
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization of peptides self-assembly by low frequency Raman spectroscopy†
Maria Ronen‡
 ac,
Basanth S. Kalanoor‡bc,
Ziv Orend,
Izhar Rone,
Yaakov R. Tischler*bc and
Doron Gerber*ac
ac,
Basanth S. Kalanoor‡bc,
Ziv Orend,
Izhar Rone,
Yaakov R. Tischler*bc and
Doron Gerber*ac
aMina and Everard Goodman Faculty of Life Sciences, Bar-Ilan University, Ramat-Gan 5290002, Israel. E-mail: Doron.Gerber@biu.ac.il
bDepartment of Chemistry, Bar-Ilan University, Ramat-Gan 5290002, Israel. E-mail: Yaakov.Tischler@biu.ac.il
cBar-Ilan Institute for Nanotechnology and Advanced Materials, Bar-Ilan University, Ramat-Gan 5290002, Israel
dDepartment of Biotechnology, Israel Institute for Biological Research, Nes-Ziona 7410001, Israel
eDepartment of Physical Chemistry, Israel Institute of Biological Research, Nes-Ziona 7410001, Israel
First published on 30th April 2018
Abstract
Low Frequency Vibrational (LFV) modes of peptides and proteins are attributed to the lattice vibrations and are dependent on their structural organization and self-assembly. Studies taken in order to assign specific absorption bands in the low frequency range to self-assembly behavior of peptides and proteins have been challenging. Here we used a single stage Low Frequency Raman (LF-Raman) spectrometer to study a series of diastereomeric analogue peptides to investigate the effect of peptides self-assembly on the LF-Raman modes. The structural variation of the diastereomeric analogues resulted in distinct self-assembly groups, as confirmed by transmission electron microscopy (TEM) and dynamic light scattering (DLS) data. Using LF-Raman spectroscopy, we consistently observed discrete peaks for each of the self-assembly groups. The correlation between the spectral features and structural morphologies was further supported by principal component analysis (PCA). The LFV modes provide further information on the degrees of freedom of the entire peptide within the higher order organization, reflecting the different arrangement of its hydrogen bonding and hydrophobic interactions. Thus, our approach provides a simple and robust complementary method to structural characterization of peptides assemblies.
Introduction
Early studies on the Low Frequency Vibrational (LFV) modes of proteins including α-chymotrypsin, lysozyme, aldolase, thyroglobulin and concanavalin A revealed broad bands that raised interest in their role and origin.1–3 Studies conducted throughout the years revealed the significance of LFV modes in the molecular mechanism of protein activity including collective modes, structural folding and stabilization, affinity and selectivity of molecular recognition, charge shifting, and interactions with water molecules.4–10 Experimental and theoretical data have revealed that LFV modes of proteins result from crystalline phonon vibration, large-scale delocalized molecular vibrations, and intra/inter-molecular domains connected by weak interactions, such as hydrogen-bond stretches, torsional vibrations, van der Waals forces, and hydrophobic interactions.11–13Due to the involvement of hydrogen bonds and their collective nature, LFV modes are sensitive to the structural morphology of the protein.10 Thus, several approaches have been taken in order to identify the functional vibrations and assign specific bands in the low frequency range to different amino acid sequences and secondary structures. These approaches included studying the contribution of individual L-amino acids, short peptides with different sequences, and different secondary structures of the same amino acids sequence.10,13–23 These studies hypothesized that every specific peptide sequence examined had a unique (“fingerprint”) spectrum. However, despite the sensitivity of LFV spectra toward protein structural morphology, efforts to assign specific secondary structures to LFV spectra were unsuccessful.10
Similar challenges were encountered when attempting to assign bands in the low frequency range to specific features of peptide and protein self-assembly including that of partially denatured proteins with altered tertiary structures. Two major methods were used to study protein structural organization in the low frequency range: Terahertz Time-Domain Spectroscopy (THz-TDS) and FT-IR spectroscopy using a synchrotron light source. These methods were used to study thermal denaturation and aggregation of CP43 protein, heat-induced gels of β-lactoglobulin, polyomavirus capsid protein VP1, aggregative states of insulin, lysozyme fibrils, fibrillar state of concanavalin A, lysozyme, insulin and BSA fibrils.24–29 The THz spectra of the aggregated/fibrillar proteins revealed shifts in the THz band and its width (CP43 protein, β-lactoglobulin, concanavalin A), and increases in absorbance (insulin, lysozyme, and BSA fibrils) explained by a greater degree of light scattering.24–30 Thus, neither distinct peaks nor general characteristics were observed for self-assembled products.
Until recently, LF-Raman studies have been challenging because of the experimental difficulties of measuring the low frequency Raman spectra. However, new advances in ultra-narrow-band notch filter technology with volume holographic gratings (VHG) enable the generation of high-quality, low-frequency Raman spectra using a relatively compact, easy-to-use and cost-effective system.31
Here we describe an approach for studying and characterizing the effect of peptides self-assembly on the LFV modes, using the single-stage Raman spectrometer and VHG notch filters. Our approach is based on model peptides composed of amino acids with L-configuration and their structural diastereomers, each of which includes two amino acids with D-configuration incorporated at different locations within the primary sequence. The model peptides were designed as a short minimalist sequence of 12-mer amino acids composed solely of leucine (hydrophobic) and lysine (hydrophilic) amino acids, either in periodicity that favors the formation of an amphipathic α-helix or a scrambled analogue that favors the formation β-sheet structures.32,33 Site-specific introduction of D-amino acids into the primary all-L sequence promotes local distortion in the peptides structure interfering with their native conformation.
Generally, the physicochemical properties associated with the peptide sequence such as hydrophobicity, net charge, and propensity to form a β-sheet are believed to interfere with intermolecular interactions thus significantly affecting self-assembly kinetics.34–36 As specifically designed, all diastereomers used in this study and their all-L-amino acid parental peptides share their respective primary sequence and hydrophobicity-to-charge ratio. Thus, the site-specific introduction of D-amino acids allowed us to specifically manipulate intermolecular interactions to study the effect of peptides self-assembly on the LFV modes. The LFV spectra obtained from a single stage LF-Raman spectrometer were complemented by molecular fingerprint modes (MFM) Raman spectroscopy, Dynamic Light Scattering (DLS), and Transmission Electron Microscopy (TEM) providing insight into the manifestation of peptides self-assembly phenomena.
