DOI:
10.1039/C8RA00797G
(Paper)
RSC Adv., 2018,
8, 17060-17072
Effect of Rhizoma Paridis saponin on the pain behavior in a mouse model of cancer pain†
Received
26th January 2018
, Accepted 13th April 2018
First published on 9th May 2018
Abstract
Rhizoma Paridis saponins (RPS) as active parts of P. polyphylla Smith var. yunnanensis has been used as an anti-cancer drug in traditional Chinese medicine. In this study, RPS was first found to demonstrate a potent effect on markedly reducing the pain induced by cancer. Therefore, the aim of this study was to further explore the analgesic effect of RPS and its possible reaction pathway on H22 hepatocarcinoma cells inoculated in the hind right paw of mice. Cancer-induced pain model mice were randomly divided into 5 groups (n = 10) and orally administered with RPS (50–200 mg kg−1) for 2 weeks. On the last day of treatment, the pain behavior of mice was measured using hot-plate test and open field test, and brain tissues were sampled for detection of biochemical indices, malondialdehyde (MDA), superoxide dismutase (SOD), prostaglandin E2 (PGE2), serotonin (5-HT) and β-endorphin (β-EP). Moreover, the concentrations of NF-κB and IL-1β in the blood serum were measured by ELISA reagent kits. In addition, naloxone, the non-selective antagonist of opioid receptors, was used to identify the opioid receptors involved in RPS's action. It has been found that RPS alleviates cancer pain mainly via the suppression of inflammatory pain induced by oxidative damage, such as decreasing MDA and PGE2 levels, renewing activity of SOD, as well as increasing 5-HT and β-EP in the brain and suppressing the expression of NF-κB and IL-1β in the serum in a concentration-dependent manner. Overall, the current study highlights that RPS has widespread potential antinociceptive effects on a mouse model of chronic cancer pain, which may be associated with the peripheral nervous system and the central nervous system.
Introduction
Pain is the first symptom of cancer in 30–50% of patients, and it is estimated that at least 70–90% of cancer sufferers will eventually experience chronic pain during their illness.1 Malignant, long-lasting pain is a frequent symptom in cancer patients that impedes antineoplastic therapy and critically diminishes the health condition of patients.2 Thus, effective therapy involving various pharmacological and non-pharmacological interferences for treating pain has been extensively explored in both basic research and clinical trials.
According to WHO guidelines, opioids are the mainstay analgesics for moderate to severe pain management. The analgesic effect of opiate drugs occurs through the stimulation of opioid receptors in the central nervous system and the peripheral nervous system. In addition to producing analgesia by exerting their analgesic action in the central nervous system, classical opioids simultaneously cause the onset of undesirable side effects, such as tolerance, nausea or sedation, which are recognized as significant burdens for cancer patients.3 Moreover, many patients suffer the pain via the peripheral nervous system induced by inflammation during the period of tumor growth.
Reactive oxygen species scavengers (ROS) are natural byproducts of cellular metabolism with important physiological and pathophysiological functions.4 However, excessive ROS levels due to increased ROS production and/or decreased antioxidant defense ability leads to lipid peroxidation, protein oxidation, and nucleic acid oxidation.5 When generated beyond the normal physiological range, excessive ROS can cause oxidative stress. Recently, increasing evidence had supported the involvement of ROS in pathological pain induced by cancer. Mechanical allodynia in rats was effectively attenuated by the intrathecal injection of a ROS scavenger, indicating that excessive ROS levels may be closely related to cancer pain development, probably through the peripheral nervous system.6 Serotonin (5-HT) and β-endorphin (β-EP) in the brain play critical roles in behavioral responses to nociceptive mechanisms.7,8 The serotonergic system modulates nociception mainly via descending serotonergic pathways that run principally from rostroventromedial medulla through the dorsolateral funiculus.9,10 It is known that several different subtypes of spinal 5-HT receptors have been proposed to mediate the antinociceptive effects of 5-HT. β-Endorphin is normally expressed in neurons and it is reported that they specifically activate μ-opioid receptors localized in post-synaptic neurons and modulate pain transmission and transduction.11
To date, opioid analgesics remain the most efficacious pharmacological agents for the treatment of moderate to severe pain, but their therapeutic benefit is often hampered by the development of analgesic tolerance and hyperalgesia.12 Non-steroidal anti-inflammatory drugs (NSAID) are also used to manage pain, but they also have side effects, such as cardiovascular risks and gastrointestinal diseases.13,14 Medicinal plants are among the potential adjuvant agents that could also be evaluated in this therapeutic approach. In fact, phytotherapy offers valid support to conventional medicine in a number of diseases, and this also applies to the management of pain. Paris polyphylla is the dried rhizome of P. polyphylla Smith var. yunnanensis, which has been widely used for the treatment of hemostasis, abscess, snake bites, abnormal uterine bleeding, tumors, analgesia and fractures in clinical practice for thousands of years in China.15,16 Rhizoma Paridis saponins (RPS) as active parts of P. polyphylla Smith var. yunnanensis has been used as an anti-cancer drug in traditional Chinese medicine.17–19 Furthermore, previous studies showed that Rhizoma Paridis saponins display anti-inflammatory, antioxidant and antinociceptive effects in rodents.20,21 However, the underlying mechanisms by which Rhizoma Paridis saponins reduce the pain induced by cancer are not well understood yet.
A plethora of animal models showing local signs of cancer-induced hypersensitivity have been developed, which accurately mimic cancer pain and produced by the H22 hepatocarcinoma cells implantation into the hind paw.22,23 Chronic cancer pain model was established to evaluate the analgesic effect of the oral administration of RPS. We studied the analgesic effect on the pain threshold using the hot-plate test, and we also evaluated the behavioural characteristics of mice by utilizing the open field test in the cancer pain model. Finally, in order to define whether the analgesic effect of RPS on cancer pain is related to the central nervous system or/and peripheral nervous system, we evaluated the influence of RPS on the biochemical changes (SOD, MDA, PGE2, β-EP and 5-HT) induced by cancer pain in brain tissue. These studies point to the analgesic effect of RPS and the relevance of central and peripheral analgesia in cancer pain treatment.
Materials and methods
Materials
Dried rhizomes of P. polyphylla Smith var. yunnanensis were purchased from Yunnan Province, China, and identified by Prof. Wenyuan Gao (Tianjin University, China). Rhizoma Paridis saponins (RPS) were prepared in Tasly Holding Group Co., Ltd. (Tianjin, China) and the method was similar to that previously reported.24 RPS was suspended in distilled water. Tramadol hydrochloride (INN) was obtained from Kunming Pharmaceutical Group Co., Ltd. (Yunnan, China). ELISA assay kits for the determination of superoxide dismutase (SOD) activity, malondialdehyde (MDA), prostaglandin E2 (PGE2), serotonin (5-HT) and β-endorphin (β-EP) were obtained from Nanjing Jiancheng Bioengineering Research Institute (Nanjing, China).
Cell culture
H22 hepatocarcinoma cells (H22) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cancer cells that were collected from the ascites of tumor-bearing mice after three generations were suspended in 0.9% sodium chloride with a concentration of 6 × 107 cells per mL.
Animals
Kunming mice, typically weighing 18 to 22 g for females, were purchased from the Institute of Tianjin Laboratory Animal Center, Tianjin. Mice were kept at 25 ± 1 °C under a 12 h light/dark cycle condition, while providing free access to food (standard pellet diet) and water ad libitum. All experimental protocols were approved by the Animal Ethics Committees of the Faculty of Medicine, Tianjin University, Tianjin, China, and carried out in accordance with “Principles of Laboratory Animal Care and Use in Research” (State Council of China, 1988).
Cancer pain model establishment
The general method of preparation and implantation of melanoma cells has been published previously.25 Briefly, H22 hepatoma cells, which were collected from the ascites of tumor-bearing mice, were suspended in 0.9% sodium chloride. To establish the hind paw cancer pain model, 20 μL H22 hepatoma cells (6 × 107 cells per mL) were subcutaneously implanted into the plantar region of the right hind paw using a 30-gauge needle. Control mice received an equivalent volume of normal saline (NS) injections in right hind paws.
Determination of paw volume
Tumor growth was determined as previously described.26 Paw thickness and width were measured with an electronic caliper with 0.1 mm accuracy. Paw volume was calculated in the tumor-bearing and intact paws according to the following formula: V = (t × w2) × 0.52, where V is the paw volume, t is the paw thickness and w is the paw width.
Experimental design
Experiments were conducted on post-inoculation day 14 when mice exhibited a stable decrease in paw licking latency. Analgesic effect was assessed using the hot-plate test. In total, 60 qualified mice were randomly divided into 6 groups of 10 mice each: control group (control), model group (model), positive group (INN, 10 mg kg−1, i.p.), high-dose RPS group (RPS-H, once daily, 200 mg kg−1, i.g.), middle-dose RPS group (RPS-M, once daily, 100 mg kg−1, i.g.), and low-dose RPS group (RPS-L, once daily, 50 mg kg−1, i.g.). All drugs were dissolved in 0.9% (w/v) NaCl. Tramadol hydrochloride (INN) was injected intraperitoneally (i.p.). Mice in the control and model groups were administered NS, once daily for 14 days. The pain behavior of each mouse was determined before and after treatment. On the last day of treatment, the pain behavior of mice was measured, and specimens were sampled for testing.
Hot-plate test
The hot-plate test was performed with a plate of pure aluminium slab that was mated to thin high-surface heating elements, which provided an even temperature on the entire test area (Socrel DS-35, Ugo Basile, Comerio, Italy). The heating plate was kept at a constant temperature of 55 ± 0.5 °C. Mice were confined by a clear removable Perspex hood. The latencies for paw licking of the hind paw or jumping were recorded for each animal. Mice that displayed jumping during the baseline testing were discharged, while jumping was accepted only in the test latency evaluation. The analgesic efficacy of the cancer pain model was evaluated using paw licking latency. An 80 s cut-off was used to prevent tissue damage. The reaction times (in seconds) were registered immediately before the beginning of the baseline latency and 10 min after the; immediately thereafter, the animals were evaluated by the open field test.
Effects on open field test
An open field (36 cm × 36 cm) was constructed by enclosing an area of tiled floor with 40 cm-high plywood planks, whose floor was divided into 9 equal squares by black lines. The animals were carefully placed at the center of the open field and behaviors in 5 min were recorded. Their movement trajectories were recorded by a video camera and analyzed using a tracking software (TM-Vision, Stoelting, Wood Dale, IL). Immediately thereafter, the animals were anaesthetised with ethyl ether and decapitated. The brains were removed and stored at −80 °C until required for assay.
Determination of brain tissue biomarkers
After evaluating by the open field test, the mice were used for the analyses of brain tissue biochemical parameters. In brief, mice were sacrificed, and brain tissues were rapidly excised, weighed (0.5 g of each tissue), and homogenized with 2.0 mL of iced normal saline. The interstitial fluid was prepared by centrifugation at 1000g at 4 °C for 15 min. The supernatant was tested to determine the levels of SOD activities, MDA, PGE2, β-EP and 5-HT. The biomarkers were tested following the recommended procedures provided in the kits.
Measurement of blood NF-κB and IL-1β levels by ELISA assay
Fresh blood samples were collected from the tail veins of cancer pain model mice. Blood serum was separated by centrifugation at 3000 rpm for 20 min. The concentrations of NF-κB and IL-1β in the blood serum were measured by ELISA reagent kits (BD Bioscience, Franklin Lakes, NJ, USA), as detailed by the manufacturer.
Histology
For examination of the primary hind paw tumor and potential metastasis into lungs, all mice were euthanatized with 100 mg kg−1 intraperitoneal pentobarbital sodium (Nembutal, Polfa, Poland). Paws were removed and post fixed in Bouin's and 1% paraformaldehyde in 0.1 M PBS solution, respectively. Fixed tissues were embedded in paraffin. Sections (5 mm) were mounted onto Superfrost Plus microscope slides (Fisher Scientific, Pittsburgh, PA) and stained with hematoxylin and eosin. Histopathologic examination was performed under a light microscope (Nikon, Tokyo, Japan).
