
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolydopamine-coated graphene nanosheets as efficient electrocatalysts for oxygen reduction reaction†
Dan-Dan Wanga,
Xiuli Gaob,
Lianming Zhaob,
Jin Zhou a,
Shuping Zhuoa,
Zifeng Yanb and
Wei Xing*b
a,
Shuping Zhuoa,
Zifeng Yanb and
Wei Xing*b
aSchool of Chemical Engineering, Shandong University of Technology, Zibo 255049, P. R. China
bSchool of Science, State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Qingdao 266580, P. R. China. E-mail: xingwei@upc.edu.cn
First published on 30th April 2018
Abstract
Polydopamine-modified graphene (G-PDA) materials were synthesized by in situ polymerization of a dopamine monomer on the surface of graphene oxide. X-ray photoelectron spectroscopy (XPS) has confirmed that new N-containing functional groups are formed during the synthesis process, which result in the excellent electrocatalytic activity of the composite towards ORR in terms of onset potential, number of electron transferred and limiting current density. The electrocatalytic activity of the optimized G-PDA sample is better than N-doped graphene and comparable to the commercial 20 wt% Pt/C catalyst. Furthermore, compared with the Pt-based catalysts, the G-PDA showed superior stability and methanol resistance, which favored its practical applications in fuel cells.
1 Introduction
The growing global energy demand and environmental pollution of traditional energy resources pose severe challenges to human health, energy regeneration, and environmental protection.1–4 One promising solution is fuel cell technology because of their high efficiency and environmental friendliness. It is well known that fuel cells require a catalyst for oxygen reduction reaction (ORR) in the cathode. Traditionally, platinum (Pt) has been identified as the best ORR catalyst. Whereas, the high cost,5,6 few reserves7 and weak durability8,9 of Pt seriously impede their large-scale applications. Thus, exploring non-precious metals or even metal free catalysts is highly desirable.Actually, many studies have demonstrated that heteroatom-doping can effectively enhance ORR activity of carbon. Those heteroatoms include nitrogen,10–14 boron,15,16 sulfur,17,18 phosphorous19,20 and their mixtures.21–23 Among these heteroatom-doped carbons, the nitrogen-doped carbon materials, such as nitrogen-doped graphene,24–26 nitrogen-doped nanotubes27,28 and nitrogen-doped carbon spheres,29,30 have been mostly investigated due to their easy preparation and high ORR activity.31 For example, Zhang et al. prepared the N-doped mesoporous graphene by a chemical vapor deposition (CVD) using CH4 and NH3 as the carbon source and nitrogen source, respectively. The prepared N-doped graphene exhibited superior ORR activity with onset potential of 0.81 V vs. RHE to its counterpart without N-doping of 0.76 V.32 Ruoff et al. prepared nitrogen-doped graphene by annealing of polyaniline or polypyrrole/reduced graphene oxide composite. The product displays an onset potential of up to −0.18 V vs. Ag/AgCl, which is a little bit smaller than that of 20% Pt/C.33 Wong et al. prepared N-doped graphene by thermal annealing graphene oxide with melamine. The results show that the ORR undergo by a one-step four-electron pathway, and the onset potential is up to −0.10 V vs. Ag/AgCl that is comparable to that of 20 wt% Pt/C (−0.03 V vs. Ag/AgCl).34 Chen et al. synthesized nitrogen-doped carbon nanotubes (N-CNT) by a CVD method using ethylenediamine as nitrogen source. The half-wave potential of the N-CNT is −0.15 V vs. Ag/AgCl and the limiting current density is −4.91 mA cm−2, which are similar to those of the commercial Pt/C catalyst.35 Wang et al. prepared pyridinic nitrogen-rich carbon nanospheres by direct annealing of polydopamine derivative. The product shows superior ORR activity with a onset potential of 0.98 V and a high half-wave potential of 0.88 V, which are comparable to the 20 wt% Pt/C.36 Up to date, the N-containing carbonaceous ORR catalysts reported previously are all N-doped carbons, where the N atoms exist in the carbon lattice. Besides, most the N-doped carbon catalysts were prepared by high energy-consuming processes, such as chemical vapor deposition37 in the presence of a nitrogen source, thermal annealing with ammonia38 or pyrolysis of a nitrogen-containing precursor (such as melamine, polypyrrole, and pyridine)39–41 at high temperatures.
Herein, we propose a novel polydopamine (PDA)/graphene catalyst with high performance towards ORR by in situ polymerization of dopamine monomer on the surface of graphene oxide. This catalyst distinguished itself with the previous N-doped carbon catalysts in the following two points: (1) the N atoms in the catalyst do not exist in the carbon lattice, but in the polymer (polydopamine); (2) the preparation avoids annealing or pyrolysis at high temperatures, which is a quite energy-saving process. Electrochemical measurements show that the optimized PDA/graphene composite has better catalytic activity towards ORR than N-doped graphene and comparable to the commercial 20 wt% Pt/C catalyst. Furthermore, compared with the Pt-based catalysts, the composite catalyst shows superior stability and methanol resistance, which highlights its practical applications in fuel cells.