Materials and methods
Diastereomeric model peptides design
12-mer K–L model peptides (K and L for lysine and leucine, respectively) and their diastereomers were synthesized by solid phase synthesis by Cellmano Biotech Ltd. (Hefei, China). Each peptide was analyzed by analytical HPLC and determined by mass spectrometry analysis with purity ranging between 95.29% to 99.69%. For stock solutions, each peptide was dissolved in HPLC-grade water to a final concentration of 25 mg ml−1, incubated for 1 hour at RT and stored at −20 °C until used.Sample preparation
For Raman measurements, the peptide stock solutions were diluted to a final concentration of 12.5 mg ml−1 (in 50% acetonitrile and 0.05 M HCl in HPLC-grade water), briefly sonicated and allowed to equilibrate by 60–90 min incubation at room temperature. Next, 3 μl of each sample was spotted onto a glass slide in two consecutive layers. Since Raman spectroscopy requires high concentrations of studied molecules, between the layers, the samples were allowed to dry completely at room temperature. However, the use of air-dried spots of biomolecules retains enough water to keep the molecules in a hydrated environment. The measurements were carried out at room temperature (293 K). For each sample, the spectral data were collected in the range of 0–250 cm−1 and 1600–1700 cm−1 for LFV and molecular fingerprint modes (MFM), respectively.Reproducibility of data was checked by repeating each experiment in triplicates using fresh samples. For every sample, spectra were collected from three randomly selected areas and averaged. No appreciable variation of band shapes during the experiments was noticed, indicating preservation of the sample integrity during the measurements. In order to estimate the contribution of buffer solutions, we carried out the LFV and MFM Raman spectrum of HPLC-grade water and the dilution buffer (50% acetonitrile and 0.05 M HCl in HPLC-grade water). The buffer solutions were allowed to dry completely at room temperature. No measurable peaks were obtained for the dried traces in the spectral region of interest; this concludes that the detected signals belong to the samples alone (Fig. S1, in the ESI†).
Raman measurement apparatus
The Raman system used for the measurements is a home-built apparatus which consists of several components as explained earlier.31 The schematic setup of the system along with the positioning of the filters in the apparatus is shown in Fig. S2, in the ESI.† The Raman signal was collected with a confocal configuration using a 50 μm diameter fiber to define the confocal pinhole, and spectrally resolved in a 0.5 meter long imaging spectrometer (SP-2500i from Princeton Instruments). An EM-CCD (ProEm16002: eXcelon3, 1600 × 200 pixels) was used as the camera for the spectrometer. The Raman measurements were carried out with a grating of 1800 grooves per mm. The integration time for spectral collection was 60 s per acquisition. Each spectrum was recorded by accumulating 5 frames, yielding an overall integration time of 300 s. The objective lens (100×) and confocal aperture used (50 μm) provide for an approximate 1 μm2 (X, Y) and 2 μm (Z) sample volume together with strong attenuation of fluorescence. Laser power at the sample was 60 mW. The Raman frequencies were calibrated using a thin film polystyrene sample and a silicon wafer). All experiments were conducted at room temperature (293 K).Data analysis
In order to resolve overlapping bands and calculate peak centers, the spectra were processed using PEAKFIT software (Jandel Scientific, San Rafael, CA) in the 0–210 cm−1 and 1600–1700 cm−1 sections for LFV and amide I spectral regions, respectively. Due to a broad residual Rayleigh scattering at low Raman wave numbers, quadratic function baseline correction was applied in the LFV region only. For the both spectral regions, second-derivative spectra accompanied by 5% Savitzky–Golay smoothing were calculated to identify the position of the component bands. Next, the resulted wave numbers were used as initial parameters for curve fitting with Gaussian component peaks. Finally, peak centers, bandwidths, and amplitudes of the peaks were varied until a good agreement between the calculated sum of all components and the experimental spectra was achieved (r2 > 0.99).The Raman scattering intensity I(![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) ) was converted into a Raman absorption by multiplying by ||[1 − exp(−h||/kT)] as proposed by Lund et al.37 k is Boltzmann constant, T temperature used in the measurement, h is Planck's constant.
) was converted into a Raman absorption by multiplying by ||[1 − exp(−h||/kT)] as proposed by Lund et al.37 k is Boltzmann constant, T temperature used in the measurement, h is Planck's constant.
Multivariate analysis of the spectral data was performed using MATLAB and Statistics Toolbox Release 2013b, the MathWorks, Inc., Natick, Massachusetts, United States. Principal component analysis (PCA) was used as a multivariate analytical technique, to explore different clusters in the collection of LF-Raman spectra from the various peptides. In PCA, each PC is a linear combination of the original features (spectral features in our case).38 PCs are calculated from the covariance matrix of the data in such a way that the first PC accounts for the maximum variance in the original data set. The second PC is orthogonal, namely uncorrelated, to the first and accounts for most of the remaining variance, and so on. The score values derived from PCA analysis represent the projections of the spectra onto the principal components. A score plot is a common graphical approach for visualization of the existence of different groups within the data set.