Involvement of opioid receptors in RPS antinociceptive effect
The involvement of the opioid system in the antinociceptive effect of RPS was studied as previously described.27 The mice subjected to intraplantar injection of 20 μL formalin (2.5%, i.pl.) were pretreated with either a systemic (i.p.) injection of saline (10 mL kg−1) or naloxone (a nonselective opioid receptor antagonist, 1 mg kg−1) 30 min before RPS (200 mg kg−1, i.g.) or morphine (2.5 mg kg−1, s.c.) treatments. The nociceptive response was evaluated as the time spent licking the injected paw during 5 min and 30 min after the treatments with RPS and morphine.
Statistical analysis
Statistical analyses were conducted using SPSS 17.0 software. The results are expressed as the mean ± standard error (SE) in tables and are indicated by the vertical bars in figures. Differences between the groups were determined by one-way analysis of variance (ANOVA) and Student's t-test. Correlations were expressed using the Pearson's product-moment correlation coefficient. A probability value (P) less than 0.05 was considered significant.
Results
Tumor growth
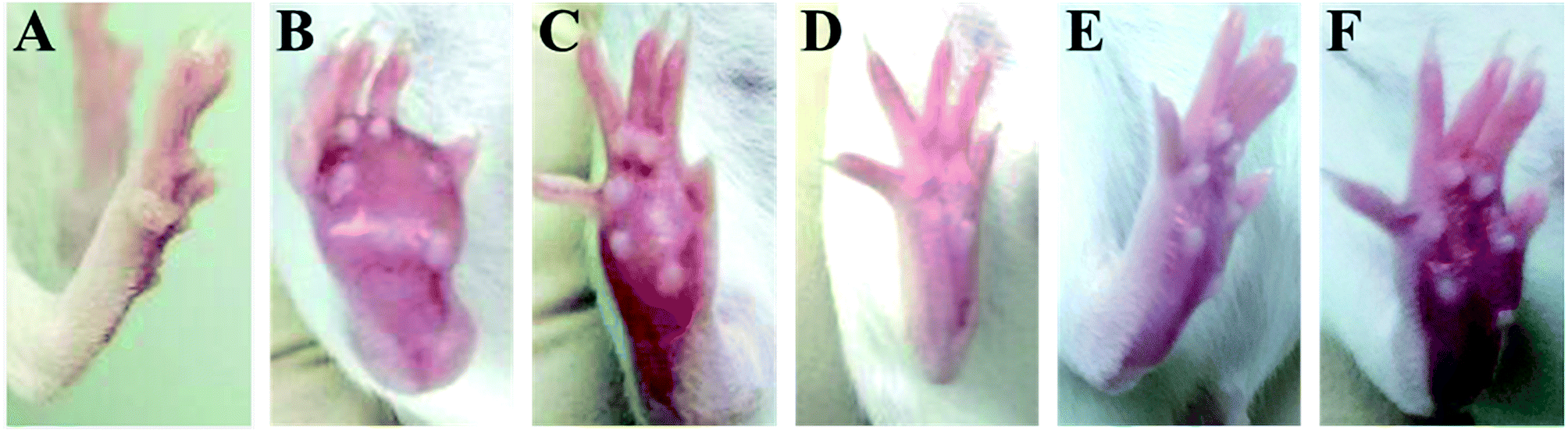
The time-course of tumor growth in mice inoculated with H22 hepatoma cells is shown in Fig. 1. The paw volume significantly differed in the tumor-bearing paw, compared to the NS-injected paw (P < 0.001). On day 9 post implantation, there was a significant increase in the volume of the paw inoculated with H22 cells (model: 112.38 ± 13.94 vs. control: 26.55 ± 2.18; P < 0.001, Fig. 1). The paw volume of the model group inoculated with H22 cells further increased significantly until day 14 when animals were sacrificed. Moreover, the present results showed that the paw volume was significantly lower in the RPS-treated group than that in the model group (P < 0.05 and P < 0.01). Mice from the model group developed large-sized tumors (Fig. 2) much earlier than the control mice. Furthermore, as shown in Table 1, RPS with different doses exhibited significant inhibition effects on tumor weights of paws inoculated with H22 cells, which were 0.36 ± 0.13 g, 0.32 ± 0.15 g and 0.21 ± 0.07 g (P < 0.05 and P < 0.01). Groups of RPS-L, RPS-M, and RPS-H demonstrated increased ratios of inhibition rates, which were 49.33%, 53.86% and 69.47%, respectively. These results indicated that RPS had significant inhibition effect (P < 0.05) on the growth of tumors in mice with evident dose–effect relationship.
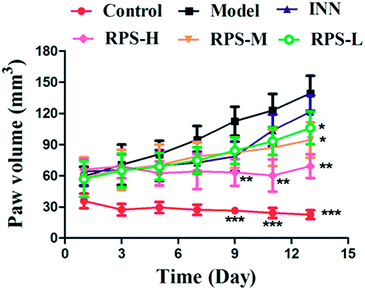 |
| | Fig. 1 Time-course of tumor growth in H22 hepatocarcinoma cells inoculated and normal saline. Each value represents the mean ± SE. Compared with the model group using one-way ANOVA with Student's t-test, *P < 0.05, **P < 0.01, ***P < 0.01. | |
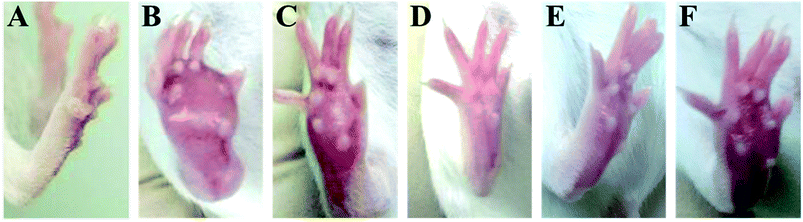 |
| | Fig. 2 The picture of H22 tumor after subcutaneous inoculation with H22 hepatoma cells into the plantar region of the hind right paw. (A) Control group, (B) model group, (C) INN group, (D) RPS-H group, (E) RPS-M group, and (F) RPS-L group. | |
Table 1 The inhibitory rate and paw weight in cancer pain model H22 hind paw of micea
| Groups |
Paw weight (g) |
Inhibition rate (%) |
| Each value represents the mean ± SE. Compared with the model group, *P < 0.05, **P < 0.01, ***P < 0.01. |
| Control |
0.15 ± 0.01*** |
— |
| Model |
0.70 ± 0.23 |
— |
| INN |
0.48 ± 0.13* |
31.24 |
| RPS-H |
0.21 ± 0.07*** |
69.47 |
| RPS-M |
0.32 ± 0.15** |
53.86 |
| RPS-L |
0.36 ± 0.13** |
49.33 |
Analgesic effect of RPS in cancer pain model
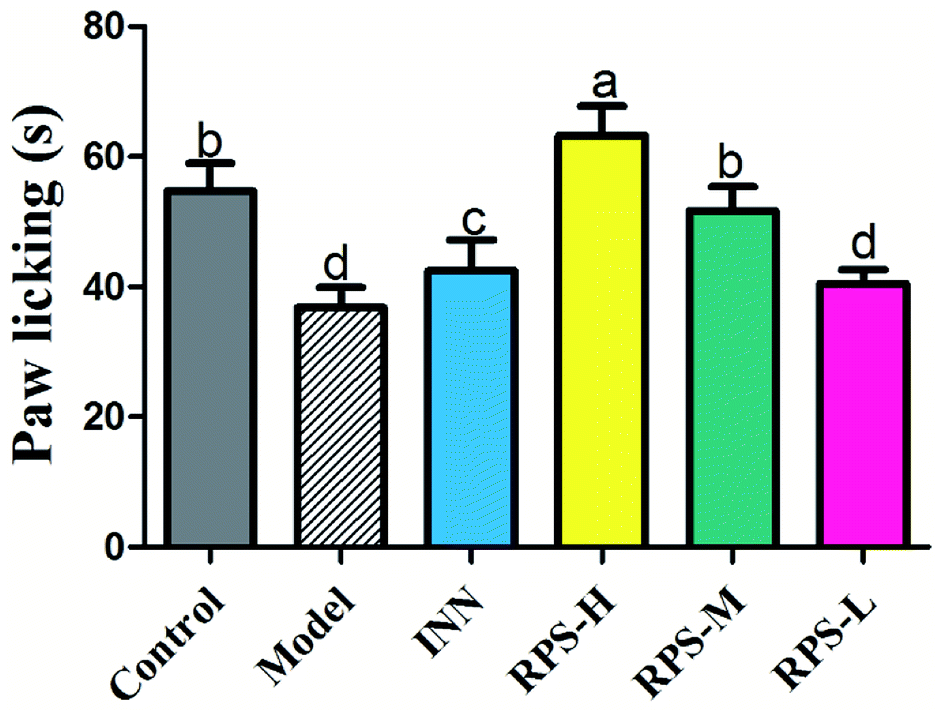
As shown in Fig. 3, there was a dose-dependent increase in the analgesic effect in the tumor-bearing paw (P < 0.001 and P < 0.01, Fig. 3) after the oral administration of three doses of RPS (50–200 mg kg−1). Mice injected with the highest dose of 200 mg kg−1 showed the highest total analgesic response on paw licking latency, which significantly differed from NS-injected controls (63.33 ± 10.80 vs. 36.83 ± 7.44, P < 0.001, Fig. 3). We also found that the paw licking latency of the model group inoculated with H22 cells was significantly reduced in comparison to control mice on the hot-plate test. Furthermore, the effect of positive control group on analgesia was similar to that of RPS-M, but weaker than that of RPS-H. These results indicated that the thermally-induced pain on H22-inoculated mice was a successful model for cancer pain and RPS had a distinct analgesic effect with a dose–effect relationship.
