
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSAR study on N2,N4-disubstituted pyrimidine-2,4-diamines as effective CDK2/CDK9 inhibitors and antiproliferative agents†
Liandong Jing a,
Yanbo Tanga,
Masuo Gotob,
Kuo-Hsiung Leebc and
Zhiyan Xiao*a
a,
Yanbo Tanga,
Masuo Gotob,
Kuo-Hsiung Leebc and
Zhiyan Xiao*a
aBeijing Key Laboratory of Active Substance Discovery and Druggability Evaluation, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, China. Tel: +86-10- 63189228E-mail: xiaoz@imm.ac.cn
bNatural Products Research Laboratories, UNC Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, North Carolina 27599-7568, USA
cChinese Medicine Research and Development Center, China Medical University and Hospital, Taichung, Taiwan
First published on 27th March 2018
Abstract
Cyclin-dependent kinases (CDKs) are pivotal kinases in cell cycle transition and gene transcription. A series of N2,N4-diphenylpyrimidine-2,4-diamines were previously identified as potent CDK2/CDK9 inhibitors. To explore the SAR of this structural prototype, twenty-four novel N2,N4-disubstituted pyrimidine-2,4-diamines were designed and synthesized. Among them, twenty-one compounds exhibited potent inhibitory activities against both CDK2/cyclin A and CDK9/cyclin T1 systems, and the most potent CDK2 and CDK9 inhibitors, 3g and 3c, showed IC50 values of 83 nM and 65 nM respectively. Most of these compounds displayed significant inhibition against the tested tumor cell lines in the SRB assay, and in particular, remained active against the triple-negative breast cancer (TNBC) cell line MDA-MB-231. Flow cytometer analysis of compounds 2a, 2d and 3b in MDA-MB-231 cells indicated that these compounds induced cell cycle arrest in G2/M phase. Docking studies on compound 3g were performed, which provided conducive clues for further molecular optimization.
Introduction
Cyclin-dependent kinases (CDKs) are a family of serine–threonine protein kinases. Upon binding to their regulatory partner cyclins, CDKs can be activated and play critical roles in cell cycle transition and gene transcription. Up to now, 20 CDKs and 29 cyclins have been identified from the human proteome. These CDKs and cyclins form a myriad of different heterodimeric complexes and modulate a variety of physiological and pathological processes.1,2 As a representative member in cell-cycle regulation, CDK2 plays a key role in the control of G1-S phase transition when in complex with cyclin E, while CDK2–cyclin A complex is required for the transition of the S phase.3,4 Different from CDK2, the transcriptional CDK9 appears to have a minimal effect on cell cycle transition.5 In contrast, CDK9/cyclin T is involved in the regulation of RNA transcription as the catalytic subunit of positive transcription elongation factor (P-TEFb).6,7The recognition of the pivotal roles of CDKs has stimulated extensive research interests on CDK inhibitors in recent two decades.8,9 A number of structural diverse CDK inhibitors are currently under active clinical investigations for the treatment of cancer (Fig. 1A),10,11 and four inhibitors have received approval from the U. S. Food and Drug Administration (FDA) (Fig. 1B). Flavopiridol (alvocidib) is the first CDK inhibitor approved by FDA. As a pan-CDK inhibitor competing with ATP, alvocidib was granted an orphan drug designation in the E. U. for the treatment of chronic lymphocytic leukemia (CLL) in 2007.12 It was later approved for the treatment of acute myeloid leukemia (AML) in both the US and the EU.12 The advent of selective CDK4/6 inhibitors symbolizes an important breakthrough in the translational research of CDK inhibitors,13,14 and has led to the successive approvals of palbociclib from Pfizer, ribociclib from Novartis and abemaciclib from Eli Lilly by the FDA in the recent three years.12 These therapeutic drugs selectively target the cyclin D-CDK4/6 axis and have made significant contributions to the treatment of hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative breast cancer.
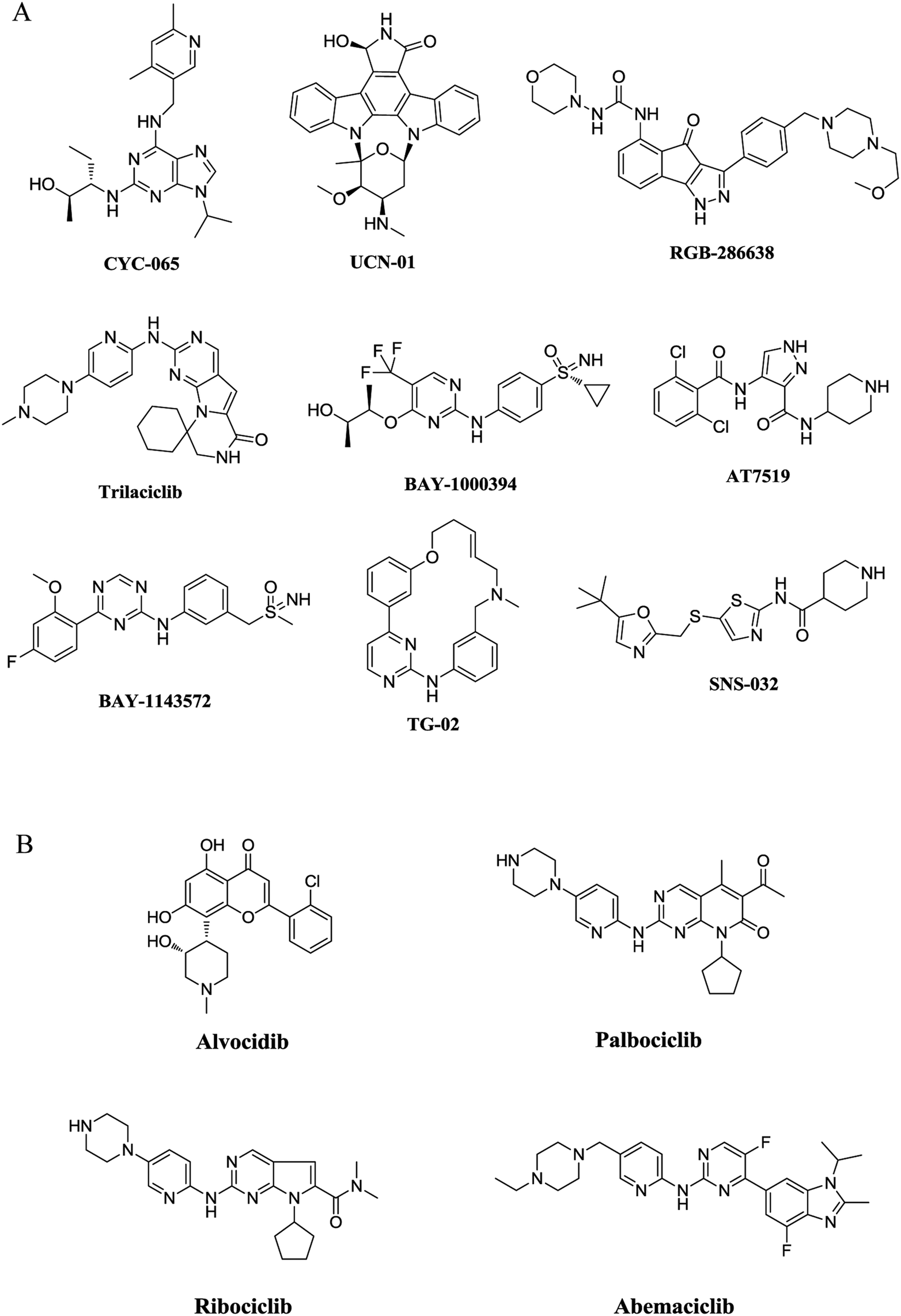 | ||
| Fig. 1 Representative structures of CDK inhibitors. (A) Representative structures of CDK inhibitors in clinical trials. (B) Structures of CDK inhibitors approved by FDA. | ||
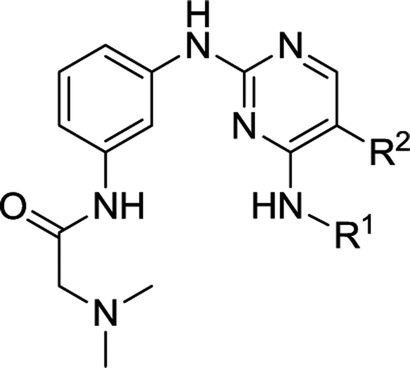
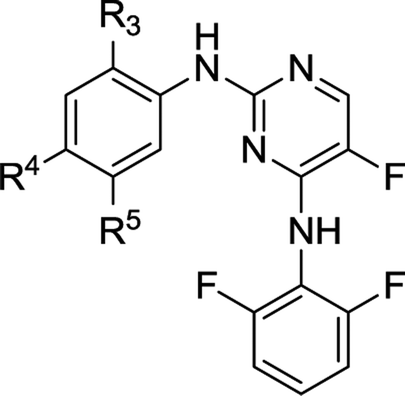
As representative cell cycle regulatory CDK and transcriptional CDK, CDK2 and CDK9 have been widely investigated for their potent inhibitors. In our previous efforts, a 3D-QSAR pharmacophore derived from a diverse set of known CDK9 inhibitors was constructed.15 Guided by the pharmacophoric features revealed and structural requirements of the ATP sites of CDK2 and CDK9, a series of novel N2,N4-diphenylpyrimidine-2,4-diamines were designed and synthesized, and compound 1 (Fig. 2) was identified as the most potent inhibitor against CDK2 and CDK9.16 We report herein the exploration of structure–activity relationships (SAR) on compound 1. Accordingly, twenty-four novel N2,N4-disubstituted pyrimidine-2,4-diamines were prepared and evaluated.
