
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNMR-based metabolomic analysis of the effects of alanyl-glutamine supplementation on C2C12 myoblasts injured by energy deprivation†
Zhiqing Liu a,
Caihua Huangb,
Yan Liu*a,
Donghai Lin*a and
Yufen Zhaoa
a,
Caihua Huangb,
Yan Liu*a,
Donghai Lin*a and
Yufen Zhaoa
aCollege of Chemistry and Chemical Engineering, The Key Laboratory for Chemical Biology of Fujian Province, MOE Key Laboratory of Spectrochemical Analysis & Instrumentation, Xiamen University, Xiamen 361005, China. E-mail: dhlin@xmu.edu.cn; stacyliu@xmu.edu.cn; Tel: +86-592-218-6078 Tel: +86-592-218-5610
bExercise and Health Laboratory, Xiamen University of Technology, Xiamen 361024, China
First published on 30th April 2018
Abstract
The dipeptide alanyl-glutamine (Ala-Gln) is a well-known parenteral nutritional supplement. The Ala-Gln supplementation is a potential treatment for muscle-related diseases and injuries. However, molecular mechanisms underlying the polyphenic effects of Ala-Gln supplementation remain elusive. Here, we performed NMR-based metabolomic profiling to analyze the effects of Ala-Gln, and the free alanine (Ala) and glutamine (Gln) supplementations on the mouse myoblast cell line C2C12 injured by glucose and glutamine deprivation. All the three supplementations can promote the differentiation ability of the injured C2C12 cells, while only Ala-Gln supplementation can facilitate the proliferation of the injured cells. Ala-Gln supplementation can partially restore the metabolic profile of C2C12 myoblasts disturbed by glucose and glutamine deprivation, and exhibits more significant effects than Ala and Gln supplementations. Our results suggest that Ala-Gln supplementation can promote MyoD1 protein synthesis, upregulate the muscle ATP-storage phosphocreatine (PCr), maintain TCA cycle anaplerosis, enhance the antioxidant capacity through promoting GSH biosynthesis, and stabilize lipid membranes by suppressing glycerophospholipids metabolism. This work provides new insight into mechanistic understanding of the polyphenic effects of Ala-Gln supplementation on muscle cells injured by energy deprivation.
Introduction
Skeletal muscle is one of the most dynamic and plastic tissues of the human body. In humans, skeletal muscle comprises approximately 40% of total body weight and contains 50–75% of all body proteins.1 Almost all activities of the body are completed by skeletal muscle contraction, and glucose is the main energy to maintain skeletal muscle contraction.2 As a consequence, skeletal muscle is the largest organ system in terms of energy storage and consumption by the human body. The alternations of skeletal muscle play critical roles in some common diseases and physiological conditions. Glucose depletion is inevitable in exhaustive exercise,3 which might result in muscle injury and dysfunction. Moreover, muscle wasting, hypoglycemia and muscle energy deficit are common phenomena in cachexia and sarcopenia,4,5 probably leading to poor prognosis, increased oxidative stress and skeletal muscle dysfunction.6–8 Since muscle mass plays a key role in the recovery from critical illness, muscle strength and function is central to the recovery process. As a crucial part in muscle growth and repair, the functions of C2C12 myoblast are quite important. It has been reported that glucose deprivation could inhibit proliferation and differentiation of C2C12 myoblast.9 Down-regulation of protein levels were also observed in C2C12 cells with glucose deprivation.10 Furthermore, glucose deprivation could also result in cessation of cell metabolic activity, reduction in cell viability and promotion of cell death.11 Thus, developments of energy supplements are quite essential for injured muscle cells in certain conditions, especially in case of injury and dysfunction induced by energy deprivation.As well known, glutamine (Gln) is the most abundant free amino acid in the human body. Glutamine is not only the important precursor of glucose as the dominant energy source,12,13 but also the precursor of many other amino acids, such as aspartate, glutamate, proline and alanine. Therefore, glutamine is widely used as an essential nutrient in muscle recovery from injury and catabolism. However, the efficacy of glutamine remains controversial due to its limited solubility, poor thermal stability and incapable long-term preservation.14 Hence, the development of more stable glutamine substitutes is imminently needed for clinical applications.
Compared with glutamine, the dipeptide alanyl-glutamine (Ala-Gln) has higher solubility in aqueous solution, higher thermal stability, and is more conducive to long-term storage. Thus, the applications of Ala-Gln are being explored and developed as a stable glutamine derivative. It has been reported that the Ala-Gln treatment could increase the ATP production and stimulate mTOR activation in enterocytes.15 Furthermore, Ala-Gln supplementation improves intestinal epithelium damage by promoting the proliferation and migration in vitro.14 In addition, Ala-Gln could be up-taken by intestinal mucosal cells through peptide transporter 1 (PepT1),16 and the GeneAtlas dataset shows that PepT1 also exists in mouse skeletal muscle. Previous works have proven that Ala-Gln supplementation improves glutamine availability, mitigates muscle damage and attenuates oxidative stress.17–19 It was demonstrated that oral supplementation with Ala-Gln could improve redox defense and attenuate markers of muscle damage in the trained mice through the heat shock protein pathways.20 However, studies on the Ala-Gln treatment have rarely addressed the underlying metabolic mechanisms. Given both the importance of Ala-Gln in muscle metabolisms and its protective effects, mechanistic understanding of the polyphenic effects of Ala-Gln supplementation will be particularly helpful for further exploring Ala-Gln applications in the treatments of muscle-related diseases and injuries.
In recent years, cell metabolomic analysis has been developed as a powerful, convenient and efficient platform to systematically clarify the molecular mechanisms underlying nutritional supplementation. Metabolites, as the downstream products of gene transcription, its alternations may reflect overall metabolic changes of cells intuitively. Hence, quantitative cell metabonomic analysis can be used to evaluate the effects of nutritional interventions on muscle cells and reveal underlying molecular mechanisms. Until now, the studies of Ala-Gln supplementation have been conducted primarily through in vivo animal experiments,17–19 but rarely performed in vitro cell experiments by using metabonomic analysis.
On the other hand, as one of the three primary detection techniques used in metabolomic analysis, high-resolution 1H-nuclear magnetic resonance (NMR) spectroscopy is particularly appropriate for investigating compositions of biofluid and cells as a wide range of metabolites can be quantified simultaneously ‘without prejudice’.21 Moreover, NMR-based metabolomic analysis has the requisite reproducibility and sensitivity for quantitatively characterizing variations in metabolite concentrations.22 Thus, NMR-based cell metabolomic analysis has been applied extensively to reveal molecular mechanisms underlying biological functions of cells, and also mechanistically understand pharmacological effects on cells.
In the present work, we performed NMR-based metabolomics analysis to explore metabolic responses of Ala-Gln supplementation on the mouse myoblast cell line C2C12 (a model of activated satellite cells) injured by glucose and glutamine deprivation. For obtaining a mechanistic understanding of the advantages of Ala-Gln supplementation over free Ala and Gln supplementations, we also conducted metabolomic analysis to address the effects of both Ala and Gln supplementations on injured C2C12 cells. Our results highlight the differences in the effects and mechanisms between the Ala-Gln, Ala and Gln supplementations. Our work may be of benefit to promotion of the Ala-Gln applications for treatments of muscular diseases and injuries.
Materials and methods
Cell culture
The murine skeletal muscle myoblast cell line C2C12 was obtained from the China Center for Typical Culture Collection (CCTCC). Alanine and glutamine were purchased from Sangon Biotech (China). Ala-Gln was synthesized according to the previous method (WO 03/106481A1), and its purity was characterized by 1D 1H-NMR spectroscopy (Fig. S1†). Commercial culture Dulbecco's modified Eagle's medium with (DMEM) and without (DMEM-NGG) glucose and glutamine (Gibco, USA) were supplemented with 10% (v/v) fetal bovine serum (FBS, Gemini, USA), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. Cell culture was carried out at 37 °C in a humidified atmosphere of 5% CO2.C2C12 cells were divided into five groups: (1) the Nor group (n = 6), cultured in DMEM for 12 h, then replaced with fresh DMEM for another 12 h; (2) the NGG group (n = 5), cultured in DMEM-NGG for 12 h, then replaced with fresh DMEM-NGG for another 12 h; (3) the Ala-Gln/Ala/Gln groups (n = 6 for each group), cultured in DMEM-NGG for 12 h and then cultured in fresh DMEM-NGG supplied with 3 mM Ala-Gln/Ala/Gln for another 12 h.