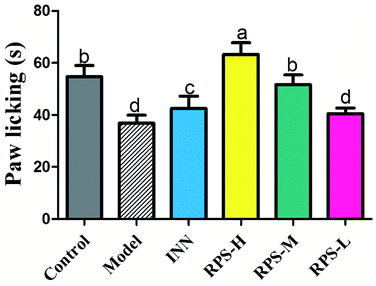 |
| | Fig. 3 Analgesic effect of three dosages of RPS on established tumor-bearing paw model in hot-plate test. Each value is represented as the mean ± SE. Values within treatment groups having different letters are significantly different, as indicated by one-way ANOVA and Student's t-test. Letters a–d, means with the same letter is not significantly different (p < 0.05). | |
Effects on open field test
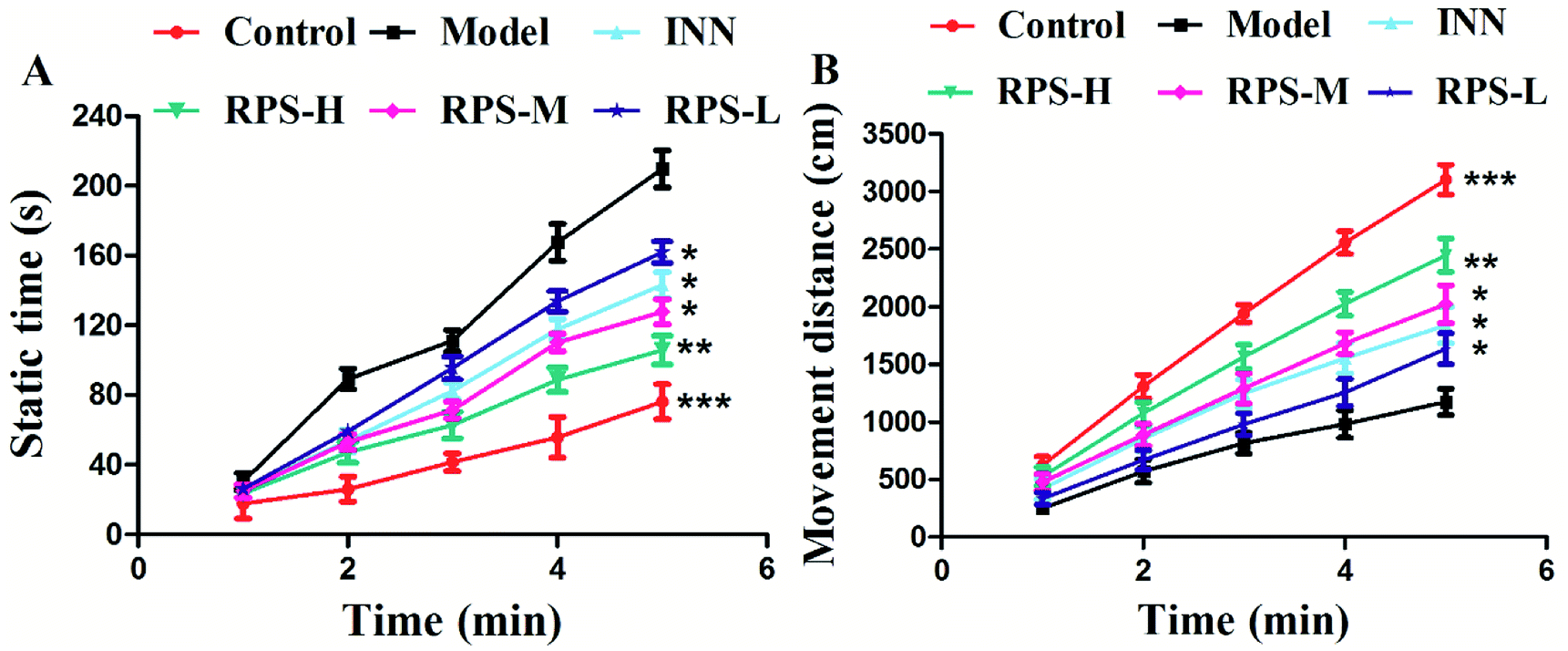
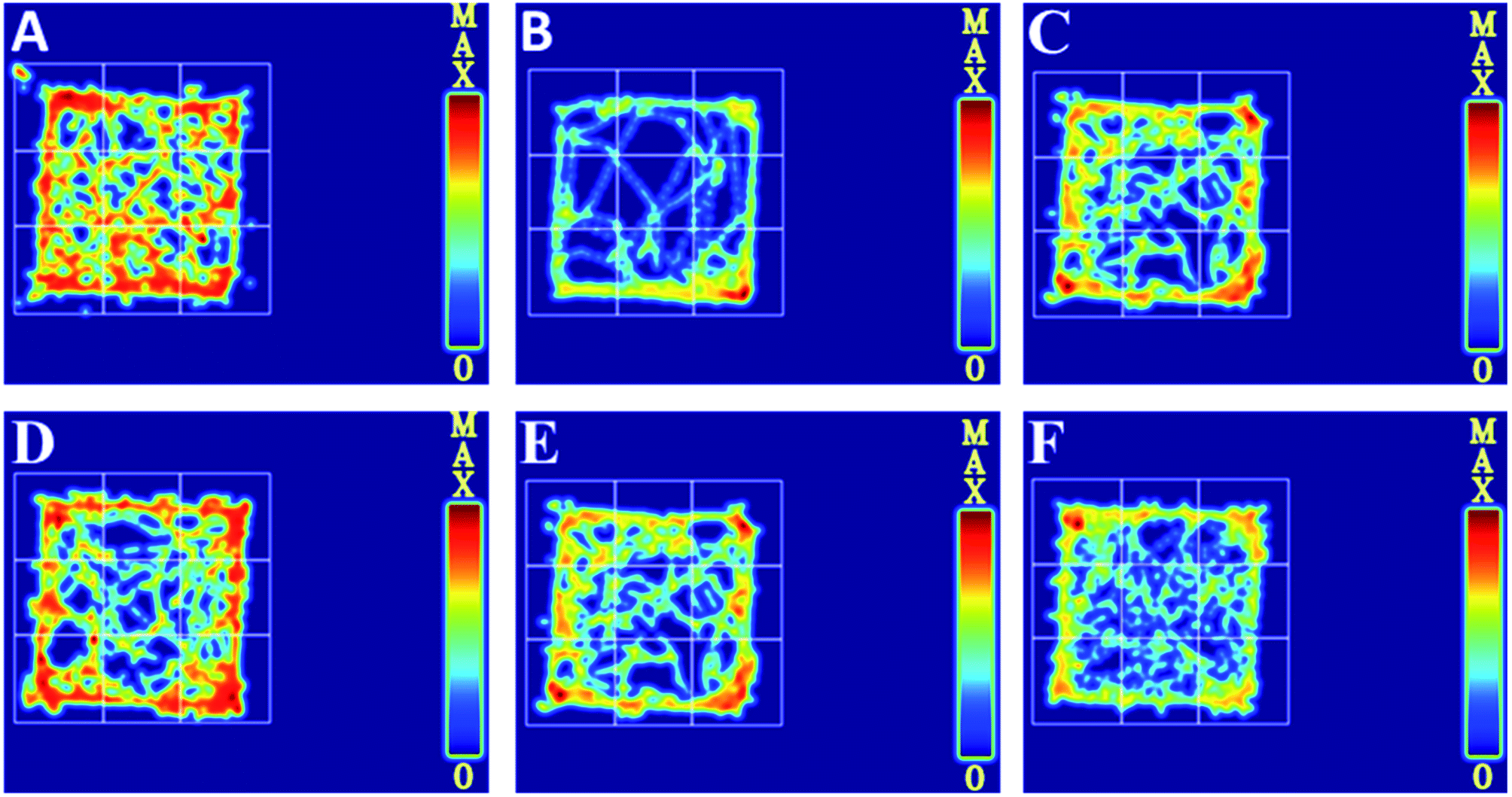
To further evaluate whether RPS was effective in attenuating cancer pain, an open field test related to pain-behaviors was performed. In the open field test, mice inoculated with H22 cancer cells in the hind paw exhibited significant changes in static times and movement distances when compared to the mice in the control groups (Fig. 4A and B). The static time of the model group increased and the movement distance reduced during the second to fifth minutes of observation relative to the control group. A reduction in static time was also observed when the animals were treated with three doses of RPS (50–200 mg kg−1, i.g.). With these doses, movement distances increased in the third to fifth minutes in comparison to the model group (Fig. 4B). Among rats treated with the higher dose of RPS, although the movement distances initially reduced, the values subsequently remained elevated. In addition, movement distances of the positive group also remained elevated in the third to fifth minutes relative to the model group (Fig. 4B), but the effect was weaker than that of the group treated with 200 mg kg−1 RPS. The movement track of mice is exhibited in Fig. 5. The animal model was well established for the evaluation of drug efficacy on cancer pain, as evidenced by static time and movement distance during the open field test. These results indicated that the administration of RPS increased the activity level of cancer pain mice, particularly in the group treated with a dose of 200 mg kg−1.
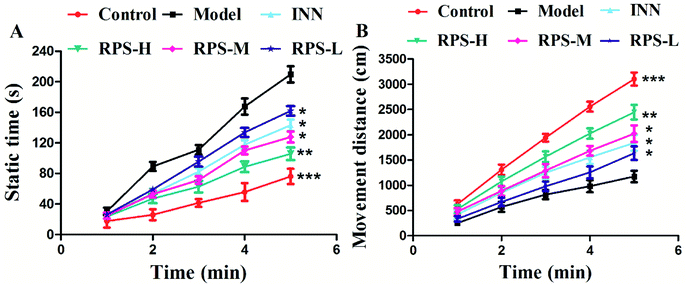 |
| | Fig. 4 Effect of RPS on the open field test of the H22 cell inoculated cancer pain model. (A) Static time of mice. (B) Movement distance of mice. Each value represents the mean ± SE. Compared with the model group using one-way ANOVA with Student's t-test, *P < 0.05, **P < 0.01, ***P < 0.01. | |
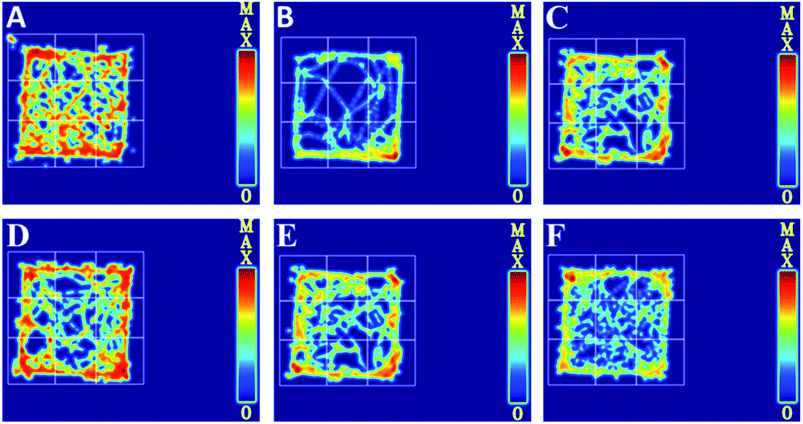 |
| | Fig. 5 Movement track in cancer pain model H22 hind paw of mice, as recorded in an open field test. (A) Control group, (B) model group, (C) INN group, (D) RPS-H group, (E) RPS-M group, (F) RPS-L group. | |
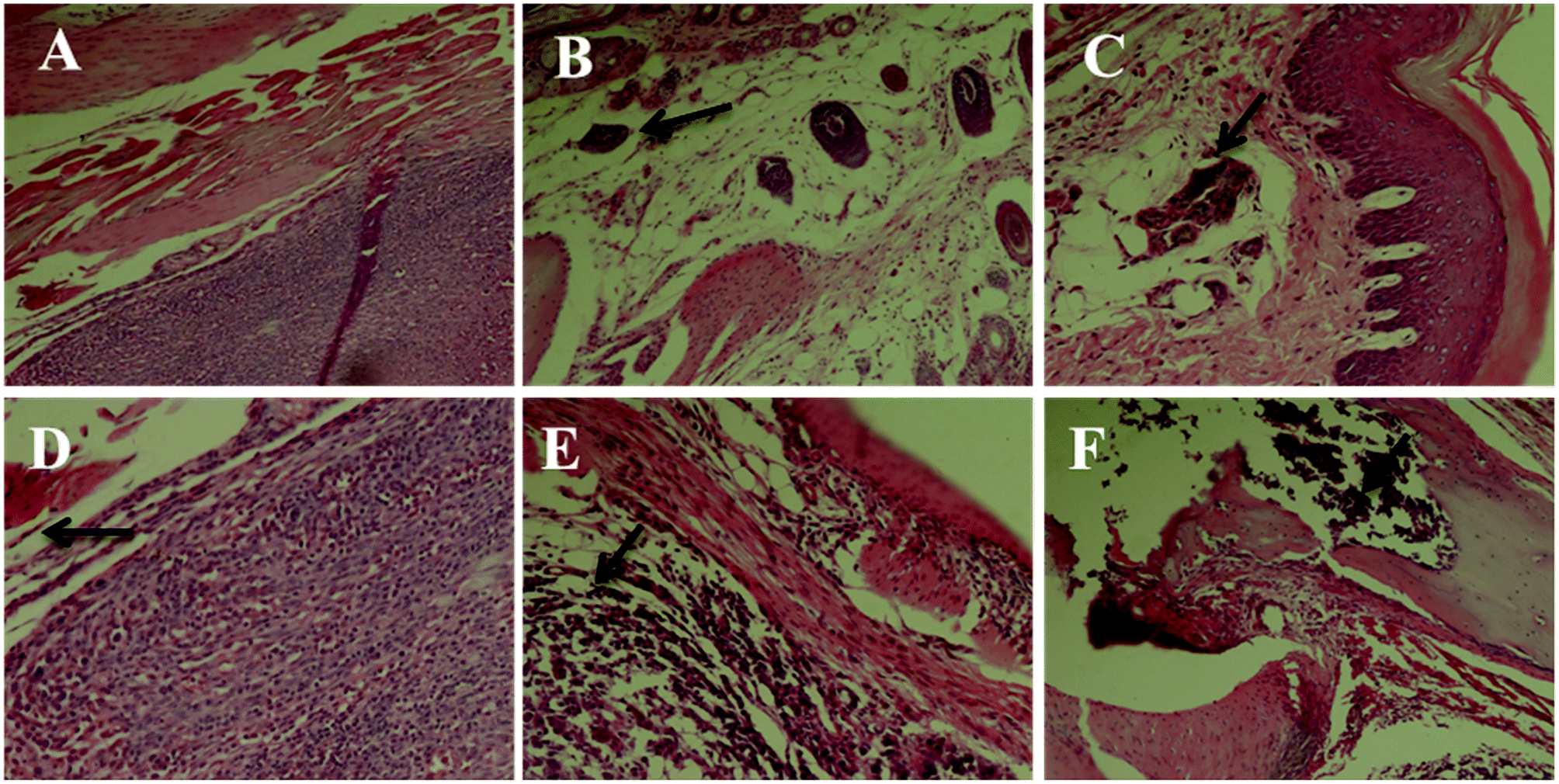
Paw histopathology
We first performed a morphological and histological study to confirm carcinoma formation. At 14 days after surgery, the transplanted carcinoma caused swelling and severe necrosis. Hematoxylin and eosin (HE) staining of the carcinoma-bearing paw showed the bone marrow cavity of the paw filled with tumor cells and a destroyed muscle matrix (Fig. 6).