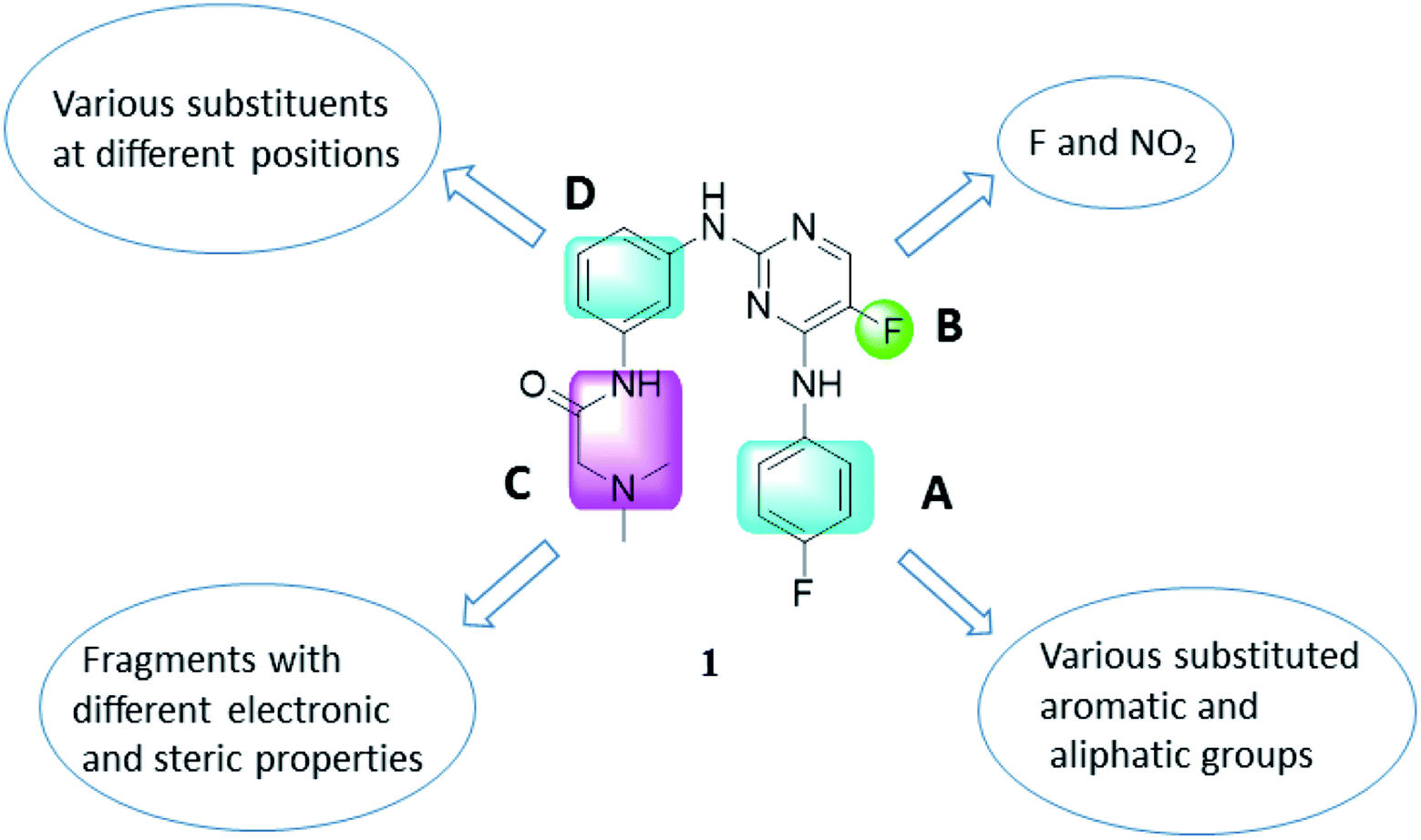 | ||
| Fig. 2 Molecular optimization based on compound 1. | ||
Results and discussion
Molecular design
As illustrated in Fig. 2, a stepwise molecular optimization was applied to the scaffold of compound 1, and the molecular areas A, B, C and D in 1 were modified to explore SAR. For area A, various substituted aromatic and aliphatic groups were introduced to investigate their effects on kinase inhibitory activity (Table 1, 2a–2h). It was also attempted to replace the fluoro atom in area B by nitro group (Table 1, 2i). Previous molecular docking suggested a change of Lys89 (CDK2) to Gly122 (CDK9) in the sub-pocket occupied by the N,N-dimethyl glycine moiety in area C.16 This observation implied that CDK2/CDK9 selectivity might be sensitive to the size and charge status in this molecular area. Therefore, various fragments with different electric and steric properties were extracted from known CDK inhibitors and grafted to area C (Table 2, 3a–3e, 3m–3o).17 Various substituents were also introduced at different positions of the benzene ring in area D (Table 2, 3f–3l). Such molecular optimization was expected to provide preliminary SAR on the prototype structure of 1. For all the molecular areas, electrophilic moieties were avoided intently to maintain the reversible nature of the inhibitors.
|
||||
|---|---|---|---|---|
| Compd. | R1 | R2 | IC50a (μM) | |
| CDK2/cyclin A | CDK9/cyclin T1 | |||
| a The IC50 value was interpolated from dose–response data with six different concentrations. Each test was performed in duplicate. Data represent the average of duplicate measurements.b The data were cited from ref. 16. | ||||
| 1 | 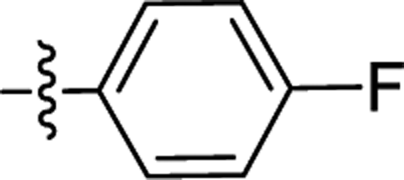 |
F | 1.71 | 0.78b |
| 2a | 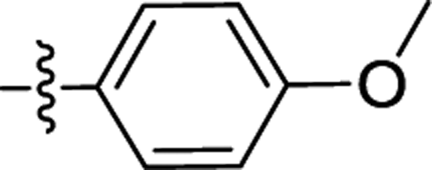 |
F | 0.32 ± 0.28 | 0.30 ± 0.01 |
| 2b | 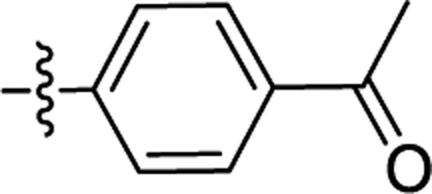 |
F | 0.78 ± 0.04 | 0.39 ± 0.08 |
| 2c | 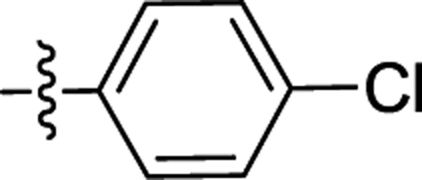 |
F | 0.91 ± 0.11 | 1.10 ± 0.02 |
| 2d | 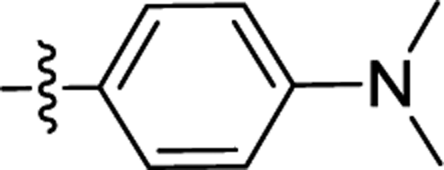 |
