
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaleimide end-functionalized poly(2-oxazoline)s by the functional initiator route: synthesis and (bio)conjugation†
Gabriela Gil Alvaradejoab,
Mathias Glassnerc,
Richard Hoogenboom c and
Guillaume Delaittre*ab
c and
Guillaume Delaittre*ab
aInstitute of Toxicology and Genetics (ITG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 176344, Eggenstein-Leopoldshafen, Germany. E-mail: guillaume.delaittre@kit.edu
bMacromolecular Architectures, Institute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), 76128 Karlsruhe, Germany
cSupramolecular Chemistry Group, Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281 S4, 9000 Ghent, Belgium
First published on 5th March 2018
Abstract
The synthesis of poly(2-ethyl-2-oxazoline)s with a maleimide group at the α chain end was carried out from new sulfonate ester initiators bearing a furan-protected maleimide group. The conditions of the polymerization were optimized for 50 °C using conventional heating (in contrast to microwave irradiation) to counteract the thermal lability of the cycloadduct introduced to protect the maleimide double bond. At this temperature, a tosylate variant was found to be unable to initiate the polymerization after several days. The controlled polymerization of 2-ethyl-2-oxazoline with a nosylate derivative was, however, successful as shown by kinetic experiments monitored by gas chromatography (GC) and size-exclusion chromatography (SEC). Poly(2-ethyl-oxazoline)s of various molar masses (4500 < Mn < 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) with narrow dispersity (Đ < 1.2) were obtained. The stability of the protected maleimide functionality during the polymerization, its deprotection, and the reactivity of the deprotected end group by coupling with a model thiol molecule were proven by 1H NMR spectroscopy and electrospray ionization mass spectrometry (ESI-MS). Finally, the conjugation of maleimide-functionalized poly(2-oxazoline) to a model protein (bovine serum albumin) was demonstrated by gel electrophoresis and MALDI-ToF mass spectrometry.
000 g mol−1) with narrow dispersity (Đ < 1.2) were obtained. The stability of the protected maleimide functionality during the polymerization, its deprotection, and the reactivity of the deprotected end group by coupling with a model thiol molecule were proven by 1H NMR spectroscopy and electrospray ionization mass spectrometry (ESI-MS). Finally, the conjugation of maleimide-functionalized poly(2-oxazoline) to a model protein (bovine serum albumin) was demonstrated by gel electrophoresis and MALDI-ToF mass spectrometry.
Introduction
The synthesis of poly(2-alkyl/aryl-2-oxazoline)s (PAOx) by polymerization of 2-oxazolines was first described in the 1960s.1–4 However, it is only in recent years that interest in these polymers has strongly increased, thanks to their potential in biomedical applications5–7 and the thermoresponsive properties of certain PAOx.8 Particularly, poly(2-methyl-2-oxazoline) (PMeOx) and poly(2-ethyl-2-oxazoline) (PEtOx) share properties such as biocompatibility, low toxicity, and antifouling behavior with poly(ethylene glycol) (PEG), the gold standard for stealth polymers.9,10 The most important advantage of these PAOx over PEG is that their synthesis does not require a complex experimental setup (e.g., no handling of gaseous monomer), while allowing the tailoring of the functionalities at the chain ends or the side chains through the substituent at the 2-position of the 2-oxazoline ring.11 In addition, there is an increasing concern regarding the use of PEG in some life sciences applications due to its degradation mode12,13 and potential immunogenicity.14,15Polymerization of 2-oxazolines can proceed, under appropriate conditions, via a living cationic ring-opening (CROP) mechanism, which results in a controlled length of the polymer, narrow molar mass distributions, and the introduction of specific end groups by initiation and termination.16 Taking advantage of the CROP mechanism, several functionalization strategies of PAOx are available (Scheme 1).11,17 Besides specific monomer design, end-functionalized PAOx can be obtained using functional initiators and terminating agents.18 While many reports on end functionalization are based on a terminating strategy, the use of functional initiators may be more convenient in terms of stoichiometry – 1 functional initiator molecule ideally leads to 1 polymeric chain – while terminating agents are usually introduced in (large) excess. Common initiators for CROP are alkyl halides,19,20 or alkyl sulfonates, including tosylates,21 nosylates,22 and triflates.21 A number of initiators that add a specific functionality to the polymeric chain or allow further functionalization have been reported. Among these functionalities are allyl,23 amine,24 hydroxyls,25 or carboxylic acid,26 sometimes requiring protecting group strategies.26
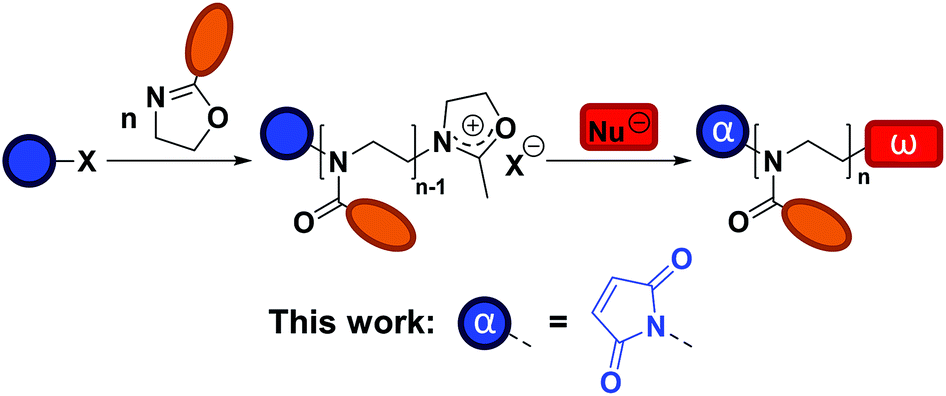 | ||
| Scheme 1 General strategies for the functionalization of PAOx and aim of the current work. | ||
In this work, our aim is to develop a strategy for the introduction of a maleimide moiety at the α chain end of PAOx (see Scheme 1). The maleimide moiety can undergo radical copolymerization27 and may engage in a range of coupling reactions, including Diels–Alder reactions,28–31 dipolar cycloadditions,32 and Michael-type additions.33 This versatility makes it a suitable group for post-polymerization modification (PPM), protein conjugation, synthesis of block copolymers, (reversible) network formation, or surface functionalization. Some examples of maleimide end-functionalized polymers have been reported,34–38 including poly(2-oxazoline)s. In the latter case, Schacher and co-workers introduced the maleimide end group by PPM at the ω chain end, by first capping with sodium azide and then performing azide–alkyne cycloaddition with an alkyne-functionalized protected maleimide, which had been synthesized in three steps with an overall yield of 9%, including a column chromatography step.37 A second approach to add a maleimide functionality at the ω chain end was recently reported by Luxenhofer and co-workers,38 where furan-protected maleimide was directly used as terminating agent for the CROP of AOx. The capping agent was obtained without complex purification, in a single step with a yield of 45%. Yet, the termination efficiency was reported to be 55%, and it was followed by a deprotection step (approx. 85%) to yield the maleimide-bearing PAOx. Using a functional initiator to introduce the maleimide at the α chain end offers an interesting alternative, as the maleimide moiety – provided no transfer reactions occur – will be present in all chains from the start, while giving a new possibility of creating maleimide-based bifunctional (hetero)telechelic PAOx by terminating the reaction with a functional nucleophile. In the current study, we present the straightforward synthesis of novel maleimide-containing CROP initiators and their application to the synthesis of α-maleimido-PAOx. We finally demonstrate the reactivity of the maleimide end group in small molecule coupling as well as in the formation of protein–PAOx conjugates.