Cell proliferation assay and western blotting
The five groups of C2C12 cells were cultured in 96-well plates at density of a 5 × 103 per well for 24 h as described above. Each cell sample in 100 μL of the corresponding culture medium was added 20 μL of CellTiter 96 AQueous solution (MTS, Promega, USA). After incubation for 3 h, colored MTS products were detected by absorbance at 490 nm on a microplate reader (BioTek, USA) to evaluate cell proliferation rate. In addition, C2C12 myoblasts were washed by PBS three times to remove the dead cells and fixed with 4% paraformaldehyde for 20 min at room temperature. Then, cell morphological images of five groups were taken randomly on an inverted microscope (Nikon, Japan).The cell lysates were harvested using commercial cell lysis buffer (Sangon Biotech, China) according to the instruction. Aliquots volume cell lysate were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were blocked with 5% non-fat powdered milk and followed by transferred to PVDF membranes (GE, USA). Subsequently, PVDF membranes were incubated overnight at 4 °C with primary antibodies: anti-MyoD1 (Abcam, UK) and anti-Actin (Abcam, UK). After incubation with the secondary antibody for 1 h at room temperature, MyoD1 and actin were visualized by using the commercial enhanced chemiluminescence reagent (ECL, Beyotime, China). The density was detected by ImageJ (National Institutes of Health, USA), and the ratio of MyoD1/actin was used to represent the relative level of MyoD1.
Extraction of intracellular metabolites and samples preparation
Cell intracellular extracts were prepared following a direct quenching method described previously.23 To minimize the influence of the carryover of culture medium, cells were quickly rinsed thrice by phosphate-buffered saline (PBS, pH 7.4). The residual PBS was removed immediately by vacuum suction. Subsequently, cells were quenched by using 3 mL of HPLC grade methanol before being gently detached from the culture dish using a cell scraper (Costar, Mexico). The methanol solution containing quenched cells was pipetted into a 15 mL centrifuge tube for extraction. A mixture of methanol, chloroform and water in the volume ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:2.85 was applied in a dual phase extraction for extracting intracellular metabolites. Standing for 30 min on the ice, the mixture was centrifuged at 12000g for 15 min at 4 °C. Finally, only the aqueous phase was lyophilized and subjected to NMR-based metabolomic analysis.
:4:2.85 was applied in a dual phase extraction for extracting intracellular metabolites. Standing for 30 min on the ice, the mixture was centrifuged at 12000g for 15 min at 4 °C. Finally, only the aqueous phase was lyophilized and subjected to NMR-based metabolomic analysis.
The aqueous cell extract powder was resolved in 550 μL of phosphate buffer [50 mM, pH 7.4, 10% D2O, 0.01 mM sodium 3-(trimethylsilyl)propionate-2,2,3,3-d4 (TSP) and 0.02% NaN3], vortexed and then centrifuged at 12000g for 15 min at 4 °C. Aliquots of the supernatant were transferred into 5 mm NMR tubes.
NMR measurements and data preprocessing
All NMR measurements were performed at 298 K on a Bruker Avance III 850 MHz NMR spectrometer (Bruker Bio Spin, Germany). One-dimensional (1D) 1H NMR spectra were recorded using the pulse sequence NOESYPR1D [(RD)-90°-t1-90°-τm-90°-ACQ] with water suppression. A spectral width of 20 ppm was used, and a total of 128 transients were collected into 64k data points, giving an acquisition time of 1.93 s, using an additional relaxation delay (RD) of 4 s. Chemical shifts correction were referenced to the methyl group of TSP at 0 ppm. The two-dimensional (2D) 1H–13C heteronuclear single quantum coherence (HSQC) spectrum was acquired with a spectral width of 10 ppm in the 1H dimension and 110 ppm in the 13C dimension, a data matrix of 1024 × 128 points, and a relaxation delay of 1.5 s. The 2D total correlation spectroscopy (TOCSY) spectrum was acquired with a spectral width of 10 ppm in both 1H dimensions, a data matrix of 2048 × 256, and a relaxation delay of 1.5 s.Phase correction, baseline correction and resonance alignment were carried out for all 1D NMR spectra using the MestReNova 9.0 software (Mestrelab Research S.L., ESpain). 1D 1H spectral region of δ 9.5 to 0.0 was segmented into bins with a width of 0.002 ppm for further statistical analysis. The water region of δ 5.2 to 4.5 was excluded to eliminate distortion from the residual water resonance in all 1D spectra. The remaining spectral integrals for each spectrum were normalized by the TSP spectral integral and the cell number. NMR resonances of metabolites were assigned by using a combination of the Chenomx NMR Suite (version 8.1, Chenomx Inc., Edmonton, Canada), and the Human Metabolome Data Base (HMDB, http://www.hmdb.ca/) as well as relevant literatures.24,25 In addition, 2D 1H–13C HSQC and 1H–1H TOCSY spectra were used to confirm the assigned metabolites.