2 Experimental methods
2.1 Materials and apparatus
Flake graphite and potassium hydroxide (KOH) was obtained from Sinopharm Chemical Reagent. Dopamine hydrochloride (98%) and Tris(hydroxylmethyl) aminomethane hydrochloride (Tris–HCl, 99%) were purchased from Aladdin Chemical Reagent. The commercial Pt/C 20 wt% was obtained from Johnson Matthey Co. Ltd, which is commonly used as a standard in the ORR test. All reagents were of analytical grade and used as received without further purification. Deionized water was used as solvent for all experiments.Morphology of the samples was observed by field-emission scanning electron microscope (SEM, JEOL JSM-6700F). X-ray diffraction (XRD) measurements were obtained on a D/Max 2500V/PC diffractometer using Cu Kα radiation with wavelength of 0.15405 nm. The surface chemical state of the samples was measured by X-ray photoelectron spectroscopy (XPS, ESCALAB MK II VG Co., UK) using Al Kα radiation. Fourier transform infrared (FTIR) spectra were recorded on a Hitachi EPI Infrared Spectrophotometer using a KBr disk with an active material/KBr mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50. Raman spectra were conducted on a confocal laser micro Raman spectrometer (LABRAM-HR, JY Co.).
:50. Raman spectra were conducted on a confocal laser micro Raman spectrometer (LABRAM-HR, JY Co.).
2.2 Preparation of graphene oxide aerogel (GOA)
The graphite oxide was prepared according to the modified Hummer's method.42,43 The graphene oxide hydrogel was prepared according to Worsley's method.44 In a typical synthesis, the graphite oxide was dispersed in deionized water and sonicated to get homogeneous suspension of graphene oxide (2 mg mL−1). The suspension was then aged at 85 °C for 72 h. The resulting graphene hydrogel was washed with deionized water for several times to remove impurities and then lyophilized to obtain GOA.2.3 Preparation of PDA/graphene composite
The PDA/graphene composite was prepared by using the following procedure: 40 mg of graphite oxide was dispersed in deionized water and the suspension was dispersed by sonication for 30 min (2 mg mL−1). Then a certain amount of dopamine hydrochloride was added and the mixture was stirred at room temperature for 30 min. Subsequently, 15 μL of Tris–HCl solution (pH = 8.5) was added and the mixture was stirred vigorously at room temperature for another 30 min, and then aged at 85 °C for 72 h. The obtained product was washed with deionized water several times to remove impurities and then the wet hydrogel was lyophilized. The dried product is designated as G-PDA-m, where m represents the concentration of dopamine in the solution. For example, G-PDA-0.5 denotes that the composite is prepared in the dopamine solution with concentration of 0.5 mg mL−1.For comparison, G-PDA-1 was also carbonized at different temperatures in Ar atmosphere, and the carbonized products are denoted as C-G-PDA-t, where t represents the carbonization temperature.
2.4 Electrochemical measurements
The preparation of the working electrode was as follows: 1 mg of active material was added into 0.68 mL of deionized water, 0.3 mL of ethanol and 20 μL of 5 wt% Nafion solution, which was dispersed by ultrasonication for 30 min to obtain a homogeneous suspension. Subsequently, 5.65 μL of this suspension was transferred onto a rotating disk electrode to be fully dried.The ORR activity of all samples was tested in a conventional three-electrode cell using a CHI 760E electrochemical workstation (Shanghai Chenhua Instrument Co., China) equipped with a rotating electrode setup. Ag/AgCl electrode and Pt wire were used as the reference electrode and counter electrode, respectively. The RDE (rotating disk electrode) and rotating ring disk electrode (RRDE) measurements were obtained in an O2-saturated 0.1 M KOH aqueous solution at a scan rate of 10 mV s−1 with various rotation speeds (400–2500 rpm).
In the process of oxygen catalytic reduction, the electron transfer number (n) and kinetic current density (Jk) were calculated according to the Koutecky–Levich (K–L) equation:

| B = 0.2nFCo(Do)2/3v−1/6 |
485 C mol−1), Co is the bulk concentration of O2 (1.2 × 10−6 mol cm−3), Do is the diffusion coefficient of O2 (1.9 × 10−5 cm2 s−1), and ν is the kinetic viscosity of the electrolyte (0.01 cm2 s−1). Here, the constant 0.2 is adopted when the rotating rate is expressed in rpm.
Hydrogen peroxide yields and the number of transferred electrons were calculated using the following equation:

where the collection efficiency (N) was determined to be 0.412 in an Ar atmosphere using 10 mM K3[Fe(CN)6].