Experimental section
Materials
Maleic anhydride (99%, Bernd Kraft), furan (99%, Acros), ethanolamine (99%, Acros), 4-toluenesulfonyl chloride (99%, Acros), 4-nitrobenzenesulfonyl chloride (97%, Sigma-Aldrich), pyridine (99%, Roth), tetramethylammonium hydroxide (25 wt% in methanol, Acros), bovine serum albumin (BSA; Sigma), toluene (Fisher), petroleum ether (Acros), ethanol (99.8%, Acros), acetone (Fisher), tetrahydrofuran (THF; VWR), diethyl ether (99.5%, Roth), and dichloromethane (DCM; VWR) were used as received. Acetonitrile (MeCN; Sigma-Aldrich) was dried in a solvent purification system (JC Meyer) before use as a polymerization solvent. 2-Ethyl-2-oxazoline (EtOx; Aldrich) was distilled over barium oxide and stored under argon.General characterization
All polymer solutions and samples were prepared in a VIGOR Sci-Lab SG 1200/750 Glovebox System with a water concentration ≤0.1 ppm.:2 v/v) containing sodium trifluoroacetate (0.14 μg L−1).000 to 80000 Da. The laser intensity was set to 4700. The spectra obtained represent the average of laser shots taken by an automatic scheme measuring spectra over the whole spot.Synthesis of maleimide-functionalized initiators
1H NMR (CDCl3, 500 MHz, δ): 6.58 (s, 2H), 5.47 (s, 2H), 3.18 (s, 2H) ppm.
1H NMR (CDCl3, 500 MHz, δ): 6.53 (s, 2H), 5.29 (s, 2H), 3.76–3.79 (m, 2H), 3.70–3.72 (m, 2H), 2.90 (s, 2H) ppm.
:1 v/v). After filtration and drying under reduced pressure, 3 was obtained as white crystals (1.10 g, 63%).1H NMR (CDCl3, 500 MHz, δ): 7.75–7.73 (d, 2H), 7.31–7.33 (d, 2H), 6.50 (s, 2H), 5.22 (s, 2H), 4.17 (t, 2H), 3.73 (t, 2H), 2.84 (s, 2H), 2.42 (s, 3H) ppm.
13C NMR (CDCl3, 125 MHz, δ): 21.75, 37.77, 47.63, 65.51, 80.94, 128.05, 129.99, 132.74, 136.61, 145.15, 175.86 ppm.
ESI-MS (m/z): [M − Na+] calc.: 386.067, found: 386.066 amu.
:1 v/v). After filtration and drying under reduced pressure, 4 was recovered as a white powder (0.55 g, 29%).1H NMR (CDCl3, 500 MHz, δ): 8.38–8.40 (d, 2H), 8.09–8.11 (d, 2H), 6.52 (s, 2H), 5.24 (s, 2H), 4.32 (t, 2H), 3.80 (t, 2H), 2.88 (s, 2H) ppm.
13C NMR (CDCl3, 125 MHz, δ): 37.09, 47.11, 67.49, 80.35, 125.04, 129.32, 136.47, 140.11, 150.78, 176.12 ppm.
ESI-MS (m/z): [M − Na+] calc.: 417.036, found: 417.035 amu.
Cationic ring-opening polymerization of 2-ethyl-2-oxazoline
The furan-protected maleimide initiators were dissolved in MeCN in glass vials at different monomer-to-initiator ratios ([M]/[I] = 20, 40, and 60) at a monomer concentration of 1.5 M. The vials were capped under inert conditions in a glove box. The capped vials were heated to 50 °C with constant stirring under inert atmosphere. Samples for GC and SEC analysis were taken periodically using a syringe, quenched by the addition of water, and injected without further purification in order to monitor kinetics and molar mass distributions. For further experiments, the polymerization was terminated by addition of tetramethylammonium hydroxide.39 After evaporation of the solvent under reduced pressure and dissolving the residue in DCM, the polymer was precipitated in diethyl ether, centrifuged, separated, and dried under vacuum. The dry product was analyzed by DMAc SEC, ESI-MS, and 1H NMR spectroscopy.Deprotection of the maleimide end group (Mal-PEtOx)
In a 25 mL round-bottom flask, 100 mg of a furan-protected maleimide end-functionalized polymer FurMal-PEtOx (Mn,SEC = 6500 g mol−1, Đ = 1.20, Mn,NMR = 4400 g mol−1) was dissolved in toluene (10 mL). This solution was then heated to 110 °C for 4 h. After evaporation of the solvent under reduced pressure and dissolving the residue in DCM, the product was precipitated in diethyl ether, centrifuged, separated, and dried under vacuum. The dry product Mal-PEtOx was analyzed by DMAc SEC, ESI-MS, and 1H NMR spectroscopy.Model Michael addition with benzyl mercaptan
In a 25 mL round-bottom flask, deprotected Mal-PEtOx (30 mg) was dissolved in DCM (10 mL), then mixed with a 10-fold excess of benzylmercaptan and 0.1 equivalent of triethylamine. The mixture was stirred overnight at room temperature. After evaporation of the solvent under reduced pressure and dissolving the residue in DCM, the product was precipitated in diethyl ether, centrifuged, separated, and dried under vacuum. The dry product was analyzed by ESI-MS and 1H NMR spectroscopy.Bioconjugation with BSA
Three solutions of different polymer-to-protein molar ratios were prepared (5, 10, and 15). A solution of 100 mM PBS was degassed by bubbling nitrogen for 30 min. 3 mL of oxygen-free water was transferred via a degassed syringe into a sealed vial equipped with a rubber septum, containing 40 mg of BSA and either 18.75, 37.5, or 56.5 mg of MalPEtOx (Mn,SEC = 10600 g mol−1, Đ = 1.13, Mn,NMR = 5500 g mol−1) stored under a nitrogen atmosphere. After 18 h, several aliquots were taken for SDS-PAGE. For the sample with the polymer-to-protein ratio of 5:1, preparative FPLC was carried out and a relevant fraction was analyzed by MALDI-ToF mass spectrometry.