F | 0.94 ± 0.43 | 0.32 ± 0.05 |
| 2e | 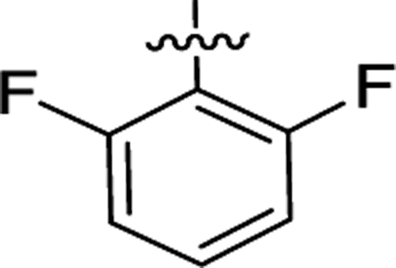 |
F | 0.24 ± 0.04 | 0.095 ± 0.004 |
| 2f | 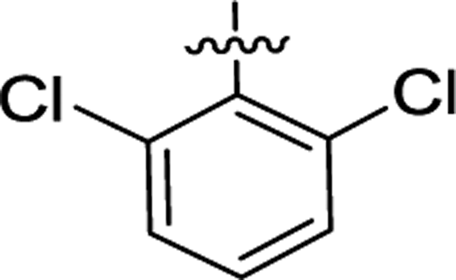 |
F | 1.60 ± 0.11 | 0.57 ± 0.12 |
| 2g | 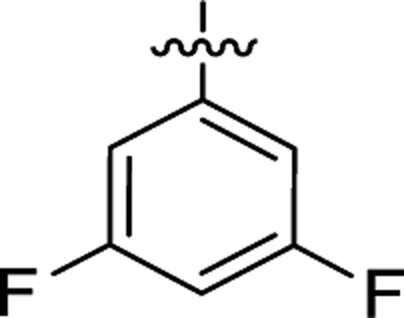 |
F | 0.57 ± 0.05 | 0.49 ± 0.27 |
| 2h | 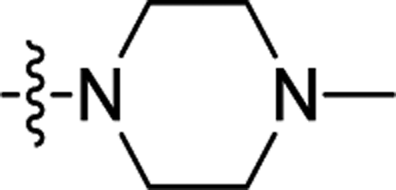 |
F | 5.60 ± 0.53 | 2.20 ± 0.32 |
| 2i | 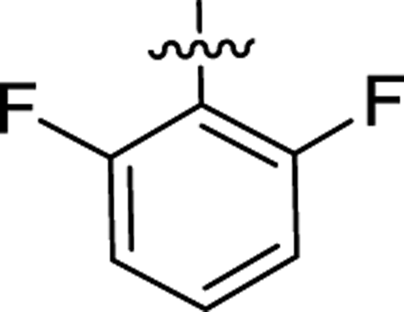 |
NO2 | 0.78 ± 0.12 | 0.48 ± 0.08 |
|
|||||
|---|---|---|---|---|---|
| Compd. | R3 | R4 | R5 | IC50a (μM) | |
| CDK2/cyclin A | CDK9/cyclin T1 | ||||
| a The IC50 value was interpolated from dose–response data with six different concentrations. Each test was performed in duplicate. Data represent the average of duplicate measurements.b Not determined due to the lack of significant inhibition at the concentration of 10 μM. | |||||
| 2e | H | H | 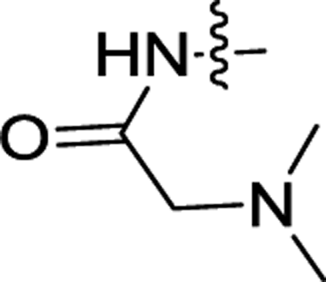 |
0.24 ± 0.04 | 0.095 ± 0.004 |
| 3a | H | H | 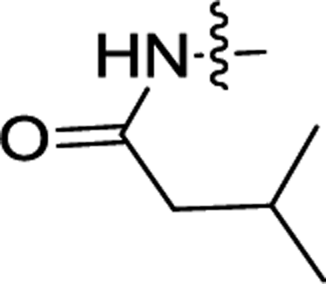 |
1.10 ± 0.15 | 0.48 ± 0.22 |
| 3b | H | H | 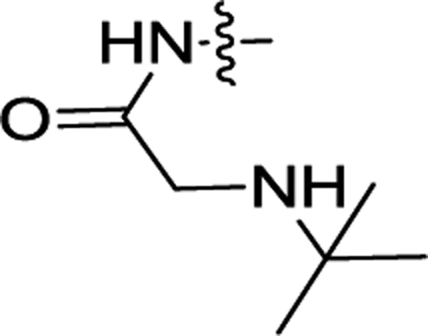 |
0.38 ± 0.06 | 0.08 ± 0.001 |
| 3c | H | H | 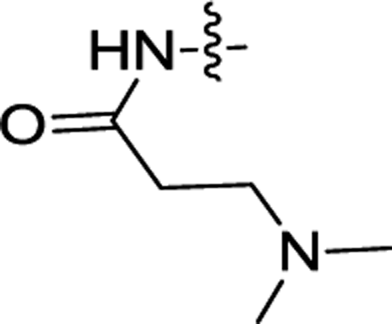 |
0.30 ± 0.07 | 0.065 ± 0.023 |
| 3d | H | H | 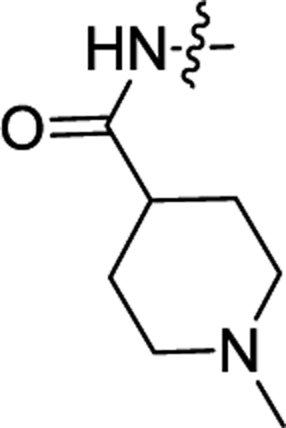 |
0.13 ± 0.02 | 0.66 ± 0.02 |
| 3e | H | H | 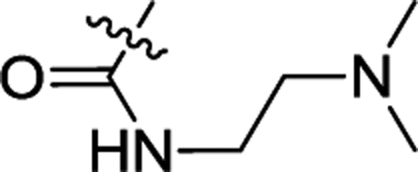 |
0.75 ± 0.14 | 0.23 ± 0.01 |
| 3f | F | H | 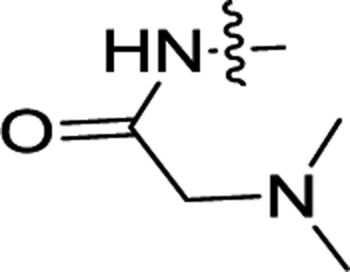 |
NDb | ND |
| 3g | H | F | 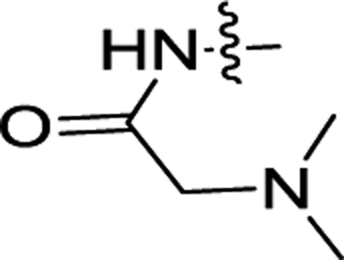 |
0.083 ± 0.035 | 0.10 ± 0.01 |
| 3h | OCH3 | H | 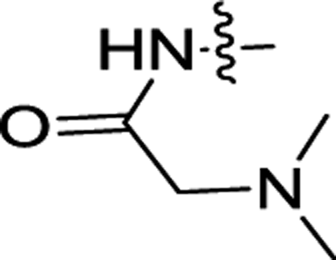 |
ND | ND |
| 3i | H | OCH3 | 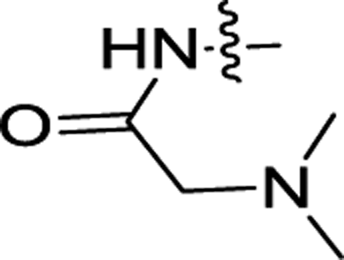 |
0.57 ± 0.08 | 0.19 ± 0.02 |
| 3j | H | CH3 | 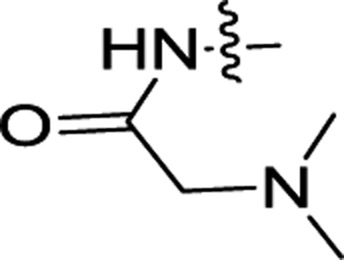 |
2.90 ± 0.06 | 0.16 ± 0.02 |
| 3k | H | 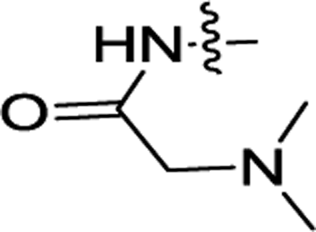 |
H | 0.44 ± 0.04 | 0.083 ± 0.002 |
| 3l | 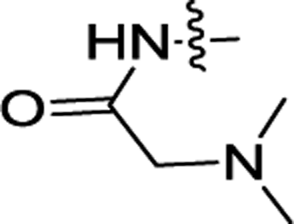 |
H | H | ND | ND |
| 3m | H | H | 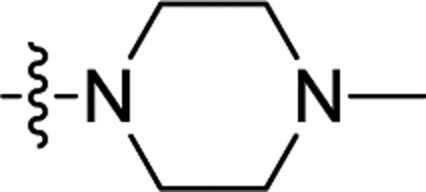 |
0.47 ± 0.04 | 0.290 ± 0.002 |