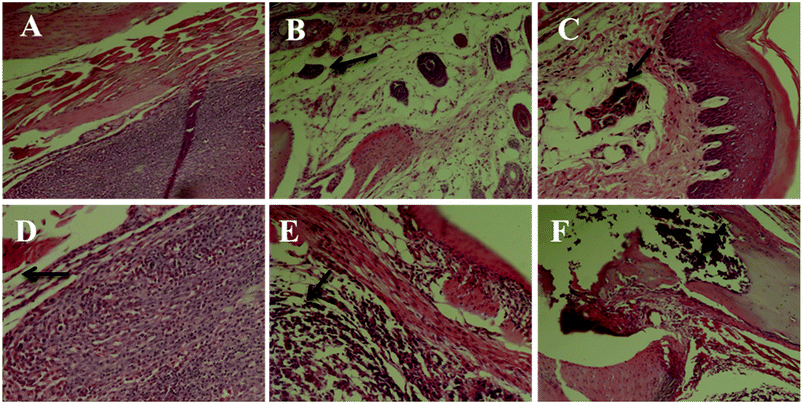 |
| | Fig. 6 Histomorphological micrograph of the hematoxylin and eosin (HE) stained paws of the control and carcinoma-bearing rats (scale bar = 10 μm). (A) Control group, (B) model group, (C) INN group, (D) RPS-H group, (E) RPS-M group, and (F) RPS-L group. | |
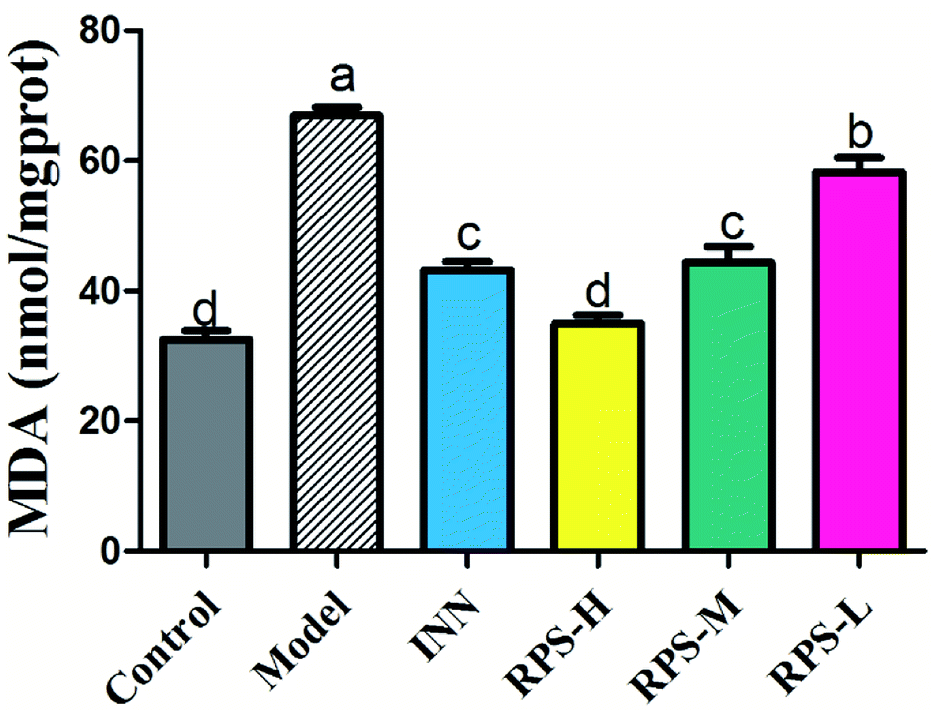
Effects on MDA of brain tissue
To explore whether RPS would affect endogenous MDA, we utilized an ELISA Kit to quantitatively analyze the level of MDA in brain tissue. The results showed that MDA expression was significantly increased in the carcinoma-bearing mice when compared to that in the control group (P < 0.01). As shown in Fig. 7, the levels of MDA were significantly reduced by the treatment with RPS-H, RPS-M and RPS-L when compared to the model group (P < 0.01), and exhibited a dose-dependent relationship. A similar pattern was also found in the INN group.
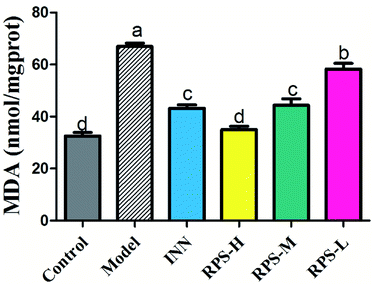 |
| | Fig. 7 Effects of three dosages of RPS and positive drug (INN) on the expression of MDA in brain tissues. Each value represents the mean ± SE from three independent experiments (n = 6 per group). Values within treatment groups having different letters (a–d) are significantly different and vice versa, as indicated by one-way ANOVA and Student's t-test (p < 0.05). | |
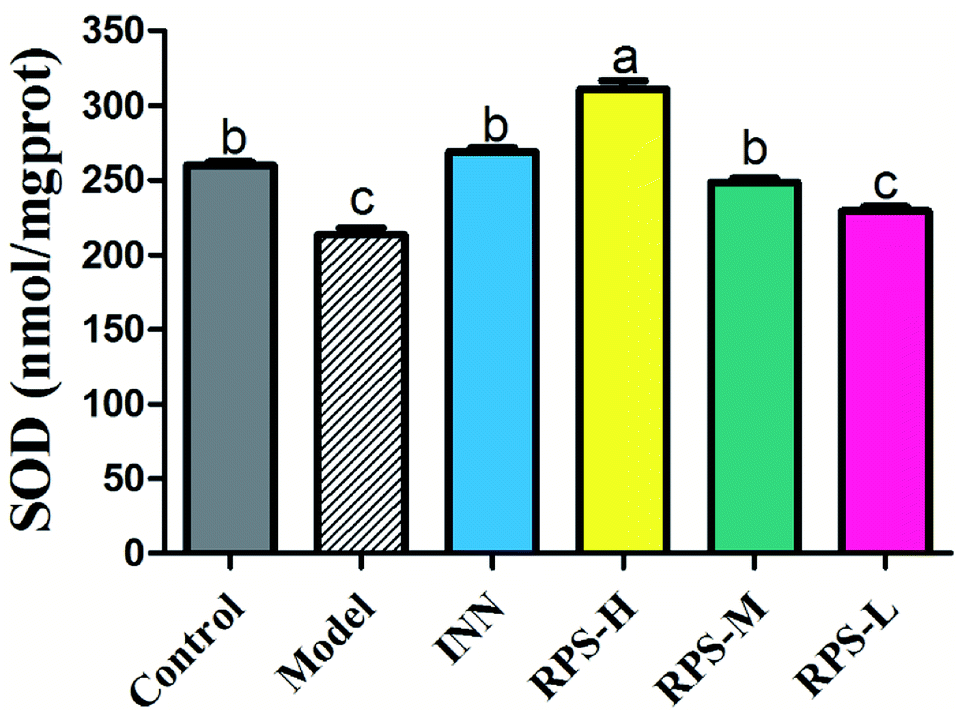
Effects on SOD of brain tissue
We next investigated whether RPS could regulate the activation of SOD. The present results indicated that the model group's SOD content was significantly reduced when compared to that of the control group (P < 0.01, Fig. 8), which indicated that the tumor may have induced the decrease in SOD content. After the administration of RPS-H and RPS-M, the activity of SOD was drastically up-regulated (P < 0.01).
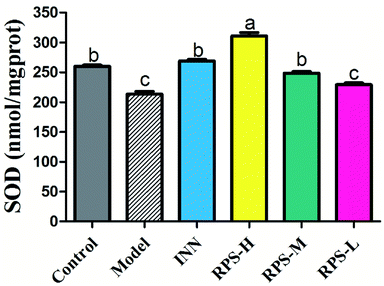 |
| | Fig. 8 Effects of three dosages of RPS and positive drug (INN) on the expression of SOD in brain tissues. Each value represents the mean ± SE from three independent experiments (n = 6 per group). Values within treatment groups having different letters (a–c) are significantly different and vice versa, as indicated by one-way ANOVA and Student's t-test (p < 0.05). | |
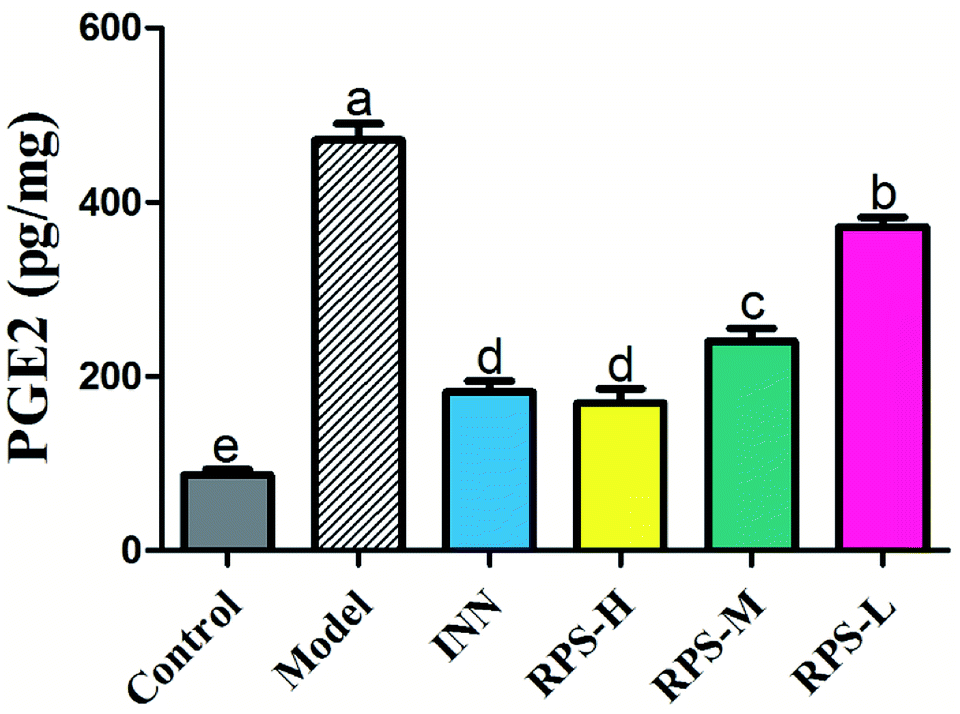
Effects on PGE2 of brain tissue
The results presented in Fig. 9 show the effect of RPS on the PGE2 level in cancer pain model. As shown in Fig. 9, the levels of PGE2 in the model group were significantly higher than that in the control group. However, the increase in PGE2 level in brain tissue was attenuated by the treatment of RPS (50–500 mg kg−1, i.g.). The positive control INN was also able to significantly reduce the concentration of PGE2 (P < 0.001). We also found that the tumor growth induced a distinct decrease in the PGE2 level, which was significantly reversed by RPS in a dose-dependent manner. Previous reports demonstrated that the treatment of esophageal cancer cells with RPS significantly down-regulated the expression of prostaglandin E2 in a dose-dependent manner, which is consistent with the result of the present research.28
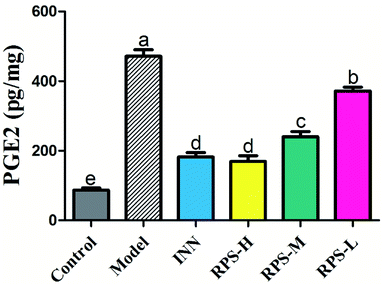 |
| | Fig. 9 Effects of three dosages of RPS and positive drug (INN) on the expression of PGE2 in brain tissues. Each value represents the mean ± SE from three independent experiments (n = 6 per group). Values within treatment groups having different letters (a–e) are significantly different and vice versa, as indicated by one-way ANOVA and Student's t-test (p < 0.05). | |
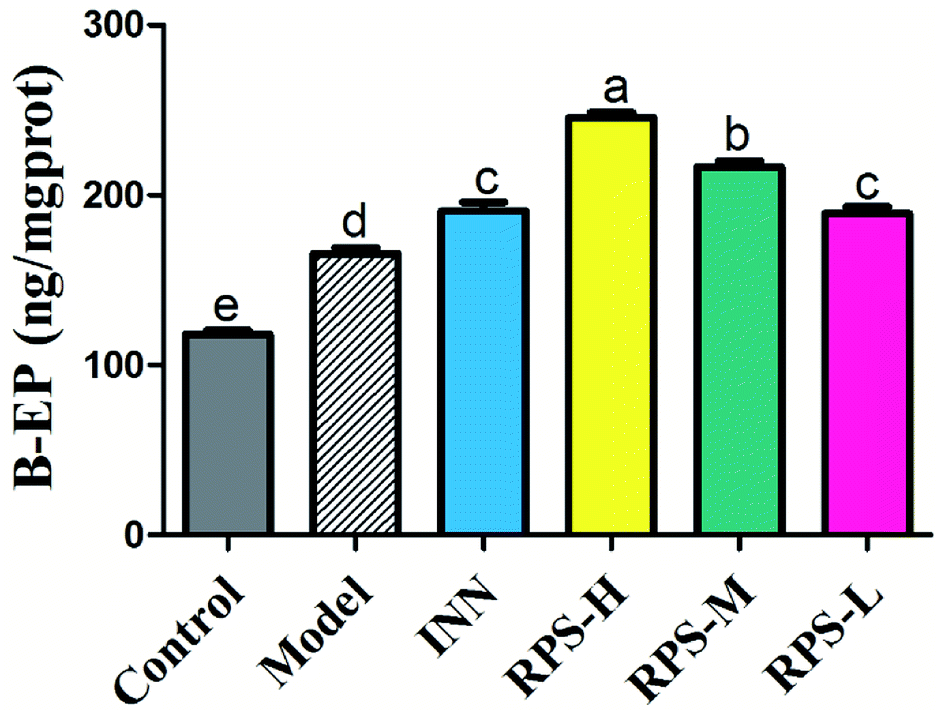
Effects on β-EP of brain tissue
β-EP is normally expressed in neurons, astrocytes and microglia. The stimulatory effect of tumor growth on β-endorphin expression was tested in the cancer pain model. Cerebral homogenates of control and model groups were obtained for β-EP measurements. Furthermore, the groups of cancer pain model mice received INN or RPS (50–200 mg kg−1) and 1 h later, their cerebral homogenates were obtained for β-EP measurement. As exhibited in Fig. 10, the levels of β-EP significantly increased in the model group compared to that in control mice. Based on previous data, we inferred that the significant increase in β-endorphin in the brain was due to the release of β-endorphin after stress and inflammation induced by cancer.29,30 Administration of RPS (50–200 mg kg−1) could significantly increase β-EP levels in brain tissue by 1.14, 1.31 and 1.48 times (P < 0.01, P < 0.001). In addition, treatment with INN in cancer pain mice also significantly altered the β-EP release (190.68 ± 13.60 vs. 118.13 ± 6.18, P < 0.01).