Results and discussion
Synthesis of protected maleimide initiators
Protection of the maleimide is necessary during CROP to avoid nucleophilic attacks and other potential side reactions. In the current study, it was decided to employ alkyl sulfonate-type initiators because they typically lead to faster polymerizations via exclusive cationic propagation21 and are readily synthesized from alcohol derivatives in one step, typically with purification by simple recrystallization.40 While tosylate derivatives are significantly more popular, the utility of nosylate initiators was recently demonstrated.41 Here, both types of sulfonates were synthesized and characterized for the CROP of 2-ethyl-2-oxazoline. Scheme 2 depicts the synthetic strategy followed to obtain the initiators. In this contribution, maleic anhydride is used as the starting material to incorporate the activated ene group. Prior to further functionalization, the latter was protected through a classic Diels–Alder cycloaddition with furan. The cycloadduct 1 was subsequently ring-opened with ethanolamine and dehydrated, to yield the hydroxylated N-substituted protected maleimide 2. The corresponding tosylate 3 and nosylate 4 were finally obtained by nucleophilic substitution of 2 with tosyl and nosyl chloride, respectively. The overall yields for these 3-step syntheses were of 17.5 and 8.1%, for 3 and 4, respectively. It is to note that the starting materials are extremely cheap and that no chromatographic step is required throughout the entire synthesis, in contrast to the aforementioned synthetic strategies for maleimide end-functional PEtOx (vide supra).37,38 Analysis of both initiators by 1H and 13C NMR spectroscopy, as well as ESI-MS confirmed that the targeted structures were obtained with high purity (Fig. S1–S6†). Dissolution tests revealed a limited solubility for the nosylate initiator (FurMalNos 4) in MeCN, the most commonly employed solvent for the CROP of 2-oxazolines.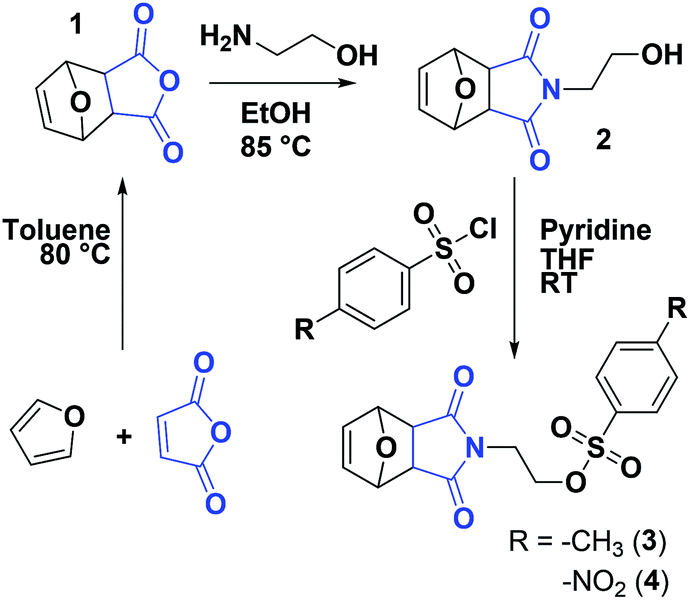 | ||
| Scheme 2 Synthetic route for maleimide-functionalized tosylate and nosylate initiators for the CROP of 2-oxazolines. | ||
Polymerization
Our interest was to produce water-soluble poly(2-oxazoline)s with a view on applications in life sciences, such as antifouling surface treatment and protein conjugation. In the current study, 2-ethyl-2-oxazoline (EtOx) was chosen as the monomer (Scheme 3). Despite a lower water-solubility due to the existence of an LCST at approx. 60 °C, PEtOx is usually more convenient than PMeOx which possesses a low solubility in other solvents, including in the monomer itself.34 It has been reported that very fast CROP of various 2-oxazolines, including EtOx, can be achieved by microwave-assisted polymerization in acetonitrile at 140 °C.42 However, such a high temperature is not suitable here as it would lead to deprotection of the maleimide group by cycloelimination. Therefore, significantly lower temperatures were directly employed for the preliminary experiments. Unfortunately, microwave-assisted polymerization at 80 °C was found to already trigger the loss of the maleimide protecting group (Fig. S7†). In addition, polymerization tests carried out at 80 °C using conventional heating showed broader molar mass distributions (Đ = 1.32–1.41), indicating the loss of control over the polymerization (Fig. S8†). Consequently, to prevent the deprotection of the maleimide group and favor a controlled polymerization, the polymerization temperature was further lowered to 50 °C using conventional heating.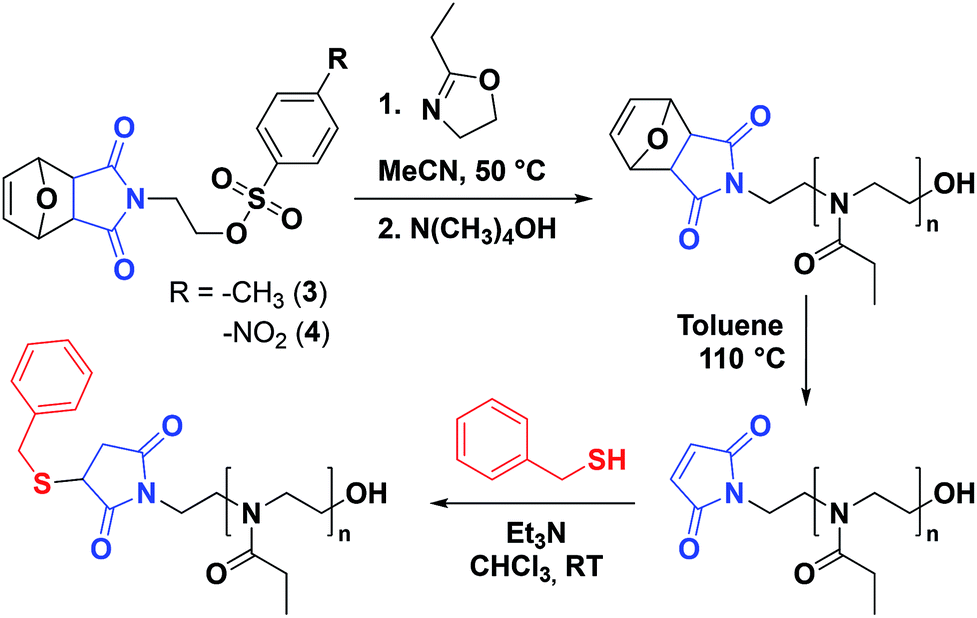 | ||
| Scheme 3 Synthetic route for the synthesis of α-maleimido-PEtOx by polymerization of EtOx using functional initiators 3 and 4 and retro-Diel–Alder cycloelimination, followed by Michael addition with a model small molecule. | ||
A first set of kinetics of EtOx polymerization was carried out in MeCN with [EtOx] = 4 M, in the case of FurMalTos 3, and [EtOx] = 1.5 M for FurMalNos 4. The latter monomer concentration is significantly lower than what is typically encountered in the literature,43 due to the aforementioned limited solubility of FurMalNos 4 in MeCN. It has been previously reported that optimum monomer concentrations are usually established around 4 M.43
An initial assessment of the ability of the two new protected maleimide-functionalized sulfonic esters to initiate the polymerization at 50 °C was carried out by targeting a degree of polymerization (DP) of 50 at full conversion, that is [EtOx]/[initiator] = 50. After 74 hours, a conversion of 72% was achieved with the FurMalNos 4, whereas for the FurMalTos 3 less than 10% was obtained (Fig. S9†). In a recent study on CROP of 2-oxazolines in MeCN at 80 °C, it was reported that, apart from methyl tosylate, initiation with alkyl tosylates typically leads to slow initiation and low propagation rates due to low electrophilicity and relatively low leaving group ability, respectively.41 However, the reactivity of ethyl nosylate is sufficiently high to quantitatively initiate in the first instants of the polymerization. Therefore, subsequent investigations were exclusively conducted with FurMalNos 4 as initiator. Importantly, at the end of the polymerization, the survival of the furan-maleimide cycloadduct could be observed by 1H NMR spectroscopy (Fig. 2).