| 3n | H | H | 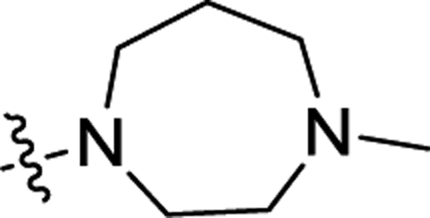 |
0.95 ± 0.27 | 1.6 ± 0.1 |
| 3o | H | CH3 | 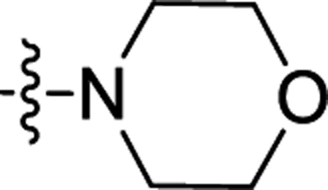 |
5.60 ± 0.03 | 0.74 ± 0.04 |
Chemistry
The general synthetic route for compounds 2a–2i was shown in Scheme 1. The key intermediate 6 was produced from the starting material 4 by reacting with N,N-dimethylglycine hydrochloride in the presence of 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIEA), and catalytic hydrogenation by Pd/C. The other key intermediate 8 was prepared by regio-selective substitution of compound 7 with appropriate amines.16 Cross-coupling between 6 and 8 under microwave irradiation afforded the target compounds 2a–2i.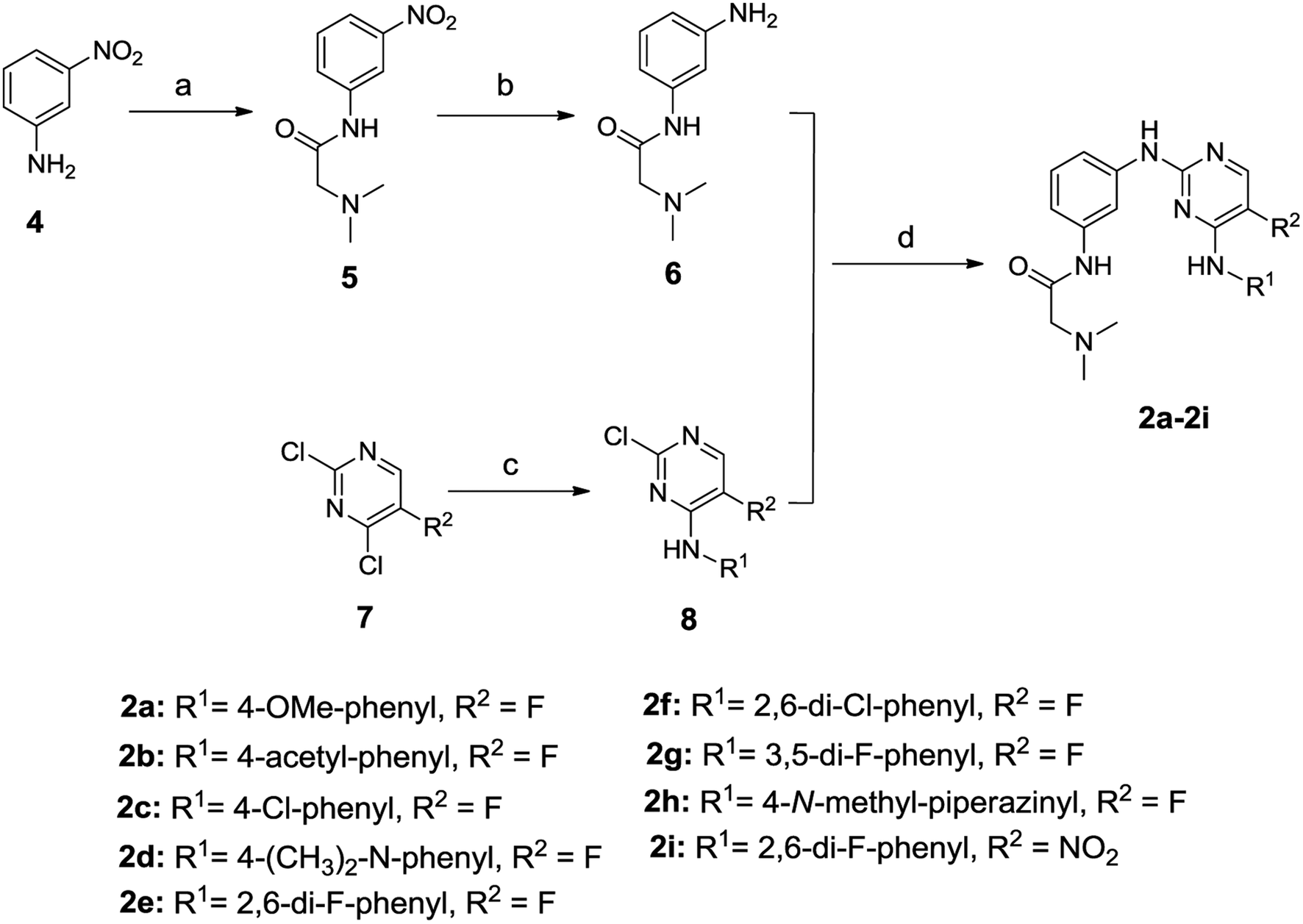 | ||
| Scheme 1 Synthesis of compounds 2a–2i. *Reagents and conditions: (a) N,N-dimethylglycine hydrochloride, HATU, DIEA, dichloromethane, 63%; (b) Pd/C, H2, CH3OH/ethyl acetate, 95%; (c) appropriate amine, MeOH/H2O, 35%; or Et3N/EtOH, 48–74%; (d) Pd(OAc)2, xantphos, Cs2CO3, dry THF, N2, microwave irradiation, 20–57%. | ||
The synthetic routes to compounds 3a–3l are illustrated in Scheme 2. The intermediates 9, 11 and 13 were prepared through a C–N cross-coupling reaction between 8e and appropriate amines. Catalytic hydrogenation of compounds 9 and 11 was performed to afford compounds 10 and 12. The target compounds 3a–3l were synthesized by condensation between appropriate amines and acids in the presence of HATU and DIEA.
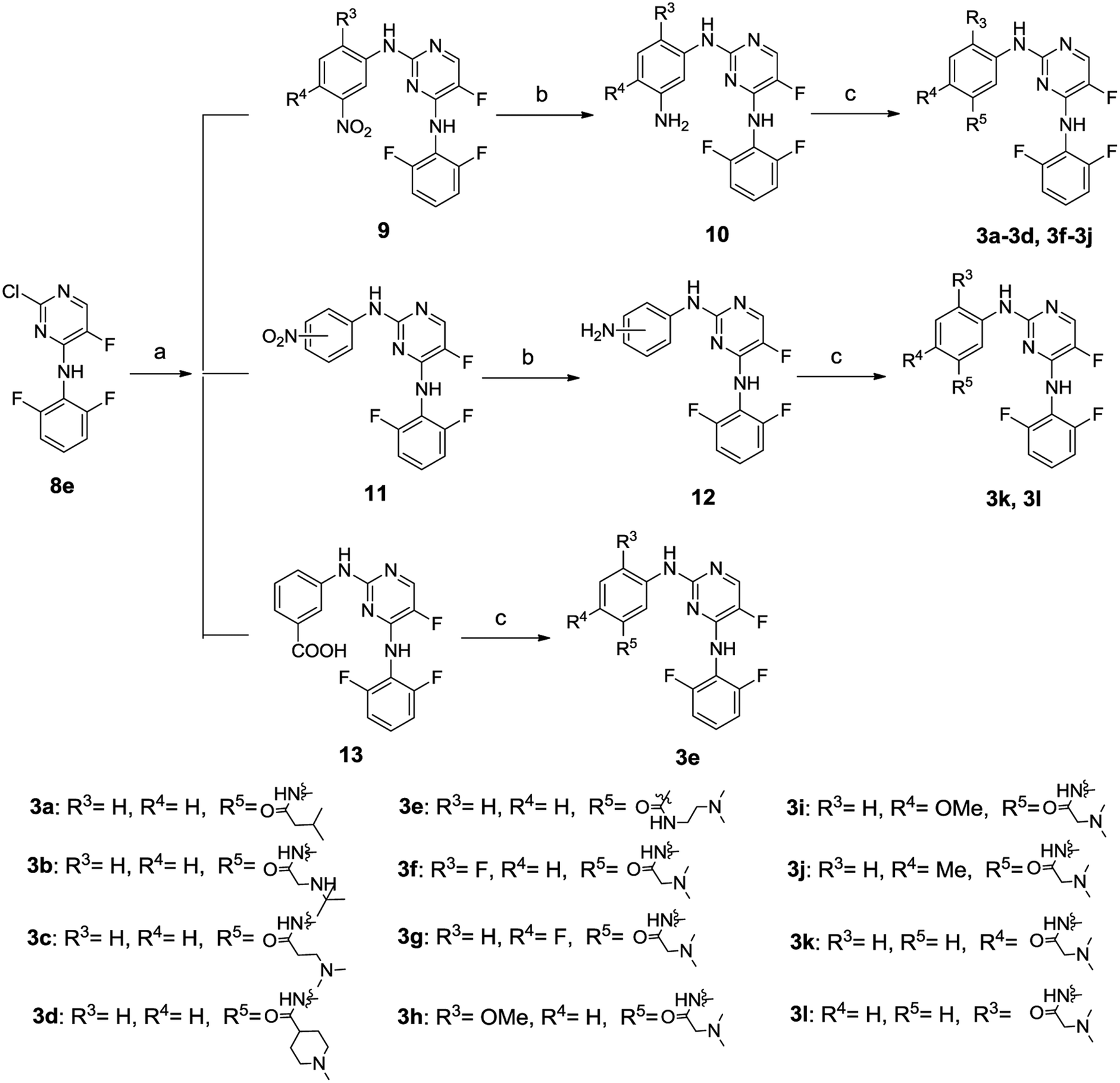 | ||
| Scheme 2 Synthesis of compounds 3a–3l. *Reagents and conditions: (a) appropriate amine, X-phos, Pd2(dba)3, K2CO3, dry toluene, t-butanol, N2, microwave irradiation, 49–70%; (b) Pd/C, H2, CH3OH/ethyl acetate, 73–95%; (c) appropriate amine, HATU, DIEA, DMF, 22–74%. | ||
The synthesis of compounds 3m–3o is outlined in Scheme 3. The intermediate 16 was prepared through successive cross-coupling and catalytic hydrogenation, which was subsequently subjected to C–N cross-coupling to provide compounds 3m–3o.