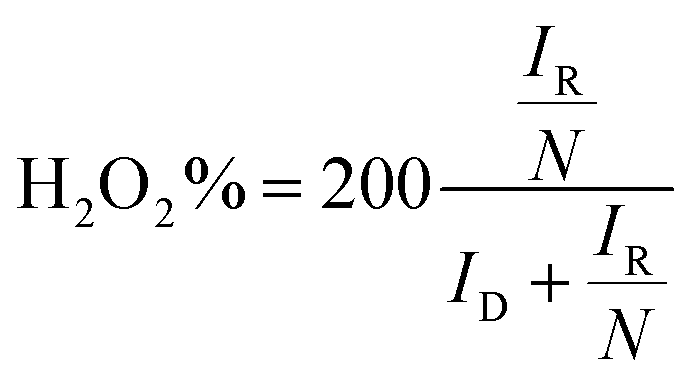
3 Results and discussion
3.1 Morphology and structure of G-PDA-m materials
The morphology of the as-prepared G-PDA-m were observed by SEM. As shown in Fig. 1a, GOA shows three-dimensional network structure with gauzy and folded graphene sheets cross-linked with each other. With the presence and increase of PDA, the corrugated nanosheets become flat gradually possibly due to the presence of the PDA that is adsorbed on the surface of graphene oxide (Fig. 1b–d). However, when there is too much PDA present, the aggregation of G-PDA nanosheets occurs, as is shown in Fig. 1e and f. | ||
| Fig. 1 SEM images of (a) GOA, (b) G-PDA-0.5, (c) G-PDA-1, (d) G-PDA-2, (e) G-PDA-3, (f) G-PDA-4. | ||
X-ray diffraction (XRD) was conducted to investigate the crystal structure of the prepared samples (Fig. 2). A sharp peak at 2θ = 10° was assigned to the [002] plane of GO with a d-spacing of 0.95 nm (Fig. 2a). After the addition of the dopamine, the peak (2θ = 9.3°) decreased observably and a new broad diffraction peak (2θ = 25°) with an interlayer spacing of (∼0.356 nm) appeared, corresponding to the [002] plane of graphite (Fig. 2b).
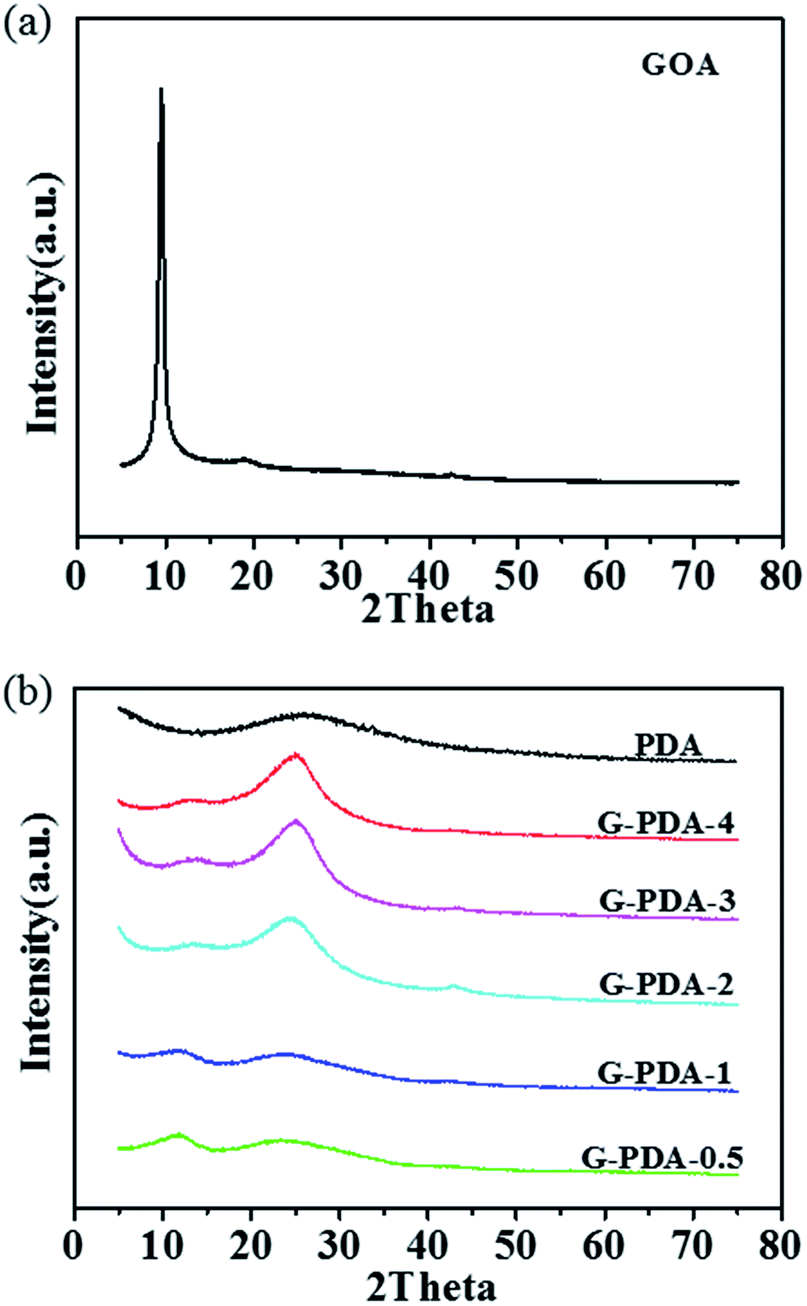 | ||
| Fig. 2 XRD patterns of (a) GOA and (b) G-PDA-m and PDA products. | ||
This means that GO are partially reduced to rGO during the polymerization of dopamine. As the increase amount of dopamine, the intensity of peak at 2θ = 9.3° was decreasing and the peak at 2θ = 25° was increasing gradually, indicating that more dopamine dosage results in deeper reduction of GO. At the same time, the peak (2θ = 9.3°) becomes very broad, and shifts to higher angles with increasing dopamine. This is because the oxygen-functional groups in the interlayer spacing was gradually removed in the process of GO reduction.45,46
Fourier transform infrared (FTIR) spectroscopy was used to reveal the functional groups contained in the as-prepared materials. As shown in Fig. 3a, the GOA showed typical bands at 1450, 1666, 1600 and 1080 cm−1, which were attributed to the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching, CO carboxyl stretching, asymmetric and symmetric C–O stretching, respectively. The bands at 2943 and 2826 cm−1 were attributed to the stretching vibration of C–H, which may be formed by the combination of proton (H+) and carbon defects on the edge of graphene during the hydrothermal process.47 For G-PDA nanocomposites, the same bands could be found, and two new-bands occur at 1000 cm−1 and 3260 cm−1, which are due to the C–N and N–H groups in the PDA. The above information suggest the successful combination of PDA and GO. Comparing the as-prepared catalysts with different PDA/GO mass ratio, its easy to found that the curves are similar, and the bands due to C–N and N–H stretching vibrations became stronger with the increase of PDA content (Fig. 3b).