Further polymerizations varying monomer-to-initiator ratios were carried out in order to evaluate the possibility of producing α-maleimido-PEtOx of various molar masses (Fig. 1). As previously observed,21 with all other parameters remaining constant, the lower the monomer-to-initiator ratio, the faster the polymerization (Fig. 1A). All kinetics exhibited a similar trend, i.e., a rather slow polymerization with two distinct regimes: (i) a slow non-linear increase of ln(1/(1 − conversion)) with time at the beginning of the polymerization and (ii) a linear behavior indicating a constant concentration of propagating species. The kp values corresponding to this second regime are collated in Table S1† and appear to be independent of the initial monomer-to-initiator concentration, as observed in previous studies.21 The two-stage nature of the polymerization can be explained by a relatively slow initiation reaction resulting in a slow build-up of the concentration of propagating species. In comparison to previously reported polymerizations, which were initiated with a nosylate of similar structure (ethyl nosylate) and did not exhibit slow initiation,41 this phenomenon can certainly be attributed to the different polymerization conditions (lower temperature and monomer concentration).
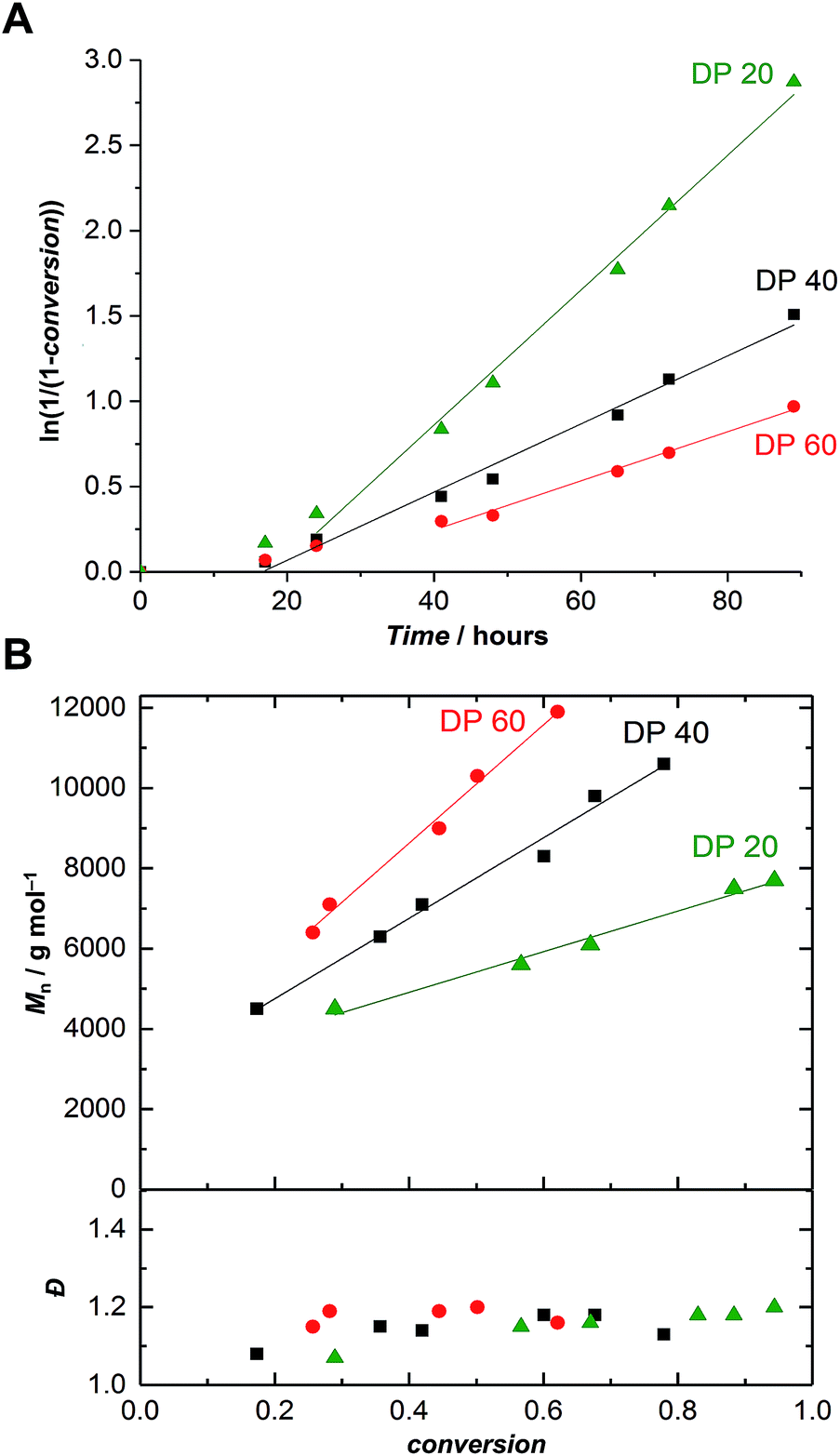 | ||
| Fig. 1 (A) First-order kinetic plots and (B) evolution of the number-average molar mass and dispersity vs. conversion for the CROP of EtOx initiated by FurMalNos 4 at various targeted degrees of polymerization (DP = [EtOx]/[FurMalNos]) in MeCN at 50 °C. [EtOx] = 1.5 M. Please refer to Fig. S11–S13† for corresponding SEC traces. | ||
Satisfactorily, with increasing conversion, all SEC traces shifted progressively towards lower retention times (Fig. S11–S13†) and number-average molar masses (Mn) increased linearly, while dispersity values remained confined between 1.1 and 1.2 (Fig. 1B), pointing at a polymerization with living character. This demonstrates that FurMalNos enables the synthesis of well-defined polymers despite the slow initiation reaction. The fact that the molar masses are significantly higher than the theoretical ones further underpins the slow initiation reaction. However, it should be noted that the discrepancy between theoretical and experimental Mn values is also due to the fact that the latter are calculated relative to poly(methyl methacrylate) standards which results in an overestimation.44 1H NMR spectroscopy was used to calculate molar masses for final samples, after purification. The end group signals from the protected maleimide moiety found at 5.2 and 6.5 were considered and compared to those corresponding to the side chain and backbone of the PEtOx (Fig. 2A).