Data analysis
Multivariate data analysis was performed by using the SIMCA-P software (Version 12.0, Umetrics AB, Umeå, Sweden). Normalized integrals were scaled by Pareto scaling to increase the magnitude of low-level metabolites without significant amplification of noise.26 In order to observe the trend of metabolic profiles and outliers, unsupervised principle component analysis (PCA) was conducted. Moreover, hierarchical cluster analysis (HCA) with Pearson distance measure and Ward clustering algorithm was performed on the normalized NMR data using the MetaboAnalyst webserver (Version 3.0, http://www.metaboanalyst.ca/) as a measure of similarity to evaluate metabolic profile clustering.Subsequently, supervised partial least-squares discriminant analysis (PLS-DA) was conducted to obtain better separation between metabolic profiles.27 Significant metabolites were identified from the PLS-DA loading plot, which were significantly responsible for the difference between metabolic profiles. Two criteria were used to identify significant metabolites: VIP ≥ 1; either 0.602 ≤ |r| ≤ 0.735 (significant) or |r| ≥ 0.735 (very significant).
Univariate data analysis was also performed on the levels of the identified metabolites for the five groups. Levels of the metabolites were calculated based on their NMR integrals relative to the integral of TSP with a certain concentration and the proton number ratios of the metabolites to TSP. Comparison of metabolite levels among the groups were calculated by one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparisons test in GraphPad Prism (version 6, GraphPad software). Differential metabolites were identified by Student's t-test based on the identified significant metabolites, and P < 0.05 was considered statistically significant.
Characteristic metabolites were finally determined by a combination of the significant metabolites identified from the PLS-DA loading plot and differential metabolites identified from Student's t-test analysis.
Results
Effects on myoblast cells proliferation rate and differentiation ability
We assessed the cell proliferation rate to determine the optimal concentration of Ala-Gln by treating C2C12 myoblasts with varying concentrations of Ala-Gln (0.5–8.0 mM). The cells exhibited a better proliferate ability when treated with 3 mM Ala-Gln. For statistical comparisons of the NGG group with the other four groups, cell numbers per a defined area were counted for each group: NGG, 221.0 ± 1.4; Nor, 417.5 ± 4.9; Ala, 221.5 ± 3.5; Gln, 219.5 ± 2.1; Ala-Gln, 242.5 ± 3.6. As shown in Fig. 1A and B, the NGG treatment significantly inhibited C2C12 proliferation resulting in C2C12 cell injury. Both Ala and Gln treatments did not show statistically significant difference from the NGG treatment, whereas the Ala-Gln treatment markedly attenuated the inhibition effect from energy deprivation. Furthermore, MTS cell proliferation assay also exhibited that the NGG treatment inhibited C2C12 proliferation, while the Ala-Gln treatment promoted C2C12 proliferation (Fig. 1C). Absorbance at wavelength of 490 nm showed that the Ala-Gln treatment partially alleviated the inhibition of cell proliferation resulting from energy deprivation, while either the Ala treatment or the Gln treatment did not show the same effect.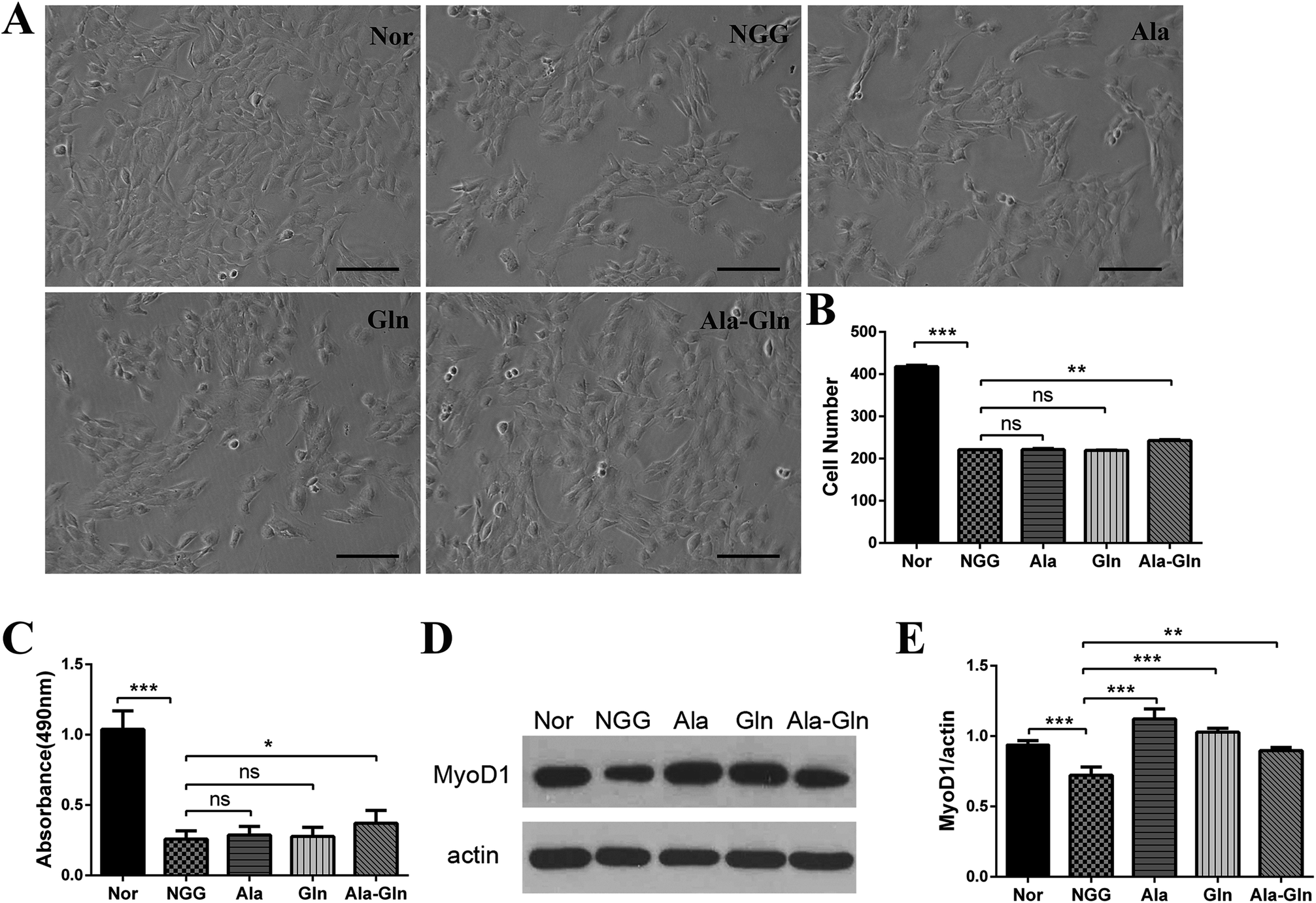 | ||
| Fig. 1 Effects of different treatments on proliferation and differentiation abilities of C2C12 myoblast cells. (A) Cell morphologies after being fixed in 4% paraformaldehyde (scale bar: 100 μm). (B) Cell numbers corresponding to panel (A). (C) MTS cell proliferation assay. (D) Western blot analysis showing the expression of MyoD1. The anti-actin antibody was used to standardize the amount of protein in each lane. (E) Statistical analysis corresponding to panel (D). *P < 0.05, **P < 0.01, ***P < 0.001. | ||