 | ||
| Scheme 3 Synthesis of compounds 3m–3o. *Reagents and conditions: (a) appropriate amine, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), Pd(OAc)2, Cs2CO3, dry toluene, N2, reflux, 60–72%; (b) Pd/C, H2, CH3OH/ethyl acetate, 80–89%; (c) Tris(dibenzylideneacetone) dipalladium (0) (Pd2(dba)3), X-phos, K2CO3, toluene, t-butanol, N2, microwave irradiation, 34–51%. | ||
CDK2/cyclin A and CDK9/cyclin T1 inhibition
All the target compounds were initially tested against CDK2/cyclin A and CDK9/cyclin T1. The inhibitory activities of these compounds in terms of IC50 values were shown in Tables 1 and 2. Most of the compounds showed significant inhibition against both CDK2/cyclin A and CDK9/cyclin T1 systems, and were more potent than the prototype structure 1. As demonstrated in Table 1, various substituents on the phenyl ring in R1 were generally well accommodated (2a–2g vs. 1), whereas the variation in substitution patterns (e.g. type, position and number of substituents) did lead to a slight fluctuation in the inhibitory activity (2a vs. 1, 2e vs. 2g, 2e vs. 1). Among the substitution patterns investigated, 2,6-difluorophenyl (2e) seemed to be optimal for R1, and compound 2e exhibited IC50 values of 0.24 μM and 0.095 μM against CDK2/cyclin A and CDK9/cyclin T1, respectively. However, the introduction of aliphatic fragment 4-methylpiperazin-1-yl at R1 caused a moderate decrease in the inhibitory activity (2h vs. 1). Decreased inhibition was also observed when the fluoro atom in area B was replaced by nitro group (2e vs. 2i).For compounds in Table 2, the optimal 2,6-difluorophenyl substitution at R1 (2e) was preserved, and SAR exploration in area D, in particular the substitution patterns of the phenyl ring, was performed. At a first glance of data in Table 2, it is surprising to find that the tolerance for substitution at R3 (C-2′ of the phenyl) was rather poor, and any substituent other than a hydrogen atom might result in dramatic loss of inhibitory activity (3f vs. 1 and 3g, 3h vs. 3i, 3k vs. 3l). This is probably due to an interference posed by the substituent to the key hydrogen bonding interaction between the N-1 atom/2-amino and the hinge region of the kinase. In contrast, substitution at R4 showed no significant effects on the inhibitory activity (2e vs. 3i and 3j), and the exchange of substituents at R3 and R4 had little influence on kinase inhibition (2e vs. 3k). As expected, a range of substituents with different electronic and steric properties, which were extracted from known CDK inhibitors, were generally well accepted at R5 of the C5′ position (3a–3e, 3m, 3n vs. 2e, 3j vs. 3o). Interestingly, the incorporation of a methyl at R4 in 3j resulted in moderate subtype selectivity (about 18-fold for CDK9 over CDK2), which implied that modification at R4 might be an attemptable approach to achieve subtype selectivity. It was previously suggested that the orientation of the morpholino ring at R5 might be responsible for the selectivity to CDK9 over CDK2.16 Methyl was introduced at R4 intently (3o), and it is assumed to affect the orientation of the morpholino ring at R5 so as to alter the subtype selectivity. However, compound 3o displayed only 7.5-fold selectivity towards CDK9, which is less selective than its congener without methyl substitution. The biological data listed in Tables 1 and 2 revealed obvious SAR trends for this compound class, and such results implied specific inhibition against the targets.
Inhibitory activities against tumor cell lines
All the compounds that showed significant inhibition in the enzymatic assays were further tested against the tumor cell lines, including MDA-MB-231, MCF-7, A549, KB and KB-vin in the sulforhodamine B (SRB) assay. The inhibitory activities of the target compounds in terms of GI50 values were demonstrated in Table 3. Most of the tested compounds displayed potent antiproliferative activities against the five human tumor cell lines and showed GI50 values at micromolar or even submicromolar level. Nevertheless, there was no obvious correlation between molecular enzymatic inhibition and cellular cytotoxicity, which might mainly owe to the variation in cellular concentration caused by different physicochemical properties. Notably, these compounds were not only active against the vincristine-resistant cell line KB-vin, but also the triple-negative breast cancer (TNBC) cells MDA-MB-231, which is a clinically aggressive form generally unresponsive to chemotherapy.| Compd. | GI50a (μM) | ||||
|---|---|---|---|---|---|
| MDA-MB-231 | MCF-7 | A549 | KB | KB-vinb | |
| a The GI50 values are expressed as means ± SD (standard deviation) of three independent measurements.b Cell lines: MDA-MB-231 (human breast carcinoma cell line), MCF-7 (Michigan Cancer Foundation-7), A549 (lung cancer), KB (nasopharyngeal carcinoma) and KB-vin (vincristine-resistant KB subline).c No significant inhibition against tumor cell growth was observed under the concentration of 10 μM.d The data were cited from ref. 16. | |||||
| 1 | — | — | 1.18 ± 0.13 | 1.26 ± 0.05 | 1.81 ± 0.60 |
| 2a | 1.20 ± 0.06 | 0.67 ± 0.06 | 0.72 ± 0.01 | 0.57 ± 0.01 | 2.54 ± 0.01 |
| 2b | 5.80 ± 0.08 | 3.80 ± 0.24 | 2.33 ± 0.03 | 2.20 ± 0.37 | 5.99 ± 0.26 |
| 2c | 6.77 ± 0.38 | 3.94 ± 0.31 | 2.12 ± 0.26 | 2.03 ± 0.32 | 3.90 ± 0.07 |
| 2d | 1.64 ± 0.15 | 2.83 ± 0.03 | 1.89 ± 0.19 | 3.08 ± 0.16 | 3.20 ± 0.18 |
| 2e | 3.98 ± 0.16 | 4.49 ± 0.03 | 1.31 ± 0.07 | 4.78 ± 0.38 | 2.54 ± 0.26 |
| 2f | 12.99 ± 0.38 | 6.02 ± 0.51 | 3.47 ± 0.44 | 7.04 ± 0.08 | 4.54 ± 0.04 |
| 2g | 4.64 ± 0.06 | 0.67 ± 0.03 | 0.71 ± 0.06 | 0.56 ± 0.08 | 1.66 ± 0.16 |
| 2h | >10c | >10 | >10 | >10 | >10 |
| 2i | 5.94 ± 0.33 | 10.23 ± 0.03 | 4.90 ± 0.45 | 5.73 ± 0.35 | 5.98 ± 0.24 |
| 3a | >10 | 5.97 ± 0.19 | 0.72 ± 0.04 | 0.59 ± 0.05 | 1.66 ± 0.16 |
| 3b | 2.07 ± 0.12 | 2.73 ± 0.48 | 1.31 ± 0.18 | 3.27 ± 0.13 | 2.36 ± 0.33 |
| 3c | 3.24 ± 0.05 | 2.65 ± 0.40 | 1.20 ± 0.05 | 2.31 ± 0.11 | 6.34 ± 0.54 |
| 3d | 3.30 ± 0.23 | 6.16 ± 0.14 | 1.09 ± 0.03 | 2.60 ± 0.45 | 7.11 ± 0.23 |
| 3e | 5.42 ± 0.35 | 6.73 ± 0.23 | 0.72 ± 0.01 | 3.82 ± 0.11 | 11.64 ± 0.04 |
| 3g | 5.51 ± 0.16 | 3.98 ± 0.26 | 0.69 ± 0.002 | 3.89 ± 0.06 | 0.98 ± 0.13 |
| 3i | 4.05 ± 0.14 | 1.88 ± 0.12 | 0.70 ± 0.002 | 2.87 ± 0.24 | 1.09 ± 0.06 |
| 3j | 5.31 ± 0.19 | 2.37 ± 0.62 | 1.15 ± 0.08 | 0.60 ± 0.04 | 0.88 ± 0.01 |
| 3k | 6.01 ± 0.08 | 4.48 ± 0.39 | 3.04 ± 0.18 | 3.80 ± 0.34 | 4.52 ± 0.01 |
| 3m | 2.84 ± 0.67 | 7.76 ± 0.52 | 1.25 ± 0.08 | 0.40 ± 0.09 | 3.70 ± 0.53 |