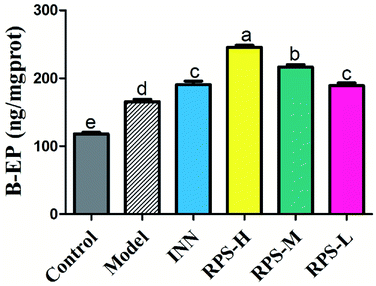 |
| | Fig. 10 Effects of three dosages of RPS and positive drug (INN) on the expression of β-EP in brain tissues. Each value represents the mean ± SE from three independent experiments (n = 6 per group). Values within treatment groups having different letters (a–e) are significantly different and vice versa, as indicated by one-way ANOVA and Student's t-test (p < 0.05). | |
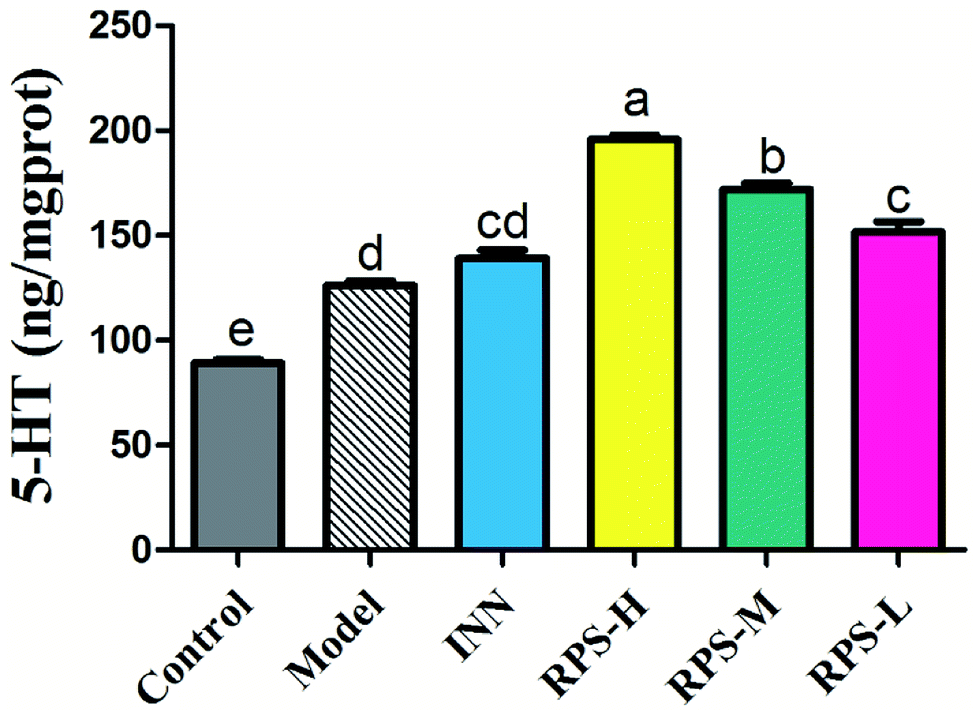
Effects on 5-HT of brain tissue
Serotonin (5-HT) in the brain and spinal cord plays a critical role in behavioral responses to environmental stressors and nociceptive mechanisms. To investigate the effect of RPS on the central nervous system in cancer-induced pain model, the concentration of 5-HT was measured by ELISA kits. Compared to the control group, the concentration of 5-HT in the model group significantly increased (P < 0.01). Moreover, the oral administration of RPS (50–200 mg kg−1) dose-dependently increased the level of 5-HT in brain tissue (P < 0.01 and P < 0.05, Fig. 11). However, the intraperitoneal injection of INN did not significantly alter the expression of 5-HT (the relative 5-HT expression value of 138.99 ± 10.81 vs. 126.22 ± 5.38).
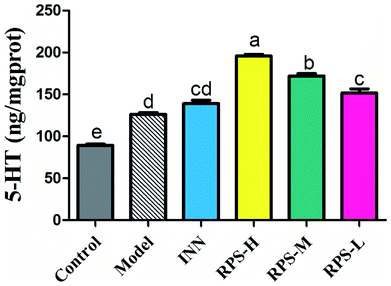 |
| | Fig. 11 Effects of three dosages of RPS and positive drug (INN) on the expression of 5-HT in brain tissues. Each value represents the mean ± SE from three independent experiments (n = 6 per group). Values within treatment groups having different letters (a–e) are significantly different and vice versa, as indicated by one-way ANOVA and Student's t-test (p < 0.05). | |
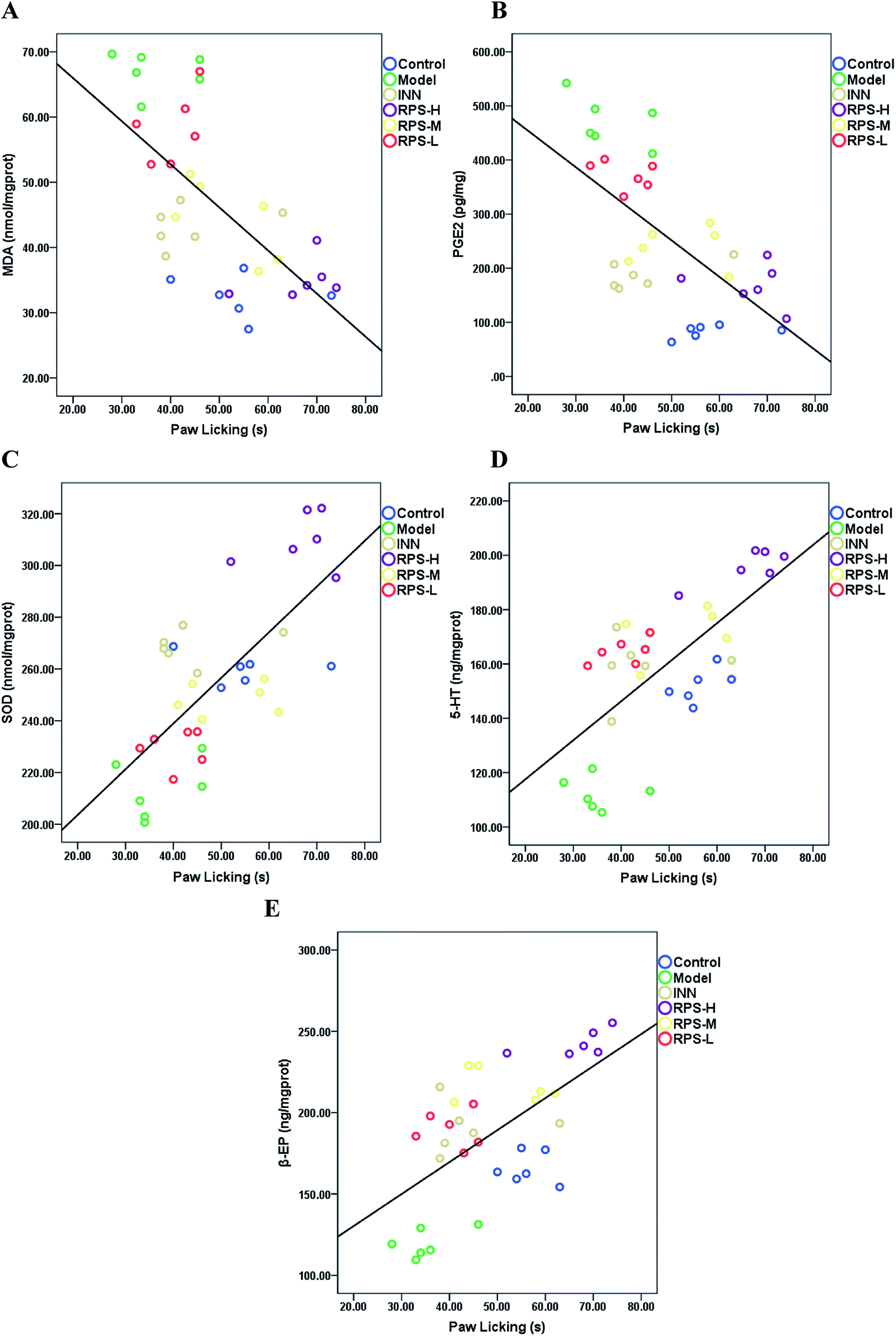
Correlation
Pearson correlation coefficients for the relationship between paw licking latency of the hot-plate test and biochemical parameters (SOD activities, MDA, PGE2, β-EP and 5-HT level) in brain tissue are presented in Fig. 12. Significant negative correlations were identified between paw licking latency and MDA level (r = −0.605, P < 0.001, Fig. 12A) and PGE2 level (r = −0.637, P < 0.001, Fig. 12B), while significant positive correlations were observed between paw licking latency and SOD activity (r = 0.706, P < 0.001, Fig. 12C), 5-HT level (r = 0.685, P < 0.001, Fig. 12D), and β-EP level (r = 0.565, P < 0.01, Fig. 12E).