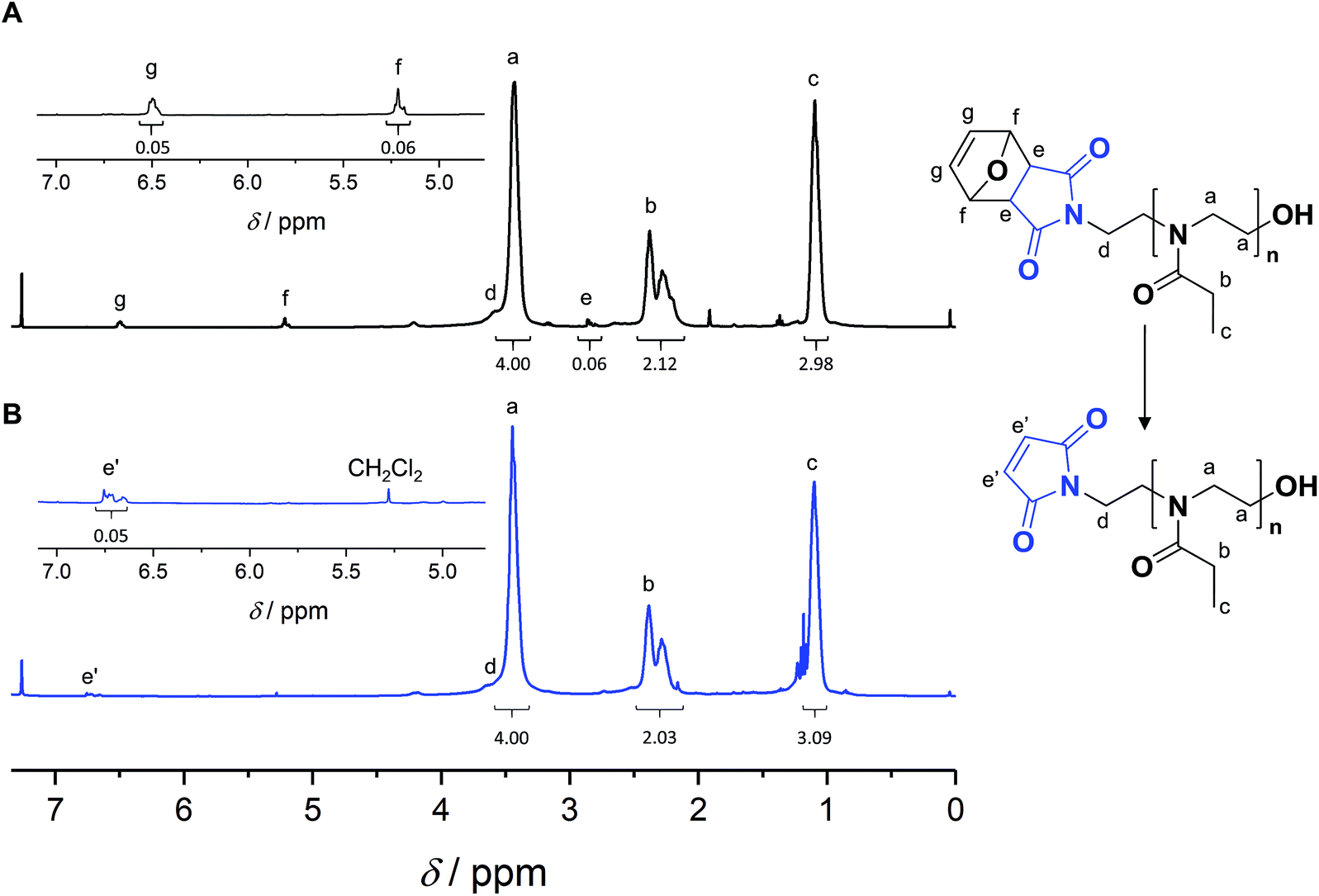 | ||
| Fig. 2 NMR spectra of a poly(2-ethyl-2-oxazoline) FurMal-PEtOx obtained through FurMalNos initiation ([EtOx]/[FurMalNos]) = 20; Mn,SEC = 6500 g mol−1, Đ = 1.20; Mn,NMR = 4400 g mol−1 before (A) and after (B) deprotection of the maleimide group by retro-Diels–Alder cycloelimination. | ||
In contrast to the SEC results, the values obtained were significantly closer to the theoretical ones. For instance, for a α-furan-maleimide-PEtOx sample prepared with [EtOx]/[FurMalNos] = 40, after 78% conversion, Mn,SEC = 10600 g mol−1 while Mn,NMR = 6100 g mol−1. This latter value, although significantly closer, is still higher than the theoretical value. This fact may corroborate the slow initiation which would result in a higher effective monomer-to-initiator ratio throughout a significant part of the polymerization. Yet, the linear trend observed after 40 hours of polymerization in all cases suggests another cause. The difference between the expected and NMR calculated degrees of polymerization of the resulting polymer would lead to an initiator efficiency of 53% or more. We can assume that non-negligible chain transfer occurs, leading to new chains which do not carry the functional group. However, for many applications, e.g., surface functionalization, this would not be an issue, as non-reactive polymer can be easily washed away.
Deprotection and reactivity assessment
A polymer FurMal-PEtOx obtained (Mn,SEC = 6500 g mol−1, Đ = 1.20; Mn,NMR = 4400 g mol−1) was purified to confirm the nature of the end groups and was subsequently employed for coupling reactions. 1H NMR spectroscopy and ESI-MS were used to assess the stability of the furan-protected maleimide throughout the polymerization and to allow the monitoring of further modifications (Fig. 2 and 3). The 1H NMR spectrum of FurMal-PEtOx displays the expected peaks for the 6 methine protons (e–g) of the furan-maleimide Diels–Alder cycloadduct (Fig. 2A). Furthermore, no signal corresponding to vinyl protons of deprotected maleimide could be found (expected δ at approx. 6.7 ppm). The stability of the protecting group was further corroborated by ESI-MS with the presence of a major population corresponding to chains possessing the initiating group and a hydroxyl moiety as end groups (Fig. 3A and S14†). The only major side-product is a proton-initiated PEtOx (see Fig. S14† for full assignment), which is seen in most PAOx reports and originates from a chain transfer reaction via β-elimination.45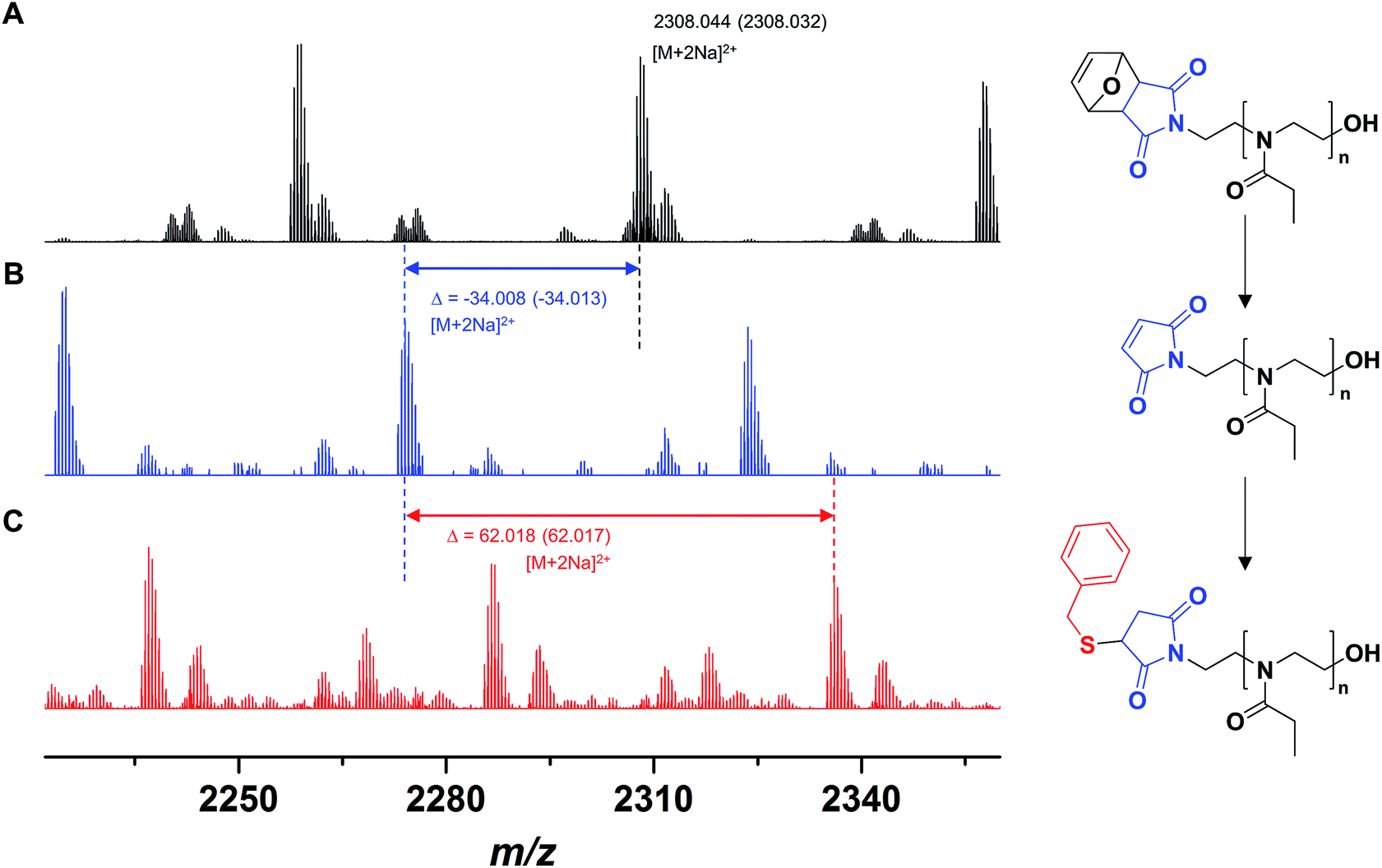 | ||
| Fig. 3 ESI-MS spectra of (A) FurMal-PEtOx as obtained after CROP of EtOx initiated by FurMalNos, (B) Mal-PEtOx obtained after thermal treatment of FurMal-PEtOx, and (C) the Michael addition product of Mal-PEtOx with benzyl mercaptan (BzSMal-PEtOx). For a more extensive assignment of the spectra, refer to Fig. S14.† | ||
Straightforward thermal treatment in solution at 110 °C for 4 h yielded the deprotected α-functional Mal-PEtOx. As shown in the 1H NMR spectrum, the characteristic oxanorbornene signals of the cycloadduct quantitatively vanished (Fig. 2B), leaving new peaks (e') corresponding to vinylic protons of an electron-deficient structure. The quantitative survival of the maleimide group is evident from the similar integration values obtained from the corresponding signals. To assess chain-end transformation, mass spectrometry is one of the most potent methods. While the original peaks assigned to FurMal-PEtOx disappeared, a new set of peaks with an unambiguous shift of about 34 amu (doubly charged species) corresponding to the loss of a furan molecule was observed after thermal treatment, with essentially no by-product (Fig. 3B and S14†).
