152, 336 CrossRef CAS.
- B. Yang, Y. Li, L. Wei, C. Huang, J. Wang, Z. Tian, R. Yang, L. Sheng, Y. Zhang and F. Qi, Proc. Combust. Inst., 2007, 31, 555 CrossRef.
- B. Shukla, A. Miyoshi and M. Koshi, J. Am. Soc. Mass Spectrom., 2010, 21, 534 CrossRef CAS PubMed.
- T. Yang, D. S. N. Parker, B. B. Dangi, R. I. Kaiser and A. M. Mebel, Phys. Chem. Chem. Phys., 2015, 17, 10510 RSC.
- R. I. Kaiser, D. S. Parker, M. Goswami, F. Zhang, V. V. Kislov, A. M. Mebel, J. Aguilera-Iparraguirre and W. H. Green, Phys. Chem. Chem. Phys., 2012, 14, 720 RSC.
- D. S. Parker, F. Zhang, Y. S. Kim, R. I. Kaiser, A. Landera and A. M. Mebel, Phys. Chem. Chem. Phys., 2012, 14, 2997 RSC.
- D. S. N. Parker, B. B. Dangi, R. I. Kaiser, A. Jamal, M. N. Ryazantsev, K. Morokuma, A. Korte and W. Sander, J. Phys. Chem. A, 2014, 118, 2709 CrossRef CAS PubMed.
- D. S. N. Parker, F. Zhang, Y. S. Kim, R. I. Kaiser, A. Landera, V. V. Kislov, A. M. Mebel and A. G. G. M. Tielens, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 53 CrossRef CAS PubMed.
- T. Yang, L. Muzangwa, R. I. Kaiser, A. Jamal and K. Morokuma, Phys. Chem. Chem. Phys., 2015, 17, 21564 RSC.
- R. I. Kaiser, B. B. Dangi, T. Yang, D. S. N. Parker and A. M. Mebel, J. Phys. Chem. A, 2014, 118, 6181 CrossRef CAS PubMed.
- D. S. Parker, B. B. Dangi, R. I. Kaiser, A. Jamal, M. Ryazantsev and K. Morokuma, J. Phys. Chem. A, 2014, 118, 12111 CrossRef CAS PubMed.
- T. Yang, L. Muzangwa, D. S. N. Parker, R. I. Kaiser and A. M. Mebel, Phys. Chem. Chem. Phys., 2015, 17, 530 RSC.
- A. Raj, M. J. Al Rashidi, S. H. Chung and S. M. Sarathy, J. Phys. Chem. A, 2014, 118, 2865 CrossRef CAS PubMed.
- S. M. Lee, S. S. Yoon and S. H. Chung, Combust. Flame, 2004, 136, 493 CrossRef CAS.
- S. S. Yoon, D. H. Anh and S. H. Chung, Combust. Flame, 2008, 154, 368 CrossRef CAS.
- S. S. Yoon, S. M. Lee and S. H. Chung, Proc. Combust. Inst., 2005, 30, 1417 CrossRef.
- J. Y. Hwang, W. Lee, H. G. Kang and S. H. Chung, Combust. Flame, 1998, 114, 370 CrossRef CAS.
- A. Raj, I. D. C. Prada, A. A. Amer and S. H. Chung, Combust. Flame, 2012, 159, 500 CrossRef CAS.
- F. Liu, X. He, X. Ma, Q. Zhang, M. J. Thomson, H. Guo, G. J. Smallwood, S. Shuai and J. Wang, Combust. Flame, 2011, 158, 547 CrossRef CAS.
- J. H. Choi, B. C. Choi, S. M. Lee, S. H. Chung, K. S. Jung, W. L. Jeong, S. K. Choi and S. K. Park, J. Mech. Sci. Technol., 2015, 29, 2259 CrossRef.
- B. C. Choi, S. K. Choi, S. H. Chung, J. S. Kim and J. H. Choi, Int. J. Auto. Tech., 2011, 12, 183 CrossRef.
- K. L. McNesby, A. W. Miziolek, T. Nguyen, F. C. Delucia, R. R. Skaggs and T. A. Litzinger, Combust. Flame, 2005, 142, 413 CrossRef CAS.
- D. H. Kim, J. A. Mulholland, D. Wang and A. Violi, J. Phys. Chem. A, 2010, 114, 12411 CrossRef CAS PubMed.
- D. Wang, A. Violi, D. H. Kim and J. A. Mulholland, J. Phys. Chem. A, 2006, 110, 4719 CrossRef CAS PubMed.
- V. V. Kislov and A. M. Mebel, J. Phys. Chem. A, 2008, 112, 700 CrossRef CAS PubMed.
- J. A. Mulholland, M. Lu and D. H. Kim, Proc. Combust. Inst., 2000, 28, 2593 CrossRef CAS.
- M. Lu and J. A. Mulholland, Chemosphere, 2004, 55, 605 CrossRef CAS PubMed.
- R. I. Kaiser, D. S. N. Parker and F. Zhang, J. Phys. Chem. A, 2012, 116, 4248 CrossRef CAS PubMed.
- N. Hansen, S. J. Klippenstein, J. A. Miller, J. Wang, T. A. Cool, M. E. Law, P. R. Westmoreland, T. Kasper and K. Kohse-Hőinghaus, J. Phys. Chem. A, 2005, 110, 4376 CrossRef PubMed.
- L. Wei, L. Yang, J. Chen, X. Wang, H. Li, Y. Zhu, L. Wen, C. Xu, J. Zhang, T. Zhu and W. Wang, Air Qual., Atmos. Health, 2015, 9, 823 Search PubMed.
- Y. Matsukawa, K. Ono, K. Dewa, A. Watanabe, Y. Saito, Y. Matsushita, H. Aoki, K. Era, T. Aoki and T. Yamaguchi, Combust. Flame, 2016, 167, 248 CrossRef CAS.
- B. Öktem, M. P. Tolocka, B. Zhao, H. Wang and M. V. Johnston, Combust. Flame, 2005, 142, 364 CrossRef.
- A. Ciajolo, R. Barbella, A. Tregrossi and L. Bonfanti, Symp. (Int.) Combust., [Proc.], 1998, 27, 1481 CrossRef.
- A. Santamaría, F. Mondragon, A. Molina, N. D. Marsh, E. G. Eddings and A. F. Sarofi, Combust. Flame, 2006, 146, 52 CrossRef.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1992, 97, 9173 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern, A. G. Baboul and J. A. Pople, Chem. Phys. Lett., 1999, 314, 101 CrossRef CAS.
- D. C. Young, Computational chemistry: a practical guide for applying techniques to real-world problems, John Wiley & Sons Inc., 2001 Search PubMed.
- B. Temelso, C. D. Sherrill, R. C. Merkle and R. A. Freitas Jr, J. Phys. Chem. A, 2006, 110, 11160 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- B. B. Dangi, S. Maity, R. I. Kaiser and A. M. Mebel, J. Phys. Chem. A, 2013, 117, 11783 CrossRef CAS PubMed.
- M. Wei, T. Zhang, S. Li, G. Guo and D. Zhang, Can. J. Chem., 2017, 95, 816 CrossRef CAS.
- A. W. Jasper and N. Hansen, Proc. Combust. Inst., 2013, 34, 279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01449c |
| This journal is © The Royal Society of Chemistry 2018 |