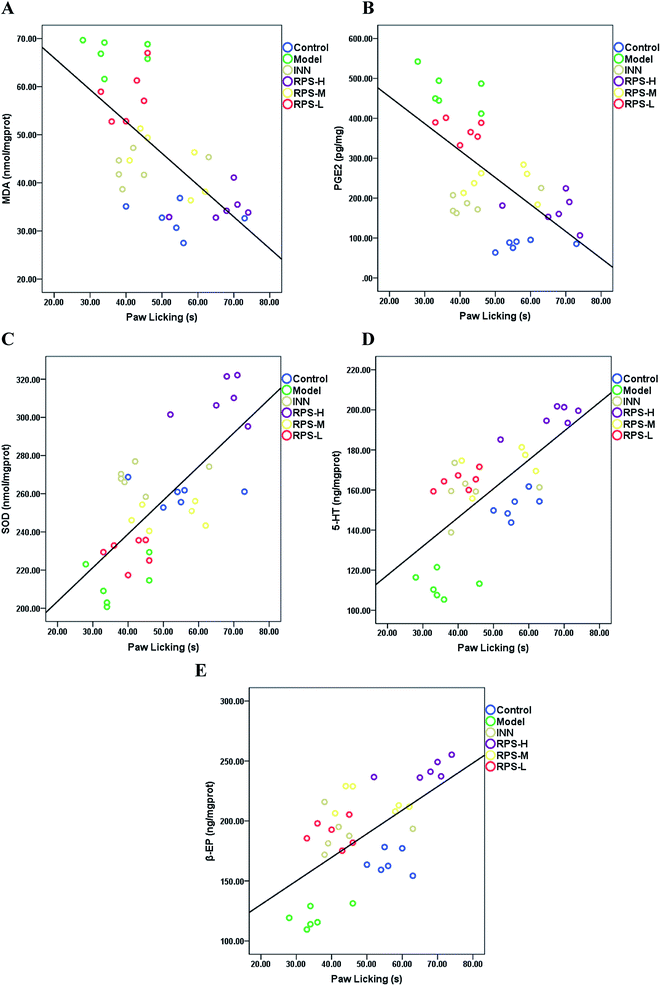 |
| | Fig. 12 The relationship between the paw licking latency of the hot-plate test and (A) MDA level, (B) PGE2 level, (C) SOD activity, (D) 5-HT level and (E) β-EP level are shown. The values of r and p (Pearson's test), and linear fit (black line) were also obtained. | |
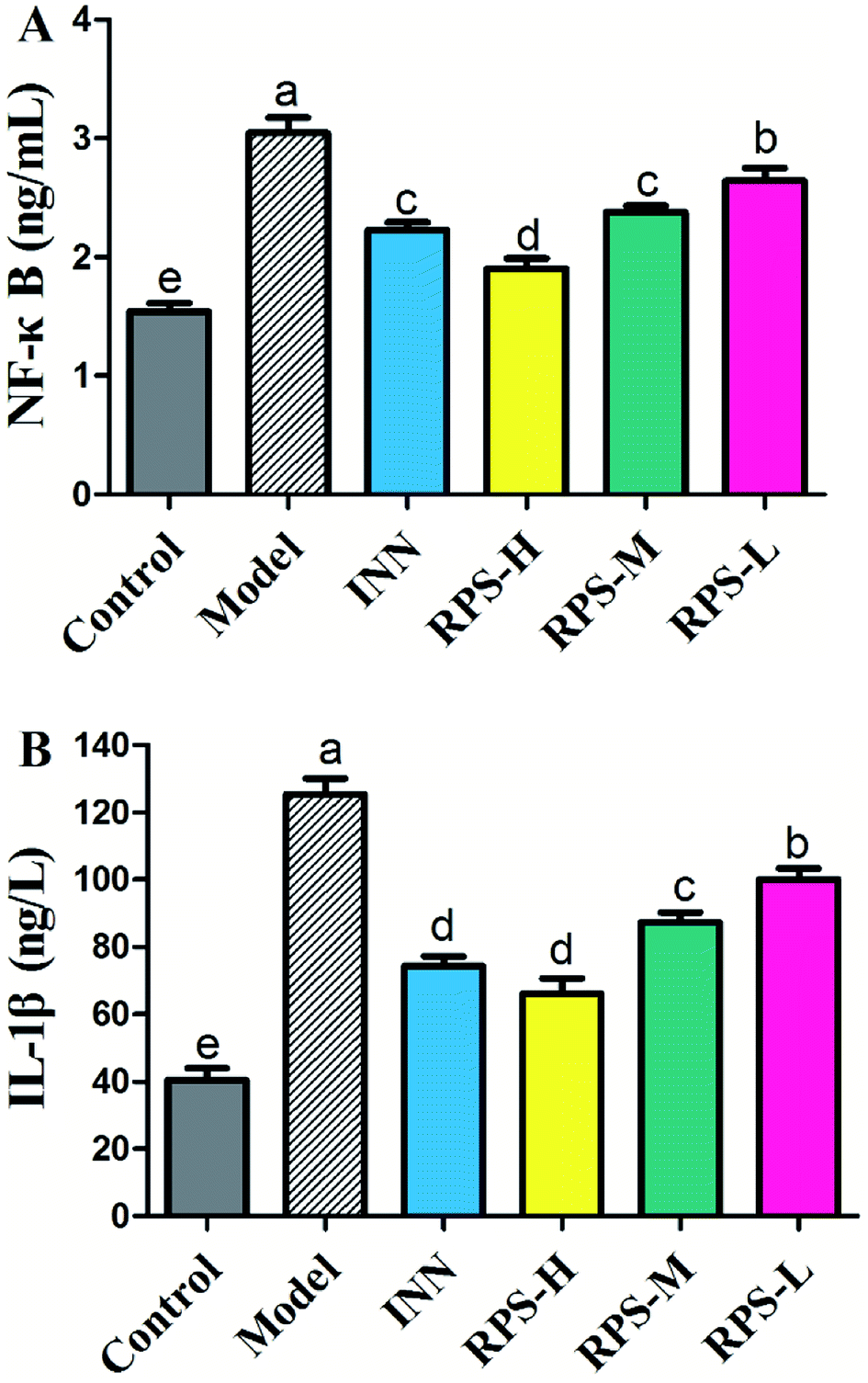
Effects on serum NF-κB and IL-1β
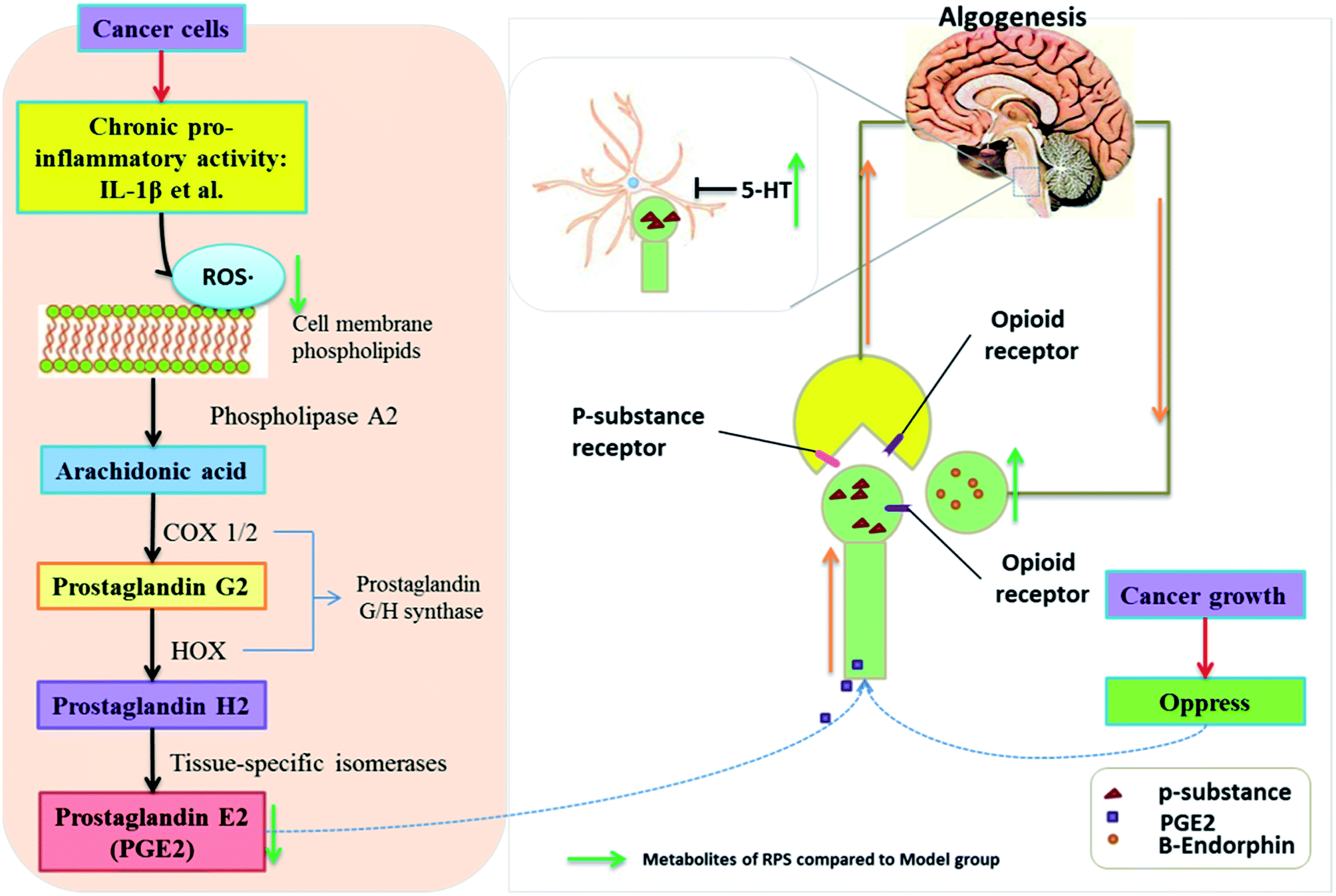
To confirm the influence of inflammation and oxidative stress on the analgesic effect of RPS in cancer pain model, blood NF-κB and IL-1β levels were measured by ELISA assay (Fig. 14). The results demonstrated that compared to control mice, the model group expressed markedly increased levels of NF-κB and IL-1β (P < 0.001), which was consistent with the previous study,31 which provided a reference method and stimulated the idea of pain research. Furthermore, treatment with 50–200 mg kg−1 RPS on established tumor-bearing paw model almost completely suppressed the expression of NF-κB and IL-1β (P < 0.01 and P < 0.001) in a concentration-dependent manner. In addition, treatment with tramadol HCl (INN, 10 mg kg−1, i.p.) in cancer pain mice also significantly down-regulated the release of NF-κB and IL-1β (P < 0.01). Using the established model of cancer pain in the hind paw, we found that RPS (50–200 mg kg−1) could down-regulate the expression of NF-κB and IL-1β in a concentration-dependent manner. NF-κB and IL-1β released from activated microglia and consequently sensitized the neurons in the spinal dorsal horn by altering the excitatory or inhibitory synaptic transmission, contributing to pain facilitation.32 It is thus possible that RPS produced antinociception by the blockade of proinflammatory cytokine expression via the inhibition of NF-κB and IL-1β activation.