C stretching, CO carboxyl stretching, asymmetric and symmetric C–O stretching, respectively. The bands at 2943 and 2826 cm−1 were attributed to the stretching vibration of C–H, which may be formed by the combination of proton (H+) and carbon defects on the edge of graphene during the hydrothermal process.47 For G-PDA nanocomposites, the same bands could be found, and two new-bands occur at 1000 cm−1 and 3260 cm−1, which are due to the C–N and N–H groups in the PDA. The above information suggest the successful combination of PDA and GO. Comparing the as-prepared catalysts with different PDA/GO mass ratio, its easy to found that the curves are similar, and the bands due to C–N and N–H stretching vibrations became stronger with the increase of PDA content (Fig. 3b).
 | ||
| Fig. 3 FTIR analyses of the (a) PDA, G-PDA-1 and GOA and (b) G-PDA-m; (c) Raman analyses of the PDA, G-PDA-m and GOA. | ||
The Raman spectra are commonly used to reveal the structure of carbon skeleton in the materials. As shown in Fig. 3c, the two typical peaks at approximate 1354 cm−1 and 1600 cm−1 are consistent with the characteristic D and G bands, respectively, for the GOA. The D band was associated with structural defects and partially disordered structures on the graphene nanosheets. The G band, as an index of the degree of graphitization, is in connection to the E2g vibration mode of sp2 carbon domains. Generally, the intensity ratio of the D band to G band (ID/IG) is used to evaluate the degree of defects for graphene-based materials. In contrast, D band is almost invisible and G band is prominent for PDA, indicating that most carbon atoms in PDA are sp2 carbon. Of special interest is the fact that ID/IG of G-PDA (1.00–1.02) was higher than that of GOA (0.99), suggesting that PDA may strongly interact with GO in the composite and induce more carbon defects.
The surface chemical composition and elemental chemical state of the as-prepared samples were characterized by XPS. As shown in Fig. 4a, C and O elements were present in all the three samples of GOA, PDA and G-PDA-1, but N element only existed in the G-PDA-1 and PDA but not in GOA, confirming the successful combination of PDA and GO using in situ polymerization of dopamine. The C1s spectrum of G-PDA-1 (Fig. 4b) can be deconvolved into five peaks with binding energies at about 284.8, 286.0, 286.8, 287.8 and 288.9 eV, corresponding to C–C, C–N, C–O, CO, and O–CO species, respectively. The N1s spectrum of G-PDA-1 (Fig. 4c) can be deconvolved into four peaks at about 398.7, 400.3, 401.4 and 402.4 eV, attributable to pyridinic N (6.84%), pyrrolic N (59.8%), graphitic N (20.51%) and oxidized N (12.82%), respectively. In contrast, the N1s spectrum of PDA showed only one peak at about 400.3 eV, which is attributed to the pyrrolic N (Fig. 4d). Combined with the results of Raman analyses, it can be deduced that the strong interaction between GO and PDA generated both carbon defects and various types of N-containing functional groups that may show activity towards ORR.