The reactivity of the maleimide group of Mal-PEtOx was then assessed by Michael addition with a model thiol (benzyl mercaptan), catalyzed by triethylamine. Again, the peak population corresponding to the precursor polymer disappeared and a new main population appeared with a shift of approx. 62 amu (double charges), which matches perfectly with the addition of a benzyl mercaptan molecule and proves that the maleimide at the α end of the polymeric chain is reactive (Fig. 3C and S14,† BzSMal-PEtOx). Two by-products can also be observed and have so far not been identified. However, this might be avoided with optimization of the Michael addition conditions, which is not the subject of the current study. The integrity of the polymer backbone during the successive transformations could be confirmed by SEC, where no significant difference in the molar mass distributions of FurMal-PEtOx, Mal-PEtOx, and BzSMal-PEtOx was observed (see Fig. S15;† Mn = 6500–7000 g mol−1, Đ = 1.20–1.27).
Bioconjugation
The therapeutic use of proteins and peptides is a rapidly expanding area of research. One of its limitations, however, is the delivery of protein-based drugs and other biologically active proteins to their desired targets.34 Conjugation of PEG to proteins – so-called PEGylation – has been a popular approach to overcome these constraints and give the conjugates favorable pharmacokinetic properties.46 However, due to the aforementioned issues related to PEG, the search for alternatives is vivid. PAOx have been discussed as a promising option for stealth polymers9 and, as such, can be used for “PAOxylation” of proteins. Examples include the conjugation of PEtOx and PMeOx to proteins such as catalase,47,48 bovine serum albumin,48 ribonuclease,48 uricase,48 insulin,48 and granulocyte colony stimulating factor.49Considering the relatively low abundance of cysteine in natural proteins, maleimide-functionalized polymers call for quasi-site-specific conjugation compared to the often employed coupling strategies making use of lysine residues. The thiol addition to the activated double bond in aqueous conditions works best at neutral or mild alkaline pH (7.5–8.5).50 A conjugation experiment was performed to attach the functional Mal-PEtOx (Mn,NMR = 6100 g mol−1) to bovine serum albumin (BSA). BSA was chosen as a well-known model protein possessing one free cysteine exposed at its surface (Cys-34 residue) and available for conjugation.51 The experiment was carried out overnight with three different polymer-to-protein molar ratios (5, 10, and 15) in 100 mM phosphate buffer at pH 7.6. Under these pH conditions the conjugation via the amino group of lysine is avoided, ensuring only attachment to the free cysteine present in BSA.52 A first characterization was performed using SDS-PAGE electrophoresis (Fig. 4A and B). All lanes corresponding to the incubation of BSA with Mal-PEtOx (Fig. 4A, last three lanes) show a rather similar shift in the protein signal towards higher molar masses, which indicates successful attachment of the polymer to the protein. The reason for the broadening of the BSA band is related to the fact that only about 55 to 70 percents of commercial BSA contain the free cysteine residue.34,51,53 Importantly, no shift occurred when non-deprotected FurMal-PEtOx was incubated with BSA in identical conditions (Fig. 4A and B, 4th lane from the left and green trace, respectively). To further prove the formation of the bioconjugate, a preparative chromatographic separation was carried out and a sample collected at lower retention time than the BSA eluting time (highest intensity considered) was analyzed by MALDI-ToF mass spectrometry. The spectrum of the conjugate sample (Fig. 3C, bottom) shows that the sample is a mixture of two species. First, a signal corresponding to the mass of pure BSA is observed, which was expected since not all BSA molecules contain a free thiol available for conjugation. Satisfactorily, a second population at higher m/z with a shift corresponding approximately to the mass of Mal-PEtOx can be observed. It also exhibits a significantly broader distribution than the protein, as expected from a synthetic polymer–protein conjugate.