| 3n | 4.77 ± 0.27 | 7.16 ± 0.54 | 0.71 ± 0.01 | 4.44 ± 0.42 | 0.95 ± 0.001 |
| 3o | 7.00 ± 0.06 | 3.75 ± 0.28 | 0.85 ± 0.01 | 0.98 ± 0.05 | 0.84 ± 0.02 |
| FVPd | — | — | 0.14 ± 0.01 | 0.16 ± 0.01 | 0.18 ± 0.01 |
| Paclitaxel (nM) | 7.76 ± 0.39 | 8.78 ± 0.79 | 5.98 ± 0.40 | 4.94 ± 0.21 | 1518.36 ± 141.54 |
Effects on cell cycle progression in MDA-MB-231 cells
To evaluate their effects on the cell cycle progression, compounds 2a, 2d and 3b, which were the most potent compounds against MDA-MB-231 cells, were subjected to flow cytometry following protocols described previously.18 As depicted in Fig. 3, compounds 2a, 2d and 3b imposed obvious effects on the cell cycle progression under a concentration of five-fold IC50. It seemed that these compounds inhibited the onset of mitosis and induced cell cycle arrest at G2/M. Such observations implied that they might target proteins critical for the onset of mitosis. While CDK2 is generally regarded as a G1/S and S phase regulator, cell cycle arrest at the G2/M has been previously reported for CDK1 inhibition.19 This observation, in combination with the comparable inhibitory activities against both CDK2 and CDK9 systems for these compounds suggested that these compounds were likely to be pan-CDK inhibitors and multiple CDKs could be involved in the cells growth inhibition induced by compounds 2 and 3. In addition, as a privileged scaffold for kinase inhibitors, the pyrimidine scaffold were also found in inhibitors against other kinases, such as aurora, ALK, PI3K.20–22 Taking together, these inhibitors were likely to exert inhibitory effects against tumor cells via non-selective inhibition against multiple CDKs and even members of other kinase families.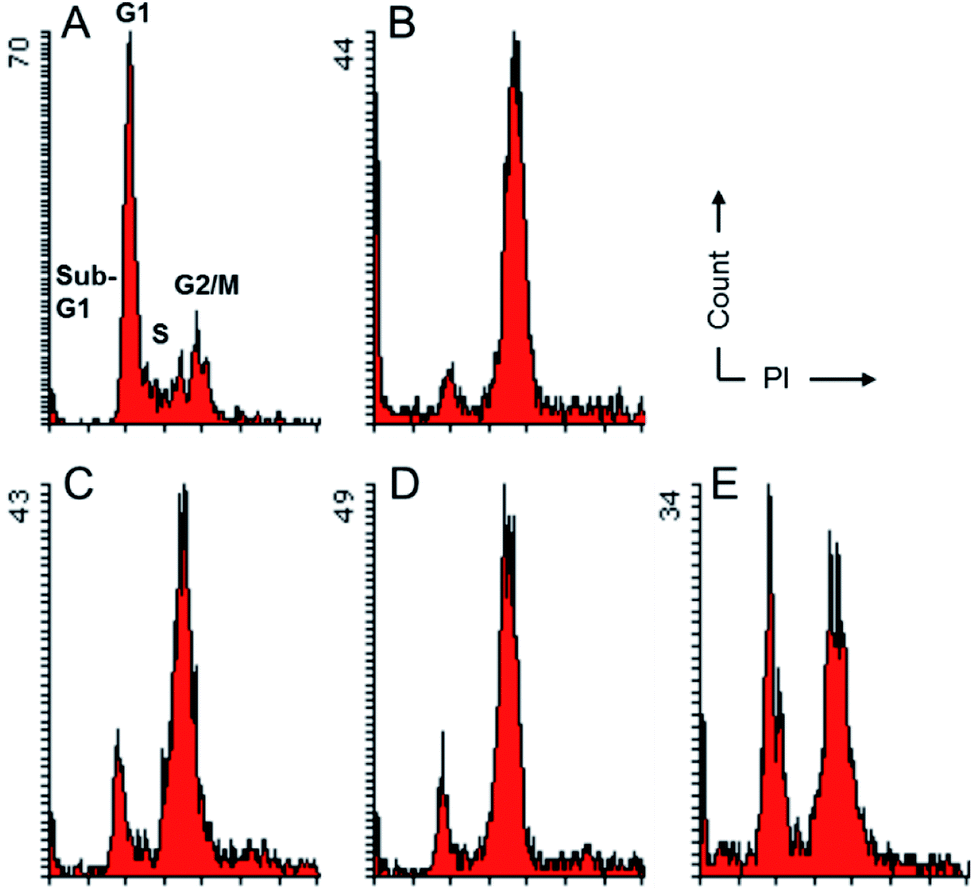 | ||
| Fig. 3 Effects of compounds 2a, 2d and 3b on cell cycle progression. MDA-MB-231 cells were treated for 24 h with compounds at a concentration five-fold GI50. Cells were fixed and stained with propidium iodide (PI) followed by cell cycle analysis using a flow cytometer. DMSO (A) and 200 nM combretastatin A-4 (CA-4) (B) were used as negative and positive control, respectively, for induction of cell cycle arrest in G2/M. Compounds 2a at 6.0 μM (C), 2d at 8.0 μM (D) or 3b at 10.5 μM (E) were used. | ||
Molecular modeling
Due to the structural similarity between compound 3g and the inhibitor co-crystalized, the 3D structures of 4BCP (CDK2) and 4BCG (CDK9) were selected for docking studies on compound 3g to explore its interaction modes with both CDK2 and CDK9 (Fig. 4). As illustrated by Fig. 4A, compound 3g situated in the ATP-binding site of CDK9, where the N,N-dimethyl glycine moiety occupied the outside sub-pocket, and the N-1 atom as well as 2-amino moiety on the pyrimidine ring interacts with the hinge residue Cys106 through hydrogen bonding. The 2,6-difluorophenyl moiety protruded toward the inside sub-pocket, and π–π stacking interactions were monitored between 2,6-difluorophenyl and Phe103, as well as between the pyrimidine ring and Phe105. Besides, hydrophobic interactions between 3g and residues Ala46 and Leu156 were detected in the binding pocket. As depicted by Fig. 4B, 3g adopted a similar pose in the ATP-binding site of CDK2. The N,N-dimethyl glycine moiety occupied the outside pocket and 2,6-difluorophenyl moiety situated inside. Hydrogen bonding was also observed between N-1 atom/2-amino moiety of 3g and the hinge residue Leu83. Besides, hydrophobic interactions between 3g and residues Val18, Ala31 and Leu134 were detected in CDK2. The insights gained from docking studies might guide further molecular design of potent CDK inhibitors. Whereas, the almost identical interaction modes of compound 3g with either CDK9 or CDK2 implied severe challenges to achieve subtype selectivity.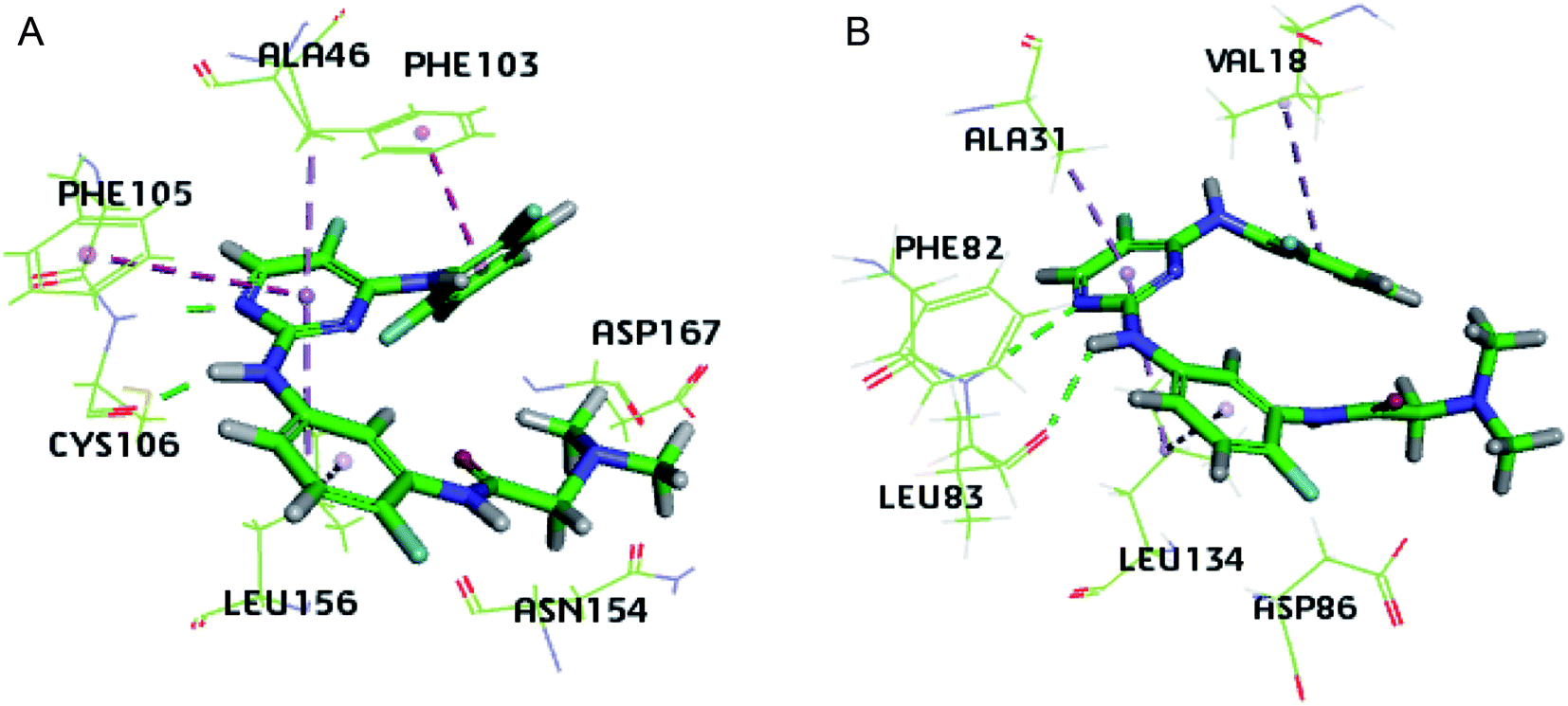 | ||
| Fig. 4 Interaction modes of 3g with CDK9 (A) and CDK2 (B) as suggested by the docking study. Green dashed lines indicate hydrogen bonds, deep purple dashed lines represent π–π stacking interactions and hydrophobic interactions are shown as light purple dashed lines. | ||
Experimental
Chemistry
Chemical reagents and solvents were obtained from commercial sources and were used without further purification. When necessary, solvents were dried and/or purified by standard methods. All reactions were monitored by thin-layer chromatography with preparative silica gel F254 plates purchased from Merck, Inc. Flash chromatography was performed using either a glass column packed with silica gel (200–300 mesh) or pre-packed silica gel cartridges by a combi-flash machine from Teledyne Isco. The purity of all target compounds assayed was identified by thin-layer chromatography (silica gel F254) and NMR spectra. In particular, the purity of the most active compounds, 2a, 2d and 3b, was further determined by HPLC (ESI†). 1H-NMR spectra were recorded on Varian Mercury 300 MHz or 400 MHz spectrometer. 13C-NMR spectra were recorded on Varian Mercury 400 MHz or 500 MHz spectrometer. Chemical shifts are reported in parts per million (δ) relative to internal tetramethylsilane (TMS) standard. Coupling constants are given in hertz. All melting points were determined on Yanaco melting point apparatus. Mass spectra were taken in HR-ESI mode on Exactive Plus™ LC/MSD Orbitrap from Thermo Fisher Scientific. Reactions under microwave irradiation were performed in a Biotage Initiator microwave reactor in sealed vessels.Preparation of 3-(dimethylamino-acetamino) aniline (6)
3-Nitroaniline 4 (276 mg, 2 mmol) and N,N-dimethylglycine hydrochloride (419 mg, 3 mmol) were dissolved in CH2Cl2. HATU (1.14 g, 3 mmol) and DIEA (1.29 g, 10 mmol) were then added. The reaction mixture was stirred at room temperature overnight. After the solvent was evaporated, the residue was dissolved in ethyl acetate and then washed with saturated sodium bicarbonate solution. The organic layer was concentrated and the crude material was purified by column chromatography on silica gel, eluted with petroleum ether/ethyl acetate (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford compound 5 as a yellow solid (280 mg, 63%). Compound 5 (280 mg, 1.26 mmol) was dissolved in the mixture of methanol and ethyl acetate (8 mL, volume ratio = 1:1), Pd/C (10% Pd, 28 mg) was then added. The mixture was stirred under hydrogen atmosphere for 6 h at room temperature. Pd/C was filtered by diatomite and the solvents were removed under reduced pressure to afford compound 6 as a gray solid (230 mg, 95%).