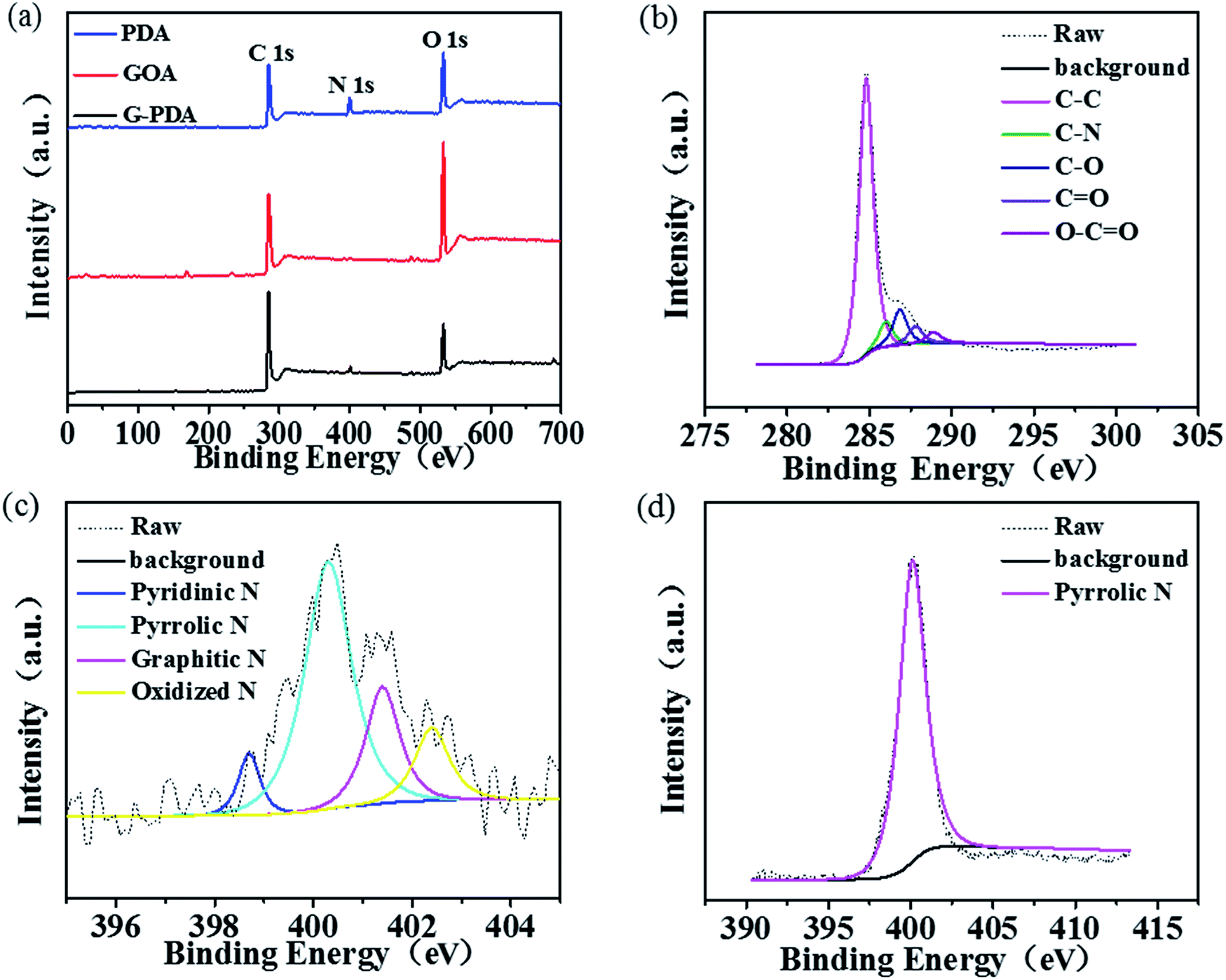 | ||
| Fig. 4 (a) XPS spectra of GOA, PDA and G-PDA-1; (b) C 1s of G-PDA-1; (c) N 1s of G-PDA-1 (d) N 1s of PDA. | ||
3.2 Electrocatalytic activity of the as-prepared samples towards ORR
The catalytic activity of the samples towards ORR was assessed by linear sweep voltammograms (LSV) in O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1 between 0 and +1.0 V vs. RHE. For comparison, the LSVs of different catalysts with a rotation rate of 1600 rpm were shown in Fig. 5a, Fig. S1 and S2.† According to limiting current densities, the onset potential for G-PDA-1 was around +0.88 V vs. RHE, which was higher than the pure GOA (+0.74 V vs. RHE), PDA (+0.74 V vs. RHE) and C-G-PDA-800 (+0.76 V vs. RHE), but still lower than 20 wt% Pt/C (+0.96 V vs. RHE). Moreover, G-PDA-1 displayed the most positive half-wave potential (the potential at which the current is half of the limiting current) among all the other samples prepared, and is almost the same as the 20 wt% Pt/C, further confirming that the G-PDA-1 had excellent electrocatalytic activity towards ORR. The possible interpretation for the outstanding electrochemical performance of G-PDA-1 is mainly because of the carbon defects and various types of N-containing functional groups. The defects change the charge density, resistance in charge transfer, and hydrophilicity of the composite, which may increase its electrical conductivity and interaction with oxygen.48 Moreover, the various types of N-containing functional groups will provide more active sites for ORR.2,49 | ||
| Fig. 5 (a) LSV curves for G-PDA-1, C-G-PDA-800, GOA, PDA and 20 wt% Pt/C in O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1; (b) the corresponding Koutecky–Levich plots at different potentials; (c) the rotating-disk voltammograms of G-PDA-1 in O2-saturated 0.1 M KOH with a sweep rate of 10 mV s−1 at various rotation speeds; (d) electron-transfer numbers and H2O2 yields of G-PDA-1, GOA, PDA and 20 wt% Pt/C with a rotation rate of 1600 rpm. | ||