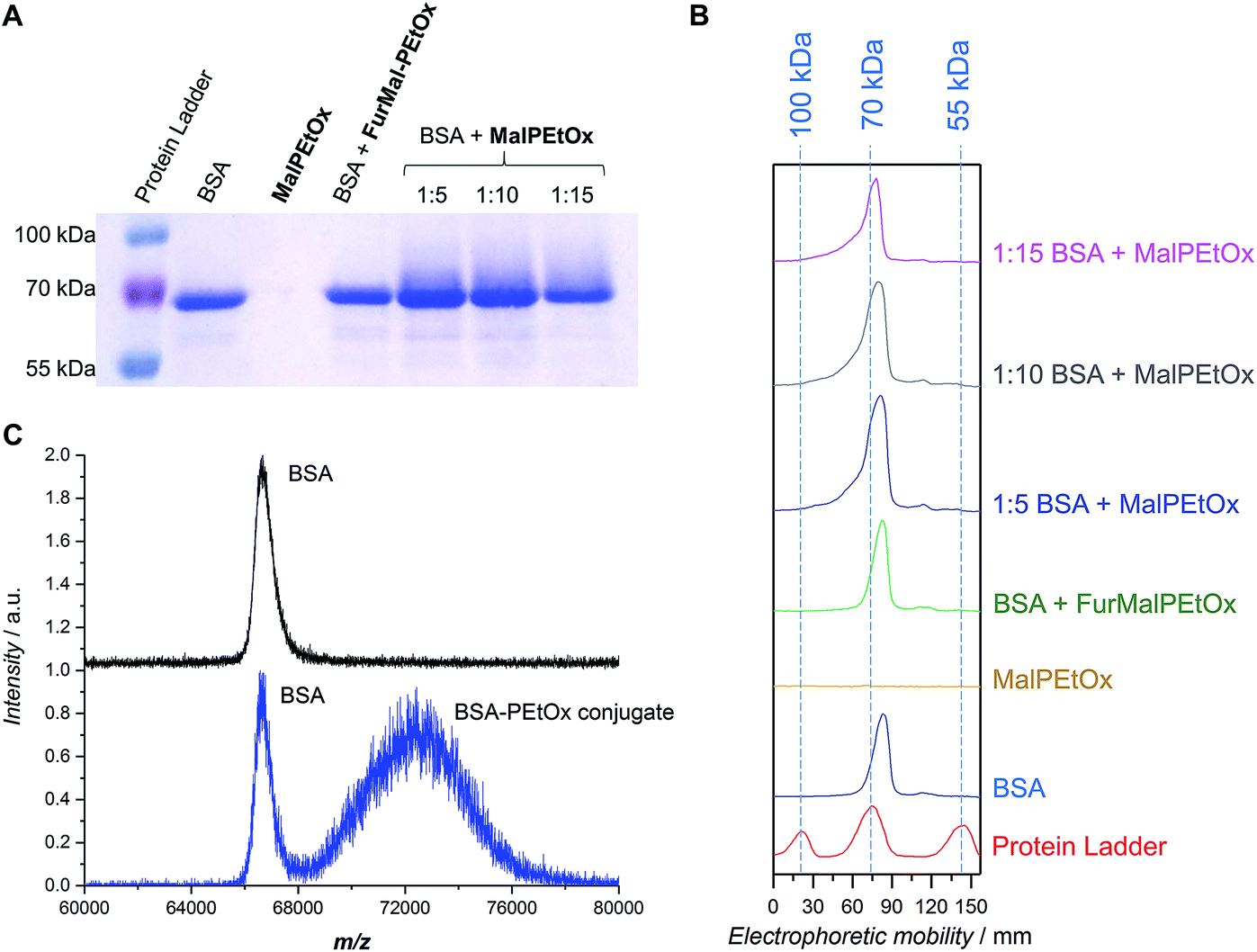 | ||
| Fig. 4 Protein conjugation experiment in 100 mM phosphate buffer at pH 7.6 for 18 hours at room temperature using three different [Mal-PEtOx]/[BSA] ratios. (A) SDS-PAGE gel stained with Coomassie blue. (B) Intensity of the signals from each lane as a function of electrophoretic mobility. (C) MALDI-ToF spectra of (top) BSA and (bottom) the fraction collected after preparative FPLC of the PEtOx-BSA conjugate [Mal-PEtOx]/[BSA] 5:1. | ||
Conclusions
A novel, efficient route for the synthesis of maleimide end-functionalized poly(2-alkyl-2-oxazoline)s has been established using a nosylate initiator bearing a maleimide moiety protected in the form of a furan-based Diels–Alder adduct. While the limited solubility of the initiator requires polymerization at relatively low monomer concentrations, molar masses relevant for bioconjugation and surface functionalization could be obtained. Although the functionalized initiator route can lead to the presence of non-maleimide functionalized chains via H transfer, the combination of a straightforward synthesis with the availability of the ω chain end for further functionalization makes this route particularly attractive for advanced application in life sciences, for instance for the development of bifunctional linkers for surface biofunctionalization or for the construction of artificial protein dimers, where in both cases excess, non-reacted polymer can easily be removed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. D. thanks the German Federal Ministry of Education and Research (BMBF) for current funding in the frame of the Molecular Interaction Engineering program (Biotechnologie 2020+; Grant No. 031A095C). G.G.A. thanks the Mexican National Council for Science and Technology (Conacyt) for a doctoral research scholarship. The authors acknowledge support by the Deutsche Forschungsgemeinschaft and the Open Access Publishing Fund of the Karlsruhe Institute of Technology. R. H. is grateful to Ghent University and FWO for financial support. Josefine Morgenstern and Prof. Jürgen Hubbuch (MAB-BLT, KIT), as well as Frank Kirschhöfer, Boris Kühl, and Dr Gerald Brenner-Weiss (IFG, KIT) are acknowledged for providing access to and support on preparative FPLC and MALDI-ToF, respectively. Prof. Barner-Kowollik (ITCP, KIT) is thanked for his constant support.Notes and references
- D. A. Tomalia and D. P. Sheetz, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 2253–2265 CrossRef CAS.
- A. Levy and M. Litt, J. Polym. Sci., Part B: Polym. Lett., 1967, 5, 871–879 CrossRef.
- W. Seeliger, E. Aufderhaar, W. Diepers, R. Feinauer, R. Nehring, W. Thier and H. Hellmann, Angew. Chem., Int. Ed., 1966, 5, 875–888 CrossRef CAS PubMed.
- T. Kagiya, T. Maeda, K. Fukui and S. Narisawa, J. Polym. Sci., Part B: Polym. Lett., 1966, 4, 441–445 CrossRef CAS.