As the MyoD1 protein is one of the markers associated with skeletal muscle cell differentiation, we thus measured its cellular level to evaluate the differentiation ability of C2C12 myoblast (Fig. 1D and E). The comparison of relative densities of the protein bands exhibited that, the NGG treatment significantly inhibited the MyoD1 expression, thus affecting C2C12 cell differentiation. Ala, Gln and Ala-Gln treatments all significantly enhanced MyoD1 expressions of the injured C2C12 myoblasts. Interestingly, MyoD1 level under Ala-Gln treatment was close to the normal while Ala and Gln treatments enhanced the MyoD1 than normal.
NMR spectra of aqueous extracts of C2C12 myoblasts
Fig. 2 illustrates the average 1D 1H NMR spectra (850 MHz) recorded on aqueous extracts derived from the five groups of C2C12 cells. A total of 46 metabolites were assigned and summarized in Table 1. Visual inspection of the spectra shows that the levels of most metabolites are distinctly different among the five groups. The resonance assignments of the metabolites were confirmed by using 2D 1H–13C HSQC and 1H–1H TOCSY spectra (Fig. S2–5†).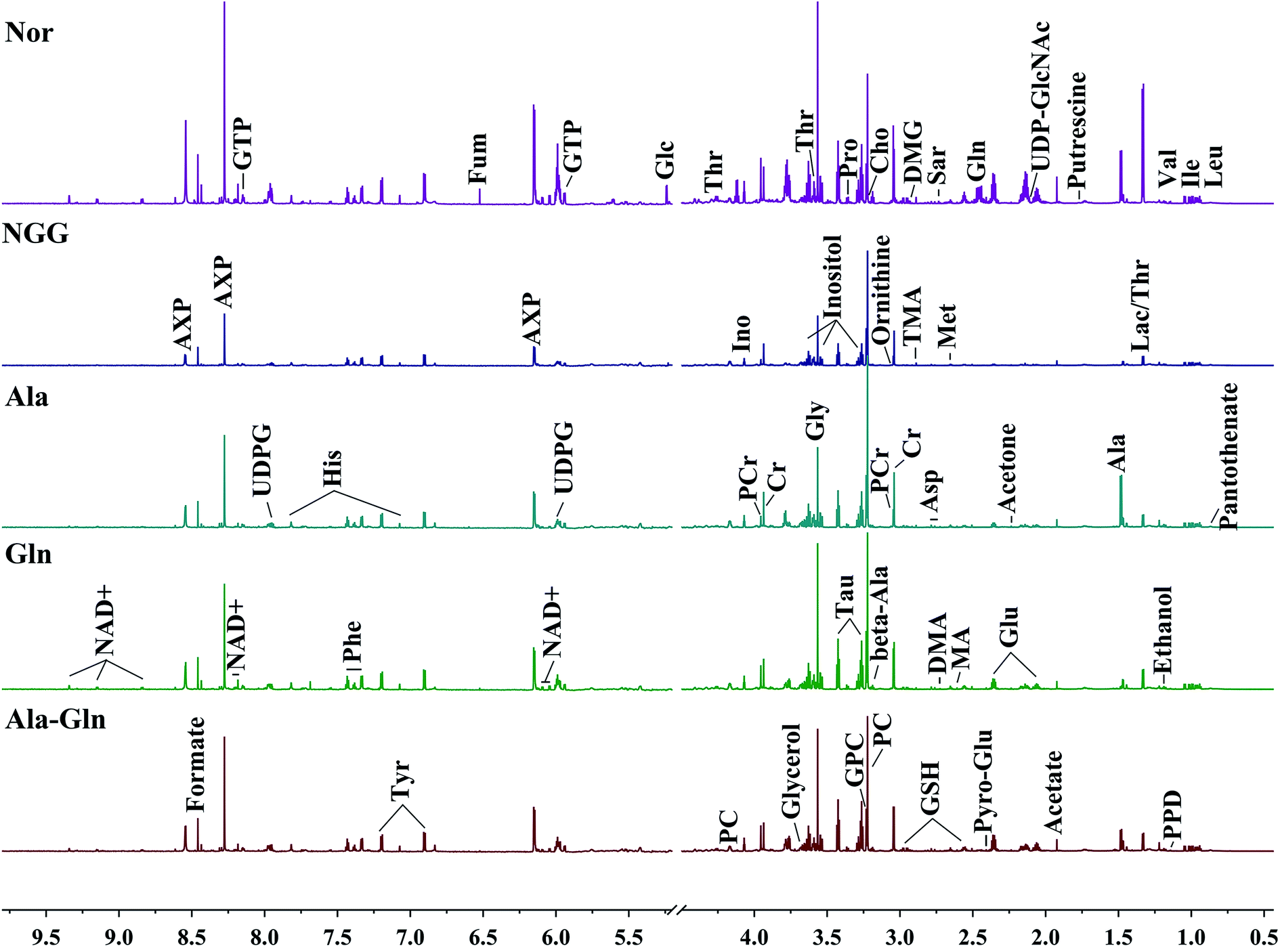 | ||
| Fig. 2 Average 850 MHz 1H NMR spectra recorded on aqueous extracts derived from five groups of C2C12 cells. Vertical scales were kept constant in all the 1H spectra. Spectral regions of 0.5–4.5 ppm and 5.2–9.5 ppm are showed, and the water region of 4.5–5.2 ppm was removed. The region of 5.2–9.5 ppm has been magnified 10 times compared to another region of 0.5–4.5 ppm for the purpose of clarity. Identified metabolites are shown in Table 1. | ||
| No. | Metabolites | δ 1H (ppm) and multiplicity | Moieties |
|---|---|---|---|
| a Multiplicity: s, singlet; d, double; t, triplet; q, quartet; m, multiple; dd, double of double. | |||
| 1 | Pantothenate | 0.88 (s), 0.92 (s) | CH3, CH3 |
| 2 | Isoleucine (Ile) | 0.94 (t), 1.01 (d), 1.21 (m), 1.42 (m), 2.00 (m), 3.67 (d) | δ-CH3, γ-CH3, half γ-CH2, half γ-CH2, β-CH, α-CH |
| 3 | Leucine (Leu) | 0.96 (d), 0.97 (d), 1.69 (m), 1.70 (m), 1.73 (m), 3.73 (m) | α-CH3, α-CH3, γ-CH, β-CH2, α-CH |
| 4 | Valine (Val) | 0.99 (d), 1.05 (d), 2.26 (m), 3.60 (d) | γ-CH3, γ-CH3, β-CH, α-CH |
| 5 | Propanediol (PPD) | 1.13 (s), 3.43 (dd), 3.53 (dd), 3.9 (m) | CH3, half CH2, half CH2, CH |
| 6 | Ethanol | 1.17 (t), 3.65 (q) | β-CH3, CH2 |
| 7 | Threonine (Thr) | 1.30 (d), 3.58 (d), 4.24(m) | γ-CH2, β-CH |
| 8 | Lactate (Lac) | 1.33 (d), 4.11 (q) | β-CH3, α-CH |
| 9 | Alanine (Ala) | 1.47 (d), 3.78 (q) | β-CH3, α-CH |
| 10 | Putrescine | 1.76 (m), 3.04 (m) | 2CH2, 2N–CH2 |
| 11 | Ornithine | 1.9 (m), 3.0 (t) | β-CH2, N–CH2 |
| 12 | Acetate (Ace) | 1.91 (s) | CH3 |
| 13 | Methionine (Met) | 1.98 (m), 2.13 (s), 2.17 (m), 2.66 (dd), 3.78 (m) | δ-CH3, γ-CH2, β-CH2 |
| 14 | Proline (Pro) | 1.99 (m) | γ-CH2 |
| 15 | Pyroglutamate (Pyro-Glu) | 2.05 (m), 2.39 (d), 2.51 (m), 4.18 (dd) | β-CH, γ-CH2, β-CH, α-CH |
| 16 | UDP-GlcNAc | 2.07 (s), 5.5 (q), 6.0 (dd), 7.94 (d), 8.3 (d) | CH3, CH, 2CH, CH, NH |
| 17 | Glutamate (Glu) | 2.08 (m), 2.12 (m), 2.34 (m), 2.37 (m), 3.75 (m) | Half β-CH2, half β-CH2, half γ-CH2, half γ-CH2, α-CH |
| 18 | Glutamine (Gln) | 2.13 (m), 2.45 (m), 3.77 (t) | γ-CH2, β-CH2, α-CH |
| 19 | Glutathione (GSH) | 2.15 (m), 2.55 (m), 2.96 (m), 3.77 (m), 4.56 (m) | β-CH2, γ-CH2, CH2-SH, α-CH &CH2–NH, CH–NH |
| 20 | Acetone | 2.22 (s) | 2CH3 |
| 21 | Beta-alanine (Beta-Ala) | 2.54 (t), 3.17 (t) | CH2, CH2 |
| 22 | Methylamine (MA) | 2.59 (s) | N–CH3 |
| 23 | Aspartate (Asp) | 2.68 (dd); 2.81 (dd); 3.90 (dd) | β-CH2; α-CH |
| 24 | Dimethylamine (DMA) | 2.72 (s) | CH3 |
| 25 | Sarcosine (Sar) | 2.74 (s), 3.6 (s) | N–CH3, α-CH2 |
| 26 | Trimethylamine (TMA) | 2.88 (s) | CH3 |
| 27 | Dimethylglycine (DMG) | 2.9 (s), 3.71 (s) | N-(CH3)2, α-CH2 |
| 28 | Creatine (Cr) | 3.04 (s), 3.93 (s) | N–CH3, α-CH2 |
| 29 | Phosphocreatine (PCr) | 3.05 (s), 4.05 (s) | N–CH3, CH2 |
| 30 | Tyrosine (Tyr) | 3.05 (dd), 3.19 (dd), 6.92 (d), 7.19 (d) | Half β-CH2, half β-CH2, β-CH, α-CH |
| 31 | Phenylalanine (Phe) | 3.12 (dd), 3.30 (dd), 3.99 (dd), 7.33 (d), 7.37 (t),7.43 (t) | α-CH, half β-CH2, half β-CH2, α-CH, β-CH, γ-CH |
| 32 | Choline (Cho) | 3.21 (s), 3.51 (dd), 4.04 (t) | N–(CH3)3, N–CH2, CH2OH |
| 33 | Phosphocholine (PC) | 3.22(s), 3.60(t), 4.18(m) | N–(CH3)3, N– CH2, CH2OH |
| 34 | sn-Glycero-3-phosphocholine (GPC) | 3.23 (s), 3.60 (dd), 3.68 (dd), 3.87 (m), 3.94 (m), 4.33 (m) | N–(CH3)3, half 1CH2, 2CH2, half 2CH2, half 3CH2, half 3CH2, 1CH2 |
| 35 | Taurine (Tau) | 3.24 (t), 3.41 (t) | 1CH2, 2CH2 |
| 36 | Inositol (Ino) | 3.28 (t), 3.53 (dd), 3.63 (t), 4.07 (t) | 2CH, 4,6CH, 1,3CH, 5CH |
| 37 | Glucose (Glc) | β(3.24 (dd), 3.48 (t), 3.90 (dd)), α(3.54 (dd), 3.71 (t), 3.72 (dd), 3.83 (m)) | β(H2, H3, H5), α(H2, H3, H6) |