:1) to afford compound 5 as a yellow solid (280 mg, 63%). Compound 5 (280 mg, 1.26 mmol) was dissolved in the mixture of methanol and ethyl acetate (8 mL, volume ratio = 1:1), Pd/C (10% Pd, 28 mg) was then added. The mixture was stirred under hydrogen atmosphere for 6 h at room temperature. Pd/C was filtered by diatomite and the solvents were removed under reduced pressure to afford compound 6 as a gray solid (230 mg, 95%).
Preparation of 2-chloro-5-fluoro-N-substituted-pyrimidin-4-amine and 2-chloro-N-substituted-5-nitropyrimidin-4-amine (8)
2,4-Dichloro-5-fluoropyrimidine (7a: R2 = F, 334 mg, 2 mmol), p-anisidine (246 mg, 2 mmol) were mixed in 3 mL ethanol, and triethylamine (304 mg, 3 mmol) was then added. The mixture was stirred for 5 h at room temperature. The solvent was removed under reduced pressure and the crude material was washed by ethyl ether to give 8a as a gray solid (374 mg, 74%). Compounds 8b–8d and 8h were synthesized from different starting materials with procedures similar to that of 8a.Compound 7a (1.67 g, 10 mmol) and 2,6-difluoroaniline (1.29 g, 10 mmol) were dissolved in the mixture of methanol and H2O (4:1). The mixture was stirred at room temperature for 48 h, and the precipitation was filtered and dried to afford compound 8e as a white solid (900 mg, 35%). 8f and 8g were synthesized from different starting materials with procedures similar to that of 8e. Compound 8i was synthesized from 2,4-dichloro-5-nitro-pyrimidine (7b: R2 = NO2) using procedure similar to that of 8e.
General procedure for the preparation of (2)
Compound 6 (96 mg, 0.5 mmol), compound 8a (126 mg, 0.5 mmol), xantphos (29 mg, 0.05 mmol), Pd(OAc)2 (6 mg, 0.025 mmol) and Cs2CO3 (489 mg, 1.5 mmol) were added to anhydrous THF (2 mL). Nitrogen was bubbled through the reaction mixture for 5 min, and then the reaction vessel was sealed and heated under microwave irradiation to 130 °C for 25 min. The mixture was filtered and the filtrate was concentrated under vacuum. The crude product was purified by silica gel chromatography, and eluted with petroleum ether/ethyl acetate (6:1) to give 2a as a white solid (104 mg, 50%).
Compounds 2b–2i were synthesized from different starting materials using the same procedure as described for 2a.
Preparation of N4-(2,6-difluorophenyl)-5-fluoro-N2-(substituted-phenyl)pyrimidine-2,4-diamine (9) (11) (13)
Compound 8e (518 mg, 2 mmol), 3-nitroaniline (276 mg, 2 mmol), X-phos (95 mg, 0.2 mmol), Pd2(dba)3 (92 mg, 0.1 mmol), and Cs2CO3 (1.95 g, 6 mmol) were added to toluene (2 mL) and tertiary butanol (2 mL). Nitrogen was bubbled through the reaction mixture for 5 min, and then the reaction vessel was sealed and heated under microwave irradiation to 120 °C for 25 min. The mixture was filtered and the filtrate was concentrated under vacuum. The crude product was washed by petroleum ether and DCM (4:1) to provide 9a as a yellow solid (350 mg, 49%) for the next step.
Compounds 9b–9d, 9f–9j, 11, 13 were prepared from 8e and other different starting materials with the same procedure as described for 9a.
Preparation of N4-(2,6-difluorophenyl)-5-fluoro-N2-(substituted-phenyl)pyrimidine-2,4-diamine (10) (12)
Compound 9a (150 mg, 0.42 mmol) was dissolved in the mixture of methanol and ethyl acetate (6 mL, 1:1), Pd/C (10% Pd, 15 mg) was then added. The mixture was stirred under hydrogen atmosphere for 6 h at room temperature. Pd/C was filtered by diatomite and the solvents were removed under reduced pressure to afford compound 10a as a gray solid (100 mg, 73%). Compounds 10b–10d, 10f–10j and 12 were prepared from different starting materials with the same procedure as described for 10a.
General procedure for the preparation of (3a–3l)
Compound 10a (100 mg, 0.3 mmol) and isopentanoic acid (34 mg, 0.33 mmol) were dissolved in 3 mL DMF. HATU (137 mg, 0.36 mmol) and DIEA (194 mg, 1.5 mmol) were then added. The reaction mixture was stirred at room temperature overnight. After the solvent was evaporated, the residue was dissolved in ethyl acetate and then washed with saturated sodium bicarbonate. The organic layer was concentrated and the crude material was purified by column chromatography on silica gel to afford compound 3a as a white solid (65 mg, 52%).Compounds 3b–3l were prepared from respective starting materials with the same procedure as described for 3a.
General procedure for the preparation of (16)
Compound 14m (1.08 g, 5 mmol), morpholine (1.31 g, 15 mmol), BINAP (150 mg, 0.24 mmol), Pd(OAc)2 (50 mg, 0.22 mmol), and Cs2CO3 (3.25 g, 10 mmol) were added to toluene(10 mL). Nitrogen was bubbled through the reaction mixture for 5 min, and then the mixture was heated to 120 °C under atmosphere for 8 h. The mixture was filtered and the filtrate was concentrated under vacuum. The crude product was purified by column chromatography on flash silica gel, and eluted with petroleum ether/ethyl acetate (50:1) to afford compound 15m as a yellow solid (800 mg, 72%).
Compound 15m (260 mg, 1.17 mmol) was dissolved in the mixture of methanol and ethyl acetate, Pd/C (10% Pd, 26 mg) was then added. The mixture was stirred under hydrogen atmosphere for 4 h at room temperature. Pd/C was filtered by diatomite and the solvents were removed under reduced pressure to afford compound 16m as a gray solid (200 mg, 89%).
Compounds 16n and 16o were prepared from other starting materials with the same procedure as described for 16m.
General procedure for the preparation of (3m–3o)
Compound 16m (134 mg, 0.7 mmol), 8e (181 mg, 0.7 mmol), X-phos (33 mg, 0.07 mmol), Pd2(dba)3 (32 mg, 0.035 mmol), and K2CO3 (290 mg, 2.1 mmol) were added to toluene (1 mL) and tertiary butanol (1 mL). Nitrogen was bubbled through the reaction mixture for 5 min, and then the reaction vessel was sealed and heated under microwave irradiation to 125 °C for 25 min. The mixture was filtered and the filtrate was concentrated under vacuum. After concentration, the crude product purified by column chromatography on flash silica gel, eluted with DCM/MeOH (80:1) to afford compound 3m as a white solid (150 mg, 51%).
Compounds 3n and 3o were prepared from 16n and 16o respectively with the same procedure as described for 3m.