Peptides self-assembly characterization
The self-assembly properties of the model peptides and their diastereomers were evaluated using Transmission Electron Microscope (TEM) and Dynamic Light Scattering (DLS) techniques. For both measurements, the peptide stock solutions were diluted to a final concentration of 2.5 mg ml−1 (in 50% acetonitrile and 0.05 M HCl in HPLC-grade water), briefly sonicated and allowed to equilibrate by 60–90 minutes incubation at room temperature.Transmission electron microscope (TEM)
TEM allows for visual observation of the self-assembly products. The examinations of the model peptides and their diastereomers were performed with a JEM-1400 (JEOL Ltd, Tokyo, Japan) analytical transmission electron microscope operating at 120 kV using a 15 nm probe size. Samples (50 μl each) were mounted on Formvar support film of 200 mesh carbon-coated copper grids (Proscitech GSCU200CH-100). Uranyl acetate was applied to negatively stain the grids.Dynamic light scattering (DLS)
Mean particle size, particle size distribution and intensity were assessed at room temperature by Vasco™ Particle Size Analyser (Cordouan Technologies, France) using 200 μl of each sample applied directly in the center of the chamber with a pipette. Additionally, the signal was interpreted in terms of an autocorrelation function using the NanoQ technology software algorithm. Generally, a correlator compares the signal measured at a time t0 with very short time delays (dt) and the obtained autocorrelation function describes how fast or slow the intensity changes. Small particles deliver fast intensity changes whereas large particles result in a much slower variation. In order to ensure the accuracy and reproducibility of the data, the measurements were performed at least in triplicates using fresh samples. No measurable particles were detected within the dilution buffer.Results and discussion
Peptide design
Two groups of 12-mer peptides were synthesized in order to examine the effect of peptides self-assembly on their LFV modes. Each group includes one model peptide composed solely of L amino acids of hydrophobic amino acid leucine (L) and positively charged amino acid lysine (K) and a set of its diastereomeric analogous each include two amino acids of D-configuration, incorporated at different locations within the primary sequence. The two groups differ in periodicity of K/L amino acids with one group that favors the formation of an ideal amphipathic α-helix secondary structure while the second represents a scrambled sequence of hydrophobic and hydrophilic amino acids. Due to the strong dependence of self-assembly pathways on the presence of hydrophobic amino acids (such as leucine), each group was designed to maintain the same amino acid composition and hydrophobicity-to-charge ratio.39 Point mutations, involving the insertion of D-amino acids, were used in order to induce local distortions in the native peptide structural arrangement. The sequences, abbreviations and Schiffer–Edmundson helical wheel projection of the peptides are listed in Table 1.40| Peptide abbreviation | Sequencea | Helical wheelb |
|---|---|---|
| a The position of the amino acids in D configuration is bolded and underlined.b The black shading represents hydrophilic amino acids (lysine); otherwise, the amino acids are hydrophobic (leucine). | ||
| AM-K4L8 | KLLLKLLLKLLK | 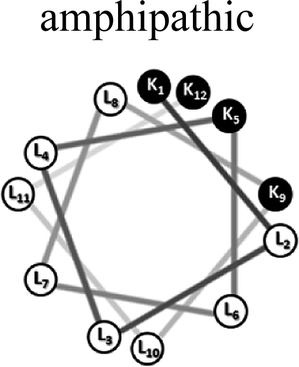 |
| AM-[D]-K1,12 | KLLLKLLLKLLK | |
| AM-[D]-L2,11 | KLLLKLLLKLLK | |
| AM-[D]-K5,9 | KLLLKLLLKLLK | |
| AM-[D]-L4,10 | KLLLKLLLKLLK | |
| SC-K4L8 | LLKLKLLKLLKL | 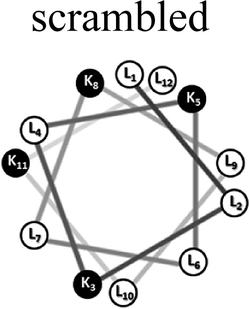 |
| SC-[D]-L1,12 | LLKLKLLKLLKL | |
| SC-[D]-K3,8 | LLKLKLLKLLKL | |
| SC-[D]-L4,9 | LLKLKLLKLLKL | |
The effect of D-amino acids on the model peptides self-assembly
First, we tested the effect of D-amino acids on the peptides structural morphology using TEM and DLS techniques. Fig. 1 represents the structural patterns of the amphipathic and scrambled model peptides and their diastereomeric analogues obtained with TEM. The data demonstrate that under our experimental conditions, different diastereomers exhibit diverse morphology, depending on the presence and the location of D-amino acids. Thus, AM-K4L8 and AM-[D]-K1,12 peptides undergo self-assembly to form dense mat of networked rod-like fibrils, typical for β-sheet forming structures. Long individual fibrils are readily observed with diameters of 5–6 nm, whereas SC-K4L8 presents morphology of thicker and much shorter rod-like fibrils with a lower tendency to associate into fiber networks. AM-[D]-L4,10 and SC-[D]-L4,9 both aggregate to form rod-like crystalline structures. The remainder of the diastereomers undergo self-assembly into a variety of microstructures including bundles of thin, parallel-arranged fibrils (AM-[D]-L2,11), tape-like structures (AM-[D]-K5,9), and amorphous aggregates (SC-[D]-L1,12 and SC-[D]-K3,8).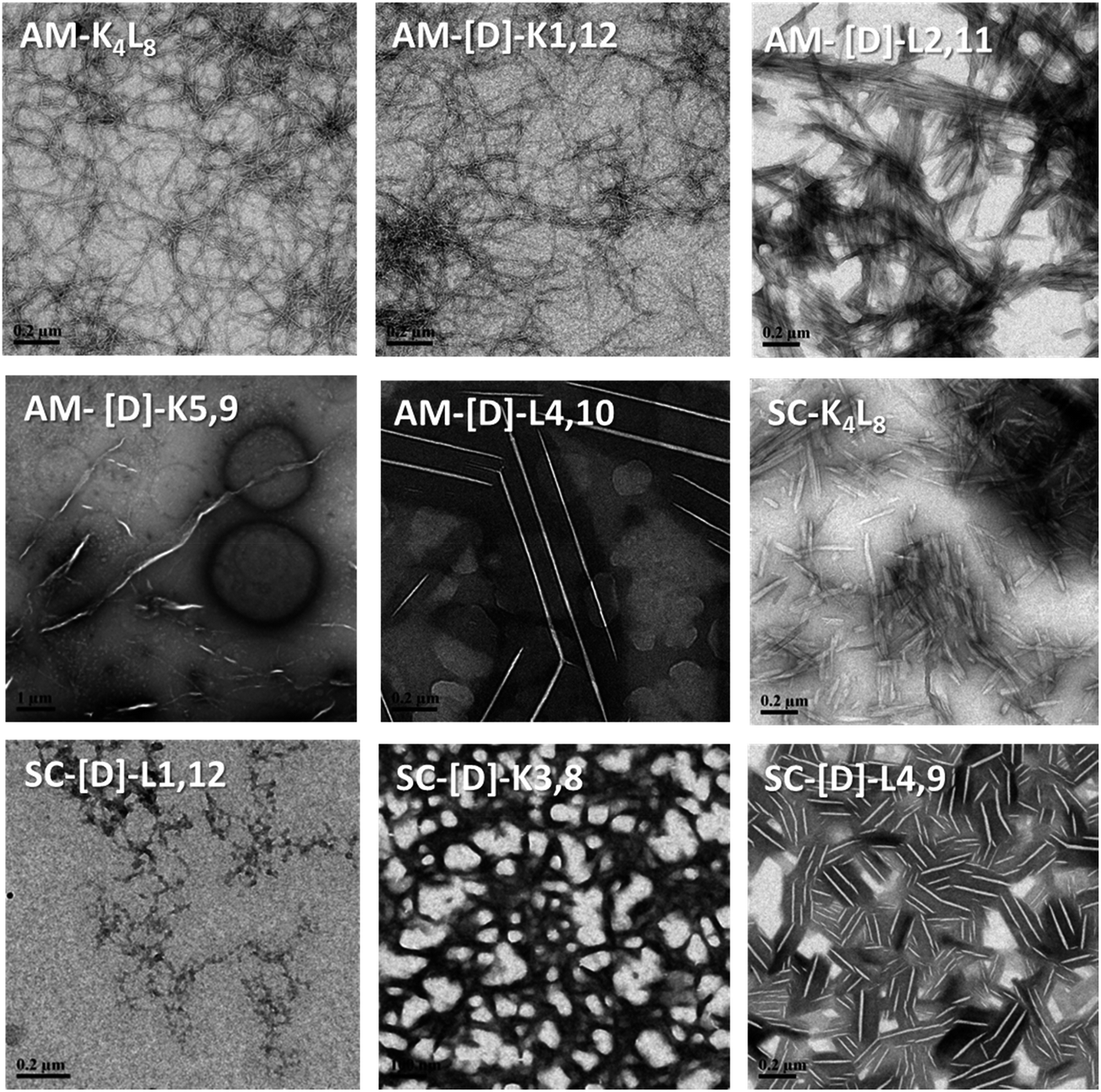 | ||
| Fig. 1 TEM images of the structural morphology of the amphipathic and scrambled sets of diastereomeric peptides. The TEM images were obtained with a JEM-1400 operated at 120 V. The measurements were performed in triplicates using 2.5 mg ml−1 of each sample. | ||