| 38 | Glycerol | 3.55 (dd), 3.64 (dd), 3.77 (m) | Half 1CH2, half 3CH2, 2CH |
| 39 | Glycine (Gly) | 3.57 (s) | α-CH2 |
| 40 | UDP-glucose (UDPG) | 5.62 (dd), 6.0 (m) | CH, 2CH |
| 41 | GTP | 5.92 (d), 8.1 (s) | CH, CH |
| 42 | NAD+ | 6.03 (d), 6.08 (s), 8.16 (s), 8.20 (m), 8.41 (s), 8.82 (d), 9.13 (d), 9.32 (s) | NH2, NH2(CO), δ-CH, β-CH, 2CH, γ-CH, α-CH |
| 43 | AXP | 6.14 (d), 8.27 (s), 8.58 (s) | NH2, δ-CH, 2CH |
| 44 | Fumarate (Fum) | 6.51 (s) | CH |
| 45 | Histidine (His) | 7.06 (s), 7.85 (s) | 5CH, 2CH |
| 46 | Formate | 8.46 (s) | CH |
Multivariate data analysis for exploring cellular metabolic profiles
An unsupervised PCA model with the first two components (PC1, PC2) was conducted to overview the normalized NMR integral data set. The PCA scores plots show that the NGG treatment distinctly changed the cellular metabolic profile compared with the Nor group (Fig. 3A and B). The Ala, Gln and Ala-Gln groups could partially restore the metabolic profiles along the PC1. The metabolic profile of the Ala-Gln group is somewhat closer to that of the Nor group compared to those of Ala and Gln groups (Fig. 3A–E).. In addition, HCA was performed to confirm the reliabilities of the PCA models (Fig. 3F). Obviously, the Nor, NGG, Ala, Gln and Ala-Gln groups are classified as five distinct clusters, supporting the scores plots of the PCA models.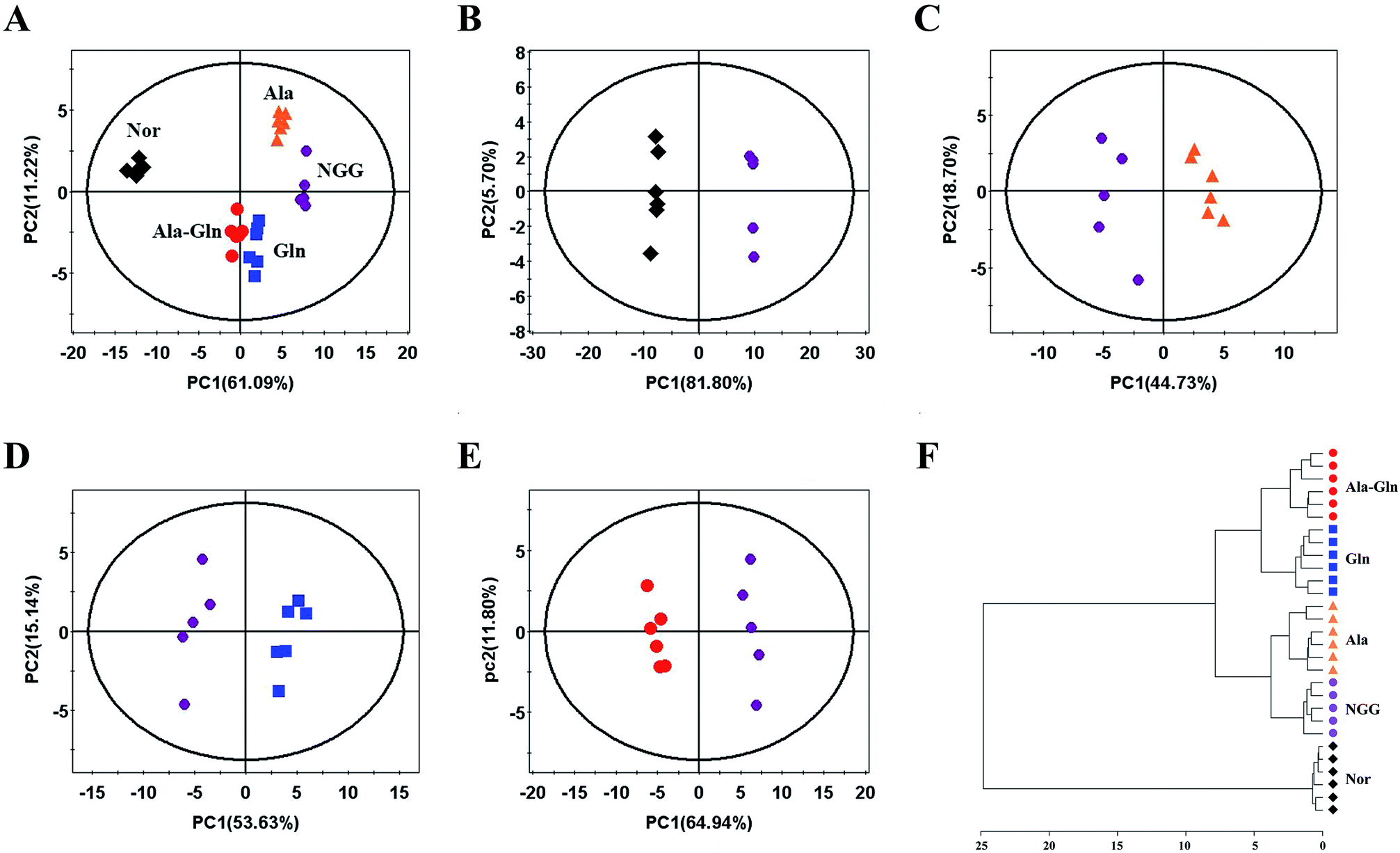 | ||
| Fig. 3 Pattern recognition analysis of 1D 1H NMR spectra recorded on aqueous extracts derived from five groups of C2C12 cells. (A) PCA scores plot of the five groups of cells. (B–E) PCA scores plots for NGG vs. Nor (B), Ala vs. NGG (C), Gln vs. NGG (D), Ala-Gln vs. NGG (E). (F) Hierarchical cluster analysis for the five groups of cells. | ||
Identification of characteristic metabolites
PLS-DA models were conducted to obtain better separations between the NGG group and other four groups (Fig. 4A, C, E and G). Significant metabolites were identified from the PLS-DA loading plots (data not shown), and differential metabolites were identified by Student's t-test analysis. Characteristic metabolites were then identified by a combination of the significant metabolites and differential metabolites (Fig. 4B, D, F and H).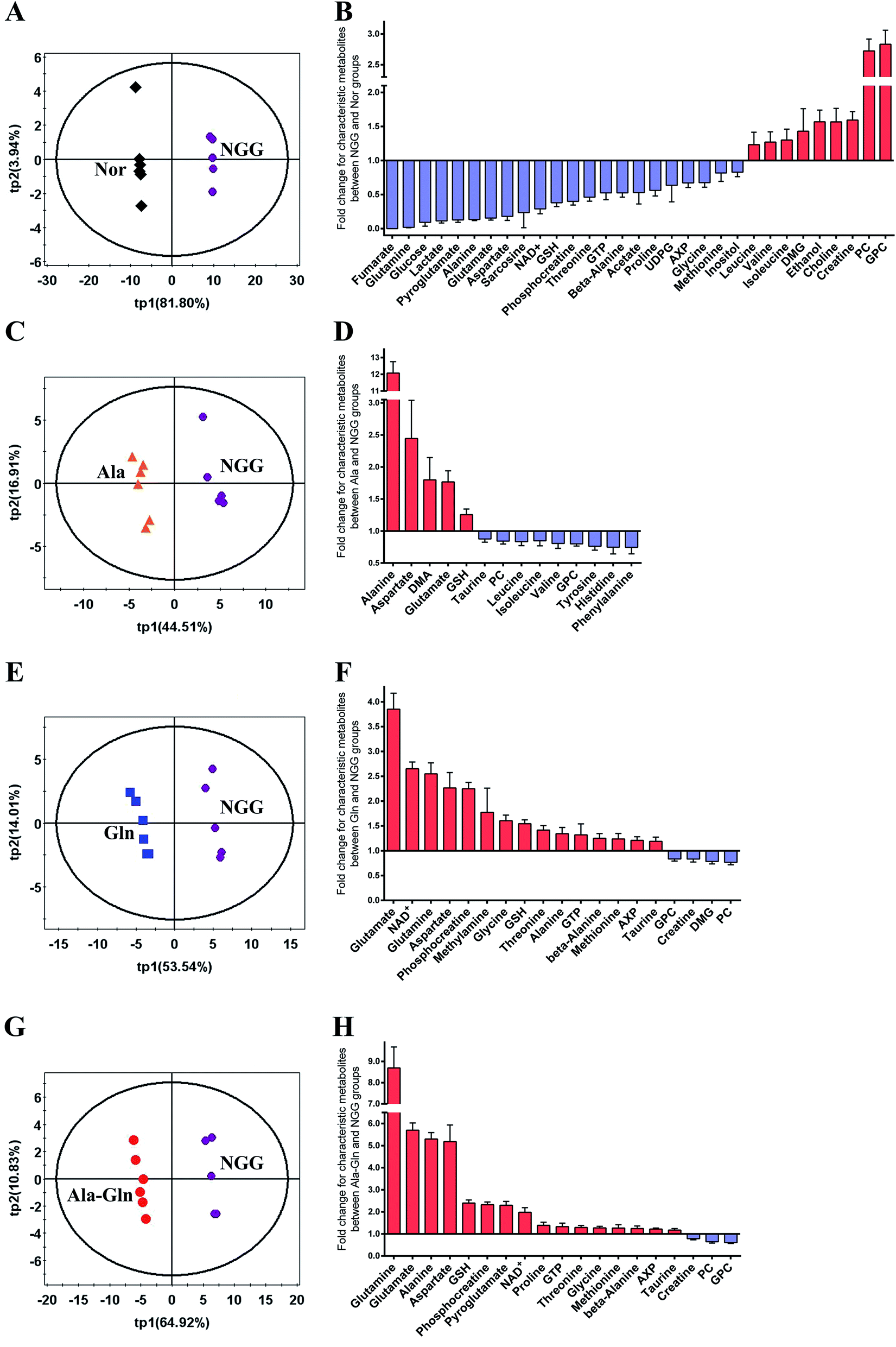 | ||
| Fig. 4 PLS-DA analysis for identifying significant metabolites primarily responsible for discriminations of metabolic profiles between the NGG group and other four groups. (A, C, E and G) PLS-DA scores plots for NGG vs. Nor (A), Ala vs. NGG (C), Gln vs. NGG (E), Ala-Gln vs. NGG (G). Differential metabolites were identified by student's t-test analysis based on the significant metabolites. Thereafter, characteristic metabolites were identified by a combination of the significant metabolites and differential metabolites. (B, D, F and H) Fold changes of characteristic metabolite levels for comparisons between NGG vs. Nor (B), Ala vs. NGG (D), Gln vs. NGG (F), Ala-Gln vs. NGG (H). | ||
For the quantitative comparison between NGG and Nor groups, 31 characteristic metabolites were identified. The fold changes (FCs) of these metabolite levels are showed in Fig. 4B. The NGG group displays distinctly decreased levels of glutamine (glucose and energy precursor), glucose (primary energy source of skeletal muscle) and glucose metabolism-related metabolites (alanine, lactate, NAD+, fumarate). The energy storage PCr was significantly down-regulated, whereas creatine (Cr) was markedly up-regulated. Moreover, the energy metabolism-related molecule AXP (adenine mono/di/tri phosphate) was also decreased. Additionally, energy deprivation increased the levels of branched-chain amino acids (BCAAs, valine, leucine and isoleucine) resulting from protein degradation. Substrates of TCA cycle anaplerosis, such as aspartate, glutamine, glutamate, threonine, glycine and sarcosine, were declined significantly. Levels of GSH and its related metabolites (glycine, glutamate, pyroglutamate, methionine and glutamine) also showed decline trends. Simultaneously, choline, GPC, PC (glycerophospholipid metabolism-related) were up-regulated dramatically, which were associated with cell membrane stability. Other characteristic metabolites including GTP, beta-alanine, acetate, proline, UDP-glucose (UDPG) and inositol were down-regulated in the NGG group. These results demonstrate that the cellular responses to the NGG treatment reflect not only the changes of cellular glucose and glutamine levels, but also the overall metabolic alterations in C2C12 myoblasts.