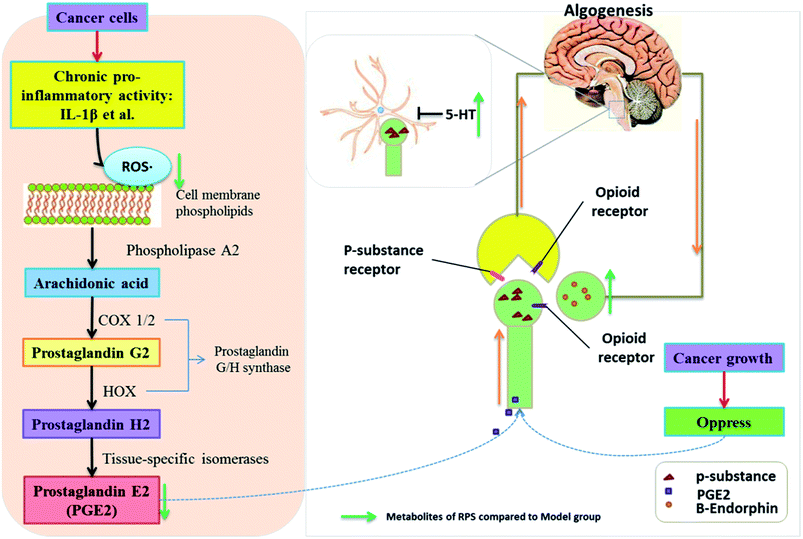 |
| | Fig. 13 The mechanism of analgesic effect of RPS on cancer-induced pain model mice. | |
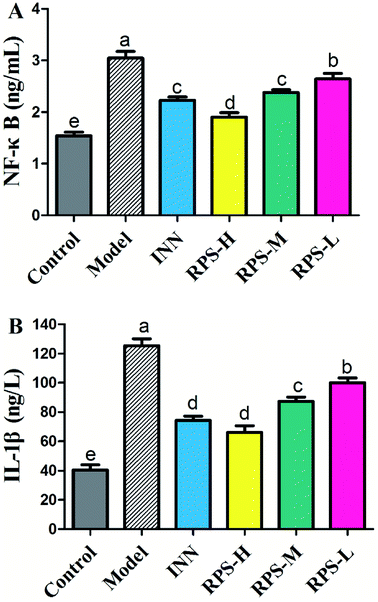 |
| | Fig. 14 Effects of three dosages of RPS and positive drug (INN) on the serum levels of NF-κB and IL-1β in established tumor-bearing paw model. (A) ELISA-based assay with plate-adhered oligonucleotides containing an NF-kB consensus binding sequence. (B) ELISA-based assay with plate-adhered oligonucleotides containing an IL-1β consensus binding sequence. Data represent mean ± SE from three independent experiments (n = 6 per group). Values within treatment groups having different letters (a–e) are significantly different and vice versa, as indicated by one-way ANOVA and Student's t-test (p < 0.05). | |
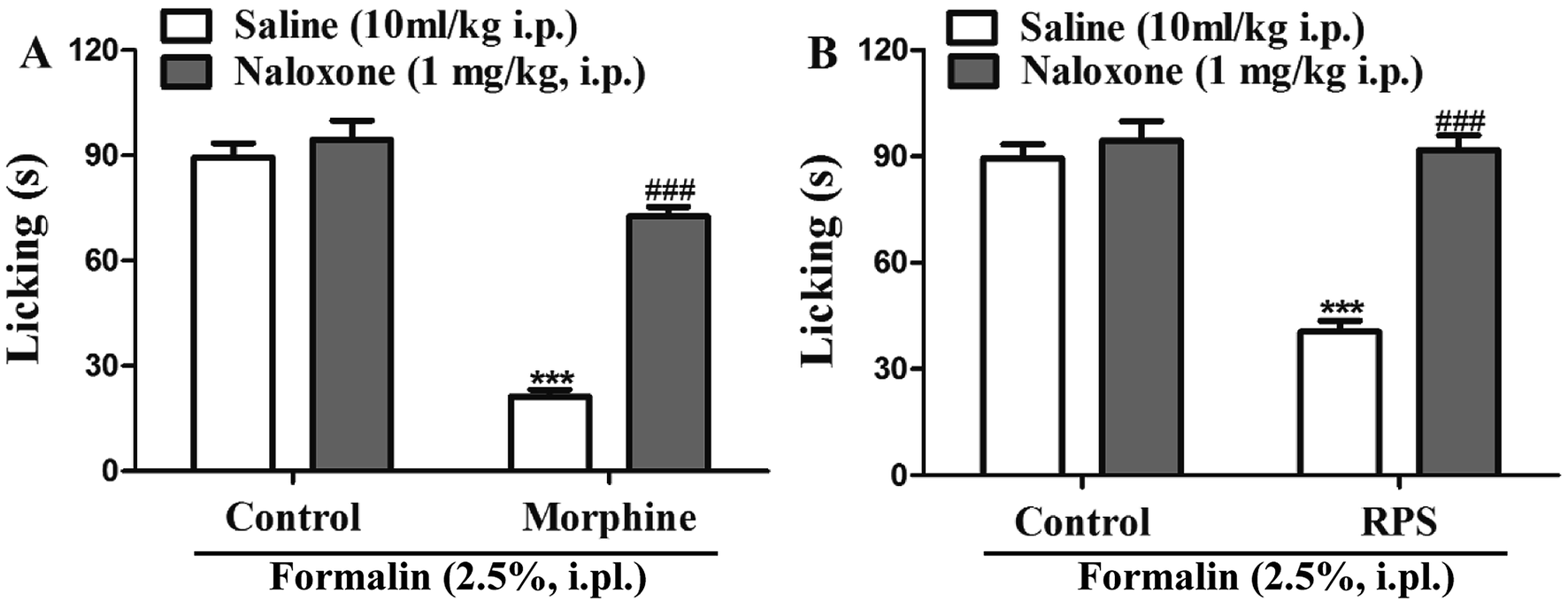
Involvement of opioid receptors in the systemic antinociceptive action of RPS
In order to identify the opioid receptors involved in RPS's action, naloxone (a non-selective antagonist of opioid receptors) was used. The antinociception induced by morphine (nonselective opioid receptor agonist, used as the positive control) was reversed by the pre-treatment with naloxone (Fig. 15A). Similarly, as shown in Fig. 15B, administration of naloxone (1 mg kg−1, i.p.) significantly antagonized the antinociceptive effects of RPS in comparison to the control group, after the intraplantar injection of formalin. The opioid hypothesis was preliminarily investigated by attempting to reverse analgesia after the administration of naloxone (opioid antagonist).33 The results showed that the pre-administered naloxone was able to counteract the antinociception of RPS, clearly demonstrating an important involvement of RPS in the endogenous opioid receptors' activation to produce analgesia.
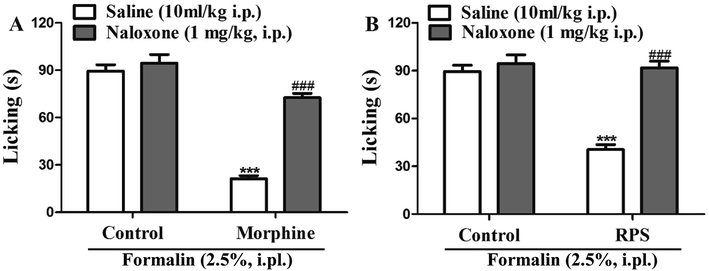 |
| | Fig. 15 Opioid receptors mediate RPS-induced antinociception. The systemic preadministration of naloxone (1 mg kg−1, i.p.), an opioid receptor antagonist, 30 min before pretreatment with morphine ((A) 2.5 mg kg−1, s.c.) and RPS ((B) 200 mg kg−1, i.g.) significantly reverses the antinociceptive effects of these treatments, when compared to the control group, after intraplantar injection of formalin (2.5%, i.pl.) in mice. Each column represents the mean of 6 mice, and the vertical lines indicate the SE. ***P < 0.001 as compared with control group or ###P < 0.001 when comparing treatment with naloxone + treatment (one-way ANOVA followed by and Student's t-test). | |
Discussion
Numerous studies have found that long-lasting stress and inflammation increases an individual's pain during cancer, which is related to the central nervous system and the peripheral nervous system.34 Because of its multifaceted character, cancer-induced pain remains inadequately controlled, thus posing a challenge for modern medicine. Among traditional systems of medicine, traditional Chinese medicine (TCM) is the most extraordinary system with unique theories and practice in promoting people's health and alleviating diseases for thousands of years. Our results demonstrated that the administration of Rhizoma Paridis saponins (RPS), the active parts of P. polyphylla Smith var. yunnanensis, exhibited a dose-dependent analgesic effect in cancer pain, as evidenced by the hot-plate test, and prevented the development of tumor in size and in histopathology. In addition, the results of biochemical factors suggested the possible involvement of the peripheral nervous system and central nervous system in the observed analgesic effect.35,36 The mechanisms and pathways responsible for the analgesic effect of RPS are depicted in Fig. 13.
Rhizoma Paridis saponins (RPS), as active parts of P. polyphylla Smith var. yunnanensis, has been used as an anti-cancer drug in traditional Chinese medicine for a long time.17–19 In the animal model of skin cancer pain presented in this study, mice developed robust thermal hypersensitivity restricted to the tumor-bearing paw that was correlated with the tumor growth (Fig. 1 and 3 and Table 1). In our research, oral administration of RPS (50–200 mg kg−1) could delay the growth of tumor, which reduced the stress induced by the tumor. This observation was consistent with those reported in previous studies where RPS was identified as the main effective component of Rhizoma Paridis with antitumor properties.37,38 To evaluate the analgesic effect of RPS, the hot-plate test and the open field test were performed in the present research. The results indicated that RPS exhibited a dose-dependent analgesic effect after oral administration, which was had a significant positive correlation with the tumor inhibition rate of RPS. Furthermore, the activity level of cancer pain mice significantly increased after treating with RPS, which was also positively correlated with the tumor inhibition rate. Thus, based on present results, it may be hypothesized that the anti-tumor effect of RPS, which is related to stress, may account for the analgesic effect induced by cancer.
Several hypotheses can be formed to explain RPS's analgesic effect demonstrated in the hot-plate test, which was related to the possible involvement of peripheral nervous system and central nervous system. Inflammation and neuropathic pain represent major pain categories and involve increased afferent signaling (peripheral sensitization), increased pain transmission (spinal sensitization), and altered central pain connectivity (integration and modulation).39 Pharmacological studies have demonstrated that IL-1β induces a dose-related increase in PGE2 concentration, which is mediated by COX-2 enzyme in the peripheral inflammation system.40 Related biological factors in brain tissue that contribute to the pain development during cancer, such as MDA, SOD, PGE2, NF-κB, IL-1β, β-EP and 5-HT, were measured in this research. These biological factors are involved with peripheral nervous system and central nervous system.6,40,41
Superoxide dismutase (SOD), the most studied antioxidant enzyme, is ubiquitous in all organisms. Recently, SOD was shown to be involved in cancer pain states. An acute inflammatory process consists of inflammation mediators, including neutrophil-derived reactive oxygen species (ROS), which play an important role in pathogenesis of many diseases, such as local or systemic inflammatory disorders, diabetes, atherosclerosis, cancer, and neurodegenerative diseases.42,43 ROS generation is induced by growth factors, cytokines, and tumor promoters.44 MDA can injure the quadriceps femoris cells, induce lipid peroxidation, attack nucleic acids and proteins, and finally disrupt the biological functions of the cell.45 The natural cellular antioxidant enzyme SOD is accepted as one of the most important physiological antioxidants that mediates free radicals and prevents subsequent lipid peroxidation by increasing the rate at which they are removed.46 SOD mimetic compounds and antioxidants have exhibited potent analgesic effects in rodent pain models.6 Furthermore, extensive research has shown that the ROS-dependent COX pathway may contribute to the production of PGE2.47 However, among inflammatory factors, PGE2 is known to play the most important role in pain related to the peripheral nervous system.48 PGE2 can reduce the neuronal threshold, speed the firing and depolarization and consequently increase the number of action potentials per stimulus and reproduce the pain.49 In our study, data obtained from ELISA showed that SOD expression decreased, while MDA and PGE2 expression increased in mouse model of cancer pain. Moreover, the present results indicated that RPS treatment reduced tumor-induced oxidative and inflammation damage via the increase in SOD and the reduction in MDA and PGE2 levels. Based on existing evidence, we propose that RPS would have potential analgesic effect on the peripheral nervous system through regulated tumor-induced oxidative and inflammation damage.