To further research the oxygen reduction process occurred on G-PDA-1, LSV measurements were carried out at various rotation speeds in O2-saturated 0.1 M KOH solution. As shown in Fig. 6b and c, the Koutecky–Levich (K–L) plots (J−1 vs. ω−1/2) of G-PDA-1 at various potentials were given. Accordingly, the electron transfer number (n) is calculated to be 4.07. The H2O2 yield and the electron transfer number (n) calculated from the RRDE curves are presented in Fig. 5d. The measured H2O2 yields are around 10% over the entire potential range of 0.1 to 0.8 V vs. RHE for the G-PDA-1 catalyst. This value is significantly lower than that of the GOA or PDA catalyst, which is near 80%, and the electron transfer number was about 3.74–3.9 that is close to a commercial Pt/C catalyst (n = 3.78–3.94). This is consistent with the result obtained from the K–L plot based on the RDE measurements, indicating that G-PDA-1 favors a 4e− oxygen reduction process, while GOA electrode involves a 2e− reduction process with H2O2 as the intermediate agent.50
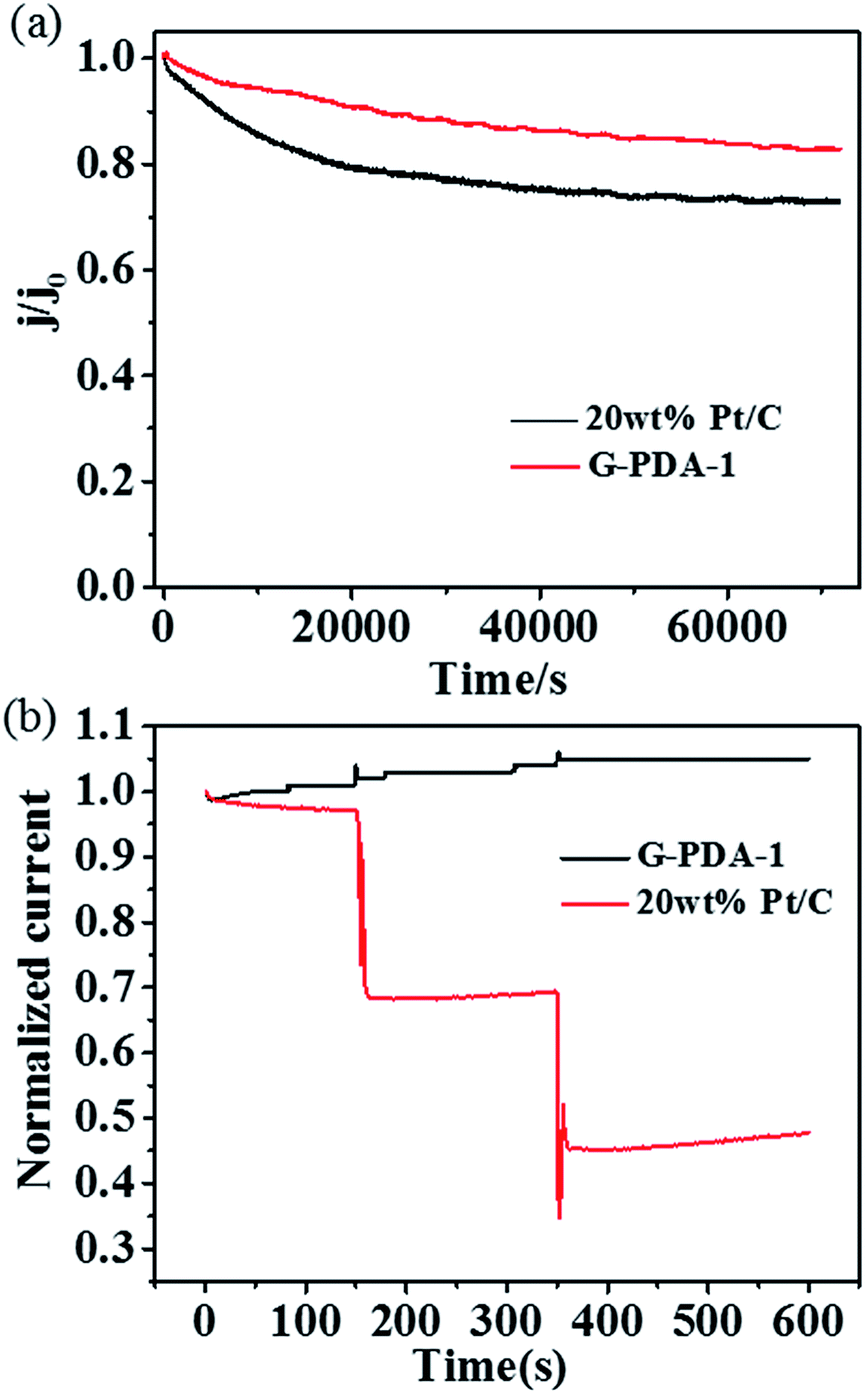 | ||
| Fig. 6 (a) Stability test of G-PDA-1 and 20 wt% Pt/C catalyst for 70000 s, (b) methanol crossover test by addition of methanol to the electrochemical cell at 150 s and 350 s. | ||
Based on the above results, we can conclude that G-PDA-1 possessed excellent electrocatalytic activity towards ORR, which can be ascribed to the following three reasons: (1) the strong interactions between GO and PDA induced the carbon defects and a variety of N-containing active sites for ORR; (2) the large surface of G-PDA nanosheets will further increase the catalytically active sites; (3) the intimate contact between GO and PDA facilitates charge-transfer, and the good conductivity of partially-reduced GO substrate facilitates electron transport.