For the quantitative comparison between Ala and NGG groups, 14 characteristic metabolites were identified, including alanine, aspartate, dimethylamine (DMA), glutamate, GSH, taurine, PC, GPC, leucine, isoleucine, valine, tyrosine, phenylalanine and histidine (Fig. 4D). As expected, Ala supplementation significantly increased the level of alanine in injured C2C12 myoblasts. In the Ala group, alanine was the metabolite with the largest FC (12.1 times). Furthermore, Ala supplementation up-regulated GSH, down-regulated BCAAs and aromatic amino acids (tyrosine, phenylalanine). In addition, the Ala treatment increased glutamate and aspartate, and decreased GPC and PC.
For the quantitative comparison between Gln and NGG groups, 19 characteristic metabolites were identified, including glutamate, glutamine, NAD+, aspartate, PCr, methylamine, glycine, GSH, threonine, alanine, GTP, beta-alanine, methionine, AXP, taurine, GPC, DMG, PC and Cr (Fig. 4F). Notably, glutamate was the metabolite with the largest FC rather than glutamine (3.85 times vs. 2.55 times), probably since the unstable glutamine was rapidly converted to glutamate. We also observed the increased levels of energy-related metabolites (NAD+, AXP) and the substrates of TCA cycle anaplerosis (alanine, aspartate, threonine). The level of PCr (FC, 2.25 times) was up-regulated while Cr (FC, 0.83 times) was down-regulated. Moreover, the Gln treatment also enhanced the levels of the antioxidants taurine, GSH and its related metabolites (glycine, methionine, glutamate, glutamine). Similarly, the Gln treatment also declined the levels of GPC and PC.
For the quantitative comparison between Ala-Gln and NGG groups, 19 characteristic metabolites were identified, including glutamine, glutamate, alanine, aspartate, GSH, PCr, pyroglutamate, NAD+, proline, GTP, threonine, glycine, methionine, beta-alanine, AXP, taurine, Cr, PC and GPC (Fig. 4H). Glutamine in the Ala-Gln group was the metabolite with the largest FC (8.69 times). Note that, glutamine in the Gln group had a smaller FC (2.55 times). Expectedly, the FC of alanine in the Ala-Gln group was higher than that in the Gln group (5.30 times vs. 1.34 times), but lower than that in the Ala group (12.1 times). Moreover, both glutamate and aspartate were considerably up-regulated in the Ala-Gln group with higher FCs than those in the Gln group (5.70 times vs. 3.85 times for glutamate; 5.18 times vs. 2.27 times for aspartate). Similarly, the Ala-Gln treatment obviously elevated the levels of energy-related metabolites (NAD+, AXP) and PCr (FC, 2.33 times), and evidently reduced the levels of Cr, GPC and PC. Furthermore, the Ala-Gln treatment also increased the levels of taurine, GSH and its relative metabolites (glycine, methionine, pyroglutamate, glutamate and glutamine). The FC of GSH in the Ala-Gln group was higher than that in the Gln group (2.40 times vs. 1.54 times).
Significantly altered metabolic pathways under NGG and Ala-Gln treatments
To visualize significant changes in the levels of characteristic metabolites induced by NGG and Ala-Gln treatments, we projected these changed metabolites onto a metabolic map based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and the webserver MetaboAnalyst (Fig. 5). From metabolic comparisons between NGG and Nor groups and Ala-Gln and NGG groups, we identified significantly altered metabolic pathways, including energy metabolism, GSH biosynthesis, glycerophospholipid metabolism, and TCA cycle anaplerotic flux. Most of characteristic metabolites in the Ala-Gln group displayed changing trends in an opposite direction to those in the NGG group, indicating that the Ala-Gln treatment could partially reverse metabolic changes caused by energy deprivation due to lack of glucose and glutamine in the culture medium. Both the distinctly changed characteristic metabolites and significantly altered metabolic pathways, provide new insights into the molecular mechanisms underlying the polyphenic effects of Ala-Gln supplementation.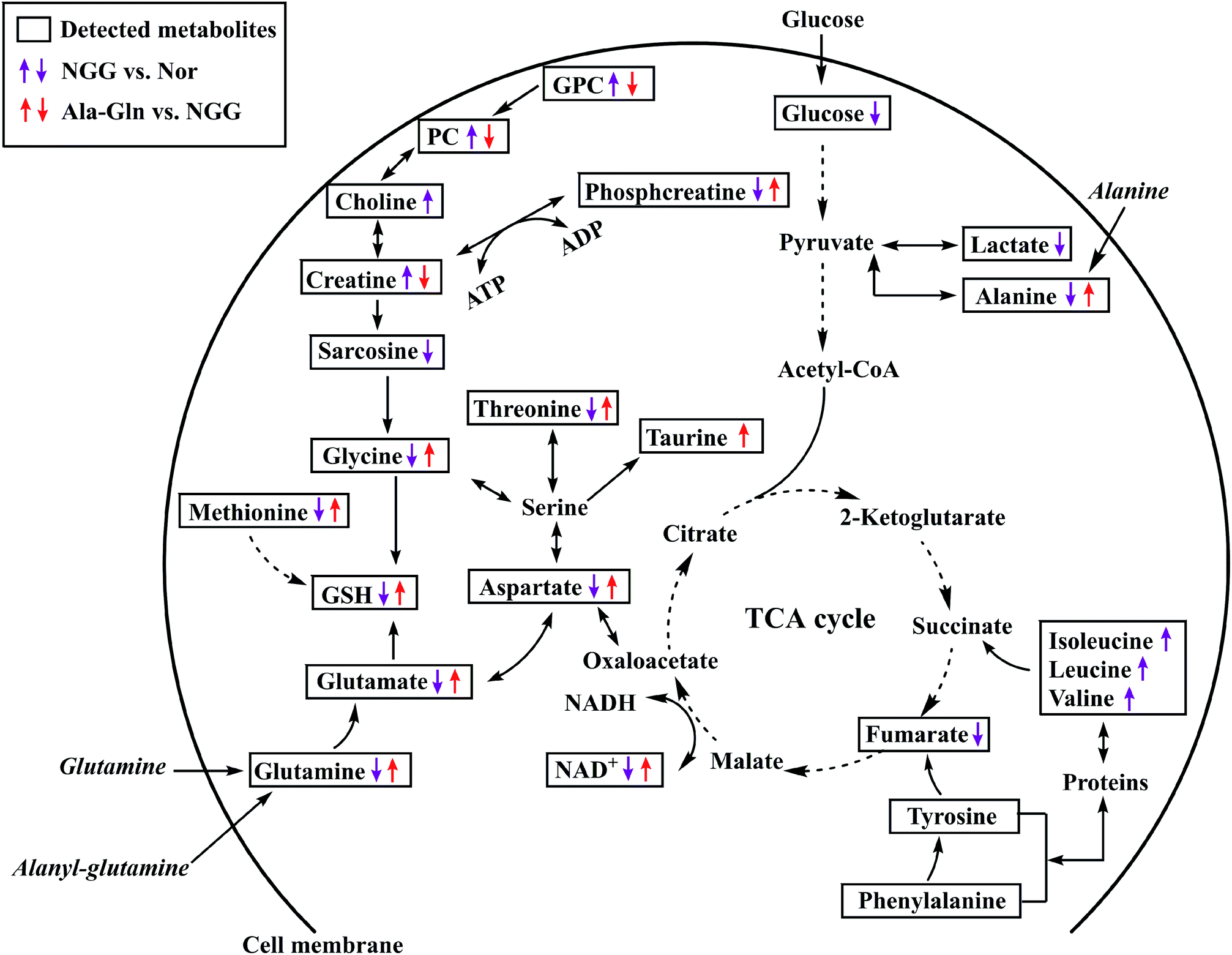 | ||
| Fig. 5 Schematic representation of significantly altered metabolic pathways of C2C12 myoblasts under NGG and Ala-Gln treatments based on the KEGG database and the MetaboAnalyst webserver. Dotted arrow indicates multiple biochemical reactions; solid arrow denotes single biochemical reaction. | ||
Discussion
Ala-Gln has been extensively recognized not only as a parenteral postoperative nutritional supplement, but also as a potential treatment for muscle-related diseases and injuries. However, the molecular mechanisms underlying the polyphenic effects of Ala-Gln supplementation remain to be systematically addressed in detail. In this work, we performed NMR-based cellular metabolomic analysis to explore the metabolic responses of injured C2C12 myoblast when treated with Ala, Gln and Ala-Gln.As well known, myogenic satellite cell is critically important in muscle growth and repair as well as the processes of adaptation to stresses including exercise, disease, injury, and aging.28 Natural functioning of satellite cells is crucial to maintain skeletal muscle homeostasis, and satellite cell proliferation is an essential step to generate sufficient number of muscle progenitors in myogenesis.29 Therefore, C2C12 myoblast proliferation is an early and pivotal process in muscle growth and repair. Furthermore, as a marker of myogenic commitment, the MyoD protein plays an important role in myogenesis.30
Based on the results from both MTS assay and western blot analysis of MyoD1, we found that glucose and glutamine deprivation has a large impact on the proliferation and differentiation ability of C2C12 myoblasts, thus causing cells injured. Even though Ala, Gln and Ala-Gln supplementations all enhanced the differentiation ability of the injured C2C12 myoblasts, only Ala-Gln supplementation can improve the proliferation ability of the injured cells (Fig. 1). These results suggest that Ala-Gln is more suitable for the treatment of injured skeletal muscle cells compared with free Ala and Gln.