In order to provide additional qualitative characterization of peptide self-assembly, the structural organization of the diastereomeric sets of peptides was monitored by DLS. Due to inherent stochasticity of self-assembly, the aggregate growth rates can vary from one experiment to another making it difficult to compare results between different quantitative measurements (Table S1, in the ESI†). Alternatively, time-dependent autocorrelation function of the fluctuated signal provides qualitative characterization of the particle mobility based on the rate of exponential decay. During measurement, a digital auto-correlator compares the signal measured at a time t0 with very short time delays (dt). As particles move, the correlation between t0 and subsequent dt signals decreases with time, from a perfect correlation to a complete decorrelation at infinite time. In the case of large particles, the signal changes slowly and the correlation persists for a long time, whereas small particles have high Brownian movement causing rapid decorrelation.
We observed a correlation between the peptide structural morphology detected with TEM and their autocorrelation function decay rate. Thus, we found that samples with networked rod-like fibrous morphology (AM-K4L8, and AM-[D]-K1,12 and SC-K4L8) exhibited the fastest exponential decay rates, whereas crystalline structures (AM-[D]-L4,10 and SC-[D]-L4,9) were characterized by minimal decay indicating large aggregates with very low mobility (Fig. S3, in the ESI†). The remaining peptides exhibited autocorrelation function decay rates to an intermediate degree. Although the exact particle size cannot be calculated due to the large variability between the measurements, the overall tendency of quantitative DLS analysis comes in agreement with the qualitative estimation based on autocorrelation function decay rates. Thus, quantitative DLS analysis revealed that each sample consist of at least two separate populations of particles characterized by different mean size and intensity. Whereas AM-K4L8 and AM-[D]-L4,10 samples consist of particles characterized by the smallest and the biggest mean size, respectively, AM-[D]-K2,11 peptide exhibits both very small and very large particles in the mixture. (Table S1, in the ESI†).
LF-Raman modes of the diastereomeric model peptides
At the next step, we studied the correlation between the obtained structural morphologies of the diastereomeric model peptides and their LF-Raman modes. Generally, LFV modes are attributed to the lattice vibrations and dependent on the structure and the intermolecular forces between the molecules.41 Within the low frequency region, certain vibrational modes are due to the lattice vibrational motion of the entire molecule, whereas other modes are strongly influenced by vibrations of the hydrogen bonds.42–46The raw LF-Raman intensity modes of the amphipathic peptide (AM-K4L8) and the set of its diastereomeric analogous are shown at Fig. 2A and are followed by a table that summarizes the peak centers of the individual bands evaluated as explained earlier.
 | ||
| Fig. 2 The raw LF-Raman intensity modes of the amphipathic (A) and scrambled (B) sets of model peptides and their diastereomeric analogous. The curves are offset for better visualization of modes intensity changes between different diastereomeric peptides in the set. For each set, the below tables summarize the peak center of each individual band. The measurements were performed in triplicates using 12.5 mg ml−1 of each sample. | ||
At a glance, the LF-Raman spectra of the diastereomeric peptides share several distinct areas for which low-frequency vibrational modes are active. However, besides the general similarity of the spectra, the relative intensity of each individual mode varies between different diastereomeric peptides in the set. Thus, the region below 90 cm−1 of the amphipathic AM-K4L8 model peptide is characterized by a single, broad mode near 60 cm−1. Through the amphipathic set of diastereomers, it can be clearly seen that along with this mode, the introduction of D-amino acids into the primer sequence resulted in the appearance of additional mode near ∼30 cm−1 with intensity that varies between different diastereomers in the set. Closer analysis revealed that there is a direct correlation between the appearance and the relative intensity of the modes in this area and the position of the D-amino acids within the primary sequence. Evidently, the clearest visual appearance of the ∼30 cm−1 mode is presented for diastereomers with D-amino acids positioned close to the center of the sequence. As the position of D-amino acids moves away from the center, this mode intensity decreases gradually and it totally disappears for AM-[D]-K1,12 peptide for which the D-amino acids are positioned at the ends. Moreover, it seems that the type of amino acid also affects the intensity of the modes, as AM-[D]-L2,11 displays sharper peak at ∼30 cm−1 than AM-[D]-K5,9, despite the larger distance from the center. We can attribute this observation to the larger effect of the hydrophobic amino acid leucine (L) both on local structural distortion and intermolecular hydrophobic interactions, as compared to the positively charged lysine (K). Similarly, the peak at ∼60 cm−1, that appears weak for AM-K4L8 and AM-[D]-K1,12 peptides, becomes more intense as the D-amino acid position moves closer to the center. Based on these results, we hypothesize that the insertion of D-amino acids close to the center of the sequence induces local lattice distortions that enable more structural degrees of freedom of the peptides resulting in wider range of lattice vibrations (phonons) as reflected in LF-Raman spectra.