In addition, the durability of G-PDA-1 and 20 wt% Pt/C was measured by holding them at +0.67 V vs. RHE for 70000 s in O2-saturated 0.1 M KOH solution (Fig. 6a). Compared with 20 wt% Pt/C, the chronoamperometric response for the G-PDA-1 exhibited much slower decay. After reaction for 70000 s, G-PDA-1 still maintains 83% of its initial current density, which surpasses 20 wt% Pt/C of about 73%. Unambiguously, the G-PDA-1 also has an excellent stability in alkaline solution, which highlights its great potentials in alkaline fuel cells. It is thought that the combination of PDA and graphene also enhanced the material's mechanical stability, which may contribute to its excellent electrocatalytic stability.51
Because methanol tolerance of cathode catalyst is the one of the major evaluating factor for direct methanol fuel cell (DMFC), chronoamperometry was used to characterize methanol tolerance of the catalysts at a constant voltage of 0.67 V vs. RHE in O2-saturated 0.1 M KOH aqueous solution (Fig. 6b). Obviously, after addition of methanol at 150 s and 350 s, two keen-edged current decrease were immediately observed for Pt/C catalyst, suggesting the poisoning of Pt/C catalyst. Whereas, no visible change was observed for G-PDA-1, indicating its excellent property of methanol tolerance and its potential application in DMFC.
4 Conclusions
In summary, we present a novel G-PDA catalyst with high activity towards ORR, which were synthesized by in situ polymerization of dopamine on the surface of GO. As evidenced by structure characterizations, the strong interaction occurs between GO and PDA, inducing a variety of N-containing active sites towards ORR. Electrochemical measurements demonstrated that G-PDA-1 exhibited excellent catalytic activity towards ORR, in terms of onset potential, number of electron transferred and limiting current density, which was comparable to the commercial 20 wt% Pt/C catalyst. Both the G-PDA catalyst proposed in this work and the energy-saving strategy used to synthesize G-PDA are very significant to developing advanced ORR catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (21476264), Taishan Scholar Foundation (tsqn20161017, ts20130929), and Fundamental Research Funds for the Central Universities (18CX05007A, 15CX08009A).Notes and references
- M. S. Balogun, W. Qiu, Y. Luo, H. Meng, W. Mai, A. Onasanya, T. Olaniyi and Y. Tong, Nano Res., 2016, 9, 2823–2851 CrossRef CAS
.
- C. Hu and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 11736–11758 CrossRef CAS PubMed
- J. B. Goodenough, Acc. Chem. Res., 2012, 46, 1053–1061 CrossRef PubMed
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459–1606479 CrossRef PubMed
- K. Li, Y. Li, Y. Wang, F. He, M. Jiao, H. Tang and Z. Wu, J. Mater. Chem. A, 2015, 3, 11444–11452 CAS
- S. Sugawara, K. Tsujita, S. Mitsushima, K. Shinohara and K. I. Ota, Electrocatalysis, 2011, 2, 60–68 CrossRef CAS
- Y. Shao, J. Liu, Y. Wang and Y. Lin, J. Mater. Chem., 2009, 19, 46–59 RSC
- W. M. Wang, Z. L. Li, Z. Zou, H. Yang and S. Feng, ECS Trans., 2008, 16, 613–619 CAS
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed
- C. Venkateswara Rao and Y. Ishikawa, J. Phys. Chem. C, 2012, 116, 4340–4346 CAS
- G. Wu, N. H. Mack, W. Gao, S. Ma, R. Zhong, J. Han, J. K. Baldwin and P. Zelenay, ACS Nano, 2012, 6, 9764–9776 CrossRef CAS PubMed
- S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593–5597 CrossRef CAS PubMed
- J. Xu, S. Lu, X. Chen, J. Wang, B. Zhang, X. Zhang, C. Xiao and S. Ding, Nanotechnology, 2017, 28, 485701 CrossRef PubMed