β-Endorphin (β-EP) is an endogenous opioid peptide neurotransmitter, which is produced from neurons in the pituitary gland and the hypothalamus; it specifically activates post-synaptic μ-opioid receptors to modulate pain transmission and transduction50,51 and induces analgesic tolerance.52,53 In this study, compared to the model group, the expression of β-EP significantly increased in brain tissue after treatment with RPS (50–200 mg kg−1). Therefore, we speculated that β-endorphin expression in brain tissue and subsequent activation of neuronal μ-opioid receptors are the mechanisms responsible for RPS's analgesic effect. Our current data provided additional evidence that Rhizoma Paridis saponins might stimulate β-endorphin expression, which is one of the transmitters for microglia to produce analgesia. Serotonin (5-HT) is involved in some central nervous system diseases, such as stress/anxiety and depression; these pathological conditions are related to changes in serotonergic activity.54 In particular, increased serotonin release was induced by the acute administration of physiological stressors in some brain areas of mice. A previous study showed that 5-HT was involved in analgesia in a neuropathic pain model.55 Our data further demonstrated that RPS may increase the spinal release of 5-HT that acts on 5-HT receptor to alleviate hyperalgesia. Opioid and serotonin pathways clearly play a key role in the anesis of pain perception through the release of endogenous opioids and 5-HT that act on specific receptors after the oral administration of RPS.
Paris polyphylla is the dried rhizome of P. polyphylla Smith var. yunnanensis, which has been widely used for the treatment of hemostasis, abscess, snake bites, abnormal uterine bleeding, tumors, analgesia and fractures in clinical practice for thousands of years in China.15,16 Surprisingly, in this study, the results indicated that the oral administration of RPS has markedly reduced the pain induced by cancer inoculated in the hind right paw of mice. Interestingly, in addition to antinociceptive mechanisms, RPS appears to modulate the activation of biochemical parameters, such as SOD, MDA, PGE2, β-EP and 5-HT, which are related to the possible involvement of the peripheral nervous system and the central nervous system in the observed analgesic effect.
Conclusion
In summary, our study provides new evidence that in a mouse model of chronic cancer pain, RPS alleviates the symptoms of cancer pain in mice, increases the activity level and raises the pain threshold according to the hot-plate test and open field test. The mechanism may involve the down-regulation of MDA, PGE2, NF-κB and IL-1β levels and the up-regulation of the activity of SOD and the expression of 5-HT and β-EP in brain tissue in the hind right paw tumor model. In addition, this peripheral and central analgesic pathway might have widespread potential for clinical use.
Conflicts of interest
We have no conflict of interest in this research.
Acknowledgements
The authors acknowledge the “Drug Creation Project 2014ZX09301307-018 from Science and Technology in China”.
References
- P. J. Christo and D. Mazloomdoost, Ann. N. Y. Acad. Sci., 2008, 1138, 278–298 CrossRef CAS PubMed.
- P. W. Mantyh, Nat. Rev. Neurosci., 2006, 7, 797–809 CrossRef CAS PubMed.
- E. Mcnicol, N. Horowicz-Mehler, R. A. Fisk, K. Bennett, M. Gialeli-Goudas, P. W. Chew, J. Lau and D. Carr, J. Pain, 2003, 4, 231 CrossRef PubMed.
- M. Valko, D. Leibfritz, J. Moncol, M. T. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS PubMed.
- P. G. Winyard, C. J. Moody and C. Jacob, Trends Biochem. Sci., 2005, 30, 453 CrossRef CAS PubMed.
- Y. Q. Zhou, D. Q. Liu, S. P. Chen, J. Sun, X. R. Zhou, H. Rittner, W. Mei, Y. K. Tian, H. X. Zhang and F. Chen, Redox Biol., 2017, 14, 391 CrossRef PubMed.
- H. Fan, T. F. Li, N. Gong and Y. X. Wang, Neuropharmacology, 2015, 101, 98 CrossRef PubMed.
- G. Vitale, R. Arletti and M. Sandrini, Life Sci., 2005, 77, 2500–2513 CrossRef CAS PubMed.
- N. Malek, M. Kostrzewa, W. Makuch, A. Pajak, M. Kucharczyk, F. Piscitelli, B. Przewlocka, V. D. Marzo and K. Starowicz, Pharmacol. Res., 2016, 111, 251–263 CrossRef CAS PubMed.
- Y. Zhang, R. X. Zhang, M. Zhang, X. Y. Shen, A. Li, J. Xin, K. Ren, B. M. Berman, M. Tan and L. Lao, Br. J. Anaesth., 2012, 109, 245–252 CrossRef CAS PubMed.
- H. Qian, X. F. Mao, H. Y. Wu, L. Hao, M. L. Sun, W. Xiao and Y. X. Wang, Brain, Behav., Immun., 2017, 62, 64 CrossRef PubMed.
- R. Benyamin, A. M. Trescot, S. Datta, R. Buenaventura, R. Adlaka, N. Sehgal, S. E. Glaser and R. Vallejo, Pain Physician, 2008, 11, 105–120 Search PubMed.
- R. Ghosh, A. Alajbegovic and A. V. Gomes, Oxid. Med. Cell. Longevity, 2015, 2015, 536962 Search PubMed.
- P. I. Hsu and T. J. Tsai, Curr. Pharm. Des., 2015, 21, 5049–5055 CrossRef CAS PubMed.
- B. Wang, S. Ji, Z. Hai, Z. Liang, L. Lei, Y. Li, G. Zhou and G. Zhang, Steroids, 2013, 78, 1164–1170 CrossRef CAS PubMed.
- G. Yang, L. Wei, P. Meng, C. Zhang, Z. Yuan, H. Pei, H. Ming and S. Gao, J. Pharm. Biomed. Anal., 2017, 145, 675–681 CrossRef CAS PubMed.
- S. Man, W. Fan, Z. Liu, W. Gao, Y. Li, L. Zhang and C. Liu, Steroids, 2014, 84, 17–21 CrossRef CAS PubMed.
- S. Man, Y. Li, W. Fan, W. Gao, Z. Liu, Y. Zhang and C. X. Liu, Steroids, 2014, 80, 1–6 CrossRef CAS PubMed.
- S. Man, Y. Li, W. Fan, W. Gao, Z. Liu, N. Li, Y. Zhang and C. Liu, Int. J. Pharm., 2013, 454, 296–301 CrossRef CAS PubMed.
- S. Man, W. Fan, W. Gao, Y. Li, Y. Wang, Z. Liu and H. Li, J. Ethnopharmacol., 2014, 151, 407–412 CrossRef CAS PubMed.
- S. Man, J. Li, W. Fan, H. Chai, Z. Liu and W. Gao, J. Steroid Biochem. Mol. Biol., 2015, 154, 62–67 CrossRef CAS PubMed.
- Q. L. Mao-Ying, K. M. Cui, Q. Liu, Z. Q. Dong, W. Wang, J. Wang, H. Sha, G. C. Wu and Y. Q. Wang, Eur. J. Pain, 2006, 10, 689 CrossRef PubMed.
- T. Andoh, K. Sugiyama, M. Fujita, Y. Iida, H. Nojima, I. Saiki and Y. Kuraishi, Biol. Pharm. Bull., 2008, 31, 520 CAS.
- S. Man, W. Gao, Y. Zhang, L. Yan, C. Ma, C. Liu and L. Huang, Steroids, 2009, 74, 1051–1056 CrossRef CAS PubMed.
- T. Chen, S. J. Yuan, X. Q. Yu, L. B. Jiao, W. Hu, W. L. Chen and B. Xie, Int. Immunopharmacol., 2016, 42, 90 CrossRef PubMed.
- X. J. Ji, S. H. Chen, L. Zhu, H. Pan, Y. Zhou, W. Li, W. C. You, C. C. Gao, J. H. Zhu and K. Jiang, Oncol. Rep., 2013, 30, 157–164 CrossRef CAS PubMed.
- F. R. Lapa, V. Gadotti, F. Missau, M. Pizzolatti, M. Marques, A. Dafré, M. Farina, A. Rodrigues and A. Santos, Basic Clin. Pharmacol. Toxicol., 2009, 104, 306–315 CrossRef CAS PubMed.
- S. Yan, S. Tian, Q. Kang, Y. Xia, C. Li, Q. Chen, S. Zhang and Z. Li, PLoS One, 2015, 10, e0131560 Search PubMed.
- L. Yh, D. Wang, Q. Yu and C. Xq, J. Clin. Anesth., 2017, 37, 123 CrossRef PubMed.
- C. G. Parsons, A. Członkowski, C. Stein and A. Herz, Pain, 1990, 41, 81 CrossRef CAS PubMed.
- A. W. Lund, T. R. Medler, S. A. Leachman and L. M. Coussens, Cancer Discovery, 2016, 6, 22 CrossRef CAS PubMed.
- Y. Kawasaki, L. Zhang, J. K. Cheng and R. Ji, J. Neurosci., 2008, 28, 5189–5194 CrossRef CAS PubMed.
- R. G. Hill, Neurosci. Lett., 1981, 21, 217–222 CrossRef CAS PubMed.
- F. S. Dhabhar, A. N. Saul, T. H. Holmes, C. Daugherty, E. Neri, J. M. Tillie, D. Kusewitt and T. M. Oberyszyn, PLoS One, 1932, 7, e33069 Search PubMed.
- A. Kumar, H. Kaur and A. Singh, Pharmacol. Rep., 2018, 70, 206–216 CrossRef PubMed.
- L. Coxon, A. W. Horne and K. Vincent, Best Pract Res Clin Obstet Gynaecol, 2018 DOI:10.1016/j.bpobgyn.2018.01.014.
- S. Man, H. Chai, P. Qiu, Z. Liu, W. Fan, J. Wang and W. Gao, Pathol., Res. Pract., 2015, 211, 948–954 CrossRef CAS PubMed.
- Z. Liu, W. Gao, S. Man, Y. Zhang, H. Li, S. Wu, J. Zhang and C. Liu, Environ. Toxicol. Pharmacol., 2014, 38, 31–40 CrossRef CAS PubMed.
- J. Sawynok and J. Liu, Eur. J. Pharmacol., 2014, 734, 114–121 CrossRef CAS PubMed.
- L. Shi, I. Smolders, V. Umbrain, M. H. Lauwers, S. Sarre, Y. Michotte, M. Zizi and F. Camu, Pain, 2006, 120, 307–314 CrossRef CAS PubMed.
- R. Al-Hasani and M. Bruchas, Anesthesiology, 2011, 115, 1363–1381 CAS.
- J. Liao, J. Deng, C. Chiu, W. Hou, S. Huang, P. Shie and G. Huang, J. Evidence-Based Complementary Altern. Med., 2012, 2012, 429320 Search PubMed.
- G. J. Huang, S. S. Huang and J. S. Deng, PLoS One, 2012, 7, e35922 CAS.
- C. H. Woo, J. H. Lim and J. H. Kim, J. Immunol., 2004, 173, 6973–6980 CrossRef CAS.
- C. Hou, J. G. Long and J. K. Liu, Med. Recapitulate, 2013, 2889–2891 CAS.
- O. M. Ighodaro and O. A. Akinloye, Alexandria Journal of Medicine, 2017 DOI:10.1016/j.ajme.2017.09.001.
- W. T. Chiu, S. C. Shen, J. M. Chow, C. W. Lin, L. T. Shia and Y. C. Chen, Neurobiol. Dis., 2010, 37, 118–129 CrossRef CAS PubMed.
- K. Ma, Q. Zhou, J. Chen, D. Du, Y. Ji and W. Jiang, Brain Res., 2008, 1209, 57 CrossRef CAS PubMed.
- H. Vanegas and H. G. Schaible, Prog. Neurobiol., 2001, 64, 327–363 CrossRef CAS PubMed.
- F. W. Bach, Dan. Med. Bull., 1997, 44, 274–286 CAS.
- M. Petraschka, S. Li, T. L. Gilbert, R. E. Westenbroek, M. R. Bruchas, S. Schreiber, J. Lowe, M. J. Low, J. E. Pintar and C. Chavkin, Neuroscience, 2007, 146, 1795–1807 CrossRef CAS PubMed.
- H. N. Bhargava, Life Sci., 1981, 29, 1945–1949 CrossRef CAS PubMed.
- J. Fichna, A. Janecka, J. Costentin and J. C. Do Rego, Pharmacol. Rev., 2007, 59, 88 CrossRef CAS PubMed.
- F. G. Graeff, F. S. Guimarães, T. G. C. S. D. Andrade and J. F. W. Deakin, Pharmacol., Biochem. Behav., 1996, 54, 129–141 CrossRef CAS.
- Z. Q. Zhao, Prog. Neurobiol., 2008, 85, 355–375 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00797g |
| ‡ These three authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Yuanxue Liu‡
abc,
Yuanxue Liu‡