The region of 90–200 cm−1, that reflects the intermolecular vibrational modes is characterized by two distinguishable spectral bands near 135 cm−1 and 170 cm−1, the relative intensities of which are also determined by the position of the D-amino acids. The LF-Raman spectrum of AM-K4L8 model peptide is characterized by a clear peak at 136 cm−1 accompanied by weak and unclear shoulder at 174 cm−1. The peaks of similar appearance and intensity are also measured for the AM-[D]-K1,12 diastereomer for which the D-amino acids are positioned at the ends of the sequence. However, there is a reversal of peak intensities for both AM-[D]-L4,10 and AM-[D]-K5,9 diastereomers, for which the ∼135 cm−1 peak weakens, whereas the second mode at ∼175 cm−1 appears as much clearer.
In order to further validate the effect of D-amino acids on the LF-Raman modes and examine the effect of amino acids sequences, we measured the Raman spectra of an additional group of peptides that composed of a scrambled analogue of K4L8 (composed of the same amino acids but in non-amphipathic periodicity) and a set of its diastereomer peptides (Fig. 2B). In according to expectations, we observed the similar trend in the appearance and the intensity of the LF-Raman modes with respect to the position of the D-amino acids.
Finally, in order to draw direct parallels to the spectral observations acquired using THz spectroscopy, we converted the Raman scattering intensity into Raman absorption (Fig. S4, in the ESI†).37 For the both sets of peptides, we can clearly observe peaks above 100 cm−1 for the Raman absorption spectra that also have similar trends as described for the Raman scattering intensity.
The designed set of diastereomeric model peptides includes D-amino acids that cause local distortions within the peptide native secondary structure that may affect their LF-Raman modes. In order to examine the effect of the D-amino acids on peptides secondary structure, the LFV modes were complemented by measuring an amide I domain in the molecular fingerprint modes (MFM) spectral region, using the same Raman apparatus. All diastereomeric model peptides display uniformity of spectrum frequencies in the MFM region with a main peak that appears at ∼1674 cm−1, which is attributed to β-sheets, as well as two smaller shoulders at ∼1663 and 1686 cm−1, which are attributed to unordered α-helixes and β-sheets, respectively (Fig. S5, in the ESI†).47 Based on these results, we can conclude that the observed variability of individual bands cannot be ascribed to differences in secondary structures between different diastereomers.
Altogether, the results of the LFV and MFM Raman spectra of the amide I band presented herein demonstrate that stereo-chemical manipulations (by the introduction of D-amino acids) can specifically and selectively manipulate molecular vibrations without changing the sequence, hydrophobicity-to-charge ratio and structure of the peptide.
Correlation between structural morphologies and LF-Raman modes
In order to correlate the structural morphology obtained with TEM/DLS with LFV bands, we broadly categorized the diastereomeric peptides into three groups based upon morphology as fibrils, crystals and aggregates with intermediate structural morphology (Fig. 3A–C). It was difficult to ensure that the laser passed through the identical amount of sample in each measurement. Therefore, we mainly investigated the relative shape changes of the LFV spectra, but not the intensity or absorbance changes. The data were normalized to the peak value of the spectrum at 25 °C.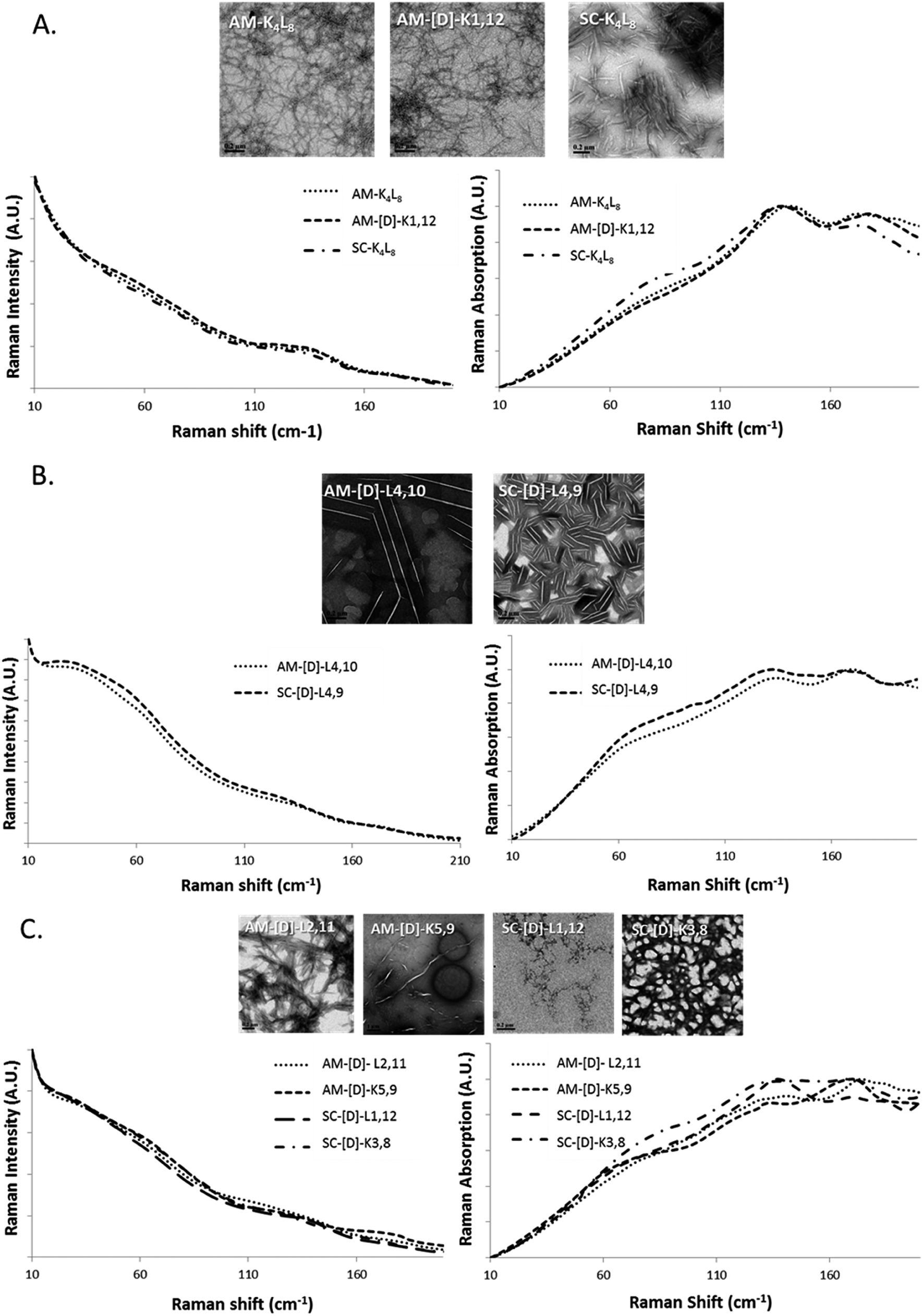 | ||
| Fig. 3 The raw LF-Raman modes of the amphipathic and scrambled sets of model peptides and their diastereomeric analogous categorized into groups according to morphology as (A) fibrils, (B) crystalline and (C) intermediate structures. For each group, the LFV modes are shown as Raman scatterings intensity (left) and Raman absorption (right) spectra. | ||