- V. R. De La Rosa, J. Mater. Sci.: Mater. Med., 2014, 25, 1211–1225 CrossRef CAS PubMed.
- N. Adams and U. S. Schubert, Adv. Drug Delivery Rev., 2007, 59, 1504–1520 CrossRef CAS PubMed.
- R. Luxenhofer, Y. Han, A. Schulz, J. Tong, Z. He, A. V. Kabanov and R. Jordan, Macromol. Rapid Commun., 2012, 33, 1613–1631 CrossRef CAS PubMed.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- M. Bauer, C. Lautenschlaeger, K. Kempe, L. Tauhardt, U. S. Schubert and D. Fischer, Macromol. Biosci., 2012, 12, 986–998 CrossRef CAS PubMed.
- R. Konradi, C. Acikgoz and M. Textor, Macromol. Rapid Commun., 2012, 33, 1663–1676 CrossRef CAS PubMed.
- B. Guillerm, S. Monge, V. Lapinte and J. J. Robin, Macromol. Rapid Commun., 2012, 33, 1600–1612 CrossRef CAS PubMed.
- B. Pidhatika, M. Rodenstein, Y. Chen, E. Rakhmatullina, A. Muehlebach, C. Acikgoz, M. Textor and R. Konradi, Biointerphases, 2012, 7, 1–15 CrossRef CAS PubMed.
- J. Ulbricht, R. Jordan and R. Luxenhofer, Biomaterials, 2014, 35, 4848–4861 CrossRef CAS PubMed.
- R. P. Garay, R. El-Gewely, J. K. Armstrong, G. Garratty and P. Richette, Expert Opin. Drug Delivery, 2012, 9, 1319–1323 CrossRef CAS PubMed.
- H. Schellekens, W. E. Hennink and V. Brinks, Pharm. Res., 2013, 30, 1729–1734 CrossRef CAS PubMed.
- B. Verbraeken, B. D. Monnery, K. Lava and R. Hoogenboom, Eur. Polym. J., 2016, 88, 451–469 CrossRef.
- E. Rossegger, V. Schenk and F. Wiesbrock, Polymers, 2013, 5, 956–1011 CrossRef.
- K. Lava, B. Verbraeken and R. Hoogenboom, Eur. Polym. J., 2015, 65, 98–111 CrossRef CAS.
- T. Saegusa, S. Kobayashi and A. Yamada, Makromol. Chem., 1976, 177, 2271–2283 CrossRef CAS.
- T. Saegusa and H. Ikeda, Macromolecules, 1973, 6, 808–811 CrossRef CAS.
- R. Hoogenboom, M. W. M. Fijten and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1830–1840 CrossRef CAS.
- P. Mueller, C. Woerner and R. Muelhaupt, Macromol. Chem. Phys., 1995, 196, 1929–1936 CrossRef CAS.
- S. Kobayashi, H. Uyama and Y. Narita, Macromolecules, 1990, 23, 353–354 CrossRef CAS.
- R. Obeid and C. Scholz, Biomacromolecules, 2011, 12, 3797–3804 CrossRef CAS PubMed.
- M. Reif and R. Jordan, Macromol. Chem. Phys., 2011, 212, 1815–1824 CrossRef CAS.
- R. M. England, J. I. Hare, P. D. Kemmitt, K. E. Treacher, M. J. Waring, S. T. Barry, C. Alexander and M. Ashford, Polym. Chem., 2016, 7, 4609–4617 RSC.
- S. Pfeifer and J.-F. Lutz, Chem.–Eur. J., 2008, 14, 10949–10957 CrossRef CAS PubMed.
- G. Delaittre, N. K. Guimard and C. Barner-Kowollik, Acc. Chem. Res., 2015, 48, 1296–1307 CrossRef CAS PubMed.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- D. J. Hall, H. M. Van Den Berghe and A. P. Dove, Polym. Int., 2011, 60, 1149–1157 CrossRef CAS.
- A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417–1425 CrossRef CAS.
- M. Dietrich, G. Delaittre, J. P. Blinco, A. J. Inglis, M. Bruns and C. Barner-Kowollik, Adv. Funct. Mater., 2012, 22, 304–312 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS PubMed.
- R. J. Pounder, M. J. Stanford, P. Brooks, S. P. Richards and A. P. Dove, Chem. Commun., 2008, 5158–5160 RSC.
- M. J. Stanford, R. L. Pflughaupt and A. P. Dove, Macromolecules, 2010, 43, 6538–6541 CrossRef CAS.
- F. Wendler, T. Rudolph, H. Görls, N. Jasinski, V. Trouillet, C. Barner-kowollik and F. H. Schacher, Polym. Chem., 2016, 7, 2419–2426 RSC.
- J. F. Nawroth, J. R. Mcdaniel, A. Chilkoti, R. Jordan and R. Luxenhofer, Macromol. Biosci., 2016, 16, 322–333 CrossRef CAS PubMed.
- V. R. de la Rosa, S. Tempelaar, P. Dubois, R. Hoogenboom and L. Mespouille, Polym. Chem., 2016, 7, 1559–1568 RSC.
- R. Hoogenboom, M. W. M. Fijten, G. Kickelbick and U. S. Schubert, Beilstein J. Org. Chem., 2010, 6, 773–783 CrossRef CAS PubMed.
- M. Glassner, D. R. D'Hooge, J. Y. Park, P. H. M. Van Steenberge, B. D. Monnery, M.-F. Reyniers and R. Hoogenboom, Eur. Polym. J., 2015, 65, 298–304 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, M. A. M. Leenen, M. A. R. Meier and U. S. Schubert, Macromolecules, 2005, 38, 5025–5034 CrossRef CAS.
- R. Hoogenboom, R. M. Paulus, M. W. M. Fijten and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1487–1497 CrossRef CAS.
- P. J. M. Bouten, D. Hertsen, M. Vergaelen, B. D. Monnery, M. A. Boerman, H. Goossens, S. Catak, J. C. M. van Hest, V. Van Speybroeck and R. Hoogenboom, Polym. Chem., 2015, 6, 514–518 RSC.
- J. M. Warakomski and B. P. Thill, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 3551–3563 CrossRef CAS.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- M. Miyamoto, K. Naka, M. Shiozaki, Y. Chujo and T. Saegusa, Macromolecules, 1990, 23, 3201–3205 CrossRef CAS.
- T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, A. Mero, G. Pasut and F. M. Veronese, Bioconjugate Chem., 2011, 22, 976–986 CrossRef CAS PubMed.
- A. Mero, Z. Fang, G. Pasut, F. M. Veronese and T. X. Viegas, J. Controlled Release, 2012, 159, 353–361 CrossRef CAS PubMed.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS PubMed.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372–15373 CrossRef CAS PubMed.
- B. Jung and P. Theato, Adv. Polym. Sci., 2013, 253, 37–70 CrossRef CAS.
- C. K. Riener, G. Kada and H. J. Gruber, Anal. Bioanal. Chem., 2002, 373, 266–276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00948a |
| This journal is © The Royal Society of Chemistry 2018 |