In response to Ala supplementation, the intracellular levels of BCAAs are significantly decreased in C2C12 myoblasts due to energy deprivation. BCAAs are primary components of proteins, and can not only stimulate protein synthesis via mTOR complex 1 (mTORC1) in muscles, but also ameliorate muscle atrophies of humans and animals, such as cancer cachexia models.31,32 A previous report has indicated that the only metabolism of tyrosine and phenylalanine in muscle is involved in protein synthesis and degradation.33 Our work shows that the Ala treatment decreases the intracellular levels of tyrosine and phenylalanine, suggesting that those two amino acids are applied for protein synthesis. Taken together, our results reveal that the Ala treatment can obviously promote the protein synthesis of the injured C2C12 myoblasts.
On the other hand, we found that energy deprivation reduces the level of alanine in the NGG group, and Ala, Gln and Ala-Gln supplementations can enhance the intracellular level of alanine. As well known, alanine is not only the glycolytic end product, but also an important intermediate of TCA cycle capable of being utilized for energy production.34 Ala-Gln, Gln and Ala supplementations can all improve energy production of the impaired C2C12 myoblasts by up-regulating the level of alanine. Nevertheless, the Ala treatment exhibits the most significant effect due to the contribution of exogenous alanine to the intracellular level of alanine. By contrast, the Ala-Gln treatment can induce the contribution of exogenous alanine to the intracellular level of alanine only after the dipeptide Ala-Gln is hydrolyzed, potentially accounting for the lower FC relative to the Ala treatment (5.30 times vs.12.1 times). Furthermore, the Gln treatment does not provide exogenous alanine to the cells, which explains the low level of alanine in the Gln group (FC, 1.34 times).
As indicated in previous reports, the mutual conversion between Cr and PCr is carried out via the PCr–Cr kinase pathway (Cr + ATP ↔ PCr + ADP + H+) for utilizing ATP in muscle cells.35,36 The enhanced level of PCr and declined level of Cr in Ala-Gln and Gln groups relative to the NGG group, suggesting that ATP is accumulated in the form of PCr. These results illustrate that Ala-Gln and Gln supplementations can ameliorate the energy deficit of injured myoblasts through increasing PCr and decreasing Cr.
When mammal cells are deprived of glucose, AMP-activated protein kinase (AMPK) is rapidly activated due to the reduced rate of glucose metabolism and the increased ratio of AMP/ATP.37,38 AMPK activation can reinforce oxidative phosphorylation for generating energy,39,40 which might promote the activity of TCA cycle anaplerotic flux and lead into a large number of amino acids entering TCA cycle anaplerosis. The metabolic comparison between NGG and Nor groups, shows that energy deprivation can dramatically decrease intracellular levels of aspartate, glutamate, glutamine, glycine and threonine as the sources of TCA cycle anaplerosis. Both Ala-Gln and Gln supplementations can partially restore the decreased levels of these amino acids resulting from energy deprivation, while the Ala treatment can only partly recover the decreased levels of aspartate and glutamate. Moreover, the Ala-Gln treatment displays the most significant effect of enhancing the levels of aspartate and glutamate compared to Gln and Ala treatments (Fig. 4). Especially, comparing the FCs of glutamine between Ala-Gln and Gln groups (8.19 times vs. 2.55 times), suggests that Ala-Gln supplementation is a preferred way to provide glutamine, as previously reported.14,41 Taken together, these results prove that Ala-Gln supplementation is more suitable for the treatment of injured myoblast cells.
As it is known, cellular reactive oxygen species (ROS) are primarily generated by the respiratory chain during oxidative phosphorylation.42,43 Energy deprivation in the NGG group remarkably decreases GSH and taurine, and also reduced glycine and methionine acting as the necessary precursors for GSH synthesis (Fig. 4B). As the most abundant nonprotein thiol in the antioxidant defense system, GSH plays an important role in maintaining the redox balance.44 Moreover, as the most abundant free amino acid in excitable tissues and cells, taurine can prevent cell damage from oxidative stress.45 As reported, the Ala-Gln treatment can reduce muscle cell damage and enhance antioxidant capacity by elevating the level of GSH.46 Our work shows that both Ala-Gln and Gln supplementations can enhance the levels of anti-oxidant related metabolites relative to the NGG treatment, as indicated by the FCs of GSH (2.40 times, 1.54 times), glycine (1.61 times, 1.27 times), methionine (1.24 times, 1.27 times) and taurine (1.17 times, 1.18 times) in injured C2C12 myoblasts. These results indicate that the Ala-Gln treatment has a more significant effect of preventing oxidative damage through promoting GSH biosynthesis compared with the Gln treatment.
On the other hand, ROS formation is an event upstream of the lipid peroxidation which results in cell membrane instability.47 As well known, membrane phospholipids are mostly composed of phosphatidylcholine. Both choline and PC are essential precursors of phosphatidylcholine, while GPC is breakdown products of phosphatidylcholine.25 As reported, the increased levels of PC and GPC indicate the activation of choline kinase and promotion of membrane breakdown by phospholipases.48 Our work shows that the NGG treatment significantly up-regulates PC and GPC, suggesting that energy deficit can attenuate the membrane stability by promoting phosphatidylcholine breakdown via ROS formation. Meanwhile, Ala-Gln, Gln and Ala treatments can decrease PC and GPC and increase GSH, implying that the three treatments enhance the stability of lipid membrane by suppression of glycerophospholipid metabolism through promoting GSH biosynthesis. Besides, the Ala-Gln group exhibits a higher intracellular level of GSH than Gln and Ala groups. This result reveals again the advantage of the Ala-Gln treatment over Gln and Ala treatments for partially restoring the metabolic profile of the C2C12 cells injured by energy deprivation.
Conclusions
This work elucidates the metabolic responses of C2C12 myoblasts injured by energy deprivation to Ala-Gln, Gln and Ala supplementations. We found that glucose and glutamine deprivation can cause cell injuries. Moreover, we observed that Ala-Gln, Gln and Ala supplementations can all promote the differentiation ability of the injured cells, but only Ala-Gln supplementation can facilitate the proliferation ability of the injured cells.The three supplementations can partially restore the metabolic profiles of the C2C12 cells disturbed by energy deprivation. More importantly, Ala-Gln supplementation shows more significant effects of treating injured myoblasts than Gln and Ala supplementations. The Ala-Gln treatment can promote protein synthesis, up-regulate ATP-storage PCr, maintain TCA cycle anaplerosis, enhance the membrane stability by suppression of glycerophospholipids metabolism through promoting GSH biosynthesis. These results shed light on the molecular mechanisms underlying the polyphonic effects of the Ala-Gln treatment on muscle cells injured by energy deprivation. This work may be helpful for further exploring the extensive use of Ala-Gln supplementation in new treatments for muscle-diseases and injuries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by grants from the Joint Research Program of Fujian Provincial Health and Family Palnning Commision and the Education Department of Fujian Province (WKJ-FJ-12), and the Xiamen Ocean Economic Innovation and Development Demonstration Project (No. 16PZP001SF16).References
- W. R. Frontera and J. Ochala, Calcif. Tissue Int., 2015, 96, 183–195 CrossRef CAS PubMed.
- J. A. Romijn, E. F. Coyle, L. S. Sidossis, A. Gastaldelli, J. F. Horowitz, E. Endert and R. R. Wolfe, Am. J. Physiol., 1993, 265, E380–E391 CAS.
- T. Matsui, H. Omuro, Y. F. Liu, M. Soya, T. Shima, B. S. McEwen and H. Soya, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6358–6363 CrossRef CAS PubMed.
- E. Hutchinson, Nat. Rev. Cancer, 2004, 4, 662 CrossRef CAS.
- S. Yae, F. Takahashi, T. Yae, T. Yamaguchi, R. Tsukada, K. Koike, K. Minakata, A. Murakami, F. Nurwidya, M. Kato, M. Tamada, M. Yoshikawa, H. Kobayashi, K. Seyama and K. Takahashi, J. Evidence-Based Complementary Altern. Med., 2012, 2012, 926–976 Search PubMed.