We found peptides that exhibited networked rod-like fibrous morphology all share similar LVF modes, as shown in Fig. 3A. In the range below 90 cm−1, broad and unclear shoulder is observed near 60 cm−1, and the modes near 30 cm−1 are missing. In the range between 90–200 cm−1, sharp and clear shoulder is observed near 135 cm−1 accompanied by additional broad mode near 175 cm−1. However, in the case of crystalline morphology, the LFV spectra are characterized by clear 30 and 60 cm−1 peaks, while between 90–200 cm−1, both 135 and 175 cm−1 peaks are well-defined, but the 135 cm−1 shoulders much broader in comparison to the fibrils (Fig. 3B). In the case of intermediate structural morphology, we observed mixed spectral features of fibrous and crystalline morphology as shown at Fig. 3C. Below 90 cm−1, there is unclear 30 cm−1 shoulder, while near 60 cm−1, the peaks are similar to those observed for the fibrils. In the 90–200 cm−1 range, both 135 and 175 cm−1 mode had each unique pattern that did not clearly belong to either fibrous or crystalline morphology.
Based on these results, we can attribute tentatively the peaks located at ∼30 cm−1 and ∼60 cm−1 to the flexibility of the whole molecule that depends on the location of D-amino acids. The closer the D-amino acids positioned to the center of the sequence, the sharper the peaks. Similarly, the peaks at ∼130 and ∼175 cm−1 can be correlated with the morphology of the aggregate (fibrillar/crystalline/amorphous).
In order to further investigate the correlation between the three structural groups (fibrils, crystals and aggregates with intermediate structural morphology) and their characteristic LF-Raman modes, the data was inspected using multivariate analysis based on principal component analysis (PCA).48 PCA enables the clustering of data into several groups, relying on a few calculated features out of the original number of spectral features. The PCA was performed on 40 samples in the spectral range 10–200 cm−1, namely 290 spectral channels, in the sampling intervals of the measurement setup. The first three principal components were used to explain 99.82% of the variance in this dataset (PC1 – 92.22%, PC2 – 6.48%, PC3 – 1.12%). Hence, we differentiated between groups in the dataset using only three features instead of the original 290 features. A common way to examine the ability of PCA to differentiate between groups is to use score plots. We have created score plots based on the first three PCs, as shown in Fig. 4. For clarity, different colors were used in the plot for fibrils, crystalline and intermediate structures. Three distinct groups are well resolved in the new 3-dimensional space of the first three PCs. This separation strengthens our conclusion that LF-Raman modes may be used to discriminate between different self-assembly organization of peptides. The actual spectral channels that dominate this discrimination may be used in the future to shed more light on the chemical features of the peptide that lead to different self-assembly organization. Further theoretical study has to be carried in order to gain detailed information in terms of molecular dynamics.
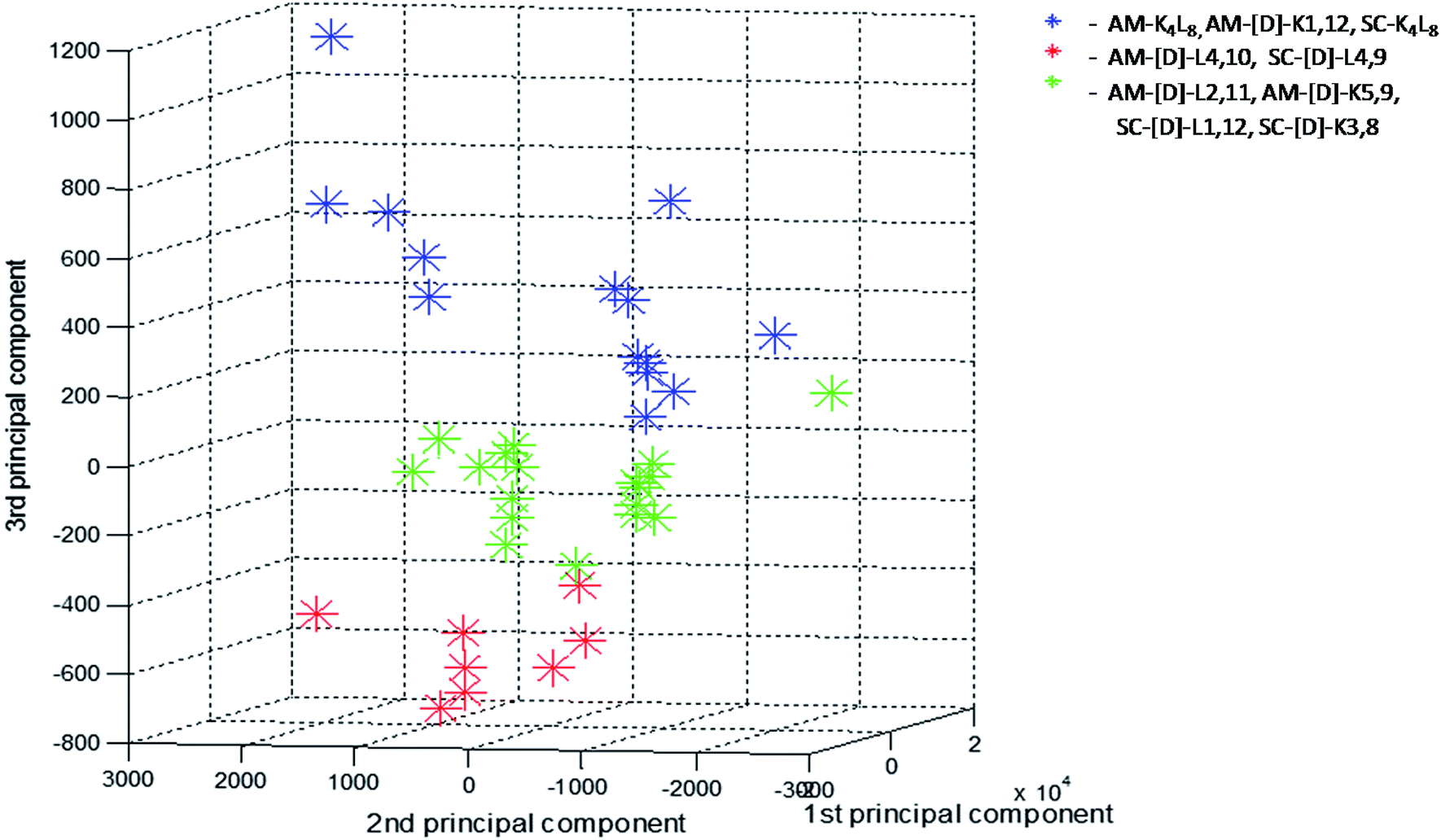 | ||
| Fig. 4 Score values of PC1 × PC2 × PC3 from PCA analysis of the LF-Raman intensity modes in the spectral range 10–200 cm−1 of the various peptides. For each peptide 3–4 spectra replicates were analyzed and presented. Peptides are grouped according to their structural morphologies: blue-fibrils, red-crystals, green-aggregates with intermediate structural morphology. | ||
Conclusions