- Y. Zhao, L. Yang, S. Chen, X. Wang, Y. Ma, Q. Wu, Y. Jiang, W. Qian and Z. Hu, J. Am. Chem. Soc., 2013, 135, 1201–1204 CrossRef CAS PubMed
- M. Chisaka, T. Iijima, Y. Ishihara, Y. Suzuki, R. Inada and Y. Sakurai, Electrochim. Acta, 2012, 85, 399–410 CrossRef CAS
- L. Zhang, J. Niu, M. Li and Z. Xia, J. Phys. Chem. C, 2014, 118, 3545–3553 CAS
- L. Chen, X. Cui, Y. Wang, M. Wang, R. Qiu, Z. Shu, L. Zhang, Z. Hua, F. Cui, C. Wei and J. Shi, Dalton Trans., 2014, 43, 3420–3423 RSC
- D. S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. S. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed
- S. K. Ramasahayam, U. B. Nasini, V. Bairi, A. U. Shaikh and T. Viswanathan, RSC Adv., 2014, 4, 6306–6313 RSC
- F. Razmjooei, K. P. Singh, M. Y. Song and J. S. Yu, Carbon, 2014, 78, 257–267 CrossRef CAS
- H. Wu, L. Shi, J. Lei, D. Liu, D. Qu, Z. Xie, X. Du, P. Yang, X. Hu, J. Li and H. Tang, J. Power Sources, 2016, 323, 90–96 CrossRef CAS
- Z. Jiang, X. Zhao, X. Tian, L. Luo, J. Fang, H. Gao and Z. J. Jiang, ACS Appl. Mater. Inter., 2015, 7, 19398–19407 CrossRef CAS PubMed
- L. Qu, Y. Liu, J. B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS
- Y. Tang, S. C. Burkert, Y. Zhao, W. A. Saidi and A. Star, J. Phys. Chem. C, 2013, 117, 25213–25221 CAS
- D. Yu, Q. Zhang and L. Dai, J. Am. Chem. Soc., 2010, 132, 15127–15129 CrossRef CAS PubMed
- Y. Li, T. Li, M. Yao and S. Liu, J. Mater. Chem., 2012, 22, 10911–10917 RSC
- X. Zhou, Z. Yang, H. Nie, Z. Yao, L. Zhang and S. Huang, J. Power Sources, 2011, 196, 9970–9974 CrossRef CAS
- L. Dai, Y. Xue, L. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed
- H. F. Wang, C. Tang and Q. Zhang, Catal. Today, 2018, 301, 25–31 CrossRef CAS
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 CAS
- Z. Lin, M. K. Song, Y. Ding, Y. Liu, M. Liu and C. P. Wong, Phys. Chem. Chem. Phys., 2012, 14, 3381–3387 RSC
- Z. Chen, D. Higgins, H. Tao, R. S. Hsu and Z. Chen, J. Phys. Chem. C, 2009, 113, 21008–21013 CAS
- Z. Yi, Z. Zhang, S. Wang and G. Shi, J. Mater. Chem. A, 2017, 5, 519–523 CAS
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed
- S. Wang, D. Yu, L. Dai, D. W. Chang and J. B. Baek, ACS Nano, 2011, 5, 6202–6209 CrossRef CAS PubMed
- K. Ai, Y. Liu, C. Ruan, L. Lu and G. M. Lu, Adv. Mater., 2013, 25, 998–1003 CrossRef CAS PubMed
- K. N. Wood, R. O'Hayre and S. Pylypenko, Energy Environ. Sci., 2014, 7, 1212–1249 CAS
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed
- M. A. Worsley, T. Y. Olson, J. R. Lee, T. M. Willey, M. H. Nielsen, S. K. Roberts, P. J. Pauzauskie, J. Biener, J. H. Satcher and T. F. Baumann, J. Phys. Chem. Lett., 2011, 2, 921–925 CrossRef CAS PubMed
- L. Guo, Q. Liu, G. Li, J. Shi, J. Liu, T. Wang and G. Jiang, Nanoscale, 2012, 4(19), 5864–5867 RSC
- X. Yu, H. Wang, L. Guo and L. Wang, Chem.–Asian J., 2014, 9(11), 3221–3227 CrossRef CAS PubMed
- L. B. Xing, S. F. Hou, J. Zhou, S. Li, T. Zhu, Z. Li, W. Si and S. Zhuo, J. Phys. Chem. C, 2014, 118(45), 25924–25930 CAS
- C. M. Parnell, B. Chhetri, A. Brandt, F. Watanabe, Z. A. Nima, T. K. Mudalige, A. S. Biris and A. Ghosh, Sci. Rep., 2016, 6, 31415 CrossRef CAS PubMed
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351(6271), 361–365 CrossRef CAS PubMed
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed
- J. Liebscher, R. Mrówczyński, H. A. Scheidt, C. Filip, N. D. Hădade, R. Turcu, A. Bende and S. Beck, Langmuir, 2013, 29(33), 10539–10548 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra01027g |
| This journal is © The Royal Society of Chemistry 2018 |