- K. J. Sanders, A. E. Kneppers, C. van de Bool, R. C. Langen and A. M. Schols, J. Cachexia Sarcopenia Muscle, 2016, 7, 5–22 CrossRef PubMed.
- H. A. Ebhardt, S. Degen, V. Tadini, A. Schilb, N. Johns, C. A. Greig, K. C. H. Fearon, R. Aebersold and C. Jacobi, J. Cachexia Sarcopenia Muscle, 2017, 8, 567–582 CrossRef PubMed.
- A. H. V. Remels, H. R. Gosker, R. C. J. Langen and A. M. W. J. Schols, J. Appl. Physiol., 2012, 114, 1253–1262 CrossRef PubMed.
- T. Nedachi, A. Kadotani, M. Ariga, H. Katagiri and M. Kanzaki, Am. J. Physiol.: Endocrinol. Metab., 2008, 294, E668–E678 CrossRef CAS PubMed.
- M. Ariga, Y. Yoneyama, T. Fukushima, Y. Ishiuchi, T. Ishii, H. Sato, F. Hakuno, T. Nedachi and S. I. Takahashi, Endocr. J., 2017, 64, 255–268 CrossRef PubMed.
- D. Gawlitta, C. W. Oomens, D. L. Bader, F. P. Baaijens and C. V. Bouten, J. Appl. Physiol., 2007, 103, 464–473 CrossRef CAS PubMed.
- Y. F. Huang, Y. Wang and M. Watford, J. Nutr., 2007, 137, 1357–1362 CrossRef CAS PubMed.
- G. Letellier, E. Mok, C. Alberti, A. De Luca, F. Gottrand, J. M. Cuisset, A. Denjean, D. Darmaun and R. Hankard, Clin. Nutr., 2013, 32, 386–390 CrossRef CAS PubMed.
- M. B. Braga-Neto, C. A. Warren, R. B. Oria, M. S. Monteiro, A. A. Maciel, G. A. Brito, A. A. Lima and R. L. Guerrant, Dig. Dis. Sci., 2008, 53, 2687–2696 CrossRef CAS PubMed.
- B. Tan, H. Liu, G. He, H. Xiao, D. Xiao, Y. Liu, J. Wu, J. Fang and Y. Yin, Amino Acids, 2017, 49, 2023–2031 CrossRef CAS PubMed.
- Y. Hu, D. E. Smith, K. Ma, D. Jappar, W. Thomas and K. M. Hillgren, Mol. Pharm., 2008, 5, 1122–1130 CrossRef CAS.
- V. F. Cruzat and J. Tirapegui, Nutrition, 2009, 25, 428–435 CrossRef CAS PubMed.
- R. Raizel, J. S. Leite, T. M. Hypolito, A. Y. Coqueiro, P. Newsholme, V. F. Cruzat and J. Tirapegui, Br. J. Nutr., 2016, 116, 470–479 CrossRef CAS PubMed.
- V. F. Cruzat, A. Bittencourt, S. P. Scomazzon, J. S. Leite, P. I. de Bittencourt Jr and J. Tirapegui, Nutrition, 2014, 30, 602–611 CrossRef CAS PubMed.
- E. R. Petry, V. F. Cruzat, T. G. Heck, J. S. Leite, P. I. Homem de Bittencourt Jr and J. Tirapegui, Life Sci., 2014, 94, 130–136 CrossRef CAS PubMed.
- J. K. Nicholson, J. C. Lindon and E. Holmes, Xenobiotica, 1999, 29, 1181–1189 CrossRef CAS PubMed.
- M. E. Bollard, E. G. Stanley, J. C. Lindon, J. K. Nicholson and E. Holmes, NMR Biomed., 2005, 18, 143–162 CrossRef CAS PubMed.
- Q. Teng, W. Huang, T. W. Collette, D. R. Ekman and C. Tan, Metabolomics, 2008, 5, 199–208 CrossRef.
- S. Lamichhane, C. C. Yde, M. S. Schmedes, H. M. Jensen, S. Meier and H. C. Bertram, Anal. Chem., 2015, 87, 5930–5937 CrossRef CAS PubMed.
- W. Xu, D. Lin and C. Huang, Acta Biochim. Biophys. Sin., 2017, 1–11, DOI:10.1093/abbs/gmx043.
- A. Craig, O. Cloareo, E. Holmes, J. K. Nicholson and J. C. Lindon, Anal. Chem., 2006, 78, 2262–2267 CrossRef CAS PubMed.
- J. Trygg, E. Holmes and T. Lundstedt, J. Proteome Res., 2007, 6, 469–479 CrossRef CAS PubMed.
- S. E. Alway, M. J. Myers and J. S. Mohamed, Front. Aging Neurosci., 2014, 6, 246 Search PubMed.
- M. Kurosaka, Y. Ogura, T. Funabashi and T. Akema, J. Cell. Physiol., 2017, 232, 1114–1122 CrossRef CAS PubMed.
- O. Kanisicak, J. J. Mendez, S. Yamamoto, M. Yamamoto and D. J. Goldhamer, Dev. Biol., 2009, 332, 131–141 CrossRef CAS PubMed.
- Y. Okimura, in Branched Chain Amino Acids in Clinical Nutrition, ed R. Rajendram, V. R. Preedy and V. B. Patel, Springer New York, New York, NY, 2015, vol. 2, ch. 4, pp. 49–63 Search PubMed.
- H. L. Eley, S. T. Russell and M. J. Tisdale, Biochem. J., 2007, 407, 113–120 CrossRef CAS PubMed.
- M. H. Vendelbo, A. B. Møller, B. Christensen, B. Nellemann, B. Frederik, F. Clasen, K. S. Nair, J. O. Lunde Jørgensen, N. Jessen and N. Møller, PLoS One, 2014, 9, e102031 Search PubMed.
- M. C. Matrka, M. Watanabe, R. Muraleedharan, P. F. Lambert, A. N. Lane, L. E. Romick-Rosendale and S. I. Wells, PLoS One, 2017, 12, e0177952 Search PubMed.
- V. Saks, J. Physiol., 2008, 586, 2817–2818 CrossRef CAS PubMed.
- T. Wallimann, M. Wyss, D. Brdiczka, K. Nicolay and H. Eppenberger, Biochem. J., 1992, 281, 21–40 CrossRef CAS PubMed.
- P. de Lange, M. Moreno, E. Silvestri, A. Lombardi, F. Goglia and A. Lanni, FASEB J., 2007, 21, 3431–3441 CrossRef CAS PubMed.
- D. G. Hardie, Genes Dev., 2011, 25, 1895–1908 CrossRef CAS PubMed.
- M. M. Mihaylova and R. J. Shaw, Nat. Cell Biol., 2011, 13, 1016–1023 CrossRef CAS PubMed.
- E. Watson, L. S. Yilmaz and A. J. Walhout, Annu. Rev. Genet., 2015, 49, 553–575 CrossRef CAS PubMed.
- C. V. da Rosa, S. C. Azevedo, R. B. Bazotte, R. M. Peralta, N. C. Buttow, M. M. Pedrosa, V. A. de Godoi and M. R. Natali, PLoS One, 2015, 10, e0143005 Search PubMed.
- J. Dan Dunn, L. A. Alvarez, X. Zhang and T. Soldati, Redox Biol., 2015, 6, 472–485 CrossRef CAS PubMed.
- M. Maryanovich and A. Gross, Trends Cell Biol., 2013, 23, 129–134 CrossRef CAS PubMed.
- X. Ren, L. Zou, X. Zhang, V. Branco, J. Wang, C. Carvalho, A. Holmgren and J. Lu, Antioxid. Redox Signaling, 2017, 27, 989–1010 CrossRef CAS PubMed.
- X. Shao, Z. Hu, C. Hu, Q. Bu, G. Yan, P. Deng, L. Lv, D. Wu, Y. Deng, J. Zhao, R. Zhu, Y. Li, H. Li, Y. Xu, H. Yang, Y. Zhao and X. Cen, Toxicol. Appl. Pharmacol., 2012, 260, 260–270 CrossRef CAS PubMed.
- W. F. Alves, E. E. Aguiar, S. B. Guimaraes, A. R. da Silva Filho, P. M. Pinheiro, S. Soares Gdos and P. R. de Vasconcelos, Annals of Vascular Surgery, 2010, 24, 461–467 CrossRef PubMed.
- H. Lee, E. H. Ko, M. Lai, N. Wei, J. Balroop, Z. Kashem and M. Zhang, Mol. Immunol., 2014, 58, 151–159 CrossRef CAS PubMed.
- P. K. Valonen, J. L. Griffin, K. K. Lehtimaki, T. Liimatainen, J. K. Nicholson, O. H. Grohn and R. A. Kauppinen, NMR Biomed., 2005, 18, 252–259 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. DOI: 10.1039/c8ra00819a |
| This journal is © The Royal Society of Chemistry 2018 |