Under various conditions, peptides and proteins alter from their soluble forms into an aggregated/fibrillar form. Such self-assembly can give rise to pathological conditions ranging from neurodegenerative disorders to systemic amyloidosis.34 On the other hand, molecular self-assembly is a promising strategy for the construction of assemblies of controlled morphology such as peptide and protein-based hydrogels for biomedical applications and peptide nanotubes for inorganic deposition of scaffolds.49,50 Current methods to examine higher-order protein structures including fluorescence, light scattering, electron microscopy, and atomic-force microscopy provide an incomplete picture of the structural interactions.29 In this study, we used LF-Raman spectroscopy to investigate a series of biomolecules in the low frequency range, to gain insight into the structural morphology of peptide self-assembly products. Combining the diastereomeric peptide approach with the single-stage Raman spectrometer and VHG notch filters, we distinguished between different structural morphologies of the diastereomers and observed discrete and consistent peaks after self-assembly, as supported also by PCA analysis of the spectra.The self-assembly tendencies of the peptides examined in this study was mainly driven by their overall hydrophobicity. Amide I domain data showed that despite the different location of D-amino acids in the diastereomers, the aggregates were predominantly organized in β-sheet secondary structures. Thus, the LFV modes we observed in the 90–200 cm−1 band are probably the result of countless intermolecular hydrogen bonding and hydrophobic interactions that hold the β-sheet peptides together, while the modes below 90 cm−1 characterize the flexibility or the degrees of freedom of each peptide within the aggregate. However, at these vibrational energies, there is significant mixing of external and internal motions and thus, the observed vibrational features may originate from far more complex motions. The current work has demonstrate a new way to investigate the self-assembly of peptides. However, a more intensive study is required to characterize and assign the peaks.
Evidently, LF-Raman spectroscopy has the ability to detect structural changes in protein assemblies by providing information regarding the different arrangement of hydrogen bonding and hydrophobic interactions, density of aggregates and their higher order organization. Additionally, this method provides an affordable and robust means to study protein assemblies and simultaneously monitor secondary and higher order structures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
DG thanks European Research Council, ERC-STG grant number 309600 (DG) for funding the research.References
- K. G. Brown, S. C. Erfurth, E. W. Small and W. L. Peticolas, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 1467–1469 CrossRef CAS.
- L. Genzel, F. Keilmann, T. P. Martin, G. Winterling, Y. Yacoby, H. Fröhlich and M. W. Makinen, Biopolymers, 1976, 15, 219–225 CrossRef CAS PubMed.
- P. C. Painter, L. E. Mosher and C. Rhoads, Biopolymers, 1982, 21, 1469–1472 CrossRef CAS PubMed.
- R. H. Austin, M. K. Hong, C. Moser and J. Plombon, Chem. Phys., 1991, 158, 473–486 CrossRef CAS.
- G. Acbas, K. A. Niessen, E. H. Snell and A. G. Markelz, Nat. Commun., 2014, 5, 3076 Search PubMed.
- E. Balog, T. Becker, M. Oettl, R. Lechner, R. Daniel, J. Finney and J. C. Smith, Phys. Rev. Lett., 2004, 93, 28103 CrossRef PubMed.
- L. Xie, Y. Yao and Y. Ying, Appl. Spectrosc. Rev., 2014, 49, 448–461 CrossRef CAS.
- K.-C. Chou, Biophys. Chem., 1988, 30, 3–48 CrossRef CAS PubMed.
- K.-C. Chou, Biophys. J., 1984, 45, 881 CrossRef CAS PubMed.
- Y. El Khoury, A. Trivella and P. Hellwig, IEEE Trans. Terahertz Sci. Technol., 2010, 3, 183–191 Search PubMed.
- D. K. George and A. G. Markelz, in Terahertz Spectroscopy and Imaging, Springer, 2013, pp. 229–250 Search PubMed.
- N. N. Brandt, A. Y. Chikishev, A. V Kargovsky, M. M. Nazarov, O. D. Parashchuk, D. A. Sapozhnikov, I. N. Smirnova, A. P. Shkurinov and N. V. Sumbatyan, Vib. Spectrosc., 2008, 47, 53–58 CrossRef CAS.
- M. Zhang, D. Wei, M. Tang, C. Shi, H. Cui and C. Du, Mol. Simul., 2016, 42, 398–404 CrossRef CAS.