Kinase assays
The enzymatic assays against CDK2/cyclin A and CDK9/cyclin T1 were performed by following experimental protocols published previously. The assays were completed through contract services provided by Cerep Drug Discovery Services Co. Ltd.CDK2/cyclin A kinase assay
The kinase assay against CDK2/cyclin A was performed by following experimental protocols described in the ref. 23 The compounds were mixed with the enzyme (2.28 ng) in a buffer containing 40 mM Hepes/Tris(pH 7.4), 0.8 mM EGTA/Tris, 8 mM MgCl2, 1.6 mM DTT and 0.008% Tween 20. Thereafter, the reaction was initiated by adding 100 nM of the substrate Ulight-CFFKNIVTPRTPPPSQGK-amide (MBP) and 10 μM ATP, and the mixture was incubated for 15 minutes at room temperature. For the control basal measurements, the enzyme was omitted from the reaction mixture. Following incubation, the reaction was stopped by adding 13 mM EDTA. After 5 minutes, the anti-phospho-MBP antibody labeled with europium chelate was added. After 60 more minutes, the fluorescence transfer was measured at λex = 337 nm, λem = 620 nm and λem = 665 nm using a microplate reader (Envision, Perkin Elmer). The enzyme activity was determined by dividing the signal measured at 665 nm by that measured at 620 nm (ratio). The results were expressed as a percent inhibition of the control enzyme activity. The compounds were tested in each experiment at six concentrations to obtain an inhibition curve from which the IC50 values were calculated.CDK9/cyclin T1 kinase assay
The kinase assay against CDK9/cyclin T1 was performed by following experimental protocols described in the ref. 24 The compounds were mixed with the enzyme (21.72 ng) in a buffer containing 40 mM Hepes/Tris(pH 7.4), 0.8 mM EGTA/Tris, 8 mM MgCl2, 1.6 mM DTT and 0.008% Tween 20. Thereafter, the reaction was initiated by adding 100 nM of the substrate Ulight-CFFKNIVTPRTPPPSQGK-amide (MBP) and 10 μM ATP, and the mixture was incubated for 90 minutes at room temperature. For control basal measurements, the enzyme was omitted from the reaction mixture. Following incubation, the reaction was stopped by adding 13 mM EDTA. After 5 minutes, the anti-phospho-MBP antibody labeled with europium chelate was added. After 60 more minutes, the fluorescence transfer was measured at λex = 337 nm, λem = 620 nm and λem = 665 nm using a microplate reader (Envision, Perkin Elmer). The enzyme activity was determined by dividing the signal measured at 665 nm by that measured at 620 nm (ratio). The results were expressed as a percent inhibition of the control enzyme activity. The compounds were tested in each experiment at six concentrations to obtain an inhibition curve from which the IC50 values were calculated.SRB assay
The following five human tumor cell lines were used in the assay: MDA-MB-231 (human breast carcinoma cell line), MCF-7 (Michigan Cancer Foundation-7), A549 (lung cancer), KB (nasopharyngeal carcinoma) and KB-vin (vincristine-resistant KB subline). All cell lines were obtained from the Lineberger Comprehensive Cancer Center (UNC-CH) or from ATCC (Rockville, MD) and were cultured in RPMI-1640 medium supplemented with 25 mM HEPES, 0.25% sodium bicarbonate (Gibco), 10% fetal bovine serum (Corning), and 100 μg mL−1 streptomycin, 100 IU mL−1 penicillin, and 0.25 μg mL−1 amphotericin B (Corning).The cytotoxicity assay was performed by following experimental protocols described in the ref. 25 Freshly trypsinized cell suspensions were seeded in 96-well microtiter plates at densities of 4000–11000 cells per well with compounds added from DMSO-diluted stock. After 3 days in culture, attached cells were fixed in cold 10% trichloroacetic acid and then stained with 0.04% sulforhodamine B (SRB). The absorbance at 515 nm was measured using a microplate reader (ELx800, BioTek) operated by Gen5 software (BioTek) after solubilizing the bound dye with 10 mM Tris. The mean IC50 is the concentration of agents that reduces cell growth by 50% under the experimental conditions and it is the average from three independent determinations.
Cell cycle analysis
The flow cytometry was performed by following protocols described previously.18 Briefly, MDA-MB-231 cells were treated for 24 h with compound at the concentration of five-fold of GI50. Cells were fixed and stained with propidium iodide (PI) (BD Biosciences) followed by analysis of cell cycle using flow cytometer (LSRFortessa, BD Biosciences).Molecular modeling
The structure of compound 3g was generated and molecular docking was performed with the Discovery Studio 2017 software package (Accelrys, San Diego, USA). Due to the structural similarity between compound 3g and the inhibitor co-crystalized, the 3D structures of CDK2 (4BCP) and CDK9 (4BCG) were selected and obtained from the Protein Data Bank for docking study. The docking calculation was carried out with the CDOCKER protocol. In the process of CDOCKER docking, the pose cluster radius was set to “0.5”. Other parameters were used as default.Conclusion
In conclusion, twenty-four N2,N4-disubstituted pyrimidine-2,4-diamines derivatives were synthesized and their structure–activity relationship was investigated. Most of the compounds demonstrated potent inhibition against both CDK9 and CDK2. Compounds 3g and 3c were identified as the most potent CDK2 and CDK9 inhibitors, and showed IC50 values of 83 nM and 65 nM respectively. Furthermore, most compounds showed significant inhibition against tumor cells, including the triple-negative breast cancer cell line MDA-MB-231. These compounds also imposed obvious effects on cell cycle progression in MDA-MB-231 cells, and induced cell cycle arrest in G2/M phase. Preliminary SAR analysis and molecular docking of 3g provided new clues for further structural optimization of this compound class.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the financial support from National Natural Science Foundation of China (81172985 and 81261120391) and CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-3-009) awarded to Z Xiao. Partial support was also supplied from NIH grant CA177584 awarded to K. H. Lee.Notes and references
- M. Malumbres, E. Harlow, T. Hunt, T. Hunter, J. M. Lahti, G. Manning, D. O. Morgan, L.-H. Tsai and D. J. Wolgemuth, Nat. Cell Biol., 2009, 11, 1275–1276 CrossRef CAS PubMed.
- S. Lim and P. Kaldis, Development, 2013, 140, 3079–3093 CrossRef CAS PubMed.
- A. Marais, Z. Ji, E. S. Child, E. Krause, D. J. Mann and A. D. Sharrocks, J. Biol. Chem., 2010, 285, 35728–35739 CrossRef CAS PubMed.
- Y.-J. Chen, C. Dominguez-Brauer, Z. Wang, J. M. Asara, R. H. Costa, A. L. Tyner, L. F. Lau and P. Raychaudhuri, J. Biol. Chem., 2009, 284, 30695–30707 CrossRef CAS PubMed.
- J. Garriga, S. Bhattacharya, J. Calbo, R. M. Marshall, M. Truongcao, D. S. Haines and X. Grana, Mol. Cell. Biol., 2003, 23, 5165–5173 CrossRef CAS PubMed.
- J. Garriga and X. Graña, Gene, 2004, 337, 15–23 CrossRef CAS PubMed.
- D. H. Price, Mol. Cell. Biol., 2000, 20, 2629–2634 CrossRef CAS PubMed.
- J. Cicenas and M. Valius, J. Cancer Res. Clin. Oncol., 2011, 137, 1409–1418 CrossRef CAS PubMed.
- C. Di Giovanni, E. Novellino, A. Chilin, A. Lavecchia and G. Marzaro, Expert Opin. Invest. Drugs, 2016, 25, 1215–1230 CrossRef CAS PubMed.
- V. Malínková, J. Vylíčil and V. Kryštof, Expert Opin. Ther. Pat., 2014, 25, 1–18 Search PubMed.
- U. Asghar, A. K. Witkiewicz, N. C. Turner and E. S. Knudsen, Nat. Rev. Drug Discovery, 2015, 14, 130–146 CrossRef CAS PubMed.
- Thomson Reuters Integrity, https://integrity.thomson-pharma.com/integrity/xmlxsl/pk_home.util_home, accessed Janaury 2018.
- S. L. Sammons, D. L. Topping and K. L. Blackwell, Curr. Cancer Drug Targets, 2017, 17, 637–649 CrossRef CAS PubMed.
- C. C. O'Sullivan, Expert Opin. Pharmacother., 2016, 17, 1657–1667 CrossRef PubMed.
- C. Fang, Z. Xiao and Z. Guo, J. Mol. Graphics Modell., 2011, 29, 800–808 CrossRef CAS PubMed.
- J. Gao, C. Fang, Z. Xiao, L. Huang, C. H. Chen, L. T. Wang and K. H. Lee, MedChemComm, 2014, 6, 444–454 RSC.
- H. Galons, N. Oumata, O. Gloulou and L. Meijer, Expert Opin. Ther. Pat., 2013, 23, 945–963 CrossRef CAS PubMed.
- K. Nakagawagoto, A. Oda, E. Hamel, E. Ohkoshi, K. H. Lee and M. Goto, J. Med. Chem., 2015, 58, 2378–2389 CrossRef CAS PubMed.
- L. T. Vassilev, C. Tovar, S. Chen, D. Knezevic, X. Zhao, H. Sun, D. C. Heimbrook and L. Chen, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10660–10665 CrossRef CAS PubMed.
- I. Aliagasmartin, B. Dan, L. Corson, J. Dotson, J. Drummond, C. Fields, O. W. Huang, T. Hunsaker, T. Kleinheinz and E. Krueger, J. Med. Chem., 2009, 52, 3300–3307 CrossRef CAS PubMed.
- Y. Wang, G. Zhang, G. Hu, Y. Bu, H. Lei, D. Zuo, M. Han, X. Zhai and P. Gong, Eur. J. Med. Chem., 2016, 123, 80–89 CrossRef CAS PubMed.
- J. Q. Zhang, Y. J. Luo, Y. S. Xiong, Y. Yu, Z. C. Tu, Z. J. Long, X. J. Lai, H. X. Chen, Y. Luo and J. Weng, J. Med. Chem., 2016, 59, 7268–7274 CrossRef CAS PubMed.
- L. Meijer, A. Borgne, O. Mulner, J. P. Chong, J. J. Blow, N. Inagaki, M. Inagaki, J. G. Delcros and J. P. Moulinoux, Eur. J. Biochem., 1997, 243, 527–536 CAS.
- G. De Falco and A. Giordano, J. Cell. Physiol., 1998, 177, 501–506 CrossRef CAS PubMed.
- M. V. B. Reddy, Y. C. Shen, E. Ohkoshi, K. F. Bastow, K. Qian, K. H. Lee and T. S. Wu, Eur. J. Med. Chem., 2012, 47, 97–103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01440j |
| This journal is © The Royal Society of Chemistry 2018 |