- K. Yamamoto, K. Tominaga, H. Sasakawa, A. Tamura, H. Murakami, H. Ohtake and N. Sarukura, Biophys. J., 2005, 89, L22–L24 CrossRef CAS PubMed.
- A. Matei, N. Drichko, B. Gompf and M. Dressel, Chem. Phys., 2005, 316, 61–71 CrossRef CAS.
- T. M. Korter, R. Balu, M. B. Campbell, M. C. Beard, S. K. Gregurick and E. J. Heilweil, Chem. Phys. Lett., 2006, 418, 65–70 CrossRef CAS.
- C. S. Ponseca Jr, O. Kambara, S. Kawaguchi, K. Yamamoto and K. Tominaga, J. Infrared, Millimeter, Terahertz Waves, 2010, 31, 799–809 CrossRef.
- C. S. Ponseca Jr, O. Kambara, S. Kawaguchi and K. Tominaga, in Infrared, Millimeter, and Terahertz Waves, 2009. IRMMW-THz 2009. 34th International Conference on, IEEE, 2009, pp. 1–2 Search PubMed.
- K. Siegrist, C. R. Bucher, C. Pfefferkorn, A. Schwarzkopf and D. F. Plusquellic, in ACS symposium series, Oxford University Press, 2007, vol. 963, pp. 280–296 Search PubMed.
- D. F. Plusquellic, K. Siegrist, E. J. Heilweil and O. Esenturk, ChemPhysChem, 2007, 8, 2412–2431 CrossRef CAS PubMed.
- B. Fanconi, Biopolymers, 1973, 12, 2759–2776 CrossRef CAS PubMed.
- J. F. Rabolt, W. Wedding and K. W. Johnson, Biopolymers, 1975, 14, 1615–1622 CrossRef CAS PubMed.
- K. Siegrist, C. R. Bucher, I. Mandelbaum, A. R. Hight Walker, R. Balu, S. K. Gregurick and D. F. Plusquellic, J. Am. Chem. Soc., 2006, 128, 5764–5775 CrossRef CAS PubMed.
- F. Piccirilli, G. Schirò, V. Vetri, S. Lupi, A. Perucchi and V. Militello, Biophys. Chem., 2015, 199, 17–24 CrossRef CAS PubMed.
- H. A. Zakaria, B. M. Fischer, A. P. Bradley, I. Jones, D. Abbott, A. P. J. Middelberg and R. J. Falconer, Appl. Spectrosc., 2011, 65, 260–264 CrossRef CAS PubMed.
- R. Liu, M. He, R. Su, Y. Yu, W. Qi and Z. He, Biochem. Biophys. Res. Commun., 2010, 391, 862–867 CrossRef CAS PubMed.
- R. J. Falconer, H. A. Zakaria, Y. Y. Fan, A. P. Bradley and A. P. J. Middelberg, Appl. Spectrosc., 2010, 64, 1259–1264 CrossRef CAS PubMed.
- Y. Qu, H. Chen, X. Qin, L. Li, L. Wang and T. Kuang, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 1614–1618 CrossRef CAS PubMed.
- G. M. Png, R. J. Falconer and D. Abbott, IEEE Trans. Terahertz Sci. Technol., 2016, 6.1, 45–53 CrossRef.
- R. J. Falconer and A. G. Markelz, J. Infrared, Millimeter, Terahertz Waves, 2012, 33, 973–988 CrossRef CAS.
- B. S. Kalanoor, M. Ronen, Z. Oren, D. Gerber and Y. R. Tischler, ACS Omega, 2017, 2, 1232–1240 CrossRef CAS PubMed.
- Z. Oren, J. Hong and Y. Shai, J. Biol. Chem., 1997, 272, 14643–14649 CrossRef CAS PubMed.
- H. Xiong, B. L. Buckwalter, H.-M. Shieh and M. H. Hecht, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 6349–6353 CrossRef CAS.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- K. F. DuBay, A. P. Pawar, F. Chiti, J. Zurdo, C. M. Dobson and M. Vendruscolo, J. Mol. Biol., 2004, 341, 1317–1326 CrossRef CAS PubMed.
- A.-M. Fernandez-Escamilla, F. Rousseau, J. Schymkowitz and L. Serrano, Nat. Biotechnol., 2004, 22, 1302–1306 CrossRef CAS PubMed.
- P. A. Lund, O. Faurskov Nielsen and E. Praestgaard, Chem. Phys., 1978, 28, 167–173 CrossRef CAS.
- I. T. Jolliffe, Principal Component Analysis, Springer New York, New York, NY, 1986 Search PubMed.
- S. E. Blondelle and R. A. Houghten, Biochemistry, 1992, 31, 12688–12694 CrossRef CAS PubMed.
- M. Schiffer and A. B. Edmundson, Biophys. J., 1967, 7, 121 CrossRef CAS PubMed.
- P. J. Larkin, M. Dabros, B. Sarsfield, E. Chan, J. T. Carriere and B. C. Smith, Appl. Spectrosc., 2015, 68, 758–776 CrossRef PubMed.
- K.-C. Chou, Biophys. J., 1985, 48, 289 CrossRef CAS PubMed.
- S. Dörr, U. Schade and P. Hellwig, Vib. Spectrosc., 2008, 47, 59–65 CrossRef.
- L. I. Maklakov and S. V. Aksakova, Russ. Chem. Rev., 1997, 66, 375–388 CrossRef.
- R. Hielscher and P. Hellwig, ChemPhysChem, 2010, 11, 435–441 CrossRef CAS PubMed.
- Y. El Khoury, A. Trivella, J. Gross and P. Hellwig, ChemPhysChem, 2010, 11, 3313–3319 CrossRef CAS PubMed.
- S. U. Sane, S. M. Cramer and T. M. Przybycien, Anal. Biochem., 1999, 269, 255–272 CrossRef CAS PubMed.
- S. Nasser, A. Hédoux, A. Giuliani, C. Le Floch-Fouéré, V. Santé-Lhoutellier, I. de Waele and G. Delaplace, J. Sci. Food Agric., 2018, 98, 2243–2250 CrossRef CAS PubMed.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- C. Valéry, F. Artzner and M. Paternostre, Soft Matter, 2011, 7, 9583 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01232f |
| ‡ Equally contributed authors. |
| This journal is © The Royal Society of Chemistry 2018 |